
Optimization of lipid assisted polymeric nanoparticles for siRNA delivery and cancer immunotherapy†
Song
Lin‡
a,
Houjin
Jing‡
a,
Xiaojiao
Du
*b,
Xianzhu
Yang
 *a and
Jun
Wang
a
*a and
Jun
Wang
a
aSchool of Biomedical Sciences and Engineering, South China University of Technology, Guangzhou International Campus, Guangzhou, Guangdong 511442, P. R. China. E-mail: yangxz@scut.edu.cn
bSchool of Medicine, South China University of Technology, Guangzhou 510006, PR China. E-mail: duxjz@scut.edu.cn
First published on 12th March 2024
Abstract
To date, five siRNA-based medications have received clinical approval and have demonstrated remarkable therapeutic efficacy in treating various diseases. However, their application has been predominantly limited to liver-specific diseases due to constraints in siRNA delivery capabilities. In this study, we have developed a siRNA delivery system utilizing clinically approved mPEG-b-PLGA, a cationic lipid, and an ionizable lipid. We optimized this system by carefully adjusting their mass ratios, resulting in highly efficient gene silencing. Furthermore, the optimized nanoparticle formulation, which encapsulates siRNA targeting CD47, induces a robust immune response. This response effectively suppresses the progression of melanoma tumors by blocking this critical immune checkpoint.
1. Introduction
Small interfering RNA (siRNA) exhibits a remarkable ability to selectively and precisely silence the expression of specific genes through the RNA interference (RNAi) mechanism.1 This holds great promise for therapeutic interventions across a wide range of diseases. At present, five siRNA medications, namely patisiran, givosiran, inclisiran, lumasiran, and vutrisiran, have already received clinical approval,2 with several RNAi-based therapeutics currently undergoing clinical trials.3 Among them, patisiran4 and givosiran5 stand out as groundbreaking siRNA-based medications, making significant advancements in the treatment of genetic and rare diseases. These drugs serve as exemplars of the potential of siRNA technology, demonstrating its ability to precisely target the underlying causes of complex genetic disorders and offering hope for improved quality of life.The success of these approved siRNA-based therapies can be primarily attributed to the lipid nanoparticle (LNP) technology and GalNAc delivery technology.6,7 This is because siRNA, being a large, negatively charged, hydrophilic macromolecule, cannot penetrate target cells on its own.8 Despite the significant advancements achieved, it is important to note that both of the approved siRNA delivery systems were primarily designed for liver-specific delivery. Given this limitation, there is an urgent need to develop alternative clinically available delivery systems for siRNA therapy.9–18
Previously, we utilized a poly(ethylene glycol)-b-poly(D,L-lactide) and cationic lipid combination to encapsulate siRNA,19–23 employing a double emulsion–solvent evaporation technique. This approach yielded exceptional siRNA loading efficiency, exceeding 95%, owing to its nanoconfined loading mechanism.24 Moreover, our formulation exhibited the ability to efficiently deliver siRNA into various types of target cells, leading to the effective silencing of disease-related genes. In our latest study, we took a step further by refining the optimized formulation, which we refer to as iCLAN. This was achieved by carefully adjusting the mass ratios of mPEG-b-PLGA, the cationic lipid, and the ionizable lipid (as illustrated in Scheme 1). Our findings revealed that the incorporation of ionizable lipids led to a significant enhancement in the siRNA silencing efficiency, surpassing even the performance of the commercial transfection reagent Lipofectamine RNAiMAX. Notably, the optimized nanoparticle carrying siCD47 demonstrated an ability to induce a robust immune response. This immune response effectively suppressed the progression of melanoma tumors by blocking this crucial immune checkpoint. These promising results underscore the potential of our iCLAN formulation as an advanced siRNA delivery system, offering a valuable tool for gene silencing and cancer immunotherapy.
 | ||
| Scheme 1 Schematic illustration of the preparation of iCLAN. mPEG-b-PLGA, poly(ethylene glycol)-b-poly(lactic-co-glycolic acid); the cationic lipid DOTAP: 1,2-dioleoyloxy-3-trimethylammonium-propane; the ionizable lipid DLin-MC3-DMA: (6Z,9Z,28Z,31Z)-heptatriacont-6,9,28,31-tetraene-19-yl 4-(dimethylamino)butanoate. The iCLAN nanoparticles were formed by adding an aqueous solution of siRNA into an organic solution of mPEG-b-PLGA, DOTAP, and DLin-MC3-DMA under vigorous shaking with a vortex. | ||
2. Materials and methods
2.1. Materials
DOTAP, DLin-MC3-DMA, SM-102 and ALC-0315 were all purchased from AVT (Shanghai, China). mPEG-b-PLGA with a molecular weight of 2000 for both PEG and PLGA blocks was provided free of charge by Guangzhou Kelan Biotechnology Co. (Guangdong, China). Fetal bovine serum (FBS), trypsin–EDTA solution, Dulbecco's modified Eagle's medium (DMEM), RPMI 1640 medium and other reagents for cell culture were purchased from Gibco (Grand Island, USA). Cy5-labeled siRNA, GFP siRNA (siGFP), CD47 siRNA (siCD47) and negative siRNA (siNC) were purchased from BeiXin (Suzhou, China) and the sequences were as follows: siGFP (sense) 5′-CAAGCUGACCCUGAAGUUCTT-3′, (antisense) 5′-GAACUUCAGGGUCAGCUUGTT-3′, siCD47 (sense) 5′-GGACUUGGCCUCAUUGUAATT-3′, (antisense) 5′-UUACAAUGAGGCCAAGUCCTT-3′; siNC (sense) 5′-UUCUCCGAACGUGUCACGUTT-3′, and (antisense) 5′-ACGUGACACGUUCGGAGAATT-3′. Antibodies for flow cytometry analysis – BV510 anti-mouse CD45 (clone: 30-F11), PE anti-mouse CD11c (clone: N418), FITC anti-mouse CD80 (clone: 16-10A1), APC/Cy7 anti-mouse CD86 (clone: GL-1), BV421 anti-mouse CD11b (clone: M1/70), APC anti-mouse F4/80 (clone: QA17A29), and PE anti-mouse CD47 (clone: miap301) – were purchased from Biolegend (CA, USA). For the antibodies for western blot analysis, rabbit anti-CD47 antibody (DF6649) was from Affinity Biosciences (OH, USA); mouse anti-GAPDH antibody (60004-1-Ig) was from ProteinTech (IL, USA); and goat anti-rabbit IgG-HRP (BL003A) and goat anti-mouse IgG-HRP (BL001A) were from Biosharp (Hefei, China).2.2. Preparation of iCLAN nanoparticles
The lipids (ionizable lipid, DOTAP) or mPEG-b-PLGA were dissolved in ethanol or DMSO to a final concentration of 10 mg mL−1 or 150 mg mL−1, respectively. Secondly, an organic phase (28 μL), which was prepared by mixing the ionizable lipid, DOTAP and mPEG-b-PLGA at a predetermined weight ratio, and the aqueous phase of siRNA (72 μL, 0.15 OD siRNA) were mixed by vigorous vortexing and then incubated for 15 min at room temperature. The method for preparing NP loaded with siRNA is completely identical to the method for preparing iCLAN, with the only difference being that ionizable lipids are not added during the preparation of the NP. The fresh iCLAN was diluted with deionized water to 6 ng μL−1 siRNA for size and zeta potential detection using dynamic light scattering (DLS, Zetasizer Nano ZS90, Malvern, United Kingdom), and the morphology of the sample was further characterized by transmission electron microscopy (TEM, Talos L120C, Thermo Fisher, America).2.3. Cell culture and iCLAN formulation screening
B16F10 cells and B16F10-GFP cells stably expressing GFP were cultured and maintained in complete 1640 medium at 37 °C under 5% CO2. Panc02 cells and Gal3-GFP-Panc02 cells were cultured and maintained in complete DMEM medium at 37 °C under 5% CO2. For screening the iCLAN formulations, B16F10-GFP cells (5.0 × 104) were seeded in 24-well plates and incubated overnight. The cells were treated with iCLAN@siGFP at a siGFP concentration of 2 nM. Lipofectamine RNAiMAX reagent carrying 10 nM siGFP was used as the positive control. The expression of the GFP protein in the treated B16F10-GFP cells was detected using FACS (Accuri C6 plus, BD, America) after incubation for 24 h.2.4. Downregulation of CD47 gene expression after treatment with iCLAN@siCD47
B16F10 cells (5.0 × 104) were seeded in 24-well plates and incubated overnight. The cells were treated with iCLAN@siCD47 at a final concentration of siCD47 2 nM. Lipofectamine RNAiMAX Reagent carrying 10 nM siCD47 was used as the positive control. To determine the CD47 expression of the B16F10 cells, the treated B16F10 cells were collected after incubation for 24 h, washed twice with PBS, and then labeled with an anti-CD47-PE antibody for flow cytometric analysis.For assessing CD47 gene silencing in Panc02 cells, the Panc02 cells (5.0 × 104) were seeded in 24-well plates and incubated overnight. The cells were treated with iCLAN@siCD47 at a final concentration of siCD47 of 10 nM. Lipofectamine RNAiMAX reagent carrying 10 nM siCD47 was used as the positive control. The expression of CD47 at the mRNA level in Panc02 cells was examined by real-time PCR (RT-PCR) after incubation for 24 h.
2.5. In vitro toxicity of iCLAN in B16F10 cells
B16F10 cells were treated with various concentrations (10, 50, 100, 150, 200, 250, and 300 nM) of iCLAN for 24 h under standard cell culture incubation conditions. Then cell viability was assessed using cell counting kit-8 (Beyotime).2.6. The cellular uptake mechanism of iCLAN
B16F10 cells (5.0 × 104) were seeded in 24-well plates and incubated overnight. Subsequently, the cells were co-incubated with 20 μg mL−1 amiloride (Amilo), 10 μg mL−1 chlorpromazine (CPZ), or 100 μg mL−1 methyl-β-cyclodextrin (MβCD) for 1 h, followed by co-incubation with iCLAN@Cy5-siRNA for 4 h. The uptake of iCLAN@Cy5-siRNA by the cells was then assessed using flow cytometry.2.7. Stability of iCLAN nanoparticles
To study the colloidal stability of iCLAN nanoparticles, the size of the samples was monitored for 72 h during storage in deionized water at room temperature. Additionally, to evaluate the gene-silencing efficiency of iCLAN nanoparticles, B16F10 cells (5.0 × 104) were seeded in 24-well plates and incubated overnight. The cells were treated with iCLAN@siCD47 stored at 4 °C for a certain period of time at a final concentration of siCD47 10 nM. The B16F10 cells were collected after incubation for 24 h, washed twice with PBS and then labeled with an anti-CD47-PE antibody for flow cytometric analysis.2.8. Cellular uptake of iCLAN and intracellular trafficking
B16F10 cells (5 × 104) were seeded into 24-well plates and incubated overnight. The cells were then incubated at 37 °C with iCLAN@Cy5-siRNA or NP@Cy5-siRNA at a Cy5-siRNA concentration of 50 nM. After incubation for different times, the cells were washed, trypsinized, and then collected for FACS analyses (BD FACSCanto™ II). For CLSM observation, the treated cells were fixed with 4% paraformaldehyde for 10 min, and cell nuclei were stained with DAPI (BioFroxx, Germany) for CLSM (OLYMPUS SpinSR10) observation.The endosomal/lysosomal escape of iCLAN@Cy5-siRNA was visualized using CLSM. The B16F10 cells were seeded into a glass bottom cell culture dish and cultured overnight. The iCLAN@Cy5-siRNA or NP@Cy5-siRNA solution was added at a Cy5-siRNA concentration of 50 nM and incubated for 12 h. LysoTracker Green DND-26 (75 nM, MA, USA) was added to the dish and incubated at 37 °C for 30 min. The treated cells were then washed with PBS, stained with Hoechst 33342 for 5 min, and then observed using CLSM (OLYMPUS SpinSR10).
Furthermore, the endosomal/lysosomal escape of iCLAN@Cy5-siRNA was assessed in Gal3-GFP-Panc02 cells. After culturing overnight, iCLAN@siRNA or NP@siRNA was added to the dish at a siRNA concentration of 50 nM and incubated for 12 h. The cells were then stained with Hoechst 33342 for 5 min and washed twice with 1 × PBS. All samples were observed using CLSM (Zeiss LSM880).
2.9. Antitumor efficacy in xenograft tumor models
C57BL/6 mice bearing B16F10 tumors (∼30 mm3) were randomly divided into four groups and treated with PBS, iCLAN@siNC, NP@siCD47, or iCLAN@siCD47 by intravenous injection once every other day. The injection dose of siRNA was 20 μg per mouse per injection. The tumor volume was monitored using calipers.2.10. Analysis of mRNA and protein in tumor tissues
Tumor tissue samples were processed for both mRNA and protein analyses. Approximately 20 mg of the samples was homogenized in RNAiso Plus (Takara, Japan) to detect CD47 expression at the mRNA level using qRT-PCR. Another portion of the samples, approximately 30 mg, was homogenized in RIPA lysis solution for the analysis of CD47 protein levels, which was conducted using western blotting.2.11. Intratumoral lymphocyte analysis
To examine the intratumoral infiltration of immune cells, tumors were harvested after treatment, cut into small pieces, and immersed in a digestion solution (1.0 mg mL−1 collagenase type IV, 40 μg mL−1 hyaluronidase, and 40 μg mL−1 DNase I) for 15 min at 37 °C. Tumor cell suspensions were passed through a 200-mesh nylon mesh and resuspended in 40% Percoll after centrifugation to collect lymphocytes by gradient centrifugation. Then, red blood cell lysis buffer was added, and red blood cells were lysed on ice for 10 min. Then, the samples were centrifuged to collect the cells, which were resuspended in PBS. The single-cell suspension was stained with fluorophore-labeled antibodies according to the manufacturer's protocols. For analysis of M1 macrophages (CD45+CD11b+F4/80+CD86+) in tumor tissue, tumor cell suspensions were stained with BV510 anti-mouse CD45, BV421 anti-mouse CD11b, APC anti-mouse F4/80, and APC/Cy7 anti-mouse CD86 antibodies and analyzed using flow cytometry (BD FACSCanto™ II). Furthermore, the tumor-draining lymph nodes (TDLNs) were harvested after treatment. The TDLNs were stained with BV510 anti-mouse CD45, PE anti-mouse CD11c, FITC anti-mouse CD80, and APC/Cy7 anti-mouse CD86 antibodies. The frequency of mature DCs (CD45+CD11c+CD80+CD86+) in the TDLNs was then examined by flow cytometric analysis.2.12. Statistical analysis
All the data are shown as the mean ± standard deviation (SD). Statistical analysis was performed using Student's t-test (two-tailed) or one-way ANOVA with post-hoc analysis. Significant differences are indicated by *p < 0.05, **p < 0.01, and ***p < 0.001; p < 0.05 is considered statistically significant.3. Results and discussion
3.1. Preparation and screening of iCLAN nanoparticles
To prepare iCLAN nanoparticles, we added an ethanol-DMSO mixed solution of the ionizable lipid, the cationic lipid DOTAP, and the amphiphilic polymer mPEG-b-PLGA into an aqueous solution of siRNA. To achieve efficient siRNA loading, the N/P ratio of DOTAP to siRNA was first determined. As shown in Fig. 1A, siRNA could be completely loaded into iCLAN nanoparticles at an N/P ratio (DOTAP to siRNA) of 4. Subsequently, we investigated the effect of ionizable lipid DLin-MC3-DMA on siRNA loading. In the presence of the cationic lipid (N/P = 4), the addition of the ionizable lipid did not affect siRNA loading (Fig. 1B). However, without the cationic lipid, siRNA cannot be loaded at various ratios of ionizable lipid/siRNA (Fig. 1C). These results indicated that the cationic lipid played critical roles in siRNA loading, and the addition of the ionizable lipid negligibly affected the siRNA loading of the obtained nanoparticles.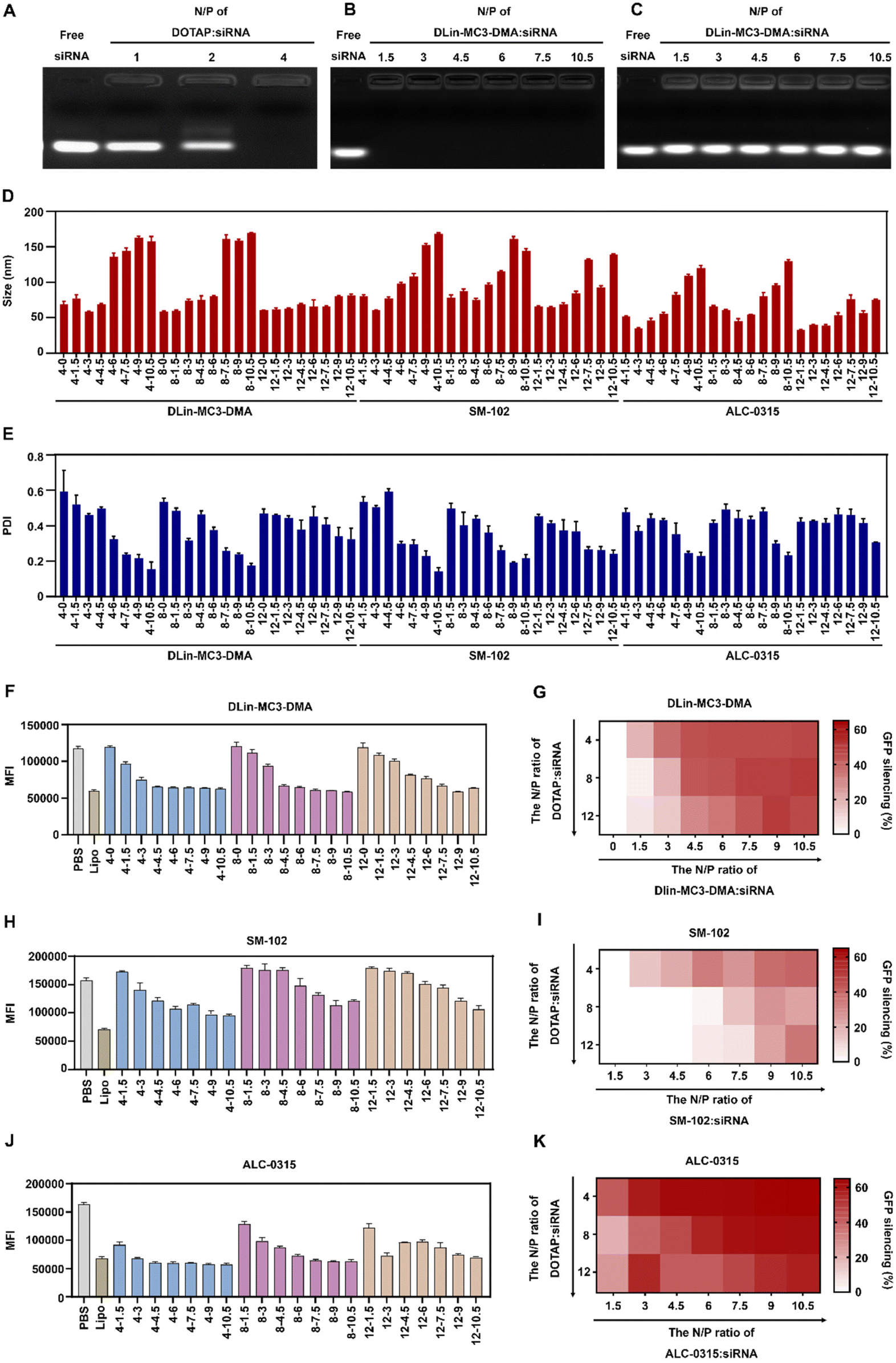 | ||
| Fig. 1 Preparation and screening of iCLAN formulations for siRNA delivery in vitro. (A) Agarose gel electrophoresis of iCLAN without ionizable lipids at various ratios of DOTAP to siRNA. (B) Agarose gel electrophoresis of iCLAN at various ratios of ionizable lipids to siRNA, and the ratios of DOTAP to siRNA were 4 in all samples. (C) Agarose gel electrophoresis of iCLAN without DOTAP at various ratios of ionizable lipids to siRNA. (D and E) Particle size (D) and polydispersity index (PDI) (E) of iCLAN@siGFP formulations. (F–K) The intracellular GFP fluorescence intensity of B16F10-GFP cells after treatment with various formulations of iCLAN@siGFP. The concentration of siGFP was 2 nM. The ionizable lipids were DLin-MC3-DMA (F and G), SM-102 (H and I), and ALC-0315 (J and K), respectively. Data are shown as the mean ± SD (n = 3). | ||
The incorporation of an ionizable lipid in our design is expected to improve the transfection efficacy of siRNA in the iCLAN system. To identify the most appropriate ionizable lipid and the optimal feeding ratio of the ionizable lipid, the iCLAN system at various N/P ratios of ionizable lipids (DLin-MC3-DMA, SM-102, and ALC-0315) and siRNA was prepared. The N/P ratios of DOTAP and siRNA were selected as 4, 8, and 12 to ensure complete siRNA loading. As shown in Fig. 1D and E, the particle size of iCLAN increases with an increase in the proportion of ionizable lipids, while the PDI of iCLAN decreases as the proportion of ionizable lipids increases.
Furthermore, siRNA targeting GFP was used to evaluate the silencing efficiencies of these formulations in B16F10-GFP cells. As shown in Fig. 1F and G, the silencing efficiencies of iCLAN@siGFP increase with an increase in the feeding ratios of DLin-MC3-DMA but decrease with an increase in the feeding ratios of DOTAP. Similarly, the ionizable and cationic lipid-dependent silencing efficiencies were also demonstrated when substituting the ionizable lipid of DLin-MC3-DMA with SM-102 (Fig. 1H and I) and ALC-0315 (Fig. 1J and K). In addition, it should be noted that the optimized iCLAN system at an siRNA concentration of 2 nM exhibited better transfection efficiency than the current commercial transfection agent Lipofectamine RNAiMAX.
To confirm the above results, CD47, a critical immune checkpoint receptor to evade macrophage phagocytosis of tumor cells, was used as another target gene to evaluate the silencing efficiencies of the iCLAN system. Similar ionizable and cationic lipid-dependent silencing efficiencies were observed in both cell lines B16F10 (Fig. 2A) and Panc02 (Fig. 2B). Among the three types of ionizable lipids, DLin-MC3-DMA showed slightly better efficiency. We also observed that ALC-0315 exhibited more toxic side effects than DLin-MC3-DMA and SM-102. Therefore, the iCLAN formulation prepared at a DLin-MC3-DMA/siRNA feeding ratio of 3 and a DOTAP/siRNA feeding ratio of 4 was used as the optimized formulation in the subsequent experiments.
 | ||
| Fig. 2 Characterization and silencing efficiencies of iCLAN@siCD47. (A) The expression of CD47 on B16F10 cells treated with different formulations of iCLAN@siCD47 at a siCD47 concentration of 2 nM. (B) Relative CD47 expression in Panc02 cells treated with different formulations of iCLAN@siCD47 at a siCD47 concentration of 10 nM. (C) TEM images of iCLAN@siCD47. Scale bars, 200 nm. (D) Size distribution of iCLAN@siCD47 in water. (E) Zeta potential of iCLAN@siCD47. (F) Colloidal stability of iCLAN@siCD47. (G and H) Silencing efficiency of iCLAN@siCD47 in B16F10 cells after storing for various times at 4 °C. (I and J) Flow cytometry analysis of B16F10 cells co-incubated with iCLAN@Cy5-siRNA after being treated with different cell uptake inhibitors. (K) The evaluation of cytotoxicity of iCLAN@siRNA. Data are shown as the mean ± SD (n = 3). | ||
Subsequently, transmission electron microscopy (TEM) was utilized to determine the morphologies of the optimized siCD47-loaded iCLAN nanoparticle (denoted as iCLAN@siCD47). As shown in Fig. 2C, iCLAN@siCD47 exhibits a compact and spherical morphology with a size ranging from 30 nm to 60 nm. The size and zeta potential of the optimized iCLAN@siCD47 nanoparticles was also detected by dynamic light scattering (DLS). The hydrodynamic diameter of iCLAN@siCD47 was approximately 60 nm (Fig. 2D) and its zeta potential was ∼30 mV (Fig. 2E). The size changes of iCLAN@siCD47 versus time are further shown in Fig. 2F, exhibiting colloidal stability within 72 h.
Furthermore, we investigated the long-term storage of the optimized iCLAN nanoparticles. The prepared iCLAN@siCD47 was stored at 4 °C for various times, and then used to transfect the B16F10 cells. As shown in Fig. 2G and H, the silencing efficiency of iCLAN@siCD47 did not show an obvious decline even after being stored at 4 °C for 30 days. The silencing efficiency of iCLAN@siCD47 was also maintained at approximately 80% at an siCD47 concentration of 10 nM.
To investigate the mechanism of cellular uptake, we first treated B16F10 cells with various inhibitors and then co-incubated the cells with iCLAN@Cy5-siRNA. The results showed that the uptake of iCLAN by cells is mainly dependent on caveolin-mediated endocytosis (Fig. 2I and J). In addition, the cytotoxicity of iCLAN@siRNA to B16F10 cells was assessed using the CCK-8 method. As shown in Fig. 2K, the cell viability showed no obvious decrease when the concentration of siRNA in iCLAN@siRNA reached 300 nM, indicating its good biocompatibility.
3.2. Cellular uptake of iCLAN and intracellular trafficking
To demonstrate the excellent gene silencing capability of iCLAN, the formulation without the ionizable lipid (denoted as NP@siRNA) was prepared as the control formulation. The cellular uptake of iCLAN@siRNA and the control formulation NP@siRNA was first determined. Cy5-labeled siRNA was used to enhance the detectability. As shown in Fig. 3A and B, B16F10 cells incubated with iCLAN@Cy5-siRNA exhibited approximately 2-fold higher siRNA fluorescence signals than NP@Cy5-siRNA at all time points, which was confirmed by the confocal images (Fig. 3C). | ||
| Fig. 3 Cellular uptake and intracellular trafficking of iCLAN. (A and B) Flow cytometry analysis of the cellular uptake of iCLAN@Cy5-siRNA or NP@Cy5-siRNA after incubation for 4 h (A) and the mean fluorescence intensity (MFI) of Cy5-siRNA in B16F10 cells after treatment with iCLAN@Cy5-siRNA or NP@Cy5-siRNA for 0.5, 1, 2 or 4 h (n = 3) (B). (C) Confocal images of B16F10 cells transfected with iCLAN@Cy5-siRNA or NP@Cy5-siRNA. Cell nuclei were counterstained with DAPI (blue). (D) Confocal images of Gal3-GFP-Panc02 cells after transfecting with iCLAN@Cy5-siRNA or NP@Cy5-siRNA. Cell nuclei were counterstained with Hoechst 33342 (blue). (E) Confocal images of B16F10 cells after transfecting with iCLAN@Cy5-siRNA or NP@Cy5-siRNA. Cell nuclei and endosomes were counterstained with Hoechst 33342 (blue) and Lysotracker™ Green DND26 (green), respectively. (F) Pearson coefficient of colocalization between Cy5-siRNA and endosomes obtained using the PSC Colocalization plugin from ImageJ (n = 9). Scale bar: 20 μm (C and E) and 10 μm (D). Data are shown as the mean ± SD. Unpaired t-test ***p < 0.001. | ||
Furthermore, the intracellular distribution and lysosomal/endosomal escape of siRNA were also determined. The tumor cell stably expressing Gal3-GFP fusion protein was employed as the model cell for incubation with iCLAN@siRNA or the control formulation NP@siRNA. Upon endosomal escape, the endosomal membrane undergoes localized rupture, and the lactose residues located on the inner leaflet of the lysosomal/endosomal membrane are exposed to specifically bind to the Gal3 domain of the Gal3-GFP fusion protein and form green fluorescence dots.25 It could be found that the number of green fluorescence dots in the iCLAN@siRNA group was significantly higher than that of the control formulation NP@siRNA (Fig. 3D), indicating that the incorporation of ionizable lipids in the iCLAN formulation can promote the lysosomal/endosomal escape of siRNA. In addition, the lysosome/endosome of tumor cells was stained with Lysotracker™ Green DND26, and siRNA in the iCLAN@siRNA or NP@siRNA was labelled with Cy5. After co-incubation for 12 h, the co-localization of both fluorescence signals was observed using confocal microscopy. The degree of co-localization of green and red fluorescence signals was lower than that of NP@siRNA without ionizable lipids (Fig. 3E), which was indicated by the Pearson correlation coefficient (Fig. 3F). Collectively, these results demonstrate that the incorporation of ionizable lipids in the iCLAN can enhance the transfection efficiency through promoting cellular uptake and lysosomal/endosomal escape.
3.3. Therapeutic activity of iCLAN@siCD47 and the mechanism
To investigate the effectiveness of iCLAN-mediated siRNA delivery in vivo, mice bearing B16F10 tumors were employed and intravenously injected with PBS, iCLAN@siNC, NP@siCD47, or iCLAN@siCD47. The injection dosage of siRNA was 20 μg per mouse every other day and a total of four injections were administered (Fig. 4A). It could be found that mice treated with iCLAN@siNC showed almost no suppression of tumor growth, while the highest inhibition of tumor growth was observed in the iCLAN@siCD47 group (Fig. 4B and C). In contrast, the anticancer activity of the control formulation NP@siCD47 was obviously lower than that of iCLAN@siCD47. The tumor tissue collected at the end of therapy was imaged (Fig. 4D) and weighed (Fig. 4E), which further confirmed the best anticancer activity of iCLAN@siCD47. In addition, we determined the CD47 expression in the collected tumor tissues of the various treatment groups. Apparently, efficient CD47 gene silencing in vivo was achieved after treatment with iCLAN@siCD47 (Fig. 4F). At the end of the treatment, hematoxylin and eosin (H&E) staining of collected tissue sections revealed no significant lesions in the iCLAN@siCD47 treatment compared to the PBS group (Fig. 4G), and the change in the body weight of these treated mice was negligible (Fig. 4H). To more comprehensively assess the safety of iCLAN@siCD47, we conducted biochemical assays, the results of which showed no significant changes in alanine transaminase (ALT), aspartate aminotransferase (AST), total protein (TP), urea, and uric acid (UA) levels between the control and iCLAN@siCD47 treated groups (Fig. 4I). Collectively, these results suggest that iCLAN@siCD47 has effective therapeutic activity and good biosafety. | ||
| Fig. 4 Antitumor activity of iCLAN@siCD47 in the B16F10 melanoma model. (A) Schematic illustration showing the treatment timelines. Mice were intravenously injected with iCLAN@siCD47 or control formulations every 2 days; the injection doses of siCD47 were 20 μg per mouse. (B and C) Individual (B) and average (C) B16F10 tumor growth curves of various groups. (D and E) Photograph (D) and weights (E) of collected B16F10 tumors of various groups at the end of therapy. (F) CD47 expression in collected tumor tissues. (G) Representative hematoxylin and eosin (H&E) staining of tissue sections from major organs of mice after treatment with PBS and iCLAN@siCD47. (H) Body weights of mice bearing B16F10 tumor when receiving various treatments. (I) Biochemical indicators of mice after treatment with PBS or iCLAN@siCD47. Data are shown as the mean ± SD; one-way ANOVA with post-hoc analysis; *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
As reported, down-regulation of the CD47 receptor in tumor cells would re-activate macrophages to recognize and phagocytize tumor cells, and elicit the macrophage-mediated anticancer immune response.26,27 To elucidate this, the immune cell populations in the collected tumor tissue were determined using multiparameter flow cytometry. The M1-type macrophages, which play a critical role in the anticancer immune effect,28 were determined. As shown in Fig. 5A, the proportion of subpopulation M1-type macrophages in the iCLAN@siCD47 treatment group was the highest, reaching 22.6%. In contrast, the proportions of M1-type macrophages were 6.3%, 6.6%, and 11.0% in the PBS, iCLAN@siNC, and NP@siCD47 groups, respectively.
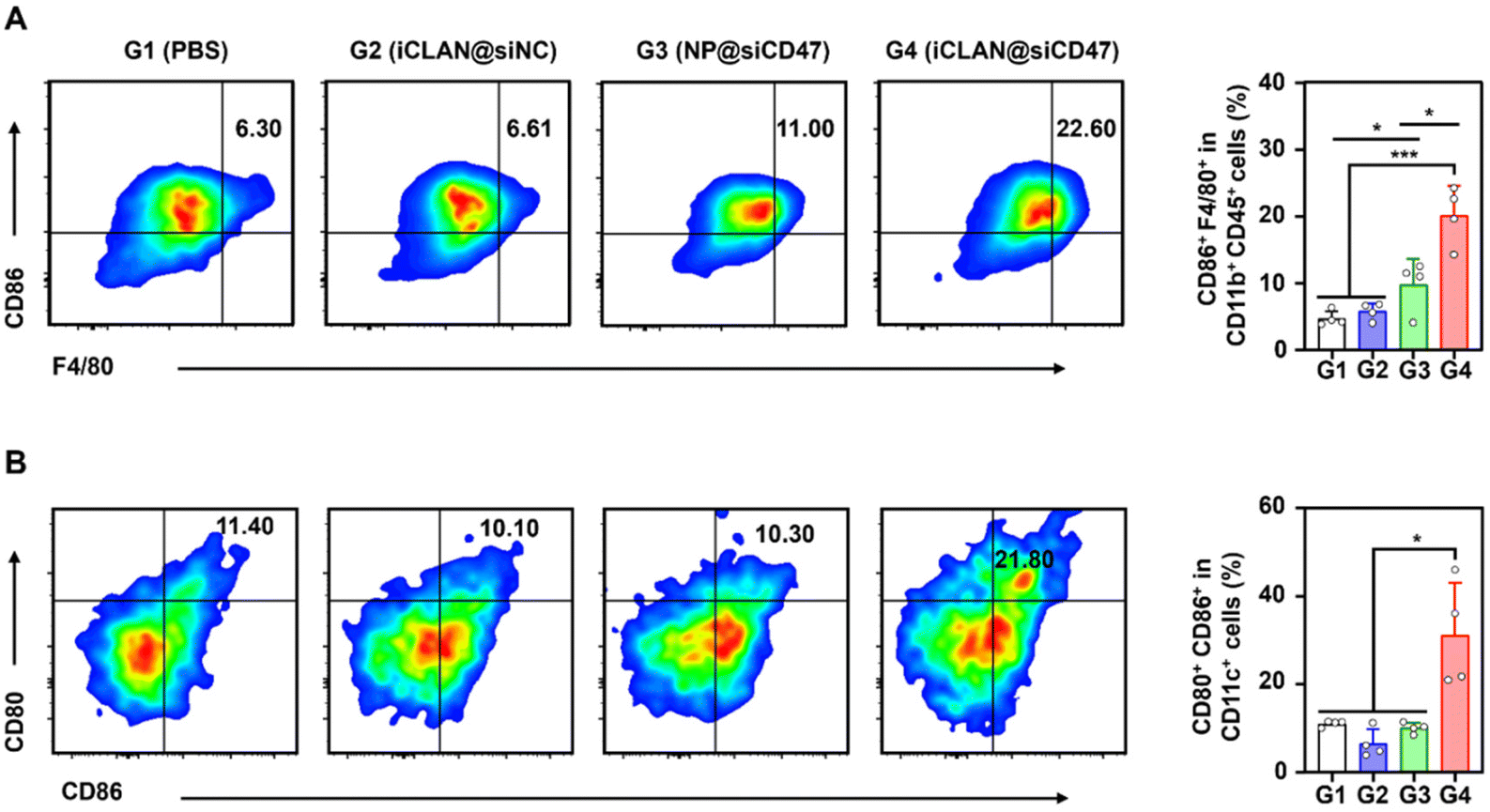 | ||
| Fig. 5 Robust anticancer immune response induced by iCLAN@siCD47 in the B16F10 melanoma model. (A) Flow cytometric analysis of M1-like macrophages in tumor tissue. (B) Flow cytometric analysis of mature DCs in the TDLNs at the end of therapy. Data are shown as the mean ± SD (n = 4), and one-way ANOVA with post-hoc analysis; *p < 0.05, **p < 0.01, and ***p < 0.001. | ||
In addition, the dendritic cell (DC)-driven anticancer immunity also critically relies on the CD47-SIRPα axis, and the proportion of DC in the TDLN post-treatment was assessed.29,30 Mice treated with iCLAN@siCD47 exhibited a notably elevated frequency of mature DCs (CD80+ CD86+), demonstrating an approximately two-fold increase compared to the mice treated with PBS, NP@siCD47 or iCLAN@siNC (Fig. 5B).
4. Conclusions
In summary, we successfully used the clinically approved mPEG-b-PLGA, the cationic lipid, and the ionizable lipid to develop iCLAN nanoparticles for siRNA delivery. Our results demonstrate that efficient siRNA loading was achieved by the cationic lipid DOTAP, while the incorporation of ionizable lipids can enhance the uptake and promote endosomal/lysosomal escape of siRNA in tumor cells. Moreover, the optimized formulation of the iCLAN nanoparticles exhibited better gene-silencing efficiency than the commercial transfection reagent Lipofectamine. In addition, the optimized nanoparticle encapsulating siRNA targeting CD47 could efficiently suppress melanoma tumor progression by blocking the key immune checkpoint. Since all the components of these nanoparticles are approved pharmaceutical excipients, this siRNA delivery system holds significant potential for clinical translation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC3401400), the National Natural Science Foundation of China (32071378, 52373137), the “Open Competition to Select the Best Candidates” Key Technology Program for Nucleic Acid Drugs of NCTIB (NCTIB2022HS01012), and the Open Project of Guangdong Province Key Laboratory of Biomedical Engineering (GDKLBEM202203). All animal studies were carried out under the guidelines evaluated and approved by the South China University of Technology Animal Care and Use Committee (approval number: 2022040).References
- R. C. Wilson and J. A. Doudna, Annu. Rev. Biophys., 2013, 42, 217–239 CrossRef CAS PubMed.
- J. Patterson, Drug Ther. Bull., 2023, 61, 72–76 CrossRef PubMed.
- E. A. Narasipura, R. V. Miller, Y. Ma and O. S. Fenton, Bioconjugate Chem., 2023, 34, 1177–1197 CrossRef CAS PubMed.
- H. Wood, Nat. Rev. Neurol., 2018, 14, 570–570 Search PubMed.
- P. Ventura, H. L. Bonkovsky, L. Gouya, P. A. Peiró, D. M. Bissell, P. E. Stein, M. Balwni, D. K. E. Anderson, C. Prker, D. J. Kuter, S. Monroy, J. Oh, B. Ritchie, J. J. Ko, Z. Hua, M. T. Sweetser and E. Sardh, Liver Int., 2022, 42, 161–172 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. V. D. Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- M. M. Janas, M. K. Schlegel, C. E. Harbison, V. O. Yilmaz, Y. Jiang, R. Parmar, I. Zlatev, A. Castoreno, H. Xu, S. S. Morskaya, K. G. Rajeev, M. Manoharan, N. D. Keirstead, M. A. Maier and V. Jadhav, Nat. Commun., 2018, 9, 723 CrossRef PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- X. Yang, S. Dou, Y. Wang, H. Long, M. Xiong, C. Mao, Y. Yao and J. Wang, ACS Nano, 2012, 6, 4955–4965 CrossRef CAS PubMed.
- X. Yang, S. Dou, T. Sun, C. Mao, H. Wang and J. Wang, J. Controlled Release, 2011, 156, 203–211 CrossRef CAS PubMed.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M. G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef CAS PubMed.
- X. Xu, J. Wu, Y. Liu, P. E. Saw, W. Tao, M. Yu, H. Zope, M. Si, A. Victorious, J. Rasmussen, D. Ayyash, O. C. Farokhzad and J. Shi, ACS Nano, 2017, 11, 2618–2627 CrossRef CAS PubMed.
- X. Xu, J. Wu, Y. Liu, M. Yu, L. Zhao, X. Zhu, S. Bhasin, Q. Li, E. Ha, J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2016, 55, 7091–7094 CrossRef CAS PubMed.
- X. You, Z. Gu, J. Huang, Y. Kang, C. C. Chu and J. Wu, Acta Biomater., 2018, 74, 180–191 CrossRef CAS PubMed.
- B. Hu, S. Kong, Y. Weng, D. Zhao, A. Hussain, Q. Jiao, S. Zhan, L. Qiu, J. Lin, M. Xie, B. Li and Y. Huang, Chin. Chem. Lett., 2023, 34, 108210 CrossRef CAS.
- Z. Huang, S. Liu, N. Lu, L. Xu, Q. Shen, Z. Huang, Z. Huang, P. E. Saw and X. Xu, Exploration, 2022, 2, 20220013 CrossRef PubMed.
- S. Guo, K. Li, B. Hu, C. Li, M. Zhang, A. Hussain, X. Wang, Q. Cheng, F. Yang, K. Ge, J. Zhang, J. Chang, X. Liang, Y. Weng and Y. Huang, Exploration, 2021, 1, 35–49 CrossRef PubMed.
- M. Huang, M. Zhang, H. Zhu, X. Du and J. Wang, Acta Pharm. Sin. B, 2022, 12, 3456–3474 CrossRef CAS PubMed.
- Y. Zhang, C. Chen, M. Su, J. Wang, C. Li and X. Yang, Nano Lett., 2024, 24, 1376–1384 CrossRef CAS PubMed.
- D. Li, Z. Cao, C. Chen, H. Li, S. He, X. Hou, M. Liang and X. Yang, Biomatrials, 2023, 302, 122339 CrossRef CAS PubMed.
- Q. Huang, M. Su, L. Zhao, Z. Zhang, Y. Zhang, X. Yang and J. Wang, Nano Today, 2023, 50, 101857 CrossRef CAS.
- L. Zhao, D. Li, Y. Zhang, Q. Huang, Z. Zhang, C. Chen, C. Xu, X. Chu, Y. Zhang and X. Yang, ACS Nano, 2022, 16, 13821–13833 CrossRef CAS PubMed.
- W. Zhang, Y. Zhang, Y. Luo, S. Chen, Q. Huang, Z. Cao, M. Liang and X. Yang, Nano Today, 2022, 43, 101418 CrossRef CAS.
- C. Xu, S. Iqbal, S. Shen, Y. Luo, X. Yang and J. Wang, Small, 2019, 15, 1900055 CrossRef PubMed.
- Y. Rui, D. R. Wilson, S. Y. Tzeng, H. M. Yamagata, D. Sudhakar, M. Conge, C. A. Berlinicke, D. J. Zack, A. Tuesca and J. J. Green, Sci. Adv., 2022, 8, eabk2855 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhang, S. Li, L. Zhao, D. Li, Z. Cao, X. Xu and X. Yang, ACS Nano, 2021, 15, 16030–16042 CrossRef CAS PubMed.
- X. Zhou, X. Liu and L. Huang, Adv. Funct. Mater., 2021, 31, 2006220 CrossRef CAS PubMed.
- D. Chen, J. Xie, R. Fiskesund, W. Dong, X. Liang, J. Lv, X. Jin, J. Liu, S. Mo, T. Zhang, F. Cheng, Y. Zhou, H. Zhang, K. Tang, J. Ma, Y. Liu and B. Huang, Nat. Commun., 2018, 9, 873 CrossRef PubMed.
- F. Zhou, B. Feng, H. Yu, D. Wang, T. Wang, Y. Ma, S. Wang and Y. Li, Adv. Mater., 2019, 31, 1805888 CrossRef PubMed.
- X. Liu, Y. Pu, K. Cron, L. Deng, J. Kline, W. A. Frazier, H. Xu, H. Peng, Y. Fu and M. M. Xu, Nat. Med., 2015, 21, 1209–1215 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3bm02071a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |