
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic understanding of microstructure formation during synthesis of metal oxide/carbon nanocomposites†
Mennatalla
Elmanzalawy
 ab,
Alessandro
Innocenti
ab,
Maider
Zarrabeitia
ab,
Nicolas J.
Peter
c,
Stefano
Passerini
abd,
Veronica
Augustyn
e and
Simon
Fleischmann
*ab
ab,
Alessandro
Innocenti
ab,
Maider
Zarrabeitia
ab,
Nicolas J.
Peter
c,
Stefano
Passerini
abd,
Veronica
Augustyn
e and
Simon
Fleischmann
*ab
aHelmholtz Institute Ulm (HIU), 89081 Ulm, Germany
bKarlsruhe Institute of Technology (KIT), 76021 Karlsruhe, Germany. E-mail: simon.fleischmann@kit.edu
cInstitute of Energy and Climate Research (IEK-2), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
dDepartment of Chemistry, Sapienza University of Rome, P. A. Moro 5, 00185 Rome, Italy
eDepartment of Materials Science & Engineering, North Carolina State University, Raleigh, NC 27606, USA
First published on 13th June 2023
Abstract
Nanocomposite materials consisting of metal oxide and carbon are of interest as electrode materials for both high rate intercalation-type and high capacity conversion-type charge storage processes. Facile synthesis processes like the pyrolysis of an organic carbon-source can yield a well-dispersed carbon phase within the metal oxide structure. Detailed understanding of the carbon formation process is required to tailor the resulting material microstructure. Herein, both the formation and the final microstructure of a molybdenum oxide/carbon nanocomposite are studied in detail. Octylamine assembled in the interlayer space of layered MoO3 serves as a carbon source. The structural changes during pyrolysis are characterized using a combination of in situ heating X-ray diffraction with simultaneous FTIR- and mass spectroscopy-coupled thermogravimetric analysis experiments. These reveal mobility and partial desorption of octylamine and interlayer water at low temperatures, octylamine decomposition and loss of long-range order at intermediate temperatures, and carbothermic reduction of molybdenum oxide at high temperatures during pyrolysis. The resulting nanocomposite mainly contains nanocrystalline MoO2 domains surrounded by a well-dispersed carbon phase, as observed with scanning transmission electron microscopy of focus-ion beam prepared cross-sectional lamellae. The electrochemical behavior is evaluated in organic, lithium-containing electrolyte for both intercalation and conversion-type reactions, showing good intercalation kinetics and a high first cycle efficiency for the conversion-type reaction.
1. Introduction
Advancing state-of-the-art electrochemical energy storage devices requires improvements in both specific power and energy.1,2 High power capability requires electrodes with both fast ionic and electronic transport properties, necessitating engineering across length scales, from the atomic scale to the macroscale. It is necessary to consider not only the active material properties (e.g., composition, electronic conductivity, ion diffusivity, and particle size), but also the particle-to-particle interactions that contribute to the overall electrode performance. For example, Luo et al. found that V2O5 electrodes with micron-scale platelets showed greater heterogeneity in the spatial variation of the state of charge than those with nanoscale platelets.3 Park et al. further showed that phase separation observed in layered metal oxides that undergo a solid-solution ion insertion is due to electro-autocatalytic reactions between particles with different states of charge.4 These recent results highlight the importance of engineering the particle-to-particle interactions and optimizing the electrode microstructure in energy storage materials.Transition metal oxides (TMOs) can electrochemically react with Li+via intercalation5 and conversion reactions.6 The exact mechanism depends on the extent of electron transfer which determines the degree of structural distortion, with intercalation reactions favored for up to ∼1 Li+/e− per transition metal and conversion reactions for >1 Li+/e− per transition metal. Many TMOs are semiconductors or insulators and require the addition of conductive carbon to the electrode for high-rate operation. Electrochemical conversion-type reactions in TMOs are further associated with large volumetric changes, leading to reduced efficiency and cycle life. One strategy to alleviate this issue is to confine TMO particles in a carbonaceous matrix to buffer volumetric expansion.7,8 Both functionalities – improved electronic percolation and volumetric buffering – can be achieved with TMO/carbon (nano)composite electrodes.9,10
Among many synthesis approaches for TMO/carbon nanocomposites, one of the most widely adopted is the addition of an organic species to the TMO particles and its subsequent carbonization.8,11 Recent works have shown that the carbonization of amine molecules added into the interlayer of layered TMOs can produce TMO/C nanocomposites with a well-dispersed carbon phase.8,12–16 In some cases, it was described that this synthesis route (and similar ones using different sources of carbon17) yielded heterostructured TMO/C12,16 (and transition metal dichalcogenide/carbon18) nanocomposites, where layers of dissimilar phases alternate. This particular microstructure results in abundant heterointerfaces which may synergistically combine beneficial electrochemical properties of the individual building blocks (redox-activity, electrical conductivity).19,20
Comparison of these studies8,12–16 reveals variations in structural properties of the resulting TMO/C materials, especially in terms of nanocomposite microstructure (i.e., phase distribution), degree of TMO reduction, and carbon hybridization. A more generalized understanding of the synthesis process is required to be able to precisely tune the resulting nanocomposite properties. Therefore, it is important to develop mechanistic insights into the microstructure evolution during carbonization. Understanding the process via in situ characterization methods, as well as microscopic analysis representative of the bulk volume of the nanocomposites can give valuable insights into the microstructure evolution. This is essential to further optimize nanocomposite electrode materials for electrochemical energy storage applications and beyond.
In this study, the microstructural changes during pyrolysis of an octylamine-functionalized molybdenum oxide are systematically analyzed. Molybdenum trioxide (α-MoO3) consists of layers of corner- and edge-sharing distorted MoO6-octahedra in an orthorhombic crystal structure, where the individual layers are held together by van der Waals forces.21 MoO3 is chosen for this study because organic molecules such as alkylamines can assemble in its interlayer space, serving as a confined carbon source.22 The resulting nanocomposite derived from pyrolysis has relevance for electrochemical applications, because molybdenum oxides can undergo intercalation and conversion reactions with lithium.23,24 The octylamine (OA) molecule is chosen for its model character, as alkylamines are linear organic molecules which are widely available with variable carbon chain length, allowing to customize the carbon-source molecule.
We employ a combination of in situ characterization and X-ray spectroscopy and electron microscopy techniques to analyze the structural and chemical changes of the molybdenum oxide/octylamine material during pyrolysis. We show that carbon formation occurs predominantly within the micron-sized product aggregates and coincides with molybdenum reduction that is accompanied by CO2 & CO release. Simultaneously, nanosized oxide grains in the range of 5–10 nm are formed within the aggregates that exhibit local variations in oxygen and carbon content. Scanning transmission electron microscopy (STEM) imaging of specimen prepared via focused ion beam (FIB) reveals the microstructure of the materials representative of their bulk volume. The obtained MoOx–C nanocomposite exhibits improved kinetics for electrochemical intercalation and an enhanced initial coulombic efficiency for the conversion reaction, depending on the employed potential range, compared to pristine molybdenum oxide samples.
2. Experimental section
2.1 Materials synthesis
Molybdenum oxide dihydrate (MoO3·2H2O) was synthesized via a modified Freedman method.25 50 grams of sodium molybdate dihydrate Na2MoO4·2H2O (VWR International) were dissolved in 100 mL of deionized H2O and slowly added to 300 mL of 5 mol L−1 HNO3 (VWR International) through a burette under continuous stirring. The solution was stirred at room temperature for about 2 weeks, then the stirring was stopped for another week, until a significant amount of yellow precipitate was visible. The precipitated MoO3·2H2O was recovered via filtration of the solution through a Büchner funnel (reaction yield around 20 g), followed by subsequent washing with about 3 L of deionized H2O and drying at room temperature for 1 week.Octylamine-functionalized molybdenum oxide (MoOx–OA) was synthesized by dispersing 2 grams of MoO3·2H2O in 57 mL of ethanol under stirring, before adding 1.43 grams (1.825 mL) of octylamine (Sigma Aldrich) to obtain a molar ratio of Mo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :OA of 1:1. The solution was stirred for 3 days at room temperature until a white precipitate formed. The precipitate was recovered by washing and centrifugation with ethanol at 2000 rpm for 5 minutes for 3 times and drying at 80 °C overnight. While in this study, a molar ratio of Mo:OA of 1:1 was chosen, variation of the ratio yields structures with similar crystallography (Fig. S1†).
:OA of 1:1. The solution was stirred for 3 days at room temperature until a white precipitate formed. The precipitate was recovered by washing and centrifugation with ethanol at 2000 rpm for 5 minutes for 3 times and drying at 80 °C overnight. While in this study, a molar ratio of Mo:OA of 1:1 was chosen, variation of the ratio yields structures with similar crystallography (Fig. S1†).
Octylamine-derived molybdenum oxide–carbon composite (MoOx–C) was synthesized by pyrolysis of MoOx–OA in a tube furnace under flowing argon atmosphere at 700 °C for 2 hours, with a heating rate of 5 K min−1, then allowed to cool to room temperature.
In order to compare the electrochemical properties to conventional molybdenum oxide, MoO3·2H2O underwent the same thermal treatment described above, but without assembly of OA in the interlayer (i.e., in the absence of the carbon source). This led to a partially reduced form of molybdenum oxide (MoO3−x).
2.2 Structural and morphological characterization
Powder X-ray diffraction (XRD) patterns of all the samples were obtained using a Bruker D8 Advance equipped with a Cu Kα1 radiation source (λ = 0.15406 nm). The patterns were recorded in Bragg–Brentano mode in the range between 2° and 60° 2θ with 0.02° step size. X-ray photoelectron spectroscopy (XPS) of MoOx–C powder was performed in a fixed analyzer transmission mode, using a monochromatic Al Kα (hν = 1.487 eV) X-ray and a Phoibos 150 XPS spectrometer (Surface Concept) equipped with a micro-channel plate and Delay Line Detector (DLD). High resolution Mo 2p, C 1s, and O 1s regions were acquired with an X-ray power source of 200 W (12 kV), pass energy of 30 eV, and 0.1 eV energy steps. The spectra analysis was carried out by CasaXPS software, using a nonlinear Shirley-type background and 70% Gaussian and 30% Lorentzian profile functions.26 Scanning electron microscopy (SEM) imaging was performed using a ZEISS Crossbeam 340 electron microscope operated at 5 kV and working distance of 7.7 mm. Raman spectra were obtained using a Renishaw inVia Raman spectrometer equipped with an Nd:YAG laser with a wavelength of 532 nm using 0.25 to 5 mW laser power. In situ heating XRD was carried out using a PANalytical Empyrean diffractometer equipped with a HTK 1200N heating stage (Anton Paar) operating under nitrogen atmosphere and replicating the heat treatment conditions during pyrolysis synthesis, while recording a diffractogram every 6 minutes. Thermogravimetric analysis coupled with simultaneous mass spectroscopy (TGA-MS) and Fourier transform infra-red spectrometry (TGA-FTIR) of the pyrolysis gas was performed using a NETZSCH TG 209 F1 Libra thermal analyzer under Ar flow at a heating rate of 10 K min−1 from 40 to 700 °C in Al2O3 crucibles loaded with ca. 20 mg of material. MS data were acquired with a QMS 403 Aëolos Quadro mass spectrometer in the 10–300 m/z range, while FTIR spectra were recorded using a Bruker Invenio spectrophotometer in absorption mode, in the range of 4500–650 cm−1 and at a resolution of 2 cm−1. Scanning transmission electron microscopy (STEM) was carried out at 200 kV and with a semi-convergence angle of 24.7 mrad in an aberration-corrected Titan G2 60-200 CREWLEY microscope (ThermoFisher Scientific, formerly FEI, Netherlands) equipped with a high-brightness field emission electron gun and a Super-X energy-dispersive X-ray spectroscopy (EDS) system. High-angle annular dark-field (HAADF) images were acquired to make use of the associated Z-number contrast with a semi-collection angle of 69–200 mrad.Samples for STEM were prepared using a dual-beam Helios NanoLab400S (ThermoFisher Scientific, formerly FEI, Netherlands) focused ion beam (FIB). Powder particles were fixated on a conventional scanning electron microscope stub using glue. Afterwards, regions of high particle density were identified, and Pt was deposited on top to make a good connection between the particles. A conventional lift-out technique was applied to transfer the thick TEM lamella onto a TEM grid. Before thinning the lamella, the side of it was again glued with Pt to maintain a good particle connection during thinning. Finally, the sample was thinned down to electron transparency with a final thinning step consisting of 5 kV ion polishing to reduce beam damage and Ga contamination as much as possible.
2.3 Electrode and electrochemical cell preparation
The electrode slurries were prepared by mixing 80 wt% active material and 10 wt% conductive carbon black (Super C65, C-NERGY) in an agate mortar, followed by the addition of 10 wt% polyvinylidene fluoride (PVDF, Solef 6020, Arkema Group) in N-methyl-2-pyrrolidone solvent (NMP, anhydrous, Sigma-Aldrich, 2 wt% of PVDF in NMP). The slurries were homogenized using a planetary mixer (Thinky, ARE-250) and then cast onto aluminum and copper foil current collectors (Wellcos Corporation) using a laboratory scale doctor blade (wet film thickness set to 60 μm). The cast electrodes were further dried in an oven set at 80 °C overnight. Electrode discs 12 mm in diameter were then punched with active material loading ranging between 0.7 to 0.9 mg cm−2, and dry coating thickness ranging between 12.0 to 13.8 μm.2.4 Electrochemical characterization
All cyclic voltammetry and galvanostatic cycling measurements were done in 2-electrode configuration coin cells, where 12 mm diameter metallic Li discs were used as a counter and reference electrode and glass microfiber (Whatman grade GF/A) as separator. Standard LP30 (1 M LiPF6 in 1:1 volume mixture of ethylene carbonate and dimethyl carbonate, Solvionic) was used as the electrolyte. Electrochemical measurements were performed in temperature-controlled chambers (Binder) at 20 °C using a potentiostat/galvanostat (Bio-Logic VMP3). The potential window was between 1.1 and 3.1 V vs. Li+/Li (for MoOx–C) or 1.5 and 3.5 V vs. Li+/Li (for MoO3−x) to study the Li+ intercalation reaction. To study the conversion reactions at more negative potentials, the electrodes were cycled between 0.01 and 3 V vs. Li+/Li.
Electrochemical impedance spectroscopy (EIS) measurements were performed in custom-made 3-electrode cells, where the components are sandwiched between two spring-loaded titanium pistons, and Li foils were used as separate counter and reference electrodes. Detailed description of the cell is found in ref. 27. Impedance spectra were recorded under open circuit potential conditions with sinusoidal potential amplitude of 10 mV, in the frequency range between 200 kHz and 100 mHz.
3. Results and discussion
3.1 Structural characterization
The synthesis of molybdenum oxide dihydrate (MoO3·2H2O, also referred to as molybdic acid) was performed via the method described by Freedman25 and the chemical self-assembly reaction of octylamine (OA) in the interlayer of molybdenum oxide was achieved by a wet chemistry synthesis to obtain MoOx–OA. This material was subsequently pyrolyzed at 700 °C for 2 hours under argon flow to form an OA-derived carbonaceous phase. The final product of this synthesis is a nanocomposite MoOx–C, as described in the Experimental section and shown in the schematic in Fig. 1A. In the following, we provide mechanistic insight into the synthesis/pyrolysis process and final product structure, pursuant of the overarching goal to understand where a carbonaceous phase forms and how the microstructure evolves in the molybdenum oxide/carbon nanocomposite material. In particular, we aim to answer whether a heterostructured microstructure forms, where in an ideal case, 2D layers of molybdenum oxide and carbon alternate throughout the bulk volume of the structure.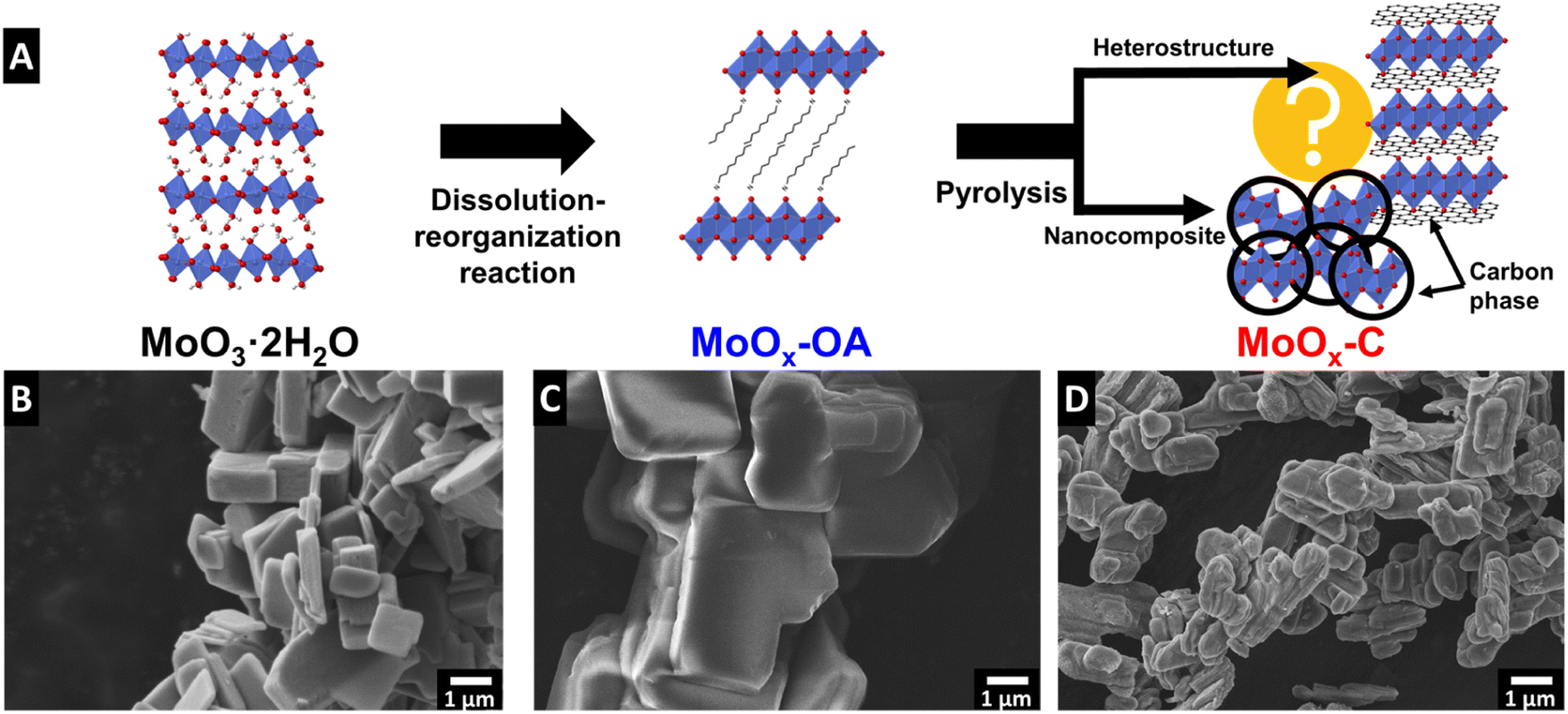 | ||
| Fig. 1 (A) Schematic illustration of the synthesis process: octylamine is assembled in the MoO3 interlayer space via a dissolution–reorganization reaction; subsequent pyrolysis yields the carbon-containing nanocomposite. SEM images of (B) molybdenum oxide dihydrate (MoO3·2H2O), (C) MoOx–OA, and (D) MoOx–C after pyrolysis in argon at 700 °C. | ||
The reaction mechanism of alkylamines with molybdic acids can be likened to that in isostructural tungstic acids, which has been described in literature to occur via a dissolution–reorganization mechanism that leads to a change in the morphology and particle size.28,29 Scanning electron micrographs of MoO3·2H2O and MoOx–OA (Fig. 1B and C) show an increased particle size after reaction with octylamine. Therefore, we hypothesize that, like in tungstic acids,28 the alkaline conditions upon octylamine addition lead to an initial dissolution of MoO3·2H2O and a subsequent reassembly of the structure with octylamine in the interlayer space of MoO3.
Subsequent pyrolysis of MoOx–OA to form MoOx–C leads to a change in particle morphology (Fig. 1D), most notably, a reduced average particle size from 7.0 ± 2.3 to 1.7 ± 0.6 μm (Fig. S2†), indicating significant densification after pyrolysis.
The Raman spectrum of MoO3·2H2O (Fig. 2A) shows the typical Raman signals of α-MoO3·2H2O, where the high intensity peaks at 934 cm−1 and 727 cm−1 are assigned to the stretching modes of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Mo and O–Mo2, respectively.30 Peaks in lower wavenumber ranges, from 400 to 200 cm−1, are assigned to lattice and deformation modes.30 After assembly of OA in the interlayer, a sharp peak appears at 896 cm−1, corresponding to Mo–O stretching in [MoO4]2−,31 suggesting the formation of molybdate as a result of OA functionalization.32 The other strong peak at 819 cm−1 is characteristic of the doubly-coordinated oxygen (O–Mo2) stretching mode.33 The Raman peak around 2882 cm−1 could be assigned to the asymmetric stretching of the methylene (CH2) group in octylamine.34 The Raman spectrum of the pyrolyzed MoOx–C shows characteristic peaks for MoO2 according to previous reports (note that a reduced laser power has to be chosen to avoid (re-)oxidation of MoO2 to MoO3 during Raman measurement35).36 This suggests that the α-MoO3 is reduced to MoO2 during pyrolysis. Successful carbon formation is confirmed by two peaks appearing in MoOx–C at 1604 and 1357 cm−1, corresponding to bond stretching of pairs of sp2-carbon (G-mode) and defect-induced breathing modes of sp2-carbon (D-mode), respectively.37 In the 2000 to 3000 cm−1 range, the absence of the G′ band, also called the 2D band, indicates that the formed carbon is highly defective.38
Mo and O–Mo2, respectively.30 Peaks in lower wavenumber ranges, from 400 to 200 cm−1, are assigned to lattice and deformation modes.30 After assembly of OA in the interlayer, a sharp peak appears at 896 cm−1, corresponding to Mo–O stretching in [MoO4]2−,31 suggesting the formation of molybdate as a result of OA functionalization.32 The other strong peak at 819 cm−1 is characteristic of the doubly-coordinated oxygen (O–Mo2) stretching mode.33 The Raman peak around 2882 cm−1 could be assigned to the asymmetric stretching of the methylene (CH2) group in octylamine.34 The Raman spectrum of the pyrolyzed MoOx–C shows characteristic peaks for MoO2 according to previous reports (note that a reduced laser power has to be chosen to avoid (re-)oxidation of MoO2 to MoO3 during Raman measurement35).36 This suggests that the α-MoO3 is reduced to MoO2 during pyrolysis. Successful carbon formation is confirmed by two peaks appearing in MoOx–C at 1604 and 1357 cm−1, corresponding to bond stretching of pairs of sp2-carbon (G-mode) and defect-induced breathing modes of sp2-carbon (D-mode), respectively.37 In the 2000 to 3000 cm−1 range, the absence of the G′ band, also called the 2D band, indicates that the formed carbon is highly defective.38
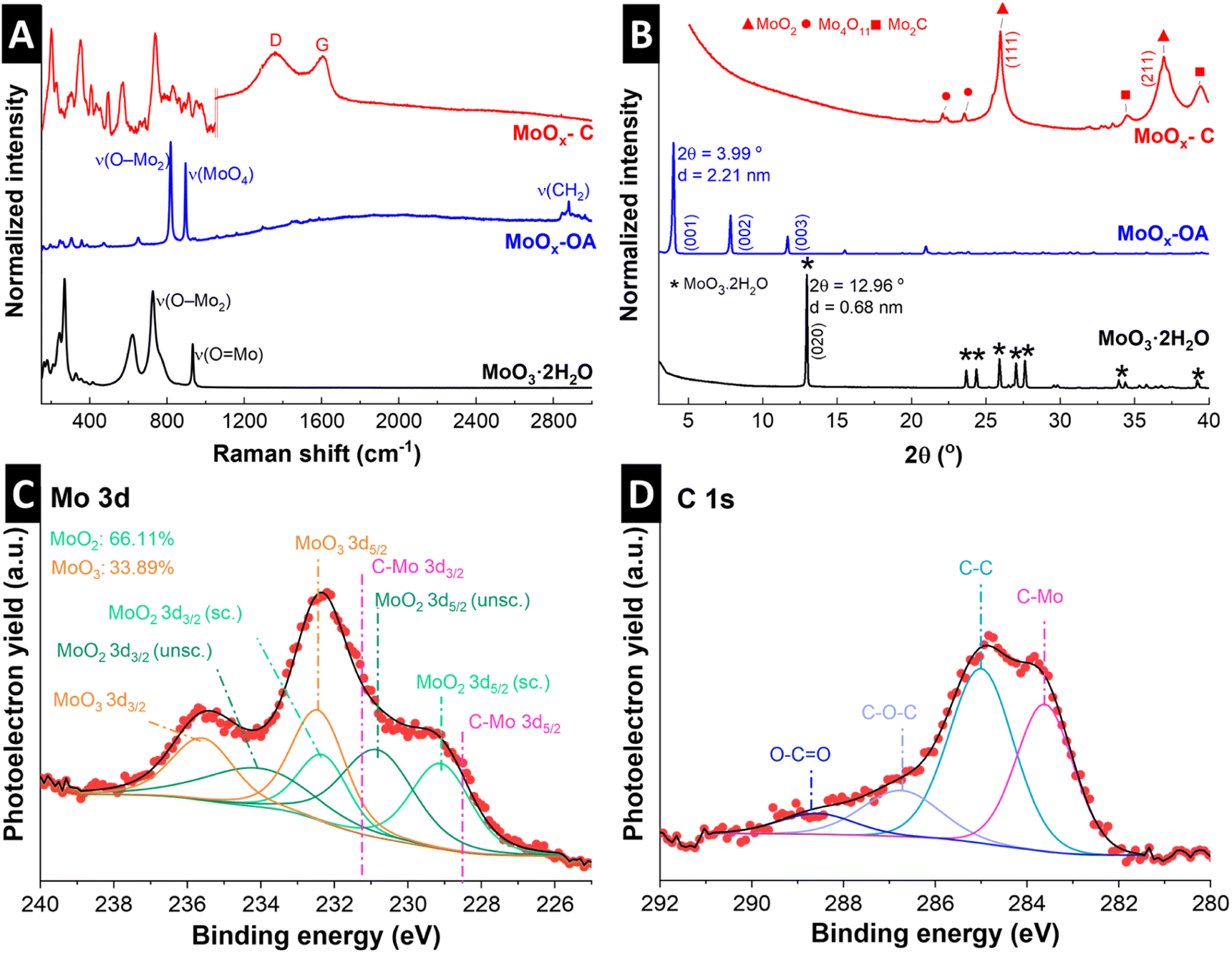 | ||
| Fig. 2 Structural characterization of hydrous molybdenum oxide (MoO3·2H2O), molybdenum oxide with octylamines assembled in the interlayer space (MoOx–OA) and pyrolyzed MoOx–C: (A) Raman spectra of all samples. Raman spectrum of MoOx–C consists of two individual measurements with different laser power: 0.25 mW between 100–1050 cm−1 to avoid oxidation of MoO2 to MoO3; 5 mW between 1050–3000 cm−1 for better resolution of carbon signals (individual full spectra given in Fig. S4†). (B) X-ray diffractograms with referenced peak positions of MoO3·2H2O (PDF 00-016-0497, asterisk), monoclinic MoO2 (PDF 00-032-0671, triangle), Mo2C (PDF 00-011-0680, square), and Mo4O11 (PDF 00-005-0337, circle). (C) Mo 3d, and (D) C 1s X-ray photoelectron spectrum regions of pristine powder MoOx–C. | ||
Fig. 2B shows powder XRD patterns of MoO3·2H2O, MoOx–OA, and MoOx–C. Diffraction peaks of the as-synthesized molybdic acid are referenced to MoO3·2H2O according to PDF 00-016-0497. Upon assembly of OA in the interlayer, a shift of the (020) peak at 12.96° to 3.99° 2θ is observed, corresponding to an expansion of the interlayer, with the d-spacing increasing from the initial 0.68 nm in MoO3·2H2O to 2.21 nm in MoOx–OA. Since the octylamine molecule has a chain length of ca. 1.2 nm,39 the large increase in interlayer spacing in MoOx–OA suggests a tilted bilayer arrangement of OA molecules between the MoOx sheets.22
After pyrolysis, the XRD pattern of MoOx–C shows peaks matching with monoclinic MoO2 reflections according to PDF 00-032-0671, in agreement with Raman measurements. Furthermore, low-intensity signals at ca. 34° 2θ and 39.4° 2θ suggest that traces of Mo2C and Mo4O11 were also formed, according to PDF 00-011-0680 and PDF 00-005-0337, respectively.
The oxidation states of Mo in the resulting nanocomposite MoOx–C were investigated by means of XPS. The analysis of Mo 3d is rather complex, and the accurate interpretation is often not well reported. In this case, the Mo 3d (Fig. 2C) region was analyzed following Scanlon et al. work, in which the Mo(IV)O2 consisted of two doublets due to the presence of both screened and unscreened electronic environment of Mo (for more detail see ref. 40).40 The Mo 3d region indicates the presence of Mo(IV) and Mo(VI) on the MoOx–C, in agreement with XRD. The formation of the latter species is highly common on Mo(IV) oxides.41 Nevertheless, the MoOx–C consists mainly of MoO2 (66% vs. 33% of Mo(VI)), as also shown in the O 1s spectrum (Fig. S3†). In addition, the C 1s region (Fig. 2D) displays the presence of Mo2C and C–C, C–O, and O–CO species, at 283.6, 285.0, and 286.7 and 288.6 eV in agreement with Raman and XRD.42,43
To understand the structural evolution of the material during the pyrolysis, we combined in situ heating XRD of MoOx–OA in an inert atmosphere (Fig. 3A and B) with thermogravimetric analysis with simultaneous mass spectroscopy (TGA-MS, Fig. 3C) and Fourier-transform infrared spectroscopy (TGA-FTIR, Fig. 3D). The results indicate that MoOx–OA undergoes three major structural transitions during pyrolysis, which can be conclusively explained in combination with the analysis of the evolving gaseous products via MS and FTIR at different temperatures:
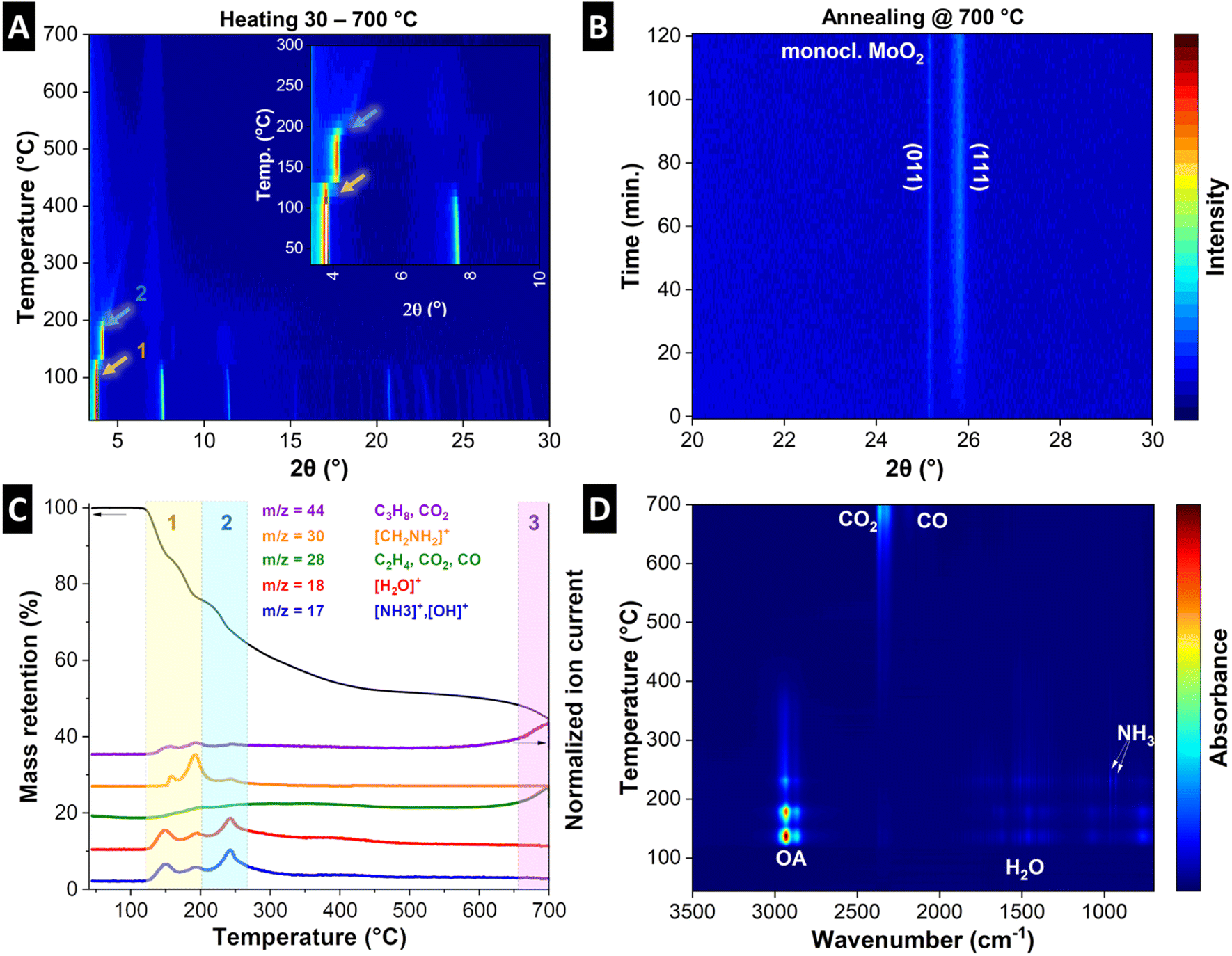 | ||
| Fig. 3 (A) In situ heating XRD patterns of MoOx–OA during the heating step of pyrolysis, with inset showing magnified view of the 2θ range 2–10° and temperature range 30–300 °C, (B) in situ heating XRD patterns of MoOx–OA during the two-hour annealing step. (C) Thermogravimetric analysis with mass spectroscopy of MoOx–OA during the heating step of pyrolysis, detecting gaseous decomposition products with various mass-to-charge ratios (m/z). Three distinct processes occurring during pyrolysis are graphically indicated by colored mass loss regions (yellow, blue, purple), also highlighted by the arrows in the in situ heating XRD diffractograms. (D) TGA with FTIR analysis of gaseous decomposition products during the heating step of pyrolysis. | ||
Stage 1: a sudden shift of the diffraction peak at ca. 3.8° 2θ, representative of the interlayer spacing, to a higher diffraction angle (ca. 4.1° 2θ) can be observed at a temperature of ca. 120 °C, indicating an initial shrinking of the interlayer spacing. In the same temperature range (120–200 °C), TGA reveals a two-step mass loss of around 24 wt%, coinciding with the evolution of gaseous species. MS signals in profiles of five mass-to-charge ratio (m/z) signals are detected, namely m/z = 17, 18, 28, 30, and 44. The signals of m/z = 17 (OH+) and 18 (H2O+) suggest the desorption of surface and loosely-bound interlayer water molecules.44 The m/z = 17 signal can also be assigned to ammonia (NH3+), which can result from the decomposition of the OA molecule. The peaks in the profile of m/z = 30 can be assigned to the base peak ion [CH2NH2]+, characteristic of β-cleavage of OA molecule.45,46 Finally, the signal at m/z = 28 can be attributed to hydrocarbons, CO2, or N2, and m/z = 44 can be attributed to hydrocarbons (C3H8) or CO2.44 FTIR results in Fig. 3D confirm the evolution of mainly molecular OA and water, and small traces of CO2 and NH3 as likely OA decomposition product in this temperature range (see the selected FTIR spectra of the pyrolysis gas compared with the typical FTIR spectra of OA, water, NH3, and CO2 in Fig. S5A and B†).47 Hence it can be concluded that loss of loosely bonded OA and water likely by evaporation are the main causes of initial interlayer shrinking, while a first onset of OA decomposition can be detected.
Stage 2: above 200 °C up to around 350 °C, a continuous shrinking of the interlayer spacing to about 1.7 nm (ca. 5° 2θ) is observed with the intensity strongly reducing before fully disappearing, suggesting complete loss of long-range crystalline order above ca. 350 °C during pyrolysis. The corresponding TGA in this region up to 350 °C detects an accumulated mass loss of ca. 43 wt% with simultaneous MS signals at m/z = 17, 18, and 44. This indicates further release of water and decomposition products of OA (NH3 and hydrocarbon fragments). From the FTIR spectra of the pyrolysis gas in this temperature range, the evolution of hydrocarbons is evident from the strong absorption between 3000 and 2800 cm−1, but it cannot be assigned univocally to a specific molecule like in the previous temperature range, due to the lack of significant fingerprints in the 1500–650 cm−1 region of the spectra (see the selected FTIR spectra of the pyrolysis gas in Fig. S5C and D†). The detection of a stronger absorption pattern typical of NH3 in the same temperature range confirms the decomposition of OA, hence the hydrocarbons detected through FTIR could be regarded as decomposition products. Simultaneously, an increase in the CO2 signal is detected in FTIR above 350 °C (Fig. S5D†), suggesting the onset of (elemental) carbon formation in the material.
Stage 3: towards the end of the heating step (above 650 °C) and during the two hours holding step at 700 °C in argon (Fig. 3B), the emergence and intensification of two diffraction peaks at 25.8 and 25.2° 2θ can be detected, indicative of the (111) and (011) signals of monoclinic MoO2. This can be conclusively linked to the last mass loss step observed above ca. 650 °C (highlighted in purple, accumulated mass loss of 58 wt%), and an MS signal at m/z = 44, which is attributed to CO2 and indicative of carbothermic reduction of MoO3 to MoO2 with carbon acting as reducing agent.48 Another strong peak at m/z = 28 can be attributed both to CO2 and CO, and the FTIR spectra at this temperature confirm the simultaneous presence of these two gases (Fig. 3D and S5E†). This is further supported by the detection of MoO2 after the pyrolysis by Raman results (Fig. 2A), XRD (Fig. 2B), and further confirmed by the electrochemical characterization (vide infra).
The stepwise transformations of MoOx–OA during pyrolysis, primarily during the heating step with several structural changes and loss of initial long range order, indicate the formation of an intricate, intraparticle microstructure. Due to the apparent mobility of OA and OA-fragments (i.e., the carbon source) during the heating step, the intraparticle arrangement of the carbon in the nanocomposite product has to be investigated carefully. For this purpose, the microstructural features of MoOx–C were analyzed using scanning transmission electron microscopy (STEM). Electron-transparent lamellae were prepared via focused ion beam (FIB) to observe regions of interest taken from the bulk of MoOx–C particles (Fig. 4A) to reveal a representative intraparticle structure. STEM images show a difference in the Z-contrast for features with a size of 5–10 nm. This indicates the formation of domains or grains in a size range of 5–10 nm with varying elemental composition, as shown in the zoomed-in view in Fig. 4B. High-resolution STEM micrographs (Fig. 4C and D) show the presence of nanocrystalline domains homogenously distributed throughout the sample with a size comparable to the sizes of differing Z-contrast. The observation suggests that carbon formation occurred primarily outside the crystalline nanodomains, which contradicts the initially hypothesized formation of a heterostructured, i.e., alternating layer-by-layer arrangement of molybdenum oxide and the carbon phase. Rather, pyrolysis of MoOx–OA yields a carbon-containing molybdenum oxide nanocomposite. The lattice fringes in Fig. 4C and D (corresponding HAADF-STEM image in Fig. S6†) show two characteristic distances with slight local variation. The larger one is around 3.5 Å and the smaller around 2.5 Å, in good agreement with the averaged values obtained from XRD for the d-spacings of the (111) plane (3.43 Å) and the (211) plane (2.43 Å) of monoclinic MoO2. Both of these planes account for the most intense reflections on the XRD diffractogram of MoOx–C (Fig. 2B), hence, the nanodomains observed in Fig. 4C and D can be assigned to monoclinic MoO2.
 | ||
| Fig. 4 Cross-sectional overview of the MoOx–C hybrid nanostructure. (A) HAADF-STEM micrograph of MoOx–C showing microstructure with non-homogeneous particle composition. (B) Magnified micrograph showing phase Z-contrast variations in MoOx–C, (C) bright field STEM micrograph taken at the area indicated in (B). The crystalline domains are highlighted and measurements of planar spacings are indicated on the image. Lattice spacings in regions 1, 2, and 3 are in good agreement with the (111), (211), and (111) planes of MoO2, respectively. (D) High resolution HAADF-STEM image of monoclinic MoO2 nanodomain. | ||
To investigate the elemental distribution, we performed energy dispersive X-ray spectroscopy (EDS). HAADF-STEM (Fig. 5A) with EDS elemental mapping of molybdenum, oxygen, and carbon (Fig. 5B–D) were recorded for FIB-prepared MoOx–C sample cross-sections. The EDS maps reveal an elemental separation with regions enriched in Mo and C and regions enriched in O. The size of these regions corresponds well with the domain/grain size identified in HAADF and high-resolution STEM micrographs (Fig. 4 and S6†). Furthermore, the EDS maps show variations of oxygen- and molybdenum-concentration in the nanosized grains of molybdenum oxide suggesting localized carbothermic reduction of the molybdenum oxide during pyrolysis. EDS spectra indicate that the carbon-concentration is higher in the areas with lower oxygen concentration (Fig. 5E), suggesting that the carbon formation process and MoO3 reduction to MoO2 are correlated. EDS line scan profiles of Mo, O, and C in MoOx–C are shown in Fig. S7,† where local variations in the concentrations of the three elements are shown. TGA measurements in oxygen reveal that the bulk carbon content of MoOx–C nanocomposites is ca. 2.7 wt% (Fig. S8†).
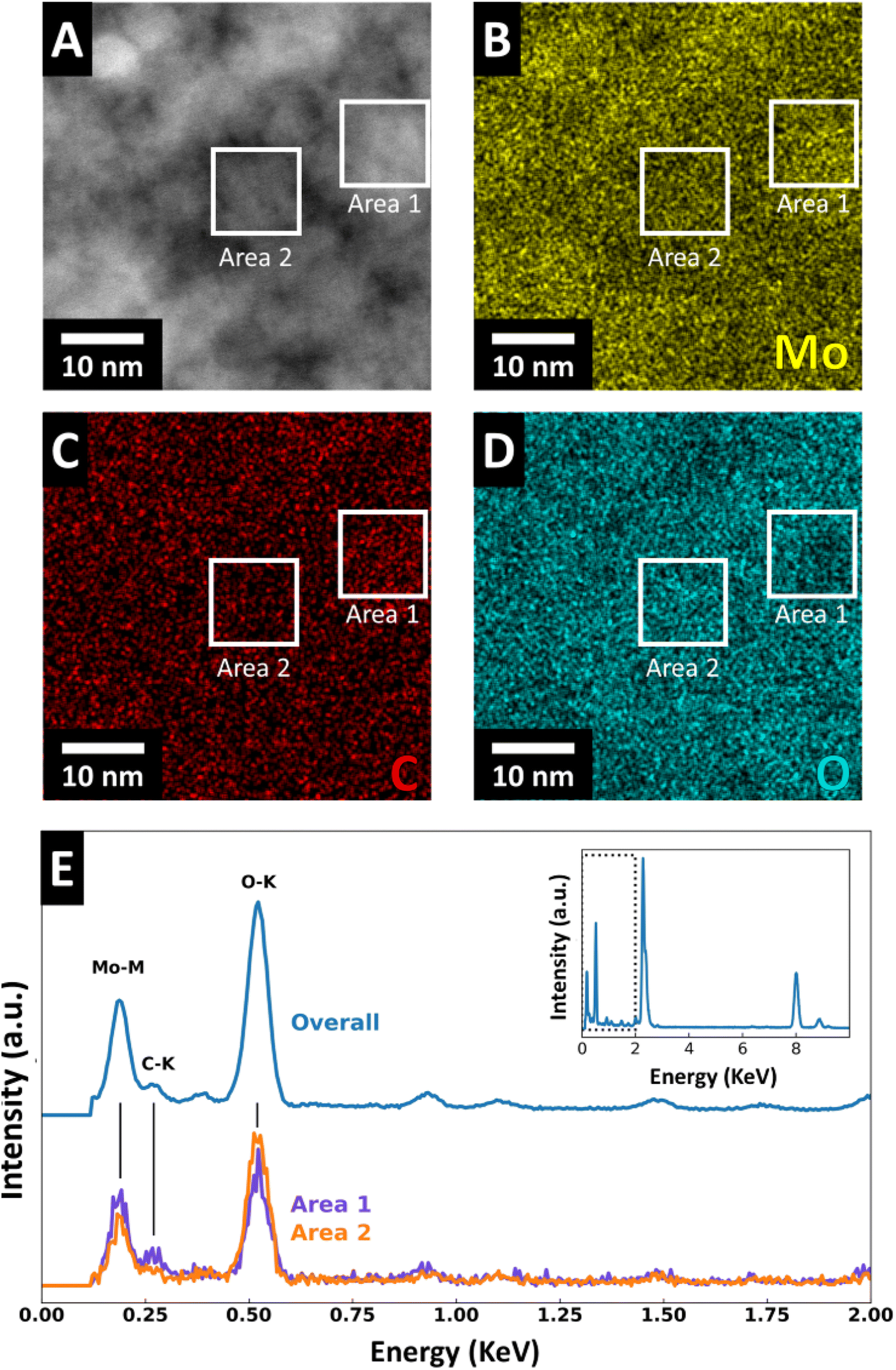 | ||
| Fig. 5 (A) HAADF-STEM micrograph showing Z-number contrast and (B–D) EDS mapping with elemental distribution of Mo, C, and O in MoOx–C. (E) Chemical analysis of the two marked areas by quantitative STEM-EDS spectral imaging analysis. | ||
3.2 Electrochemical characterization
To understand the implications of the nanocomposite microstructure in MoOx–C on electrochemical reactivity, we tested the nanocomposite as an electrode material in standard organic, lithium-containing electrolyte (LP30). Two different potential windows are chosen to establish a complete picture of the electrochemical performance of the electrode material: according to literature, cycling of molybdenum oxides in a potential range of 1–3 V vs. Li+/Li leads to reversible lithium intercalation according to:23| MoO3 + xLi+ + xe− → LixMoO3, (xmax = ∼1.5) | (1) |
| MoO2 + xLi+ + xe− → LixMoO2 (xmax = ∼0.98), | (2) |
After complete lithium intercalation, further extension of the potential range to 0.01 V leads to the conversion-type reaction during the first full reduction according to:
| LixMoO3 + (6 − x)Li+ + (6 − x)e− → Mo + 3Li2O | (3) |
| LixMoO2 + (4 − x)Li+ + (4 − x)e− → Mo + 2Li2O | (4) |
For direct comparison, the electrochemical behavior of MoO3−x, (i.e., partially reduced molybdenum oxide that underwent the same pyrolysis treatment, but without a carbon source, Fig. S10†) was studied alongside MoOx–C. This allowed for insights into the influence of the nanocomposite microstructure resulting from the pyrolysis treatment of MoOx–OA on both, the electrochemical lithium intercalation and conversion reactions.
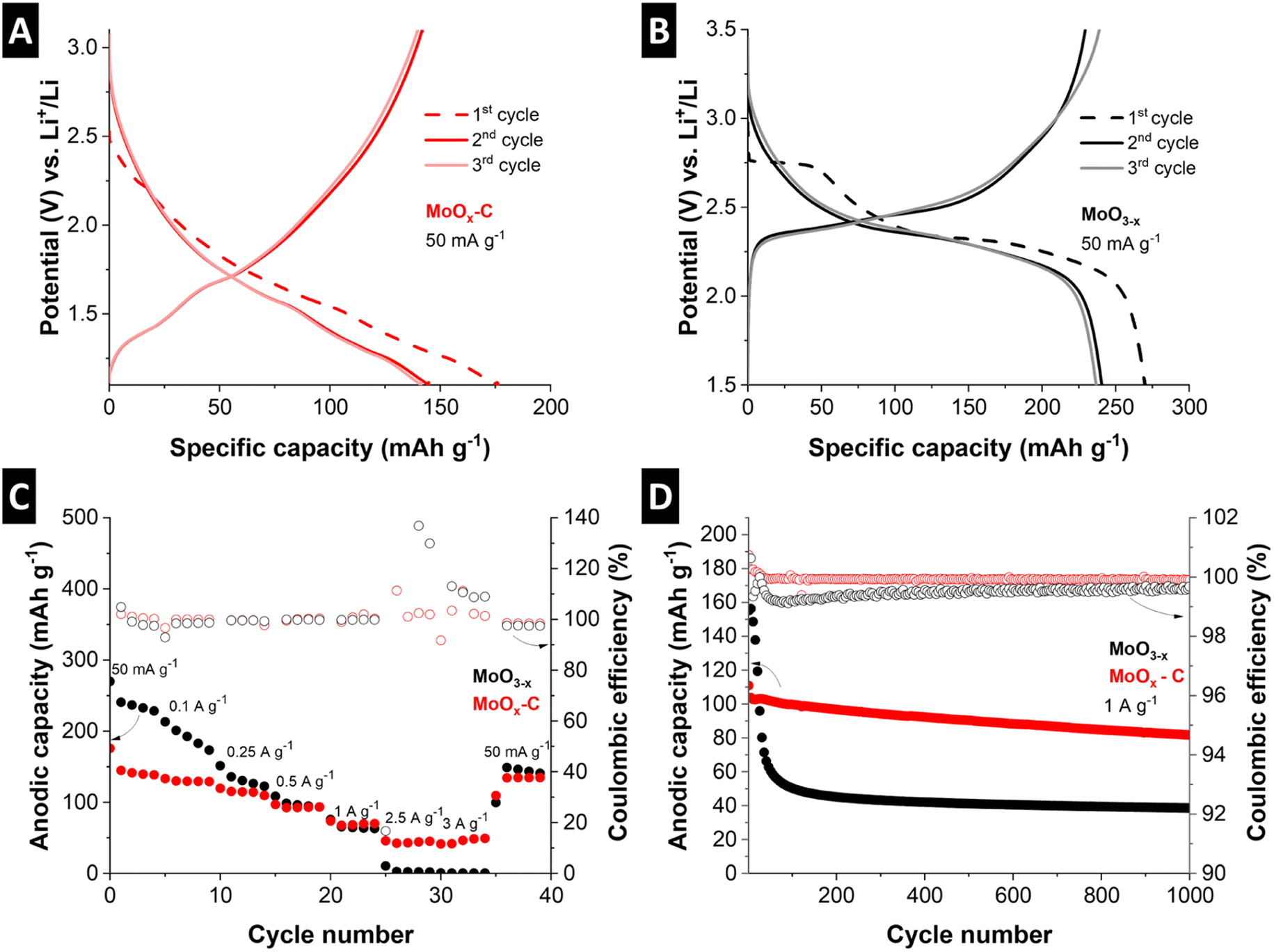 | ||
| Fig. 6 First three galvanostatic charge–discharge cycles of (A) MoOx–C and (B) MoO3−x at 50 mA g−1, (C) rate capability tests at various specific current values from 50 mA g−1 to 3 A g−1, then back to 50 mA g−1, in the voltage range 1.5 V to 3.5 V for MoO3−x and 1.1 V to 3.1 V for MoOx–C, (D) cycling stability test for MoO3−x and MoOx–C at 1 A g−1 for 1000 cycles. | ||
Fig. 6C shows a comparison of the rate capability of MoOx–C and MoO3−x at current densities ranging from 50 mA g−1 to 3 A g−1. Despite the higher initial capacity of MoO3−x, the cycling stability is poor (decrease from 213 to 173 mA h g−1 after 5 cycles at 100 mA g−1). The anodic capacity continues to deteriorate at high currents. Meanwhile, MoOx–C outperforms MoO3−x at high currents, retaining 41 mA h g−1 discharge capacity at 3 A g−1, compared to the negligible 0.6 mA h g−1 capacity for MoO3−x at the same specific current.
Fig. 6D shows cycling stability for the two materials cycled at a fixed specific current of 1 A g−1 for 1000 cycles. MoOx–C shows an initial anodic capacity of 110 mA h g−1, which stabilizes to 82 mA h g−1 after 1000 cycles, retaining ca. 75% of the initial capacity. On the other hand, MoO3−x undergoes a rapid capacity decay, with the anodic capacity dropping from 155 to 39 mA h g−1 after 1000 cycles, a loss of more than 74% of the initial anodic capacity. The averaged coulombic efficiency of MoO3−x, and MoOx–C over 1000 cycles is 99.5%, and 99.9%, respectively, demonstrating the absence of significant side reactions specifically in the nanocomposite electrode MoOx–C.
The improved rate handling behavior, as well as the improved cycling stability and coulombic efficiency of MoOx–C, can be attributed to two factors. First, the nanocomposite approach leads to a well-dispersed conductive carbon phase. This improves the electronic conductivity, as demonstrated by a significantly lower impedance as compared to MoO3−x electrodes (Fig. S11†). Second, the carbothermic reduction during the pyrolysis process in the nanocomposite led to the formation of MoO2, which was shown to exhibit improved cycling stability compared to MoO3.51
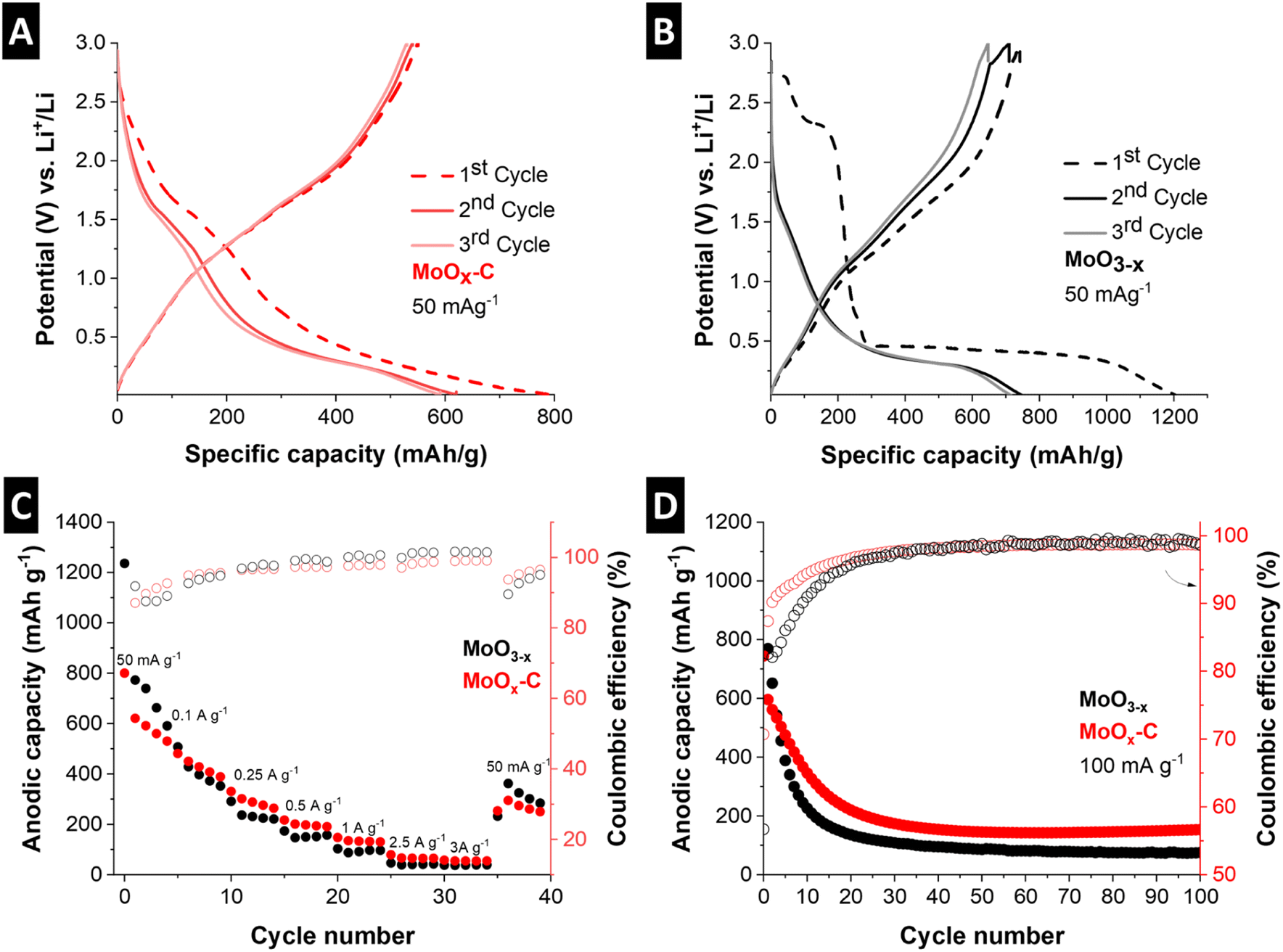 | ||
| Fig. 7 (A) Galvanostatic charge–discharge curves of MoOx–C and (B) MoO3−x at a specific current of 50 mA g−1 in the potential range of 0.01 V to 3 V vs. Li+/Li, (C) rate performance of MoO3−x and MoOx–C at rates from 50 mA g−1 to 3 A g−1, (D) stability test of MoO3−x and MoOx–C cycled at 100 mA g−1. | ||
The rate handling performance and cycling stability of the electrochemical conversion reaction in MoOx–C and MoO3−x were evaluated by galvanostatic charge–discharge at rates ranging from 50 mA g−1 to 3 A g−1 and at 100 mA g−1 for 100 cycles, respectively (Fig. 7C and D). While the nanocomposite MoOx–C outperforms the reference material, the rate handling and cycling stability performance of both materials is limited. For MoOx–C, significant capacity fading can be observed from 768 mA h g−1 to 192 mA h g−1 after 25 cycles, where the performance stabilizes. For comparison, the capacity of MoO3−x drops to 165 mA h g−1 after about 15 cycles. To analyze the improved electrochemical conversion-type charge storage performance of the MoOx–C nanocomposite, the morphology of both electrodes was evaluated via ex situ SEM after the first cathodic cycle at 0.01 V vs. Li+/Li and post mortem SEM after 39 cycles (Fig. S12†). The morphological changes of MoO3−x were more severe compared to MoOx–C. This is especially visible by the extensive particle cracking in MoO3−x already occurring after the initial reductive cycle.
Future work optimizing MoOx–C nanocomposite electrodes for conversion-type reactions needs to involve a careful investigation of the electrode microstructure after the initial cathodic cycle, for example via ex situ TEM of a FIB-prepared lamella, to gain further insights on the morphological changes occurring during the electrochemical conversion reaction.
4. Conclusion
In this work, layered molybdenum oxide with octylamine molecules assembled in its interlayer was pyrolyzed to convert the organic molecules to a carbon phase, yielding a nanocomposite MoOx–C material. The present work set its main focus on analyzing the carbon formation and microstructure evolution during pyrolysis and the precise phase distribution in the nanocomposite product.Processes occurring during pyrolysis were examined with a combination of in situ XRD, TGA-MS, and TGA-FTIR experiments. It was found that during the heating step, some loosely bound molecular OA and OA decomposition products escape from the interlayer space before being converted to elemental carbon, and a strong carbothermic reduction of the oxide takes place above 650 °C. With this knowledge, future directions towards improving the synthesis route can focus the use of more strongly bonded and/or less volatile organic molecules together with faster heating of the material during pyrolysis to obtain more localized carbon formation or a higher carbon content.
Cross-sectional specimens of the resulting MoOx–C nanocomposite prepared via FIB were analyzed to reveal the intricate intraparticle microstructure. It was found that nanoscopic domains of MoO2 are surrounded by a carbon phase, and in addition, trace amounts of Mo2C and Mo4O11 formation could be observed by XRD and XPS. Further tuning the synthesis temperature and time could thus be employed to avoid carbide formation (or to support carbide formation, if desirable).
The electrochemical properties of the resulting MoOx–C nanocomposite electrode were evaluated in detail for both, intercalation-type and conversion-type charge storage in lithium-containing, organic electrolyte. The electrode significantly outperformed the MoO3−x reference sample in terms of rate handling and stability for lithium intercalation and showed an increase in initial coulombic efficiency for the conversion reaction.
The work can be used as a guideline to obtain transition metal oxide/carbon nanocomposites with desired structural and electrochemical properties. It showcases pitfalls and opportunities arising from the pyrolysis route (organic molecule mobility, carbothermic reduction, carbide formation, etc.) which should be considered in future works.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
M. E. and S. F. acknowledge funding from the German Federal Ministry of Education and Research (BMBF) in the “NanoMatFutur” program (grant no. 03XP0423) and financial support from the Helmholtz Association. A. I. acknowledges the EU's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 860403 “POLYSTORAGE” for the funding of the position. This work contains results obtained from experiments performed at the Ernst Ruska-Centre (ER-C) for Microscopy and Spectroscopy with Electrons at the Forschungszentrum Jülich (FZJ) in Germany [https://doi.org/10.17815/jlsrf-2-106, https://doi.org/10.17815/jlsrf-2-68]. The ER-C beam-time access was provided via the DFG Core Facility Project (FZJ_IEK-2_PN1). V. A. acknowledges funding from the Fluid Interface Reactions, Structures and Transport (FIRST), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences at Oak Ridge National Laboratory under contract #DE-AC0500OR22725 with UT Battelle, LLC. All authors thank Dr Yuyoung Shin and Jaehoon Choi (both HIU) for assistance with Raman measurements.References
- P. Simon and Y. Gogotsi, Perspectives for Electrochemical Capacitors and Related Devices, Nat. Mater., 2020, 19, 1151–1163 CrossRef CAS PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Performance and Cost of Materials for Lithium-Based Rechargeable Automotive Batteries, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- Y. Luo, Y. Bai, A. Mistry, Y. Zhang, D. Zhao, S. Sarkar, J. V. Handy, S. Rezaei, A. C. Chuang, L. Carrillo, K. Wiaderek, M. Pharr, K. Xie, P. P. Mukherjee, B. Xu and S. Banerjee, Effect of Crystallite Geometries on Electrochemical Performance of Porous Intercalation Electrodes by Multiscale Operando Investigation, Nat. Mater., 2022, 21, 217–227 CrossRef CAS PubMed.
- J. Park, H. Zhao, S. D. Kang, K. Lim, C. Chen, Y. Yu, R. D. Braatz, D. A. Shapiro, J. Hong, M. F. Toney, M. Z. Bazant and W. C. Chueh, Fictitious Phase Separation in Li Layered Oxides Driven by Electro-Autocatalysis, Nat. Mater., 2021, 20, 991–999 CrossRef CAS PubMed.
- H.-S. Kim, J. B. Cook, S. H. Tolbert and B. Dunn, The Development of Pseudocapacitive Properties in Nanosized-MoO2, J. Electrochem. Soc., 2015, 162, A5083–A5090 CrossRef CAS.
- W. Tang, C. X. Peng, C. T. Nai, J. Su, Y. P. Liu, M. V. V. Reddy, M. Lin and K. P. Loh, Ultrahigh Capacity Due to Multi-Electron Conversion Reaction in Reduced Graphene Oxide-Wrapped MoO2 Porous Nanobelts, Small, 2015, 11, 2446–2453 CrossRef CAS PubMed.
- C. He, S. Wu, N. Zhao, C. Shi, E. Liu and J. Li, Carbon-Encapsulated Fe3O4 Nanoparticles as a High-Rate Lithium Ion Battery Anode Material, ACS Nano, 2013, 7, 4459–4469 CrossRef CAS PubMed.
- Y. Yao, Z. Chen, R. Yu, Q. Chen, J. Zhu, X. Hong, L. Zhou, J. Wu and L. Mai, Confining Ultrafine MoO2 in a Carbon Matrix Enables Hybrid Li Ion and Li Metal Storage, ACS Appl. Mater. Interfaces, 2020, 12, 40648–40654 CrossRef CAS PubMed.
- D. P. Dubal, O. Ayyad, V. Ruiz and P. Gómez-Romero, Hybrid Energy Storage: The Merging of Battery and Supercapacitor Chemistries, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC.
- S. Fleischmann, A. Tolosa and V. Presser, Design of Carbon/Metal Oxide Hybrids for Electrochemical Energy Storage, Chem.–Eur. J., 2018, 24, 12143–12153 CrossRef CAS PubMed.
- X. W. Lou, D. Deng, J. Y. Lee and L. A. Archer, Preparation of SnO2/Carbon Composite Hollow Spheres and Their Lithium Storage Properties, Chem. Mater., 2008, 20, 6562–6566 CrossRef CAS.
- M. Clites, R. Andris, D. A. Cullen, K. L. More and E. Pomerantseva, Improving Electronic Conductivity of Layered Oxides through the Formation of Two-Dimensional Heterointerface for Intercalation Batteries, ACS Appl. Energy Mater., 2020, 3, 3835–3844 CrossRef CAS.
- N. Norouzi, D. Omo-Lamai, F. Alimohammadi, T. Averianov, J. Kuang, S. Yan, L. Wang, E. Stavitski, D. Leshchev, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok, D. C. Bock and E. Pomerantseva, The Dopamine Assisted Synthesis of MoO3/Carbon Electrodes With Enhanced Capacitance in Aqueous Electrolyte, Front. Chem., 2022, 10, 873462 CrossRef CAS PubMed.
- N. Norouzi, T. Averianov, J. Kuang, D. C. Bock, S. Yan, L. Wang, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and E. Pomerantseva, Hierarchically Structured MoO2/Dopamine-Derived Carbon Spheres as Intercalation Electrodes for Lithium-Ion Batteries, Mater. Today Chem., 2022, 24, 100783 CrossRef CAS.
- R. Andris, T. Averianov and E. Pomerantseva, Phase Transformation and Electrochemical Charge Storage Properties of Vanadium Oxide/Carbon Composite Electrodes Synthesized via Integration with Dopamine, J. Am. Ceram. Soc., 2023, 106, 120–132 CrossRef CAS.
- G. Barim, R. Dhall, E. Arca, T. R. Kuykendall, W. Yin, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and M. M. Doeff, Heterostructured Lepidocrocite Titanate-Carbon Nanosheets for Electrochemical Applications, ACS Appl. Nano Mater., 2022, 5, 678–690 CrossRef CAS.
- H. T. Bui, D. C. Linh, L. D. Nguyen, H. I. Chang, S. A. Patil, N. K. Shrestha, K. X. Bui, T. S. Bui, T. N. A. Nguyen, N. T. Tung, S. H. Han and P. T. San, In situ Formation and Integration of Graphene into MoS2 Interlayer Spacing: Expansion of Interlayer Spacing for Superior Hydrogen Evolution Reaction in Acidic and Alkaline Electrolyte, J. Mater. Sci., 2022, 57, 18993–19005 CrossRef CAS.
- C. Zhao, X. Wang, J. Kong, J. M. Ang, P. S. Lee, Z. Liu and X. Lu, Self-Assembly-Induced Alternately Stacked Single-Layer MoS2 and N-Doped Graphene: A Novel van der Waals Heterostructure for Lithium-Ion Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 2372–2379 CrossRef CAS PubMed.
- E. Pomerantseva and Y. Gogotsi, Two-Dimensional Heterostructures for Energy Storage, Nat. Energy, 2017, 2, 17089 CrossRef CAS.
- A. Khan, J. Azadmanjiri, B. Wu, L. Liping, Z. Sofer and J. Min, Atomically Thin Nanosheets Confined in 2D Heterostructures: Metal-Ion Batteries Prospective, Adv. Energy Mater., 2021, 11, 2100451 CrossRef CAS.
- B. Yebka, C. Julien and G. A. Nazri, Electrochemical Behavior of Hydrated Molybdenum Oxides in Rechargeable Lithium Batteries, Ionics, 1999, 5, 236–243 CrossRef CAS.
- D. Chen, M. Liu, L. Yin, T. Li, Z. Yang, X. Li, B. Fan, H. Wang, R. Zhang, Z. Li, H. Xu, H. Lu, D. Yang, J. Sun and L. Gao, Single-Crystalline MoO3 Nanoplates: Topochemical Synthesis and Enhanced Ethanol-Sensing Performance, J. Mater. Chem., 2011, 21, 9332–9342 RSC.
- A. Yu, N. Kumagai, Z. Liu and J. Y. Lee, Preparation of Sodium Molybdenum Oxides by a Solution Technique and Their Electrochemical Performance in Lithium Intercalation, Solid State Ionics, 1998, 106, 11–18 CrossRef CAS.
- Y. S. Jung, S. Lee, D. Ahn, A. C. Dillon and S. H. Lee, Electrochemical Reactivity of Ball-Milled MoO3−y as Anode Materials for Lithium-Ion Batteries, J. Power Sources, 2009, 188, 286–291 CrossRef CAS.
- M. L. Freedman, Precipitation of Molybdenum(VI) in Strongly Acid Solutions, J. Chem. Eng. Data, 1963, 8, 113–116 CrossRef CAS.
- J. Walton, P. Wincott, N. Fairley and A. Carrick, Peak Fitting with CasaXPS: A Casa Pocket Book, Accolyte Science, Knutsford, UK, 2010 Search PubMed.
- D. Weingarth, M. Zeiger, N. Jäckel, M. Aslan, G. Feng and V. Presser, Graphitization as a Universal Tool to Tailor the Potential-Dependent Capacitance of Carbon Supercapacitors, Adv. Energy Mater., 2014, 4, 1400316 CrossRef.
- D. Chen, T. Li, L. Yin, X. Hou, X. Yu, Y. Zhang, B. Fan, H. Wang, X. Li, R. Zhang, T. Hou, H. Lu, H. Xu, J. Sun and L. Gao, A Comparative Study on Reactions of N-Alkylamines with Tungstic Acids with Various W-O Octahedral Layers: Novel Evidence for the “Dissolution-Reorganization” Mechanism, Mater. Chem. Phys., 2011, 125, 838–845 CrossRef CAS.
- M. A. Spencer, J. Fortunato and V. Augustyn, Electrochemical Proton Insertion Modulates the Hydrogen Evolution Reaction on Tungsten Oxides, J. Chem. Phys., 2022, 156, 064704 CrossRef CAS PubMed.
- L. Seguin, M. Figlarz, R. Cavagnat and J.-C. Lassègues, Infrared and Raman Spectra of MoO3 Molybdenum Trioxides and MoO3·xH2O Molybdenum Trioxide Hydrates, Spectrochim. Acta, Part A, 1995, 51, 1323–1344 CrossRef.
- J. Jiang, I. May, M. J. Sarsfield, M. Ogden, D. O. Fox, C. J. Jones and P. Mayhew, A Spectroscopic Study of the Dissolution of Cesium Phosphomolybdate and Zirconium Molybdate by Ammonium Carbamate, J. Solution Chem., 2005, 34, 443–468 CrossRef CAS.
- R. L. Frost, J. Bouzaid and I. S. Butler, Raman Spectroscopic Study of the Molybdate Mineral Szenicsite and Comparison with Other Paragenetically Related Molybdate Minerals, Spectrosc. Lett., 2007, 40, 603–614 CrossRef CAS.
- X. Lu, R. Wang, F. Yang, W. Jiao, W. Liu, L. Hao and X. He, Preparation of MoO3 QDs through Combining Intercalation and Thermal Exfoliation, J. Mater. Chem. C, 2016, 4, 6720–6726 RSC.
- A. M. Amorim, C. F. G. C. Geraldes and J. J. C. Teixeira, A Raman Spectroscopic Study of Molecular Interaction in Long-Chain Primary Amines Systems, J. Raman Spectrosc., 1982, 13, 56–62 CrossRef.
- M. A. Camacho-López, L. Escobar-Alarcón, M. Picquart, R. Arroyo, G. Córdoba and E. Haro-Poniatowski, Micro-Raman Study of the m-MoO2 to a-MoO3 Transformation Induced by Cw-Laser Irradiation, Opt. Mater., 2011, 33, 480–484 CrossRef.
- P. A. Spevack and N. S. McIntyre, Thermal Reduction of MoO3, J. Phys. Chem., 1992, 96, 9029–9035 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Interpretation of Raman Spectra of Disordered and Amorphous Carbon A, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. Kaniyoor and S. Ramaprabhu, A Raman Spectroscopic Investigation of Graphite Oxide Derived Graphene, AIP Adv., 2012, 2, 032183 CrossRef.
- Z. Wang, X.-D. Wen, R. Hoffmann, J. S. Son, R. Li, C.-C. Fang, D.-M. Smilgies and T. Hyeon, Reconstructing a Solid-Solid Phase Transformation Pathway in CdSe Nanosheets with Associated Soft Ligands, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17119–17124 CrossRef CAS PubMed.
- D. O. Scanlon, G. W. Watson, D. J. Payne, G. R. Atkinson, R. G. Egdell and D. S. L. Law, Theoretical and Experimental Study of the Electronic Structures of MoO3 and MoO2, J. Phys. Chem. C, 2010, 114, 4636–4645 CrossRef CAS.
- J. Baltrusaitis, B. Mendoza-Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Generalized Molybdenum Oxide Surface Chemical State XPS Determination via Informed Amorphous Sample Model, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- D. Wang, J. Wang, X. Luo, Z. Wu and L. Ye, In situ Preparation of Mo2C Nanoparticles Embedded in Ketjenblack Carbon as Highly Efficient Electrocatalysts for Hydrogen Evolution, ACS Sustainable Chem. Eng., 2018, 6, 983–990 CrossRef CAS.
- M. C. Biesinger, Accessing the Robustness of Adventitious Carbon for Charge Referencing (Correction) Purposes in XPS Analysis: Insights from a Multi-User Facility Data Review, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- F. W. McLafferty and F. Turecek, Interpretation of Mass Spectra, University Science Books, 1993 Search PubMed.
- R. D. Bowen and A. Maccoll, Low Energy, Low Temperature Mass Spectra, Org. Mass Spectrom., 1983, 18, 576–581 CrossRef CAS.
- B. Chen, J. Liu, H. Chen, W. Chen and C. Lin, Comparison of the Characteristic Mass Fragmentations of Phenethylamines and Tryptamines by Electron Ionization Gas Chromatography Mass Spectrometry, Electrospray and Matrix-Assisted Laser Desorption Ionization Mass Spectrometry, Appl. Sci., 2018, 8, 1022 CrossRef.
- W. E. Wallace, Mass Spectra, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, 2018, p. 20899 Search PubMed.
- L. Trif, F. P. Franguelli, G. Lendvay, E. Majzik, K. Béres, L. Bereczki, I. M. Szilágyi, R. P. Pawar and L. Kótai, Thermal Analysis of Solvatomorphic Decakis (Dimethylammonium) Dihydrogendodecatungstate Hydrates, J. Therm. Anal. Calorim., 2021, 144, 81–90 CrossRef CAS.
- J. Swiatowska-Mrowiecka, S. de Diesbach, V. Maurice, S. Zanna, L. Klein, E. Briand, I. Vickridge and P. Marcus, Li-Ion Intercalation in Thermal Oxide Thin Films of MoO3 as Studied by XPS, RBS, and NRA, J. Phys. Chem. C, 2008, 112, 11050–11058 CrossRef CAS.
- T. Tao, A. M. Glushenkov, C. Zhang, H. Zhang, D. Zhou, Z. Guo, H. K. Liu, Q. Chen, H. Hu and Y. Chen, MoO3 Nanoparticles Dispersed Uniformly in Carbon Matrix: A High Capacity Composite Anode for Li-Ion Batteries, J. Mater. Chem., 2011, 21, 9350–9355 RSC.
- S. Fleischmann, M. Zeiger, A. Quade, A. Kruth and V. Presser, Atomic Layer-Deposited Molybdenum Oxide/Carbon Nanotube Hybrid Electrodes: The Influence of Crystal Structure on Lithium-Ion Capacitor Performance, ACS Appl. Mater. Interfaces, 2018, 10, 18675–18684 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01230a |
| This journal is © The Royal Society of Chemistry 2023 |