
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biologically bound nickel accelerated de-polymerization of polyethylene to high value hydrocarbons and hydrogen†
Parul
Johar
 a,
Elizabeth L.
Rylott
b,
C. Robert
McElroy
a,
Avtar S.
Matharu
a and
James H.
Clark
*a
a,
Elizabeth L.
Rylott
b,
C. Robert
McElroy
a,
Avtar S.
Matharu
a and
James H.
Clark
*a
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK. E-mail: james.clark@york.ac.uk
bCentre for Novel Agricultural Products, Department of Biology, University of York, Wentworth Way, York, YO10 5DD, UK
First published on 4th November 2022
Abstract
The goal of a carbon-neutral society can be realized by utilizing a circular carbon pathway, which combines recycling, biomass utilization, carbon capture and utilization. Inspired by the potential of metal-contaminated biomass and plastic waste as valuable feed-stocks, we have developed a biologically-bound nickel catalyst (Ni-phytocat) to accelerate de-polymerization of polyethylene into high value chemicals. The synergistic effect of microwaves, together with Ni-phytocat as microwave absorbers, accelerate the catalytic de-polymerization process at low temperature (250 °C). The single step process typically takes up to 70 s to transform a sample of low-density polyethylene into liquid hydrocarbons (40–60% oil yield), hydrogen (11–30% gas yield) and filamentous carbon (25–37% solid yield), depending on varying catalyst to polymer weight ratios. The Ni-phytocat enhanced the production of C6–C12 aliphatics (up to 56% selectivity) and favored the aromatization of linear alkanes to form monocyclic aromatics (up to 33% selectivity), thereby releasing more H2 (up to 74% selectivity) as gaseous fractions. The enhancement of (de)hydrogenation, de-carboxylation and cyclization, utilizing Ni-phytocat can be established as a proof of concept to advance and enable selective transformation of polymeric consumer products, paving the way to harness complete circular chemical potential of these future feed-stocks.
Sustainability spotlightTo realize the goal of a carbon-neutral society, the closing of carbon and hydrogen cycles in plastics would be extremely important. Chemo-catalytic strategies to transform waste plastics are cost prohibitive, energy intensive and require high operating temperatures, often leading to uncontrollable product distribution with little product value other than as a low-grade fuel. Our interest is in developing a protocol to accelerate de-polymerization of plastics into valuable hydrocarbons and hydrogen without the need of added hydrogen or artificially fabricated metal catalysts. In this context, we envisioned the development of our phytocat material, an air-stable and inexpensive, biologically bound Ni catalyst prepared from Ni-rich plant biomass. The unique metal–support interaction between biologically bound Ni and the bio-carbon matrix could enable lower-energy pathways to plastic recycling. We propose a new microwave-driven de-polymerization of polyethylene, enabled by sustainable Ni0/Ni2+/bio-carbon synergistic effects. Remarkably, we achieve PE depolymerization at moderate temperatures in very short periods of time, and we can tune the process to maximise the production of hydrogen and/or smaller, fuel-suitable hydrocarbons. This product flexibility could be very important given the shifting, and politically-dependent balance in energy requirements and the emergence of the hydrogen economy. Our work emphasizes the importance of the following UN sustainable development goals: affordable and clean energy (SDG 7), industry, innovation, and infrastructure (SDG 9), climate action (SDG 13). |
Introduction
The linear approach to resource utilization has led to the accumulation of waste plastic (around 200 million metric tons generated annually) in the environment over decades.1 Thus to realize the goal of a carbon-neutral society, closing of carbon and hydrogen cycles in plastics would be extremely important.2 There exists an imbalance between the production and end-of-life treatment of plastic wastes.3 Among many plastic materials that are commercially manufactured, polyolefins (>90% of packaging materials) account for nearly half of world plastic demand, with 36% of all plastics being polyethylene (PE), the subject of this study.4 Polyethylene is designed to resist the tough conditions of high temperature and pressure, mechanical forces, and chemical corrosion, and to maintain these properties throughout their consumption. This physical and chemical stability makes PE recycling an ambitious challenge.5 The global spread of COVID-19 has further worsened the problem by increasing the use of single-use personal protective equipment (PPE) and food packaging.6 Chemo-catalytic strategies to produce high value chemicals, fuels, and monomers hold the promise of positively transforming waste plastic, which is barely addressed by mechanical recycling and incineration, into an opportunity.4,7–9 Both de-polymerization of plastics into value-added chemicals and carbonization to high value carbon materials will relieve environmental pollution and reduce demand for virgin resources.10,11 However, these methods are often cost prohibitive, energy intensive and require high operating temperatures, often leading to uncontrollable product distribution with little product value other than as a low-grade fuel.12,13 For instance, noble metal-catalyzed hydrogenolysis of polyethylene normally requires high reaction temperatures and long reaction times. Bifunctional metal/acid catalysts are an attractive option because the acid catalyst can activate C–C bonds, while the metal catalysts possess high hydrogenation ability and can inhibit catalyst coking. For example, Liu and coworkers reported a Pt/WO3/ZrO2 + HY(30) zeolite catalytic system for mild hydrocracking in low density polyethylene (LDPE) melts to produce a mixture of gasoline, diesel, and jet fuel range hydrocarbons.12 When using Pt/WO3/ZrO2 or HY(30) alone as catalyst, the conversion of LDPE is relatively low even at 250 °C. Edwards and co-workers used microwaves, together with inexpensive iron-based catalysts (FeAlOx) as microwave absorbers to convert a mixture of mechanically pulverized, real-world plastic waste into hydrogen and multi-walled carbon nanotubes.14 However, these bifunctional metal/acid catalyst-based protocols focus on the use of either additional H2 or artificial synthesis of noble and non-noble metallic species onto different supports. Our interest is in developing a protocol to accelerate de-polymerization of polyethylene into high value hydrocarbons without the need of molecular hydrogen or artificially fabricated metal catalysts.The transition to a low-carbon future has potentially increased demand for certain metals such as copper, nickel, and lithium needed for low-carbon technologies, meaning that the extractive industry has an essential role to play towards sustainable development.15 In 2019, around 55% of global nickel ore extraction took place in the species-richest tropical biome,16 suffering from massive deforestation in recent decades, with nickel mining known as a significant driver. Growing demand for essential minerals and declining quality of ores have lead to larger volumes of unused material extracted and disposed, increasing appropriation of land.17–19 There is likely enough economically exploitable primary Ni to meet increased demand; however, mining, purifying, and refining Ni metal releases greenhouse gases (7.64 kg CO2-eq. per kg), degrades the environment (including soil contamination with heavy metals and acidification of local wetlands), and presents human health concerns in nickel mining and refining workplaces.20,21 The significant proportion of nickel that comes from Russia may exaggerate this problem. An alternative method of primary Ni production which could complement mining is phytomining.22 The key steps in developing sustainable phytomining are: (i) identifying potential metal-rich soils, (ii) selecting suitable plant species, and (iii) developing valuable product recovery processes.23 The target metal-rich soils include smelter-contaminated land, ore beneficiation tailings and naturally occurring ultramafic soils. Nickel-hyperaccumulating plants uptake metal through low-selectivity cation transporters in the roots storing it inside the aerial plant biomass at >1000 mg kg−1 dry weight; concentrations higher than some commercially-mined ores.24 Despite this metal-accumulating ability, these plants might not be suitable for soils contaminated with multiple contaminants.25,26 In these environments, fast-growing, high-biomass yielding species such as willow hybrids (Salix spp.) or poplar (Populus spp.) trees can be used to extract a wide range of metals from soil.27 with the harvested biomass used as a feedstock for production of bio-energy or bio-materials.28–31 A significant added advantage of using phytomining is that it can help remediate contaminated land and make it suitable for growing crops.
In this context, we envisioned the development of our phytocat material, an air-stable and inexpensive, biologically bound Ni catalyst prepared from Ni-rich plant biomass using a simple, one-step protocol. The single step, low-temperature, microwave assisted biosynthesis of catalytically active Ni makes it cost-effective alternative to conventional catalysts, which are typically fabricated via precious metals or multi-step synthesis. Furthermore, no new resource is consumed other than the energy to produce the microwaves. We propose a new microwave-driven de-polymerization of polyethylene, enabled by sustainable Ni0/Ni2+/bio-carbon synergistic effects. Metal/metal oxide catalysts have been studied extensively for small alkane hydrocracking but the transfer of this knowledge to catalytic cracking of polyethylene is lacking.32,33 As a result of this functionality, the nickel content in our phytocats plays a significant role in the LDPE de-polymerization. Remarkably, we achieve PE depolymerization at moderate temperatures in very short periods of time, and we can tune the process to maximise the production of hydrogen and/or smaller, fuel-suitable hydrocarbons. This product flexibility could be very important given the shifting, and politically-dependent balance in energy requirements and the emergence of the hydrogen economy.
Results and discussion
Structural design of the biologically bound nickel catalyst (phytocat)
The Ni hyperaccumulating plant Odontarrhena muralis (previously Alyssum murale) and non –hyperaccumulator willow (Salix vimnalis) were used in this study. The phyto-extracted Ni present within the biomass of each species is determined by the inherent biology of that species (including the gene-encoded metal transporters and associated detoxification enzymes).27 Six-week-old, hydroponically-grown S. vimnalis rods were dosed with 100 mg kg−1 Ni for two weeks, the tissues air-dried and ground. Odontarrhena muralis was grown in Ni-rich serpentine soil. The Ni catalysts were prepared using a low temperature, microwave-assisted pyrolysis (200 W, 250 °C) to produce S. viminalis biochar (0.1 wt% Ni, termed Ni-phytocat-0.1), and O. murale biochar (2.5 wt% Ni, termed Ni-phytocat-2.5).34 The control catalyst was prepared using Ni undosed S. viminalis biochar (<0.01 wt% Ni, termed as control phytocat). Phytocat, as a good microwave absorber, aids in transferring the microwave energy leading to the in situ synthesis of catalytically active Ni0 from phyto-extracted Ni2+ (Fig. 1a) | ||
| Fig. 1 (a) Six-week-old, hydroponically grown willow (b) energy-dispersive X-ray (EDX) spectroscopy analysis of Ni-phytocat-2.5 highlighting carbon (green), nickel (blue), oxygen (red) and calcium (orange) (inset images: elemental mapping of Ni-phytocat-2.5 highlighting carbon, nickel, silicon, calcium, potassium and oxygen), scanning electron microscopy (SEM) images equipped with energy-dispersive X-ray spectroscopy (EDX) (c–e) phytocat-2.5 and recovered phytocat-2.5 respectively after de-polymerization of LDPE, (f–h) phytocat-0.1 and recovered phytocat-0.1 respectively (i–k) control phytocat and recovered control phytocat respectively. | ||
Energy-dispersive X-ray (EDX) spectroscopy analysis of the Ni phytocat revealed the presence of small amounts of oxygen, which is evident of nickel oxide formation on the particle surface due to storage in air. As can be seen from Fig. 1b, nickel nanoparticles embedded in a bio-carbon matrix of phytocat contained traces of calcium, silicon and oxygen, elements also present in the control phytocat (Fig. S1†).
Field emission scanning electron microscopy (FESEM) images of phytocat materials before and after the de-polymerization reaction were observed. They exhibited irregular spherical morphologies and high dispersion of Ni species on the surface of Ni-phytocat-2.5 (Fig. 1c and d) and relatively moderate dispersion on Ni-phytocat-0.1 (Fig. 1e and f). But the presence of Ni was not detected on the surface of irregular clusters of control phytocat (Fig. 1g and h). As shown in the FESEM images, carbon deposits on the recovered phytocat comprised carbon filaments with a diameter of ∼10 nm on phytocat-2.5. Particularly, the presence of a dense, entangled growth of filamentous carbons covering the surface of Ni-phytocat was observed. The carbon growth was irregular in morphology with hollow capsule structures on phytocat-0.1, while no such growth was observed on the surface of control phytocat. With the presence of Ni, the carbon growth was controlled to generate filamentous structure (corresponding to carbon nanotubes, CNTs) alongside some amorphous small particles. On the other hand, for control phytocat, carbon was simply deposited on the surface as amorphous small particles.
High-resolution transmission electron microscopy (HR-TEM) measurement was then employed to observe the lattice fringes corresponding to the biologically bound nickel in the phytocat materials. The HR-TEM image shown in Fig. S1† revealed irregular fringes, mainly with a planar spacing of 0.242 nm and 0.216 nm point to (111) and (200) crystal planes, respectively, of cubic Ni phase. Fig. 2d–f show the HR-TEM images of the recovered catalysts after de-polymerization of the LDPE. These images confirmed that the solid carbon deposition on the surface of the used catalyst contained filamentous carbon, and that the black nanoparticles encapsulated within the filamentous carbons (CNTs) were related to Ni0 (Fig. 2). In addition, as shown in Fig. 2d and e, there were many metal particles in the middle or top of the carbon filaments, indicating that the tip growth mode dominates the formation of CNTs for plastic waste. Furthermore, as shown in Fig. 2e and f, both filamentous and amorphous carbon was observed on phytocat-0.1 post-reaction, whereas only amorphous carbon was detected on the surface of control phytocat. The amorphous carbon might originate from the oligomerisation reaction of lower molecular weight polycyclic aromatic compounds.
 | ||
| Fig. 2 High-resolution transmission electron microscopy (HRTEM) image of (a) Ni-phytocat-2.5, (b) Ni-phytocat-0.1, (c) control phytocat, (d) recovered Ni-phytocat-2.5, (e) recovered Ni-phytocat-0.1, (f) recovered control phytocat; high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image of (g) Ni-phytocat-2.5 (h) Ni-phytocat-0.1, (i) control phytocat; (j) temperature programmed oxidation and derivative thermograms of recovered catalysts after de-polymerization of LDPE. | ||
Following the observation of the microstructural features, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were used to gain more structural insight. In the field of view of HAADF-STEM images, intensity is relative to the square of atomic number of the element.35 Therefore, the atomically dispersed Ni particles can be clearly distinguished as the brighter spots on bio-carbon. As shown in Fig. 2g, the formed Ni nanoparticles were uniformly distributed and exhibited an average particle size of 5.2 ± 1.1 nanometers. Most of the observed nano-particles were within a range of 4.5 to 7.3 nm; however, a few particles up to 11 nm were observed.
Thermogravimetric analysis (TGA) experiments were conducted to understand the thermal stability and degradation profiles of the raw plant biomass of S. viminalis and O. murale and also for the prepared phytocats. As shown in Fig. S6,† the raw biomass exhibited an initial weight loss around 370 °C. The TGA plots of Ni-phytocat-2.5, Ni-phytocat-0.1 and control phytocat, prepared using low-temperature microwave activation, indicated a weight loss of 6.1%, 7.2% and 7.5% at low temperature (T ≤ 150 °C), which may be assigned to the removal of the adsorbed water or residual solvents from the surface (Fig. S6†). This result reveals the remarkable thermal stability of phytocat, even at high temperatures (T ≤ 700 °C). Temperature programmed oxidation (TPO) was used to characterize the carbon deposits as the oxidation temperature is influenced by the nature of carbon. Fig. 2j describes the TPO profiles and the association between the measured carbon content and the oxidation peak temperatures. Oxidation at temperatures exceeding 500 °C could be assigned to graphitic coke, while oxidation at lower temperatures was attributable to amorphous or encapsulating carbon species.33,36 The high temperatures for coke combustion indicate that the formation of graphitic coke was more favored than that of amorphous coke from LDPE pyrolysis volatiles. Two stages of carbon oxidation were identified: (i) amorphous carbons (∼550 °C) and (ii) filamentous carbons (CNTs) (∼660 °C). The TPO results indicate that the carbon formed over control phytocat consists of amorphous type carbons that oxidize at lower temperature compared to filamentous carbon (phytocat-2.5 and phytocat-0.1) that oxidizes at higher temperature. The carbon with a high degree of graphitization would have a high thermal stability and would decompose at higher temperatures compared with the less graphitized/amorphous carbon.37
To obtain insight into the surface chemistry of the phytocats, the elemental composition and chemical states of biologically bound nickel in the bio-carbon matrix were analyzed using X-ray photoelectron spectroscopy (XPS) analysis. Various plant-based ligands such as histidine, nicotianamine and some organic acids form complexes with Ni2+ ions, enabling its detoxification by plants.38 Our one-step, low temperature microwave assisted pyrolysis approach leads to in situ reduction to Ni0. This chemical valence state of the naturally entrapped Ni was determined using the corresponding binding energies (eV) of Ni 2p3/2 peaks, as shown in Fig. S4.† Generally, catalysts with low nickel metallic loadings are dominated by Ni2+, along with some sequestered Ni0 sites.39 The differences in Ni0 lead to changes in the surface chemistry and consequently the catalytic activity. However, an oxide layer formed around supported Ni particles can suppress coke formation while preserving high catalytic activity.39 The peak around 852 eV is assigned to Ni0 and peaks between 855 and 861 eV are assigned to Ni2+ in the form of Ni(OH)2 and NiO.2 The surface of the phytocat consisted of both Ni0 and Ni2+. However, with increasing nickel content, there was an increase in intensity of Ni0 peak with a simultaneous decrease in intensity of Ni2+ peak. After use, XPS analysis on the recovered catalyst revealed a significant decrease in the intensity of the Ni0 peak, which may be due to increase in filamentous carbon growth on the surface of phytocat.
Deconvoluted, high-resolution C 1s XPS spectra of phytocat showed characteristic peaks for C–C (284.6 eV), C–N (285.4 eV), C–O (286.3 eV), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.2 eV), and O–CO (288.9 eV) bonds.40 The prominent peaks at 284.3–284.5 eV revealed that the majority of carbons in the phytocat were aromatic. The presence of these functional groups on the surface of phytocat could facilitate binding with Ni nano-particles (Fig. S4†). The peaks corresponding to π–π* transition (290.4 eV) were found to intensify for the recovered phytocat, which again suggests an increase in filamentous carbon growth on the surface of phytocat.
O (287.2 eV), and O–CO (288.9 eV) bonds.40 The prominent peaks at 284.3–284.5 eV revealed that the majority of carbons in the phytocat were aromatic. The presence of these functional groups on the surface of phytocat could facilitate binding with Ni nano-particles (Fig. S4†). The peaks corresponding to π–π* transition (290.4 eV) were found to intensify for the recovered phytocat, which again suggests an increase in filamentous carbon growth on the surface of phytocat.
To understand the crystal structure and phase purity of the phytocat material, X-ray diffraction (XRD) was used. Initial XRD analysis revealed Ni0 particles along with a carbon-rich phase. As shown in Fig. S4,† the diffraction peaks at 44.6°, 52.2°, and 77.3° belong to metallic Ni ([111], [200] and [220] diffraction peaks of Ni0).41 Both Ni-phytocat materials exhibits a broad graphitic (002) peak at about 25°, which belongs to the hexagonal conjugated carbon structure. This result confirms that the structure of the Ni-phytocat is relatively stable and the majority of entrapped NiO is reduced to Ni metal by the surrounding bio-carbon matrix during the microwave-assisted pyrolysis. Similarly, after use, the characteristic peaks of bio-carbon (2θ = 24.5°) and Ni0 (2θ = 44.5°, 51.9° and 76.9°) were observed for all the phytocat materials, in which the intensity of [111], [200] and [220] diffraction peaks due to Ni0 gradually increased with increasing Ni content (Fig. S4†).42 Moreover, the presence of unreacted polyethylene residue was observed over the recovered phytocat-0.1, as characterized by the peak at about 22°, whereas no such peak was observed over recovered phytocat-2.5.
The Brunauer–Emmett–Teller (BET) surface area of Ni-phytocat-2.5 and Ni-phytocat 0.1 was found to be 34.2 ± 4.5 m2 g−1 and 21.7 ± 2.4 m2 g−1 respectively and contain micro-pores and meso-pores (Fig. S7†).42 There are numerous advantages of using microwave-heating, which promotes high dispersion of Ni species, easy reduction of surface Ni species, and formation of uniformly nano-sized Ni particles.43 Our clean strategy of using a simple, one step, low-temperature microwave assisted pyrolysis resulted in the formation of an air-stable Ni phytocat, which could be tested further for accelerated de-polymerization of polyethylene.
Efficiency and degree of de-polymerization of LDPE
Random scission of the polymer chains dominated the thermal degradation of LDPE, leading to the formation of several micro molecule hydrocarbon species, such as alkenes and alkanes, alongside mono-aromatics.44 Our analysis indicates that two active sites with different functions take part in the de-hydro-cyclization step involved in the degradation reaction. One was bio-carbon or activated carbon surface sites, on which the abstraction of hydrogen atoms occurred predominantly; the other was metal sites which catalyzed desorption of the hydrogen atoms. Desorption was a slow step of the de-hydro-cyclization on the bio-carbon or activated carbon, but was enhanced by the biologically bound Ni (Ni-phytocat), resulting in an increase in the yield of aromatics.Phytocat-2.5 resulted in maximum plastic conversion into high value chemicals (up to 89%) at low temperature (250 °C), which is essential for practical implementation of a catalytic technology. The phytocat preferentially cracked the C>12 compounds formed over the biologically bound Ni active sites at first to C8–12, then to smaller C5–7 hydrocarbons, eventually producing monocyclic aromatics as its fraction in the catalyst blend increases. The bio-carbon matrix (control phytocat) alone showed lower activity for LDPE conversion (up to 71%), which is comparable to commercially used activated carbon (up to 70%). Remarkably our best phytocat maintains its activity even at quite low loadings (5% catalyst) and much better than the other catalysts (Fig. 3a).
 | ||
| Fig. 3 (a) Comparison of efficiency of various catalysts for accelerated de-polymerization of LDPE, (b) relative amounts of the gaseous molecules formed during microwave-assisted accelerated de-polymerization of LDPE using various catalysts, depth of cracking of various hydrocarbons formed during microwave-assisted accelerated de-polymerization of LDPE on the surface of (c) control phytocat or activated carbon and (d) Ni-phytocat. | ||
Fig. 3b shows the relative volume fractions of the gaseous components for catalytic de-polymerization of LDPE using different catalysts. The gaseous products were mainly H2 and CH4. Simultaneously, some C2+ hydrocarbons were also detected. There was a significant reduction in selectivity towards ethylene production using Ni-phytocat-2.5 as compared to the other catalysts. This could be due to the strong interaction of ethylene molecules with the surface of Ni-phytocat, which leads to subsequent bond breaking and enhances carbon deposition (as shown previously in Fig. 1 and 2).45 Furthermore, this could be attributed to the increasing formation of aromatics at the expense of olefins. The aromatization enhanced by Ni-phytocat also gives rise to hydrogen generation, resulting in a significant increase in hydrogen volume yield (70%), as compared to activated carbon and control phytocat (<50%). This result confirms the excellent dehydrogenation ability of NiO present on the surface of Ni-phytocat. For this reason, Ni-based catalysts have been extensively used in industry to generate both H2 and CNTs (carbon nanotubes) from catalytic reforming of plastics.11,33 The decrease in hydrogen content using control phytocat and activated carbon is due to the breakage of C–C bonds, which results in a significant increase of hydrocarbon compounds in the gaseous product. As the reaction progresses, more microwave energy input is provided, leading to an increase in C–C and C–H bond breaking and increased hydrocarbon yields. In contrast, the yield of liquids shows the opposite trend. The additional energy input also promotes aromatization reactions, forming polycyclic aromatic hydrocarbons and increasing coking. In addition, the loading amount of Ni also affects the performance of the catalyst. With increasing Ni loading, more catalytic sites are formed on the bio-carbon surface, making the process more favorable for the upgrading of liquid products rather than the loss of liquid product quality (conversion to coke). Interestingly, Ni active sites in phytocat promote the occurrence of hydrogen transfer reactions and catalyze the aromatization of long-chain olefins and alkanes.
In the TEM images of the used catalysts (Fig. 2d–f), filamentous carbon generation was observed on the metal particles, which confirmed that the metal sites can adsorb carbonaceous intermediates and catalyze C–H breakage, with carbon deposited on the metal particles and further growth, similar to the process of carbon nanotube growth on the metal sites at high temperatures in other studies.11,36 The active Ni sites can also facilitate the occurrence of hydrogen transfer reactions, where alkane cyclization produces cycloalkanes as hydrogen donors and olefins as hydrogen donors reacting to produce alkanes and aromatics. The end result of the catalytic reaction is the catalytic conversion of long-chain hydrocarbons to short-chain aliphatics and aromatics (Fig. 3c and d).
Effect of varying catalyst to polymer ratios
Experiments using catalyst blends of varying composition further confirmed the synergy between the Ni active sites and bio-carbon matrix (Fig. 4). The bio-carbon matrix forms C>17 compounds as well as C13–16 and C8–12 molecules with slight deep cracking to C5–7 and C1–4 gases but was incapable of complete conversion of the plastic at mild conditions and short reaction times. The bio-carbon or activated carbon is likely responsible for the initiation steps in the reaction network by converting large paraffins to large olefins.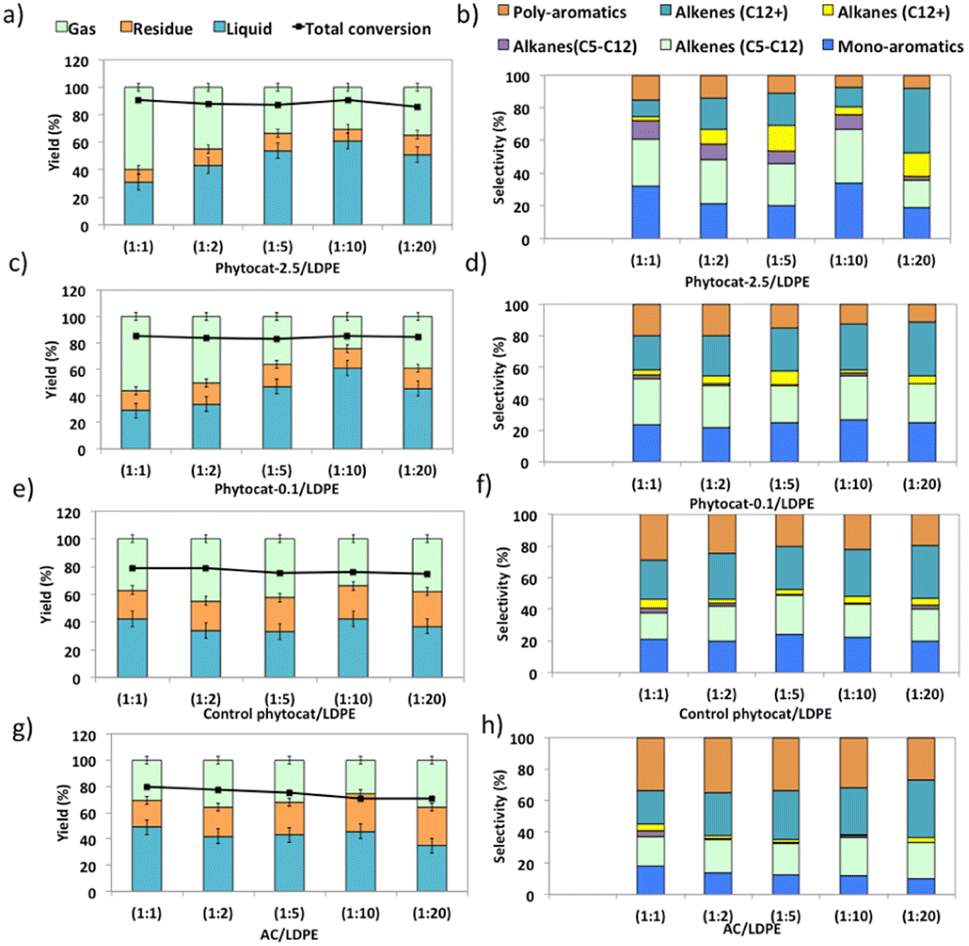 | ||
| Fig. 4 Comparison of experimental results of microwave assisted de-polymerization of LDPE using varying catalyst to polymer ratio: (a) and (b) phytocat-2.5, (c) and (d) Ni-phytocat-0.1, (e) and (f) control phytocat and (g) and (h) activated carbon. | ||
With the increase in catalyst loading, the gas and residual carbon yields increased due to an increase in vapor residence time. An increase in residence time also favored the cracking reactions, which eventually produced light gas molecules.
Overall, the yield of oil decreased with an increase in catalyst loading due to excessive cracking. The minimum yields of char (21.2 wt%) and coke (1.5 wt%) were obtained when the catalyst: polymer mass ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. The yield of aromatic hydrocarbons increased and that of long chain aliphatics decreased with an increase in catalyst loading. Yet Ni-phytocat was even affective at low catalytic loadings in the mixture, which shows its high efficiency in catalytic cracking of LDPE. The presence of Ni-phytocat also induced the enhancement of oil and gas yields by reducing the char product formation.
:10. The yield of aromatic hydrocarbons increased and that of long chain aliphatics decreased with an increase in catalyst loading. Yet Ni-phytocat was even affective at low catalytic loadings in the mixture, which shows its high efficiency in catalytic cracking of LDPE. The presence of Ni-phytocat also induced the enhancement of oil and gas yields by reducing the char product formation.
The liquid product components, grouped according to their carbon number, and the concentrations are expressed, based on the percentage areas of the corresponding compounds, in Fig. 4b, d, f and h. For most catalytic reactions, C6–C20 hydrocarbons were the main compositions in pyrolysis oils. Almost no Cn>23 products were present in the liquid even after 3 consecutive microwave cycles. Furthermore, the lack of heavier products could indicate that polymer cracking proceeds with one chain adsorbed and reacting until being completely consumed without releasing medium-sized products.46 The optimized reaction condition was found to be 10% of catalyst loading and 250 °C, at this condition, the oil was primarily composed of mono-cyclic aromatics and short chain aliphatic hydrocarbons (Fig. 4b). Utilizing Ni-phytocat proved to be effective even at low catalytic loadings, whereas with decreasing content of activated carbon, the selectivity towards mono-aromatics and short chain aliphatics declined sharply (Fig. 4h).
Effect of Ni content
The bio-carbon matrix (control phytocat) alone showed relatively low activity for LDPE conversion at 250 °C, giving mainly long chain aliphatics and poly-aromatics (an average of 39 and 29%, respectively) (Fig. 5) with a broad carbon number distribution centered at C10. Activated carbon, under the same reaction conditions, showed little LDPE conversion with extensive coking, leading to 36% solid residue (Fig. 5a and e). Using Ni-phytocat, the product distribution becomes narrower and shifted to short-chain aliphatics and mono-aromatic hydrocarbons (Fig. 5c and d). The results suggest a strong synergy between Ni0/Ni2+ and bio-carbon matrix to effectively depolymerize LPDE into higher value chemicals and fuels. Initially, the polymer undergoes random scission over bio-carbon into relatively large olefins (C12+) or alkanes. These intermediates diffuse into Ni active sites, where they crack relatively quickly into smaller C<12 alkenes and alkanes. | ||
| Fig. 5 Gas chromatography-mass spectrometry (GC-MS) characterization of oils formed on microwave-assisted pyrolysis of LDPE using (a) activated carbon, (b) control phytocat, (c) phytocat-0.1 and (d) phytocat-2.5; product yields (e) and selectivity (f) using various catalysts (1:10 catalyst to polymer ratio, by weight). | ||
Decreasing the Ni content in the phytocat resulted in a decrease of the short-chain aliphatics (C5–12) yield from 47 to 29% and an increase of the long chain aliphatics (C12+) yield from 16 to 31%. At the same time, the yield of solid residue over Ni-phytocat-0.1, having lower actives site increased. Thus, the presence of Ni active sites can significantly affect the consumption of C>12 intermediates produced by bio-carbon matrix (Fig. 5f). These results mainly leads to a slowdown of the initial polymer proceeding with a shift in selectivity toward heavier hydrocarbons. Good dispersion of Ni active sites is an essential dial for tuning product distribution from heavier fractions to low-molecular weight alkanes and alkenes, depending upon varying fractions of catalyst and polymer blends, as discussed in the previous section.
The de-polymerization experiments over these micro-porous catalysts with varying metallic dispersion indicated that the LDPE conversion to liquid and gaseous products follows the order, phytocat-2.5 > phytocat-0.1 > activated carbon > control phytocat (Fig. 5e). The product distribution reflects catalytic ability during cracking; Ni-phytocat-2.5 favors a short-chain aliphatics and mono-aromatics and Ni-phytocat-0.1 favors C1–4 gas products. Although Ni-phytocat-2.5 gives higher gas products than Ni-phytocat-0.1, it favors hydrogen production as opposed to C1–4 gas products. This selectivity arises primarily from slower diffusion of reaction intermediates over phytocat with narrower pores, leading to their secondary cracking to smaller products. Additionally, the presence of well-dispersed catalytic active sites found in Ni-phytocat-2.5 favors further aromatization of these alkanes and alkenes, thereby increasing proportions of mono-aromatics. As observed in Fig. 5, a small proportion of alkenes (C>12) were produced, providing sufficient reactants for both alkylation reaction of aromatics and the dehydrocyclization process. When activated carbon was analyzed, higher yields of poly-aromatics (naphthalene (∼20%) and pyrene (∼15%)) were obtained. Over the Ni-phytocat, most of the intermediates were confined in the bio-carbon microporous network, where larger hydrocarbons exhibited substantial diffusional limitations. So, the reaction selectivity shifts to light products.
Conventional and microwave assisted de-polymerization of LDPE
Compared with catalytic thermal pyrolysis, catalytic microwave pyrolysis gives increased gas yields and reduced oil yields (Fig. 6). | ||
| Fig. 6 Comparison of experimental results of the microwave-assisted (for 3 successive cycles to reach the set-point of 250 °C) and conventional thermal (500 °C) de-polymerization of polyethylene (low density polyethylene, LDPE) using (a) and (b) Ni-phytocat-2.5, (c) and (d) Ni-phytocat-0.1, (e) and (f) control phytocat and (g) and (h) activated carbon (1:10, catalyst: LDPE by weight). | ||
Furthermore, microwave assisted pyrolysis of LDPE resulted in a narrower carbon atoms number distribution, producing mainly mono-aromatics and short chain aliphatics. During conventional pyrolysis, polycyclic aromatics were the main compositions of the aromatic components (Fig. S8†). These polycyclic aromatics were mostly derived from the secondary reactions (e.g., alkylation, transalkylation, dehydrogenation, and condensation) of monocyclic aromatics.46,47
The synergistic influence of microwaves and the presence of Ni in the phytocat increased the gas yield from 20 wt% to 27 wt%. Besides, the addition of Ni-phytocat also decreased the carbon residue yield to 14.5 wt%, as compared to 33–34.5 wt% for activated carbon or control phytocat. The presence of Ni-phytocat promoted the primary degradation of larger fragments into smaller fragments, which reduced the possibility of coke deposition caused by larger molecules. With the increase in consecutive microwave cycles, the oil yields increased, while gas and coke yields decreased (Fig. 6a). An increase in residence time also favored the cracking reactions, which eventually produced short chain hydrocarbons alongside monocyclic aromatics. The yield of bio-oil increased from 43–45% without Ni to 61% with the Ni-phytocat.
In comparison, two major technical challenges hinder the scale-up of conventional pyrolysis technology for use in de-polymerization of plastics. The first one is the production of high molecular weight, waxy hydrocarbons and high temperature (T > 700 °C) operation for de-polymerization into lighter compounds. Secondly, the conventional high temperature catalytic cracking leads to the deactivation by coking and poisoning of the catalyst. Therefore, active sites available to the incoming reactant molecules affect the product selectivity in a conventional process. Catalyst coking also reduces the useful lifetime of the catalyst and increases the overall operational cost.
After the catalytic de-polymerization, olefins from pyrolysis were largely converted to aromatics through dehydrogenation; consequently, the hydrogen proportion in the gas products using Ni-phytocat was higher than that from the control phytocat or activated carbon (Fig. 6b–h). However, a much lower yield of ethylene was observed using the Ni-phytocat. This decrease in ethylene could be ascribed to the Diels–Alder reactions promoted by the Ni active sites, which resulted in the consumption of ethylene (Fig. 6b). Increasing hydrogen yields are important as we move towards a hydrogen economy – the conversion of plastic waste to hydrogen in a process that does not consume virgin metals and is energy efficient, is very attractive.
Possible reaction mechanism for the accelerated de-polymerization of polyethylene using phytocat
Based on the yield distribution, and chemical compositions of the products, possible reaction pathways for the de-polymerization of PE using phytocat are proposed in Fig. 7. Broadly, large amounts PE-derived large molecular weight hydrocarbons from individual catalytic microwave pyrolysis were transformed into light hydrocarbons, which formed hydrocarbon pools inside the micropores and/or on the surface of phytocats and were then ultimately converted into aromatic hydrocarbons through oligomerization and aromatization over the active sites. | ||
| Fig. 7 Mechanistic pathway for accelerated de-polymerization of low-density polyethylene utilizing synergistic interactions of microwave and Ni-phytocat. | ||
Hydrogen transfer reaction from hydrogen-rich materials (polyolefin chains) to hydrogen-deficient materials (phytocat) occurred during polyethylene and phytocat's interaction. The dehydrogenation process was enhanced by the abstraction of hydrogen from hydrogen-enriched hydrocarbons like alkanes onto the carbon surface, and thus, the production of alkenes increased substantially.
It has been reported that pure carbon support was also effective in promoting the hydrogen transfer reaction and cyclization of a straight-chain intermediate.32 Here, we show that the dehydrogenation step in hydrogen transfer was enhanced by the abundant surface functional groups on the carbon surface such as carbonyl groups, whereas the step of hydrogen release was relatively limited due to the lack of metal sites in control phytocat, leaving more hydrogen atoms on the carbon surface available for the hydrogenation of hydrogen acceptors such as alkenes. The slight decrease in aromatic content was probably caused by the restrained hydrogen transfer reaction due to the lack of metal sites.
During the initiation stage, the main chain scission (generally random scission) is activated by the energy input of microwave, producing unstable long-chain hydrocarbon radicals. The addition of Ni phytocat lowers the degradation energy of C–C bond and thus boosts the chain scission reactions. Moreover, the required energy for cracking C–H bond is also reduced by NiO, resulting in the growing formation of carbenium ions.48 Similar to Diels–Alder reactions, cyclization and aromatization reactions will also contribute to the production of aromatics.48 In this regard, cyclization between the unsaturated chain ends and the chain ends with a radical will produce cyclohexane products. Further dehydrogenation of these products over phytocat results in the generation of aromatics. Besides, a chain-end radical could also terminate by recombining with another chain-end radical and thus produce a saturated and enlarged chain.
Further addition of Ni-phytocat will lead to abstraction of hydrogen from the hydrocarbon fragments and enhancement of the production of alkenes and H2. As a result, the yield of alkanes is inhibited. Among the alkenes, the increase in diene compounds is advantageous to the production of aromatics via Diels–Alder reactions over the phytocat or activated carbon. These reactions can explain the increase in aromatic components when Ni-phytocat is used in comparison to the control phytocat or activated carbon alone. Ni active sites form more short chain olefins by promoting β-scission reactions, and olefins aromatize on active sites to form aromatic hydrocarbons. In addition, Ni active sites can adsorb carbonaceous intermediates and activate C–H bonds and catalyze hydrogen transfer reactions between cycloalkanes and olefins, upgrading them to alkanes and aromatics.33,36 Additionally, the formed dienes can be oligomerized with alkenes to form cycloalkenes, and then dehydrogenate to form aromatics or reaction with butadiene and dehydrogenate to naphthalene (as observed with control phytocat and activated carbon).48
We also compared the quantitative distribution of products obtained from pyrolysis of LDPE using other catalysts (Table S7†). Past studies have used predominantly zeolite catalysts, which have high activity and stability but conversely are relatively costly, easy to coke, and deactivate. The liquid yield of products from catalytic pyrolysis of LDPE with these zeolite catalysts is relatively low, typically 30%.46,48,49 Compared with zeolite catalyst, activated carbon is cheaper, both acidity and porosity can be simply adjusted by changing activation conditions, so that the distribution of products can be tuned. The liquid yield obtained by pyrolysis of LDPE using activated carbon can reach 75.3%, which is much higher than that from using a zeolite catalyst.50–52 Therefore, it is more advantageous to use activated carbon support to convert plastic into high value chemicals. The presence of phytocat was conducive to altering product distribution and enhancing production of aromatic hydrocarbons, together with dehydrogenation of intermediate hydrocarbons (CnHm) to generate H2 and other valuable products especially at the low catalytic temperature (250 °C) as compared to conventional technique (T > 500 °C).11
Energy efficiency of the accelerated de-polymerization of polyethylene due to synergistic effect of microwaves and Ni-phytocat
Polyethylene is almost entirely transparent to incident microwaves, and thus the resulting temperature rise is very small, yielding a relatively large temperature difference with the catalyst.48 As a result, a heat flow from the catalyst surface to the PE is generated. At the optimum temperature of 250 °C, PE reacts on the surface of the catalyst for dehydrogenation. This localized heating effect reduces energy loss, increases product selectivity, and reduces side reactions. The de-polymerization products composition can be adjusted by changing the reaction time, temperature, and concentration of the catalyst. Microwave-assisted catalysis can speed up the reaction from a few hours to a few minutes, or even a few seconds; the short reaction time inhibits many side reactions, allowing for products to be obtained with high purity and yield. Microwave heating relies on electricity to produce microwaves, but the overall process overall has a high energy utilization efficiency.43,53Oils obtained from microwave pyrolysis are different from those from conventional pyrolysis. Particularly, the synergistic use of microwaves and Ni-phytocat establishes a balance between cracking and cyclization to improve selectivity towards aromatics.
The microwaves along with phytocat were able to produce the liquid compounds with narrow distribution of carbon numbers compared to the conventional thermal pyrolysis, which resulted in the broader range (predominantly poly-aromatics and aliphatics C>12). The lower operating temperature is also advantageous.
Under microwaves, Ni-phytocat increased the heating rate to 190–200 °C min−1, depending on the amount of catalyst added to de-polymerize LDPE at fixed power of 200 W. Phytocat favors selective reaction pathways by minimizing the overall energy requirement of decomposition (Fig. 8). Hence, the energy requirement can also be reduced apart from the production of value-added chemicals.
 | ||
| Fig. 8 (a) Schematic illustration of the processes to generate high value chemicals, (b) microwave energy consumption for accelerated de-polymerization of LDPE (to reach the set point of 250 °C) using varying catalyst to polymer ratio. | ||
Conclusions
The successful demonstration of accelerated de-polymerization process based on a combination of waste-sourced Ni-phytocat and microwaves is extremely promising in recovering H2 as well as valuable hydrocarbons and filamentous carbon from plastic waste. The catalytic effect of Ni-phytocat was mainly characterized by promoting the hydrogen transfer process, which increased the yield of short chain aliphatics and aromatics at the expense of high molecular weight aliphatics and poly-aromatics. Microwave-assisted accelerated de-polymerization of polyethylene under mild reaction conditions is a promising energy-saving approach toward plastic waste-to-valuable hydrocarbons. The synergy of the Ni-phytocat and microwave driven process allows the fine-tuning of activity and selectivity to highly desirable monocyclic aromatics and low molecular weight hydrocarbon products, and H2. Because the proposed catalyst is active in rapidly de-polymerizing of different types of plastics, including the least recycled polystyrene and low density polyethylene, pre-separation of waste feed-stocks may sometimes not be required.34 The active site of Ni-phytocat can adsorb carbonaceous intermediates and catalyze C–H bond breaking; it can also catalyze the β-scission reaction to form short-chain olefins, which then enter the bio-carbon micro-pores and are directly aromatized to form aromatic hydrocarbons. Meanwhile, the Ni active site can promote hydrogen transfer reactions between cycloalkanes and olefins to increase the proportion of alkanes and aromatics in the lower molecular weight range. As the amount of Ni in the phytocat increases, the percentage of short chain alkanes and monocyclic aromatics increases. The novelty of this work is that the biologically bound Ni driven sustainable de-polymerization process allows for product-tunable decomposition of polyethylene to rapidly (at least 40 times faster than the corresponding high temperature thermally driven reaction) produce high value low molecular weight hydrocarbons with no added H2 or artificially fabricated catalyst. Serpentine soils are present in many locations around the world, and phytomining technologies, using hyperaccumulator species, are being developed to harvest the Ni; the use of phytocat to de-polymerize polystyrene presents added value to this Ni-rich plant biomass.34 The process has the potential to upcycle a range of commercial plastics into monomers or high value chemical feedstock without employing either noble metals or H2 feeds as an economically viable and environmentally sustainable alternative to the current recycling methods. Indeed hydrogen becomes a major product in these reactions.Author contributions
Parul Johar: conceptualization, investigation, analysis, writing – original draft, review and editing, resources, funding acquisition. Elizabeth L. Rylott: resources, supervision, writing – review & editing. C. Robert McElroy: supervision. Avtar S. Matharu: supervision. James H. Clark: conceptualization, supervision, writing – review & editing, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the University of York (Wild Fund Platinum Scholarship) and the Association of Commonwealth Universities (ACU) Blue Charter fellowship. We also acknowledge the assistance of Dr Baptiste Laubie and Dr Guillaume Echevarria from University of Lorraine in the field collection of hyperaccumulator species. We are grateful to receive the technical support from Mr Karl Heaton, Dr Jon Barnard and Dr Leonardo Lari.References
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6 CrossRef
.
- K. Hu, Y. Yang, Y. Wang, X. Duan and S. Wang, Chem. Catal., 2022, 2, 724–761 CrossRef
- A. Stubbins, K. L. Law, S. E. Muñoz, T. S. Bianchi and L. Zhu, Science, 2021, 373(6550), 51–55 CrossRef CAS PubMed
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef PubMed
- H. Inderthal, S. L. Tai and S. T. L. Harrison, Trends Biotechnol., 2021, 39, 12–23 CrossRef CAS PubMed
- T. M. Adyel, Science, 2020, 369, 1314–1315 CrossRef PubMed
- B. C. Vance, P. A. Kots, C. Wang, Z. R. Hinton, C. M. Quinn, T. H. Epps, L. T. J. Korley and D. G. Vlachos, Appl. Catal., B, 2021, 299, 120483 CrossRef CAS
- X. Zhao, B. Boruah, K. F. Chin, M. Đokić, J. M. Modak and H. Sen Soo, Adv. Mater., 2021, 2100843 Search PubMed
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. van Harmelen, P. de Wild, G. P. van Der Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed
- K. Sarah and R. Gloria, Science, 2021, 373, 49–50 CrossRef PubMed
- D. Yao, H. Yang, Q. Hu, Y. Chen, H. Chen and P. T. Williams, Appl. Catal., B, 2021, 280, 119413 CrossRef CAS
- S. Liu, P. A. Kots, B. C. Vance, D. Andrew and D. G. Vlachos, Sci. Adv., 2022, 7, eabf8283 CrossRef PubMed
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 4659–4679 CrossRef CAS
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri, J. Dilworth, J. Thomas, T. Xiao and P. Edwards, Nat. Catal., 2020, 3, 902–912 CrossRef CAS
- L. J. Sonter, M. C. Dade, J. E. M. Watson and R. K. Valenta, Nat. Commun., 2020, 11, 4174 CrossRef CAS PubMed
- S. Luckeneder, S. Giljum, A. Schaffartzik, V. Maus and M. Tost, Global Environmental Change, 2021, 69, 102303 CrossRef
- B. K. Sovacool, S. H. Ali, B. Morgan, R. Ben, N. Benoit, O. Julia and M. Dustin, Science, 2020, 367, 30–33 CrossRef CAS PubMed
- D. Hou, D. O'Connor, A. D. Igalavithana, D. S. Alessi, J. Luo, D. C. W. Tsang, D. L. Sparks, Y. Yamauchi, J. Rinklebe and Y. S. Ok, Nat. Rev. Earth Environ., 2020, 1, 366–381 CrossRef
- V. Maus, S. Giljum, J. Gutschlhofer, D. M. da Silva, M. Probst, S. L. B. Gass, S. Luckeneder, M. Lieber and I. McCallum, Sci. Data, 2020, 7, 289 CrossRef PubMed
- S. M. Jowitt, G. M. Mudd and J. F. H. Thompson, Communications Earth & Environment, 2020, 1, 13 Search PubMed
- A. M. Abioye and F. N. Ani, Renewable Sustainable Energy Rev., 2015, 52, 1282–1293 CrossRef CAS
- P. N. Nkrumah, G. Echevarria, P. D. Erskine and A. van der Ent, Sci. Total Environ., 2022, 827, 154092 CrossRef CAS PubMed
-
R. L. Chaney, A. J. M. Baker and J. L. Morel, in Agromining: farming for metals, Springer, 2021, pp. 1–22 Search PubMed
- L. Van der Pas and R. A. Ingle, Plants, 2019, 8, 11 CrossRef CAS PubMed
- L. Wang, D. Hou, Z. Shen, J. Zhu, X. Jia, Y. S. Ok, F. M. G. Tack and J. Rinklebe, Crit. Rev. Environ. Sci. Technol., 2020, 50, 2724–2774 CrossRef
- J. Luo, S. Qi, X. W. S. Gu, J. Wang and X. Xie, J. Cleaner Prod., 2016, 119, 25–31 CrossRef CAS
- D. Tőzsér, S. Harangi, E. Baranyai, G. Lakatos, Z. Fülöp, B. Tóthmérész and E. Simon, Environ. Sci. Pollut. Res., 2018, 25, 3275–3290 CrossRef PubMed
- B.-H. Cheng, B.-C. Huang, R. Zhang, Y.-L. Chen, S.-F. Jiang, Y. Lu, X.-S. Zhang, H. Jiang and H.-Q. Yu, Sci. Adv., 2020, 6(1), 0748 Search PubMed
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick Jr, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed
- J. M. Clomburg, A. M. Crumbley and R. Gonzalez, Science, 2017, 355, aag0804 CrossRef PubMed
- R. P. Lopes and D. Astruc, Coord. Chem. Rev., 2021, 426, 213585 CrossRef
- C. Wang, H. Han, Y. Wu and D. Astruc, Coord. Chem. Rev., 2022, 458, 214422 CrossRef CAS
- J. Jia, A. Veksha, T.-T. Lim and G. Lisak, J. Cleaner Prod., 2020, 258, 120633 CrossRef CAS
- P. Johar, E. L. Rylott, C. R. McElroy, A. S. Matharu and J. H. Clark, Green Chem., 2021, 23, 808–814 RSC
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang and J. Liu, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed
- J. C. Acomb, C. Wu and P. T. Williams, Appl. Catal., B, 2016, 180, 497–510 CrossRef CAS
- E. F. Kukovitskii, L. A. Chernozatonskii, S. G. L'vov and N. N. Mel'Nik, Chem. Phys. Lett., 1997, 266, 323–328 CrossRef CAS
- A. van der Ent, D. L. Callahan, B. N. Noller, J. Mesjasz-Przybylowicz, W. J. Przybylowicz, A. Barnabas and H. H. Harris, Sci. Rep., 2017, 7, 41861 CrossRef CAS PubMed
- B. H. Lipshutz, Adv. Synth. Catal., 2001, 343, 313–326 CrossRef CAS
- C. J. Powell, Appl.
Surf. Sci., 1995, 89, 141–149 CrossRef CAS
- W. H. Qi and M. P. Wang, J. Nanopart. Res., 2005, 7, 51–57 CrossRef CAS
- R. G. Nuzzo, L. H. Dubois, N. E. Bowles and M. A. Trecoske, J. Catal., 1984, 85, 267–271 CrossRef CAS
- J. Lin, S. Sun, D. Xu, C. Cui, R. Ma, J. Luo, L. Fang and H. Li, Chem. Eng. J., 2021, 132195 Search PubMed
- L. Fan, Y. Zhang, S. Liu, N. Zhou, P. Chen, Y. Liu, Y. Wang, P. Peng, Y. Cheng and M. Addy, Energy Convers. Manage., 2017, 149, 432–441 CrossRef CAS
- K. Hernadi, A. Fonseca, J. B. Nagy, A. Siska and I. Kiricsi, Appl. Catal., A, 2000, 199, 245–255 CrossRef CAS
- L. Dai, N. Zhou, H. Li, Y. Wang, Y. Liu, K. Cobb, Y. Cheng, H. Lei, P. Chen and R. Ruan, Sci. Total Environ., 2021, 771, 144995 CrossRef CAS PubMed
- O. Dogu, M. Pelucchi, R. Van de Vijver, P. H. M. Van Steenberge, D. R. D'hooge, A. Cuoci, M. Mehl, A. Frassoldati, T. Faravelli and K. M. Van Geem, Prog. Energy Combust. Sci., 2021, 84, 100901 CrossRef
- K. Ding, S. Liu, Y. Huang, S. Liu, N. Zhou, P. Peng, Y. Wang, P. Chen and R. Ruan, Energy Convers. Manage., 2019, 196, 1316–1325 CrossRef CAS
- J. M. Escola, J. Aguado, D. P. Serrano, A. García, A. Peral, L. Briones, R. Calvo and E. Fernandez, Appl. Catal., B, 2011, 106, 405–415 CrossRef CAS
- Y. Uemichi, Y. Makino and T. Kanazuka, J. Anal. Appl. Pyrolysis, 1989, 14, 331–344 CrossRef CAS
- A. D. Russell, E. I. Antreou, S. S. Lam, C. Ludlow-Palafox and H. A. Chase, RSC Adv., 2012, 2, 6756–6760 RSC
- Y. Uemichi, Y. Kashiwaya, A. Ayame and H. Kanoh, Chem. Lett., 1984, 13, 41–44 CrossRef
- D. Voiry, J. Yang, J. Kupferberg, R. Fullon, C. Lee, H. Y. Jeong, H. S. Shin and M. Chhowalla, Science, 2016, 353, 1413–1416 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2su00001f |
| This journal is © The Royal Society of Chemistry 2023 |