
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anaerobic biodegradation of citric acid in the presence of Ni and U at alkaline pH; impact on metal fate and speciation†
Natalie
Byrd
 *a,
Jonathan R.
Lloyd
a,
Luke T.
Townsend
d,
Joe S.
Small
a,
Frank
Taylor
b,
Heath
Bagshaw
c,
Christopher
Boothman
a,
Ilya
Strashnov
e and
Katherine
Morris
*a
*a,
Jonathan R.
Lloyd
a,
Luke T.
Townsend
d,
Joe S.
Small
a,
Frank
Taylor
b,
Heath
Bagshaw
c,
Christopher
Boothman
a,
Ilya
Strashnov
e and
Katherine
Morris
*a
aResearch Centre for Radwaste Disposal and Williamson Research Centre, Department of Earth and Environmental Sciences, The University of Manchester, Manchester M13 9PL, UK. E-mail: katherine.morris@manchester.ac.uk
bNuclear Waste Services, Pelham House, Seascale, Cumbria CA20 1DB, UK
cSchool of Engineering, The University of Liverpool, Liverpool, L69 3GQ, UK
dNucleUS Immobilisation Science Laboratory, Department of Materials Science and Engineering, The University of Sheffield, Sheffield, UK
eDepartment of Chemistry, University of Manchester, UK
First published on 24th July 2023
Abstract
Citrate is a key decontaminant used in the nuclear industry and here we explore its biogeochemical fate in the presence of Ni2+ and U(VI)O22+ under conditions relevant to low level radioactive waste (LLW) disposal. Anaerobic microcosm experiments were performed under nitrate- and sulfate-reducing conditions at between pH 9 and 10. Citrate (1 mM) was supplied as both an electron donor and a potential metal ion complexant. Incubation experiments with citrate, inoculated with nitrate- or sulfate-reducing microbial consortia, were challenged with three different concentrations of Ni: 0.01, 0.1 or 1 mM, or U: 0.005, 0.05, or 0.5 mM. The nitrate- and sulfate-reducing inocula were enriched from well characterised alkaline sediments obtained from high pH lime-workings. A multi-technique approach was adopted to characterise the aqueous geochemistry, solid phase mineralogy, and bacterial communities in each incubation system. In the 0.01 mM Ni systems citrate underwent full biodegradation under both nitrate and sulfate-reducing conditions in less than 15 days. In the sulfate-reducing experiments, 50% of the added 0.01 mM Ni(aq) was removed from solution and black solids formed; SEM and TEM analysis suggested that these were Ni-sulfides. For the higher Ni concentration incubations, no changes were observed in the nitrate-amended experiments. In the sulfate-amended experiments only citrate fermentation was observed, likely because elevated levels of Ni were toxic to nitrate- and sulfate-reducing bacteria in the inocula. Interestingly, although fermentative bacteria were key citrate degraders in the sulfate-amended experiments they did not dominate in the nitrate-amended experiments presumably due to competition from other microbes. In the U experiments, citrate degradation took place over 55 days in all systems except the 0.5 mM U/nitrate-amended incubations. In all U/sulfate-amended experiments, a dark-coloured precipitate formed and XAS analysis indicated that these solids contained reduced U(IV) with EXAFS suggesting that non-crystalline U(IV)–phosphate phases dominated. Microbial community analysis by 16S rRNA gene sequencing of endpoint samples identified fermenters and nitrate- and sulfate-reducing bacteria in the relevant incubations. Overall, findings suggest microbial degradation of citrate occurs under repository relevant conditions with Ni (at 0.01–0.1 mM) and U (at 0.005–0.5 mM) but with an inhibitory effect particularly at elevated Ni concentrations. Significantly, the work suggests that under anaerobic conditions relevant to LLW disposal, citrate undergoes biodegradation leading to the development of poorly soluble Ni sulfides and/or bioreduction of U(VI) to poorly soluble U(IV) phases. This suggests that both removal of citrate, and retention of Ni and U can occur in these environments and this information can be used to further inform development of safety cases for radioactive waste disposal.
Environmental significanceCitrate, a naturally ubiquitous ligand, is a key decontaminant in the nuclear industry and is abundant in low level radioactive waste (LLW). Citrate complexation may promote contaminant metal/radionuclide migration in the environment; even at alkaline pH levels expected in cementitious LLW repositories. Understanding potential biodegradation mechanisms of citrate under LLW disposal conditions, where contaminant species (e.g. U, Ni) are present, is important to predict the impacts of citrate in wastes and to inform development of safety cases for LLW disposal. This work shows anaerobic citrate biodegradation at elevated pH, when Ni or U(VI) are present, leading to the development of poorly soluble Ni and U(IV) phases. Overall, findings directly inform the safety case for the UK's LLW repository and are relevant to other cementitious waste repositories worldwide. |
Introduction
A key challenge in low level radioactive waste (LLW) management is to understand and predict the potential migration of metal contaminants, such as Ni and U, from the wastes. Radioactive waste management design aims to mitigate contaminant migration1 typically by generating a localised anaerobic, high pH, reducing near-field environment post closure.2–4 Here, the solubility and transport of metals may be affected by organic ligands within wastes.5–9 Candidate organic ligands from decommissioning activities (citrate, oxalate, nitrilotriacetic acid [NTA] and ethylenediaminetetraacetic acid [EDTA]) can affect metal solubility even at alkaline pH, through the formation of metal–ligand complexes.10–13 In particular, citrate may be present within typical LLW in significant quantities from nuclear decontamination operations.14–16Citric acid (2-hydroxy-1,2,3-propanetricarboxylic acid, C6H8O7) is a naturally ubiquitous and widely utilised molecule.17–19 In the nuclear industry it is used as a decontaminant due to its behaviour as a multidentate ligand. It forms aqueous complexes via its hydroxyl and/or carboxylate groups with key radioactive species including Ni and U.20–23 Metal–citrate complexes are considered relatively mobile in the environment24–30 and an understanding of potential biodegradation mechanisms of citrate under LLW disposal conditions, where U and Ni are present is important to predict the impacts of using citrate as a decontaminant in nuclear decommissioning. Citrate can be utilised as an electron donor in a range of engineered and natural environments.31–41 Recently, under alkaline conditions representative of a cementitious repository, citrate was shown to be an effective electron donor in support of anaerobic metabolism.16 Furthermore, experiments showed that Fe(III)–citrate complexation may enhance the extent of high pH (pH 11.7) Fe(III)-bioreduction.16
In the current work, we explore the fate of both Ni2+ and U(VI)O22+ in citrate biodegradation experiments as they are significant contaminants in many radioactive wastes.9 Here, 63Ni (half-life ∼100 years) is present from neutron capture (by 62Ni) and stable nickel is present from its use in e.g. corrosion resistant alloys.42,43 In addition, U is typically the most significant radionuclide by mass in radioactive wastes including LLW.9,44–47 The environmental mobility of U is influenced by its speciation and oxidation state at neutral to mildly alkaline pH. Relatively soluble U(VI)(aq) species can be (bio)reduced to poorly soluble U(IV), a key mechanism in promoting U immobilisation, including at elevated pH.48,49 By contrast, Ni is not typically redox active, but is prone to immobilisation via the precipitation of insoluble mineral phases, e.g. with sulfide or phosphate.50,51
At near-neutral pH, biodegradation of a range of metal–citrate complexes, including U, Pu, Eu, Fe(III), Ni, Co, Zn, Al, Cd, and Cu have been examined under both aerobic and anaerobic conditions.7,20,21,32,52–58 Under these conditions, it was reported that biodegradation of metal–citrate complexes was dependant on the complex formed. Authors of this work suggested that tridentate complexes involving the hydroxyl group of the citrate ligand were recalcitrant to biodegradation, whilst bidentate complexes involving only the carboxylate groups were biodegradable.20,59,60 The recalcitrance of tridentate complexes to biodegradation was attributed to their limited uptake into the cell, however potential U toxicity was not ruled out/discussed.20,53,61
Citrate is expected to form bidentate complexes with Ni(II) under circumneutral conditions, but under alkaline conditions tridentate complexation may be more favourable as the hydroxyl group may ionize to offer a further binding site.23,62 Under neutral pH conditions, citrate forms bi-nuclear complexes with U(VI), e.g. [(UO2)2(Cit)2]2−, which are recalcitrant to biodegradation by Pseudomonas fluorescens under aerobic and denitrifying conditions.20,53 Indeed, Francis et al., suggested that the [(UO2)2(Cit)2]2− species is stable up to pH 9.53 However, thermodynamic modelling by Huang et al., suggested that above pH 8, the hydroxyl complex [(UO2)3(OH)7]2− would dominate58 and this is supported by more recent thermodynamic data.58,63,64 The study by Huang et al., 1998 is one of the only studies to explore biodegradation of U–citrate complexes at alkaline pH (pH 8–9) experimentally and under aerobic conditions. The study showed the citrate biodegradation rate was higher at pH 8–9 than at pH 6–7.52 Indeed, a range of metals, including Ni, Cu, Co, Tc, U, Th and Zn are shown to form complexes with citrate, and other organic ligands (e.g. EDTA, NTA, ISA).12,65–69 Again, studies relating to biodegradation of these complexes under anaerobic, alkaline conditions are sparse.
Accordingly, further research regarding Ni/U/citrate speciation under elevated pH conditions is important to further understand Ni and U mobility in LLW disposal. Although LLW disposal facilities are expected to have a bulk pH of >11, waste form heterogeneity70,71 and biogeochemical processes16,72–74 may give rise to localised areas where pH is more neutral which may impact Ni/U/citrate speciation. In addition, evolution of the repository over time is expected to lead to more neutral pH.
In studies investigating microbial degradation of Ni– or U–citrate complexes, aerobic, citrate-degrading Pseudomonas cultures have been explored e.g., P. fluorescens and P. alcaliphila.52,62 In Ni–citrate degradation experiments with P. fluorescens and P. alcaliphila, partial degradation of bidentate [Ni(Cit)]− was observed52,60,75 and P. aeruginosa and P. putida, were shown to fully degrade citrate in the presence of the bidentate [Ni(Cit)]− complex.62 In an experimental system with P. aeruginosa, a small amount of Ni was removed from solution as Ni3(PO4)2·8H2O.62
Under anaerobic conditions, there is a paucity of information on the biodegradation of Ni–citrate complexes, especially at alkaline pH. Qian et al., found that a sulfate-reducing culture was able to degrade Ni–citrate at pH 6.8 to form nickel sulfide phases.76 In addition, other disposal relevant organic Ni complexes, such as with isosaccharinic acid (ISA), a key degradation product of alkaline cellulose hydrolysis in ILW, have been shown to be degraded anaerobically at neutral pH and again, insoluble nickel sulfide phases were formed.51
Biodegradation of U(VI)–citrate complexes has been studied in pure culture using isolates of Acinetobacter calcoaceticus, Citrobacter freundii, Pseudomonas stutzeri, P. aeruginosa, P. fluorescens and P. putida under aerobic and denitrifying conditions at neutral pH and here, the U(VI)–citrate complex was not degraded.20,53,58 Under anaerobic conditions, U(VI)–citrate was reduced to soluble U(IV)–citrate which implied that citrate complexed to U(VI) was not metabolised and overall reducing conditions in the presence of citrate did not favour precipitation of U(IV) minerals.53 U(VI)–citrate and Ni–citrate systems are poorly explored under alkaline, anaerobic conditions, or using a mixed microbial culture despite their relevance to radioactive waste disposal. Here, citrate has the potential to behave as either a complexant to enhance metal solubility or an electron donor in support of nitrate, sulfate or U(VI) bioreduction, which may promote immobilisation via the formation of poorly soluble U(IV) species (via bioreduction) or by metal biomineralisation with metabolites (e.g. with sulfides from sulfate reduction). The aim of this study was to explore the anaerobic speciation and biogeochemical fate of Ni/U/citrate complexes at alkaline pH using mixed bacterial cultures enriched from alkaline impacted sediments. Throughout, we used a multi technique approach to understand the fate of citrate, nickel and uranium in these systems in order to further inform understanding of their biogeochemical fate in low level wastes. More broadly, this work can be used to inform bioremediation strategies for sites where radioactive waste is present and/or has led to near-surface contamination plumes.77–80
Methodology
Anaerobic enrichment experiments, under nitrate- and sulfate-reducing conditions were set up to explore biogeochemical fate of Ni/citrate or U/citrate at pH 9–10. During experiments, changes in geochemistry, mineralogy and microbiology were monitored. Geochemical modelling using PHREEQC,81 with the ThermoChimie database (version 11a64), was used to support data interpretation (all modelling data are available in the ESI†). All aqueous reagents were filter sterilised prior to use. Concentrations of citrate and metals were selected to be representative of levels expected in a heterogeneous LLW repository.3Sediment inoculum for primary enrichment cultures
Sediment was collected from a high pH site in Harpur Hill, Derbyshire, UK. The site is a well characterized legacy lime works; sediments have elevated pH and are Ca-rich and the geomicrobiology is considered representative of a high pH, cementitious repository.73,82Primary enrichment cultures
Citrate oxidising cultures were enriched from the Harpur Hill sediment under nitrate- and sulfate-reducing conditions at pH 9. The primary enrichments contained 5 g of sediment, 100 mL minimal medium (9.4 mM NH4Cl, 4.3 mM K2HPO4, 4 mM NaHCO3, 10 mL L−1 vitamin and mineral mix83), 5 mM of trisodium citrate (Na3C6H5O7) as the sole carbon source and electron donor, and either NaNO3 (30 mM) or Na2SO4 (15 mM) as the electron acceptor. The pH was adjusted to pH 9 using NaOH and inoculations were performed anaerobically. Under both nitrate- and sulfate-reducing conditions, stable enrichment cultures were obtained by transferring a 1% v/v inoculum from primary enrichments into fresh, heat-sterilized medium. Typically, at least 5 subcultures were performed prior to running Ni/U experiments. The nitrate-reducing cultures were transferred into minimal medium with 5 mM citrate and 30 mM nitrate.83 The sulfate-reducing cultures were transferred into modified Postgate C medium with 5 mM citrate (no other organic electron donors included) and 30 mM sulfate.84Nickel experiments
Nitrate-reducing experiments
Three batches of minimal medium83 were prepared. To each, 1 mM trisodium citrate (Na3C6H5O7) and 10 mM NaNO3 were added. A stock solution (1 M Ni2+ in pH 3 HCl) was then added to target final Ni concentrations of 0.01, 0.1 or 1 mM and then pH was re-adjusted to pH 9–10 using NaOH. Experiments were then set up in 100 mL serum bottles under N2 using aseptic technique, and in triplicate. After equilibration for 24 hours, bottles were sampled to monitor the initial solution chemistry prior to microbial inoculation. The bottles were then inoculated using 1% v/v of the nitrate-reducing culture obtained from the primary enrichments, stored at 20 °C in the dark and periodically sampled under anaerobic, aseptic conditions. Parallel, sterile controls without microbial inoculum were also set up.Sulfate-reducing experiments
The sulfate-reducing experiments were set up in the same way as the nitrate-reducing systems, with two variations: (1) 10 mM sulfate was added instead of nitrate, (2) the sulfate-reducing enrichment culture (1% v/v) was used to inoculate the experiments.Uranium experiments
Uranium experiments were performed using washed resting cell cultures (late log phase) to prevent precipitation of uranium with components of bacterial growth medium (e.g., phosphate).Preparation of U and citrate media
TRIS buffer (20 mM TRIS; pH 9–10) containing U and citrate was prepared for nitrate and sulfate reducing experiments. Three concentrations of U were added to experiment bottles in small volumes (<300 μL) using pH 3 UO22+ stock in HCl to target concentrations of 0.005, 0.05, or 0.5 mM, U(VI). After spiking, pH was adjusted to pH 9–10 for the experiments. These are referred to as ‘low’, ‘middle’ or ‘high’ U experiments throughout. Throughout, solutions were handled to minimise carbonate ingress.Nitrate-reducing experiments
For the nitrate-reducing systems, 1 mM trisodium citrate (Na3C6H5O7), 10 mM NaNO3 and U(VI) spike were added to give a U(VI) concentration of 0.005, 0.05, or 0.5 mM. Experiments were run in 50 mL glass serum bottles in triplicate. A nitrate-reducing culture (inoculated using the same enrichment culture used in the Ni experiments) was used to grow cells to late exponential phase. These cells were collected by centrifugation, washed three times in anaerobic TRIS buffer (20 mM, pH 9–10) and this resting cell culture was added to each 50 mL bottle to give each a final OD600 = 0.2. Parallel sterile controls (with heat-sterilised cells, as sorption controls) were also set up.Sulfate-reducing experiments
Sulfate-reducing experiments were set up in the same way as the U(VI)-supplemented nitrate-reducing experiments, with two exceptions: (1) 10 mM Na2SO4 was added instead of NaNO3; (2) a washed sulfate-reducing cell culture grown in Postgate C medium,84 was added to give final OD600 = 0.2 in each bottle.Solution chemistry
The pH was measured using a Mettler Toledo SevenCompact digital meter fitted with a calibrated Fisherbrand FB68801 electrode. Concentrations of anionic species (nitrate, sulfate, citrate, and volatile fatty acids) were measured by capillary anion exchange chromatography (IC) using a Dionex ICS5000. The lower limit of detection for citrate was 0.002 mM. Total aqueous Ni and U concentrations were measured on centrifuged and acidified samples (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800g, 10 min; 2% HNO3) using inductively coupled plasma mass-spectrometry (ICP-MS; Agilent 7500cx).
800g, 10 min; 2% HNO3) using inductively coupled plasma mass-spectrometry (ICP-MS; Agilent 7500cx).
Electrospray ionisation mass spectrometry was used to examine Ni and citrate speciation at the start and end of select experiments (Thermo Q-exactive, Orbitrap mass analyser, attached to a Thermo Ultimate 3000 high performance liquid chromatography (HPLC)). Electrospray ionization was performed in both positive and negative polarity modes. The HPLC used an isocratic flow of 100% methanol for the mobile phase.
Solid phase analyses
800g, 10 minutes) solids underwent a fixing, dehydration, and dilution protocol, using glutaraldehyde and ethanol, to preserve biological structures. Preserved samples for SEM were drop-cast onto an aluminium pin stub (Zeiss, Ø12.7 mm top), dried anaerobically and gold coated before imaging on a FEI Quanta 650 FEG instrument operating at 15 kV in high vacuum mode (105 to 10−6 mbar). Energy dispersive X-ray analysis (EDS) was performed using the EDAX Gemini EDS system. Samples from the Ni experiments were also analysed by TEM (JEOL 2100+ TEM). Samples were drop cast onto a gold grid with a holey carbon film and dried anaerobically. TEM images were captured using a Gatan RIO digital camera. EDS was obtained using an Oxford Instruments Xmax 65T Detector (65 mm window size); data collection and analysis was performed using Oxford Aztec Energy software.
Spectroscopic analysis
Solid samples were analysed by X-ray absorption spectroscopy (XAS) on beamline B18 at the Diamond Light Source, Harwell, UK from the low (0.005 mM; XANES and EXAFS) and middle (0.05 mM; XANES only) U experiments. Samples were centrifuged and the resultant solids dried in an oxygen free glove box and diluted by mixing with cellulose to target a final U concentration of 1% w/w. Sample pellets were pressed under N2 and mounted on a sample holder and stored under a N2 atmosphere at −80 °C until analysis. XAS data were obtained at the Diamond Light Source on B18 using the U LIII edge in transmission or fluorescence mode with a Ge detector. The Athena and Artemis (with FEFF6) software was used for fitting and data reduction.Microbial community analysis
Post-sequencing analysis
Raw sequences were divided into samples by barcodes (up to one mismatch was permitted) using a sequencing pipeline. Quality control and trimming was performed using Cutadapt4, FastQC5, and Sickle6. MiSeq error correction was performed using SPADes7. Forward and reverse reads were incorporated into full-length sequences with Pandaseq8, and chimeras were removed using ChimeraSlayer9. OTU's were generated for the 16S rRNA gene sequences with UPARSE10. OTUs were classified by Usearch11 at the 97% similarity level, and singletons were removed. Rarefaction analysis was conducted using the original detected OTUs in Qiime12. The taxonomic assignment was performed by the RDP naïve Bayesian classifier version 2.2 13, used in combination with the Silva SSU 132 ribosomal RNA gene database 14. The OTU tables were rarefied to the sample containing the lowest number of sequences, all samples having less than 5000 sequences were removed from analyses prior to the rarefaction step. The step size used was 2000 and 10 iterations were performed at each step.Results and discussion
Anaerobic enrichment experiments were used to investigate the biogeochemical fate of Ni, U and citrate at alkaline pH. Three sets of experiments, using 0.01, 0.1 or 1 mM Ni2+ or 0.005, 0.05, or 0.5 mM U(VI)O22+, were performed under nitrate- and sulfate-reducing conditions.Nitrate-reducing experiments
Geochemical modelling (Fig. S5†) of the 0.01 mM Ni/1 mM citrate system at t = 0 predicted that 91% of the Ni was present as [Ni(Cit)]− and 9% as the [Ni(Cit)2]4− species. The ESI-MS analysis for the t = 0 aqueous sample (Fig. S7†), showed a peak at 246.9384 m/z consistent with the [Ni(Cit)]− species (246.9389 g mol −1). This peak was absent in the 162 day sample at the experimental end point indicating that, consistent with the bulk citrate measurements, [Ni(Cit)]− had been degraded. Interestingly, there was no evidence for the [Ni(Cit)2]4− species (expected at 439.9737 g mol−1) in the 0 day time point in the ESI-MS, which was predicted at low levels in the thermodynamic modelling and has been suggested as a significant species at pH 9.60,89
Despite the removal of [Ni(Cit)]− (in the low Ni systems) during incubation, and as evidenced in the ESI data, Ni remained soluble throughout incubation (Fig. 1D). Here, geochemical modelling predicted that in the end-point experiment, where HCO3− had presumably grown in from citrate degradation (4.8 mM; eqn (S1)†,16), Ni2+ would be a significant aqueous species, and that NiCO3 and Ni(OH)2 would be oversaturated (Fig. S6†). Interestingly, there was no removal of Ni in the experimental endpoints or the controls, suggesting that the Ni2+ (aq) species dominated in the nitrate-reducing incubation end points.
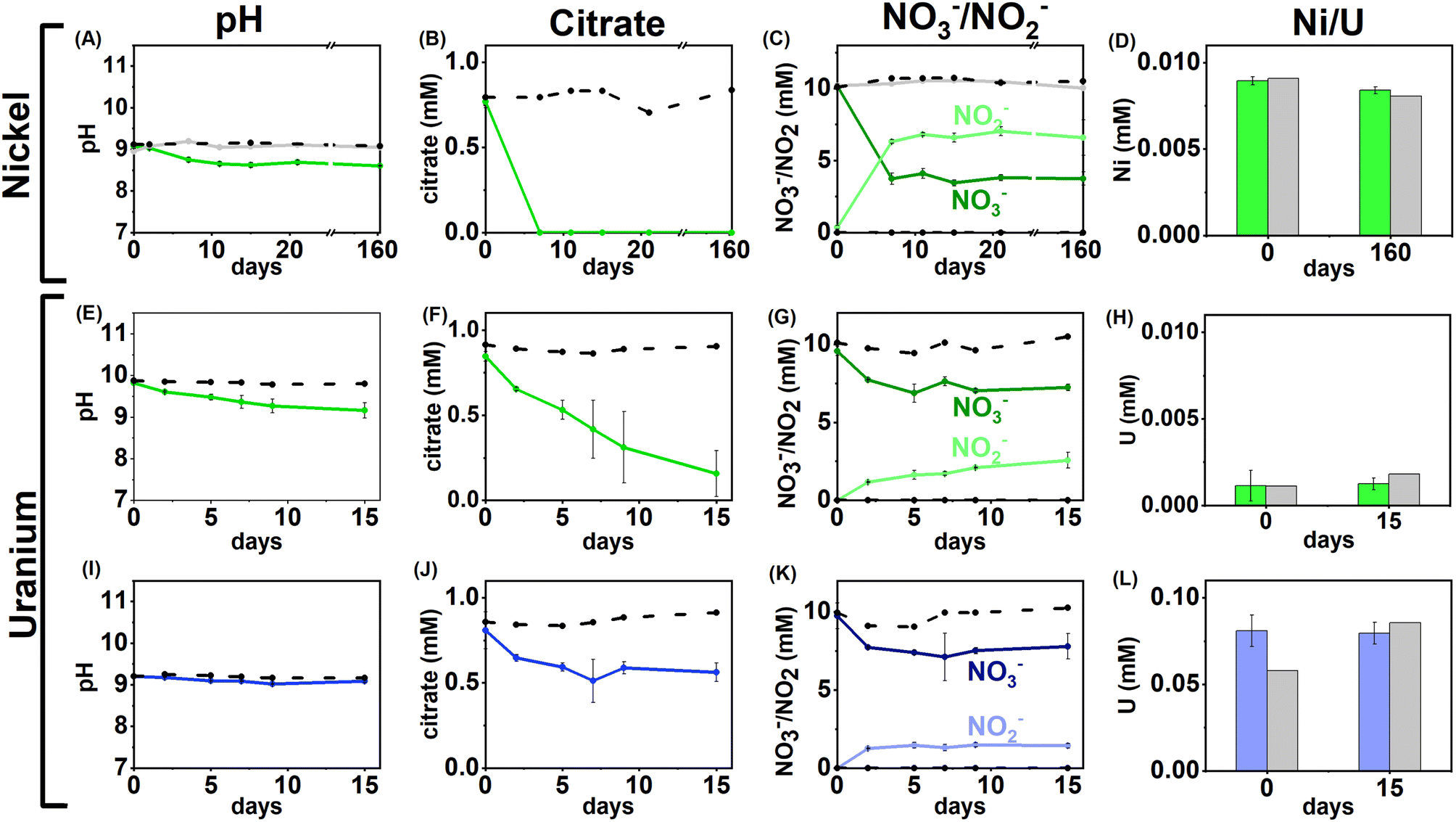 | ||
| Fig. 1 Aqueous geochemistry data for the Ni and U nitrate-reducing experiments. Top – low (0.01 mM) Ni, nitrate-reducing experiment, Bottom – low and middle U, nitrate-reducing experiments. Columns from left to right are: pH (A, E, I), citrate concentration (B, F, J), nitrate/nitrite concentration (C, G, K) and nickel/uranium concentration (D, H, L). Green lines – low Ni/U systems, blue lines – middle U experiments, dashed black lines – sterile control, grey line (Ni only) – no citrate control. Error bars represent 1σ of the triplicate samples. | ||
Overall, the data suggest that in nitrate-reducing systems with low Ni concentrations, Ni is soluble initially as Ni–citrate and as degradation of the bioavailable Ni–citrate complex occurs, it is likely present as the Ni2+ aqueous species. In the middle and high Ni inoculations there were no changes in pH, Ni, citrate and nitrate concentrations despite the presence of citrate (Fig. 1A–D, S1 & S5, Table S1†). This suggests the elevated Ni concentrations were toxic.90–92
U/nitrate reducing systems
Under nitrate-reducing conditions, the citrate concentrations fell in the low and middle U systems with 100% citrate removal (0.7 ± 0.1 mM) in the low U system, and 30% citrate removal (0.2 ± 0.1 mM) in the middle U system by day 15 (Fig. 1F and J). At the same time, reduction of nitrate to nitrite was seen in both systems (Fig. 1G and K). In the inoculated low U system, the solution pH decreased from pH 9.8 to 9.1 (Fig. 1E), whilst no significant pH change was measured in the middle U system (Fig. 1I). As observed in the Ni experiments, no VFAs were detected in either system, suggesting citrate was fully oxidised to CO2 in inoculated systems consistent with solution acidification associated with CO2 dissolution into solution.Interestingly, uranium concentrations remained constant throughout the nitrate-reducing experiment confirming citrate-driven nitrate reduction did not affect U(VI) solubility (Fig. 1H and L). This is consistent with the geochemical modelling of the starting solution which predicted a small fraction (0.03–0.1%) of the added U would form citrate complexes, and that, in the absence of carbonate, aqueous U(VI) was present as hydroxide species (including [UO2(OH)3]−, and [(UO2)3(OH)7]−) with the vast majority of citrate present as the free citrate [Cit]3− species (Fig. S5 and Table S1†). Modelling of the U solid phases at the initial time point predicted that in the absence of carbonate, there was oversaturation of mineral phases including clarkeite, metaschoepite and Na-compreignacite, in contrast to the aqueous data obtained (Fig. 4C, F and I). The increased solubility of U in some experiments compared to the thermodynamic modelling at the initial timepoint may be explained by either the dynamic nature of the experiments or in the absence of extensive U–citrate complexation, may be due to complexation by residual carbonate in solution. Geochemical modelling data for the end-point solutions in these nitrate-reducing systems, after citrate degradation to HCO3−, suggested that U(VI) would be present as aqueous U(VI)–carbonate species and that likely solid U phases (e.g., metaschoepite, clarkeite, rutherfordine and UO2(OH)2), were undersaturated (Fig. S6†).
Geochemical modelling data for the end-point solutions in these nitrate-reducing systems, after citrate degradation to HCO3−, suggested that U(VI) would be present as aqueous U(VI)–carbonate species and that likely solid U phases (e.g., metaschoepite, clarkeite, rutherfordine and UO2(OH)2), were undersaturated (Fig. S6†). Again, elevated U levels in the middle and high U experiments (0.05 mM, 0.5 mM) likely inhibited bacterial activity in the inoculum.93,94 Indeed, in the high U incubations no changes in pH, or citrate and nitrate/nitrite concentration were observed, and, in middle U incubations only modest changes (e.g., 30% citrate removal and 11% nitrate reduction) were observed when compared to the low U system (Fig. 1I–L).
Overall, the data from these nitrate-reducing experiments showed rapid citrate biodegradation can occur at elevated pH under nitrate-reducing conditions when Ni and U are present at trace levels, however citrate biodegradation may be hindered by metal toxicity where elevated concentrations of these metals are present.
Sulfate-reducing experiments
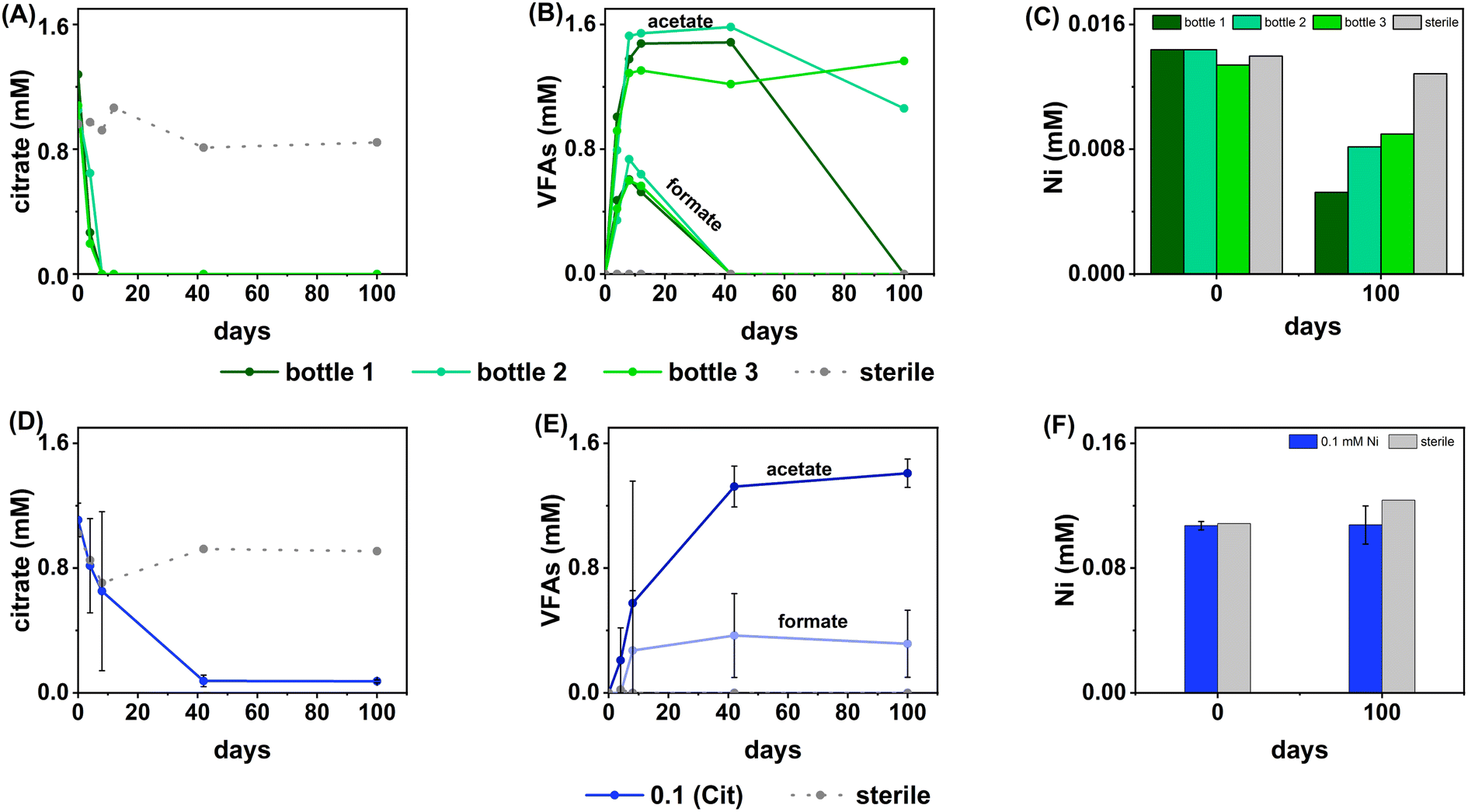 | ||
| Fig. 2 Data showing the aqueous geochemistry for the Ni, sulfate-reducing experiments. Top (green) – low (0.01 mM) Ni, Bottom (blue) – middle Ni (0.1 mM). Columns from left to right are: citrate concentration (A, D), VFA concentration (B, E), nickel concentration (C, F). Green lines – inoculated individual bottles from low Ni systems (individual bottles are plotted as there was high variability between bottles in the triplicate set), blue lines – inoculated average measurement from middle Ni experiments, dashed grey lines – sterile control. Error bars represent 1σ of the triplicate samples. | ||
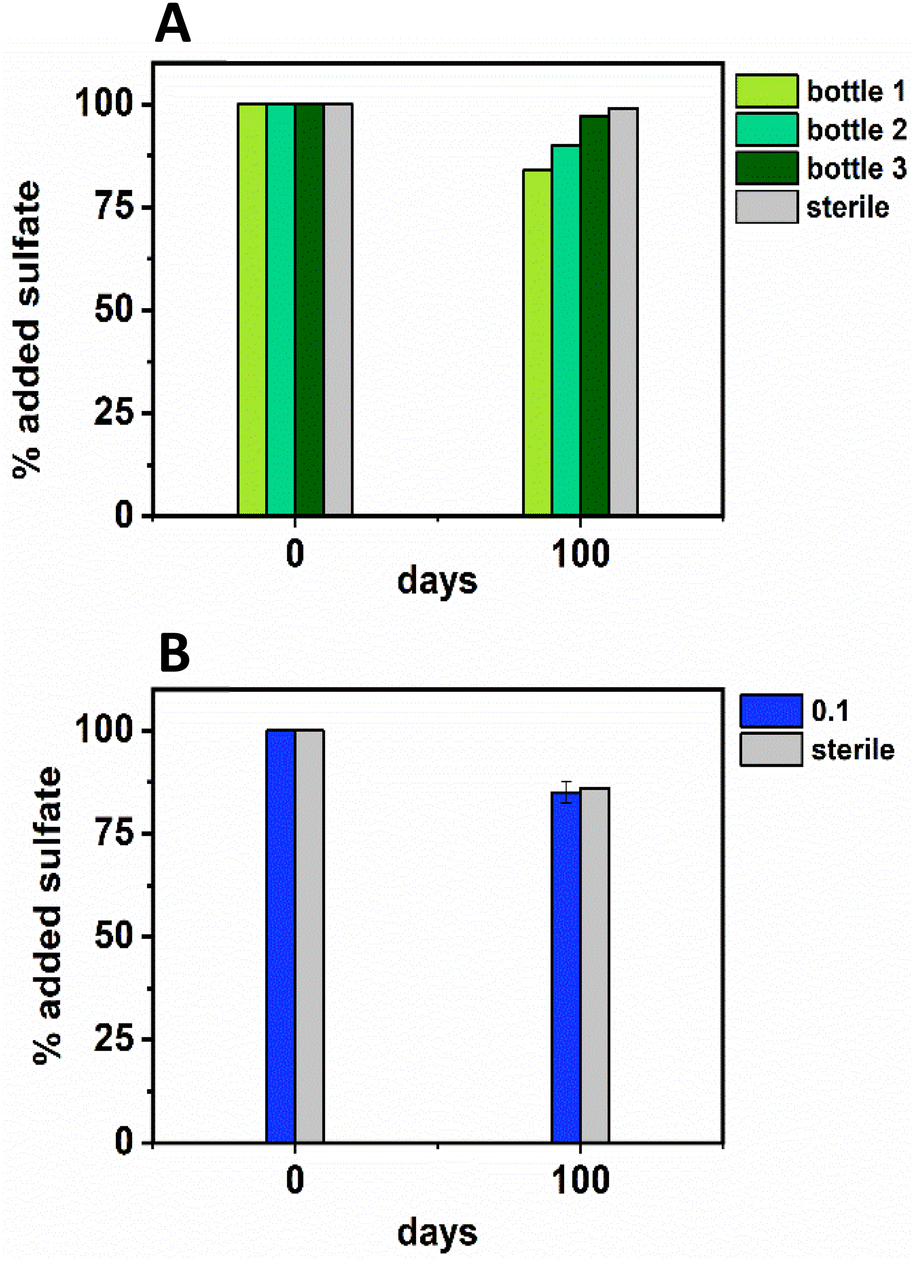 | ||
| Fig. 3 Sulfate removal data from the low Ni (0.01 mM; top, A, in green) and middle Ni (0.1 mM; bottom, B, in blue) showing the percentage removal by the experimental end point at day 100. For the low Ni experiment (green) each bottle of the triplicate is plotted (due to high variability in between bottles in the triplicate experiment), and, for the middle Ni experiment the average ± 1σ is plotted. | ||
In terms of Ni concentrations in solution, the average Ni removal for all bottles was 7.5 ± 2.0 μM (approximately 50% removal: Fig. 2C). In individual incubations the extent of Ni removal was greatest in bottles where sulfate removal was greatest with bottle 1 (0.009 mM) > bottle 2 (0.006 mM) > bottle 3 (0.004 mM; Fig. 3A). In terms of Ni solution speciation, ESI-MS confirmed that the Ni was complexed as a [Ni(Cit)]− species in the day 0 samples and that by day 100 this species was no longer present (consistent with citrate degradation). In this experiment the dominant Ni species was predicted to be Ni(CO3) (Fig. S6†), however no Ni–carbonate species were detected by ESI-MS analysis so presumably the dominant Ni(aq) species was Ni2+ (Fig. S6 and S8†).
At day 100 the removal of 7.5 ± 2 μM Ni from solution was accompanied by the formation of a black precipitate by day 42 consistent with formation of a Ni-sulfide phase.51,95,96 SEM images from the sample at 100 days showed amorphous material, presumably biomass, and EDS analysis of these regions revealed the co-location of Ni and S (Fig. S10†). High resolution TEM imaging with corresponding EDS (Fig. S11†) also confirmed the co-location of Ni and S, and selected area diffraction analysis suggested that the precipitate was amorphous. Overall these data suggest an amorphous NiS-like precipitate was forming.
In the inoculated middle Ni, sulfate-reducing systems, citrate degradation occurred but at a slower rate than the low Ni-sulfate experiment. Significant but not complete citrate removal was observed in these systems and by day 100, 0.07 ± 0.01 mM citrate remained in solution. Citrate again was fermented to acetate and formate, which peaked at 1.4 ± 0.09 mM and 0.4 ± 0.2 mM, at day 100 and 40 respectively (Fig. 2D and E). Interestingly, there was no evidence, from blackening or sulfate removal, for the development of sulfate reduction in the middle Ni concentration experiment (Fig. 3B). Here, after their increase with time, acetate and formate did not degrade further suggesting progression to sulfate reduction was inhibited. Furthermore, ESI-MS confirmed that, contrary to the low Ni experiment, the [Ni(Cit)]− species remained dominant in solution over 100 days (Fig. S9†) consistent incomplete citrate degradation (Fig. 2D) and the full retention of complexed Ni in solution (Fig. 3F). There were no observed biogeochemical changes in the high Ni systems, presumably due to Ni toxicity (Fig. S2† (ref. 90–92)).
Overall, for the low Ni experiments, the geochemical and microscopy data indicated that Ni–citrate complexes were degraded at pH 9. The data suggested a two-step mechanism where citrate was fermented to acetate and formate, which were then used to support sulfate reduction to variable degrees in the different bottles. Following sulfate reduction in the low Ni system, ESI-MS indicated Ni–citrate complexes were no longer present with aqueous phase analysis showing modest Ni2+ removal (50%) and solid phase characterisation suggesting amorphous Ni–sulfides had formed. By contrast, although significant fermentation of citrate to formate and acetate was observed in the middle Ni system, the elevated Ni concentration apparently inhibited sulfate reduction. Interestingly, this suggested in the 0.1 mM Ni experiment, that citrate fermentation was possible, but anaerobic sulfate reduction was inhibited. Here, ESI-MS analysis of the middle Ni solution phase at 100 days confirmed that the [Ni(Cit)]− complex was still present and ICP-MS showed all Ni remained soluble. Geochemical modelling of the low Ni system with 1 mM sulfide (as observed) and 0.01 mM citrate (enough to complex all added Ni), predicted oversaturation of insoluble Ni–sulfide phases would occur consistent with partial removal to nickel sulphides as observed in TEM (Table S2†).
It is interesting that fermentation occurs in sulfate-reducing systems but not in the nitrate reducing systems. At low Ni levels (0.01 mM), nitrate reducing bacteria may have outcompeted fermentative bacteria. Indeed, past work has demonstrated that nitrate reducers can outcompete fermentative bacteria, particularly where C:N ratios are low as they are in the current study.97–100 At higher Ni levels metal toxicity presumably inhibited microbial activity.
U/sulfate-reducing conditions
The inoculated low and middle U, sulfate-reducing experiments showed rapid and complete oxidation of citrate in the first 6 days of the experiment. After this, substantial acetate ingrowth was observed; 1.6 ± 0.06 mM by day 43 (low U, Fig. 4A), and 1.7 ± 0.05 mM by day 15 (middle U, Fig. 4D). These levels were then maintained up to day 55. Additionally, by day 15 formate ingrowth of 0.12 ± 0.01 mM (low U) and 0.10 ± 0.01 mM (middle U) occurred, and formate was then below the level of detection by day 55 in the middle U system. Partial and variable citrate removal (0.06–0.13 mM) was observed at 55 days in the high U triplicate experiment. After day 15 this was accompanied by production of 0.1 ± 0.01 mM acetate (no formate was detected in these samples), and these levels were maintained up to day 55 (Fig. 4G).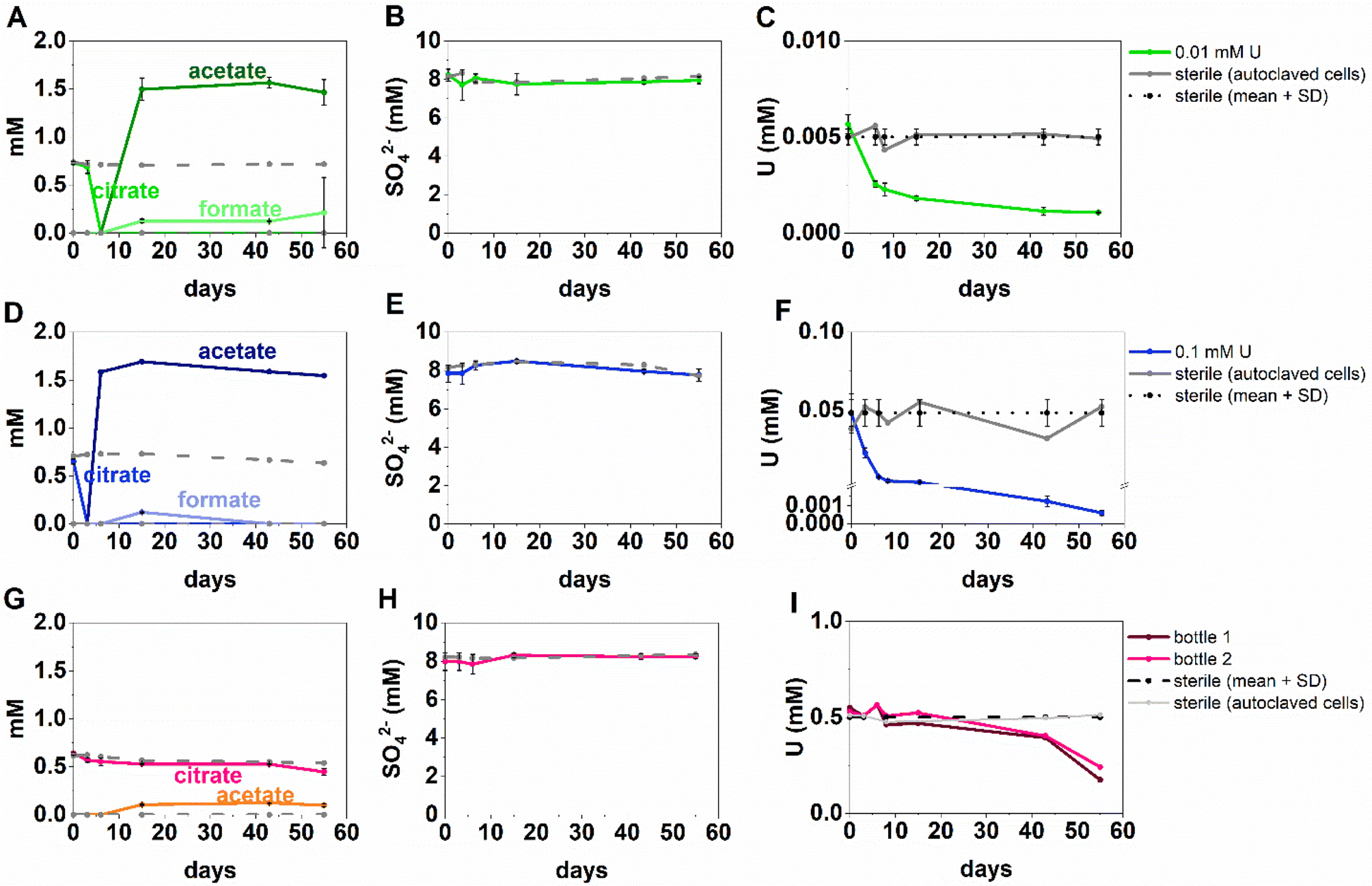 | ||
| Fig. 4 Aqueous organic acid (A, D, G), sulfate (B, E, H) and U concentration (C, F, I), data from the sulfate-reducing experiment with added U concentration of 0.005 mM U (top row, green), 0.05 mM U (middle row, blue) and 1 mM U (bottom row, pink). For the 0.5 mM U experiment only 2 out of 3 bottles in the triplicate are shown as the third bottle did not show geochemical changes. Presumably culture heterogeneity/toxicity in these “extreme” (i.e., alkaline pH) systems is impacting on the different experiment bottles. | ||
In the low (0.005 mM) and middle (0.05 mM) U incubations, dark precipitates had formed by day 12. Here, aqueous phase analysis of the low and middle U systems showed near complete removal of U from solution by day 15 (Fig. 4C and F). In the high U system, dark precipitates had formed by day 43 and by day 55, there was between 40–69% U removal (Fig. 4I). Interestingly, there was no measurable sulfate removal in these incubations over the 55 day experiment (Fig. 4B, E and H) and this suggests direct enzymatic U(VI) reduction took place, rather than a mechanism coupled to sulfate-reduction. The uranium precipitates were further analysed in the low and middle U systems by X-ray absorption spectroscopy.
Analysis of the U LIII edge XANES spectra (Fig. 5) using U(VI)–uranyl and U(IV)–uraninite standards, suggested that significant reduction from U(VI) to U(IV) had occurred in both samples (Fig. 5). EXAFS data were also available for the solids from the low concentration U experiment. Here, the relevant published literature for biogenic non-crystalline U(IV) and nanoparticulate uraninite was used to inform the best fit model for the EXAFS spectra101–104 (Fig. 5, Table S3 and S4†). Fitting initially considered biogenic uraninite and this provided a poor fit (R-factor > 0.02; Table S4†). An improved fit was achieved with a split oxygen shell of 4.5 O backscatterers at 2.31 Å and 3.5 O backscatterers at 2.49 Å, 1.5 P backscatterers at 3.14 Å, 1 P backscatter at 3.64, and 2.5 U backscatterers at 3.84 Å which was statistically significant (Table S3†). This fit suggests contributions from biogenic non-crystalline U(IV) coordinated by phosphate groups presumably present in the cell biomass in the experiment as observed in past work.101–103,105–109
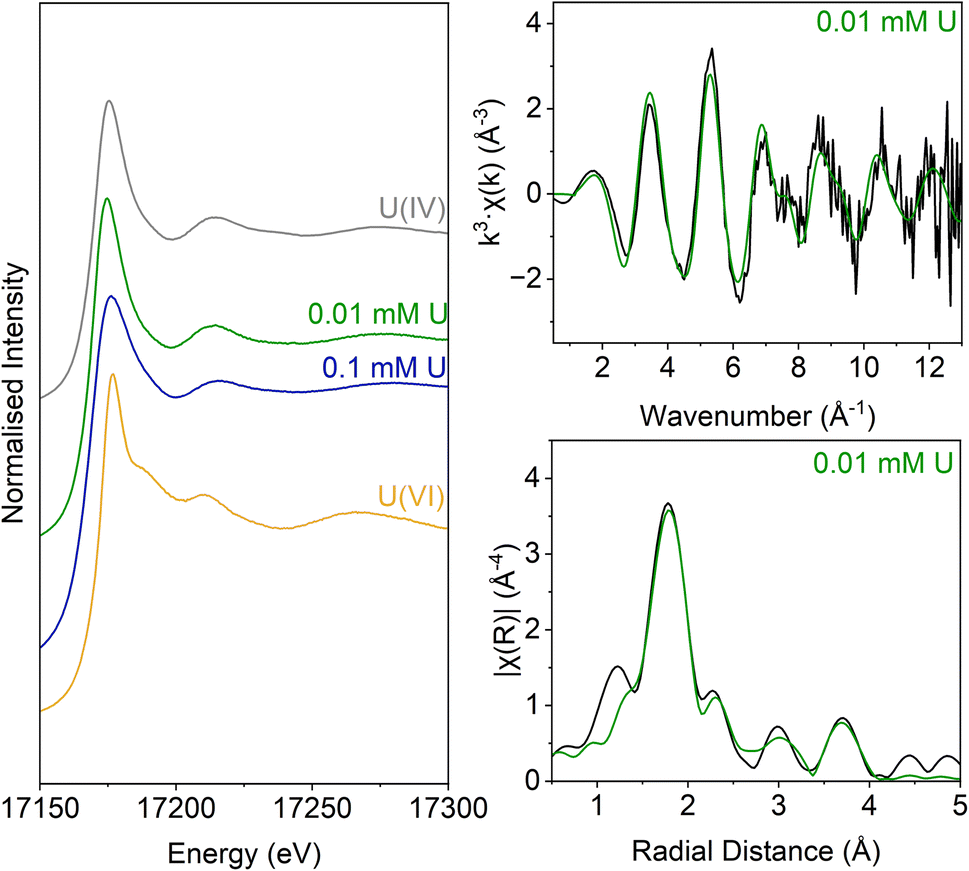 | ||
| Fig. 5 U LIII-edge spectra for solids from the low (0.005 mM) U and middle (0.05 mM) U samples. These samples are compared with U(VI) (aqueous U(VI)–triscarbonate) and U(IV) (uraninite) standards taken from previous work (Roberts et al., 2017; Townsend et al., 2020). Right: U LIII-edge XAS spectra for NB1. Top – k3-weighted EXAFS. Bottom – Fourier transform of k3-weighted EXAFS, using a Hanning window function. | ||
Nitrate-reducing experiments
At the phylogenetic class level, the initial nitrate-reducing enrichment culture was dominated by Gammaproteobacteria (87%), Alphaproteobacteria (7%) and Bacilli (6%). In the low Ni experiment, after 160 days incubation the community had a relative increase in Alphaproteobacteria (26%) and Bacilli (31%), which grew at the expense of the Gammaproteobacteria (40%). At the genus level, in the nitrate-reducing enrichment culture Pseudomonas was dominant (54.4% of sequences), followed by Achromobacter (18.7%); both genera have citrate-oxidising and nitrate-reducing members. In the Ni experiment, the endpoint sample (T160) had substantial ingrowth from the genera Anaerobacillus (29.4%) and Bosea (‘Bosea 1’ on Fig. 6; 12.8%), and again, some members of both genera reduce nitrate and oxidise citrate (e.g.37,110,111). Interestingly, at the species level, a close match for the Anaerobacillus sequence was Anaerobacillus alkaliphilus (99.6% match). This organism is reported to encode various proteins that may contribute towards Ni resistance/tolerance e.g. several metal ABC transporter proteins (e.g. NCBI Reference Sequence WP_129076357;112). In the U experiments a resting cell culture was used and this was grown using the same enrichment culture inoculum used to inoculate the Ni experiments. In turn, the community structure in the low U, day 15 samples reflected the initial enrichment culture inoculum: Gammaproteobacteria (61%), Alphaproteobacteria (34%) and Bacilli (5%). Pseudomonas was also the dominant genus in the endpoint sample from the U-experiment (60%).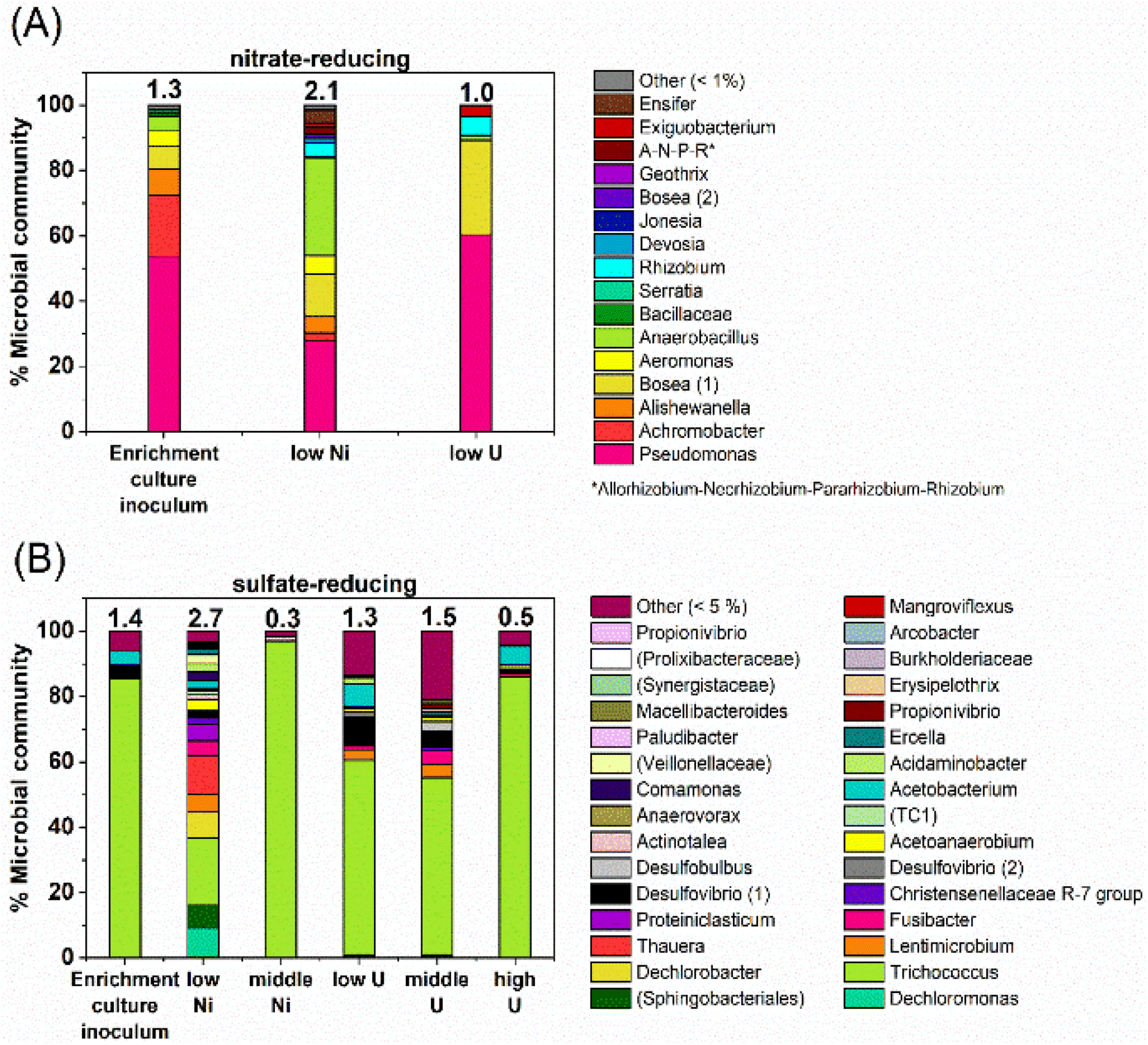 | ||
| Fig. 6 Data from 16S rRNA gene sequencing showing the microbial community, at genus-level, in end point samples from the (A) nitrate-reducing (B) sulfate-reducing experiments, in comparison to the initial enrichment culture inoculum. Where genus-level data could not be resolved, this is indicated by brackets e.g., ‘(Prolixibacteraceae)’. Numbers in brackets e.g., ‘Bosea (1)’ indicate significantly different gene sequences, presumably for different species. | ||
Sulfate-reducing experiment
At the phylogenetic class level the sulfate-reducing enrichment culture comprised mostly Bacilli (73%), Gammaproteobacteria (14%) and Clostridia (9%). The samples from the U-experiments, contained resting cells which were grown from the same sulfate-reducing enrichment culture that was used in the Ni experiments. In the endpoint samples from the U-experiments, cultures were similar to the starting inoculum; dominated by Bacilli (67–90%) and Clostridia (14%). In the 0.01 mM Ni supplemented incubations, communities changed over the duration of incubation and there was a substantial relative increase in Bacteroidia (18%), Gammaproteobacteria (33%) and Clostridia (24%). In the 0.1 mM Ni incubations, Bacilli were the dominant class in all samples (96%). Interestingly, in the 0.01 mM Ni experiments the genus level data showed increased diversity in the endpoint samples, although reasons for this remain unclear. In all other samples from sulfate-reducing experiments, diversity was constant or decreased following incubation with Ni or U (Fig. 6).In the Ni experiment, the three most dominant genera were Dechloromonas (22%), Trichococcus (19%) and an unresolved genus of the order Sphingobacteriales (10%). Sulfate-reducing bacteria were identified in the low Ni experiment and in all U experiments. These were primarily affiliated with the genus Desulfovibrio which accounted for approximately 1–11% of the communities. At the species level, the closest match to the Desulfovibrio (1) sequence (Fig. 6) was Desulfovibrio mexicanus (100% match), which is known to utilise acetate and formate as electron donors.113 In addition, organisms affiliated with the sulfate-reducing genus Desulfobulbus were also identified in the 0.05 mM supplemented U experiment (3% of the community).
In the 0.1 mM Ni systems the samples were overwhelmingly dominated by the Trichococcus genus (97% of the community), and this organism was also dominant in all of the U systems (0.005 mM U – 60%, 0.05 mM U – 55%, and 0.5 mM U – 85% of the communities). This organism was also present to a lesser extent in the 0.01 mM Ni experiment (37%). Members of the genus Trichococcus are typically fermentative, and thus, the dominance of this genus supports the suggestion that citrate was initially fermented in all sulfate-reducing systems.114
Conclusions
Overall experiments show that robust citrate biodegradation took place in the presence of low levels of Ni and U, under conditions relevant to alkaline radioactive waste repository environments. In the Ni–citrate systems, anaerobic biodegradation of [Ni(Cit)]− at alkaline pH (pH 9) occurred under nitrate- and sulfate-reducing conditions when 0.01 mM Ni was present (Fig. 1 and 2). At higher metal loadings, growth of nitrate- and sulfate-reducing bacteria may be inhibited. This may be beneficial where negative impacts of microbial metabolism are observed for example, sulfate-reduction and its impact on corrosion.115–118 In the sulfate reducing/0.01 mM Ni experiments, fermentation of citrate led to ingrowth of formate and acetate which then fuelled significant sulfate reduction (Fig. 2 and 3). Here, 50% of the added Ni (7.5 ± 2 μM) was immobilised under sulfate-reducing conditions as amorphous Ni–sulfide-like phases. Elevated Ni levels of 0.1- and 1 mM Ni were presumed to be toxic to both the nitrate- and sulfate-reducing cultures, except for members of the strictly fermentative genus Trichococcus in the 0.1 mM sulfate-reducing experiments (Fig. 2 and 6). Interestingly, ESI-MS showed the [Ni(Cit)]− complex did not degrade in the 0.1 mM Ni/sulfate-reducing experiments, even though significant, but not complete citrate removal was observed by IC (Fig. S8–S9,† 2). This suggests that sulfate-reducing bacteria were inhibited by toxicity from 0.1 mM Ni or the [Ni(Cit)]− complex. In turn, this impacted the end-state of this microcosm as NiS-like phases did not form and Ni remained largely soluble as the Ni–citrate complex.Citrate biodegradation occurred in the presence of U under nitrate- and sulfate-reducing conditions, at alkaline pH (Fig. 1 and 3). In the nitrate-reducing system 0.5 mM U was toxic to the microbes over the experimental timescale. However, in the sulfate-reducing systems citrate removal was observed in all experiments, even when 0.5 mM U was present (Fig. 3). Importantly, in all sulfate-reducing systems citrate fermentation products (acetate & formate) likely supported enzymatic U(VI) reduction which led to reductive precipitation of a nanoparticulate U(IV)–phosphate phase likely formed through reaction with phosphate associated with the biomass in the experiment (Fig. 5). This study has demonstrated that at pH 9–10 under sulfate-reducing conditions bacteria can ferment citrate, a widely used decontaminant in the nuclear industry. The citrate fermentation products can then support sulfidation in low Ni systems to partially precipitate nickel sulfides and bioreduce U(VI) to form poorly soluble non-crystalline U(IV)–phosphates. Removal of citrate by biodegradation in wastes at high pH will eliminate the potential for radionuclide-citrate complexation, and the implied elevated solubility of radionuclides, notably Ni2+, in radioactive waste disposal scenarios where citrate is present.
Author contributions
NB – conceptualization, methodology, investigation, data curation, formal analysis, visualisation, project administration, writing – original draft, writing – review & editing. JRL – conceptualization, methodology, writing – review & editing, resources, supervision, validation. LTT – XAS data curation and formal analysis, writing – review & editing. JSS – conceptualization, validation. FT – conceptualization, writing – review & editing, supervision, funding acquisition, validation. HB – TEM data acquisition and support. CB – 16S rRNA data acquisition and support. IS-mass spectrometry measurements and support. KM – conceptualization, methodology, writing – original draft, writing – review & editing, resources, supervision, validation, funding acquisition.Conflicts of interest
The authors declare that this study received funding from Low Level Waste Repository Ltd. The funder had the following involvement with the study: conceptualization, writing – review & editing, supervision, funding acquisition, validation. The funder was not involved in the collection, analysis and interpretation of data, or, the decision to submit it for publication. FT was employed by company Low Level Waste Repository Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
NERC and Low Level Waste Repository Ltd. co-funded the Ph.D. studentship to NB via the NERC Manchester and Liverpool Doctoral Training Partnership (NE/R009732/1). NB acknowledges access to the EPSRC NNUF RADER Facility (EP/T011300/1) for analyses performed in this work. We thank Don Reed (Los Alamos National Laboratory, US), we sincerely appreciate valuable comments on this document. Additional thanks to Lewis Hughes (Department of Earth and Environmental Sciences, University of Manchester) for support with SEM, and Gina Kuippers for advice. We would also like to thank the University of Liverpool for support with TEM data acquisition.Notes and references
- IAEA, Iaea Ssr-5, 2014, WS-R-1, 29 Search PubMed.
- U. R. Berner, Waste Manag., 1992, 12, 201–219 CrossRef CAS.
- LLW Repository Ltd., LLWR/ESC/R(11)10019 CAS.
- L. Duro, C. Domènech, M. Grivé, G. Roman-Ross, J. Bruno and K. Källström, Appl. Geochem., 2014, 49, 192–205 CrossRef.
- D. Read, D. Ross and R. J. Sims, J. Contam. Hydrol., 1998, 35, 235–248 CrossRef CAS.
- L. R. Van Loon, M. A. Glaus and K. Vercammen, Acta Chem. Scand., 1999, 53, 235–240 CrossRef CAS.
- A. J. Francis, C. J. Dodge and J. B. Gillow, Radiochim. Acta, 2006, 94, 731–737 CrossRef CAS.
- W. Hummel, Monatshefte fur Chemie, 2008, 139, 459–480 CrossRef CAS.
- M. J. Keith-Roach, Sci. Total Environ., 2008, 396, 1–11 CrossRef CAS PubMed.
- M. Sutton, Uranium solubility, speciation and complexation at high pH, University of Loughborough, 1999 Search PubMed.
- N. D. M. Evans, S. A. Gascón, S. Vines and M. Felipe-Sotelo, Mineral. Mag., 2012, 76, 3425–3434 CrossRef.
- M. Felipe-Sotelo, M. Edgar, T. Beattie, P. Warwick, N. D. M. Evans and D. Read, J. Hazard. Mater., 2015, 300, 553–560 CrossRef CAS.
- M. Felipe-Sotelo, J. Hinchliff, L. P. Field, A. E. Milodowski, J. D. Holt, S. E. Taylor and D. Read, J. Hazard. Mater., 2016, 314, 211–219 CrossRef CAS PubMed.
- R. Serne, A. Felmy and K. J. Cantrell, Characterization of Radionuclide-Chelating Agent Complexes Found in Low-Level Radioactive Decontamination Waste, Pacific Northwest National Laboratory, NUREG/CR-6124, PNL-8856, 1996 Search PubMed.
- IAEA, Management of low and intermediate level radioactive wastes with regard to their chemical toxicity, Vienna, 2002 Search PubMed.
- N. Byrd, J. R. Lloyd, J. Small, F. Taylor, H. Bagshaw, C. Boothman and K. Morris, Front. Microbiol., 2021, 12, 1–22 Search PubMed.
- H. A. Krebs, Biochem. J., 1940, 34, 460–463 CrossRef CAS.
- J. L. Pierre and I. Gautier-Luneau, BioMetals, 2000, 13, 91–96 CrossRef CAS PubMed.
- R. Ciriminna, F. Meneguzzo, R. Delisi and M. Pagliaro, Chem. Cent. J., 2017, 11, 1–9 CrossRef.
- A. J. Francis, C. J. Dodge and J. B. Gillow, Nature, 1992, 356, 57–59 CrossRef.
- S. C. Brooks, J. S. Herman, G. M. Hornberger and A. L. Mills, J. Contam. Hydrol., 1998, 32, 99–115 CrossRef CAS.
- A. Apelblat, Citric Acid, Springer International Publishing, Switzerland, 2014 Search PubMed.
- J. P. Glusker, Acc. Chem. Res., 1980, 13, 345–352 CrossRef CAS.
- U. Neubauer, G. Furrer, A. Kayser and R. Schulin, Int. J. Phytorem., 2000, 2, 353–368 CrossRef CAS.
- A. P. Schwab, D. S. Zhu and M. K. Banks, Chemosphere, 2008, 72, 986–994 CrossRef CAS PubMed.
- L. Duquène, F. Tack, E. Meers, J. Baeten, J. Wannijn and H. Vandenhove, Sci. Total Environ., 2008, 391, 26–33 CrossRef CAS.
- I. F. Poulsen and H. C. B. Hansen, Water. Air. Soil Pollut., 2000, 120, 249–259 CrossRef CAS.
- N. U. Yamaguchi, A. C. Scheinost and D. L. Sparks, Clays Clay Miner., 2002, 50, 784–790 CrossRef CAS.
- A. P. Schwab, Y. He and M. K. Banks, J. Environ. Eng., 2004, 130, 1180–1187 CrossRef CAS.
- X. Liang, Y. Zang, Y. Xu, X. Tan, W. Hou, L. Wang and Y. Sun, Colloids Surf., A, 2013, 433, 122–131 CrossRef CAS.
- M. J. C. Starrenburg and J. Hugenholtz, Appl. Environ. Microbiol., 1991, 57, 3535–3540 CrossRef CAS PubMed.
- A. J. Francis, C. J. Dodge, J. B. Gillow and H. W. Papenguth, Environ. Sci. Technol., 2000, 34, 2311–2317 CrossRef CAS.
- Y. V. Boltyanskaya, V. V. Kevbrin, A. M. Lysenko, T. V. Kolganova, T. P. Tourova, G. A. Osipov and T. N. Zhilina, Microbiology, 2007, 76, 739–747 CAS.
- K. K. Kim, L. Jin, H. C. Yang and S. T. Lee, Int. J. Syst. Evol. Microbiol., 2007, 57, 675–681 CrossRef CAS.
- X. W. Xu, Y. H. Wu, Z. Zhou, C. S. Wang, Y. G. Zhou, H. Bin Zhang, Y. Wang and M. Wu, Int. J. Syst. Evol. Microbiol., 2007, 57, 1619–1624 CrossRef PubMed.
- S. Fox, N. Mozes, O. Lahav, N. Mirzoyan and A. Gross, Water. Air. Soil Pollut., 2015, 226, 134 CrossRef.
- J. Switzer Blum, A. Burns Bindi, J. Buzzelli, J. F. Stolz and R. S. Oremland, Arch. Microbiol., 1998, 171, 19–30 CrossRef CAS.
- J. Switzer Blum, S. Hoeft McCann, S. Bennett, L. G. Miller, J. F. Stolz, B. Stoneburner, C. Saltikov and R. S. Oremland, Geomicrobiol. J., 2016, 33, 677–689 CrossRef CAS.
- H. Yang, P. Shao, T. Lu, J. Shen, D. Wang, Z. Xu and X. Yuan, Int. J. Hydrogen Energy, 2006, 31, 1306–1313 CrossRef CAS.
- V. M. Gámez, R. Sierra-Alvarez, R. J. Waltz and J. A. Field, Biodegradation, 2009, 20, 499–510 CrossRef.
- A. J. M. Stams, J. Huisman, P. A. Garcia Encina and G. Muyzer, Appl. Microbiol. Biotechnol., 2009, 83, 957–963 CrossRef CAS PubMed.
- D. Féron, in Nuclear Corrosion Science and Engineering, ed. D. Féron, Woodhead Publishing, 2012, pp. 31–56 Search PubMed.
- C. A. C. Sequeira, D. S. P. Cardoso, L. Amaral, B. Šljukić and D. M. F. Santos, Corros. Rev., 2016, 34, 187–200 CrossRef CAS.
- H. S. Klapper, N. S. Zadorozne and R. B. Rebak, Acta Metall. Sin., 2017, 30, 296–305 CrossRef.
- M. L. Carboneau and J. P. Adams, National Low-Level Waste Management Radionuclide Report Series, Idaho Falls, Idaho, 1995, vol. 17 Search PubMed.
- T. A. Marshall, K. Morris, G. T. W. Law, J. F. W. Mosselmans, P. Bots, H. Roberts and S. Shaw, Mineral. Mag., 2015, 79, 1265–1274 CrossRef.
- J. R. Fox, R. J. G. Mortimer, G. Lear, J. R. Lloyd, I. Beadle and K. Morris, Appl. Geochem., 2006, 21, 1539–1550 CrossRef CAS.
- L. Newsome, K. Morris and J. R. Lloyd, Chem. Geol., 2014, 363, 164–184 CrossRef CAS.
- A. J. Williamson, K. Morris, G. T. W. Law, A. Rizoulis, J. M. Charnock and J. R. Lloyd, Environ. Sci. Technol., 2014, 48, 13549–13556 CrossRef CAS.
- H. Gamsjäger and F. J. Mompean, Chem. Thermodyn., NEA Data Bank and OECD Nuclear Energy Agency, Issy-les-Moulineaux (France), 2005, p. 617 Search PubMed.
- G. Kuippers, C. Boothman, H. Bagshaw, M. Ward, R. Beard, N. Bryan and J. R. Lloyd, Sci. Rep., 2018, 8, 1–11 CAS.
- G. Joshi-Tope and A. J. Francis, J. Bacteriol., 1995, 177, 1989–1993 CrossRef CAS PubMed.
- A. J. Francis, J. Alloys Compd., 1998, 271–273, 78–84 CrossRef CAS.
- A. J. Francis and C. J. Dodge, Environ. Sci. Technol., 2008, 42, 8277–8282 CrossRef CAS.
- Y. Suzuki, T. Nankawa, T. Yoshida, T. Ozaki, T. Ohnuki, A. J. Francis, S. Tsushima, Y. Enokida and I. Yamamoto, J. Radioanal. Nucl. Chem., 2005, 266(2), 199–204 CrossRef CAS.
- R. Ganesh, K. G. Robinson, L. Chu, D. Kucsmas and G. D. Reed, Water Res., 1999, 33, 3447–3458 CrossRef CAS.
- R. Ganesh, K. G. Robinson, G. D. Reed and G. S. Sayler, Appl. Environ. Microbiol., 1997, 63, 4385–4391 CrossRef CAS PubMed.
- F. Y. C. Huang, P. V. Brady, E. R. Lindgren and P. Guerra, Environ. Sci. Technol., 1998, 32, 379–382 CrossRef CAS.
- A. J. Francis, Uranium Environ. Min. Impact Consequences, 2006, 191–197 Search PubMed.
- A. J. Francis, G. A. Joshi-Tope and C. J. Dodge, Environ. Sci. Technol., 1996, 30, 562–569 CrossRef CAS.
- A. J. Francis and C. J. Dodge, Appl. Environ. Microbiol., 1993, 59, 109–113 CrossRef CAS.
- R. A. P. Thomas, A. J. Beswick, G. Basnakova, R. Moller and L. E. Macaskie, J. Chem. Technol. Biotechnol., 2000, 75, 187–195 CrossRef CAS.
- W. Hummel, G. Anderegg, I. Puigdomenech, R. Linfeng and O. Tochiyama, Radiochim. Acta, 2005, 93, 719–725 CrossRef CAS.
- E. Giffaut, M. Grivé, P. Blanc, P. Vieillard, E. Colàs, H. Gailhanou, S. Gaboreau, N. Marty, B. Madé and L. Duro, Appl. Geochem., 2014, 49, 225–236 CrossRef CAS.
- E. Colàs, M. Grivé, I. Rojo and L. Duro, Radiochim. Acta, 2011, 99, 269–273 CrossRef.
- E. Colàs, M. Grivé and I. Rojo, J. Solution Chem., 2013, 42, 1545–1557 CrossRef.
- E. Colàs, M. Grivé, I. Rojo and L. Duro, J. Solution Chem., 2013, 42, 1680–1690 CrossRef.
- K. Vercammen, M. A. Glaus and L. R. Van Loon, Radiochim. Acta, 2001, 89, 393–401 CrossRef CAS.
- Z. A. Begum, I. M. M. Rahman, Y. Tate, Y. Egawa, T. Maki and H. Hasegawa, J. Solution Chem., 2012, 41, 1713–1728 CrossRef CAS.
- L. Abrahamsen-Mills and J. S. Small, Organic-containing nuclear wastes and national inventories across Europe, Elsevier Inc., 2021, vol. 2 Search PubMed.
- R. A. Peterson, E. C. Buck, J. Chun, R. C. Daniel, D. L. Herting, E. S. Ilton, G. J. Lumetta and S. B. Clark, Environ. Sci. Technol., 2018, 52, 381–396 CrossRef CAS.
- J. Small, C. Lennon and L. Abrahamsen, GRM Near-Field Modelling for the LLWER 2011 ESC, 2011, http://www.llwrsite.com, accessed 25 October 2017 Search PubMed.
- A. Rizoulis, H. M. Steele, K. Morris and J. R. Lloyd, Mineral. Mag., 2012, 76, 3261–3270 CrossRef.
- S. L. Smith, A. Rizoulis, J. M. West and J. R. Lloyd, Geomicrobiol. J., 2016, 33, 455–467 CrossRef CAS.
- J. Qian, D. Li, G. Zhan, L. Zhang, W. Su and P. Gao, Bioresour. Technol., 2012, 116, 66–73 CrossRef CAS PubMed.
- J. Qian, X. Zhu, Y. Tao, Y. Zhou, X. He and D. Li, Int. J. Mol. Sci., 2015, 16, 7932–7943 CrossRef CAS PubMed.
- C. Robinson, M. White-Pettigrew, S. Shaw, K. Morris, J. Graham and J. R. Lloyd, J. Nucl. Fuel Cycle Waste Technol., 2022, 20, 307–319 CrossRef.
- J. A. Schramke, E. F. U. Santillan and R. T. Peake, Appl. Geochem., 2020, 116, 104561 CrossRef CAS.
- E. Santillan, G. Chen, P. Flanders, H. Leigh, J. Major, D. Malashock, T. Peake, K. Reiser, J. Rustick, J. Schramke, D. Schultheisz, D. Stuenkel and X. Tong, The Geochemistry of the Waste Isolation Pilot Plant, 2021 Search PubMed.
- D. L. Wilson, M. L. Baker, B. R. Hart, J. E. Marra and P. E. Shoemaker, Waste Isolation Pilot Plant Technical Assessment Team Report, 2015 Search PubMed.
- D. L. Parkhurst and C. A. J. Appelo, Description of input and examples for PHREEQC version 3-A computer program for speciation, batch-reaction, one- dimensional transport, and inverse geochemical calculations, 2013, https://pubs.usgs.gov/tm/06/a43/, accessed 24 April 2020 Search PubMed.
- A. E. Milodowski, R. P. Shaw and D. I. Stewart, British Geological Survey Commissioned Report, CR/13/104 43pp, 2013 Search PubMed.
- D. R. Lovley, R. C. Greening and J. G. Ferry, Appl. Environ. Microbiol., 1984, 48, 81–87 CrossRef CAS.
- J. R. Postgate, The sulphate-reducing bacteria, Cambridge University Press, Cambridge, 2nd edn, 1984 Search PubMed.
- J. G. Caporaso, Proc. Natl. Acad. Sci., 2011, 4516–4522 CrossRef CAS.
- J. G. Caporaso, C. L. Lauber, W. A. Walters, D. Berg-Lyons, J. Huntley, N. Fierer, S. M. Owens, J. Betley, L. Fraser, M. Bauer, N. Gormley, J. A. Gilbert, G. Smith and R. Knight, ISME J., 2012, 6, 1621–1624 CrossRef CAS PubMed.
- J. J. Kozich, S. L. Westcott, N. T. Baxter, S. K. Highlander and P. D. Schloss, Appl. Environ. Microbiol., 2013, 79, 5112–5120 CrossRef CAS.
- P. L. McCarty, Biotechnol. Bioeng., 2006, 97, 377–388 CrossRef PubMed.
- G. R. Hedwig, J. R. Liddle and R. D. Reeves, Aust. J. Chem., 1980, 33, 1685–1693 CrossRef CAS.
- L. Macomber and R. P. Hausinger, in Stress and Environmental Regulation ofGene Expression and Adaptation in Bacteria, ed. F. J. de Bruijn, John Wiley & Sons Inc., First., 2016, pp. 1133–1141 Search PubMed.
- L. Macomber and R. P. Hausinger, Metallomics, 2011, 3, 1153–1162 CrossRef CAS PubMed.
- N. V. Ashley, M. Davies and T. J. Hurst, Water Res., 1982, 16, 963–971 CrossRef CAS.
- Y. Suzuki and J. F. Banfield, Geomicrobiol. J., 2004, 21, 113–121 CrossRef CAS.
- M. P. Thorgersen, W. Andrew Lancaster, X. Ge, G. M. Zane, K. M. Wetmore, B. J. Vaccaro, F. L. Poole, A. D. Younkin, A. M. Deutschbauer, A. P. Arkin, J. D. Wall and M. W. W. Adams, Front. Microbiol., 2017, 8, 1–12 Search PubMed.
- C. Buchmaier, M. Glänzer, A. Torvisco, P. Poelt, K. Wewerka, B. Kunert, K. Gatterer, G. Trimmel and T. Rath, J. Mater. Sci., 2017, 52, 10898–10914 CrossRef CAS.
- G. Barim, S. R. Smock, P. D. Antunez, D. Glaser and R. L. Brutchey, Nanoscale, 2018, 10, 16298–16306 RSC.
- E. M. van den Berg, M. P. Elisário, J. G. Kuenen, R. Kleerebezem and M. C. M. van Loosdrecht, Front. Microbiol., 2017, 8, 1–13 Search PubMed.
- C. Allison and G. T. Macfarlane, J. Gen. Microbiol., 1988, 134, 1397–1405 CAS.
- A. Hanke, J. Berg, T. Hargesheimer, H. E. Tegetmeyer, C. E. Sharp and M. Strous, Front. Microbiol., 2016, 6, 1461 Search PubMed.
- A. Chidthaisong and R. Conrad, FEMS Microbiol. Ecol., 2000, 31, 73–86 CrossRef CAS.
- J. R. Bargar, K. H. Williams, K. M. Campbell, P. E. Long, J. E. Stubbs, E. I. Suvorova, J. S. Lezama-Pacheco, D. S. Alessi, M. Stylo, S. M. Webb, J. A. Davis, D. E. Giammar, L. Y. Blue and R. Bernier-Latmani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4506–4511 CrossRef CAS.
- D. S. Alessi, J. S. Lezama-Pacheco, J. E. Stubbs, M. Janousch, J. R. Bargar, P. Persson and R. Bernier-Latmani, Geochim. Cosmochim. Acta, 2014, 131, 115–127 CrossRef CAS.
- A. Bhattacharyya, K. M. Campbell, S. D. Kelly, Y. Roebbert, S. Weyer, R. Bernier-Latmani and T. Borch, Nat. Commun., 2017, 8, 1–8 CrossRef.
- G. Kuippers, K. Morris, L. T. Townsend, P. Bots, K. Kvashnina, N. D. Bryan and J. R. Lloyd, Environ. Sci. Technol., 2021, 55(8), 4597–4606 CrossRef CAS.
- A. J. Fuller, P. Leary, N. D. Gray, H. S. Davies, J. F. W. Mosselmans, F. Cox, C. H. Robinson, J. K. Pittman, C. M. McCann, M. Muir, M. C. Graham, S. Utsunomiya, W. R. Bower, K. Morris, S. Shaw, P. Bots, F. R. Livens and G. T. W. Law, Chemosphere, 2020, 254, 126859 CrossRef CAS PubMed.
- M. I. Boyanov, K. E. Fletcher, M. J. Kwon, X. Rui, E. J. O'Loughlin, F. E. Löffler and K. M. Kemner, Environ. Sci. Technol., 2011, 45, 8336–8344 CrossRef CAS PubMed.
- G. Morin, A. Mangeret, G. Othmane, L. Stetten, M. Seder-Colomina, J. Brest, G. Ona-Nguema, S. Bassot, C. Courbet, J. Guillevic, A. Thouvenot, O. Mathon, O. Proux and J. R. Bargar, Geochemical Perspect. Lett., 2016, 2, 95–105 Search PubMed.
- R. Bernier-Latmani, H. Veeramani, E. D. Vecchia, P. Junier, J. S. Lezama-Pacheco, E. I. Suvorova, J. O. Sharp, N. S. Wigginton and J. R. Bargar, Environ. Sci. Technol., 2010, 44, 9456–9462 CrossRef CAS.
- L. Newsome, K. Morris, D. Trivedi, A. Bewsher and J. R. Lloyd, Environ. Sci. Technol., 2015, 49, 11070–11078 CrossRef CAS PubMed.
- D. G. Zavarzina, T. P. Tourova, T. V. Kolganova, E. S. Boulygina and T. N. Zhilina, Microbiology, 2009, 78, 723–731 CAS.
- R. A. Albert, M. McGuine, S. C. Pavlons, J. Roecker, J. Bruess, S. Mossman, S. Sun, M. King, S. Hong, C. E. Farrance, J. Danner, Y. Joung, N. Shapiro, W. B. Whitman and H. J. Busse, Int. J. Syst. Evol. Microbiol., 2019, 69, 1376–1383 CrossRef CAS.
- A. K. Borsodi, J. M. Aszalós, P. Bihari, I. Nagy, P. Schumann, C. Spröer, A. L. Kovacs, K. Bóka, P. Dobosy, M. Óvari, T. Szili-Kovacs and E. Tóth, Int. J. Syst. Evol. Microbiol., 2019, 69, 631–637 CrossRef CAS PubMed.
- G. Hernandez-Eugenio, M. L. Fardeau, B. K. C. Patel, H. Macarie, J. L. Garcia and B. Ollivier, Anaerobe, 2000, 6, 305–312 CrossRef CAS.
- E. V. Pikuta and R. B. Hoover, Lact. Acid Bact. Biodivers. Taxon., 2014, 9781444333, 135–145 Search PubMed.
- T. Černoušek, R. Shrestha, H. Kovářová, R. Špánek, A. Ševců, K. Sihelská, J. Kokinda, J. Stoulil and J. Steinová, Corros. Eng., Sci. Technol., 2020, 55, 127–137 CrossRef.
- F. King, M. Kolar, S. Stroes-Gascoyne, P. Bellingham, J. Chu and P. V Dawe, MRS Online Proc. Libr., 1999, 556, 1167 CrossRef CAS.
- K. Pedersen, J. Appl. Microbiol., 2010, 108, 1094–1104 CrossRef CAS.
- M. Motamedi, O. Karland and K. Pedersen, FEMS Microbiol. Lett., 1996, 141, 83–87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00061c |
| This journal is © The Royal Society of Chemistry 2023 |