
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of Eudragit® as a component of hydrogel formulations for medical devices†
David
Esporrín-Ubieto
 a,
Ana Sofía
Sonzogni
b,
Mercedes
Fernández
c,
Arantxa
Acera
dh,
Eider
Matxinandiarena
e,
Juan F.
Cadavid-Vargas
f,
Itxaso
Calafel
c,
Ruth N.
Schmarsow
g,
Alejandro J.
Müller
eh,
Aitor
Larrañaga
i and
Marcelo
Calderón
*ah
a,
Ana Sofía
Sonzogni
b,
Mercedes
Fernández
c,
Arantxa
Acera
dh,
Eider
Matxinandiarena
e,
Juan F.
Cadavid-Vargas
f,
Itxaso
Calafel
c,
Ruth N.
Schmarsow
g,
Alejandro J.
Müller
eh,
Aitor
Larrañaga
i and
Marcelo
Calderón
*ah
aPOLYMAT, Applied Chemistry Department, Faculty of Chemistry, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizabal 3, 20018 Donostia-San Sebastián, Spain. E-mail: marcelo.calderon@polymat.eu
bGroup of Polymers and Polymerization Reactors, INTEC (Universidad Nacional del Litoral-CONICET), Güemes 3450, 3000 Santa Fe, Argentina
cPOLYMAT Institute for Polymer Materials, University of the Basque Country UPV/EHU, San Sebastián, 20018, Spain
dDepartment of Cell Biology and Histology, Experimental Ophthalmo – Biology Group (GOBE, www.ehu.eus/gobe), University of the Basque Country UPV/EHU. B Sarriena, sn, 48940 Leioa, Bizkaia, Spain
ePOLYMAT, Department of Polymers and Advanced Materials: Physics, Chemistry and Technology, University of the Basque Country UPV/EHU, Paseo Manuel de Lardizábal 3, 20018 Donostia-San Sebastián, Spain
fINIFTA-CONICET-UNLP, Instituto de Investigaciones Fisicoquímicas Teóricas y Aplicadas, Diagonal 113 y 64, 1900 La Plata, Argentina
gInstitute of Materials Science and Technology (INTEMA), University of Mar del Plata and National Research Council (CONICET), Av. Cristóbal Colón 10850, 7600 Mar del Plata, Argentina
hIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
iDepartment of Mining, Metallurgy Engineering and Materials Science, POLYMAT, Faculty of Engineering in Bilbao, University of the Basque Country (UPV/EHU), Plaza Torres Quevedo 1, 48013 Bilbao, Spain
First published on 12th September 2023
Abstract
Over the last decade, significant progress has been made in developing hydrogels as medical devices. By physically cross-linking pharmaceutically approved polymers into three-dimensional matrices, we can ensure their biocompatibility and facilitate their seamless transition from the laboratory to clinical applications. Moreover, the reversible nature of their physical cross-links allows hydrogels to dissolve in the presence of external stimuli. Particularly, their high degree of hydration, high molecular weight, and superior flexibility of the polymer chains facilitate their interaction with complex biological barriers (e.g., mucus layer), making them ideal candidates for mucosal drug delivery. However, fine-tuning the composition of the hydrogel formulations is of great importance to optimize the performance of the medical device and its therapeutic cargo. Herein, we investigated the influence of different Eudragits® on the properties of hydrogels based on polyvinylpyrrolidone (PVP), polyvinyl alcohol (PVA), and polyethylene glycol (PEG), which were originally proposed as ocular inserts in previous reports. Our research aims to determine the effects that including different Eudragits® have on the structure and protein ocular delivery ability of various hydrogel formulations. Properties such as matrix stability, protein encapsulation, release kinetics, mucoadhesion, and biocompatibility have been analyzed in detail. Our study represents a guideline of the features that Eudragits® have to exhibit to endow hydrogels with good adhesion to the eye's conjunctiva, biocompatibility, and structural strength to cope with the ocular biointerface and allow sustained protein release. This work has important implications for the design of new hydrogel materials containing Eudragits® in their composition, particularly in mucosal drug delivery.
1. Introduction
Developing drug delivery systems capable of overcoming physical and biological barriers is a topic of intense research.1 Hydrogels stand out among the various polymeric materials designed to encapsulate and control cargo release in numerous applications. They are three-dimensional cross-linked networks that can retain water and respond to external stimuli such as changes in temperature or pH.2,3 Their inherent softness and elasticity ensure negligible tissue irritation. Depending on the cross-linking mechanism used in their preparation, a distinction can be made between physically and chemically cross-linked hydrogels.4 In the first case, the polymer chains are reversibly linked, and the hydrogel can eventually be dissolved. Most polymers approved by regulatory authorities can form physically cross-linked hydrogels, without the need for chemical modification of their structure. This ensures biocompatibility, increases patient compliance, reduces side effects, and shortens the path from laboratory to market.5 Consequently, several physical hydrogels are currently available in the market for drug delivery, wound dressing, personal care, tissue engineering, and contact lenses, among others.6,7Significant efforts are being made to develop hydrogels that adhere to the mucous membranes of the eye, mouth, vagina, stomach, bladder, and nose, as these are the main administration routes for therapeutic agents into the body.8 Studies have shown that the interactions between hydrogels and mucous membranes can prolong the residence time of drugs at the application site.9 However, several problems occur when physically cross-linked hydrogels are used in mucosal tissue, as they tend to hydrate and eventually dissolve or degrade, losing their integrity and mucoadhesive properties. Therefore, developing hydrogels with optimal properties matching the drug delivery or tissue healing times remains challenging. A great focus is given to preparing materials that can interact with mucin, one of the major components of the mucosa, by selecting polymers with high molecular weight. Indeed, long and flexible chains increase the entanglement between the polymers and the mucosal layer.10 For the same reason, the degree of cross-linking between the chains and the hydration of the network are carefully fine-tuned in hydrogels. Excessive cross-linking decreases flexibility and the ability for mucosal adhesion.11 To ensure that polymers have good mucoadhesive properties, they are also provided with functional groups that can form hydrogen bonds, such as hydroxyl, carboxyl, and amino groups. Likewise, polymers with charged functional groups are commonly used to increase mucoadhesion.12
Although several hydrogels based on polyvinyl alcohol (PVA), polyethylene glycol (PEG) or polyvinylpyrrolidone (PVP) have been investigated recently, many other polymers widely used in different pharmaceutical forms are promising candidates for the next generation of mucoadhesive gels.13 Eudragits® polymers, for example, are a family of versatile polyacrylate derivatives characterized by a range of aqueous solubility with minimal chemical structural changes. Therefore, they are considered promising options for developing novel drug delivery systems.14 In this sense, Eudragits® have been extensively explored in recent years for their potential applications in gastrointestinal release and as protective coatings for tablets.15 Despite these advances, there is a surprising lack of studies addressing the use of Eudragits® in hydrogel formulations.16 Recent studies have investigated the use of pH-sensitive Eudragits® to produce polymeric films containing drugs loaded into contact lens hydrogels to open up new applications for drug delivery in ophthalmology.17 In addition, Eudragits® were chemically cross-linked with acrylic acid in the presence of methylenebisacrylamide to create pH-sensitive Eudragit®-co-acrylic acid hydrogels as smart carriers for colon drug delivery.18 Moreover, non-pH-sensitive Eudragits® have been used in hydrogel formulations to improve their mucoadhesive properties. For example, Eudragit® RS and S100 have shown remarkable improvement in the mucoadhesion of hydrogels to the vaginal and buccal mucosa.19 Furthermore, some Eudragits® have been investigated to prepare physically cross-linked hydrogels for ophthalmic applications.20,21 It is worth noting that many of the reported studies focused exclusively on drug delivery without considering the effects of the mucosal layer and high humidity that are prevalent in certain areas of the body, such as the eye. Hence, a comprehensive understanding of these parameters remains unexplored and further studies are needed.
In this work, five representatives Eudragits® were studied as components of physically cross-linked hydrogels based on PVP, PVA, and PEG (Fig. 1). This study aimed to determine similarities and differences between five different hydrogel formulations in terms of their matrix stability, protein release, pore size, crystallinity, rheological properties, and cytotoxicity. Since the five hydrogels are based on non-water soluble Eudragits® (RSPO, RL100) or water-soluble ones (S100, L100, L100-55), it is hypothesized that the release kinetic of an encapsulated protein can be fine-tuned based on the selected formulations. Indeed, we found that the release profile can range from sustained to burst release, allowing for adjustment according to specific medical requirements.
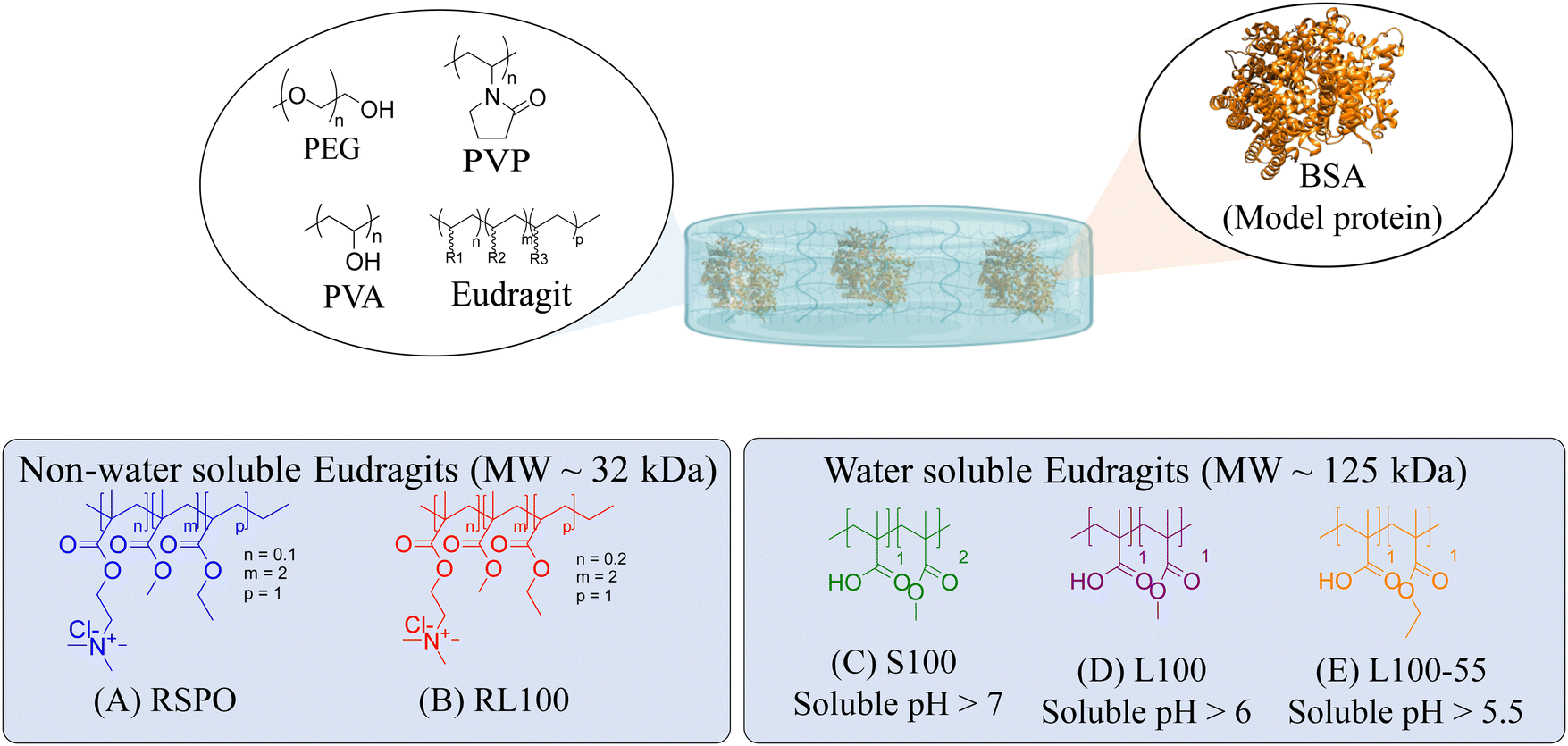 | ||
| Fig. 1 Chemical structure of the polymers that constitute the hydrogel and the model protein (Bovine Serum Albumin, BSA) encapsulated within the network. (A–E) Commercial names and chemical structures of the different Eudragits®. | ||
2. Materials and methods
The starting materials were purchased from various suppliers: Merck® (Spain) provided PEG 2 kDa, bovine serum albumin–fluorescein isothiocyanate conjugate (BSA-FITC), and phosphate buffer saline pH = 7.4. PVA 100 kDa and PVP 50 kDa were acquired from Fisher Scientific® (Spain). Evonik® (Germany) generously provided free of charge Eudragit® RSPO, RL100, S100, L100, and L100-55. 99.8% D2O NMR solvent, 99.5% glycerol, and ACS grade potassium sulphate (K2SO4) were obtained from Scharlab® (Spain). AJL Ophthalmic® (Spain) provided Balanced Salt Solution (BSS) for irrigating the tissues of the eyes. All the reagents were used as received without any further purification. Type I quality water was used for the experiments and was obtained from a purification system (18.2 MΩ cm). Thermo Fisher Scientific® (Spain) provided Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin solution (P/S) and AlamarBlue® cell viability reagent.2.1. Hydrogel preparation
Physically cross-linked hydrogel matrices were prepared by solvent casting as previously reported, with minimum modifications.20,21 For all the formulations PVP (12 wt%) and PVA (27 wt%) were mixed at 90 °C in H2O-ultra pure (100 mL) for 2 h. After cooling to room temperature (25 °C) PEG (27 wt%) and glycerol (27 wt%) were added to the mixture, followed by the addition of Eudragits® in a final concentration of 7 wt% according to Table 1 (see Results section) to produce the various hydrogels. To dissolve Eudragit® RL100, RSPO, and Eudragit® L100-55, 2 mL of acetone was used for the first case and ethanol for the remaining two. The pH of the pre-gel solutions was stabilized to 7.4 with 10 mM PBS and NaOH/HCl 1 M. The mixture was dried at 50 °C in an oven until constant weight, resulting in a final thickness of approximately 1 mm.| Matrix hydrogel component (average MW) | Chemical structures | Concentration (% w/w) |
|---|---|---|
| PVA (100 kDa) |
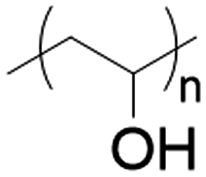
|
27 |
| PVP (50 kDa) |
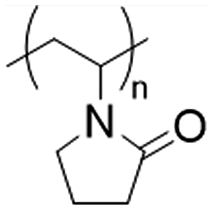
|
12 |
| PEG (2 kDa) |
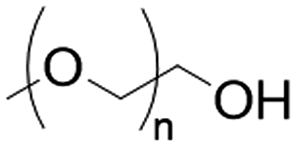
|
27 |
| Glycerol (92 Da) |
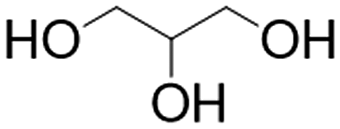
|
27 |
2.2. Dynamic oscillatory viscoelasticity
Dynamic oscillatory measurements were conducted utilizing a Tritec 2000 DMA (Triton Technology). The experimental protocol employed a consistent frequency of 1 Hz across a temperature spectrum spanning from −100 to +50 °C. The resulting curves encompassed G′, G′′, and the Tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ loss tangent. Notably, the Tanδ facilitated the determination of the glass transition temperature (Tg). Data acquisition was subjected to analysis using Microsoft Excel 2021 and GraphPad Prism 9.3.
δ loss tangent. Notably, the Tanδ facilitated the determination of the glass transition temperature (Tg). Data acquisition was subjected to analysis using Microsoft Excel 2021 and GraphPad Prism 9.3.
2.3. Hydrogels internal morphology
To observe the internal morphology of the hydrogels, their cross-sections were examined. For that, the hydrogels were cryo-fractured in liquid nitrogen into pieces and lyophilized. The resulting freeze-dried materials were then coated with gold using a Bio-Rad SC500 sputter coater for 2 min before being analyzed with a Hitachi TM3030 SEM microscope at 15 keV.2.4. Hydrogel stability under mimicked ocular conditions
The system designed to simulate eye conditions involves a 3D printed mould that holds a hydrogel sample at a 45° angle, as shown in Fig. 2. The hydrogel is exposed to the environment, and a buffer is dispensed at the upper end, flowing along the material's surface and ultimately being collected as aliquots. As the hydrogel is stabilized by physical interactions, the buffer can dissolve the polymers that form the matrix, leading to the disaggregation of the hydrogel. Given that ophthalmology is one of the potential applications for this type of material and that the hydrogel is exposed to a liquid, an isotonic salt solution (BSS) for irrigating eyes was used.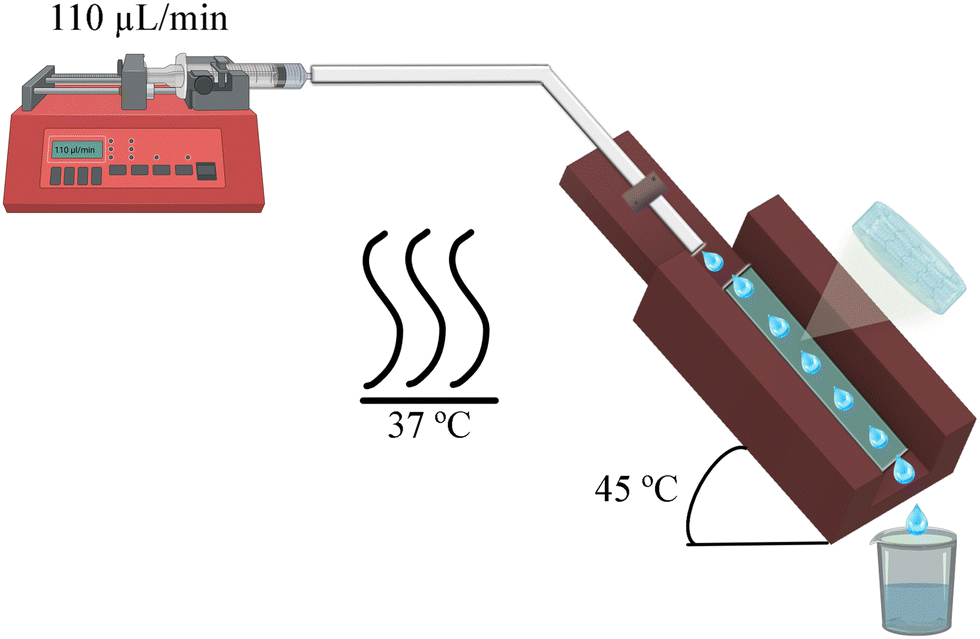 | ||
| Fig. 2 Schematic representation of the setup used to perform the stability and protein release assays. | ||
Eventhough the tear flow rate is 2.2 μL per minute in a normal eye,22 this value is too low for a laboratory experiment. Therefore, a scaled flow rate of 110 μL per min (50 times the normal rate) was used. The systems were set inside an oven maintained at a constant temperature of 37 ± 1 °C. A 1.5 × 0.5 × 0.1 cm sample was taken from each hydrogel and placed inside the 3D mould. The collected aliquots were then analyzed using a Bruker Fourier 300 NMR spectrometer with BSS/D2O 90/10 as a solvent. The data were processed with MestReNova 14.2 software with the equipment set to 1H-NMR, 128 scans, and 1 s of relaxation time in water suppress mode.
2.5. Moisture uptake and moisture content
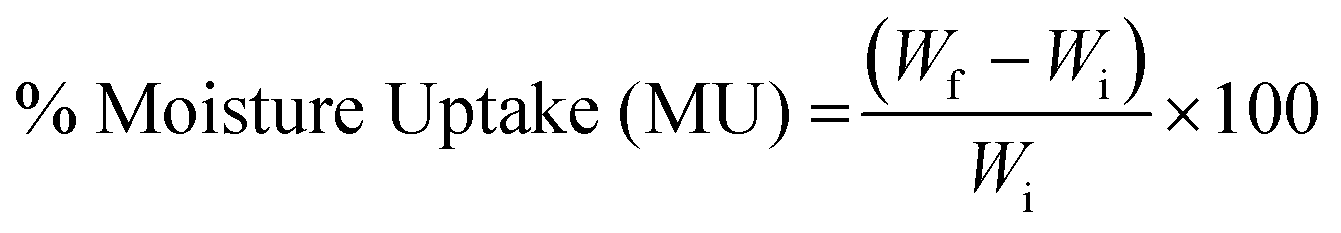 | (1) |
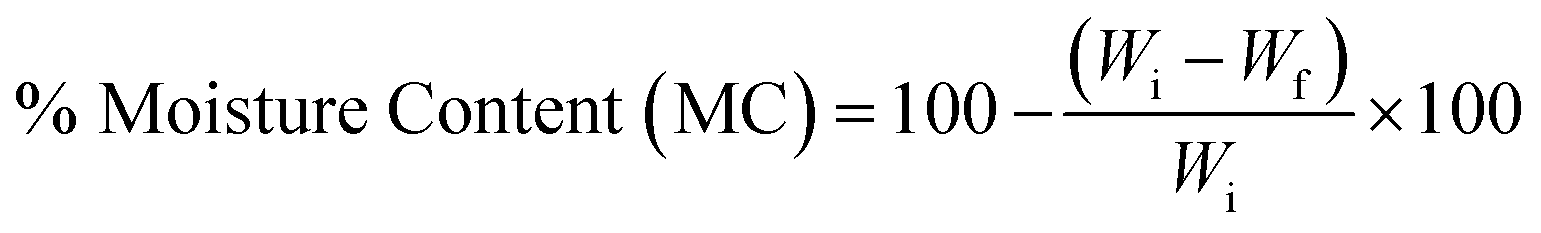 | (2) |
2.6. Bovine serum albumin as a model for encapsulation and release studies
BSA-FITC was encapsulated within the five different hydrogels. To prepare samples measuring 5.5 × 3.5 × 0.1 cm in a silicone mould, 5 mg of BSA-FITC was added to the pre-gel solution of each hydrogel. The average hydrogel's thickness was 1 mm.To conduct the release studies, a sample from each hydrogel measuring 1.5 × 0.5 × 0.1 cm was placed in the 3D mould release system, following the same methodology detailed in Section 2.3. The collected aliquots were quantified every five min by measuring the maximum absorbance of FITC at 494 nm using a NanoPhotometer NP80 from Implen®.
2.7. Mucoadhesive properties of hydrogels towards ocular conjunctiva
The adhesion capacity of the hydrogels was determined using porcine eye conjunctiva as a substrate by a probe tack adhesion test. For this purpose, axial experiments were performed using an Ares-G2 rheometer (TA Instruments) with parallel plate geometry. Porcine conjunctiva was extracted from pig eyes purchased from a local abattoir and kept frozen until use. Before testing, the tissue was hydrated with BSS, and a circular section was cut and glued to the upper plate. An approximately 800 μm thick hydrogel film was firmly attached at the bottom of the setup. The surface area for measurement (8 mm diameter upper plate) was smaller than the probe area (12 mm diameter bottom plate) to avoid hydrodynamic and bending effects during the detachment.23 The test was performed in three stages: (1) compression in which the upper plate descended at 0.1 mm s−1 until h0 = 500 μm, (2) contact period for 120 s, and (3) debonding at constant velocity (Vdeb): 0.314 mm s−1, which corresponds to a nominal strain rate of 0.6 s−1. Force (F)–displacement (h(t)) data were acquired during each experiment and transformed to stress (s)–strain curves (eqn (3)–(5)):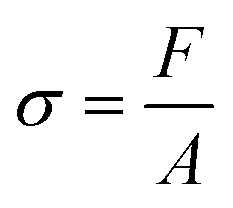 | (3) |
| ε = (h(t) − h0)/h0 | (4) |
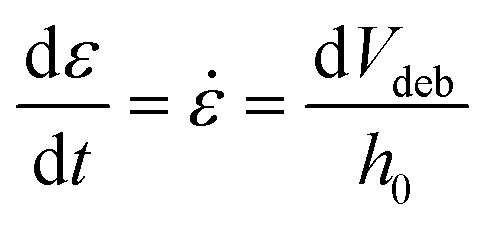 | (5) |
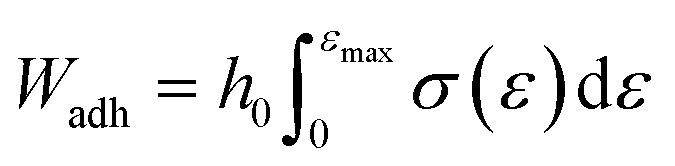 | (6) |
Preliminary adhesion tests, performed at different contact times, showed that an increase in the contact time above 120 s no longer increased the adhesion work (data not shown), so it was assumed that during this contact stage, the adhesive hydrogel-conjunctive interface was optimally established.
2.8. Thermal and X-ray diffraction characterization
2.9. Metabolic activity assessment
To investigate the cellular response to different hydrogel formulations, samples of the hydrogels (1.5 × 0.5 × 1 cm) were dissolved in 2 mL of BSS at different degrees of dissolution (25%, 50%, and 100%). The resulting solutions were mixed in a 1:1 ratio with complete media and then sterile-filtered.
To determine the cellular metabolic activity in response to the different hydrogel formulations, human fibroblasts (MRC-5, ATCC), which is a human lung fibroblast cell line supported by the ISO 10993-5 were seeded at a density of 5000 cells per well on 96-well plates with complete medium (DMEM + 10% FBS + 1% P/S) and incubated overnight to allow cell adherence to the tissue culture plastic. Then, the complete medium was aspirated and replaced by the abovementioned material solutions. The metabolic activity was determined at two different time points (i.e., 24 and 48 h) with AlamaBlue® following a previously reported protocol.24 This assay, which relies on the capacity of viable cells to reduce resazurin (i.e., the active ingredient in AlamarBlue) to resofurin, was used herein as an indicator of cell viability. In this experiment, BSS mixed with complete media (1:1 weight ratio) was used as a control.
The statistical analysis was performed using the software GraphPad Prism 9.4, a two-way ANOVA was carried out to test the variance difference among the treatments, and a post-Hoc Tukey test were run to compare all possible pairs of the means for the dissolution percentages of the hydrogels and controls, the 0.05 alpha value, and a confidence interval of 95% were set, the error bar corresponds to the SEM were n = 5 for each concentration.
3. Results and discussion
3.1. Hydrogel preparation
In this study, we attempt to analyze the influence of Eudragits® on the properties of hydrogels commonly used as ocular inserts for the controlled release of various drugs.21 For this purpose, five Eudragits® were selected based on their pH-dependent water solubility. Two of these Eudragits®, RSPO, and RL100, were included in the hydrogel formulations A and B, respectively, as pH-independent polymers. They are tri-block copolymers with an average molar mass of 32 kDa, composed of ethyl acrylate, methyl methacrylate, and a comparatively small amount of methacrylic ester with quaternary ammonium groups. The difference between the two polymers is the number of repeating units in the block containing the quaternary ammonium, with RSPO having a fraction of 0.1 units and RL100 0.2 units with respect to the ethyl acrylate monomer (see Fig. 1). The presence of the ammonium group in the polymer makes it insoluble in water, with swelling aqueous capacities, cationic charge, and non-biodegradable properties.25In addition, we also investigated water-soluble polymers, specifically Eudragit® S100 (hydrogel C), Eudragit® L100 (hydrogel D), and Eudragit® L100-55 (hydrogel E), which are soluble at pH values above 7.0, 6.0 and 5.5, respectively. These Eudragits® are diblock copolymers with methacrylic acid and ethyl acrylates repeating units. The molar ratio between the blocks differs for Eudragit® S100 and L100, being 1:2 and 1:1, respectively. Eudragit® L100-55 has a terminal ethyl group instead of a terminal methyl group in the ethyl acrylate block. These anionic polymers have an average molar mass of approximately 125 kDa.25Table 1 presents a compilation of the ingredient formulations commonly employed in creating hydrogels A–E, along with their corresponding concentrations. In Table 2, the distinct Eudragit® variations applied in each material are detailed, accompanied by their chemical structures, monomer compositions, utilized concentrations, and aqueous solubility.
| Sample | Eudragit (Average MW) | Chemical structures and monomers fractions | Concentration (% w/w) | Solubility in water |
|---|---|---|---|---|
| Hydrogel A | Eudragit RSPO (32 kDa) |
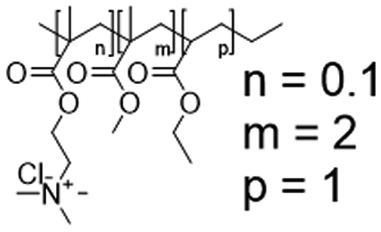
|
7 | No |
| Hydrogel B | Eudragit RL100 (32 kDa) |
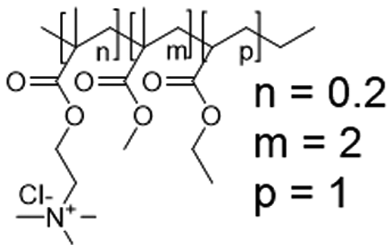
|
7 | No |
| Hydrogel C | Eudragit S100 (125 kDa) |
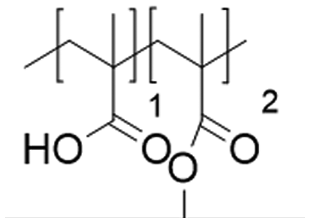
|
7 | pH > 7 |
| Hydrogel D | Eudragit L100 (125 kDa) |
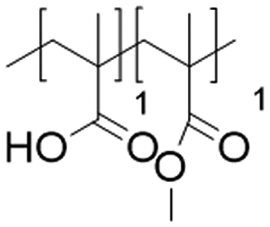
|
7 | pH > 6 |
| Hydrogel E | Eudragit L100-55 (125 kDa) |
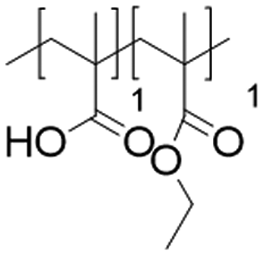
|
7 | pH > 5.5 |
It is crucial to emphasize our demonstration of the hydrogel categorization of materials A–E through dynamic viscoelasticity measurements (Section S1, ESI†). By assessing the storage (G′) and loss (G′′) moduli alongside the Tanδ, we have substantiated the presence of distinct thermal transitions. These transitions discern between the material's elastic behaviour, indicative of the hydrogel structure, and the time-dependent deformations resulting from viscous dissipation. Within this context, we have identified the glass transition temperature of hydrogels A–E within the temperature range of −19 to 1 °C. Notably, we have also established the presence of rheological moduli at temperatures below 0 °C, a compelling demonstration that unequivocally validates the hydrogel nature of these materials, as anticipated.
PVP, PVA, PEG, and glycerol were used as the main components of the hydrogels, and the respective Eudragits® were added in a proportion of 7 wt%. The pre-gel solutions were prepared as described in Section 2.1 and then dried to produce hydrogel films by solvent casting. The hydrogel matrix was constructed through the establishment of hydrogen bonds among polymeric chains, as depicted in Fig. 3. The mass of the polymers was fine-tuned during solution to obtain hydrogel films with a thickness of 1 mm. Fig. 4 shows the appearance of the hydrogels and their average thickness. Notably, different opacities were observed in the hydrogel films, suggesting that the Eudragits® might interfere with the crystallization of PVA chains during the gelation process (this phenomenon will be further discussed in Section 3.6.).
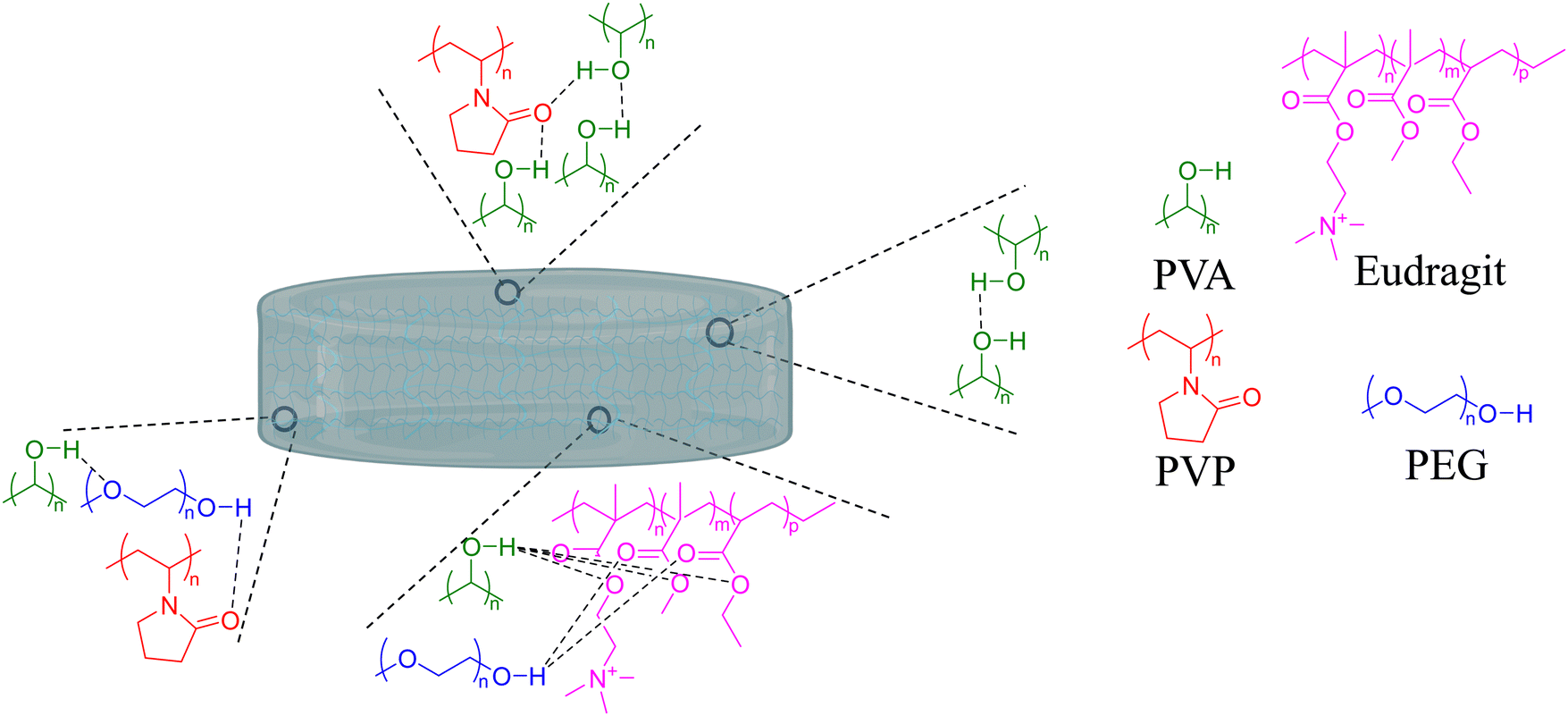 | ||
| Fig. 3 Scheme illustrating the formation of hydrogel bonds among polymeric chains, resulting in the creation of the hydrogel matrix. | ||
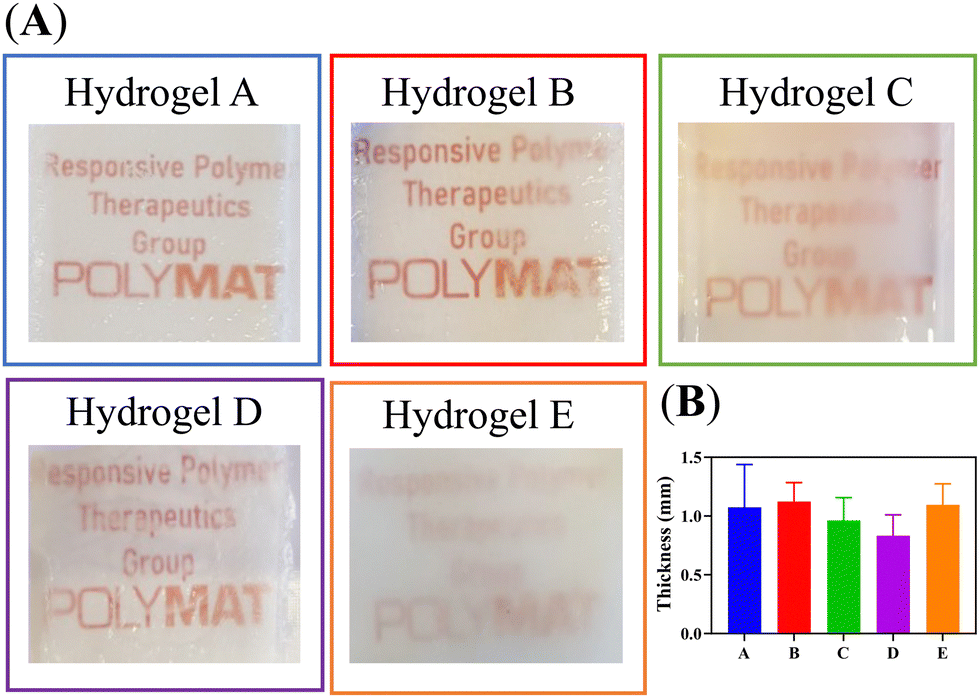 | ||
| Fig. 4 (A) Pictures of the hydrogel films. (B) Hydrogel's thickness (mm) measured with a digital calliper (N = 3). | ||
3.2. Internal structure of the hydrogels
In the preparation of hydrogels, a rational selection of polymers should be made since they are expected to influence the structure and final properties of the materials. It has been shown that the presence of hydrophobic polymers in the hydrogel composition leads to a decrease in its water swelling ability and an increase in the pore size.26 In general, the pore size of hydrogels is known to greatly affect other properties, in addition to the swelling capacity, such as mechanical properties, drug encapsulation, and cargo release kinetics.27 Therefore, to anticipate the effects of Eudragits® on the final properties of the hydrogels, we determined the pore size of the five networks in the dried state by SEM of the cross sections.Interestingly, the SEM micrographs and the histogram distribution of about 1000 pores of each hydrogel, shown in Fig. 5, suggest a strong dependence between the pore size and the type of Eudragit®. Hydrogels A and B, containing non-water-soluble Eudragits®, show wider pore size distributions than hydrogels C, D, and E, based on water-soluble Eudragits®. The average pore diameter of materials A and B ranges from 11 to 14 μm, while materials C, D, and E have smaller pore diameters with values ranging from 3 to 6 μm. These differences in pore size are directly related to the structural composition of the material in terms of the number of cross-links and physical interactions between the polymer chains. It is known that PVA-based hydrogels generally have small pores of about 1 μm because PVA forms crystals during the freeze-drying process. However, when PVA is combined with other polymers with different hydrophilicities, the number of crystals formed is affected, leading to an increase in the pore size of the hydrogels.28 We hypothesize that this effect is responsible for the variation in the pore size distribution of our materials. We anticipate these differences will also affect the matrix stability and mucoadhesion properties, which will be analyzed in detail in the following Sections (3.3 and 3.5).
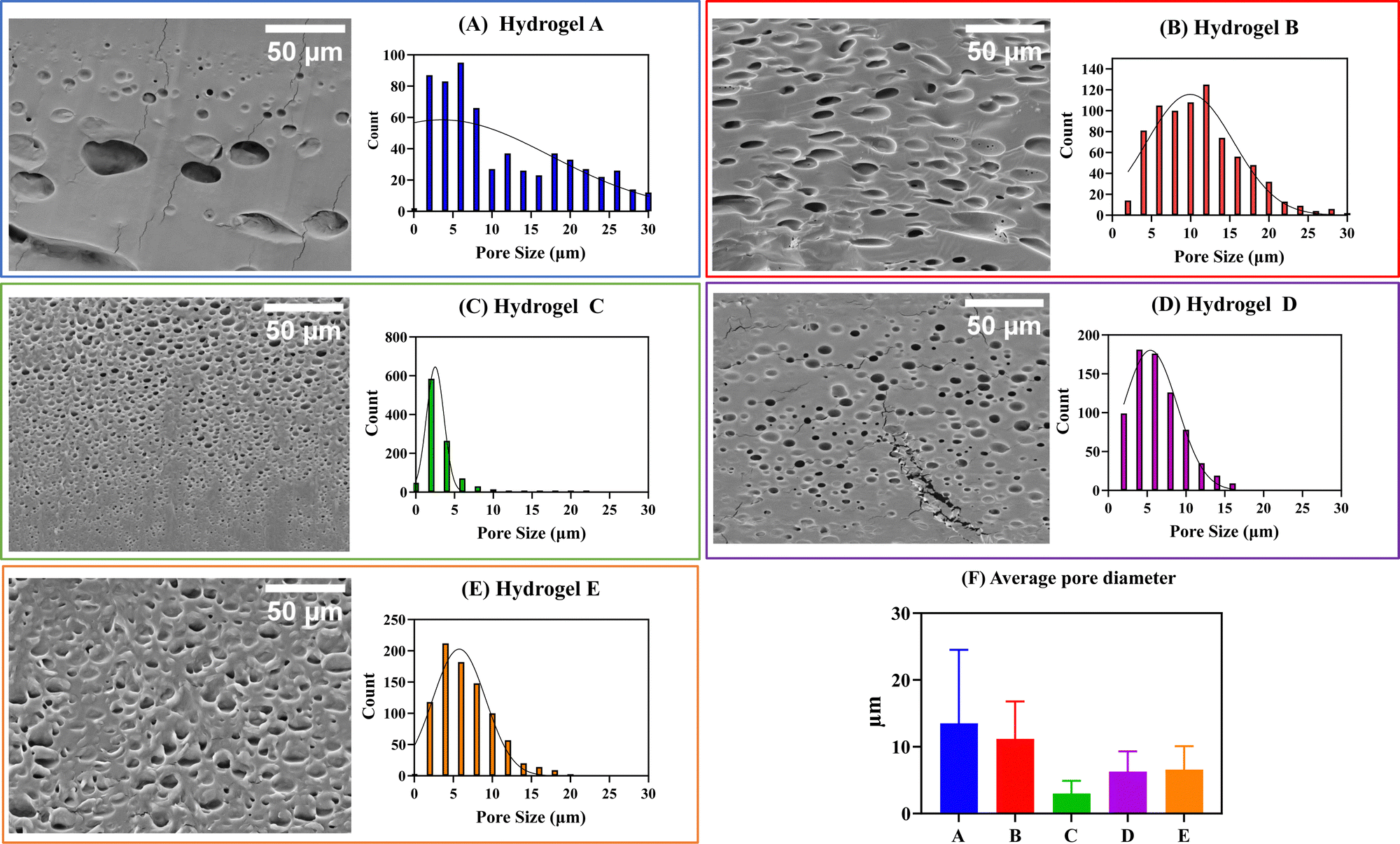 | ||
| Fig. 5 SEM pictures and histogram distribution for the pores in the transversal cut of the five hydrogels after their freeze drying. (A)–(E) SEM pictures and histograms for all the samples. Scale bar: 50 μm. (F) Summary plot with the average pore diameter for the five materials. (N > 500). | ||
3.3. Determination of the dissolution rate of the hydrogels
Considering the potential application of PVA, PVP, PEG, and glycerol-based hydrogels as ocular inserts, the effect of Eudragit® on their dissolution rate is a key factor to be evaluated.20,21 Hydrogel-based ocular inserts should ideally dissolve when placed in the eye bag, absorbing tears and slowly releasing the hydrogel matrix components and their pharmaceutical cargoes. Hydrogels that maintain their integrity over time are desirable because they ensure a reduction in dosing frequency while preserving their tissue adhesion. Therefore, we performed stability tests by incubating the hydrogels in an aqueous ocular irrigation solution (BSS) at a constant flow rate (110 μL min−1) at 37 °C. We used a 3D-printed system (Fig. 2) to hold the hydrogels during the assay, and we collected aliquots at different time intervals for NMR analysis. We correlated the signals of the 1H-NMR spectra with those corresponding to the pure components of the hydrogel formulations (Fig. S2, ESI†).Fig. 6 shows the 1H-NMR spectra of samples obtained at different dissolution times for hydrogels A and B, which are structurally similar as they contain hydrophobic Eudragits®. It should be noted that hydrogel A required 230 min for complete dissolution, while hydrogel B required only 75 min. This difference can be attributed to the relative composition of both Eudragits®; the RSPO variant contains twice the amount of quaternary amines compared to Eudragit® RL100. This fact leads to a higher hydrophobicity of hydrogel A, making this hydrogel less prone to dissolution. The signals of all polymers (PEG, PVP, PVA, and Eudragit® RSPO) can be seen together with the signal of glycerol after 5 min, indicating that they dissolve in the aqueous medium and are released from the hydrogel matrix (Fig. 6A). Similar results can be seen in the range 30–120 min. Conversely, at the end of the experiment (150–230 min), the signals from glycerol and PVP were no longer detectable. This result indicates that the degradation of the hydrogel matrix is not uniform, as the dissolution rate varies for each polymer. As a result, the number and strength of physical interactions that stabilize the matrix change when some polymers are removed. Therefore, the permeability of the matrix decreases as a function of the glycerol and PVP release as they have the fastest dissolution rate, leading to an increase in the overall stability of the matrix. The results for the dissolution of hydrogel B (Fig. 6B) support this conclusion. Since the matrix is more soluble, the total time required for its dissolution is less than for hydrogel A. During the first 5 min of dissolution of hydrogel B, signals from all polymers (PEG, PVP, PVA, and Eudragit® RL100) were observed along with the signal from glycerol. These signals were also observed in the 10–60 min spectra. At the end of the experiment (75 min), the signals from all polymers were still present, indicating that the dissolution of the hydrogel B matrix was more uniform and balanced.
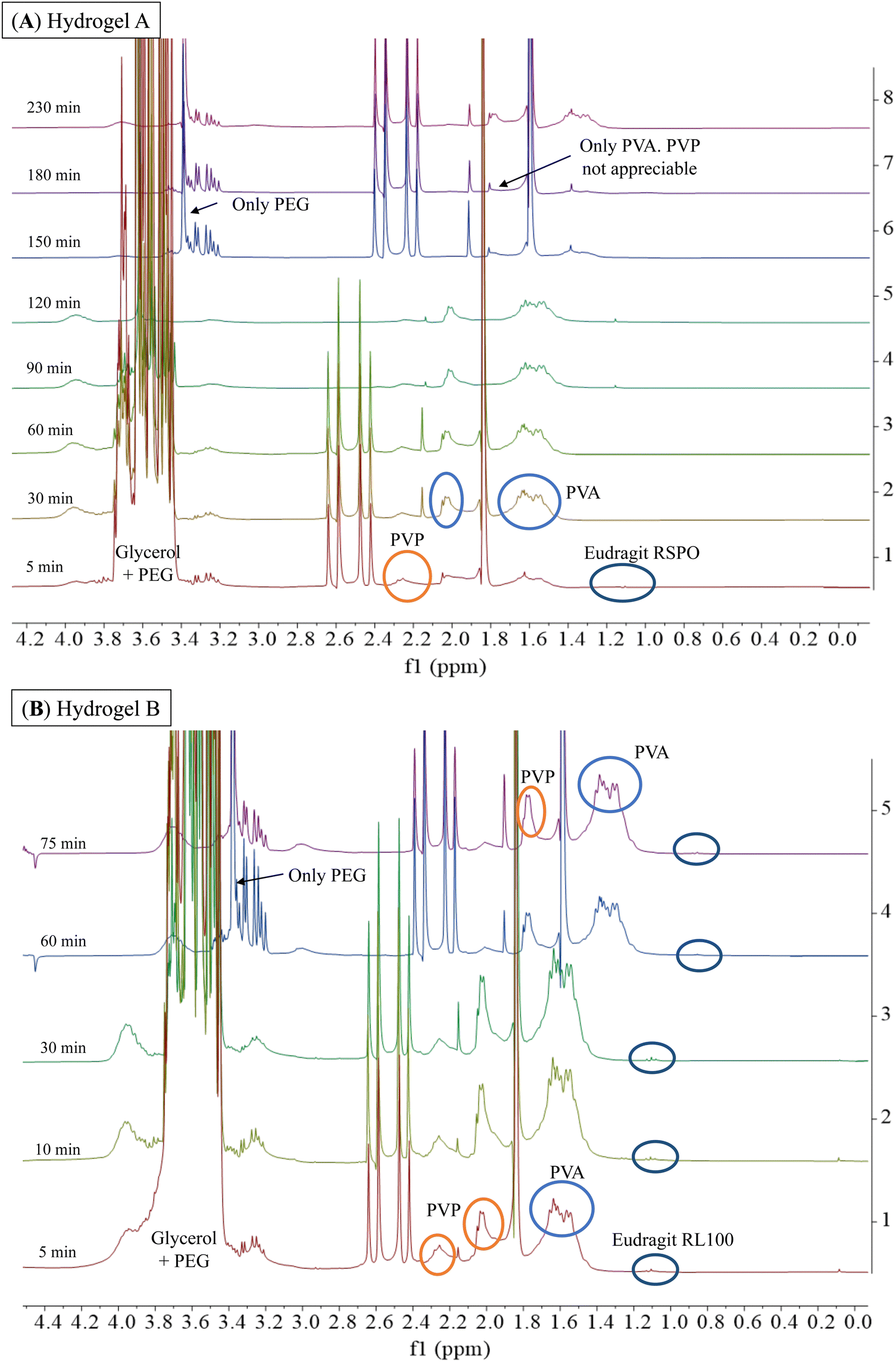 | ||
| Fig. 6 1H-NMR spectra (300 MHz, BSS/D2O 90/10) were obtained during the dissolution of hydrogel A and hydrogel B with BSS at a constant flow rate of 110 μL min−1 at 37 °C. For hydrogel A, the spectra were shifted 150 min, 180 min, and 2300 min by −0.24 ppm to improve the observation of the signals. For hydrogel B, the 60 min and 75 min spectra were shifted by −0.24 ppm. | ||
Fig. 7 shows the 1H-NMR spectra of samples obtained at different dissolution times for hydrogels C, D, and E, which are structurally similar as all contain hydrophilic Eudragits® with pH-dependent water solubility. It should be noted that hydrogel C required 145 min for complete dissolution, while hydrogels D and E required 130 min and 105 min, respectively. These results indicate that hydrogel C is the most robust among the three formulations.
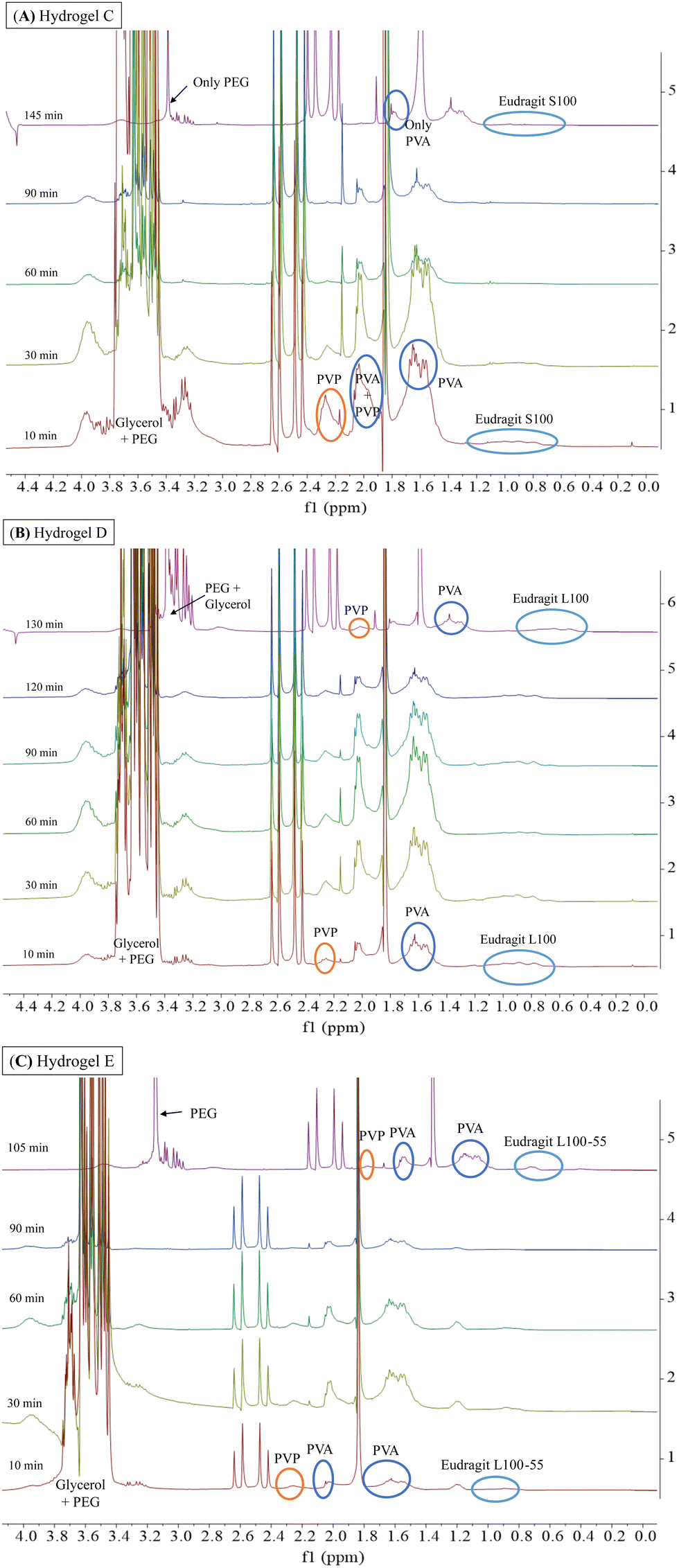 | ||
| Fig. 7 1H-NMR spectra (300 MHz, BSS/D2O 90/10) obtained during the dissolution of hydrogel C (A), hydrogel D (B), and hydrogel E (C) with BSS at a constant flow rate of 110 μL min−1 at 37 °C. For hydrogel C (A), the spectrum was shifted by −0.24 ppm for 145 min to improve the observation of the signals. For hydrogel D (B), the spectrum was shifted 130 min by −0.24 ppm and for hydrogel E (C), the spectrum was shifted 105 min by −0.24. | ||
Hydrogel C (Fig. 7A) is composed of Eudragit® S100, which is water-soluble at a pH greater than 7. During the experiment, dissolution was performed with BSS at physiological pH (pH = 7.4). As can be seen, the signals of PVP and PVA were visible during the range 10–90 min. However, at the end of the experiment (145 min), the signals of both polymers were no longer detectable, indicating uneven dissolution of the matrix. Considering that the strength and number of physical interactions changed during the dissolution process, in the absence of the PVP polymer (145 min), the stability of hydrogel C increases. This is evidenced by the longer time required for complete dissolution (145 min) compared to hydrogels D and E (130 and 105 min, respectively).
Hydrogel D and E (Fig. 7B and C) are composed of Eudragits® that are water soluble at pH above 6 and 5.5, respectively. Unlike hydrogel C, the signals corresponding to Eudragits® L100 and L100-55 exhibited consistent intensity throughout the experiments, leading to a more uniform dissolution process. The 1H-NMR spectra revealed a gradual dissolution of PVP and glycerol signals over time (10–130 min), suggesting a more evenly distributed dissolution of the matrix.
All the above data, as well as the analysis of moisture content (MC) and moisture uptake (MU) (Fig. S4, ESI†), have shown the dependence of the matrix stability on ambient humidity. Our findings in MU indicated that hydrogel C possesses a remarkable capacity to absorb a significant amount of water without compromising its structural integrity. This feature place hydrogel C as the most promising candidate for drug delivery in ophthalmology, however, further investigations should be performed.
3.4. Encapsulation and release of proteins
Hydrogels have been used to encapsulate and release various pharmaceutically active substances.29 Proteins are of particular interest, as they catalyse reactions in misfolding protein diseases (e.g., cystic fibrosis), assist in healing ocular wounds, and are fundamental components of vaccines.30 However, their lack of stability during storage and administration, as well as their difficulty in crossing biological barriers such as the mucosa and skin, calls for developing protein delivery systems.31 Therefore, we investigated the protein loading capacity of our hydrogels by encapsulating fluorescently labelled BSA (BSA-FITC) as a model cargo. Using a protein conjugated to a fluorophore allowed us to observe the homogeneity of the encapsulation in the hydrogel film. In addition, the fluorophore allows subsequent release analysis by quantification using UV-Visible and fluorescence spectrophotometry. Fig. 8 shows the appearance of the five hydrogels after BSA-FITC encapsulation and the release kinetics as a percentage of cumulative protein release over time. The orange aspect (from FITC) confirms that the protein is uniformly distributed in the hydrogel films. | ||
| Fig. 8 (A) Photos of the A–E hydrogel films containing BSA-FITC. (B) BSA-FITC release kinetics from hydrogels A–E. Results are presented as a percentage of cumulative protein release. (N = 3). | ||
To study the release kinetics of BSA-FITC from hydrogels A–E, we used the same method as in Section 3.3. As previously observed, the dissolution rate of the protein-laden hydrogels varied, indicating different protein release kinetics for each material. Table 3 shows the time in minutes, after which 50%, 80%, and 100% of BSA-FITC were released from hydrogels A–E.
| Hydrogel | 50% BSA (min) | 80% BSA (min) | 100% BSA (min) |
|---|---|---|---|
| A | 63 | 120 | 175 |
| B | 25 | 50 | 85 |
| C | 35 | 65 | 95 |
| D | 54 | 77 | 130 |
| E | 35 | 59 | 105 |
Fig. 8 and Table 3 compare the release kinetics of hydrogels A and B. Hydrogel A took more than twice longer than hydrogel B to release 50%, 80%, and 100% of the protein, probably due to the stronger physical interactions, as discussed in the previous section. The presence of the Eudragit® RSPO in hydrogel A significantly affected its release behaviour compared to the other formulation. This material exhibited a sustained release in contrast to a burst release observed in hydrogel B, which featured weaker interactions between the polymer chains.
Of the other three hydrogels, based on water-soluble Eudragits®, hydrogel C showed a burst release profile, as the protein was released around 10–25% faster than hydrogels D and E. In contrast, hydrogel D demonstrated a sustained release, while hydrogel E exhibited an intermediate behaviour. As hypothesized, it is possible to control the kinetic release profiles from sustained to burst release, depending on the specific requirements, by varying the type of Eudragit® in the formulation. It is possible to predict the hydrogel release profiles using Peppas’ model equation, which takes into account Fick's diffusion or Case-II transport.32 However, in our study, the used constant fluid flow prevented accurate comparison with the predicted results. The different outcomes obtained from the application of the model equation indicate a mixture of diffusion modes, which makes it difficult to determine the release mechanism. Therefore, no conclusive findings on the release mechanism can be presented.
A comparison between the results of the 1H-NMR dissolution rates for the empty hydrogels and the release of BSA-FITC reveals slight differences among the time required for the total dissolution of the material. In the previous section, we made an observation regarding the elution of Eudragit® and PVP in hydrogel A compared to hydrogel B. It was found that these components eluted at a faster rate in hydrogel A. Now, we have observed that hydrogel A released the total amount of protein after 175 min, while it took 230 min for the empty hydrogel to dissolve completely. In contrast, protein-laden hydrogel B achieved complete protein release in 85 min, whereas it took 75 min for the dissolution of the empty hydrogel. This significant difference in elution time between the protein and the complete dissolution of the empty hydrogel indicates that the protein exhibits strong interactions with both Eudragit® and PVP. Consequently, these interactions facilitate the protein's faster release from hydrogel A in comparison to hydrogel B. This hypothesis gains further support from hydrogels C, D, and E. For hydrogel C, the total amount of protein was released after 95 min, while it took 145 min for complete empty hydrogel dissolution under 1H-NMR studies. Notably, hydrogel C exhibited earlier elution of PVP compared to the other polymers. In contrast, hydrogels D and E required the same time for total dissolution as for the complete release of the protein. In these two materials, the signals of Eudragits® and PVP consistently eluted over time, reinforcing the theory that these two polymers have strong interactions with BSA, leading to their continuous elution.
3.5. Mucoadhesive properties of hydrogels towards ocular conjunctiva
The hydrogels developed in this work were rationally designed as ocular inserts for drug delivery, making it crucial to measure their mucoadhesion and ensure that they firmly attach to the ocular conjunctiva. Therefore, our concept aimed to leverage the adaptability of the adhesion probe method for simulating the conditions in which ophthalmic inserts are utilized. The probe tack adhesion test allowed us to assess the mucoadhesive of the material using ex vivo porcine ocular conjunctiva, eliminating the need for in vivo animal models. The objective was to identify the optimal parameters that ensure the material's adhesion to the ocular conjunctiva, considering the hydration conditions as a pivotal property to be studied. We, therefore, tested three scenarios: (A) dry hydrogel films, without pre-hydration, (B) pre-hydrating the hydrogels for 3 and 5 days at a relative ambient humidity of 95%, and (C) pre-hydrating the materials for 10 and 30 min by adding 20 μL of BSS on top of each hydrogel.Table 4 demonstrates that all dry hydrogel films exhibit negligible work of adhesion prior to the hydration (day 0). On the contrary, the hydrogels hydrated with 20 μL of BSS buffer for 10 or 30 min exhibited higher work of adhesion than those hydrated under a humid atmosphere. This demonstrates that the water absorption and swelling capacity of the hydrogels happen within minutes. Hydrogel B exhibits the highest work of adhesion, reaching 23 J m−2 after 10 min of hydration with BSS. However, after 30 min of hydration, this value remains relatively constant, slightly decreasing to 20 J m−2. Similar tendencies were observed when hydrogels were hydrated in a 95% relative humidity environment. After 3 days of hydration, the work of adhesion was 2 J m−2, but after 5 days of hydration, this value decreased to 1.5 J m−2. Hydrogel B also dissolves the fastest in the stability assay described in Section 3.3, which correlates with the decreased tendency of work of adhesion.
| Sample | Work of adhesion (J m−2) | ||||
|---|---|---|---|---|---|
| Day 0 | Day 3 | Day 5 | 10 min BSS | 30 min BSS | |
| Hydrogel A | 0.7 | 2 | 2.5 | 7.5 | 10 |
| Hydrogel B | 0.03 | 2 | 1.5 | 23 | 20 |
| Hydrogel C | 0.2 | 4 | 5 | 10.5 | 14 |
| Hydrogel D | 0.34 | 2 | 2 | 10 | 4 |
| Hydrogel E | 0.59 | 4.5 | 3 | 5 | 17.5 |
Hydrogel D exhibited a similar trend to hydrogel B in terms of the effect of hydration on its work of adhesion. As the hydrogel becomes more hydrated, its adhesive capacity decreases, with a value of 10 J m−2 after 10 min of hydration and 4 J m−2 after 30 min of hydration. However, hydrogel D had the lowest work of adhesion among the five hydrogels tested, making it the least suitable option as an ocular insert. Hydrogels A and C appear to be the most promising candidates for use as ocular inserts because of their mucoadhesive properties. Both hydrogels show a consistent increase in mucoadhesion as hydration increases. However, after 30 min of hydration, hydrogel C demonstrates a work of adhesion of 14 J m−2, whereas hydrogel A exhibits only 10 J m−2, suggesting some advantage of hydrogel C.
The hydrogel E behaves between the other hydrogels concerning mucoadhesion. After being hydrated for 10 min with BSS buffer, its work of adhesion was 5 J m−2, but it increased to 17.5 J m−2 after being hydrated for 30 min. Although this hydrogel exhibits a significant increase in mucoadhesiveness after prolonged hydration, this approach has a major drawback, as the matrix's stability could be compromised if it is hydrated for extended periods as seen by NMR dissolution and moisture uptake studies.
3.6. Thermal and X-ray diffraction characterization
The presence or absence of crystals formed during gelation can significantly affect the properties of the synthesized materials. Crystals can generate rigid structures within the gel that reduce its swelling capacity, creating a network of particles that resist deformation. Depending on the specific application, the presence or absence of crystals may be desired to tailor the final materials. Therefore, to fully understand the hydrogels’ properties, we conducted a thorough characterization using thermal and X-ray diffraction techniques to determine the presence or absence of crystals in empty and protein-laded hydrogel films. We also investigated the influence of the conjugated fluorophore on the possible appearance of crystals. Hence, 15 samples were prepared and divided into three groups. (A) Empty hydrogels A–E, named hydrogel X_1. (B) Hydrogels A–E with encapsulated BSA protein, called hydrogel X_2. (C) Hydrogel A–E with encapsulated BSA-FITC, named hydrogel X_3.DSC was employed to identify the different thermal transitions the material undergoes when the temperature increases. Fig. 9 shows heating and cooling DSC scans for hydrogel A in the three scenarios previously described. The same experiment was repeated after 24 h with a heating and cooling rate of 20 °C min−1.
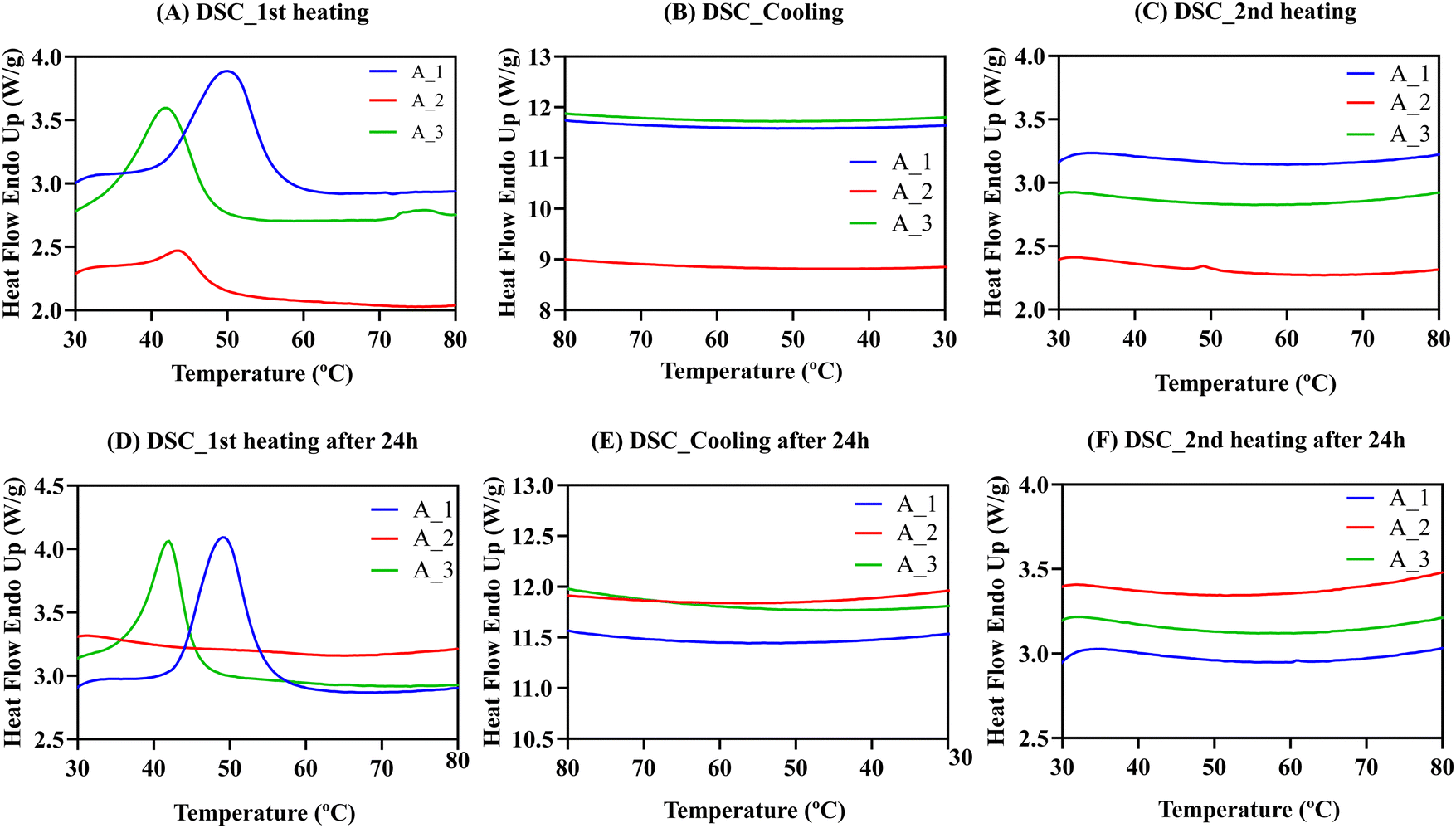 | ||
| Fig. 9 (A) First heating DSC scans for hydrogel A. A_1 empty hydrogel A. A_2 hydrogel A with BSA. A_3 hydrogel A with BSA-FITC. (B) DSC cooling scans. (C) Second heating DSC scans. (D) First heating DSC scans after 24 h. (E) DSC cooling scans for the second experiment. (F) Second heating DSC scans for the second experiment. Heating and cooling rates for all cases are 20 °C min−1. | ||
The DSC results for hydrogel A with and without BSA protein show endothermic peaks in the first heating scans in Fig. 9A. Endothermic peaks could be due to the melting of polymeric crystals within the gels, as PEG and PVA are present in the formulations, although they could also be due to the gel–sol transition.
The endothermic peak temperature in hydrogels A_2 (43.6 °C) and A_3 (42 °C) with BSA were lower than for hydrogel A_1 without BSA (50.1 °C). This result reinforces the important role that proteins play in the physical interactions of the material. However, after cooling down (Fig. 9B) and reheating the samples (Fig. 9C), no exothermic (during cooling) or endothermic (during heating) peaks were observed. It is possible that at the rate employed to perform the DSC measurements (20 °C min−1), the material cannot form crystals or form gels during cooling from 80 °C. To confirm this, the materials were stored and reheated after 24 h (Fig. 9D), and endothermic peaks were observed again for hydrogels A_1 and A_3. Thus, we concluded that physically cross-linked hydrogels take several hours to form after dissolution, so there were no transitions in the second heating scan. Data for hydrogels B–E can be found in Fig. S5 (ESI†).
Table S2 (ESI†) summarises the temperatures at which the endothermic peak (Tp) is obtained and the observed enthalpy of the endothermic transition (ΔHp) that indicates the strength of the intermolecular interactions forming the hydrogels. The enthalpies reported in Table S2 (ESI†) are very significant, between 5 and 25 J g−1. For empty hydrogel A_1, ΔHp was 24.8 J g−1. When BSA was encapsulated, ΔHp decreased to 4.2 J g−1. However, when the BSA was modified with FITC, ΔHp increased to 16.1 J g−1, indicating that the cohesive energies are influenced by the presence of the protein. Surprisingly, when FITC was added, enthalpy values increased significantly, especially compared to unmodified BSA with lower values. This result suggests that FITC may increase hydrophobic interactions between polymeric chains, strengthening the intermolecular interactions and giving higher enthalpy values, even in BSA. Similar conclusions were obtained for hydrogels B–E (Section S4.1, ESI†). We confirmed these consistent trends with thermogravimetric analysis (TGA) and Wide-angle X-ray scattering (WAXS) techniques. A comprehensive examination and analysis of these findings can be found in Sections S4.2 and S4.3 of the ESI† section.
3.7. Study of the effect of the hydrogels in MRC-5 cells
The cytocompatibility of the developed hydrogel systems was studied using the MRC-5 cell line.33 In the present study, we used a well-established cell line, obtained from a recognized culture collection, to carry out a preliminary cytotoxicity study of the developed hydrogels. The MRC-5 (CCL-171) is a human lung fibroblast cell line that is supported by the ISO 10993-5 and considered as appropriate for the biological evaluation of medical devices. The hydrogel formulations were first dissolved in BSS, with varying times required for complete dissolution (e.g., formulation A – 480 min, formulation B – 135 min, formulation C – 150 min, formulation D – 130 min, and formulation E – 135 min). Samples were acquired at different degrees of dissolution (i.e., 25 and 50%) to investigate the potential cytotoxicity of different polymer concentrations in the BSS solution. The dissolutions containing the byproducts of the hydrogel formulations were then incubated with MRC-5 cells, and their metabolic activity was measured after 24 and 48 h. As shown in Fig. 10, the metabolic activity of MRC-5 cells significantly decreased in the presence of dissolutions containing the byproducts of formulations A and B. Accordingly, the metabolic activity of MRC-5 cells after 48 h decreased from 100% to 17% and 9% for formulations A and B (100% dissolution), respectively. The cytotoxic effect was particularly pronounced for formulation B, as even a sample undergoing only 25% of its complete dissolution reduced the metabolic activity from 100% to 61% after 24 h. This fact may be attributed to the presence of quaternary amines in both formulations, (formulation A with Eudragit® RSPO having 5% of quaternary amines, while the formulation B contains Eudragit® RL100 has 10% of quaternary amines), which are associated with the cytotoxicity related to the cationic functionalities. Indeed, as recently reported in a multiparametric study, cationic functionalities not only elicit a cytotoxic response but also downregulate DNA replication and induce an inflammatory response in cells.30 The formulation D showed a mild cytotoxic effect after 24 h reducing the cell viability by 22% at the highest concentration. After 48 h, at 100% of hydrogel dissolution, the cell viability was reduced by 9% in this formulation. On the other hand, the formulation E only affected the cell viability by 10% at 48 h at the highest concentration. Interestingly, the formulation C did not show any detrimental effect on the cell viability. These observed differences can be explained by the different degrees of carboxylation among the studied formulations. Formulation D (Eudragit® L100) has a degree of carboxylation of 48%, which is similar to the one reported for formulation E (Eudragit® L100-55). In contrast, formulation C (Eudragit® S100) only has 29% of acidic groups.15 Previous studies carried out on HeLa cells showed that the polycarboxylates such as Eudragit® S100, are well tolerated by eukaryotic cells at high concentrations. Thus, we believe that the differences on the cellular viability among the formulations C, D and E, can be attributed to the number of carboxylic groups present in the Eudragit®.34 It can be concluded that these formulations C, D and E are potentially suitable for biomedical applications.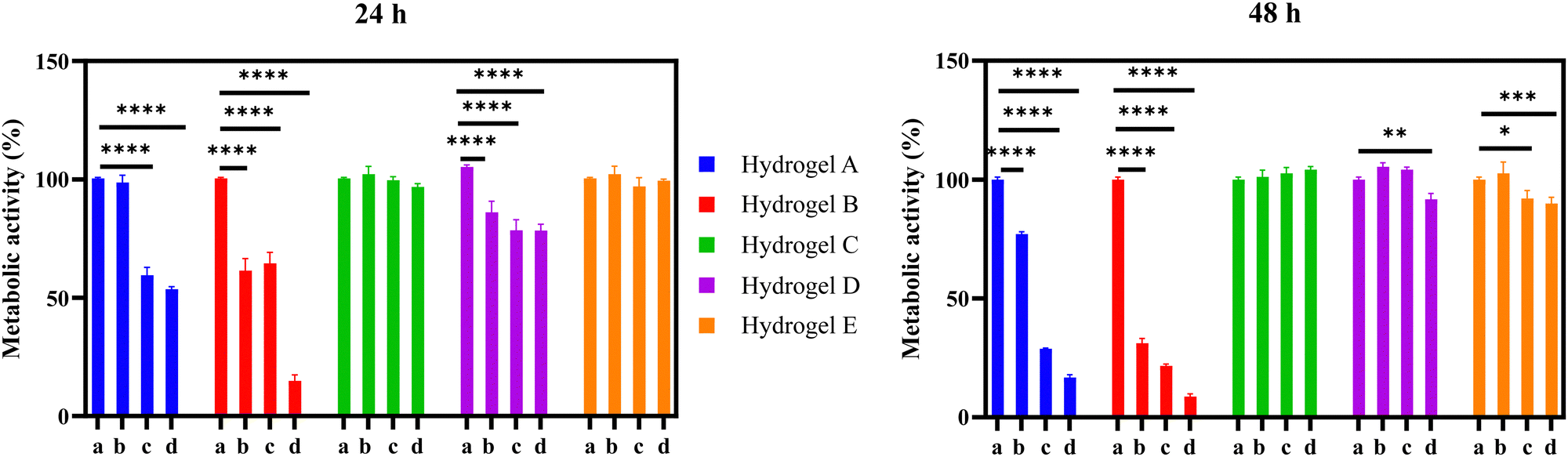 | ||
| Fig. 10 Metabolic activity of MRC-5 cells was measured after 24 and 48 h of incubation with different dissolution degrees: (a) control, 0% dissolution, (b) 25% dissolution, (c) 50% dissolution, and (d) 100% dissolution of hydrogels A–E. Asterisks denote a statistically significant difference compared with the control were ****P ≤ 0.0001, *** P ≤ 0.001, ** P ≤ 0.01, and * P ≤ 0.05. Error bar calculated as SEM were n = 5. | ||
4. Conclusions
The primary aim of this study was to explore the role of Eudragits® in the fabrication of physically cross-linked hydrogels. To this end, five distinct materials were formulated employing PVP, PVA, PEG, glycerol, and five variants of Eudragits® (RSPO, RL100, S100, L100 and L100-55) through the solvent casting method. Among these five hydrogels, hydrogel C based on Eudragit® S100 emerged as the most promising candidate as an ocular insert for protein delivery. Its remarkable physico-chemical attributes, mucoadhesive properties, and biocompatible nature distinguish it within the pool of hydrogel contenders. The synthesis of this material involved the incorporation of constituents in specific proportions: 7 wt% Eudragit® S100, 27 wt% PVA, 12 wt% PVP, 27 wt% PEG, and 27 wt% glycerol. It was prepared with high homogeneity and reproducibility, exhibited the smallest pore size, and had the best swelling ability while maintaining its integrity. In addition, hydrogel C showed a BSA burst release, which could be ideal for treatment of a wound in its acute phase, whereas the more hydrophobic formulations (hydrogels A and B) showed sustained protein release. This study not only introduced a novel methodology to quantify release under ocular conditions but also optimized a novel protocol to quantify the adhesive capacity of hydrogels toward the conjunctive of the eye using a rheometer, thereby reducing the need for in vivo animal testing. The results confirmed that hydrogel C increased its work of adhesion while increasing hydration. Finally, thermal and X-ray analysis showed that a combination of hydrophilic polymers with proteins can reduce the formation of crystals during the gelation process.In summary, Eudragits® have great potential as building blocks for the preparation of physically cross-linked hydrogels. Their rational selection enables to fine tune the hydrogel's pore size, the dissolution kinetics, and the protein release profiles, among other properties. This overall modulation enables to control the adhesion of materials to the mucus layer, yielding significant outcomes tailored to the final application.
Author contributions
D. E. U., A. S. and M. C. conceived the original idea of the project. D. E. U. synthesized the materials, performed the SEM, RMN, moisture content and uptake characterization and protein encapsulation and release experiments. M. F. S. M. and I. C. designed the rheological experiments. M. F. S. M. performed the rheological experiments. A. A. provided valuable medical needed for conceived the original idea of the project. E. M. and A. M. designed the thermal and X-ray diffraction characterization. E. M. performed the thermal characterization. R. S., E. M. and A. M. supported in the scientific discussion of the thermal and X-ray diffraction experiments. J. C. V. performed the cellular assays. A. L. assisted on the cellular assays and its scientific discussion. D. E. U. wrote the first draft of the manuscript. M. C. supervised and administered the work. M. C. and A. S. revised and improved the manuscript. All the authors revised and approved the final manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This research was funded by the Basque Government through the ELKARTEK program (KK-2019(00086), Salud carácter estratégico (2021333047), the University of the Basque Country (projects COLLAB22/05 and GIU21/033), and IKERBASQUE-Basque Foundation for Science. We also gratefully acknowledge support from the Basque Government through Grant IT1309-19 and R+D+i project PID2020-113045GB-C21 funded by MCIN/AEI/10.13039/501100011033/. A Sonzogni acknowledges financial support from CONICET, Universidad Nacional del Litoral, and the European Union Commission through RISE Horizon 2020, project 823989 IONBIKE. A Larrañaga is thankful for funds from the Basque Government, Department of Education (IT-1766-22). The authors thank UPV/EHU for technical and human support provided by SGIker. Gregor Nagel is acknowledged for scientific inputs. Matias L. Picchio, Sergio Martin-Saldaña, and Maria del Carmen Mañas Torres are acknowledged for proofreading the manuscript. Some designs were created with BioRender.com.References
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- M. Mahinroosta, Z. Jomeh Farsangi, A. Allahverdi and Z. Shakoori, Mater. Today Chem., 2018, 8, 42–55 CrossRef CAS.
- H. Shoukat, K. Buksh, S. Noreen, F. Pervaiz and I. Maqbool, Ther. Delivery, 2021, 12, 375–396 CrossRef CAS PubMed.
- M. F. Akhtar, M. Hanif and N. M. Ranjha, Saudi Pharm. J., 2016, 24, 554–559 CrossRef PubMed.
- R. C. Cooper and H. Yang, J. Controlled Release, 2019, 306, 29–39 CrossRef CAS PubMed.
- Z. Sun, C. Song, C. Wang, Y. Hu and J. Wu, Mol. Pharmacol., 2020, 17, 373–391 CAS.
- M. Kumari and A. Dhasmana, Adv. Tissue Eng. Regen. Med., 2020, 6, 20–24 Search PubMed.
- H. M. VanBenschoten and K. A. Woodrow, Adv. Drug Delivery Rev., 2021, 178, 113956 CrossRef CAS PubMed.
- V. V. Khutoryanskiy, Macromol. Biosci., 2011, 11, 748–764 CrossRef CAS PubMed.
- I. M. Yermak, V. N. Davydova and A. V. Volod’ko, Mar. Drugs, 2022, 20, 522 CrossRef CAS PubMed.
- S. Roy, K. Pal, A. Anis, K. Pramanik and B. Prabhakar, Des. Monomers Polym., 2009, 12, 483–495 CrossRef CAS.
- R. S. Dave, T. C. Goostrey, M. Ziolkowska, S. Czerny-Holownia, T. Hoare and H. Sheardown, J. Controlled Release, 2021, 336, 71–88 CrossRef CAS PubMed.
- K. Kumar, N. Dhawan, H. Sharma, S. Vaidya and B. Vaidya, Artif. Cells, Nanomed., Biotechnol., 2014, 42, 274–283 CrossRef CAS PubMed.
- S. Singh Neelam, S. Arora and Y. Singla, Asian J. Pharm. Clin. Res., 2015, 8, 1–6 Search PubMed.
- A. Nikam, P. R. Sahoo, S. Musale, R. R. Pagar, A. C. Paiva-Santos and P. S. Giram, Pharmaceutics, 2023, 15, 587 CrossRef CAS PubMed.
- S. Thakral, N. K. Thakral and D. K. Majumdar, Expert Opin. Drug Delivery, 2013, 10, 131–149 CrossRef CAS PubMed.
- Q. Zhu, C. Liu, Z. Sun, X. Zhang, N. Liang and S. Mao, Eur. J. Pharm. Biopharm., 2018, 128, 220–229 CrossRef CAS PubMed.
- S. Mahmood, M. A. Buabeid, K. Ullah, G. Murtaza, A. Mannan and S. A. Khan, Curr. Drug Delivery, 2019, 16, 548–564 CrossRef CAS PubMed.
- P. D. S. Chaves, L. A. Frank, A. G. Frank, A. R. Pohlmann, S. S. Guterres and R. C. R. Beck, Am. Assoc. Pharm. Sci., 2018, 19, 1637–1646 CAS.
- Z. Jafariazar, N. Jamalinia, F. Ghorbani-Bidkorbeh and S. A. Mortazavi, Iran. J. Pharm. Res., 2015, 14, 23–31 CAS.
- S. Mirzaeei and M. Alizadeh, J. Rep. Pharm. Sci., 2017, 6, 123–133 CAS.
- E. C. Kim, S. H. Doh, S. Y. Chung, S. Y. Yoon, M. S. Kim, S. K. Chung, M. C. Shin and H. S. Hwang, Acta Ophthalmol., 2017, 95, e314–e322 CAS.
- J. Macron, B. Bresson, Y. Tran, D. Hourdet and C. Creton, Macromolecules, 2018, 51, 7556–7566 CrossRef CAS.
- E. Marin, C. Tapeinos, S. Lauciello, G. Ciofani, J. R. Sarasua and A. Larrañaga, Mater. Sci. Eng., C, 2020, 117, 111349 CrossRef CAS PubMed.
- S. Singh Neelam, S. Arora and Y. P. Singla, Asian J. Pharm. Clin. Res., 2015, 8, 1–6 Search PubMed.
- X. Xue, Y. Hu, S. Wang, X. Chen, Y. Jiang and J. Su, Bioact. Mater., 2022, 12, 327–339 CrossRef CAS PubMed.
- S. M. LaNasa, I. T. Hoffecker and S. J. Bryant, J. Biomed. Mater. Res. Part B Appl. Biomater., 2011, 96B, 294–302 CrossRef CAS PubMed.
- Y. Zhang, L. Ye, M. Cui, B. Yang, J. Li, H. Sun and F. Yao, RSC Adv., 2015, 5, 78180–78191 RSC.
- J. Maitra and V. K. Shukla, Am. J. Polym. Sci., 2014, 4, 25–31 Search PubMed.
- A. Gallud, M. Delaval, P. Kinaret, V. S. Marwah, V. Fortino, J. Ytterberg, R. Zubarev, T. Skoog, J. Kere, M. Correia, K. Loeschner, Z. Al-Ahmady, K. Kostarelos, J. Ruiz, D. Astruc, M. Monopoli, R. Handy, S. Moya, K. Savolainen, H. Alenius, D. Greco and B. Fadeel, Adv. Sci., 2020, 7, 1–18 CrossRef PubMed.
- X. Xia, M. Yang, Y. Wang, Y. Zheng, Q. Li, J. Chen and Y. Xia, ACS Nano, 2012, 6, 512–522 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 37–42 CrossRef CAS.
- J. P. Jacobs, C. M. Jones and J. P. Baille, Nature, 1970, 227, 168–170 CrossRef CAS PubMed.
- T. Yadavalli, S. Mallick, P. Patel, R. Koganti, D. Shukla and A. A. Date, ACS Infect. Dis., 2020, 6, 2926–2937 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb01579c |
| This journal is © The Royal Society of Chemistry 2023 |