
Photo and redox active carbon nitride – dual functionality for energy storage†
Rohit
Mann
 and
Deepa
Khushalani
*
and
Deepa
Khushalani
*
Materials Chemistry Group, Department of Chemical Sciences, Tata Institute of Fundamental Research, Homi Bhabha Rd., Colaba, Mumbai, India 400005. E-mail: khushalani@tifr.res.in
First published on 13th September 2023
Abstract
The ability to directly capture and store solar energy in an electrical form within a single structure is an intriguing concept and if feasible, could be an ideal approach to overcome the intermittency issue of solar energy. Recently, ionothermal-treated carbon nitride has shown this unique dual functionality. However, the current understanding of the distinctive material is minimal and as such, presented here are data that give insight into how the dual function can be viably modulated without in fact altering the structure. Importantly, it has been shown that there is redox behavior in ionic carbon nitrides thus providing impetus for battery-like storage capability. Furthermore, the work demonstrates that by simply altering either the ions or the pH of the electrolyte, both the potential and storage capability of the photo-excited electron can be augmented. The former can be modulated over ca. 700 mV and the latter can be increased 4 times when Na ions are replaced by K ions. This work provides insight into ionic carbon nitrides and their seminal properties that have thus far not been realized in other conventional semiconductors.
Introduction
Towards a targeted approach in alleviating the intermittency issue of solar energy, a variety of methodologies have been adopted, such as the synthesis of solar fuels, where predominantly solar energy can be converted to chemical energy, and the ensuing products can then be used at will. This process can be done either through photocatalysis1,2 or through photo-electrocatalysis.3 In addition, analogously, since ideally electrical energy is the type that is most desired, a variety of approaches have also been pursued not only for sequential capturing and storing solar energy (via coupling a photovoltaic device to a battery/capacitor) but more intriguingly for direct capture and storage of the energy in one device, i.e. in the form of ‘solar battery.’ This refers to the ability to store an excited state (with sufficient potential and electron density) and release it, on demand, in the form of an electrical impulse. Salunke and co-workers recently summarized routes that enable conventional Li-ion batteries to be modified to have the capability to be photo-rechargeable.4 New device configurations involving additional electrodes (that are photoactive) have been detailed that allow, in principle, for excited electrons to be transferred (from the photoactive layer) to the redox active Li-based electrode. Considering the finite supply of Li and also the requirement to use more sustainable forms of materials, it is imperative that viable alternatives be developed. As such, concurrently, in a tangential approach, Lotsch and co-workers have recently detailed a substantially different route to charge capture and storage by utilizing a material that is not well understood thus far: a 2D ionic carbon nitride structure.5,6It needs to be noted that as far back as 2008,7 a synthetic approach of treating conventional carbon nitride precursors (dicyandiamide) with a eutectic treatment of LiCl/KCl was found to lead to the formation of a unique semiconductor – one that is photoactive and also has ionic groups, namely in the form of a negatively charged framework (where the charge predominantly resides on nitrogen) and was balanced by K+ ions that are occluded in either the open channels or within the trapped layers of a disordered polyheptazine structure.8 It must be highlighted that conventional neutral carbon nitride is ubiquitous, with a myriad of reports detailing its use in various applications with and without structural modifications (especially involving doping).9 However, the ionothermal treatment (i.e. treating with a eutectic treatment), as first detailed by Bojdys and co-workers,7 is the sole manner currently of introducing an ionic functionality into a carbon nitride polymeric backbone. Depending on the conditions of synthesis and precursors used, various condensed forms of carbon nitrides can be formed (for example, the structure can consist of triazine units).10,11 However, the data that has yielded the most promising energy capture and storage aspects involves a more porous framework consisting of polyheptazine units.
In 2018, Lotsch and co-workers published a seminal report detailing the observation of long-term charge storage on this unique ionic semiconductor material.6 This material shows a novel property in that upon light irradiation (and in the presence of hole scavengers and the absence of O2), a state can be achieved that involves ‘trapped’ electrons which can be used as reducing equivalents to drive chemical reactions (even after the light is switched off) or be converted into electricity. Over the years, work on these frameworks has gathered momentum in elucidating the structural and electronic constituents that govern the long-lived ‘blue excited state.'12–14
Based on the existing literature, it can be inferred that (a) an ionic carbon nitride framework is crucial, (b) a porous M-PHI framework (M refers to cations such as K+) needs to form where the porosity is dictated by low-angle peaks in XRD (ca. 8–10 °C 2θ),8 (c) the pores are essential as they contribute to ionic conductivity (movement of charge balancing ions, M+),15 (d) excited state electronic properties could be dictated by this charge balancing M+ ions by perhaps a ‘polaronic effect’ (e) HER studies (either during light radiation or subsequent to light radiation which is referred to as ‘dark’ photocatalysis) provide data which dictates that Na+ ions are preferred as they contribute to delayed PL of the excited state, and concurrently show improved HER production due to charge stabilization by trapping. This is assisted by the presence of Na+.15 This latter point has been used to justify not only the use of the excited trapped state for aiding chemical reduction reactions but also to provide an electrical impulse when employed as a ‘solar battery.’5
It must be stated that the material stores only one charge carrier, the excited electron. Furthermore, despite the aforementioned summary, what is still not understood is how and which properties of M-PHI allow it to show this unique dual functionality. Insight into how the excited anionic radical is maintained is needed, and what are the ramifications of the nature of the electrolyte on the charge storage capability also need to be elucidated. Currently, there is a lack of consensus as to the importance of forming this ‘blue radical’ state as Wu and co-workers recently reported a short-range ordered K+-PHI which, even though it does not form an excited blue state, still is able to show prolonged storage and dark photocatalysis for an HER reaction.16 In addition, is the storage capacitive or faradaic in behavior that needs to be clearly elucidated in order to provide fundamental insights into the origins of the dual functionality.
Towards addressing these various questions, presented here are the electrochemical and photo-electrochemical studies performed on a single M+PHI sample (F-PCN). Our data showcases that by modulating external experimental parameters, the photo-response can be modified to augment charge storage from minutes to hours – this has been done without altering the M+PHI structure. Importantly, even the potential at which the photo-excited electrons are stored can be modulated. This has allowed further insight into the nature of the excited state, and the work demonstrates that the material is entirely solar rechargeable, does not require an external bias (but has similar behavior when an external voltage is utilized), and yields a similar amount of charge each time in a recyclable manner.
Results and discussion
Detailed below is a comparative study of a conventional polymeric carbon nitride (PCN) (formed from thermal treatment of urea) and also a functionalized polymeric carbon nitride (F-PCN) which has been formed via a eutectic melt treatment (KCl and LiCl) of PCN (see ESI† for synthesis details). Fig. 1a shows the representative structure of both materials. FTIR data shown in Fig. S1† showcases that PCN and F-PCN contain the building block heptazine unit from the peak centered at 805 cm−1 (breathing mode of heptazine) and the peak in the region of 1300–1700 cm−1.17 The important aspect to note is that the eutectic treatment gives additional ionized functionalities ((C–N−–C) K+/Li+ and (–N−–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) K+/Li+) to the framework. The peak centered in FT-IR data at 994 cm−1, 1155 cm−1, and 2177 cm−1 confirm the presence of these charged moieties.18,19 The ICP-MS data (Table S1†) and XPS data (Fig. S2 and S3†) confirm the presence of the K+, Li+ ions, and a negatively charged framework.20–22 The materials have also been characterized through 13C solid state NMR. The given NMR data in Fig. S4† is done in CP (cross polarization) mode to improve the signal intensity. It is observed both the materials have two types of carbon atoms present, the peaks at 155 ppm and 162 ppm correspond to the heptazine carbons and as such confirm the presence of heptazine as the building block unit, not triazine. The SEM images for both materials are shown in Fig. S5.† The image showcases no substantial variation in the morphology of PCN when it is treated with the eutectic mixture to form F-PCN. Both appear to be irregular granular structures. Further, the XRD pattern (Fig. S6†) shows that the eutectic mixture treatment provided new ordering to the structure. The peak centered at 8.1° corresponds to the in-plane porosity of size ca. 1.1 nm.8
N) K+/Li+) to the framework. The peak centered in FT-IR data at 994 cm−1, 1155 cm−1, and 2177 cm−1 confirm the presence of these charged moieties.18,19 The ICP-MS data (Table S1†) and XPS data (Fig. S2 and S3†) confirm the presence of the K+, Li+ ions, and a negatively charged framework.20–22 The materials have also been characterized through 13C solid state NMR. The given NMR data in Fig. S4† is done in CP (cross polarization) mode to improve the signal intensity. It is observed both the materials have two types of carbon atoms present, the peaks at 155 ppm and 162 ppm correspond to the heptazine carbons and as such confirm the presence of heptazine as the building block unit, not triazine. The SEM images for both materials are shown in Fig. S5.† The image showcases no substantial variation in the morphology of PCN when it is treated with the eutectic mixture to form F-PCN. Both appear to be irregular granular structures. Further, the XRD pattern (Fig. S6†) shows that the eutectic mixture treatment provided new ordering to the structure. The peak centered at 8.1° corresponds to the in-plane porosity of size ca. 1.1 nm.8
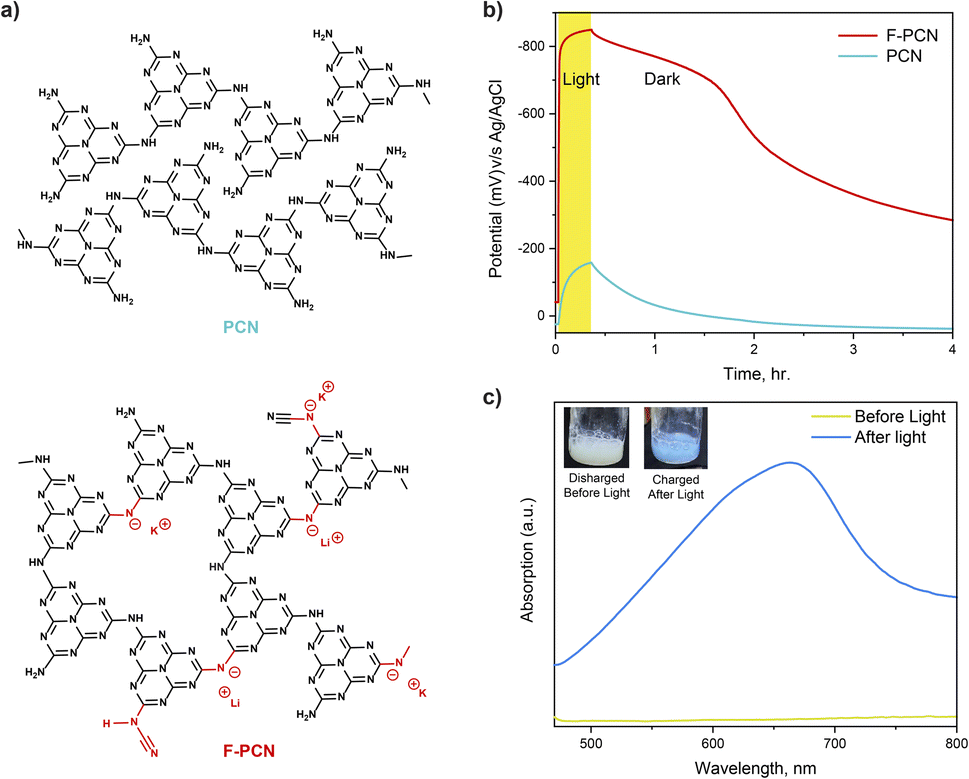 | ||
| Fig. 1 (a) Representative structure of PCN and F-PCN (b) OCP (Open Circuit Potential) measurement for PCN and F-PCN in 0.1 M Na2SO4 and 5 mM 4-MBA (c) absorption spectra of radical blue species, inset are the photographs of F-PCN in a discharged state (before irradiation) and charged state (after irradiation). | ||
With the aforementioned structural understanding of the materials, photo-electrochemical measurements have been carried out, Fig. 1b. The measurement demonstrates that upon irradiation with 1.5 AM 1 sun for 20 minutes (under an inert atmosphere and in the presence of 0.1 M Na2SO4 and 5 mM 4-MBA (4-methylbenzyl alcohol–hole quencher)), F-PCN potential rises to a reductive potential of ca. −850 mV v/s Ag/AgCl. The potential rise after light excitation is due to the combined effect of the excited charge carrier generation, separation, and accumulation. The measurement shows the OCP (Open Circuit Potential) is retained at −780 mV v/s Ag/AgCl (87% of the original value) for a further 90 minutes, even when the light is switched off. Subsequently, it decreases till it reaches equivalent to the potential in the dark. In comparison, for PCN, the photo potential obtained, upon irradiation for also 20 min, is only −186 mV. After 90 minutes of light being switched off, it retained only 5% of its photo potential. It needs to be noted that what is being observed for the OCP measurement under light-off conditions is the retention of the excited charge state, and simultaneously through a slow self-discharge process, the material loses the high potential due to percolation of contaminants (such as O2) that fulfill the thermodynamic conditions for electron quenching. Interestingly, this alternation of low (discharged) and high potential states (charged) in F-PCN is accompanied by a color change. As seen in the inset of Fig. 1c, a conversion from yellow to blue is observed for the ‘discharged state’ to ‘photo-charged state,’ respectively. In order to put this data in perspective with other conventional semiconductors, identical experiments were carried out with TiO2 and ReS2 (Fig. S7†), which have a band gap of 3.2 eV and 1.5 eV, respectively. The semiconductors were selected to ensure that 4-methyl benzyl alcohol (4-MBA) could quench the excited hole. It is observed that both the inorganic semiconductors (which consist of neutral, relatively crystalline structures) do not have the ability to retain the high potential state. From this, it can be surmised that the eutectic treatment provides a pivotal alteration in the F-PCN structure and gives rise to a unique property that allows prolonged retention of the ‘photo-charged state.’ This state can be ascribed to a radical anion species (considering the excited hole is quenched by 4-MBA, which can also be visually observed by the blue color).
To gain further insight, cyclic voltammetry (Fig. 2a) was performed on PCN and F-PCN under an inert atmosphere and in 0.1 M Na2SO4 and 5 mM 4-MBA electrolyte. F-PCN shows a visible reversible redox feature which is not observed in the case of PCN. From this observation, it can be inferred that F-PCN has moieties that can undergo viable redox reactions. Hence the melt salt treatment introduces a redox ability in F-PCN, which is not present in PCN. F-PCN shows a reducing feature at −1200 mV v/s Ag/AgCl and an oxidation feature at ca. 470 mV. The potential window-dependent measurement shown in Fig. S8† confirms that these reduction and oxidation features are complementary and correspond to the same redox moieties. Interestingly, the material changes its color from yellow to blue once the applied potential reaches the reduction peak potential. It can be inferred that the reduction feature corresponds to the radical anion species formation. Therefore, the excited state radical anion species can also be generated by applying a bias.
 | ||
| Fig. 2 (a) Cyclic voltammetry curve for PCN and F-PCN in 0.1 M Na2SO4 and 5 mM 4-MBA, scan rate 5 mV s−1 (b) charging F-PCN for 20 min with three different external biases in Na2SO4 and 4-MBA electrolyte (c) charging F-PCN in Na2SO4 and 4-MBA with different intensity lamp (1.5 AM). All the measurements are done under inert conditions. | ||
Interestingly, when three different fixed external potentials were applied for 20 minutes (light was NOT irradiated on the sample), and subsequently, the OCP measurements were performed as shown in Fig. 2b, it can be observed that the radical anion formation and charge storage is potential-dependent, and the charged state forms only when the applied potential is more than −850 mV v/s Ag/AgCl. This potential threshold value can be corroborated by CV measurements, where the reduction peak onset is also ca. −850 mV and is centered at ca. −1200 mV. Therefore, it confirms the radical anion species can also be formed by reducing F-PCN electrically. F-PCN reaches the reduced state only when the potential is above −850 mV v/s Ag/AgCl. Additionally, it does not matter how much more potential is applied, the material eventually holds on to a specific potential range of ca. −800 mV only. Along the same lines, even when different light intensities were used to charge the material (Fig. 2c). A higher photo potential rise can be observed with higher intensity, as more photon flux leads to more charge carrier generation. However, the potential that F-PCN holds is the same for all the intensities. It reinforces the realization that charging and discharging of F-PCN are purely redox events akin to what occurs in a battery. The formation of the blue species (radical anion) is analogous to forming a reduced state. This occurs through a redox route while discharging reverts the stored electron through a redox event (CV displays an oxidation event, and the color reverts to yellow). It can therefore be stated that as long as either a photo-activated or an electrically activated potential is applied to F-PCN, and as long as the reduction potential of the radical is achieved, F-PCN will be activated to a ‘blue radical anion’ state, which is akin to a reduced state.
To further understand this redox process, EIS (Electrical Impedance Spectroscopy) measurements were performed at a potential of −1.3 V v/s Ag/AgCl for both the PCN and F-PCN samples. The data is shown in Fig. S9a† and the curves have been fitted with the equivalent circuit (inset Fig. S9c and d†) and the values of the elements in the equivalent circuits have been tabulated in Table S4.†Rs refers to the solution resistance, Rct is the charge transfer resistance and Q is the constant phase element (CPE) due to the pseudocapacitive behavior. The diameter of the semicircle obtained in the Nyquist plot is predominantly a reflection of the charge transfer resistance (Rct). The smaller semicircle is indicative of better charge transfer kinetics. From the data given, it can be seen that PCN has a higher Rct compared to F-PCN. Considering PCN is not easily reduceable, the charge transfer process being observed in EIS is most likely emanating from a water reduction process, but as the impedance is high, the extent of this chemical reaction is minimal. In the case of F-PCN, the charge transfer resistance might correspond to the reduction of the F-PCN structure (also observed by CV measurements noted above). Interestingly, it was observed that Rct increased upon repeating EIS measurements with repeated cycles suggesting that this reduction reaction of F-PCN is hindered, see Fig. S9e,† however, if the EIS measurements were done after oxidizing the reduced material then Rct is found to return to a lower value immediately, clearly suggesting that for F-PCN there is a reduction process that occurs that can be reversed via a oxidation step, see Fig. S9f.†
For further insight into the nature of this reduced radical anion, UV-vis absorption, EPR, electrochemical and photo-electrochemical measurements on this species were performed. Fig. 1c shows that the radical has an absorption from 500 nm to 700 nm with maxima at 665 nm. In addition, there is a shoulder also at ca. 625 nm. The EPR of the solid blue sample, shown in Fig. 3a, confirms the presence of an unpaired electron species, which is not observed before light irradiation (yellow F-PCN). The more interesting observation is that a similar radical (with the same asymmetrical profile) can also be formed by irradiating the yellow sample but now without the presence of any quencher (4-MBA) under ambient conditions (Fig. 3b). The formed radical can be probed in the time scale of steady state EPR measurement, and interestingly this suggests that the excited carrier is sufficiently long-lived to be probed with EPR and, most likely, the excited electron and hole are not bound to each other, and in fact an exciton pair is not generated. However, in the case of PCN, light irradiation does not lead to the formation of this radical, even in the presence of the quencher.
 | ||
| Fig. 3 (a) EPR (Electron Paramagnetic Resonance) spectra of blue solid (photo-charged sample) in an inert environment (b) EPR (Electron Paramagnetic Resonance) spectra recorded during light irradiation on only pristine F-PCN (yellow solid) under ambient condition. (c) Photoluminescence (PL) of PCN and F-PCN suspended in water (at 300 nm excitation). (d) Time-resolved photoluminescence for PCN and F-PCN in water (at 295 nm excitation). | ||
Photophysical studies were performed to understand further the nature of the electronic modifications that the eutectic mixture treatment enforces onto PCN. The absorption measurements show that F-PCN has an additional absorption (ca. 450 nm) in the visible range (compared to PCN). The additional absorption comes from the charged moieties because as these moieties are removed from the structure through protonation (see Synthesis section in ESI and Fig. S10 and Table S5†), F-PCN loses this additional absorption as shown in Fig. S11.† It suggests that this augmented absorption could be crucial for the dual functionality observed in F-PCN. Furthermore, the steady-state PL measurement for PCN and F-PCN shows (Fig. 3c) that the PL intensity decreases substantially. It indicates that perhaps recombination ability decreases for F-PCN, and the excited state is more ‘trapped.’ This correlates with the EPR observation that, in F-PCN, the electron and hole are most likely not bound to each other, and the radical anion is long-lived. In comparison, solid F-PCN steady-state emission spectra are narrower than solid PCN emission spectra (Fig. S11†), suggesting that the F-PCN emissive states are more discrete. Time-resolved PL measurement (Fig. 3d) shows that the average lifetime for F-PCN is less compared to PCN. Interestingly, the Stokes shift (Fig. S11†) is less for F-PCN, suggesting a reduced energy gap between the excited and ground states of the fluorophore's electronic system. This may indicate a more closely spaced energy level structure or a stronger interaction between the excited and ground states.
A combination of OCP and chrono-potentiometry measurements were done to probe the charge/discharge capability of F-PCN (Fig. 4b). It needs to be noted that the discharging profile shows a plateauing at a fixed potential range of ca. −800 mV, and this profile is akin to a battery discharge profile rather than a capacitor discharge profile. It suggests that F-PCN shows faradaic behavior and charge storage is battery-adjacent as opposed to pseudocapacitive, as has been alluded to recently by Lotsch and co-workers.6
 | ||
| Fig. 4 (a) OCP measurement on F-PCN in the presence of different electrolytes (M2SO4 and 4-MBA) (b) Photo charging of F-PCN in the presence of different electrolytes for 20 minute (OCP) and subsequently electrical discharging with 2 mA g−1. All the measurements are done in an inert condition. | ||
It is essential to understand the factors that could contribute to augmenting the charge storage capability of F-PCN. Some factors that can intuitively be considered are the increasing number of excited charge carriers generated (i.e. molar absorptivity of the semiconductor) and, based on observations given above, concurrently the number of redox moieties (reducible functional groups synthesized onto F-PCN) should also contribute to increased charge storage. Presented here is another interesting observation derived from an earlier study from our group, which showcased that electrolyte ions play a pivotal role in altering the charge separation ability of F-PCN.23
This is due to the direct consequence of the ionic groups that are readily available to interact with cations in the electrolyte electrostatically. So despite keeping the number of redox moieties the same (i.e., the same F-PCN sample was evaluated), charge separation ability was found to be modulated by simply replacing Na2SO4 in the electrolyte with K2SO4 shown in Fig. S12,† where the photocurrent for F-PCN in the presence of K+ ions was almost 3 times that observed in the presence of Na+ ions. It needs to be noted that CV data (Fig. S13†) was similar whether Na+ or K+ ions were present; hence the type of redox moieties was not perturbed. OCP measurements (Fig. 4a) also showed that F-PCN could retain its excited potential (in the dark) for a prolonged period, almost 4 times longer, when the K+ cations were used instead of Na+. Since higher photocurrent was observed for the F-PCN in the presence of K+ ions than Na+ ions, this lends to the theory that more excited electrons are available, which should lead to more radical anions on F-PCN. The charging/discharging profile (Fig. 4b) shows that in the case of K+ ion, the additional charge carrier that can be extracted is still in the range of −800 mV potential (which was also observed for Na+). This means that the charge is trapped in similar redox moieties, albeit, now due to the presence of K+ ions, it can be inferred that additional redox moieties are now being accessed, leading to more stored charge – almost 4 times more than in the presence of Na+ ions (Fig. S14†). These sites were perhaps not being reduced when Na+ ions were present. Consequently, a longer discharge time and faster charging are observed for F-PCN in the presence of K2SO4 during the OCP measurement. This data suggests that despite using the same synthesized material, by simply changing the cation of the electrolyte, more of the redox sites of F-PCN can be accessed, leading to prolonged storage of the excited state.
Another aspect of the redox reaction was probed by altering the pH of the electrolyte during a CV measurement. The measurement shown in Fig. 5a is focused on how the reduction potential is altered when the pH of the electrolyte changes. The CV data suggests that as the pH of the electrolyte increases, the redox feature shifts towards a higher reducing potential. The shift in the reduction peak is ca. 700 mV when the pH is changed from 1 to 13, which correlates with the shift one would expect based on the Nernst equation due to the involvement of a proton in the reduction reaction. This value is calculated to be ca. 710 mV. The direction of the shift suggests that a PCET (Proton Coupled Electron Transfer) process is operating during the reduction reaction. Furthermore, the peak shift comfortably matches the potential of the accumulated electron shown in OCP measurements (Fig. 5b) at different pH values. Fig. 5b displays the increase in OCP as the pH increases. This interestingly provides an unusual handle to generate and store a photo-excited charge carrier over a substantially large potential range: – ca. 700 mV (without synthetically altering the structure). However, it must be noted that the material's capability to hold on to high potential decreases with an increase in pH, as with pH 13, there is hardly any prolonged retention observed.
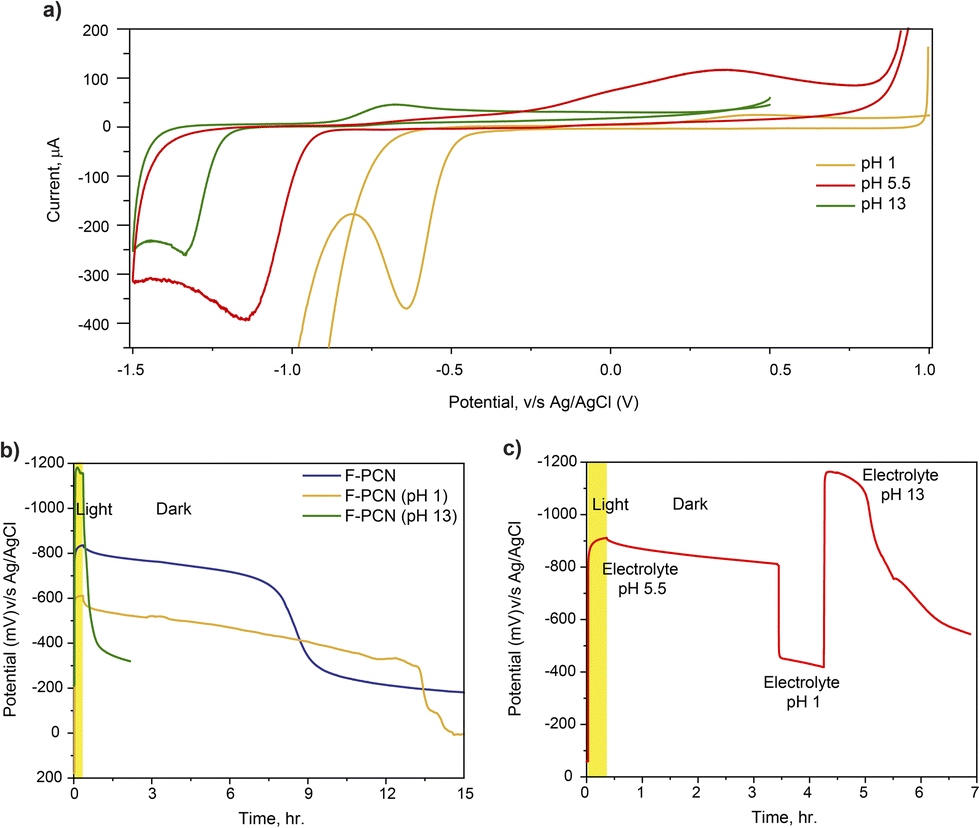 | ||
| Fig. 5 (a) Cyclic voltammetry on F-PCN at different pH of the electrolyte (M2SO4 and 4-MBA) (b) OCP measurement of F-PCN at different pH of the electrolyte (M2SO4 and 4-MBA) (c) OCP measurement on F-PCN, pH of electrolyte changed from 5.5 to 1 after 3 h of dark and again the pH to 13 after 4 h of dark. | ||
An intriguing extension is shown in Fig. 5c, where the OCP could be modulated now by varying the pH of the electrolyte at will. During photo-charging, the electrolyte was kept at a lower pH of 5.5. However, during the ‘retention’ step, the electrolyte pH was subsequently changed to 13. In this manner, with the help of pH, the potential at which the charge carriers (that were initially generated) were found to be altered at a later stage simply by changing the pH. This has ramifications in that aside from simply just storing the photo-excited electron, in principle, this augmented potential can further be utilized in a reaction to drive more challenging reactions in which species that need higher reduction potentials can be viably reduced.
Finally, one vital aspect of studying when exploiting any material for photo charging is the stability of the structure while it reversibly charges/discharges. Fig. 6 shows that the variation in OCP values upon light on and off conditions is cyclable. It was observed that F-PCN could return to a low potential state and, through light irradiation, could be augmented back to a high potential state. Also, very consistently, a similar amount of charge can be extracted from the material each time. It defines the intrinsic stability of the material even when it accumulates a large number of highly reducing photo-excited electrons.
 | ||
| Fig. 6 F-PCN cyclability (multiple time 20 minute photo-charging and subsequently electrical discharging with a current density of 2 mA g−1) using 0.1 M K2SO4 and 5 mM 4-MBA electrolyte. | ||
Based on all the data presented above, some aspects need to be highlighted, especially when F-PCN's structure is compared with other conventional semiconductors, as to why this particular structure seems to provide such a plethora of intriguing observations. We believe that because F-PCN is a non-crystalline polymer structurally, it can stabilize a photo-activated reduced state, and structural flexibility allows this deformation to the reduced state. In principle, crystalline semiconductors with well-resolved unit cells ideally would not be able to allow for this malleability. In addition, this structural flexibility could also be the main reason for the fast charging response observed in the OCP measurement. The material can be easily cycled between the reduced and oxidized states without activity loss. The synthesis of this polymer involves relatively facile controlled combustion of urea (in this manuscript, but other precursors can also be employed as has been detailed in other literature citations) followed by eutectic melt treatment. It seems that there is a probability of donor–acceptor entities embedded in the F-PCN matrix in which perhaps the acceptor states are localized entities that can be considered as deep traps but whose levels can be accessed and/or modulated by external environmental parameters such as the electrolytes' pH and ions. Current work is underway to elucidate the exact chemical structure of these donor–acceptor states.
Conclusions
In summary, the study showcases that an ionothermal treatment of carbon nitride provides a seminal change to the conventional carbon nitride structure that principally introduces the ability for redox behavior. As such, the study demonstrates that photo and redox activity are crucial for having the dual functionality of harvesting and storing solar energy simultaneously. It has been shown via electrochemical and photo-electrochemical studies that subsequent to photoexcitation, F-PCN can store the photo-excited electron in the form of a reduced acceptor state. Furthermore, the work suggests a pathway, via changing the pH of the electrolyte, to store the excited charge carrier over a range of potential (700 mV). Finally, the work demonstrates that simply changing the ions of the electrolyte (from Na+ to K+) can augment the storage of photo-excited electrons 4 times for the same period of irradiation. The study provides insight through which the energy density (by increasing the potential window and number of stored charge carriers) can be enhanced for the ionothermal-treated carbon nitride.Author contributions
RM performed all the synthesis and measurements. DK conceptualized and supervised the work. The manuscript was written with the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Prof. Jyotishman Dasgupta and Mr Dnyaneshwar P. Avhad for helping us with the EPR measurements. Mr Vilas J. Mhatre for the XRD measurements. Prof. Ankona Datta and Ms Deepika S. Furtado for the elemental (CHN) measurements. Dr Malay Patra and Mr Devidas A. Jadhav for their assistance with ICP-MS. DAE is gratefully acknowledged for funding this work through grant No. 19P0147.References
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- S. Chen, D. Huang, P. Xu, W. Xue, L. Lei, M. Cheng, R. Wang, X. Liu and R. Deng, J. Mater. Chem. A, 2020, 8, 2286–2322 RSC.
- A. D. Salunke, S. Chamola, A. Mathieson, B. D. Boruah, M. de Volder and S. Ahmad, ACS Appl. Energy Mater., 2022, 5, 7891–7912 CrossRef CAS.
- A. Gouder, F. Podjaski, A. Jiménez-Solano, J. Kröger, Y. Wang and B. V. Lotsch, Energy Environ. Sci., 2023, 16, 1520–1530 RSC.
- F. Podjaski, J. Kröger and B. V. Lotsch, Adv. Mater., 2018, 30, 1705477 CrossRef PubMed.
- M. J. Bojdys, J. O. Müller, M. Antonietti and A. Thomas, Chem.–Eur. J., 2008, 14, 8177–8182 CrossRef CAS PubMed.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- G. Zhang, L. Lin, G. Li, Y. Zhang, A. Savateev, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., 2018, 130, 9516–9520 CrossRef.
- L. Lin, C. Wang, W. Ren, H. Ou, Y. Zhang and X. Wang, Chem. Sci., 2017, 8, 5506–5511 RSC.
- Y. Markushyna, P. Lamagni, C. Teutloff, J. Catalano, N. Lock, G. Zhang, M. Antonietti and A. Savateev, J. Mater. Chem. A, 2019, 7, 24771–24775 RSC.
- A. Savateev, B. Kurpil, A. Mishchenko, G. Zhang and M. Antonietti, Chem. Sci., 2018, 9, 3584–3591 RSC.
- V. W. hei Lau, D. Klose, H. Kasap, F. Podjaski, M. C. Pignié, E. Reisner, G. Jeschke and B. V. Lotsch, Angew. Chem., Int. Ed., 2017, 56, 510–514 CrossRef PubMed.
- J. Kröger, F. Podjaski, G. Savasci, I. Moudrakovski, A. Jiménez-Solano, M. W. Terban, S. Bette, V. Duppel, M. Joos, A. Senocrate, R. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Adv. Mater., 2022, 34, 2107061 CrossRef PubMed.
- P. S. Wu, T. J. Lin, S. S. Hou, C. C. Chen, D. L. Tsai, K. H. Huang and J. J. Wu, J. Mater. Chem. A, 2022, 10, 7728–7738 RSC.
- A. Schwarzer, U. Böhme and E. Kroke, Chem.–Eur. J., 2012, 18, 12052–12058 CrossRef CAS PubMed.
- G. Zhang, G. Li, Z. A. Lan, L. Lin, A. Savateev, T. Heil, S. Zafeiratos, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2017, 56, 13445–13449 CrossRef CAS PubMed.
- D. C. Bradley and M. H. Gitlitz, J. Chem. Soc. A, 1969, 980–984 RSC.
- E. Alwin, W. Nowicki, R. Wojcieszak, M. Zieliński and M. Pietrowski, Dalton Trans., 2020, 49, 12805–12813 RSC.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 1700555 CrossRef PubMed.
- H. Ou, C. Tang, X. Chen, M. Zhou and X. Wang, ACS Catal., 2019, 9, 2949–2955 CrossRef CAS.
- R. Mann and D. Khushalani, ACS Appl. Mater. Interfaces, 2023, 15, 1219–1226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta03947a |
| This journal is © The Royal Society of Chemistry 2023 |