
Solar energy driven C–C bond cleavage in a lignin model compound with a D–π–A organic dye-sensitized photoanode†
Saerona
Kim‡
a,
Hyeong Cheol
Kang‡
b,
Chun
Chu
c,
Shuya
Li
a,
Kicheon
Yoo
b,
Udani Kaushalya
Wijethunga
a,
Weiwei
Zheng
 c,
Chang Geun
Yoo
de,
Benjamin D.
Sherman
f,
Jae-Joon
Lee
*b and
Gyu
Leem
*ae
c,
Chang Geun
Yoo
de,
Benjamin D.
Sherman
f,
Jae-Joon
Lee
*b and
Gyu
Leem
*ae
aDepartment of Chemistry, State University of New York College of Environmental Science and Forestry, Syracuse 13210, New York, USA. E-mail: gyleem@esf.edu
bDepartment of Energy and Materials Engineering, Research Center for Photoenergy Harvesting & Conversion Technology (phct), Dongguk University, Seoul 04620, Republic of Korea. E-mail: jjlee@dongguk.edu
cDepartment of Chemistry, Syracuse University, Syracuse 13244, New York, USA
dDepartment of Chemical Engineering, State University of New York College of Environmental Science and Forestry, Syracuse 13210, New York, USA
eThe Michael M. Szwarc Polymer Research Institute, 1 Forestry Drive, Syracuse 13210, New York, USA
fDepartment of Chemistry and Biochemistry, College of Science and Engineering, Texas Christian University, Fort Worth 76129, Texas, USA
First published on 16th March 2023
Abstract
The high bond dissociation energy of C–C σ-bonds presents a challenge to chemical conversions in organic synthesis, polymer degradation, and biomass conversion that require chemoselective C–C bond cleavage at room temperature. Dye-sensitized photoelectrochemical cells (DSPECs) incorporating molecular organic dyes could offer a means of using renewable solar energy to drive these types of energetically demanding chemoselective C–C bond cleavage reactions. This study reports the solar light-driven activation of a bicyclic aminoxyl mediator to achieve C–C bond cleavage in the aryl-ether linkage of a lignin model compound (LMC) at room temperature using a donor–π-conjugated bridge–acceptor (D–π–A) organic dye-based DSPEC system. Mesoporous TiO2 photoanode surfaces modified with 5-[4-(diphenylamino)phenyl]thiophene-2-cyanoacrylic acid (DPTC) D–π–A organic dye were investigated along with a bicyclic aminoxyl radical mediator (9-azabicyclo[3,3,1]nonan-3-one-9-oxyl, KABNO) in solution with and without the presence of LMC. Photophysical studies of DPTC with KABNO showed intermolecular energy/electron transfer under 1 sun illumination (100 mW cm−2). Under illumination, the D–π–A type DPTC sensitized TiO2 photoanodes facilitate the generation of the reactive oxoammonium species KABNO+ as a strong oxidizing agent, which is required to drive the oxidative C–C bond cleavage of LMC. The photoelectrochemical oxidative reaction in a complete DSPEC with KABNO afforded C–C bond cleavage products 2-(2-methoxyphenoxy)acrylaldehyde (94%) and 2,6-dimethoxy-1,4-benzoquinone (66%). This process provides a first report utilizing a D–π–A type organic dye in combination with a bicyclic nitroxyl radical mediator for heterogeneous photoelectrolytic oxidative cleavage of C–C σ-bonds, modeled on those found in lignin, at room temperature.
Introduction
Lignin is one of the most abundant natural aromatic resources on Earth and is recognized as a potential feedstock for alternative fuels and chemicals.1 Lignin valorization requires a cost effective means of driving the energy intensive conversion of lignin to value-added monomeric chemicals (e.g., vanillin, catechol, and phenol) that are important raw materials for the production of bioplastics, adhesives, and pharmaceuticals.2–9 In addition, lignin is reported as a precursor for a polymeric composite that can be used for various energy applications.10–13 Phenolic aromatic units are abundant in lignin and connected by aliphatic C–C and/or C–O σ-bonds, which are the targeted linkages for most lignin degradation strategies (Fig. 1a).14 In the past few years, C–C and/or C–O bond cleavages in lignin model compounds or real lignin have been performed using various reaction methods including retro-aldol reaction,2,15 catalytic aerobic oxidation,3,4,14,16,17 hydrogenation,5TEMPO-mediated oxidation (TEMPO = 2,2,6,6-tetramethyl-1-piperidine N-oxyl),6 electrocatalytic oxidation,7 and photocatalytic oxidation/reduction.8,18 Among these, we first reported the photoelectrocatalytic oxidation of lignin model compounds and real lignin using a polypyridyl based Ru(II) complex chromophore coated mesoporous TiO2 photoanode with a hydrogen atom transfer mediator (e.g., N-hydroxyphthalimide) in a dye-sensitized photoelectrochemical cell (DSPEC) under solar light illumination at room temperature.19 Traditionally, DSPECs have targeted solar-driven water oxidation/reduction for the production of solar fuels in aqueous media.20–22 Notably, Ru polypyridyl complexes have been extensively studied with co-catalysts for water oxidation or reduction reactions, solar energy conversion, and solar fuel production.23–25 However, these Ru based complexes share the drawback of high materials cost and the limited ability to tune the HOMO and LUMO energy levels as compared to organic dyes.26 Metal-free organic dyes containing a donor–π-conjugated bridge–acceptor (D–π–A) structure offer promising alternatives as the photocatalyst for the light-driven oxidative C–C bond cleavage of lignin in the DSPEC system because of their high molar absorption coefficient, tunable redox potentials, and facile synthesis.27 To our knowledge, oxidative cleavage of C–C bond in lignin or related model compounds in a DSPEC at room temperature has not been explored to date using D–π–A type organic chromophores.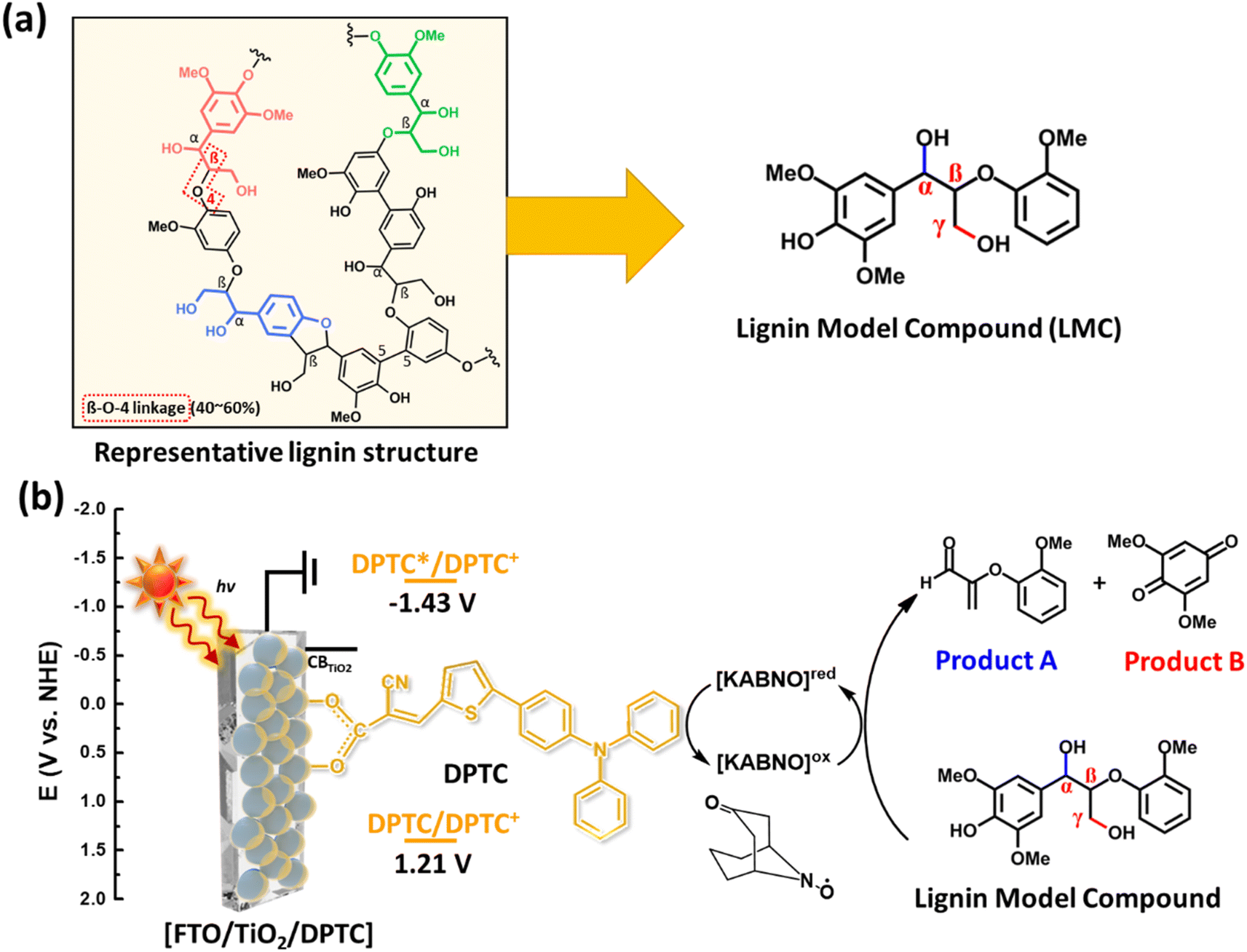 | ||
| Fig. 1 (a) Schematic representative structure of real lignin (left) and a dimeric lignin model compound (LMC) containing abundant aryl ether linkage (right), and (b) illustration of DPTC anchored on mesoporous structured TiO2 for solar light-driven oxidative cleavage of LMC in combination with a bicyclic nitroxyl mediator, 9-azabicyclo[3,3,1]nonan-3-one-9-oxyl (KABNO). | ||
Direct electrochemical oxidative C–O and/or C–C bonds cleavage of lignin model species has been widely studied.28–30 However, this approach requires high applied bias to deliver sufficient overpotential (e.g., 1.45 V vs. SCE) resulting in the formation of unwanted oxidation and degradation products.31 Electrolysis in the presence of a nitroxyl mediator (e.g., TEMPO) provides an alternative approach that lowers by several hundred mV the required overpotential compared with the direct oxidation of the lignin model compound or real lignin degradation.32,33 For example, the Stahl research group found that monocyclic TEMPO and its derivatives facilitated the oxidation of secondary benzylic alcohol or aliphatic primary alcohol groups in lignin under sufficient electrochemical bias.34 In addition, the use of bicyclic nitroxyls (e.g., 2-azaadamantane N-oxyl and KABNO) showed significant oxidation activity for primary and/or secondary alcohol oxidation due to their favorable catalytic properties.35–37 However, bicyclic nitroxyls species have not been as extensively studied in a DSPEC system even though they have been known as effective alcohol oxidation agents since the 1960s.35,38 To complement these dark electrochemical and electrocatalytic approaches, our study presents the next step in lowering the electrical energy (e.g., < 0.5 V vs. Ag/Ag+) needed to achieve chemoselective C–C bond cleavage of lignin by harnessing solar energy at room temperature using an organic dye-based DSPEC incorporating a bicyclic nitroxyl mediator. As shown in Fig. 1b, this article describes the use of D–π–A type organic dye-sensitized TiO2 photoanodes in combination with a bicyclic nitroxyl mediator, 9-azabicyclo[3,3,1]nonan-3-one-9-oxyl (KABNO), to carry out C–C bond cleavage of aryl-ether linkages in a phenolic lignin model compound (LMC) under simulated solar illumination at room temperature. The 5-[4-(diphenylamino)phenyl]thiophene-2-cyanoacrylic acid (DPTC) D–π–A organic dye featuring a triphenylamine (TPA) donor, cyanoacrylic acceptor moiety, and carboxylic anchoring group was chosen as a typical D–π–A typed dye, which is widely used in dye-sensitized solar cells because of its high molar extinction coefficients in the visible range.39 In addition, the redox potential of KABNO is a good match for the HOMO level of DPTC as shown in Fig. 1b. The KABNO•/+ couple exhibits a relatively high redox potential compared to other N-oxyl species of 200 mV vs. Fc+/Fc in acetonitrile with 0.1 M LiClO4.40Fig. 1b gives a schematic overview of the solar-light-driven selective C–C bond cleavage process studied here involving 1-(4-hydroxy-3,5-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol as the LMC, the DPTC-sensitized TiO2 photoanode (FTO/TiO2/DPTC), and solution dissolved KABNO under 1 sun illumination. This phenolic LMC structure contains the most abundant aryl ether linkage type found in native lignin.
Experimental section
Materials
5-[4-(Diphenylamino)phenyl]thiophene-2-cyanoacrylic acid was purchased from Dyenamo. Fluorine-doped tin oxide glass was purchased from Hartford Glass Co., Inc. Tetrabutylammonium hexafluorophosphate (TBAPF6) was purchased from TCI. 9-Azabicyclo[3,3,1]nonan-3-one-9-oxyl was purchased from Sigma-Aldrich. The LMC, 1-(4-hydroxy-3,5-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1,3-diol, was purchased from SY Innovation and used without further purification or modification.Fabrication of DPTC-immobilized mesoporous TiO2 photoanode (FTO/TiO2/DPTC)
The photoanodes were fabricated and modified according to previous studies.19,41,42 All FTO glass was cleaned by sonicating it with detergent, distilled water, and the mixture of acetone, isopropyl alcohol, and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio), separately, in an ultrasonic bath for 20 min. The mesoporous structured TiO2 layer was prepared by doctor blading a TiO2 paste (approximately 20 nm TiO2) onto a clean FTO glass slide followed by annealing at 500 °C for 30 min.43–45 Then, after cooling down to 80 °C, the annealed TiO2 films (FTO/TiO2) were immersed in a 0.5 mM DPTC solution in anhydrous ethanol for 4 h at room temperature. The FTO/TiO2/DPTC films were rinsed with anhydrous ethanol, dried, and stored in a sealed vial prior to use.
:1:1 ratio), separately, in an ultrasonic bath for 20 min. The mesoporous structured TiO2 layer was prepared by doctor blading a TiO2 paste (approximately 20 nm TiO2) onto a clean FTO glass slide followed by annealing at 500 °C for 30 min.43–45 Then, after cooling down to 80 °C, the annealed TiO2 films (FTO/TiO2) were immersed in a 0.5 mM DPTC solution in anhydrous ethanol for 4 h at room temperature. The FTO/TiO2/DPTC films were rinsed with anhydrous ethanol, dried, and stored in a sealed vial prior to use.
Characterization methods
UV-visible absorption spectra of FTO/TiO2/DPTC films were collected using a Thermo Scientific Evolution 220 UV-vis spectrometer. Emission spectra measurement was performed using an Edinburgh FLS 980 steady state fluorometer. The AFM images of bare FTO/TiO2 and FTO/TiO2/DPTC films were obtained with tapping mode of 0.7 Hz using the MultiMode-8 and RTESP-300 tip (Bruker Co.). All cleavage products were characterized by NMR, GC-FID (Shimadzu 2010 GC), and GC-MS (Q Exactive GC Orbitrap GC-MS/MS with TriPlus RSH autosampler). 1H-NMR, 13C-NMR, and 2D 1H–13C heteronuclear single quantum coherence (HSQC) NMR were used for the structural analysis of LMC before and after photoelectrochemical reaction by using a Bruker AVANCE III HD 800 MHz equipped with TCI Cryo probe. The HSQC NMR spectra were processed using Bruker TopSpin 4.1.4 software. After the completion of the reaction, all reaction mixtures were precipitated by a 1:1 mixture of ethyl acetate and hexane to remove the TBAPF6 electrolyte. Then, the products were dissolved in CDCl3 with dimethyl sulfoxide (DMSO) as an internal standard for NMR analysis. The conversion yield of the cleavage products was obtained by GC-FID fitted with a 30 m × 0.25 mm i.d. capillary column and AOCI205 autosampler. Helium gas was used as a carrier gas and the GC-FID system was held at 65 °C for 3 min. The temperature was ramped from 65 to 300 °C with a heating rate of 10 °C min−1, and kept at 300 °C for 5 min. The concentration of each product was determined based on the product calibration curve with the internal standard DMSO. The cleavage products were confirmed by GC-MS. The GC-MS system was held at 40 °C for 3 min, and then the temperature was gradually increased from 40 to 300 °C with a heating rate of 10 °C min−1, and then held at 300 °C for 20 min.
Electrochemical measurements
All electrochemical measurements were performed in acetonitrile solution with 0.1 M TBAPF6 as a supporting electrolyte using a single compartment three-electrode cell including a Pt wire counter electrode, a Ag/Ag+ quasi reference electrode, and FTO/TiO2/DPTC films as the working electrode. Voltammograms were recorded at a scan rate of 50 mV s−1 and potentials were reported with ferrocene/ferrocenium (Fc/Fc+) as an external reference after calibration of the Ag/Ag+ quasi reference. Photocurrent-time trace measurements were conducted at light intervals of 60 s on/off with an applied voltage of +0.4 V vs. Ag/Ag+ under 1 sun illumination (100 mW cm−2). Light irradiation was provided by an AM1.5 solar simulator (Abet Technologies). The temperature in the photoanode was maintained at ambient temperature by cooling with forced air using an electric fan.Photoelectrocatalytic oxidation of LMC
All lignin model compound (LMC) degradation studies were performed using an LSH-7320 ABA LED Solar Simulator (ORIEL) with 1 sun AM1.5 G (100 mW cm−2) light intensity for 24 hours under an applied voltage of +0.4 V vs. Ag/Ag+. The light intensity was measured using a THORLABS PM400 optical power meter with a S415C thermal power sensor head. The electrolyte solution contained 3 mM KABNO and 8 mM LMC in 20 mL of acetonitrile with 0.1 M TBAPF6. A specifically designed Teflon cell suitable for back-illumination was used to assemble all the organic dye-sensitized DSPECs.Results and discussion
The photophysical properties of DPTC were studied in the presence or absence of KABNO in acetonitrile solution. The UV-visible absorption and emission spectra of DPTC show a broad absorption and emission with peak maximum at λ ≈ 385 and 525 nm, respectively (Fig. 2a). It is noted that the absorption approximately between 380 to 500 nm corresponds to the charge transfer from the triphenylamine donor to the cyanoacrylic acid acceptor unit.46,47 The steady-state emission intensity of DPTC decreases as the concentration of KABNO increases from 0 to 1.2 mM (Fig. 2b), which indicates that the emission from the photoexcited DPTC was quenched by the presence of KABNO. On the basis of emission quenching in Fig. 2b, the Stern–Volmer quenching constant value obtained approximately Ksv ∼5.5 × 102 M−1. This emission quenching implies that intermolecular energy/electron transfer between the photoexcited DPTC and KABNO occurs. This could result in the formation of oxidized KABNO+ following photoexcited charge transfer between DPTC* and KABNO.48 On the basis of this quenching, we expect that photoexcited electron from DPTC* is injected into the TiO2 photoanode generating DPTC+ which then drives the formation of oxoammonium KABNO+ by interfacial hole transfer in the DSPEC.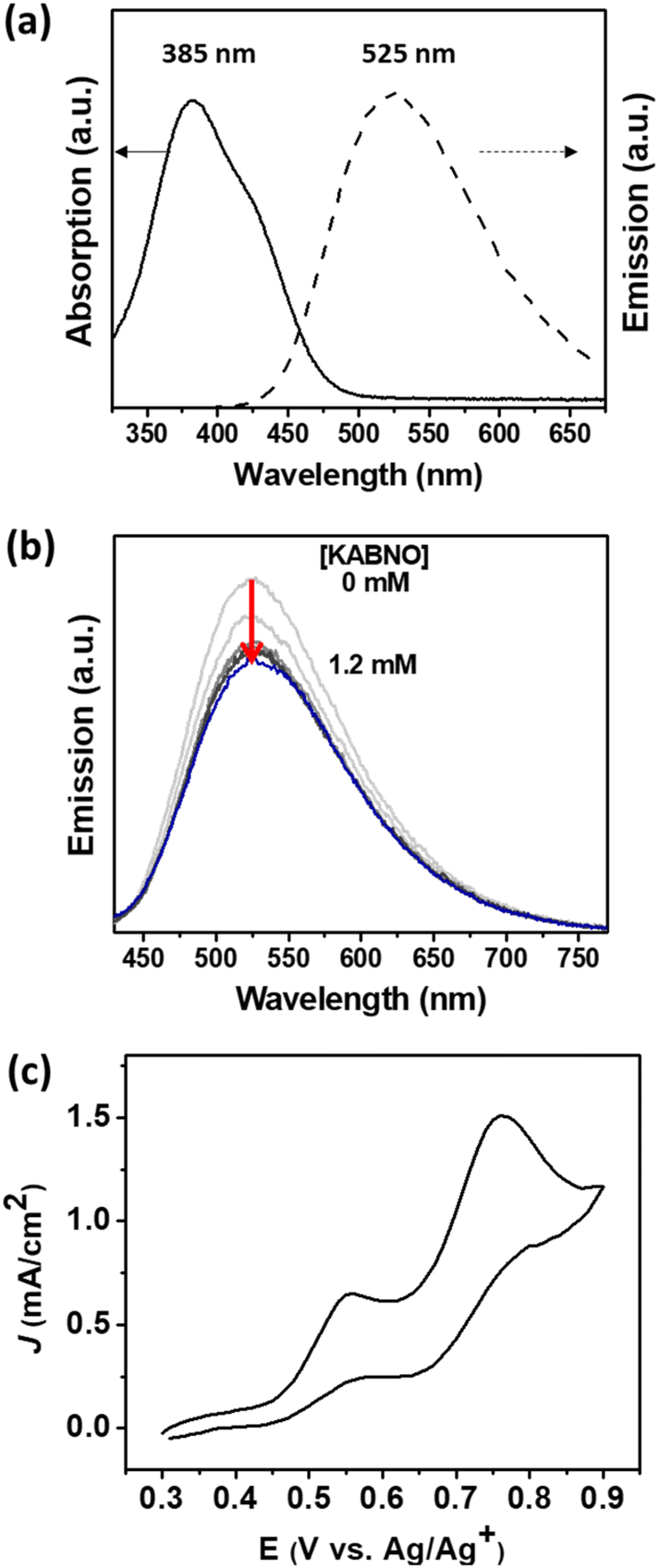 | ||
| Fig. 2 (a) UV-vis absorption (solid) and emission (dash) spectra of DPTC in acetonitrile. Emission spectrum was obtained with excitation at λex = 380 nm. (b) Emission quenching for DPTC with KABNO (conc. = 0–1.2 mM). (c) CV of KABNO and LMC in 0.1 M TBAPF6 acetonitrile solution. The experiment was performed using a glassy carbon working electrode, Pt counter electrode, and Ag/Ag+ reference electrode in the dark. | ||
Cyclic voltammetry (CV) was carried out in acetonitrile to study the relative redox potentials of KABNO and LMC using a glassy carbon working electrode. KABNO showed stable redox behavior with E1/2 = 0.54 V vs. Ag/Ag+ (Fig. S1a†); however, an irreversible anodic wave was observed for LMC with Epa = 0.76 V vs. Ag/Ag+ (Fig. S1b†) which is consistent with other studies.31,49 The addition of 3 mM of LMC to the KABNO solution results in both increased current at the anodic peak of the KABNO wave (Epa = 0.56 V vs. Ag/Ag+, Fig. 2c) and the loss of the cathodic wave for KABNO on the return scan. Both observations are consistent with the catalytic oxidation of LMC by KABNO+ in solution after its formation by interfacial electron transfer.31,32,37,50,51 These results reveal that KABNO+ can effectively oxidize the alcohol groups in LMC, and it demonstrates the importance of nitroxyl mediators (i.e., KABNO) in lowering the activation energy for converting the LMC to the corresponding ketone/aldehyde as reported in previous studies.52
To fabricate a DPTC-sensitized photoanode, mesoporous TiO2 films with a thickness of ∼6 μm were deposited on FTO substrates.42 The DPTC dye was adsorbed onto the FTO/TiO2 films (FTO/TiO2/DPTC) from 0.5 mM ethanol solution at room temperature. Fig. 3a shows the absorption spectrum with λmax ≈ 410 nm of the FTO/TiO2/DPTC surface corresponds to the charge transfer from a TPA donor to cyanoacrylic acceptor unit. This maximum absorption band of DPTC at the interface of the FTO/TiO2 film displays a similar absorption band with ∼420 nm in ethanol solution (Fig. S2†). This absorption band is shifted to a shorter wavelength (i.e., blue-shift) compared to DPTC in ethanol solution due to the adsorption on the TiO2 surface.19 The plane view topographies of the bare FTO/TiO2 and FTO/TiO2/DPTC films were further characterized by atomic force microscopy (AFM) (Fig. S3†). AFM analysis of the FTO/TiO2/DPTC films revealed a smoother surface with roughness, Rrms = 24 nm (Rrms: root mean square), compared to that of the FTO/TiO2 films (Rrms = 28 nm). These results indicate that the DPTC dye is successfully anchored onto the TiO2 surface.
 | ||
| Fig. 3 (a) UV-vis spectrum of FTO/TiO2/DPTC (inset: picture of a FTO/TiO2/DPTC electrode). (b) CVs of FTO/TiO2/DPTC (black), the same with 3 mM KABNO (blue), and the mixture of 3 mM KABNO and LMC (red) in 0.1 M TBAPF6 acetonitrile. | ||
To investigate the electrochemical response of FTO/TiO2/DPTC films in the absence or presence of KABNO and LMC, CVs were performed in 0.1 M TBAPF6 acetonitrile solution, as shown in Fig. 3b. The CV of FTO/TiO2/DPTC exhibits steady redox behavior, with a redox couple at E1/2 = 0.76 V vs. Ag/Ag+ (black voltammogram). In the presence of KABNO, the voltammogram shows corresponding redox potential peaks at E1/2 = 0.50 V vs. Ag/Ag+ (blue voltammogram), while the mixture of KABNO and LMC in acetonitrile solution exhibits increased anodic current at a high applied bias > 1.2 V vs. Ag/Ag+ (red voltammogram). The increase in peak current for the KABNO anodic wave in the red voltammogram and the absence of cathodic peaks for KABNO and DPTC on the return scan are consistent with the oxidizing equivalents generated ultimately resulting in the oxidation of LMC.52 In other words, the formation of KABNO+ occurs at 0.64 V vs. Ag/Ag+; however, the cathodic peak for the reduction of KABNO+ back to KABNO expected at 0.37 V vs. Ag/Ag+ does not appear because of the reaction of KABNO+ with LMC in solution. As a control experiment and for further comparison, a CV of the FTO/TiO2/DPTC film was performed with only LMC in solution as shown in Fig. S4.†
The photocurrent transient experiments with FTO/TiO2/DPTC in the presence of KABNO and/or LMC in solution are shown in Fig. 4. Increasing the KABNO concentration from 0, 1.5, 3.0, to 5.0 mM in 0.1 M TBAPF6 acetonitrile solution resulted in increasing photocurrent densities of 10, 53, 133, and 197 μA cm−2, respectively (Fig. 4a). The increased photocurrents with increasing [KABNO] suggest that light-driven electron transfer from KABNO to oxidized DPTC+ dye occurs following charge injection from DPTC* to the conduction band of mesoporous TiO2. According to the photocurrent experiments, the total consumed charge passed during the photocurrent measurements showed 1.5, 5.8, 14.9, and 19.7 mC with increasing [KABNO] of 0, 1.5, 3.0, and 5.0 mM KABNO, respectively (Fig. 4b). The linear increase in charge passed shows that DPTC dye is capable of generating the activated KABNO+ as a strong oxidizing mediator for lignin oxidation under visible light illumination. To probe LMC oxidation in this DSPEC system, 3 mM of LMC was added to the solution in the presence of KABNO under the same photoelectrochemical conditions. Interestingly, the photocurrent significantly increased up to ∼603 μA cm−2 (Fig. 4c). As a control experiment, the photocurrent was measured for FTO/TiO2/DPTC with 3 mM LMC and no KABNO (Fig. S5†). With LMC in solution, the photocurrents do increase compared to blank electrolyte (no LMC and KABNO). The lower observed photocurrent (∼210 μA cm−2) with only 3 mM LMC in solution compared to that of the mixture of LMC and KABNO (603 μA cm−2) demonstrates that the maximum photocatalytic activity requires the presence of both LMC and KABNO. The substantial increase in photocurrent is attributed to the effective oxidation of LMC by the oxoammonium species KABNO+ generated at the FTO/TiO2/DPTC surface under 1 sun illumination. Interestingly, reproducible current spikes are observed in the first 30 s of illumination in the presence of both KABNO and LMC. To gain further insight into transient spikes in Fig. 4c, we performed additional experiments with varying concentration of KABNO to understand better the conditions that cause the current spikes. In Fig. 4d, the KABNO concentration is varied from 1.5 to 6.0 mM while [LMC] is constant. Fig. 4e shows a magnified view of the photocurrents between 5 and 6 min. Based on these observations, the transient photocurrent peaks appear when the LMC concentration is the same or higher than [KABNO]. Upon close inspection of the current spike at the 6.0 mM KABNO condition (blue), a growth in photocurrent is still observed following the start of illumination, but peaks at a shorter time (∼5 s) after the start of illumination. As shown in Fig. 4e, the photocurrent transients can be characterized by four stages: (I) photocurrent onset and initial decay, (II) delayed increase in photocurrent reaching a peak, (III) a second decay period following the peak, and (IV) steady state photocurrent at longer (>30 s) illumination times. While these states are most obvious in the 1.5 mM and 3.0 mM experiments (black and red), the 6.0 mM KABNO (blue) shows similar behavior but with shorter periods for each stage. We interpret the behavior of the photocurrent in each stage as reflective of the reductants present in the double layer. Initially (stage I), only KABNO is present and the photocurrent decays as a diffusional gradient establishes under illumination. As the concentration of oxoammonium increases from the oxidation of KABNO, the reaction with LMC produces reductants not initially present either by the generation of KABNOH or the intermediates formed after reaction of the LMC with KABNO+. This increase in reductant concentration in the double layer as a result of chemical reaction of the photochemically generated KABNO+ causes an increase in the photocurrents (stage II) that peak and gradually decline as all the LMC in the double layer is consumed (stage III). After sufficient time (stage IV), diffusional gradients of KABNO and LMC establish and the system reaches a steady state that, in the presence of equal or greater concentration of KABNO, depends on the bulk concentration of LMC (compare 3.0 mM (red) and 6.0 mM (blue) data sets in Fig. 4e).
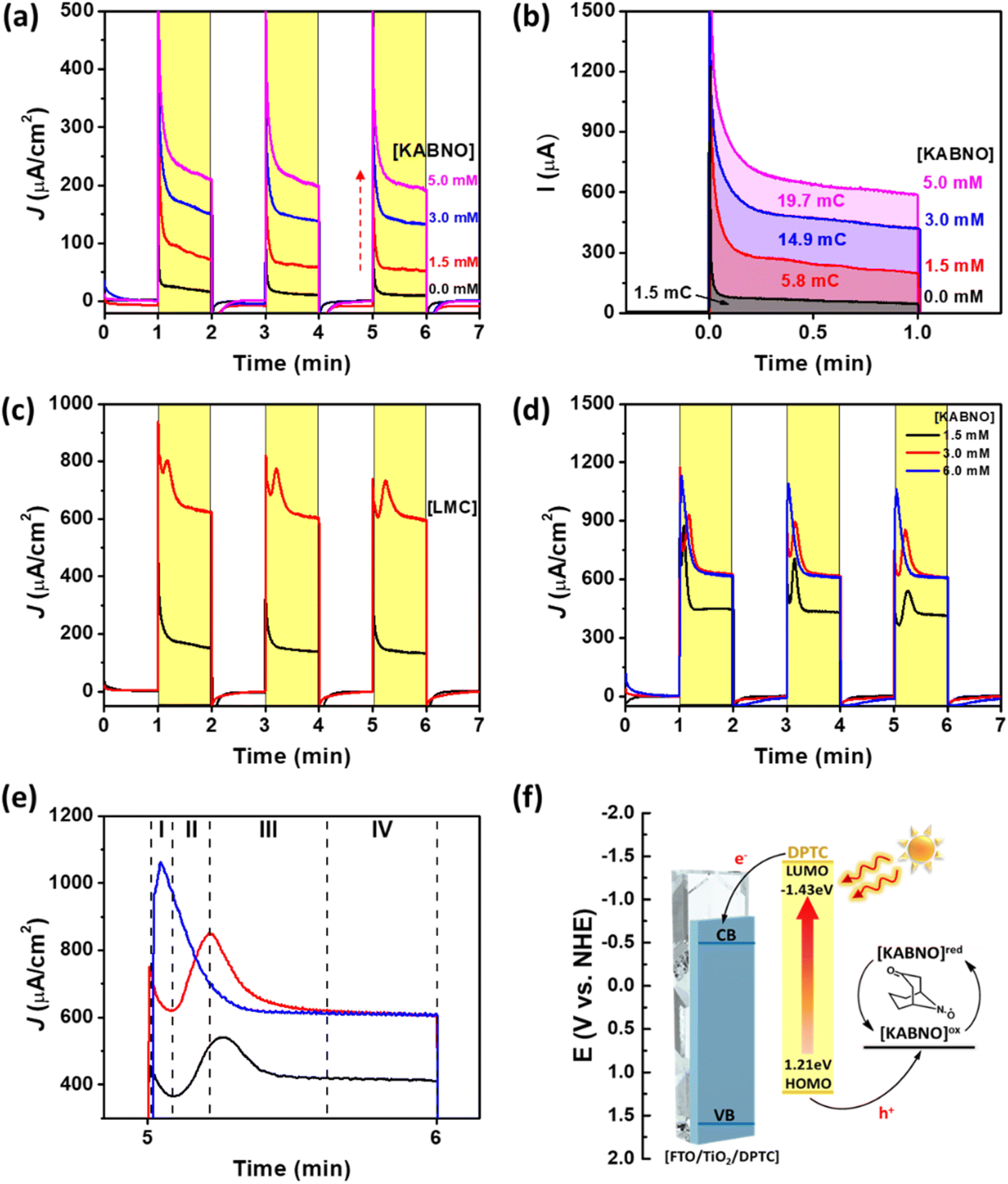 | ||
| Fig. 4 (a) Photocurrent-time trace of FTO/TiO2/DPTC (black) with different concentration of KABNO (red: 1.5 mM, blue: 3.0 mM, and pink: 5.0 mM). (b) Chronoamperograms of the FTO/TiO2/DPTC electrode in the presence of increasing concentration of KABNO from under illumination from time 0 to 1 min. Photocurrent-time trace of FTO/TiO2/DPTC (c) with 3 mM KABNO (black) and the mixture of 3 mM KABNO and LMC (red), (d) with different concentration of KABNO (black: 1.5 mM, red: 3.0 mM, and blue: 6.0 mM) at 3 mM LMC, and (e) the photocurrent densities with the period of between 5 and 6 min. (f) Energy diagram of the mesoporous TiO2, DPTC, and KABNO components of system. CB and VB indicate the conduction band and valence band potential of TiO2, respectively. All photocurrent-time trace experiments performed in 0.1 M TBAPF6 acetonitrile solution with an applied bias of 0.4 V vs. Ag/Ag+ under 1 sun illumination. | ||
While the number of N-oxyl species near the electrode is constant, the reaction of one equivalent KABNOH (R2NOH) at the electrode to give KABNO+ delivers two electron equivalents. Only KABNO should be present at the start of illumination, and the formation of some amount of KABNOH as a result of reactions in solution with LMC and KABNO+, and then the oxidation of KABNOH to KABNO+ at the photoanode, could explain the growth in photocurrent immediately following the start of illumination. Taken together, these results demonstrate the effective photoelectrochemical transformation of LMC proceeding by the following steps: (i) light absorption and photoexcited electron injection from DPTC* to the conduction band of TiO2, (ii) hole transfer between oxidized DPTC+ and KABNO to form the oxoammonium species KABNO+ in solution, (iii) oxidation of LMC by KABNO+ ([KABNO]ox), and (iv) regeneration of KABNO ([KABNO]redFig. 4f) and the C–C cleaved products A and B.
In line with our previous studies, the presence of a chromophore, nitroxyl mediator, and light are all required to perform the effective photocatalytic oxidation of LMC at room temperature.19,48,53 To better understand the products formed during the photocatalytic oxidation of LMC, a 24 h continuous illumination experiment with LMC and KABNO in acetonitrile with 0.1 M TABPF6 was performed. The photocatalytic conversion products were identified using GC-MS and NMR analyses. Cleavage products 2-(2-methoxyphenoxy)acrylaldehyde A and 2,6-dimethoxy-1,4-benzoquinone B were obtained after the 24 h reaction at room temperature based on the results of GC-MS analysis (Fig. S6†) and 1H and 13C NMR (Fig. S7†). These products were further confirmed with two-dimensional 1H–13C heteronuclear single quantum coherence (2D-HSQC) NMR analysis (Fig. 5a and b). In the aliphatic region, the β-aryl ether linkage of LMC was identified at δC/δH 72.27/4.94 ppm (Aα), δC/δH 86.66/4.15 ppm (Aβ), and δC/δH 60.14/3.91–3.67 ppm (Aγ). These peaks significantly decreased after the 24 h illumination. This observation indicated C–C and/or C–O bond cleavage of the β-aryl ether linkage of LMC. For further elucidation of the degradation of LMC, the chemical shifts in the aromatic region were identified. The chemical shifts of G2 (δC/δH 111.57/6.93 ppm), G5 (δC/δH 120.27/6.95 ppm), G6 (δC/δH 123.49/7.05 ppm), and S2/6 (δC/δH 102.23/6.62 ppm) were observed with the original LMC prior to illumination. However, after the reaction, most of these chemical shifts were not detected. Instead, new chemical shifts from the cleavage products A and B appeared at A1 (δC/δH 121.09/6.95 ppm), A2 (δC/δH 112.93/6.99 ppm), A5 (δC/δH 126.24/7.18 ppm), A6 (δC/δH 122.05/7.04 ppm), A′ (δC/δH1/δH2 107.34/5.26/5.07 ppm), B2/6 (δC/δH 107.34/5.85 ppm), and the aldehyde peak (δC/δH 186.91/9.44 ppm). The characteristic signals of A and B are consistent with our previous report.52 The yields of products A and B were quantitatively monitored by GC-FID with DMSO as an internal standard (Fig. S8†). As Fig. 5c presents, after 24 h illumination at room temperature, yields of 94% for A and 66% for B were obtained. This result is consistent with our previous study where, under aerobic conditions, product B could be further oxidized by the oxoammonium form of KABNO+ which decreased the measured yield.52 The trace amount of oxidized LMC (1-(4-hydroxy-3,5-dimethoxyphenyl)-2-(2-methoxyphenoxy)propane-1-one) was also observed at δC/δH 106.48/7.41 ppm (oxidized S2/6) and δC/δH 84.35/5.35 ppm (shifted Aβ due to the oxidized OH) as shown in the inset in Fig. S9.† Thus, the above NMR and GC characterizations of the cleavage products after illumination give strong evidence for the selective Caryl–Cα bond cleavage of LMC. We were unable to conduct recyclability tests on the same FTO/TiO2/DPTC photoanode after reaction due to the degradation of the DPTC dye at the TiO2 interface.
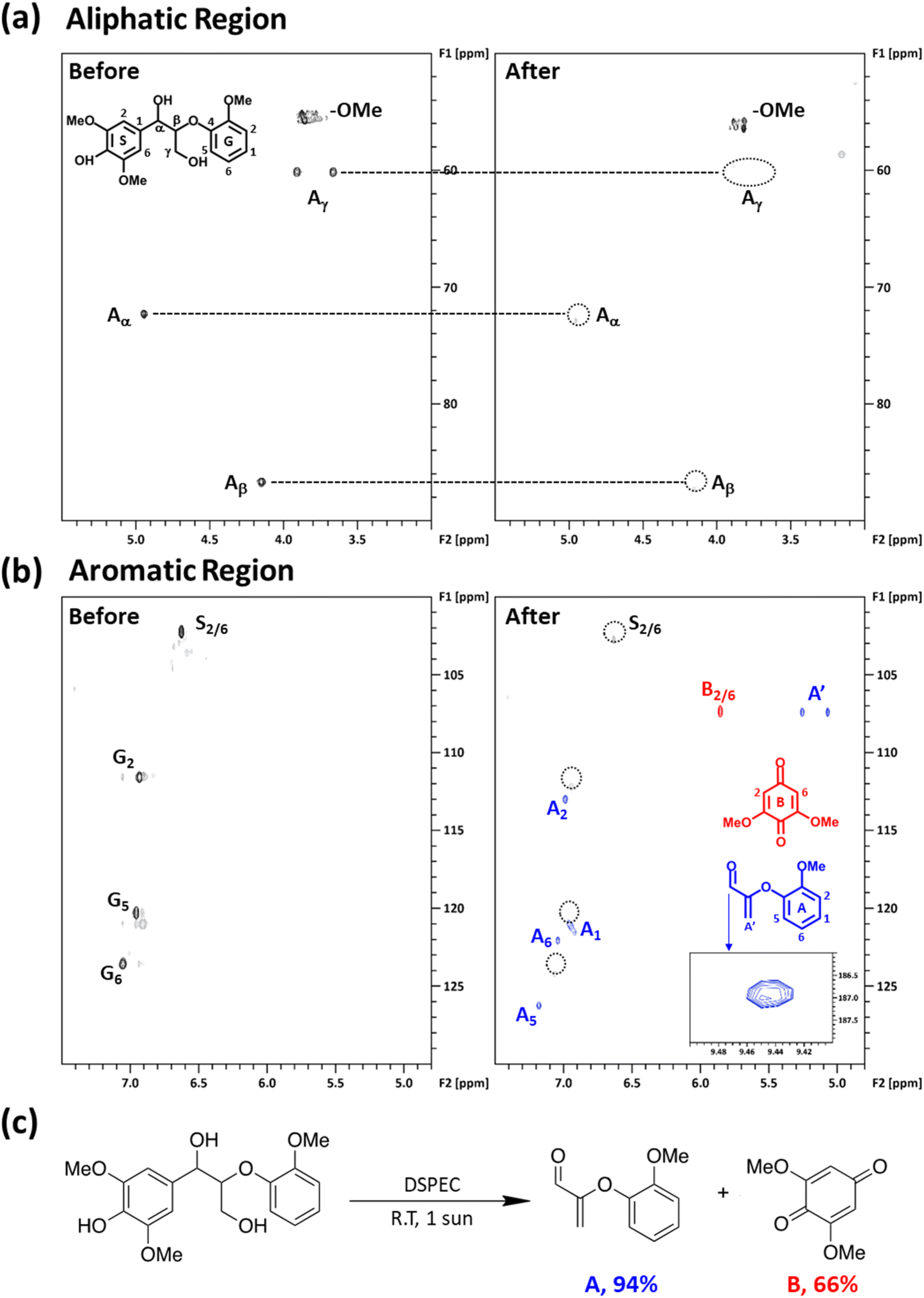 | ||
| Fig. 5 (a) Aliphatic and (b) aromatic region of 2D HSQC NMR spectra of the LMC before (left) and after (right) the photoelectrolysis reaction. (c) Photoelectrocatalytic C–C bond cleavage reaction of a phenolic lignin model compound at the FTO/TiO2/DPTC incorporating KABNO at room temperature. The yields for A and B were determined by GC-FID analysis of the reaction mixtures relative to dimethyl sulfoxide (DMSO) as an internal standard. | ||
Based on the proposed mechanism presented for Caryl–Cα cleavage in aryl ether linkages of a phenolic LMC in a related DSPEC system,43Fig. 6 illustrates the possible mechanism using the DPTC-sensitized photoanode and KABNO in the presence of LMC at room temperature under 1 sun illumination. The brief overall sequence is as follow: (1) the DPTC deposited on the surface of photoanode forms an excited-state DPTC*, which is sufficiently reducing to sensitize the TiO2 surface after absorption of visible light; (2) electron injection from DPTC* to the conduction band of TiO2 yields DPTC+ and TiO2 (e−); (3) DPTC+ oxidizes KABNO (R2-NO˙) to form KABNO+ (R2-N+ = O) and reform the ground state DPTC; (4) one equivalent of KABNO+ preferentially oxidizes the primary alcohol (Cγ–OH) of LMC to give Int 1 which leads to the formation of product B; (5) abstraction of the phenolic hydrogen atom by R2-NO˙ gives the possible LMC intermediate radical resonance structures Int 2 and Int 3; (6) reaction of intermediate Int 3 with an additional equivalent of KABNO and KABNO+ would then result in the formation of pentacyclic intermediate Int 4; (7) dissociation of Int 4 with the consumption of 1 equivalent of KABNO+ and reformation of KABNO results in Caryl–Cα cleavage and formation of products A and B.
 | ||
| Fig. 6 Proposed mechanism for oxidative C–C bond cleavage to give products A and B in the DSPEC system by using the FTO/TiO2/DPTC photoanode and KABNO (R2NO˙) in the presence of LMC. | ||
Conclusion
Organic dyes have been widely used in dye-sensitized solar cells as an alternative to transition metal complex chromophores. This study confirmed visible light-driven chemoselective Caryl–Cα bond cleavage in a phenolic LMC at room temperature by incorporating the bicyclic nitroxyl mediator KABNO and D–π–A type organic dye (DPTC). We investigated intermolecular charge transfer between KABNO and DPTC in solution and at the interface of FTO/TiO2. The FTO/TiO2/DPTC photoanode generates significant photocurrent density in the presence of KABNO and LMC under visible light illumination in acetonitrile electrolyte. This D–π–A type organic dye-based photoelectrochemical process with KABNO requires lower applied bias compared to related electrochemical or electrocatalytic approaches and this avoids overoxidation or uncontrolled degradation of the LMC. As a result, LMC was selectively oxidized in the DSPEC. We anticipate that this system can be further improved and adapted by considering the use of other organic dyes and by tuning the relative redox levels of the various components in the DSPEC. Future work will focus on the preparation of more efficient and chemically stable donor-linker-acceptor components and modifying the dye structure to better match its redox potential to that of the nitroxyl mediator.Author contributions
S. K., and H. C. K. performed experiments and contributed equally to this project. G. L., and J.-J. L. conceived the idea and provided overall supervision. C. C., S. L., K. Y., and U. K. W. contributed to data acquisition and analysis. W. Z., C. G. Y., and B. D. S. added conceptual contributions and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is supported in part by an award from the USDA National Institute of Food and Agriculture, McIntire Stennis project (No. 1026335) and the NYS Department of Economic Development (DED) through the Syracuse Center of Excellence in Environmental and Energy Systems (No. C200183). W. Z. acknowledges support from NSF CAREER grant (Award Number CHE-1944978) and NSF IUCRC Phase I grant (Award Number: 2052611). B. D. S. thanks the Welch Foundation for support of this work through award number P-2044-20200401. J.-J. Lee acknowledge the financial support by the Korean National Research Foundation, funded by the Ministry of Science, ICT & Future Planning (NRF-2021R1A2C2094554).References
- S. Li, K. Davis and G. Leem, ACS Symp. Ser. Am. Chem. Soc., 2021, 1377, 97–121 CAS.
- C. S. Lancefield, L. W. Teunissen, B. M. Weckhuysen and P. C. A. Bruijnincx, Green Chem., 2018, 20, 3214–3221 RSC.
- C. Díaz-Urrutia, B. Sedai, K. C. Leckett, R. T. Baker and S. K. Hanson, ACS Sustainable Chem. Eng., 2016, 4, 6244–6251 CrossRef.
- W. D. do Pim, F. G. Mendonça, G. Brunet, G. A. Facey, F. Chevallier, C. Bucher, R. T. Baker and M. Murugesu, ACS Appl. Mater. Interfaces, 2021, 13, 688–695 CrossRef CAS.
- L. Dong, L. Lin, X. Han, X. Si, X. Liu, Y. Guo, F. Lu, S. Rudić, S. F. Parker, S. Yang and Y. Wang, Chem, 2019, 5, 1521–1536 CAS.
- M. Wang, J. Lu, X. Zhang, L. Li, H. Li, N. Luo and F. Wang, ACS Catal., 2016, 6, 6086–6090 CrossRef CAS.
- L. Ma, H. Zhou, X. Kong, Z. Li and H. Duan, ACS Sustainable Chem. Eng., 2021, 9, 1932–1940 CrossRef CAS.
- H. Liu, H. Li, J. Lu, S. Zeng, M. Wang, N. Luo, S. Xu and F. Wang, ACS Catal., 2018, 8, 4761–4771 CrossRef CAS.
- Q. Zhang, N. K. Gupta, M. Rose, X. Gu, P. W. Menezes and Z. Chen, Chem. Catal., 2023, 3, 100470 CrossRef CAS.
- J. Wang, Y. Deng, Z. Ma, Y. Wang, S. Zhang and L. Yan, Green Chem., 2021, 23, 5120–5128 RSC.
- S. Trano, F. Corsini, G. Pascuzzi, E. Giove, L. Fagiolari, J. Amici, C. Francia, S. Turri, S. Bodoardo, G. Griffini and F. Bella, ChemSusChem, 2022, 15, e202200294 CrossRef CAS PubMed.
- J. L. Espinoza-Acosta, P. I. Torres-Chávez, J. L. Olmedo-Martínez, A. Vega-Rios, S. Flores-Gallardo and E. A. Zaragoza-Contreras, J. Energy Chem., 2018, 27, 1422–1438 CrossRef.
- A. Khan, V. Nair, J. C. Colmenares and R. Gläser, Top. Curr. Chem., 2018, 376, 20 CrossRef.
- S. K. Hanson, R. Wu and L. A. P. Silks, Angew. Chem., Int. Ed., 2012, 51, 3410–3413 CrossRef CAS PubMed.
- T. vom Stein, T. den Hartog, J. Buendia, S. Stoychev, J. Mottweiler, C. Bolm, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2015, 54, 5859–5863 CrossRef CAS PubMed.
- S. Gazi, W. K. Hung Ng, R. Ganguly, A. M. Putra Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC.
- G. Zhang, B. L. Scott, R. Wu, L. A. P. Silks and S. K. Hanson, Inorg. Chem., 2012, 51, 7354–7361 CrossRef CAS PubMed.
- L. J. Mitchell and C. J. Moody, J. Org. Chem., 2014, 79, 11091–11100 CrossRef CAS PubMed.
- S. Li, Z.-J. Li, H. Yu, M. R. Sytu, Y. Wang, D. Beeri, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Energy Lett., 2020, 5, 777–784 CrossRef CAS.
- G. Leem, B. D. Sherman and K. S. Schanze, Nano Convergence, 2017, 4, 37 CrossRef PubMed.
- B. D. Sherman, M. V. Sheridan, K.-R. Wee, S. L. Marquard, D. Wang, L. Alibabaei, D. L. Ashford and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 16745–16753 CrossRef CAS.
- G. Leem, B. D. Sherman, A. J. Burnett, Z. A. Morseth, K.-R. Wee, J. M. Papanikolas, T. J. Meyer and K. S. Schanze, ACS Energy Lett., 2016, 1, 339–343 CrossRef CAS.
- D. F. Bruggeman, T. M. A. Bakker, S. Mathew and J. N. H. Reek, Chem. - Eur. J., 2021, 27, 218–221 CrossRef CAS.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- S. Zhang, H. Ye, J. Hua and H. Tian, EnergyChem, 2019, 1, 100015 CrossRef.
- Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032–4080 CrossRef CAS.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC.
- Z. Fang, J. E. Jackson and E. L. Hegg, ACS Sustainable Chem. Eng., 2022, 10, 7545–7552 CrossRef CAS.
- X. Du, H. Zhang, K. P. Sullivan, P. Gogoi and Y. Deng, ChemSusChem, 2020, 13, 4318–4343 CrossRef CAS.
- M. Garedew, D. Young-Farhat, S. Bhatia, P. Hao, J. E. Jackson and C. M. Saffron, Sustainable Energy Fuels, 2020, 4, 1340–1350 RSC.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- I. Bosque, G. Magallanes, M. Rigoulet, M. D. Kärkäs and C. R. J. Stephenson, ACS Cent. Sci., 2017, 3, 621–628 CrossRef CAS PubMed.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS.
- M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, J. Am. Chem. Soc., 2006, 128, 8412–8413 CrossRef CAS PubMed.
- S. Hamada, T. Furuta, Y. Wada and T. Kawabata, Chemoselective Oxidation by Electronically Tuned Nitroxyl Radical Catalysts, Angew. Chem., Int. Ed., 2013, 52, 8093–8097 CrossRef CAS.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef CAS PubMed.
- L. N. Grigor'eva, A. Y. Tikhonov, K. A. Lomanovich and D. G. Mazhukin, Molecules, 2021, 26, 3050 CrossRef PubMed.
- D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550–9556 CrossRef CAS PubMed.
- M. B. Lauber and S. S. Stahl, ACS Catal., 2013, 3, 2612–2616 CrossRef CAS.
- G. Leem, Z. A. Morseth, E. Puodziukynaite, J. Jiang, Z. Fang, A. T. Gilligan, J. R. Reynolds, J. M. Papanikolas and K. S. Schanze, J. Phys. Chem. C, 2014, 118, 28535–28541 CrossRef CAS.
- G. Leem, Z. A. Morseth, K.-R. Wee, J. Jiang, M. K. Brennaman, J. M. Papanikolas and K. S. Schanze, Chem.–Asian J., 2016, 11, 1257–1267 CrossRef CAS PubMed.
- N. C. D. Nath, S. Sarker, A. J. Saleh Ahammad and J.-J. Lee, Phys. Chem. Chem. Phys., 2012, 14, 4333 RSC.
- N. C. D. Nath, J. C. Kim, K. P. Kim, S. Yim and J.-J. Lee, J. Mater. Chem. A., 2013, 1, 13439 RSC.
- G. I. Lee, N. C. D. Nath, S. Sarker, W. H. Shin, A. J. S. Ahammad, J. K. Kang and J.-J. Lee, Phys. Chem. Chem. Phys., 2012, 14, 5255 RSC.
- A. Venkateswararao, K. R. J. Thomas, C.-P. Lee, C.-T. Li and K.-C. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 2528–2539 CrossRef CAS.
- A. Tiwari and U. Pal, Int. J. Hydrogen Energy, 2015, 40, 9069–9079 CrossRef CAS.
- S. Li, E. W. Shuler, D. Willinger, H. T. Nguyen, S. Kim, H. C. Kang, J.-J. Lee, W. Zheng, C. G. Yoo, B. D. Sherman and G. Leem, ACS Appl. Mater. Interfaces, 2022, 14, 22799–22809 CrossRef CAS PubMed.
- H. Jiang, Y. Cheng, Z. Wang, Z. Bai, Y. Tang, Y. Sun, P. Wan and Y. Chen, J. Electrochem. Soc., 2021, 168, 016504 CrossRef CAS.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- C. Yang, S. Maldonado and C. R. J. Stephenson, ACS Catal., 2021, 11, 10104–10114 CrossRef CAS.
- S. Li, S. Kim, A. H. Davis, J. Zhuang, E. W. Shuler, D. Willinger, J.-J. Lee, W. Zheng, B. D. Sherman, C. G. Yoo and G. Leem, ACS Catal., 2021, 11, 3771–3781 CrossRef CAS.
- S. Li, S. Park, B. D. Sherman, C. G. Yoo and G. Leem, Chem. Commun., 2023, 59, 401–413 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of additional characterization methods and data. See DOI: https://doi.org/10.1039/d3se00194f |
| ‡ S. K. and H. C. K. are equally contributed to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |