
Unifying CO2-to-fuel and biomass valorization over a metal-free 2D carbon nitride-fullerene heterostructure: a solar-driven chemical circular economy†
Deepak Kumar
Chauhan
,
Anjali
Verma
,
Ayushi
Jain
 ,
Neha
Saini
,
Pankaj Kumar
Prajapati
,
Chandan
Bera
and
Kamalakannan
Kailasam
*
,
Neha
Saini
,
Pankaj Kumar
Prajapati
,
Chandan
Bera
and
Kamalakannan
Kailasam
*
Institute of Nano Science and Technology (INST), Knowledge City, Sector-81 Manauli, SAS Nagar, 140306 Mohali, Punjab, India. E-mail: kamal@inst.ac.in; kkamal17@gmail.com
First published on 21st August 2023
Abstract
Photocatalytic CO2 photoreduction integrated with biomass oxidation is highly attractive to produce fuels and fine chemicals. Herein, for the first time, we manifested CO2 photoreduction to CO (>95% sel.) in synergy with biomass-based alcohol oxidation via a metal-free fullerene/2D carbon-nitride (C60/TUCN) semiconductor under solar simulated light. We observed that the composite, 5-C60/TUCN showed the highest CO2 to CO (selectivity >95%) conversion efficiency along with lignin biomass model substrate p-methoxybenzyl alcohol (p-MBA) oxidation to p-methoxybenzyaldehyde (p-MBAL) under solar simulated light with an excellent CO production rate of 8.92 mmol h−1 g−1 and p-MBAL production rate of 0.65 mmol h−1 g−1. The apparent quantum yield (AQY) of CO evolution was determined to be 3.38% at λ = 450 nm. DFT calculations confirmed that the C60 loading improved the activation and reduction of CO2 to CO while considerably lowering the formation barrier of COOH* intermediates. Besides, the accelerated separation and charge transfer kinetics of TUCN after C60 modification was confirmed from EPR, PL, and photocurrent studies. 13CO2 labeling experiments and EPR studies established the mechanistic pathway of the CO2 reduction reaction. Thus, the current study showed an excellent proof-of-concept for upscaling CO2 and biomass synergistically into solar fuels and fine chemicals, featuring a sustainable approach to boost the overall (bio)-chemical economy.
Photocatalytic carbon cycling is the most appealing and offers a green methodology to convert CO2 to fuels and chemical feedstock by effective utilization of the abundant renewable solar light.1 A carbon neutral cycle of coupling CO2 reduction with H2O oxidation, mimicking natural photosynthesis, i.e. artificial photosynthesis (AP) is an ideal green process.2 Over the past few decades AP has gained huge scientific interest and insights to convert CO2 into the corresponding gaseous products (for example, CO, CH4, and C2H4) and liquid products (for example, HCOOH, CH3OH, and C2H5OH) with the assistance of a wide range of photocatalysts including metal oxides,3,4 chalcogenides,5–7 organic semiconductors,8–10 organic-inorganic heterostructures,11,12 metal complexes,13 quantum dots,14 and chalcogenide-metal(oxide) heterostructures.15
However, the thermodynamically and kinetically sluggish H2O oxidation half-reaction could be replaced by an organic oxidation reaction to make use of photo-excited holes for purposeful oxidation processes. Photocatalytic CO2 reduction integrated with organic transformation offers the most fascinating process that features effective utilization of photogenerated electrons and holes to meet the criteria of sustainability and chemical economy overwhelmingly.16
To date few photocatalysts including CdS–TiO2,17 Cu2O/Cu,18 Cu/TiO2,19 Ag/TiO2,20 CdSe/CdS QDs,21 CdSQD/NC,22 Pd/TiO2,23 and CsPbBr3/Cs4PbBr6 (ref. 24) have shown the successful coupling strategy for CO2 photoreduction along with the organic oxidation process. However, the above mentioned photocatalytic systems encompass the use of metals, ultra-violet light absorption, toxicity and poor stability. To address these challenges the choice of photocatalysts must be highly anticipated.
Layered two-dimensional (2D) graphitic carbon nitride (g-C3N4),25 a metal-free photocatalyst turned out to be a wonderful choice; in particular with a moderate band gap (2.7 eV) it has become a promising photocatalyst for CO2 reduction. The improvements in the photocatalytic activity of bulk g-C3N4 by means of facile charge transfer are highly desirable and could efficiently be promoted by making 2D layered g-C3N4 structure and (non)-metal doping to it. Moreover, coupling 2D g-C3N4 with other organic functional materials including fullerene (C60), carbon nanotubes (CNTs), and graphene offers an excellent methodology towards facile charge transfer and advancing the photocatalytic process, and is much more benign compared to a metal-free system.
The dual photoredox process for coupling selective CO2 photoreduction with an organic oxidation process over 2D g-C3N4 based semiconductors is still in its infancy. Recently, Neumann's group synthesized a three-component heterojunction consisting of g-C3N4 as the semiconductor, polyoxometalate (POM, H3PW12O40) functioning as an electron shuttle to promote electron–hole pair separation, and an Re complex as the CO2 reduction co-catalyst.26 A cascade study was initiated by the dehydrogenation of cyclohexene to 1,3-cyclohexadiene (C6H8, two electron dehydrogenation product) and benzene (C6H6, four-electron dehydrogenation product) integrated with the reduction of CO2 to CO. Furthermore, Xue and co-workers prepared a biphasic photocatalyst, i.e. pyrene (Py) functionalized polymeric g-C3N4 (Py-PCN)27 for photocatalytic reduction of CO2 in the aqueous solution with simultaneous oxidation of alkene (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in the organic phase.
C) in the organic phase.
As an advancement towards the above discussed dual redox process, herein, for the first time we report a simple photocatalytic system in a metal-free fullerene/TUCN (C60/TUCN; TUCN = thiourea derived carbon nitride) organic semiconductor to drive CO2 photoreduction process integrated with biomass alcohol oxidation under solar simulated light. Here, we utilized C60 as the co-catalyst28 over 2D TUCN due to its excellent electron capture ability, which is beneficial for the CO2 reduction process and for achieving better charge separation ability. In our initial experiments, we found that amid all, 5 wt% of C60 on TUCN (5-C60/TUCN composite) showed efficient CO2 to CO conversion along with lignin biomass model substrate para-methoxybenzyl alcohol (p-MBA) oxidation to para-methoxybenzylaldehyde (p-MBAL) under solar simulated light (100 mW cm−2) with an excellent rate of 8.92 mmol h−1 g−1 (CO selectivity of >95%) and 0.65 mmol h−1 g−1, respectively.
Moreover, the state-of-the-art photocatalyst was compared with the existing literature having different photocatalytic systems; especially main emphasis was given to metal-free g-C3N4 based photocatalysts as shown in Table S1.† The current strategy offers a new horizon for the dual photo-redox process to achieve an efficient CO2 reduction process to produce solar fuels and biomass oxidation to fine chemicals, thus featuring a sustainable approach to boost the overall (bio)-chemical economy.
Herein, Scheme S1† demonstrates the schematic illustration of the C60/TUCN synthesis. Moreover, the detailed procedure for the C60/TUCN heterostructure synthesis is provided in the ESI† under the Experimental section. It is pertinent to mention that based on superior photocatalytic performance, the 5-C60/TUCN composite was characterized and presented all throughout the manuscript.
The crystal structure of the layered TUCN and 5-C60/TUCN composite was analyzed by XRD (Fig. 1a). The XRD pattern of TUCN clearly showed a strong diffraction peak at 27.3° and weak diffraction peak at 13.1°. The diffraction peaks at 13.1° and 27.3° correspond to the (100) plane of the in-plane ordering in a tri-s-triazine sheet and the (002) plane of the inter-planar stacking of the conjugated tri-s-triazine sheets, respectively.29 Compared with TUCN, the XRD pattern of 5-C60/TUCN exhibited diffraction peaks at 10.7, 17.7, 20.7, 21.78, 30.8, and 32.9 corresponding to the (111), (220), (311), (222), (422), and (511) planes, respectively, which can be attributed to the cubic phase of C60.30,31 Moreover, the intensity and position of the peak at 27.3° did not show any changes relative to that of bare TUCN, manifesting that immobilization of C60 on the layered structure of TUCN did not influence the crystal structure of TUCN.
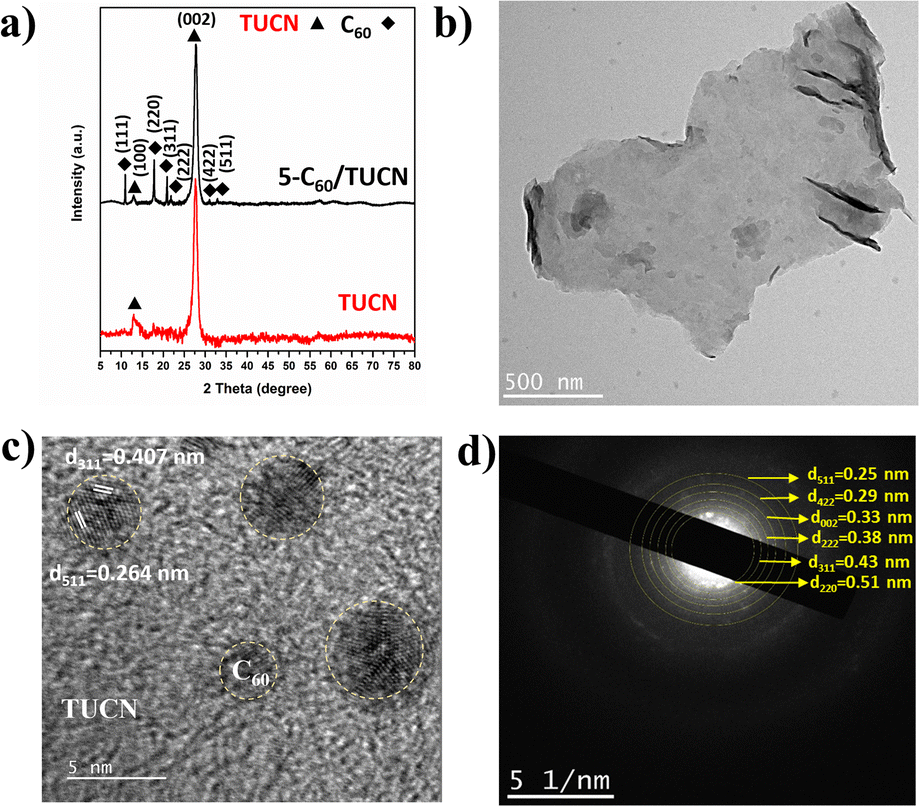 | ||
| Fig. 1 (a) XRD patterns of TUCN and 5-C60/TUCN, and (b) TEM image, (c) HR-TEM image, and (d) TEM-SAED pattern of 5-C60/TUCN. | ||
The morphology of TUCN and the 5-C60/TUCN composite was investigated via Fe-SEM (Fig. S1†) and TEM analysis (Fig. 1b–d). As observed from Fig. 1b, 5-C60/TUCN showed a 2D sheet-like structure where C60 is immobilized on the TUCN sheets. Moreover, the high-resolution image of 5-C60/TUCN exhibited lattice fringes with a d spacing of 0.407 nm and 0.264 nm corresponding to the (311) and (511) planes of crystalline C60 (Fig. 1c), which was consistent with the XRD results. The crystallinity of 5-C60/TUCN was further proved by the TEM-SAED pattern (Fig. 1d). It was observed that small brighter spots of crystalline C60 were distributed over the surface of TUCN. Concentric rings of C60 planes (220), (311), (222), (422), and (511), were found to be closely surrounded by the TUCN (002) plane, revealing the formation of a composite with strong interfacial contact between C60 and TUCN. These results were found to be in good agreement with the XRD results.
The N2 adsorption–desorption isotherm analysis indicated that TUCN and 5-C60/TUCN displayed a type IV isotherm with narrow H3 hysteresis loops, indicating the development of a mesoporous structure in the form of parallel plates (Fig. S2a†).32,33 Based on the BET analysis, the surface area of TUCN and 5-C60/TUCN was found to be identical i.e. 28 m2 g−1 and 26 m2 g−1, respectively. This significantly demonstrated that the incorporation of C60 into the TUCN network did not affect the surface area, which indicated that it is not the main factor promoting the photocatalytic performance, while the enhanced light absorption and fast charge carrier transportation abilities helped to improve the photocatalytic activity.34
The surface compositions of the material were scrutinized via XPS analysis. The XPS survey spectrum of TUCN and 5-C60/TUCN showed that the surface of the material contains C, N, and O elements (Fig. S2b†). High resolution C 1s spectra of TUCN showed three peaks at 288.4, 286.8, and 284.8 eV which corresponded to the existence of the N–CN structure of tri-s-triazine, C–NHx groups, and adventitious carbon (C–C) used to calibrate the binding energies, respectively. The C 1s peaks of 5-C60/TUCN were found to be quite consistent with those of TUCN (Fig. S2c†), indicating that the existence of C60 over the surface of TUCN and did not affect the C 1s electronic structure of TUCN. Fig. S2d† illustrates that the high resolution N 1s spectrum could be deconvoluted into four peaks. In TUCN, an intense peak at 398.5 eV showed the existence of an sp2-hybridized pyridinic nitrogen atom linked with a carbon atom (CN–C), and the peaks at 399.8 eV and 400.8 eV are attributed to the tertiary N–(C)3 and –NHx functional groups in the carbon nitride core, i.e. the tri-s-triazine motif, respectively. A weak N 1s peak at 404.1 eV was also observed which basically originated from π-excitation.35 The N 1s peaks of 5-C60/TUCN were also found to be completely consistent with those of TUCN. It is evident that C60 deposition over TUCN led to the enhancement in the carbon “C” content that was verified by using the XPS N/C ratio.30 The decrease in the N/C ratio from TUCN (1.18) to 5-C60/TUCN (1.03) underpinned the high “C” content over the surface of 5-C60/TUCN (Table S2†). This explicitly showed the formation of a heterostructure between C60 and TUCN. Moreover, the clear evidence of the enhanced “C” content over the surface of 5-C60/TUCN was further investigated by elemental CHNS analysis. The % of the elemental composition and N/C ratio are highlighted in Table S3.† The decreased N/C ratio from TUCN (1.96) to 5-C60/TUCN (1.65) significantly manifested the C60 loading as “C” content in 5-C60/TUCN.
Of note, the optical and electronic properties of TUCN and 5-C60/TUCN were examined to overlay the difference in properties which is responsible for the photocatalytic CO2 reduction and biomass alcohol oxidation. UV-vis diffuse reflectance absorption spectroscopy demonstrated broad and visible light absorption by TUCN and 5-C60/TUCN. As shown in Fig. 2a, bare TUCN exhibited an absorption edge at 435 nm, which was assigned to the electronic transition from N 2p to C 2p orbitals of TUCN. On the other hand, the light absorption ability of 5-C60/TUCN in the visible region becomes more prominent, suggesting that C60 loading enhanced the light absorption of TUCN.31 Moreover, the Kubelka–Munk function equation was used to calculate the Tauc plot for the optical band gap (Eg) energy estimation. The Eg of TUCN and C60/TUCN was found to be 2.81 eV and 2.73 eV, respectively (Fig. 2a: inset).
 | ||
| Fig. 2 (a) Diffuse reflectance UV vis spectra of TUCN and 5-C60/TUCN; inset (Tauc plot), (b) band energy diagram of TUCN and 5-C60/TUCN, (c) PL spectra of TUCN and 5-C60/TUCN, and (d) TRPL spectra of TUCN and 5-C60/TUCN. | ||
Mott–Schottky (MS) analysis was carried out to determine the conduction band (CB) position of TUCN and 5-C60/TUCN via flat-band potential (Vfb) estimation (Fig. S2a and b†). The positive slope of the plots demonstrated the typical characteristics of n-type semiconductors. It is worth mentioning that Vfb generally lies 0.1 V below the CB of n-type semiconductors.35 The Vfb of TUCN and 5-C60/TUCN was estimated to be −1.10 V and −1.20 V. On this account, the CB values of TUCN and 5-C60/TUCN were estimated to be −1.20 V and −1.30 V (vs. Ag/AgCl). On the NHE scale, the CB values were calculated to be −1.0 V and −1.10 V for TUCN and 5-C60/TUCN by using the ENHE = EAg/AgCl + 0.197 V equation. Moreover, the valence band (VB) positions of TUCN (+1.81 V) and 5-C60/TUCN (+1.63 V) were estimated by combining the obtained band gap from the Tauc plot with the relative CB positions from the EVB = ECB + Eg equation. The relative electronic band structures of TUCN and 5-C60/TUCN are shown in Fig. 2b.
The charge-transfer ability of TUCN and 5-C60/TUCN was investigated by using photoluminescence (PL), time-resolved photoluminescence (TRPL), transient photocurrent responses (TPR), and the Nyquist plot. The recorded PL spectra of 5-C60/TUCN demonstrated a prominent decrease in the PL intensity compared with that of TUCN, favoring a low rate of photo-generated charge carrier recombination or feasible charge transfer in 5-C60/TUCN (Fig. 2c). The TRPL spectra were collected to estimate the prominent interfacial transition of the photogenerated charge carriers and average lifetime 〈τ〉 of photogenerated charge carriers (Fig. 2d). Comparatively highest 〈τ〉 for 5-C60/TUCN (4.9 ns) to that of TUCN (4.3 ns) is observed, revealing that 5-C60/TUCN could furnish a longer time for photogenerated charge carriers to take part in the surface photoreaction, thereby amplifying its photocatalytic performance.36 Moreover, enhanced interfacial charge carrier separation and migration was also measured by TPR study under five periodic on/off (30 s) light cycles (Fig. S3c†), which exhibited significant and repeatable photocurrent responses. Interfacial catalytic charge transfer kinetics of the material was examined with the help of the electrochemical impedance spectroscopy (EIS)-Nyquist plot (Fig. S3d†). A decrease in the semicircle arc radius of 5-C60/TUCN compared to that of TUCN manifested the decrease in facial charge transfer resistance. This promoted the charge transfer efficiency on the catalyst interface of C60 and TUCN. Invariably, the above results manifested that existence of the 2D layered structure of 5-C60/TUCN promoted facile charge separation and migration.
Adsorption behavior of CO2 over the surface of pthe hotocatalyst was recognized by using CO2-temperature-programmed desorption (CO2-TPD) analysis (Fig. S4†). Basically, the 5-C60/TUCN surface nitrogen active sites (–NH2 dangling groups) act as electron donors (Lewis base) for CO2 molecules (Lewis acid). Therefore, a CO2-TPD analysis was employed to quantify the desorbed CO2 molecules over the surface of 5-C60/TUCN upon heating, which is regarded as explicit evidence of the structural basicity of the TUCN core. It is evident that the 5-C60/TUCN sample demonstrated two broad desorption peaks from 90–130 °C and 220–245 °C, manifesting physical and chemical adsorption of CO2 molecules on weak basic sites of TUCN (–NH2 groups).22
Obviously, the large negative CB potential of 5-C60/TUCN could feasibly promote the reduction of CO2 to CO via the two electron reduction process.37 Moreover, the VB potential of 5-C60/TUCN was utilized for p-MBA oxidation (lignin model substrate) to the corresponding aldehyde (p-MBAL) in synergy with CO production. The illustrative diagram of the photocatalytic reaction setup is shown in Scheme S2.† The photocatalytic performance of different photocatalysts under a solar simulator (AM 1.5G, 100 mW cm−2) was tested and is summarized in Table 1.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Light | Conv.b % | p-MBAL sel.b % | Production rate (mmol h−1 g−1) CO p-MBAL | |
| a Reaction conditions: catalyst (10 mg), CO2 purging for 30 min. p-MBA (0.1 mmol), 3 mL ACN, reaction time 6 h, room temperature (25 °C) and light source: solar simulator (AM 1.5G, 100 mW cm−2). b The conversion and selectivity were obtained by GC-MS. c In the absence of light. d Without a photocatalyst. e In the absence of co-catalyst, C60. N.A. – not applicable. | ||||||
| 1 | 2.5-C60/TUCN | ✓ | 20 | >99 | 3.24 | 0.33 |
| 2 | 5-C60/TUCN | ✓ | 32 | >99 | 9.80 | 0.65 |
| 3 | 7.5-C60/TUCN | ✓ | 25 | >99 | 7.61 | 0.42 |
| 4 | 10-C60/TUCN | ✓ | 21 | >99 | 4.80 | 0.35 |
| 5c | 5-C60/TUCN | × | N.A. | N.A. | N.A. | N.A. |
| 6d | × | ✓ | N.A. | N.A. | N.A. | N.A. |
| 7e | TUCN | ✓ | <5 | >99 | 0.10 | 0.04 |
It was observed that among all C60/TUCN composites, the 5-C60/TUCN composite showed the highest dual photo redox activity for simultaneous CO2 reduction and p-MBA oxidation to produce CO and p-MBAL in a synergistic manner (Table 1, entry 1–4). In the case of 5-C60/TUCN, production rates of CO and p-MBAL oxidation rates were found to be 8.92 (mmol h−1 g−1) and 0.65 (mmol h−1 g−1), respectively. The selectivity for CO and p-MBAL was found to be >95% and >99%, respectively. Furthermore, the requisite conditions such as light, photocatalyst, and C60 co-catalyst were found to be indispensable parameters to favor the photocatalytic performance (Table 1, entry 4–7). Moreover the liquid products were identified by GC-MS analysis (Fig. S5†). Meanwhile, along with the major gaseous CO product (>95% sel.), comparatively, a very low amount of CH4, and H2 was also generated which was confirmed by GC analysis (Fig. S6†). The photocatalytic activity of 5-C60/TUCN was further tested for CO2 reduction to CO in synergy with different biomass alcohol oxidations under similar reaction conditions as listed in Table S4.†
The photocatalytic efficiency for CO production was evaluated in terms of the apparent quantum yield (AQY). The AQY of CO evaluation was estimated to be 3.38% at λ = 450 nm.
It was shown that all the biomass based alcohols showed good photocatalytic activity under a CO2 atmosphere, thus promoting the CO2 reduction rate for selective production of CO. p-MBA and veratryl alcohol displayed almost similar and highest % conversion of the alcohols to the corresponding aldehydes, while HMF showed quite a sluggish % conversion among all the biomass based alcohols with only a conversion of 7% into the corresponding aldehyde i.e. diformylfuran (DFF).
Moreover, it should be noted that the moderate oxidation ability of the photogenerated holes (h+) over the TUCN surface can prevent the formation of non-selective ˙OH radicals due to the less positive VB potential of 5-C60/TUCN (EVB + 1.63 V). In addition, non-aqueous acetonitrile as a solvent also prevents the generation of ˙OH via h+. Therefore, high selectivity >99% of the oxidized products was observed as corroborated through liquid mixture analysis with the help of the GC-MS technique (Table S4†).
The recyclability/stability of 5-C60/TUCN was checked subsequently by performing five cyclic runs. The dual photo redox performance over 5-C60/TUCN indicated that the production rates of CO and p-MBAL were slightly decreased to 7.8 mmol h−1 g−1 and 0.49 mmol h−1 g−1, respectively, after five runs (Fig. S7a†). Moreover, the selectivity of p-MBAL (>99%) remained constant throughout all cycling runs. On comparing the XRD and TEM analysis of 5-C60/TUCN before and after the cycle, no significant changes were observed in the crystal structure and surface morphologies of 5-C60/TUCN, respectively (Fig. S7b and c†).
A plausible mechanism for dual photo redox performance of 5-C60/TUCN illustrating the coupling strategy of CO2 reduction to CO with p-MBA oxidation by effective utilization of electrons and holes could be underlined by performing some control experiments (Table S5†). Under an argon (without CO2) atmosphere, a trace amount of H2 and only 5% conversion of p-MBA to p-MBAL were observed (Table S5,† entry 1), implying that CO could be realized from CO2 photoreduction. To corroborate this, an isotopic labeling experiment was performed by using a 13CO2 gas source instead of 12CO2 to authenticate the origin of the produced CO. Gas chromatography-mass spectrometry (GC-MS) analysis distinctly showed the observed signal of 13CO with an m/z value of 29 (Fig. S8†) in the reaction system, manifesting that the detected CO originated from CO2.
Moreover, when an electron scavenger (AgNO3) was added to the reaction medium saturated with a CO2 atmosphere, no gaseous CO species were observed, implying that the photogenerated electrons were utilized by AgNO3 and were barely available for the CO2 photoreduction (Table S5,† entry 2). Although in the case of IPA, the CO production rate was enhanced comparatively (good electron donor) but p-MBA observed mere oxidation/conversion to p-MBAL which could be ascribed to the excellent hole scavenging ability of IPA (Table S5,† entry 3).
On comparison, the more negative Vfb value of 5-C60/TUCN (−1.10 V) than that of TUCN (−1.0 V), enables significant band bending that eventually facilitates the transport of photogenerated electrons efficiently to drive the CO2 reduction reaction. In addition, the electron transfer mechanism involved in CO2 photoreduction to CO over the surface of the 5-C60/TUCN photocatalyst was verified by electron paramagnetic resonance (EPR), demonstrating the excellent structure–activity relationship between the 5-C60/TUCN photocatalyst and CO2 molecules. Under dark and inert conditions, no peak of the products was observed. Moreover, when the 5-C60/TUCN photocatalyst was exposed to visible light and an inert atmosphere, a new EPR signal appeared at g = 2.004, which was attributed to the surface –NH2 amino groups. Interestingly, when the EPR spectra of the 5-C60/TUCN photocatalyst were recorded under a CO2 atmosphere, a comparatively reduced EPR signal appeared at g = 2.004, manifesting a strong interaction between the adsorbed CO2 species (Lewis acid) and the surface active sites (–NH2 groups, Lewis base) of the 5-C60/TUCN photocatalyst (Fig. S9†).
This significantly entailed that the strong CO2 adsorption over the surface of the 5-C60/TUCN photocatalyst was also confirmed via CO2-TPD analysis. The reduced EPR signal under light irradiation facilitated easy interfacial charge transfer from the photocatalyst, promoting the facile photoreduction process of the CO2 molecule to CO.38,39
To further elucidate the interfacial charge transfer in the optimized 5-C60/TUCN heterostructure, the density of states (DOS) was computed, as shown in Fig. S10,† and the band gap of 5-C60/TUCN was calculated to be 2.02 eV, which was found to be lower than the band gap of TUCN (2.82 eV), as calculated by the GGA method. Notably, the band gap of TUCN (2.82 eV) matched well with the experimental result (2.81 eV); however, the band gap of 5-C60/TUCN (2.02 eV) was found to be lower than the experimental result (2.73 eV). This might be due to the delocalization of the electron density with an inaccuracy of the HSE06 hybrid functional, causing underestimation of the band gap using this method.40 The band gap reduction after the heterostructure formation between C60 and TUCN resulted in facile charge carrier generation, and facilitates efficient charge separation and migration. Fig. S11† illustrates the optimized geometric structures of the TUCN monolayer, the C60 molecule, and their heterostructure with a top view and side view, respectively.
Theoretical studies demonstrated that in the 5-C60/TUCN heterostructure, due to the weak van der Waals interaction of C60 with 2D TUCN,41 a strong bend occurred in the 2D layer of TUCN, implying a strong strain in the TUCN monolayer (Fig. S11c†). This may alter the atomic structure, leading to change in its electronic properties, which seemed to be favorable for facile charge transfer42 which in turn, resulted in enhancement of the photocatalytic activity of the 5-C60/TUCN photocatalyst.
Moreover, the elementary steps of CO2 photoreduction selectively to CO on TUCN and the 5-C60/TUCN surface were further revealed by DFT calculations. The energies directly obtained from DFT and calculated Gibbs free energies (ΔG) are listed in Tables S6 and S7.† As shown in Fig. 3a, the calculated ΔG of CO2-to-CO conversion involves the following steps: (i) first, the CO2 molecules are adsorbed on the photocatalyst surface; (ii) generation of a COOH* intermediate through a proton–electron pair transfer process on the CO2 adsorbed surface. Since the ΔG for this step is uphill by 1.43 eV for TUCN and 1.19 eV for 5-C60/TUCN, this result significantly corroborated that the CO2 reduction reaction catalyzed by 5-C60/TUCN is more favorable than that catalyzed by TUCN. Afterward, (iii) the reaction intermediate (COOH*) further couples with a proton–electron pair to generate H2O and CO* detached from the surface to generate the CO product which is characterized by the release of the energy.
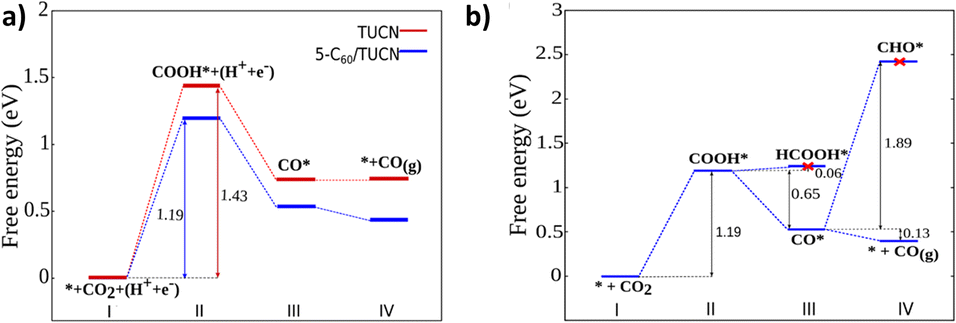 | ||
| Fig. 3 (a) Calculated free energy diagram corresponding to the reaction path followed by the CO2 conversion on TUCN and 5-C60/TUCN and (b) key steps of CO2 photoreduction to CO for the 5-C60/TUCN system. | ||
Additionally, the photoreduction process of CO2 for selective CO production over CH4 and CH3OH could further be verified via the thermodynamic free energy (ΔG) barrier. As shown in Scheme 1, four reaction paths are proposed for CO2 photoreduction on the surface of 5-C60/TUCN, where * denotes the catalytically active sites. The transformation of COOH* species into CO* is easily realized compared to that of COOH* into HCOOH* (path-1) over 5-C60/TUCN due to the downhill free energy profiles of COOH* to CO* as shown in Fig. 3b. The free energy change of CO* to CO (path-2) is downhill by 0.13 eV while in contrast, the formation of CHO* demands an input energy of 1.89 eV. This is the key selectivity step between path-2 and path-3 & 4. This result indicates that CO is a more favorable product of the CO2 reduction reaction than CH4 and CH3OH over the 5-C60/TUCN surface, which was elucidated in the experimental analysis described above.
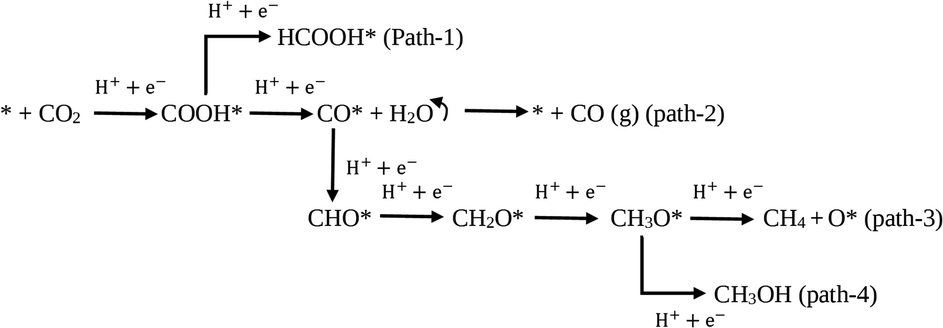 | ||
| Scheme 1 Proposed reaction paths for CO2 reduction on the surface of 5-C60/TUCN. | ||
In the synergistic photo-redox process, it is challenging to maintain the stoichiometry of the photoredox products. Herein, it was observed that the CO production rate was found to be multifold higher than the oxidation rate of the alcohols to the corresponding aldehydes. The multifold CO production rate over the p-MBAL production rate could be substantially induced by C60, as it acts as an excellent electron acceptor in the ground state and can accept, reversibly, up to six electrons.43 Moreover, the moderate HOMO–LUMO energy gap (∼1.5 eV) of C60 separates a maximally degenerate and fully occupied HOMO (5-fold) from a degenerate LUMO (3-fold). Particularly, in the presence of a suitable electron-donor or reducing agent, the electron transfer and reduction reaction can be accelerated efficiently.44 Therefore, in the present photo-redox process, excited state electron generation over C60 coupled with surplus electron transfer from the CB of TUCN to C60 resulted in a high CO production rate, in the presence of p-MBA as the electron-donor or reducing agent. Notably, the other possible reason is that the large difference between e− and h+ migration rates leads to the accumulation of photogenerated h+ in the bulk and makes the h+ short-lived.45,46 Therefore, a comparatively low production rate of p-MBAL was observed during the photoredox process.
From the above results, we proposed the following mechanistic pathway as shown in Scheme 2. First, upon light excitation charge carriers (e−/h+) were generated over the photocatalyst surface. The e− got excited from the VB of TUCN to its CB, leaving h+ at the VB of TUCN. The C60 co-catalyst acted as an electron trapper, and e− from the CB of TUCN transferred immediately to C60. In contrast, the photogenerated h+ over the surface of the photocatalyst was exploited feasibly to promote the oxidation of p-MBA to p-MBAL.
 | ||
| Scheme 2 Plausible mechanistic illustration of dual photoredox CO2 reduction to CO coupled with p-MBA oxidation over the 5-C60/TUCN photocatalyst. | ||
The discussion made above reinforced the excellent potency of the C60 molecule towards the photoredox process and such an ability significantly provided a proof-of-concept for the effective utilization of e−/h+ pairs synergistically for solar fuel generation and organic transformations over a metal-free surface.
Conclusions
In summary, we successfully demonstrated the first ever coupling strategy of CO2 reduction to CO with biomass based aromatic alcohol oxidation under solar simulated light irradiation (AM 1.5G, 100 mW cm−2) over the metal-free 5-C60/TUCN photocatalyst by effective utilization of e− and h+, respectively. Deposition of C60 over 2D TUCN not only promoted an efficient charge transfer ability as observed in PL, TRPL and TPR studies, but also offered an excellent CO production rate (8.92 mmol h−1 g−1) and p-MBA oxidation rate (0.65 mmol h−1 g−1), synergistically. Maximum 3.38% AQY was achieved for CO evolution at λ = 450 nm. 13CO2 labelling experiments, EPR studies and DFT calculations established the mechanistic pathway of the CO2 reduction reaction. This work thus provides an insight for the fabrication of metal-free organic semiconductors with carbon nitrides for solar energy utilization which in turn, achieves the sustainable dual photoredox process simultaneously for up scaling CO2 to solar fuels coupled with biomass conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Central Research Facility, IIT Delhi Sonepat campus, for the EPR facility.References
- S. Yao, J. He, F. Gao, H. Wang, J. Lin, Y. Bai, J. Fang, F. Zhu, F. Huang and M. Wang, J. Mater. Chem. A, 2023, 11, 12539–12558 RSC.
- S. Yoshino, T. Takayama, Y. Yamaguchi, A. Iwase and A. Kudo, Acc. Chem. Res., 2022, 55, 966–977 CrossRef CAS PubMed.
- B. Pan, J. Qin and C. Wang, ed. X. Wang, M. Anpo, X. Fu, Current Developments in Photocatalysis and Photocatalytic Materials New Horizons in Photocatalysis, Elsevier, 2020, pp. 77–87 Search PubMed.
- Y. Cui, X. Lian, L. Xu, M. Chen, B. Yang, C. Wu, W. Li, B. Huang and X. Hu, Materials, 2019, 12(2), 276 CrossRef CAS PubMed.
- S. Adabala and D. P. Dutta, J. Environ. Chem. Eng., 2022, 10, 107763 CrossRef CAS.
- M. T. Leng Lai, C. W. Lai and J. C. Juan, in Micro and Nano Technologies, ed. M. M. B. T.-C.-B. N. as P. Khan, Elsevier, 2021, pp. 295–306 Search PubMed.
- Y. Wang, B. Ren, J. Zhen Ou, K. Xu, C. Yang, Y. Li and H. Zhang, Sci. Bull., 2021, 66, 1228–1252 CrossRef CAS PubMed.
- H. L. Nguyen and A. Alzamly, ACS Catal., 2021, 11, 9809–9824 CrossRef CAS.
- Y. Zhang, H. Liu, F. Gao, X. Tan, Y. Cai, B. Hu, Q. Huang, M. Fang and X. Wang, EnergyChem, 2022, 4, 100078 CrossRef CAS.
- W. Zhan, H. Gao, Y. Yang, X. Li and Q.-L. Zhu, Adv. Energy Sustainability Res., 2022, 3, 2200004 CrossRef CAS.
- P. K. Prajapati, N. Saini, D. K. Chauhan and K. Kailasam, J. Mater. Chem. A, 2023, 11, 385–400 RSC.
- N. Ahmad, C.-F. Jeffrey Kuo, M. Mustaqeem, M. K. Hussien and K.-H. Chen, Mater. Today Phys., 2023, 31, 100965 CrossRef CAS.
- H. Kumagai, Y. Tamaki and O. Ishitani, Acc. Chem. Res., 2022, 55, 978–990 CrossRef CAS PubMed.
- H.-L. Wu, X.-B. Li, C.-H. Tung and L.-Z. Wu, Adv. Mater., 2019, 31, 1900709 CrossRef PubMed.
- X. Li, Z. Wang, J. Zhang, K. Dai, K. Fan and G. Dawson, Mater. Today Phys., 2022, 26, 100729 CrossRef CAS.
- L. Yuan, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Angew. Chem., Int. Ed., 2021, 60, 21150–21172 CrossRef CAS PubMed.
- G. Song, F. Xin, J. Chen and X. Yin, Appl. Catal., A, 2014, 473, 90–95 CrossRef CAS.
- Y. Chen, M. Wang, Y. Ma, Y. Li, J. Cai and Z. Li, Catal. Sci. Technol., 2018, 8, 2218–2223 RSC.
- T. Yang, Q. Yu and H. Wang, Catal. Lett., 2018, 148, 2382–2390 CrossRef CAS.
- L. Wang, X. Zhang, L. Yang, C. Wang and H. Wang, Catal. Sci. Technol., 2015, 5, 4800–4805 RSC.
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS.
- F. Wang, T. Hou, X. Zhao, W. Yao, R. Fang, K. Shen and Y. Li, Adv. Mater., 2021, 33, 2102690 CrossRef CAS.
- R. Zhang, H. Wang, S. Tang, C. Liu, F. Dong, H. Yue and B. Liang, ACS Catal., 2018, 8, 9280–9286 CrossRef CAS.
- G.-X. Dong, W. Zhang, Y.-F. Mu, K. Su, M. Zhang and T.-B. Lu, Chem. Commun., 2020, 56, 4664–4667 RSC.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- H. Yu, E. Haviv and R. Neumann, Angew. Chem., Int. Ed., 2020, 59, 6219–6223 CrossRef CAS PubMed.
- X. Gong, S. Yu, M. Guan, X. Zhu and C. Xue, J. Mater. Chem. A, 2019, 7, 7373–7379 RSC.
- M. Chen, R. Guan and S. Yang, Adv. Sci., 2019, 6, 1800941 CrossRef PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- L. Song, T. Li and S. Zhang, J. Phys. Chem. C, 2017, 121, 293–299 CrossRef CAS.
- X. Chen, K. Deng, P. Zhou and Z. Zhang, ChemSusChem, 2018, 11, 2444–2452 CrossRef CAS PubMed.
- M. O. Fuentez-Torres, F. Ortiz-Chi, C. G. Espinosa-González, M. Aleman, A. Cervantes-Uribe, J. G. Torres-Torres, M. K. Kesarla, V. Collins-Martínez, S. Godavarthi and L. Martínez-Gómez, Top. Catal., 2021, 64, 65–72 CrossRef CAS.
- X. Song, Q. Yang, M. Yin, D. Tang and L. Zhou, RSC Adv., 2018, 8, 7260–7268 RSC.
- J. Cheng, C. Li, Z. Yu and H. Liu, J. Colloid Interface Sci., 2023, 629, 739–749 CrossRef CAS PubMed.
- D. K. Chauhan, V. R. Battula, A. Giri, A. Patra and K. Kailasam, Catal. Sci. Technol., 2022, 12, 144–153 RSC.
- P. Sharma, S. Kumar, O. Tomanec, M. Petr, J. Zhu Chen, J. T. Miller, R. S. Varma, M. B. Gawande and R. Zbořil, Small, 2021, 17, 2006478 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- D. Sun, Y. Fu, W. Liu, L. Ye, D. Wang, L. Yang, X. Fu and Z. Li, Chem. – Eur. J., 2013, 19, 14279–14285 CrossRef CAS PubMed.
- W. Tu, Y. Yang, C. Chen, T. Zhou, T. Li, H. Wang, S. Wu, Y. Zhou, D. O'Hare, Z. Zou and R. Xu, Small Struct., 2023, 4, 2200233 CrossRef CAS.
- J. Xue, M. Fujitsuka and T. Majima, ACS Appl. Mater. Interfaces, 2019, 11, 40860–40867 CrossRef CAS PubMed.
- Q. Li, L. Xu, K.-W. Luo, W.-Q. Huang, L.-L. Wang, X.-F. Li, G.-F. Huang and Y.-B. Yu, Phys. Chem. Chem. Phys., 2016, 18, 33094–33102 RSC.
- G. Gao, Y. Jiao, F. Ma, Y. Jiao, E. Waclawik and A. Du, J. Catal., 2015, 332, 149–155 CrossRef CAS.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed.
- L. Brunet, D. Y. Lyon, E. M. Hotze, P. J. J. Alvarez and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4355–4360 CrossRef CAS PubMed.
- X. Zhang, P. Wang, X. Lv, X. Niu, X. Lin, S. Zhong, D. Wang, H. Lin, J. Chen and S. Bai, ACS Catal., 2022, 12, 2569–2580 CrossRef CAS.
- Q. Liu, S. Wang, W. Mo, Y. Zheng, Y. Xu, G. Yang, S. Zhong, J. Ma, D. Liu and S. Bai, Small, 2022, 18, 2104681 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04455f |
| This journal is © The Royal Society of Chemistry 2023 |