
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ detection of reactive oxygen species spontaneously generated on lead acid battery anodes: a pathway for degradation and self-discharge at open circuit†
Abdelilah
Asserghine
 a,
Aravind
Baby
ab,
Seth T.
Putnam
a,
Peisen
Qian
a,
Elizabeth
Gao
c,
Huimin
Zhao
d and
Joaquín
Rodríguez-López
*a
a,
Aravind
Baby
ab,
Seth T.
Putnam
a,
Peisen
Qian
a,
Elizabeth
Gao
c,
Huimin
Zhao
d and
Joaquín
Rodríguez-López
*a
aDepartment of Chemistry, University of Illinois Urbana-Champaign, 600 S Mathews Ave., Urbana, IL 61801, USA. E-mail: joaquinr@illinois.edu
bDepartment of Materials Science and Engineering, University of Illinois Urbana-Champaign, 1304 W Green St., Urbana, IL 61801, USA
cU.S. Army Corps of Engineers, ERDC Construction and Engineering Research Laboratory (CERL), Champaign, IL 61822, USA
dDepartment of Chemical and Biomolecular Engineering, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA
First published on 18th October 2023
Abstract
Prospects for refurbishing and recycling energy storage technologies such as lead acid batteries (LABs) prompt a better understanding of their failure mechanisms. LABs suffer from a high self-discharge rate accompanied by deleterious hard sulfation processes which dramatically decrease cyclability. Furthermore, the evolution of H2, CO, and CO2 also poses safety risks. Despite the maturity of LAB technologies, the mechanisms behind these degradation phenomena have not been well established, thus hindering attempts to extend the cycle life of LABs in a sustainable manner. Here, we investigate the effect of the oxygen reduction reaction (ORR) on the sulfation of LAB anodes under open circuit (OC). For the first time, we found that the sulfation reaction is significantly enhanced in the presence of oxygen. Interestingly, we also report the formation of reactive oxygen species (ROS) during this process, known to hamper cycle life of batteries via corrosion. Electron spin resonance (ESR) and in situ scanning electrochemical microscopy (SECM) unambiguously demonstrated the presence of OH˙ and of H2O2 as the products of spontaneous ORR on LAB anodes. High temporal resolution SECM measurements of the hydrogen evolution reaction (HER) during LAB anode corrosion displayed a stochastic nature, highlighting the value of the in situ experiment. Balancing the ORR and HER prompts self-discharge while reaction of the carbon additives with highly oxidizing ROS may explain previously reported parasitic reactions generating CO and CO2. This degradation mode implicating ROS and battery corrosion impacts the design, operation, and recycling of LABs as well as upcoming chemistries involving the ORR.
1. Introduction
Despite their technology dating back to the nineteenth century, lead-acid batteries (LABs) remain one of the most popular secondary batteries on the market due to their mature chemistry, proven safe operation, and inexpensive production.1 LABs continue to make up approximately 70% of the rechargeable battery market and have a variety of applications in automotive, photovoltaics, and grid level energy storage.2–4 LABs contain porous Pb anodes with carbon additives and PbO2 cathodes immersed in a 4.2 M H2SO4 electrolyte.5 A primary drawback of LABs is that they have a short lifespan of around 300 cycles, thus demanding frequent replacements.6 Disposal of failed LABs produces around 2.46 million tons of waste annually, most of which is Pb, a toxic heavy metal.7,8 Recycling of LABs is possible through pyrometallurgical and hydrometallurgical processes, but these methods are energy intensive, expensive, and create environmental issues.8–10 On the other hand, sustainable refurbishing LABs eliminates disassembly/reassembly processes and could minimize the latter issues.11 Thus, understanding the sources of electrode passivation and materials degradation becomes critical in determining refurbishment strategies.The main source of LAB failure is the accumulation of redox inactive crystals of PbSO4 on both the anode and the cathode during discharge. Additionally, even without cycling, LABs suffer from a fast self-discharge rate of about 10 to 15% in only 24 h, which is much higher than other battery technologies.12 Self-discharge of LABs strongly depends on the composition of the anode,13–17 and the temperature of storage.18–20 Self-discharge occurs when the LABs are stored in the charged state leading to the spontaneous formation of PbSO4 crystals on the anode (eqn (1) and (2)) and cathode (eqn (3) and (4)) surfaces.
Reported determinant reactions of the anode:18–21
Oxidation:
| Pb + H2SO4 → PbSO4 + 2H+ + 2e− | (1) |
Reduction:
| 2H+ + 2e− → H2↑ | (2) |
Reported determinant reactions of the cathode:18–21
Oxidation:
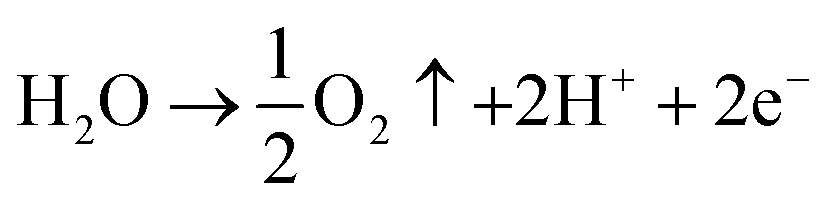 | (3) |
Reduction:
| PbO2 + H2SO4 + 2H+ + 2e− → PbSO4 + 2H2O | (4) |
During self-discharge, CO2, CO, O2, and H2 gases have been detected at open circuit (OC) conditions.18,19,22–24 Particularly, CO and H2 are regarded as flammable gases, which threaten the operational safety and pose the risk of explosions.25 Extensive research is underway to enhance the stability of LABs by changing the composition of the negative electrodes to minimize the rate of self-discharge and suppress the gas emissions.26–33 However, in order to improve the performance of LABs, the origins of the rapid self-discharge and the insidious side reactions should be explored.
Previous reports suggest that O2 gas can be produced from the spontaneous oxidation of water at the positive electrode (eqn (3)).18 Additionally, it can also diffuse from the atmosphere into the electrolyte, particularly in flood-type LABs. Meanwhile, H2 gas is generated on the anodes from the spontaneous reduction of protons (H+) (HER) by Pb (eqn (2)).18 On the other hand, the origins of CO2 and CO evolution are not well-established. A likely source could be the carbon additives in the LAB anode. These are used for increasing electrode conductivity and suppressing the occurrence of hard sulfation.34,35 Thus, the possible mechanism of CO2 and CO evolution could be the direct oxidation of the carbon additives in the commercial Pb (Pb–C) anodes, but this requires an oxidative potential >1.3 V vs. RHE.36 To consolidate these observations, we hypothesized that carbon and Pb could be directly oxidized by the reactive oxygen species (ROS) generated from the ORR on the Pb–C anodes (eqn (5)–(8)).37 It is well known that ROS such as H2O2, O2˙−, and OH˙ possess high oxidation powers (1.78 V, 0.64 V, and 2.8 V vs. RHE, respectively).38
| O2 + e− → O2˙− | (5) |
| O2 + 2H+ + 2e− → H2O2 | (6) |
| H2O2 + H+ + e− → H2O + OH˙ | (7) |
| O2˙− + 2H+ + e− → H2O2 | (8) |
2. Materials and methods
2.1. Materials and reagents
Pb sheets (0.03-inch thickness, 99.8%) and potassium nitrate (KNO3, 99%) were purchased from Fisher chemicals. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO, 97%) was purchased from Sigma Aldrich. Graphite rods (3 mm diameter, 99.9995%) were purchased from Alfa Aesar. Pb–C anodes used in the studies were procured from 6N2-2A batteries manufactured by Powersonic (Fig. S1A–C†). Pt (99.9%) and Au (99.9%) wires of diameter 25 μm was obtained from Goodfellow. H2SO4 and HNO3 were supplied by Macron.2.2. Scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX)
SEM was performed on a Hitachi S4800 at a typical accelerating voltage of 10 kV. EDX was performed in the same system with an Oxford Instruments Ultim Max 100 mm2 large silicon area drift detector. A typical accelerating voltage of 20 kV with a working distance of 15 mm was maintained for the EDX measurements.2.3. X-ray diffraction (XRD)
XRD was performed using the Rigaku Miniflex 600 powder XRD system in the scan range 10–70° with Cu-Kα radiation and a wide area scintillation counter. Standard operating conditions of 40 kV and 15 mA were used with a scan rate of 0.02° s−1. The intensities of all peaks are normalized to the Pb peak at 31.4°, which is the most intense peak in most of the samples. For elucidating and comparing the formation of PbSO4, we use the (011) peak. It has a sufficiently high intensity, and the region is devoid of any peaks from Pb oxides.2.4. Electron Spin resonance (ESR)
5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) was utilized as a radical spin trap for the detection of radical species. We employed DMPO as a trapping molecule due to its capability to capture various possible oxygenated radicals, including (OH˙, OOH˙, and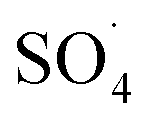 ). This assay is very specific to the type of radicals formed, providing a distinct fingerprint for each of the formed adducts (Fig. S2†). ESR experiments were performed by soaking various electrodes at OCP in solution of 5 mL of 100 mM DMPO and 5 mL of 0.5 M of H2SO4. Two independent experiments were performed by soaking the Pb–C anode in 0.5 M H2SO4 + 100 mM DMPO under aerated (purging O2) and deaerated (purging Ar) conditions for 1 hour each. Aliquots of the DMPO and electrolyte solutions were transferred to a Wilmad Glass quartz flat cell. The spectra were acquired at room temperature (∼298 K) with an EMXPlus X-band instrument. The measurements conditions were: center field = 3480 G; sweep width = 100 G; sweep time = 30 seconds; averages = 8 scans; microwave power = 20 mW; receiver gain = 30 dB; modulation amplitude = 1 G; microwave frequency = 9.783382 GHz.
). This assay is very specific to the type of radicals formed, providing a distinct fingerprint for each of the formed adducts (Fig. S2†). ESR experiments were performed by soaking various electrodes at OCP in solution of 5 mL of 100 mM DMPO and 5 mL of 0.5 M of H2SO4. Two independent experiments were performed by soaking the Pb–C anode in 0.5 M H2SO4 + 100 mM DMPO under aerated (purging O2) and deaerated (purging Ar) conditions for 1 hour each. Aliquots of the DMPO and electrolyte solutions were transferred to a Wilmad Glass quartz flat cell. The spectra were acquired at room temperature (∼298 K) with an EMXPlus X-band instrument. The measurements conditions were: center field = 3480 G; sweep width = 100 G; sweep time = 30 seconds; averages = 8 scans; microwave power = 20 mW; receiver gain = 30 dB; modulation amplitude = 1 G; microwave frequency = 9.783382 GHz.
2.5. Scanning electrochemical microscopy
All SECM measurements were conducted using the CHI-920D scanning electrochemical microscope. Au and Pt ultramicroelectrodes (UMEs) were prepared following an established procedure from literature.41 An Au or Pt wire of diameter 25 μm is thermally sealed into a borosilicate glass capillary and the sealed end is sharpened conically on a polishing wheel. The tip of the probe is then polished over a felt pad with aqueous alumina slurries of successively decreasing particle sizes till 0.05 μm. An Ag/AgCl/3 M KCl electrode connected to an agar salt bridge (0.1 M KClO4) was used as the reference electrode with a graphite rod as the counter electrode.3. Results and discussion
To study the effect of ORR on the sulfation of the Pb–C anode, we used ex situ SEM and XRD characterization. The Pb–C surface prior to immersion in H2SO4 is porous (Fig. 1A) and composed of Pb crystals and C as determined by the corresponding SEM and EDX spectra (Fig. S1†). Upon immersing the anodes in Ar-saturated 4.2 M H2SO4 for 1 h, the surface was covered with small needle-shaped crystals (Fig. 1B, yellow arrows) interspersed by some larger polygonal crystals. However, when the immersion is performed in O2-saturated 4.2 M H2SO4 for 1 h, we observed that the polygonal crystals are larger in size and more in number compared to the sample immersed in the Ar-saturated solution (Fig. 1C, green arrows). Existing battery literature and our own experiments with discharging LABs (Fig. S3†) indicate that these are PbSO4 crystals formed by oxidation of Pb during battery discharge.23 | ||
| Fig. 1 SEM images and XRD of Pb–C anodes. SEM images of (A) fresh Pb–C anodes (B) Pb–C anodes immersed in Ar-saturated 4.2 M H2SO4 for 1 h, (C) Pb–C anodes immersed in O2-saturated 4.2 M H2SO4 for 1 h. X-ray diffractograms of (D) growth of PbSO4 and Pb oxide peaks with immersion of Pb–C anodes in 4.2 M H2SO4 in the presence of Ar and O2, (E) comparison of peak intensities of PbSO4 (110) across the immersed Pb–C anodes. The peaks are denoted as follows: α = Pb, β = Pb3O4, γ = α-type PbO2, and δ = fluorite-type PbO2. Peaks denoted by planes belong to PbSO4. | ||
To confirm our observations, we used XRD to identify the products of the surface reactions happening on Pb–C anodes. Interestingly, XRD peaks belonging to PbSO4 and Pb oxides appear when the Pb–C anodes are immersed in H2SO4 (Fig. 1D) under both conditions. The relative intensity of the PbSO4 (011) peak (vs. the Pb (111) peak) is 0.09 after immersing the Pb–C anode in Ar-saturated H2SO4 for 1 h (Fig. 1E, blue curve), showing that significant amounts of PbSO4 are formed on the surface. The relative intensity grows to 0.13 when the immersion is performed in O2-saturated H2SO4 (Fig. 1E, red curve). This reiterates the observations from the SEM. Nevertheless, even in the absence of O2, there is some PbSO4 observed on the Pb–C anodes. We theorize that this could be caused by HER (eqn (1) and (2)) or due to the galvanic corrosion of Pb by C, a ubiquitous additive in LAB anodes. When Pb is in electrical contact with C, a galvanic cell is established, with Pb acting as the anode since C is more noble than Pb. This drives the oxidation of Pb into Pb2+, which then combines with SO42− ions in the electrolyte to form PbSO4. To test this idea, we performed the same experiments using strips of pure Pb (Fig. S4†). We observed the formation of some PbSO4 under argon conditions underlining that HER is likely the dominant factor as no C was present. Furthermore, we observed a large increase in the intensity of the PbSO4 peaks, confirming that the presence of O2 aggravates self-sulfation.
To confirm that HER is occurring, H2 gas was monitored in situ using SECM operated in the Substrate Generation/Tip Collection mode (SG/TC) using a 12.5 μm Pt UME as an SECM probe biased at 0.1 V for H2 oxidation as shown in Fig. 2A.44–47Fig. 2B illustrates that the recorded currents at the close vicinity of the Pb–C anode (50 μm from the Pb surface) are significantly high, in the range of 1 to 12 nA. On the other hand, the currents recorded in the bulk, far away from the surface, are very small, around 0.1 nA. This underlines that HER is happening on the Pb surface. In fact, the recorded currents at the surface are generated as transient peaks, possibly due to the intermittent formation of H2 bubbles on the Pb surface (Fig. 2B). Such transient peaks have been reported in collision electrochemistry in the case of nanoparticle or nanobubble collisions,48–53 but not in the LAB literature, to the best of our knowledge. Such observations highlight the relevance of the SECM in situ experiment in characterizing the LAB corrosion process at OC.
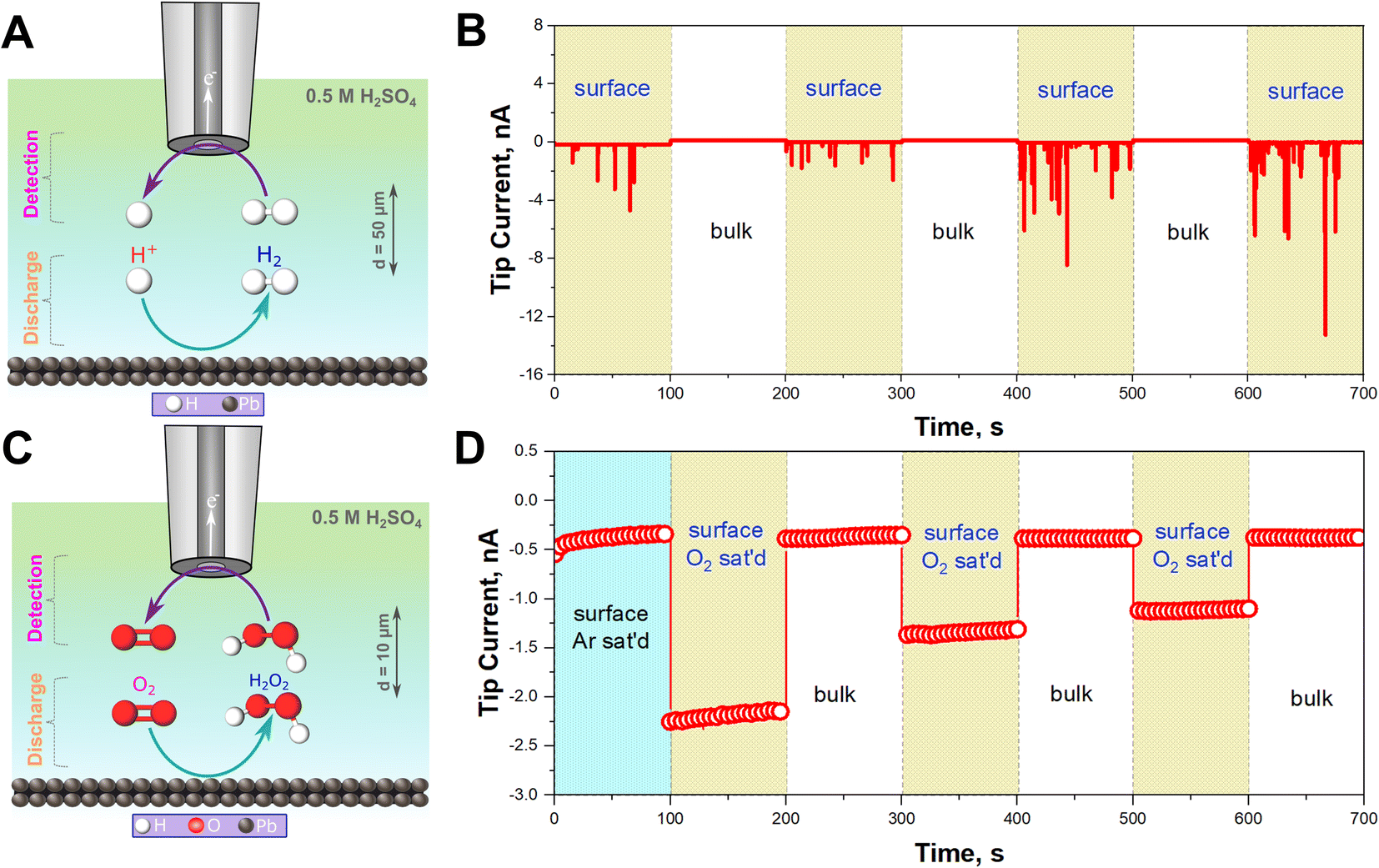 | ||
| Fig. 2 SG/TC SECM experiments for detecting H2 and H2O2. (A) Schematic of detection of H2 produced on the Pb–C anode surface at the Pt UME SECM probe under Ar saturated conditions. (B) Chronoamperograms (current vs. time curves) recorded at the 12.5 μm Pt UME biased at 0.1 V vs. Ag/AgCl/3 M KCl near the surface (50 μm) and in the bulk for three cycles (1000 μm) in 0.5 M H2SO4. (C) Schematic of detection of H2O2 produced on the Pb–C anode surface at the Pt UME SECM probe under O2 saturated conditions. (D) Chronoamperograms recorded at the Pt UME SECM probe biased at 1.1 V vs. Ag/AgCl/3 M KCl in Ar-saturated condition near the surface (50 μm) and in O2 saturated condition and near the surface (50 μm) and in the bulk (1000 μm) for three cycles in 0.5 M H2SO4. | ||
In aerated conditions, the SEM and XRD experiments showed the formation of larger PbSO4 crystals. We hypothesize that this is due to the additional contribution of ORR on the Pb–C anode. ORR on most non-noble metals produces H2O2.54 Thus, we targeted the detection of substrate-generated ROS in situ (eqn (5)–(8)) on the Pb–C anode surface under OC conditions. Using the SECM, we detected H2O2 at the close vicinity of the Pb–C anode (10 μm from the surface) using a 12.5 μm Pt UME as an SECM probe biased at 1.1 V for H2O2 oxidation as shown in Fig. 2C.37–40 We observed that the current on the Pb–C surface under Ar conditions is about −0.5 nA (Fig. 2D), whereas under O2-saturated conditions, the current raises to −2.25 nA. After retracting the Pt UME to the bulk (1000 μm from the Pb surface), the measured current drops to that observed under Ar. This highlights the significant production of H2O2 from the Pb surface, with an estimated concentration of ∼0.3 mM (Fig. S5†).
In addition, it is possible that the resulting H2O2 can be reduced on the Pb–C anode surface, leading to the formation of OH˙ radicals (eqn (7)). However, the direct detection of the radicals is very challenging due to their short lifetime of a few nanoseconds. Therefore, we added DMPO as a trapping molecule that forms a stable adduct with OH˙ radicals (Fig. 3A).55–57 The [DMPO−OH]˙ radical adduct can then be detected ex situ using ESR spectroscopy. The data recorded with ESR are shown in Fig. 3B. Under deaerated conditions, no paramagnetic signal was observed because no ORR is expected to take place on the Pb–C anode surface due to the limited concentration of O2 (Fig. 3B, black curve). This agrees with the SECM experiment which did not detect H2O2 under Ar-saturated conditions. On the other hand, the ESR recorded in aerated conditions illustrates the splitting feature (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 integration ratio) for the [DMPO−OH]˙ adduct (Fig. 3B, red curve). With the easy spin simulation (Fig. 3B, blue curve), the following parameters are obtained: aN = 14.88 G (equal to 41.81 MHz), aH = 15.02 G (equal to 42.02 MHz) which correspond perfectly with the reported hyperfine coupling constants of [DMPO−OH]˙.55–57 We also observed a similar feature in the case of pure Pb strips (Fig. S6†).
:2:2:1 integration ratio) for the [DMPO−OH]˙ adduct (Fig. 3B, red curve). With the easy spin simulation (Fig. 3B, blue curve), the following parameters are obtained: aN = 14.88 G (equal to 41.81 MHz), aH = 15.02 G (equal to 42.02 MHz) which correspond perfectly with the reported hyperfine coupling constants of [DMPO−OH]˙.55–57 We also observed a similar feature in the case of pure Pb strips (Fig. S6†).
 | ||
| Fig. 3 Detection of [DMPO−OH]˙. (A) Schematic for DMPO binding with OH˙ to produce [DMPO−OH]˙ which is then detected at the Au UME probe in an SG/TC SECM experiment. (B) ESR spectra recorded in 0.5 M H2SO4 + 100 mM DMPO after soaking Pb–C anodes for 1 h in Ar and O2 saturated conditions with the corresponding Easy Spin simulation fitting. (C) Cyclic voltammograms performed on the 12.5 μm Au UME probe from 0 to 1.1 V vs. Ag/AgCl/3 M KCl at a scan rate of 20 mV s−1 near the surface (10 μm) of Pb–C anode immersed in 0.5 M H2SO4 + 10 mM DMPO and in the solution bulk (1000 μm). | ||
In addition to ESR, we also electrochemically detected the [DMPO−OH]˙ adduct in situ using SECM. The SG/TC mode was used, enabling us to detect the [DMPO−OH]˙ adduct at the close vicinity of the Pb–C anode surface under OC conditions. Herein, a 12.5 μm Au UME was used as the SECM probe positioned at about 10 μm from the Pb–C anode surface immersed in 0.5 M H2SO4 + 10 mM DMPO (Fig. 3A). The Pb–C anode was maintained at OC potential and a cyclic voltammogram (CV) was recorded at the UME (Fig. 3C, red curve). The expected redox wave at ∼0.8 V corresponding to [DMPO−OH]˙ adduct oxidation was observed.39 Retraction of the tip showed that this signal was absent in the bulk solution. The SECM experiments agree with the ESR measurements, proving that OH˙ radicals are formed on the Pb–C anode surface. Additional control experiments (Fig. S7†) were performed to further confirm that the redox wave at 0.8 V is caused by [DMPO−OH]˙.
The SECM and ESR experiments highlight that ORR on Pb–C form H2O2 and OH˙. Normally, the formation of OH˙ on metals and semiconductors occurs at a very positive potential >1 V vs. RHE,39 but in the case of Pb, we found that OH˙ forms spontaneously at OC. The presence of ROS is regarded as detrimental to battery systems, since they have a high oxidation power which leads to the degradation of various components in the battery.58–61 In the present study, ROS formation opens an avenue to explain the reported formation of CO2 and CO in LABs.19,23 Here, spontaneous formation of OH˙ occurs on the Pb–C anode surface at OC (Fig. 4, initial stage). Subsequently, there is a high chance that the carbon in the Pb–C anodes can directly be oxidized by the OH˙ (Fig. 4, final stage). The electrochemical oxidation of carbon to CO2 and CO in aqueous solutions happens at a potential higher than 1.3 V vs. RHE.36 The significantly higher oxidation potential of OH˙ at 2.8 V vs. RHE will be sufficient to oxidize carbon via the following reactions (eqn (9)–(12):62
| OH˙ + C → CO + H2O | (9) |
| 4OH˙ + C → CO2 + 2H2O | (10) |
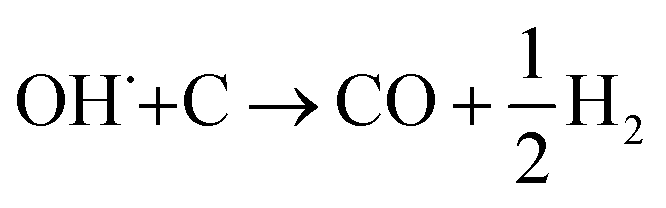 | (11) |
| 2OH˙+ C→ CO2 + H2 | (12) |
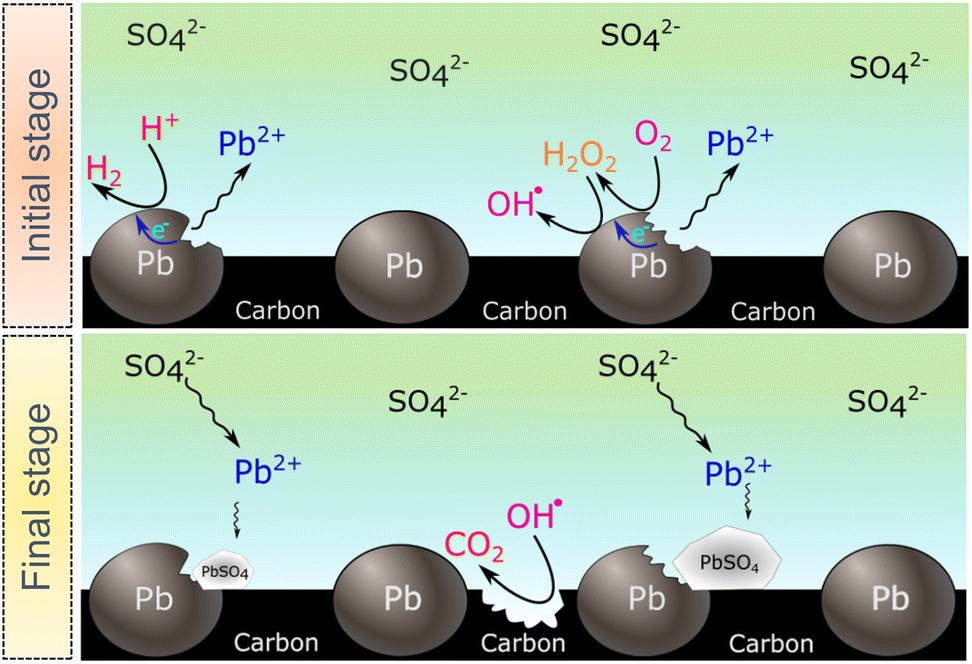 | ||
| Fig. 4 Proposed mechanism for carbon additive degradation and accelerated sulfation in the presence of ROS. Showing the HER and ORR taking places on the Pb particles at the initial stage, followed by the sulfation process and carbon binder degradation by the OH˙ forming CO2. | ||
4. Conclusion
In this paper, we investigated the effect of ORR on the sulfation of Pb–C anodes. We found that in the absence of O2, sulfation is still happening due to HER, but the formed PbSO4 crystals are relatively small. On the other hand, when O2 is present, the sulfation is enhanced and large PbSO4 crystals are observed (>5 μm). This implies that ORR plays a significant role in the sulfation mechanism. In addition, using ESR and SECM unambiguously demonstrated for the first time the presence of OH˙ and of H2O2 as the products of spontaneous ORR on Pb–C anode. Our experimental findings may explain the origin of the parasitic reactions reported in the LABs, particularly the evolution of CO2 and CO which can happen from the direct oxidation of the carbon additives in the Pb–C anode by ROS. The subsequent degradation of the carbon additives will eventually be detrimental to the battery due to the loss of conductivity. In addition, ROS may enhance the self-discharge of the anode forming PbSO4 and accelerate electrode grid corrosion. Till now, most of the research on reducing gas evolution in LAB systems have focused on suppressing HER on the Pb–C anode. But our findings suggest that O2 plays a much more significant role in the sulfation mechanism. Hence, quantifying ROS generation should be considered to improve future LAB anodes and minimize self-sulfation for enhanced performance. Better performing LABs with longer cycle life can have implications in lead waste reduction.11,15,63 However, our results and methodology are not only applicable to LABs. Monitoring ORR-related processes at air-metal batteries is crucial to understand the formation of reactive species that cause interfacial passivation due to interactions with organic components. The methods developed here to detect ROS at OC are relevant for elucidating general mechanistic aspects of battery electrode corrosion, where the intentional58–61 or adventitious presence of O2 could have a determining impact on performance and lifetime of batteries beyond LABs.Data availability
All the data supporting this article have been uploaded as part of the ESI.†Author contributions
Abdelilah Asserghine (conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization), Aravind Baby (methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization), Seth T Putnam (investigation, writing – review & editing), Peisen Qian (formal analysis), Elizabeth Gao (project administration), Huimin Zhao (validation, writing – review & editing, funding acquisition), Joaquín Rodríguez-López (conceptualization, methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization, supervision, funding acquisition).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the U.S. Army Construction and Engineering Research Laboratory under cooperative agreements (W9132T2120001). Sample preparation and characterization were carried out in part in the Materials Research Laboratory Central Research Facilities, University of Illinois, and the George L. Clark X-ray Facility and 3M Materials Laboratory, University of Illinois.Notes and references
- P. P. Lopes and V. R. Stamenkovic, Science, 2020, 369, 923 CrossRef CAS PubMed.
- K. Yanamandra, D. Pinisetty, A. Daoud and N. Gupta, J. Indian Inst. Sci., 2022, 102, 295 Search PubMed.
- J. Y. Yong, V. K. Ramachandaramurthy, K. M. Tan and N. Mithulananthan, Renewable Sustainable Energy Rev., 2015, 49, 365 CrossRef.
- S. F. Tie and C. W. Tan, Renewable Sustainable Energy Rev., 2013, 20, 82 CrossRef.
- G. J. May, A. Davidson and B. Monahov, J. Energy Storage, 2018, 15, 145 CrossRef.
- Z. P. Cano, D. Banham, S. Ye, A. Hintennach, J. Lu, M. Fowler and Z. Chen, Nat. Energy, 2018, 3, 279 CrossRef.
- Z. Sun, H. Cao, X. Zhang, X. Lin, W. Zheng, G. Cao, Y. Sun and Y. Zhang, Waste Manage., 2017, 64, 190 CrossRef CAS PubMed.
- W. Zhang, J. Yang, X. Wu, Y. Hu, W. Yu, J. Wang, J. Dong, M. Li, S. Liang, J. Hu and R. V. Kumar, Renewable Sustainable Energy Rev., 2016, 61, 108 CrossRef CAS.
- X. Zhang, L. Li, E. Fan, Q. Xue, Y. Bian, F. Wu and R. Chen, Chem. Soc. Rev., 2018, 47, 7239 RSC.
- S. Tan, D. J. Payne, J. P. Hallett and G. H. Kelsall, Curr. Opin. Electrochem., 2019, 16, 83 CrossRef CAS.
- Z. T. Gossage, F. Guo, K. O. Hatfield, T. A. Martin, Q. Tian, E. J. Gao, A. Kumar, J. Rodríguez-López and H. Zhao, J. Electrochem. Soc., 2020, 167, 120537 CrossRef CAS.
- I. Buchmann, Batteries in a Portable World - A Handbook on Rechargeable Batteries for Non-Engineer, Cadex Electronics Inc., 2017 Search PubMed.
- S. Arun, K. U. V. Kiran and S. Mayavan, J. Energy Storage, 2020, 32, 101763 CrossRef.
- D. G. Enos, S. R. Ferreira, H. M. Barkholtz, W. Baca and S. Fenstermacher, J. Electrochem. Soc., 2017, 164, A3276 CrossRef CAS.
- P. T. Moseley, J. Power Sources, 2009, 191, 134 CrossRef CAS.
- L. T. Lam, N. P. Haigh, C. G. Phyland and A. J. Urban, J. Power Sources, 2004, 133, 126 CrossRef CAS.
- X. Zou, Z. Kang, D. Shu, Y. Liao, Y. Gong, C. He, J. Hao and Y. Zhong, Electrochim. Acta, 2015, 151, 89 CrossRef CAS.
- D. Berndt, J. Power Sources, 2001, 100, 29 CrossRef CAS.
- R. Bullock, J. Power Sources, 2010, 195, 4513 CrossRef.
- P. Rüetschi and R. T. Angstadt, J. Electrochem. Soc., 1958, 105, 555 CrossRef.
- T. Kuhn, The Electrochemistry of Lead, Academic Press, 1979 Search PubMed.
- A. Singh, K. R. Ansari, I. H. Ali, M. Younas and B. Gupta, J. Energy Storage, 2023, 57, 106272 CrossRef.
- D. Pavlov, Lead-Acid Batteries: Science and Technology, Elsevier, 2011 Search PubMed.
- T. Lam, O. V. Lim, N. P. Haigh, D. A. J. Rand, J. E. Manders and D. M. Rice, J. Power Sources, 1998, 73, 36 CrossRef.
- D. Brzezińska, Energies, 2018, 11, 2086 CrossRef.
- A. Deyab, RSC Adv., 2015, 5, 41365 RSC.
- A. Deyab, J. Power Sources, 2018, 390, 176 CrossRef.
- R. K. Ghavami, F. Kameli, A. Shirojan and A. Azizi, J. Energy Storage, 2016, 7, 121 CrossRef.
- P. M. Witantyo, L. Amalia, L. Noerochim, H. Setyawan, A. Shahab and S. Suwarno, Int. J. Electrochem. Sci., 2021, 16, 21082 CrossRef.
- L. Dong, C. Chen, J. Wang, H. Li, H. Zheng, W. Yan, J. C.-Y. Jung and J. Zhang, RSC Adv., 2021, 11, 15273 RSC.
- M. A. Deyab and Q. Mohsen, Sci. Rep., 2021, 11, 1 CrossRef PubMed.
- P. Vanýsek, P. Bača and J. Zimáková, J. Energy Storage, 2021, 44, 103246 CrossRef.
- K. B. Dönmez, M. Gençten and Y. Şahin, Ionics, 2018, 24, 3655 CrossRef.
- W. Zhang, J. Yin, H. Lin, K. Lu, F. Feng and X. Qiu, Curr. Opin. Electrochem., 2021, 30, 100802 CrossRef CAS.
- J. Yin, H. Lin, J. Shi, Z. Lin, J. Bao, Y. Wang, X. Lin, Y. Qin, X. Qiu and W. Zhang, Electrochem. Energy Rev., 2022, 5, 1 Search PubMed.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32 CrossRef CAS.
- B. Halliwell and J. M. Gutteridge, Free Radicals in Biology and Medicine, Oxford University Press, USA, 2015 Search PubMed.
- K. Krumova and G. Cosa, in Singlet Oxygen: Applications in Biosciences and Nanosciences, Royal Society of Chemistry, 2016, vol. 1, ch 1, pp. 1–21 Search PubMed.
- J. S. Barroso-Martínez, A. I. B. Romo, S. Pudar, S. T. Putnam, E. Bustos and J. Rodríguez-López, J. Am. Chem. Soc., 2022, 144, 18896 CrossRef PubMed.
- M. Sánchez-Sánchez, J. Rodríguez-López and A. J. Bard, Anal. Chem., 2008, 80, 3254 CrossRef PubMed.
- Y. Shen, M. Träuble and G. Wittstock, Anal. Chem., 2008, 80, 750 CrossRef CAS PubMed.
- K. Eckhard and W. Schuhmann, Electrochim. Acta, 2007, 53, 1164–1169 CrossRef CAS.
- Y. Zhao, J. S. Adams, A. Baby, M. L. Kromer, D. W. Flaherty and J. Rodríguez-López, ACS Sustainable Chem. Eng., 2022, 10, 17207 CrossRef CAS.
- A. Asserghine, M. Medvidović-Kosanović, A. Stanković, L. Nagy, R. M. Souto and G. Nagy, Sens. Actuators, B, 2020, 321, 128610 CrossRef CAS.
- D. Filotás, B. M. Fernández-Pérez, L. Nagy, G. Nagy and R. M. Souto, Sens. Actuators, B, 2020, 308, 127691 CrossRef.
- U. M. Tefashe, M. E. Snowden, P. D. Ducharme, M. Danaie, G. A. Botton and J. Mauzeroll, J. Electroanal. Chem., 2014, 720, 121 CrossRef.
- A. Asserghine, M. Medvidović-Kosanović, L. Nagy, R. M. Souto and G. Nagy, Sens. Actuators, B, 2020, 320, 128339 CrossRef CAS.
- A. Sekretareva, Sens. Actuators Rep., 2021, 3, 100037 CrossRef.
- S. J. Kwon and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 7102 CrossRef CAS PubMed.
- A. Robinson, Y. Liu, M. A. Edwards, N. J. Vitti, S. M. Oja, B. Zhang and H. S. White, J. Am. Chem. Soc., 2017, 139, 16923 CrossRef PubMed.
- H. M. Kim and J. H. Park, Chemosensors, 2023, 11, 112 CrossRef.
- H. J. Yang, H. Kwon, B. K. Kim and J. H. Park, Electrochim. Acta, 2019, 320, 134620 CrossRef CAS.
- H. Ren and M. A. Edwards, Curr. Opin. Electrochem., 2021, 25, 100632 CrossRef CAS PubMed.
- F. Jaouen and J. P. Dodelet, J. Phys. Chem. C, 2009, 113, 15422 CrossRef CAS.
- R. Buettner, Free Radical Res. Commun., 1993, 19, s79 CrossRef PubMed.
- A. I. B. Romo, M. P. Carepo, P. Levín, O. R. Nascimento, D. E. Díaz, J. Rodríguez-López, I. E. León, L. F. Bezerra, L. Lemus and I. C. Diógenes, Dalton Trans., 2021, 50, 11931 RSC.
- P. Bilski, K. Reszka, M. Bilska and C. F. Chignell, J. Am. Chem. Soc., 1996, 118, 1330 CrossRef CAS.
- V. P. Houchins and V. Viswanathan, ACS Energy Lett., 2020, 5, 1893 CrossRef.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 1 Search PubMed.
- E. Mourad, Y. K. Petit, R. Spezia, A. Samojlov, F. F. Summa, C. Prehal, C. Leypold, N. Mahne, C. Slugovc, O. Fontaine, S. Brutti and S. A. Freunberger, Energy Environ. Sci., 2019, 12, 2559 RSC.
- L. Schafzahl, N. Mahne, B. Schafzahl, M. Wilkening, C. Slugovc, S. M. Borisov and S. A. Freunberger, Angew Chem. Int. Ed. Engl., 2017, 56, 15728 CrossRef CAS PubMed.
- M. F. R. Mulcahy and B. C. Young, Carbon, 1975, 13, 115 CrossRef CAS.
- D. Pavlov and P. J. Nikolov, Electrochem. Soc., 2012, 159, A1215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04736a |
| This journal is © The Royal Society of Chemistry 2023 |