
Polyprodrugs for tumor chemotherapy: from molecular structure to drug release performance
Peng
Liu

State Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, People's Republic of China. E-mail: pliu@lzu.edu.cn; Fax: +86 931 8912582; Tel: +86 931 8912582
First published on 20th September 2023
Abstract
Polyprodrugs have been recognized as promising carrier-free drug delivery systems (DDSs) for tumor chemotherapy in recent years, showing distinct superiority in comparison with the conventional polymer prodrugs. In the present work, the structure–property relationship of polyprodrugs was explored from the perspective of molecular structure, by discussing the effects of the conjugations and linkers on their drug content and drug releasing performance, including drug releasing rate and drug releasing selectivity, as well as the anti-tumor performance of the released drugs. Moreover, the future challenges in the design of polyprodrug-based DDSs with high anti-tumor efficacy were also highlighted.
1. Introduction
Polymer prodrugs have been widely investigated for tumor chemotherapy over the last few decades.1–6 The hydrophilic or hydrophobic drugs (cisplatin, doxorubicin (DOX), camptothecin (CPT), paclitaxel (PTX), etc.) were conjugated on the end-groups or side-groups of the polymers with different topological structures: linear, branched or hyperbranched (dendritic) ones, as shown in Scheme 1. The conjugation could be cleaved by intracellular stimuli, higher acidity, higher glutathione (GSH) level, higher reactive oxygen species (ROS) level, or specific enzymes.7–9 Furthermore, the polymer chains endow the facile self-assembly of the polymer prodrugs into micelles with a certain diameter to achieve passive targeting via the enhanced permeability and retention (EPR) effect.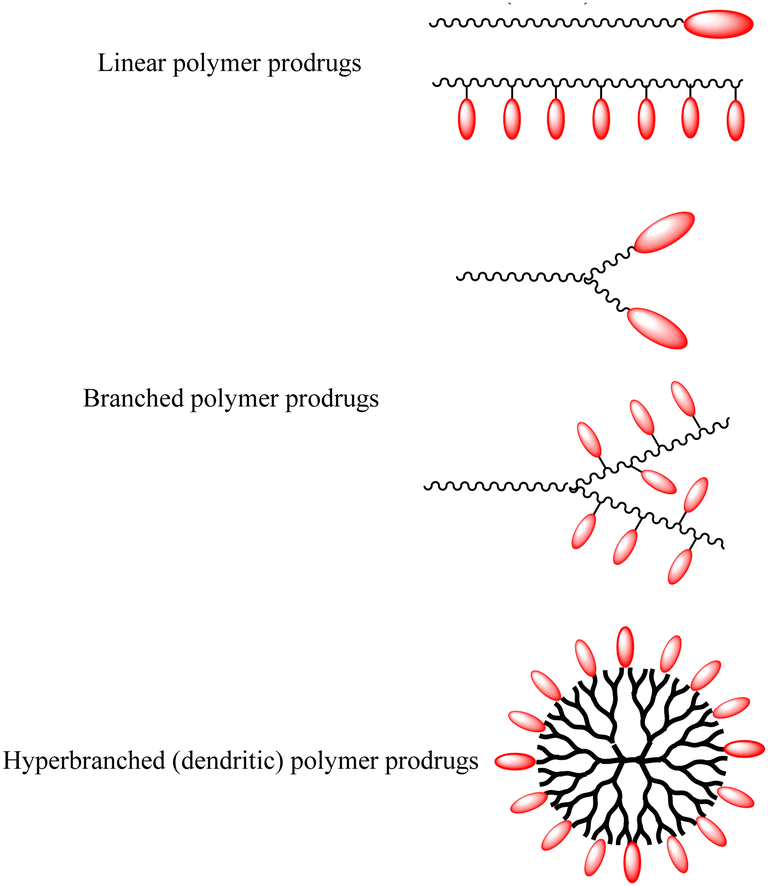 | ||
| Scheme 1 Conventional polymer prodrugs. | ||
The premature drug leakage in blood circulation from the polymer prodrugs could be minimized to a certain degree in comparison with the drug delivery systems (DDSs) via non-covalent drug-loading, showing better selective drug releasing performance. However, the drug molecule was conjugated onto the polymer with one labile conjugation, which could only be triggered by the corresponding stimulus, thus showing a single stimulus responsiveness. Due to the characteristics of the dynamic covalent bonds,8 the labile conjugation is usually not stable, and the conjugated drug would be released once the labile conjugation was broken. Therefore, the premature drug leakage could hardly be ignored.
As a powerful candidate, polyprodrugs could solve the above problem perfectly,10,11 by comprising the drug molecules as structural units. For this purpose, there should be two functional groups in the parent drugs at least. By coupling with different functional groups in the linkers (small molecules or polymers), linear or branched polyprodrugs could be obtained via conjugation such as imine, amide or ester groups (Scheme 2). As a structural unit in the polyprodrug, the drug molecule could be released only when all of the two or three conjugations were cleaved simultaneously. As a result, the premature drug leakage could be minimized efficiently. An external stimulus such as UV12,13 and intracellular signals could be used to trigger the degradation of the polyprodrugs to release the drug. Comparatively speaking, more intelligent drug release could be obtained via the latter one, owing to its excellent control via smart targeting.
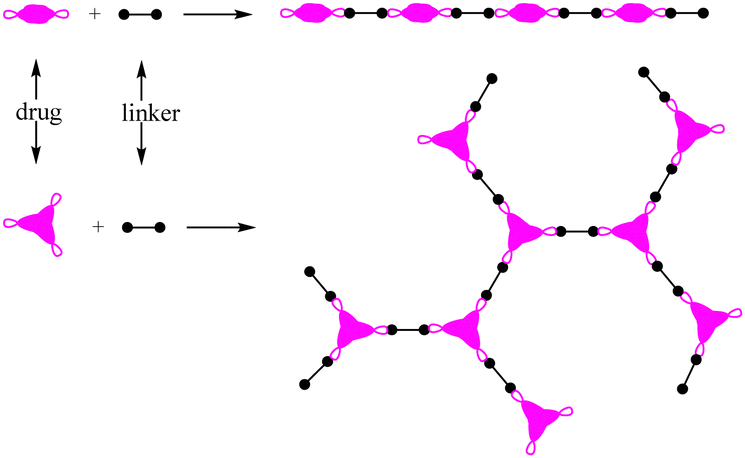 | ||
| Scheme 2 Linear or branched polyprodrugs conjugated with two or three functional groups on the parent drugs. | ||
Commonly, polyprodrugs are synthesized by coupling the functional groups on the parent drugs with various dynamic covalent bonds by step polymerization via condensation or addition, therefore endowing stimuli-triggered degradation and drug release performance for on-demand tumor chemotherapy. Owing to the excellent drug release performance because of the unique molecular structure, polyprodrugs have been intensely developed in recent years. In this work, the structure–property relationship of polyprodrugs was discussed, focusing on the effect of the molecular structure of polyprodrugs on their drug content and drug releasing performance. It is expected to open new ideas for the design of polyprodrug-based DDSs with high anti-tumor efficacy.
2. Structural analysis
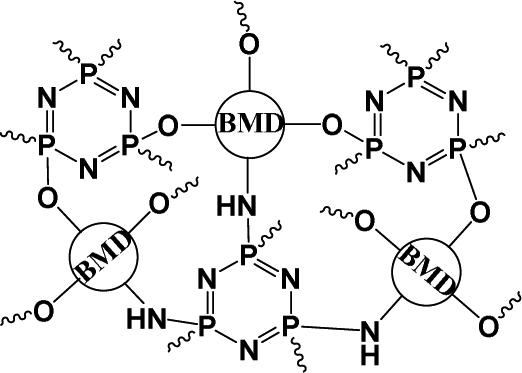
In the polyprodrugs, the drug molecules have been comprised into the polymer chains as structural units by conjugating with dynamic covalent bonds. As shown in Scheme 2, linear or branched polyprodrugs could be obtained by conjugating two or three functional groups on the parent drugs. In the branched polyprodrugs, it is hard to avoid the crosslinking reaction. The 3-dimensional structure of the branched or crosslinked ones makes it difficult to control the diameter of the final DDSs. Therefore, the reaction conditions should be strictly controlled to avoid the formation of bulky products, mainly controlling the feeding ratio of the monomers and reaction degree.14–17The chain length of the linear polyprodrugs could be easily controlled by adjusting the feeding ratio of the monomers. It could be split into three main units: drug unit, conjugation and linker, as shown in Scheme 3. The cleavable conjugation could be located between the drug unit and the linker (Scheme 3A) or in the linker (Scheme 3B). To endow self-assembly and improve the dispersibility, polyprodrugs are usually PEGylated by introducing a non-toxic and non-immunogenic polyethylene glycol (PEG) block.18–24 This could also decrease immunogenicity, reduce proteolysis and renal excretion, and prolong the blood circulation life.25 The controlled drug release performance was mainly dependent on the stimuli-triggered degradation of the polyprodrugs, which was determined by their molecular structures, specifically, the conjugations and linkers. Such effects will be discussed comprehensively below.
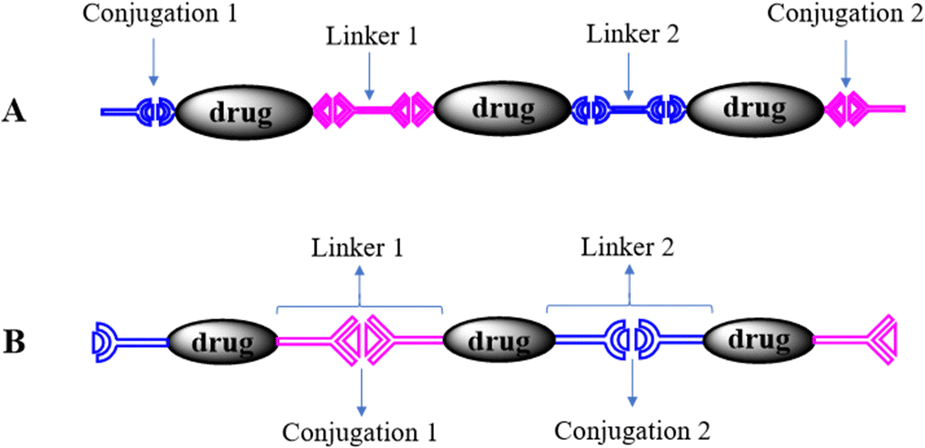 | ||
| Scheme 3 Structural analysis of the linear polyprodrugs. | ||
3. Effect of linkers
The effect of the linkers is relatively simple. Firstly, a certain length is usually required to avoid the steric hindrance of the parent drugs during the polymerization. However, it also can't be too long. A long linker decreases the drug content in the polyprodrugs, meaning that high doses or repetitive administration should be conducted.Another key factor is the hydrophilic–hydrophobic property of the linkers. Most of the chemotherapeutic drugs are hydrophobic. When they are linked with a hydrophobic linker, a completely hydrophobic polyprodrug is synthesized. After formulation as nanomedicine, the highly hydrophobic core prevents the diffusion and access of the stimuli, such as H+ ions, GSH or ROS, which are water-soluble. Therefore, a slow drug release would be resulted, because the degradation of the polyprodrug nanoparticles could only occur on their surface.
PEG with a certain length has been used as the hydrophilic linker in the design of polyprodrugs.26 The hydrophilic block could endow the self-assembly of the amphiphilic polyprodrugs to achieve the desired nanomedicine with proper diameter. The amphiphilic polyprodrugs with longer PEG blocks could self-assemble into core–shell nanoparticles with aggregates of drug units as hydrophobic cores, while the longer PEG blocks act as the hydrophilic brushes on their surfaces. This structure is expected to improve the systemic circulation time and decrease the immunogenicity of the nanomedicines.27 On the other hand, the shorter PEG blocks might be imbedded into the inner of the nanomedicines, facilitating the diffusion and access of the stimuli and subsequently accelerating the drug release.
4. Effect of conjugations
The conjugation is the most important key part in the polyprodrugs, which determines the degradability of the polyprodrugs and the degraded products (namely the released drug). To be specific, the stimuli-responsiveness and the responsive sensitivity of the polyprodrugs are controlled by the kinds of conjugations used. While the structure and site determine the structure and anti-tumor efficacy of the released drug.4.1. Triggered signal for the conjugations
The same conjugation could be used to conjugate the parent drugs into a polymer chain, which exhibited a single stimulus responsiveness, i.e. acidity (Table 1), reduction (Table 2), or oxidation (Table 3). Furthermore, two kinds of conjugations triggered by the same stimulus but with different responsive sensitivity could be used to tune the drug release rate.| Polyprodrugs | Drug content | Drug leakage | Drug release | Ref. |
|---|---|---|---|---|
|
89.5% | 7% (pH 7.4, 96 h) | 23% (pH 5.5, 96 h) | 14 |
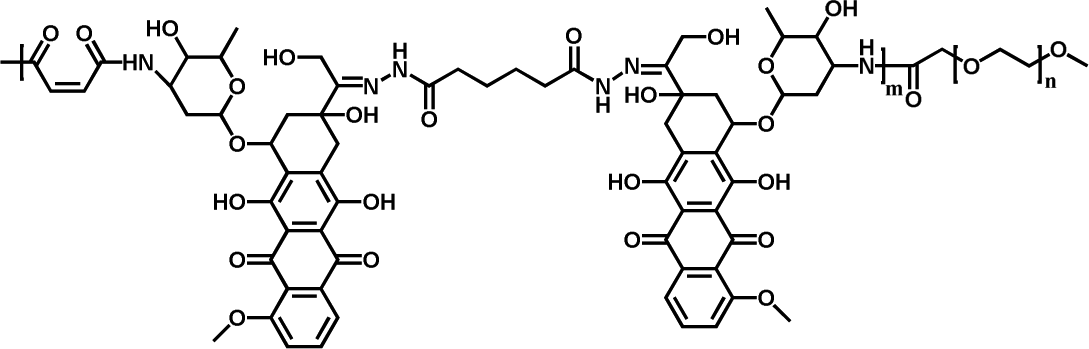
|
58.14% | 32.3% (pH 7.4, 56 h) | 84.5% (pH 5.0, 56 h) | 15 |
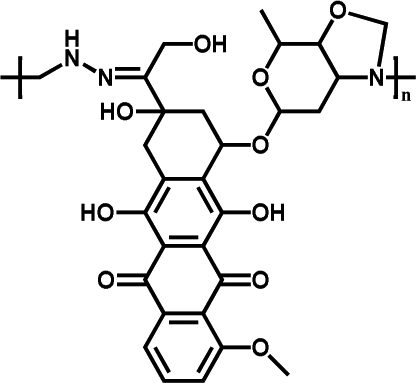
|
75.42% | 4.39% (pH 7.4, 60 h) | 39.83% (pH 5.0, 60 h) | 20 |
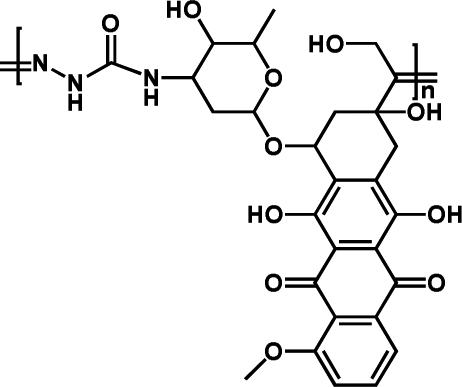
|
92.45% | 12.9% (pH 7.4, 10 h) | 100% (pH 5.0, 10 h) | 28 |
|
1.67 mmol g−1 | 6% (pH 7.4, 60 h) | 47% (pH 5.0, 60 h) | 29 |
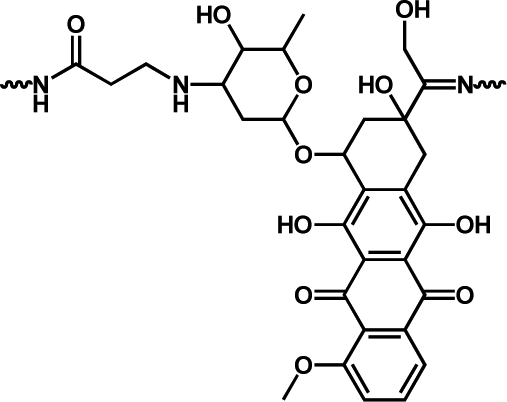
|
1.60 mmol g−1 | 1.02% (pH 7.4, 108 h) | 30.9% (pH 5.0, 108 h) | 30 |
| Polyprodrugs | Drug content | Drug leakage | Drug release | Ref. |
|---|---|---|---|---|
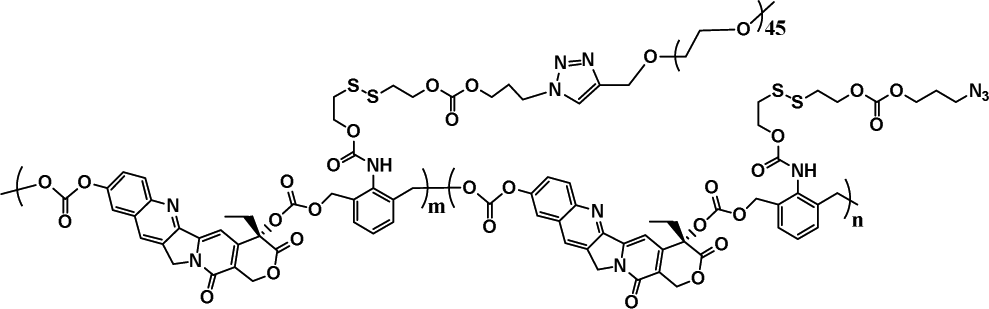
|
18% | ∼7% (without DTT, 48 h) | 40% (5 mM DTT, 48 h) | 18 |
| 90% (20 mM DTT, 48 h) | ||||
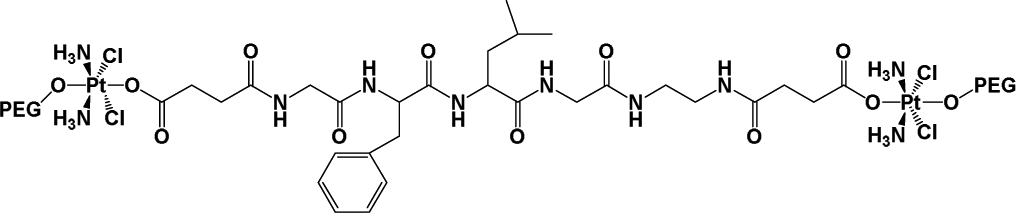
|
24.7% | 15% (pH 7.4, 36 h) | 90% (pH 5.5, 10 mM GSH, 5 × 10−8 M papain, 36 h) | 19 |
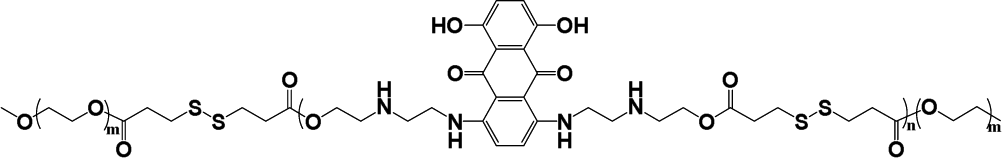
|
— | <10% (10 μM GSH, 24 h) | 90% (10 mM GSH, 24 h) | 22 |

|
12% | 35% (without GSH, 96 h) | 100% (10 mM GSH, 24 h) | 26 |
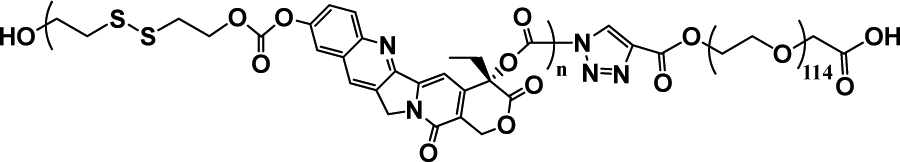
|
36.7% | 20% (20 μM GSH, 24 h) | 65% (10 mM GSH, 2 h) | 31 |

|
63% | 9% (10 μM GSH, 24 h) | 90% (10 mM GSH, 24 h) | 32 |
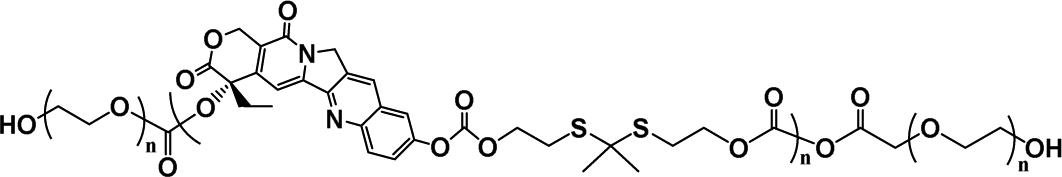
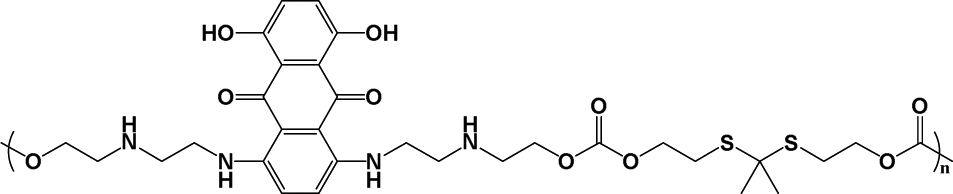
Because of the similar acidity in the intracellular microenvironment in the normal cells and the tumor cells (pH ∼ 5), such acid-triggered polyprodrugs could only minimize the premature drug leakage in the blood (pH 7.4), but not differentiate the normal cells and the tumor cells. Therefore, tumor-specific chemotherapy could not be achieved with the acid-triggered polyprodrugs, similar to the acid-triggered polymer prodrugs.
However, the premature drug leakage and the drug release were still assessed in the simulated normal physiological media (without GSH and H2O2 or with a very low level) and the simulated tumor intracellular microenvironment (10 mM GSH or 100 μM H2O2), respectively. Nevertheless, the drug release in the normal cells (2 mM GSH) was not considered. It is a promising strategy to achieve real tumor-specific chemotherapy without toxic side effects on normal cells by controlling the reduction/oxidation-responsive sensitivity of such redox-triggered degradable polyprodrugs.
| Polyprodrugs | Drug content | Drug leakage | Drug release | Ref. |
|---|---|---|---|---|
|
56.4% | 20.6% (pH 7.4, 72 h) | 36.8% (pH 5.0, 10 mM GSH, 72 h) | 16 |
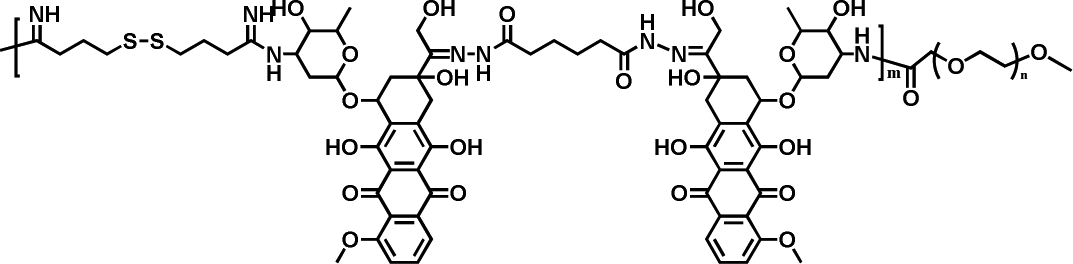
|
78.6% | 2.0% (pH 7.4, 10 μM GSH, 60 h) | 25.5% (pH 5.0, 10 mM GSH, 60 h) | 23 |

|
61.1% | 1.5% (pH 7.4, 10 μM GSH, 58 h) | 18.9% (pH 5.0, 10 mM GSH, 58 h) | 24 |
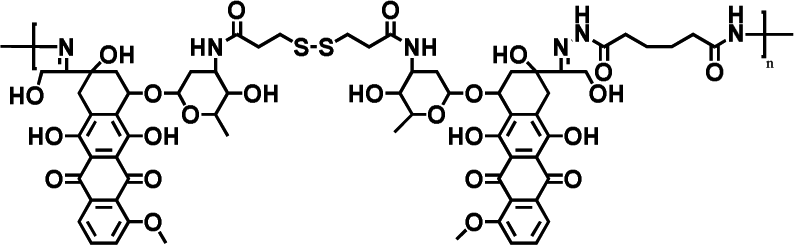
|
78% | 0.6% (pH 7.4, 10 μM GSH, 60 h) | 44% (pH 5.0, 10 mM GSH, 60 h) | 35 |
4.2. Conjugating sites
Another controlling factor is the site of the conjugations in the polyprodrugs. They could be contained in the linkers as shown in Scheme 3A or placed between the drug unit and the linker as shown in Scheme 3B. Different drug release behaviors were caused by the different sites of the conjugations. In the works with the molecular structure as shown in Scheme 3A, the cleavable conjugations were located between the drug unit and the linker. The parent drug could be released after stimuli-triggered cleavage of the conjugations.As for the polyprodrugs with the molecular structure as shown in Scheme 3B, the cleavable conjugations were in the linker. Only the derivatives of the parent drug were released after the acid-triggered cleavage of the acid-labile conjugations,29,30 reduction-triggered cleavage of the disulfide conjugations20,22,23,35 or oxidation-triggered cleavage of the thioketal conjugations.33 The derivatives could be easily revealed by the high performance liquid chromatography (HPLC) analysis. That is to say, part of the linker remained on the released drug, which could not transform into the parent drug because of the stable covalent bonds between them. Due to the derivation, the solubility of the released drug decreased. The drug release behavior could be categorized as a dissolution-controlled mode, which could be revealed by the drug release profiles in the presence of a surfactant, such as Tween, showing a much faster drug release in comparison with the case without surfactant. This phenomenon indicated that the stimuli-triggered degradation of the polyprodrugs was faster than the dissolution of the released drugs with lower solubility. Lower anti-tumor efficacy was usually obtained in comparison with the parent drugs, due to its molecular structure and subsequently lower solubility and pharmacological activity.36
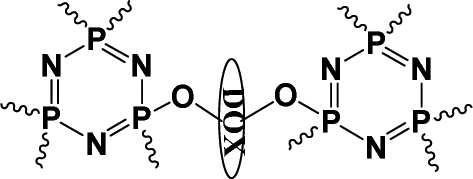
On the other hand, this characteristic might be a powerful approach to achieve a higher anti-tumor efficacy by delivering a high-performance chemotherapeutic drug with a well-designed polyprodrug. For example, doxazolidine (Doxaz) has been reported as a proposed active DOX metabolite to cross-link DNA. It could inhibit the growth of cancer cells at 1–4 orders of magnitude lower concentration than DOX and be taken up by multidrug-resistant tumor cells 3- to 4-fold better than DOX.37 However, its hydrolysis half-life was about 16 min at pH 7.5 and 3 min in human serum at 37 °C, restricting its practical application. To solve this problem, the Liu group designed acid-labile poly(doxazolidine) (P(Doxaz)) with Doxaz as structural units by a facile condensation polymerization of amino-hydrazone modified DOX (DOX-hyz) and formaldehyde.28 The P(Doxaz) was quite stable in pH 7.4 PBS, while completely degraded within 9.5 h and released high anti-tumor chemotherapeutic agent (Doxaz) in the simulated tumor intracellular microenvironment (Scheme 4).
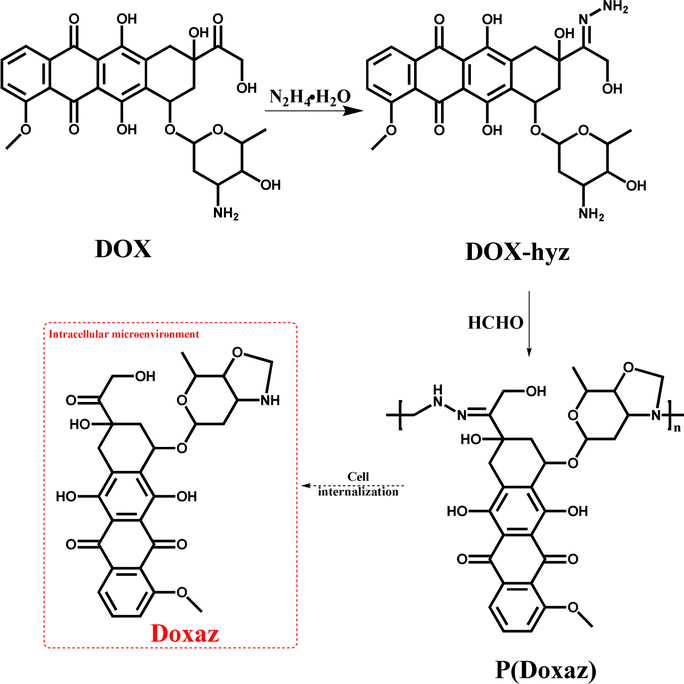 | ||
| Scheme 4 Synthesis and intracellular degradation of P(Doxaz).28 | ||
Most recently, the reaction of cyclizing into the proximate carbonyl group has been reported to transform thiolated derivatives into the parent drugs (Scheme 5),38,39 which could be used to release the parent after cleaving the reducible-sensitive disulfide18,24,26,31,32 or oxidation-sensitive thioketal conjugation.21 With such a trace-less linker, the parent drug (10-hydroxycamptothecin, HCPT) could be released from the reduction- or oxidation-triggered degradable polyprodrugs (Scheme 6).
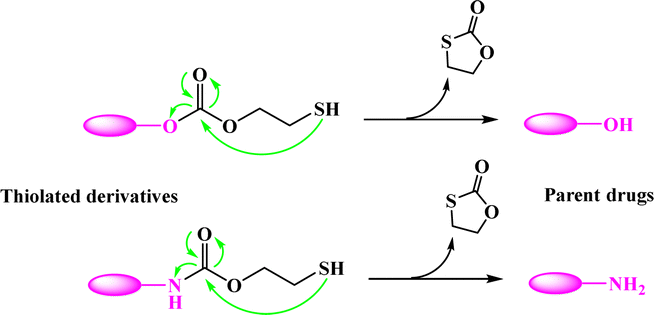 | ||
| Scheme 5 Transformation of thiolated derivatives into parent drugs. | ||
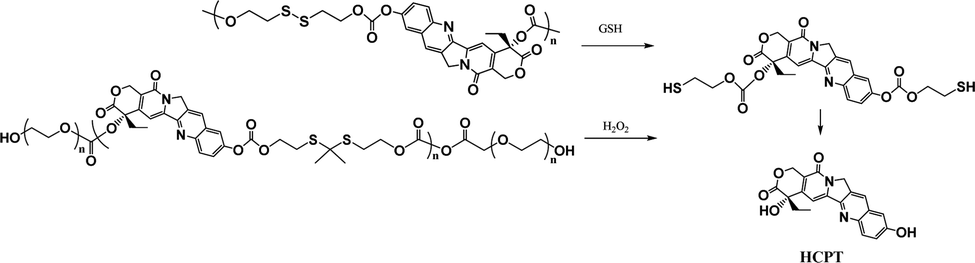 | ||
| Scheme 6 Reduction- or oxidation-triggered degradation of polyprodrugs to release the parent drug.21,32 | ||
Based on the trace-less linker, the Liu group reported an acid/reduction co-triggered degradable amphiphilic copolyprodrug with both acid-labile conjugation and reduction-cleavable conjugation adjacent to the same drug unit for the first time. The parent drug, DOX, could be released after the tumor intracellular degradation of the proposed amphiphilic copolyprodrug (PDOXSS-ADH-PEG) (Scheme 7), and an improved tumor-specific anti-tumor efficacy was achieved.24
 | ||
| Scheme 7 Tumor intracellular acid/reduction co-triggered degradation of the amphiphilic copolyprodrug (PDOXSS-ADH-PEG).24 | ||
5. Future challenges
As discussed above, the premature drug leakage and drug release were only assessed in simulated normal physiological media and a simulated tumor intracellular microenvironment, respectively, in the reported works. To avoid the toxic side effects on normal cells, the drug release should also be assessed in normal cells. By tuning the responsive sensitivity of the conjugation, an exceptional anti-tumor efficacy is expected with a high drug release in the tumor cells but a very low drug release in the normal cells, exhibiting an excellent tumor-selective drug release. Otherwise, tumor-targeted groups should be introduced to promote the internalization of the DDSs in the tumor cells.Besides the molecular structure of the polyprodrugs, their aggregation structure should be considered. Especially for the ones containing DOX or CPT, except for the usually considered hydrophobic interaction, the π–π stacking between the drug units might influence the self-assembly structure of the polyprodrug nanoparticles, and therefore, affect the drug release performance. Besides, because the polyprodrugs could degrade step-by-step to release the chemotherapeutic drugs, their degree of polymerization might affect the degradation and drug release rates. Moreover, the degree of polymerization of the rigid polyprodrug blocks would also influence their aggregation structure, as a result, influencing its degradation and drug release rates.
Finally, combination chemotherapy by co-administrating chemotherapeutic drugs has attracted more and more interest owing to its synergistic/additive action in comparison with the conventional mono-chemotherapy.40 It can also sensitize the cancer response to drugs, modulate different signaling pathways in tumor cells, and lower each drug dose to reduce side effects. The design of polyprodrugs containing two kinds of chemotherapeutic drugs for combination chemotherapy should be the next promising topic in the field.
6. Conclusions and prospects
Polyprodrugs have been recognized as having promising potential for carrier-free DDSs in tumor chemotherapy, owing to their lower premature drug leakage in comparison with the conventional polymer prodrugs. Their drug release performance could be tuned via molecular design, by selecting the conjugation and linker. Furthermore, the stability of the high anti-tumor chemotherapeutic agents could be improved in the form of polyprodrugs. Thus, better anti-tumor chemotherapeutic efficacy could be achieved with all aspects of the molecular design of polyprodrugs. However, they have a long way to go before clinical application, with genuine tumor selectivity. Besides, their aggregation structure should also be considered in formulation as nanomedicines. And better anti-tumor efficacy is expected by designing polyprodrugs containing two kinds of chemotherapeutic drugs for combination chemotherapy. Such discussions are expected to open new ideas in the design of novel polyprodrugs for smarter tumor chemotherapy.Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Hoste, K. de Winne and E. Schacht, Int. J. Pharm., 2004, 277, 119 CrossRef CAS PubMed.
- J. Khandare and T. Minko, Prog. Polym. Sci., 2006, 31, 359 CrossRef CAS.
- D. Dutta, W. D. Ke, L. C. Xi, W. Yin, M. Zhou and Z. S. Ge, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 12, e1585 Search PubMed.
- M. L. Girase, P. G. Patil and P. P. Ige, Int. J. Polym. Mater. Polym. Biomater., 2020, 69, 990 CrossRef CAS.
- X. Dong, R. K. Brahma, C. Fang and S. Q. Yao, Chem. Sci., 2022, 13, 4239 RSC.
- K. K. Yang, Z. Q. Yang, G. C. Yu, Z. H. Nie, R. B. Wang and X. Y. Chen, Adv. Mater., 2022, 34, 2107434 CrossRef CAS PubMed.
- C. A. Blencowe, A. T. Russell, F. Greco, W. Hayes and D. W. Thornthwaite, Polym. Chem., 2011, 2, 773 RSC.
- P. Theodosis-Nobelos, D. Charalambous, C. Triantis and M. Rikkou-Kalourkoti, Curr. Drug Delivery, 2020, 17, 542 CrossRef PubMed.
- A. Nguyen, R. Bottger and S. D. Li, Biomaterials, 2021, 275, 120955 CrossRef CAS PubMed.
- K. K. Yang, Z. Q. Yang, G. C. Yu, Z. H. Nie, R. B. Wang and X. Y. Chen, Adv. Mater., 2022, 34, 2107434 CrossRef CAS PubMed.
- F. Seidi, Y. J. Zhong, H. N. Xiao, Y. C. Jin and D. Crespy, Chem. Soc. Rev., 2022, 51, 6652 RSC.
- D. F. Zhou, J. S. Guo, G. B. Kim, J. Z. Li, X. S. Chen, J. Yang and Y. B. Huang, Adv. Healthcare Mater., 2016, 5, 2493 CrossRef CAS PubMed.
- Y. F. Zhang, Q. Yin, L. C. Yin, L. Ma, L. Tang and J. J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 6435 CrossRef CAS PubMed.
- S. L. Hou, S. S. Chen, Y. Dong, S. Gao, B. S. Zhu and Q. H. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 25983 CrossRef CAS PubMed.
- N. Zhou, N. Zhang, Z. Zhi, X. N. Jing, D. M. Liu, Y. P. Shao, D. Q. Wang and L. J. Meng, J. Mater. Chem. B, 2020, 8, 10540 RSC.
- D. Q. Wang, N. Zhou, N. Zhang, Z. Zhi, Y. P. Shao, L. J. Meng and D. M. Yu, Colloids Surf., B, 2021, 200, 111573 CrossRef CAS PubMed.
- X. Duan, J. X. Chen, S. Wu, D. Y. Shao and J. Kong, Chem. – Asian J., 2018, 13, 939 CrossRef CAS PubMed.
- K. M. Cai, J. Yen, Q. Yin, Y. Liu, Z. Y. Song, S. Lezmi, Y. F. Zhang, X. J. Yang, W. G. Helferich and J. J. Cheng, Biomater. Sci., 2015, 3, 1061 RSC.
- W. H. Wang, G. H. Liang, W. J. Zhang, D. Xing and X. L. Hu, Chem. Mater., 2018, 30, 3486 CrossRef CAS.
- J. G. Li, X. M. Li, P. W. Xie and P. Liu, Int. J. Pharm., 2021, 609, 121142 CrossRef CAS PubMed.
- Q. Y. Meng, H. Hu, X. D. Jing, Y. Sun, L. P. Zhou, Y. W. Zhu, B. Yu, H. L. Cong and Y. Q. Shen, J. Controlled Release, 2021, 340, 102 CrossRef CAS PubMed.
- D.-B. Cheng, X.-H. Zhang, S.-Y. Chen, X.-X. Xu, H. Wang and Z.-Y. Qiao, Adv. Mater., 2022, 34, 202109528 Search PubMed.
- X. M. Li, J. Zhang and P. Liu, Int. J. Pharm., 2021, 606, 120941 CrossRef CAS PubMed.
- X. M. Li and P. Liu, J. Mater. Chem. B, 2022, 10, 2926 RSC.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315 CrossRef CAS PubMed.
- X. L. Zhang, M. K. Zhang, M. Q. Wang, H. Peng, Q. Hua, L. W. Ma, B. Y. Wang and H. Wei, Bioconjugate Chem., 2018, 29, 2239 CrossRef CAS PubMed.
- J. S. Suk, Q. G. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Rev. Drug. Delivery, 2016, 99, 28 CrossRef CAS PubMed.
- J. G. Li, X. M. Li and P. Liu, Mol. Pharmaceutics, 2020, 17, 710 CAS.
- X. M. Li, X. M. Zhao and P. Liu, Eur. Polym. J., 2021, 161, 110820 CrossRef CAS.
- X. M. Li and P. Liu, Mater. Sci. Eng., C, 2021, 128, 112317 CrossRef CAS PubMed.
- X.-D. Xu, Y.-J. Cheng, J. Wu, H. Cheng, S.-X. Cheng, R.-X. Zhuo and X.-Z. Zhang, Biomaterials, 2016, 76, 238 CrossRef CAS PubMed.
- S. L. Li, P. E. Saw, C. H. Lin, Y. Nie, W. Tao, O. C. Farokhzad, L. Zhang and X. D. Xu, Biomaterials, 2020, 234, 119760 CrossRef CAS PubMed.
- X. D. Xu, P. E. Saw, W. Tao, Y. J. Li, X. Y. Ji, S. Bhasin, Y. L. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed.
- T. Sun and C. Jiang, Adv. Drug Delivery Rev., 2023, 196, 114773 CrossRef CAS PubMed.
- J. G. Li and P. Liu, Macromol. Rapid Commun., 2018, 39, 1800381 CrossRef PubMed.
- Q. Song, X. Wang, Y. Q. Wang, Y. Q. Liang, Y. X. Zhou, X. N. Song, B. He, H. Zhang, W. B. Dai, X. Q. Wang and Q. Zhang, Mol. Pharmaceutics, 2016, 13, 190 CrossRef CAS PubMed.
- G. C. Post, B. L. Barthel, D. J. Burkhart, J. R. Hagadorn and T. H. Koch, J. Med. Chem., 2005, 28, 7648 CrossRef PubMed.
- X. L. Hu, J. M. Hu, J. Tian, Z. S. Ge, G. Y. Zhang, K. F. Luo and S. Y. Liu, J. Am. Chem. Soc., 2013, 135, 17617 CrossRef CAS PubMed.
- Y. Q. Wang, Z. H. Wang, B. L. Chen, Q. Q. Yin, M. J. Pan, B. Zhang, Y. Yan, Z. J. Jiang, Q. Zhang and Y. G. Wang, Nano Lett., 2021, 21, 4371 CrossRef CAS PubMed.
- S.-Y. Qin, Y.-J. Cheng, Q. Lei, A.-Q. Zhang and X.-Z. Zhang, Biomaterials, 2018, 171, 178 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |