
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSortase mediated protein ubiquitination with defined chain length and topology†
Nicole R.
Raniszewski
 ,
Jenna N.
Beyer
,
Myles I.
Noel
and
George M.
Burslem
*
,
Jenna N.
Beyer
,
Myles I.
Noel
and
George M.
Burslem
*
Department of Biochemistry and Biophysics, Department of Cancer Biology and Epigenetics Institute, Perelman School of Medicine, University of Pennsylvania, PA 19104, USA. E-mail: George.Burslem@Pennmedicine.upenn.edu
First published on 7th February 2024
Abstract
Ubiquitination is a key post-translational modification on protein lysine sidechains known to impact protein stability, signal transduction cascades, protein–protein interactions, and beyond. Great strides have been made towards developing new methods to generate discrete chains of polyubiquitin and conjugate them onto proteins site-specifically, with methods ranging from chemical synthetic approaches, to enzymatic approaches and many in between. Previous work has demonstrated the utility of engineered variants of the bacterial transpeptidase enzyme sortase (SrtA) for conjugation of ubiquitin site-specifically onto target proteins. In this manuscript, we’ve combined the classical E1/E2-mediated polyubiquitin chain extension approach with sortase-mediated ligation and click chemistry to enable the generation of mono, di, and triubiquitinated proteins sfGFP and PCNA. We demonstrate the utility of this strategy to generate both K48-linked and K63-linked polyubiquitins and attach them both N-terminally and site-specifically to the proteins of interest. Further, we highlight differential activity between two commonly employed sortase variants, SrtA 5M and 7M, and demonstrate that while SrtA 7M can be used to conjugate these ubiquitins to substrates, SrtA 5M can be employed to release the ubiquitin from the substrates as well as to cleave C-terminal tags from the ubiquitin variants used. Overall, we envision that this approach is broadly applicable to readily generate discrete polyubiquitin chains of any linkage type that is accessible via E1/E2 systems and conjugate site-specifically onto proteins of interest, thus granting access to bespoke ubiquitinated proteins that are not currently possible.
Introduction
The conjugation of ubiquitin, a 76-amino acid protein, to protein sidechains is one of the most common protein post-translational modifications (PTMs) with over 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 sites reported across the proteome.1 The process of ubiquitination (also commonly referred to as ubiquitylation), where the ε-amino group on a lysine (K) sidechain forms an isopeptide linkage with the C-terminal glycine of ubiquitin, is enacted by a cascade of E1 activating enzymes, E2 conjugating enzymes, and E3 ligase enzymes, and is removed by a class of proteases called deubiquitinating enzymes (DUBs).2 Ubiquitin itself can act as a substrate via one of its own seven lysines to generate polyubiquitin chains, which have varying topologies and in turn biological roles in the cell. For example, K48-linked polyubiquitin is the canonical signal for proteasomal degradation of proteins,3 K63-linked polyubiquitin facilitates the cellular DNA damage response,4 and some atypical chains (such as K6-linked polyubiquitin) have biological roles that are not yet fully understood.5 While many strides have been made in the field towards the generation of ubiquitinated substrates with enzymatic,6 fully-synthetic,7,8 and various semi-synthetic methods9,10 available, there are still challenges to generating site specific, custom polyubiquitinated proteins in a robust, high yielding, and modular manner.
000 sites reported across the proteome.1 The process of ubiquitination (also commonly referred to as ubiquitylation), where the ε-amino group on a lysine (K) sidechain forms an isopeptide linkage with the C-terminal glycine of ubiquitin, is enacted by a cascade of E1 activating enzymes, E2 conjugating enzymes, and E3 ligase enzymes, and is removed by a class of proteases called deubiquitinating enzymes (DUBs).2 Ubiquitin itself can act as a substrate via one of its own seven lysines to generate polyubiquitin chains, which have varying topologies and in turn biological roles in the cell. For example, K48-linked polyubiquitin is the canonical signal for proteasomal degradation of proteins,3 K63-linked polyubiquitin facilitates the cellular DNA damage response,4 and some atypical chains (such as K6-linked polyubiquitin) have biological roles that are not yet fully understood.5 While many strides have been made in the field towards the generation of ubiquitinated substrates with enzymatic,6 fully-synthetic,7,8 and various semi-synthetic methods9,10 available, there are still challenges to generating site specific, custom polyubiquitinated proteins in a robust, high yielding, and modular manner.
In addition to the challenges that exist for the robust generation of custom polyubiquitin chains, there are limitations to the current strategies for the site-specific attachment of polyubiquitin chains onto proteins of interest. Commonly employed approaches utilize chemistry-driven methods such as native chemical ligation (NCL)8,11,12 and thiol-alkene conjugation13,14 and endopeptidase strategies.15 Recently, the Lang group demonstrated a methodology to site-specifically ubiquitinate proteins utilizing the bacterial transpeptidase enzyme sortase A (SrtA) from Staphylococcus aureus.16 Sortase was first discovered as a key enzyme involved in bacterial cell wall assembly and has been posited to be a potential target for antimicrobial therapies,17,18 but has since also been employed in countless bioconjugation applications including self-cleaving peptides and proteins,19,20 antibody–drug conjugate assembly,21 and many more.22 Broadly, sortase utilizes its own catalytic cysteine residue to catalyze the transfer of a peptide bearing an LPXTG motif onto an N-terminal glycine-bearing peptide substrate (Scheme 1). Sortase A from Staphylococcus aureus in particular has been a prominent target for bioengineering efforts, with labs designing mutants that catalyze the transpeptidation in the absence of calcium,23 improving the catalytic activities,24 and engineering mutants with different substrate specificities.25,26
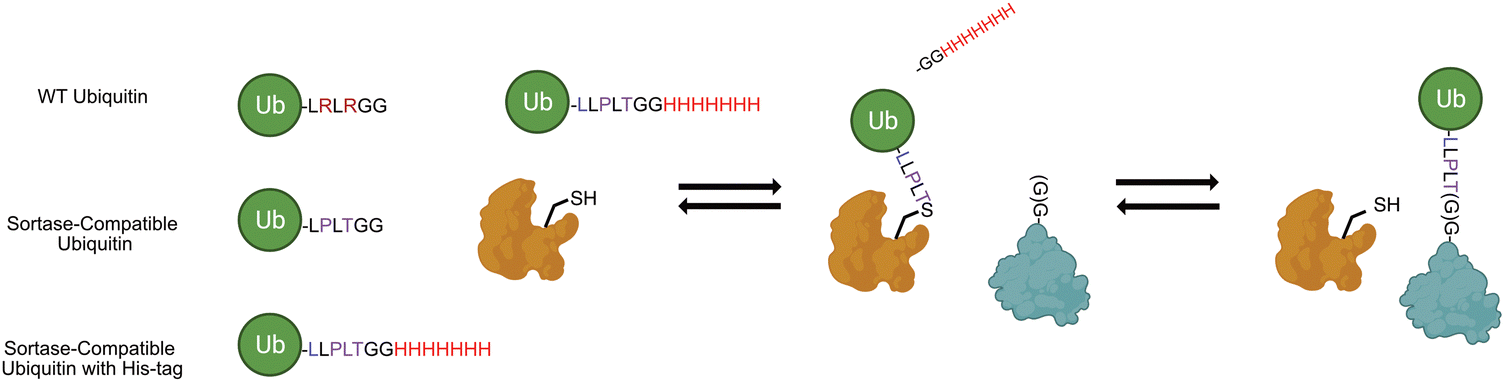 | ||
| Scheme 1 Engineered sortase bacterial transpeptidase can be used in combination with ubiquitin mutants bearing C-terminal sortase motif and His-tag (UbKXSrt-His). | ||
The approach outlined by Fottner et. al. enables expedient preparation of ubiquitinated substrates utilizing two point mutations in the C-terminal tail of ubiquitin which also renders it resistant to DUB protease activity (Scheme 1). We theorized that we could we employ this sortase system coupled with a classic E1/E2 chain extension approach to readily generate polyubiquitin chains of varying length in vitro, then subsequently select for sortase compatible chains by enrichment of a C-terminal His-tag on ubiquitin. Through this work, we successfully purified several unique polyubiquitin chains in a facile manner and conjugated them to both the N-terminal glycine and a specified internal position of substrate protein sfGFP. Further, we demonstrated the utility of this approach to remove ubiquitin from these same substrates in a sortase-dependent manner, demonstrating the modularity of this system while highlighting differential activities between SrtA 5M and SrtA 7M, two commonly employed sortase variants. Finally, to demonstrate the generality of this work, we attached K63 and K48-linked polyubiquitin to DNA replication protein proliferating cell nuclear antigen (PCNA) at position 164, a heavily modified position required for DNA translesion synthesis.27
Results and discussion
Generating sortase compatible polyubiquitin chains of discrete linkage type and length
To begin constructing the polyubiquitin chains, we first generated ubiquitin mutants which contained the C-terminal sortase recognition motif LLPLTGG (including an additional leucine insertion, reported to help sortase accessibility,16 as well as a C-terminal His-tag to aid in chain purification, notated as UbKXSrt-His). To remove ambiguity of the linkage types generated and limit chain extension, we generated these variants as single-lysine mutants of ubiquitin to serve as the C-terminal (proximal) building blocks in the polyubiquitin chain extension experiments. We hypothesized that these proximal building blocks could be used in combination with a linkage specific E1/E2 system to generate His-tagged and sortase compatible polyubiquitin chains, which could then be easily separated from the wild-type chains by immobilized metal affinity chromatography (IMAC) and purified before conjugation onto model proteins (Scheme S1, ESI†).To generate polyubiquitin chains, we generated a UbK48Srt-His construct that is proximally blocked and can only be chain extended outward from lysine 48. We reacted wild type ubiquitin (Ub WT) and UbK48Srt-His in a 3:1 ratio with UbK48Srt-His as the proximal ubiquitin to drive chain extension. For tri ubiquitin and higher order polyubiquitin chains, we found that using purified polyubiquitin chains of Ub(n-1) in lieu of monomeric Ub WT (Fig. S4, ESI†) yielded more favourable results during chain extension. As anticipated, we observed two species for each multimer of ubiquitin (Fig. 1A) corresponding to WT and Srt-compatible isoforms. Upon IMAC enrichment we were able to completely separate the His-tagged polyubiquitin species from the non-His-tagged species (Fig. 1A), thus providing facile access to substrates for sortase mediated conjugations. Previous work reports that polyubiquitin chains of different lengths can be easily purified from one another based on size and charge by cation exchange chromatography,6,28 though when we applied this to our workflow, we found that presence of the C-terminal His-tag altered the elution profiles of the chains and ultimately hindered the cation exchange purification. To combat this challenge, we explored the use of SrtA to exchange the C-terminal His-tag for a shorter motif. Pleasingly, by using SrtA 5M29 in the presence of an excess of a diglycine peptide, we were able to drive trans-amidation from the –GGHHHHHHH to –GG, restoring the original sortase recognition motif (–LLPLTGG), rather than hydrolysis (which would result in the production of sortase-incompatible UbK0-LLPLT). Indeed, upon treatment with SrtA 5M at 37 °C, we observed near complete cleavage of the His-tag from UbK48Srt-His to give UbK48Srt species in as little as 30 minutes (Fig. 1B and Fig. S1, ESI†). Interestingly, we also tested the hydrolysis and trans-amidation via the calcium-independent mutant sortase SrtA 7M23 but did not see any significant hydrolysis, even after 16 hours (Fig. S2, ESI†).
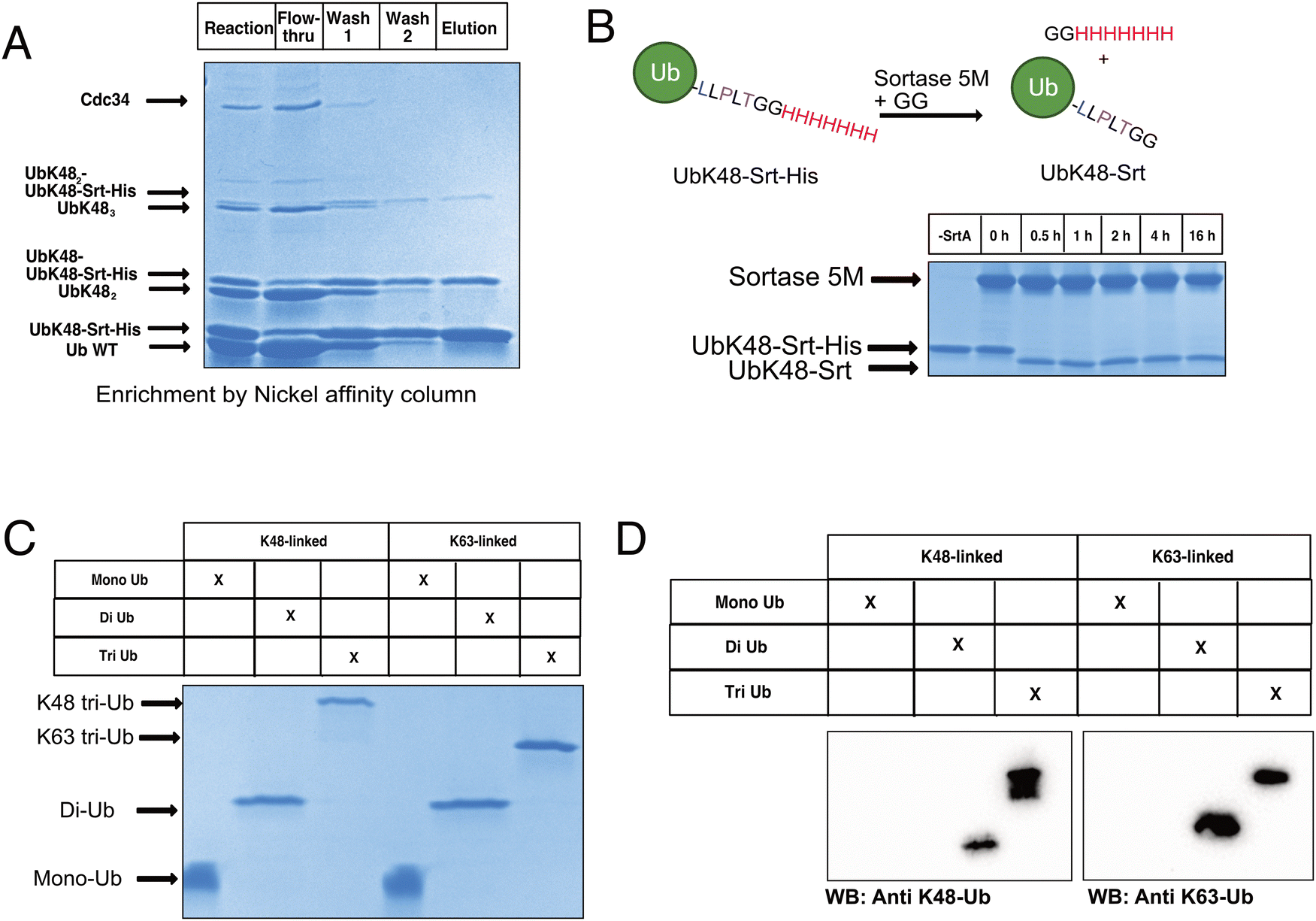 | ||
| Fig. 1 Generating sortase compatible polyubiquitin. (A) SDS-PAGE gel stained with Coomassie Blue showing IMAC purification of His-tagged polyubiquitin chains. (B) SrtA 5M-mediated cleavage of the C-terminal His-tag on UbKXSrt. (C) SDS-PAGE gel stained with Coomassie showing purification of mono, di, and triubiquitin bearing C-terminal sortase recognition motif post-cation exchange chromatography. (D) Western blots of di and triubiquitin using linkage-specific antibodies (K48-specific on left and K63-specific on right). | ||
Thus following enzymatic synthesis, IMAC and C-terminal editing, different chains could be readily separated from one another by length via cation exchange chromatography (Fig. 1C, D and Fig. S5, ESI†) with purified chains yielding 1–4% on average, which is consistent with literature reports.28 To demonstrate the generalizability of this system, we also generated K63-linked ubiquitin oligomers via the same approach (Fig. 1C, D and Fig. S5, ESI†) by using different E1/E2 enzymes.
Generating N-terminally ubiquitinated GFP
Having access to multiple sortase compatible ubiquitin chains, we sought to test the sortase mediated conjugation to a model protein with an N-terminal glycine. For this application, we cloned a superfolder green fluorescent protein bearing a V1G mutation (sfGFP-V1G) to use as the acceptor substrate. We then utilized lysine-free ubiquitin variant UbK0Srt and performed conjugation reactions at 37 °C with SrtA 7M and observed formation of the monoubiquitinated sfGFP (UbK0Srt-sfGFP) species in as little as 30 minutes, with the highest amount of UbK0Srt-sfGFP present at 16 hours (Fig. 2A and B).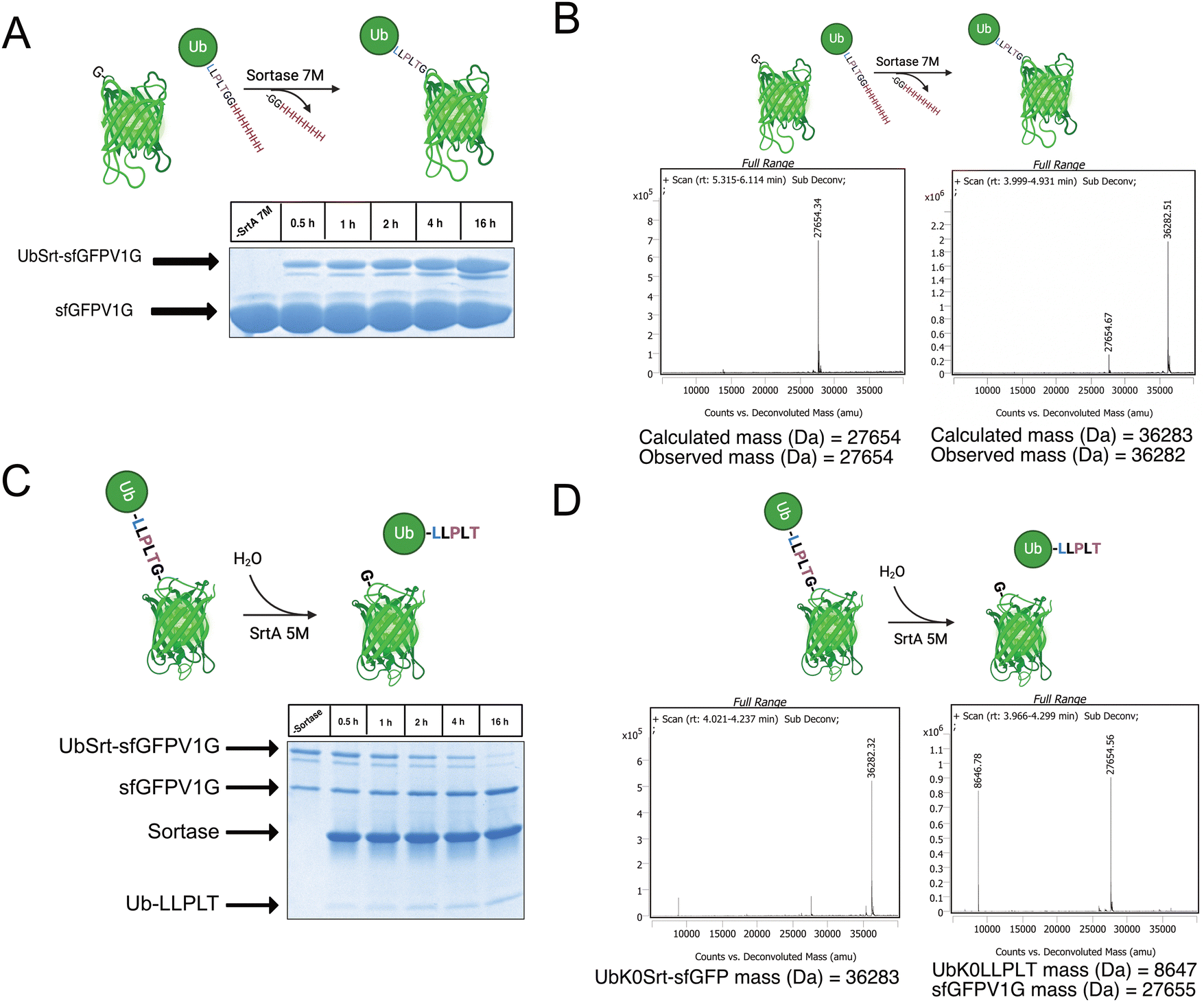 | ||
| Fig. 2 Generation of N-terminally mono and polyubiquitinated sfGFP. (A) Schematic and SDS-PAGE gel stained with Coomassie Blue showing SrtA 7M-mediated ubiquitination of sfGFPV1G over 0.5–16 hours. (B) Deconvoluted intact mass spectrometry data demonstrating N-terminal attachment of UbK0Srt to sfGFPV1G. (C) SDS-PAGE gel stained with Coomassie showing SrtA 5M-mediated hydrolysis of UbK0Srt-sfGFPV1G. (D) Deconvoluted QTOF-LC/MS data demonstrating SrtA 5M-mediated hydrolysis of UbK0Srt-sfGFPV1G to UbK0LLPLT and sfGFPV1G. | ||
Following the generation of UbK0Srt-sfGFP, we were curious about the reversibility of this reaction. After reaction of SrtA 5M with UbK0Srt-sfGFP, we observed slower hydrolysis of UbK0Srt-sfGFP than the hydrolysis of the His-tag from UbK48Srt-His (Fig. 1B), with gradual hydrolysis leading to near-complete cleavage of UbK0Srt-sfGFP after 16 hours (Fig. 2C and D). When the cleavage assays were conducted in the presence of 1 mM GG and GGG peptides, we observed a faster rate of cleavage (estimated half-life ∼2 h) by SrtA 5M for both sets of GG/GGG peptides (Fig. S3, ESI†), while we observed no cleavage of the UbK0Srt-sfGFP species after 16 h when using SrtA 7M (Fig. S2, ESI†). Further, when cleaving with SrtA 5M in the presence of GG peptide, we observed near-quantitative production of the original UbK0Srt species by LC/MS (Fig. S1, ESI†). This provided useful insight into the use of the two sortase variants for control of our experimental system.
Generating internally polyubiquitinated sfGFP
Since most ubiquitination events occur at internal lysines, we sought to use our approach to ubiquitinate an internal position of sfGFP. By using genetic code expansion (GCE) to site-specifically install para-Azido Phenylalanine (pAzF)30 into sfGFP (Fig. 3B, left), we were able to then perform strain-promoted azide–alkyne cycloaddition (SPAAC)31 with Gly-Gly-Gly-Dibenzocyclooctyne (GGG-DBCO) (Fig. 3B, right) to provide a sortase substrate as a defined internal position. Our initial trial with sfGFP-150pAzF-DBCO-GGG demonstrated that SrtA 7M could readily deposit UbK48Srt onto the internal tri-glycyl site (Fig. 3C). We then combined our sortase compatible ubiquitin chains with defined length and topology (Fig. 1C and D) with our SPAAC approach to yield internally ubiquitinated proteins bearing the desired chain type and length (Fig. 3C, D and Fig. S6, ESI†).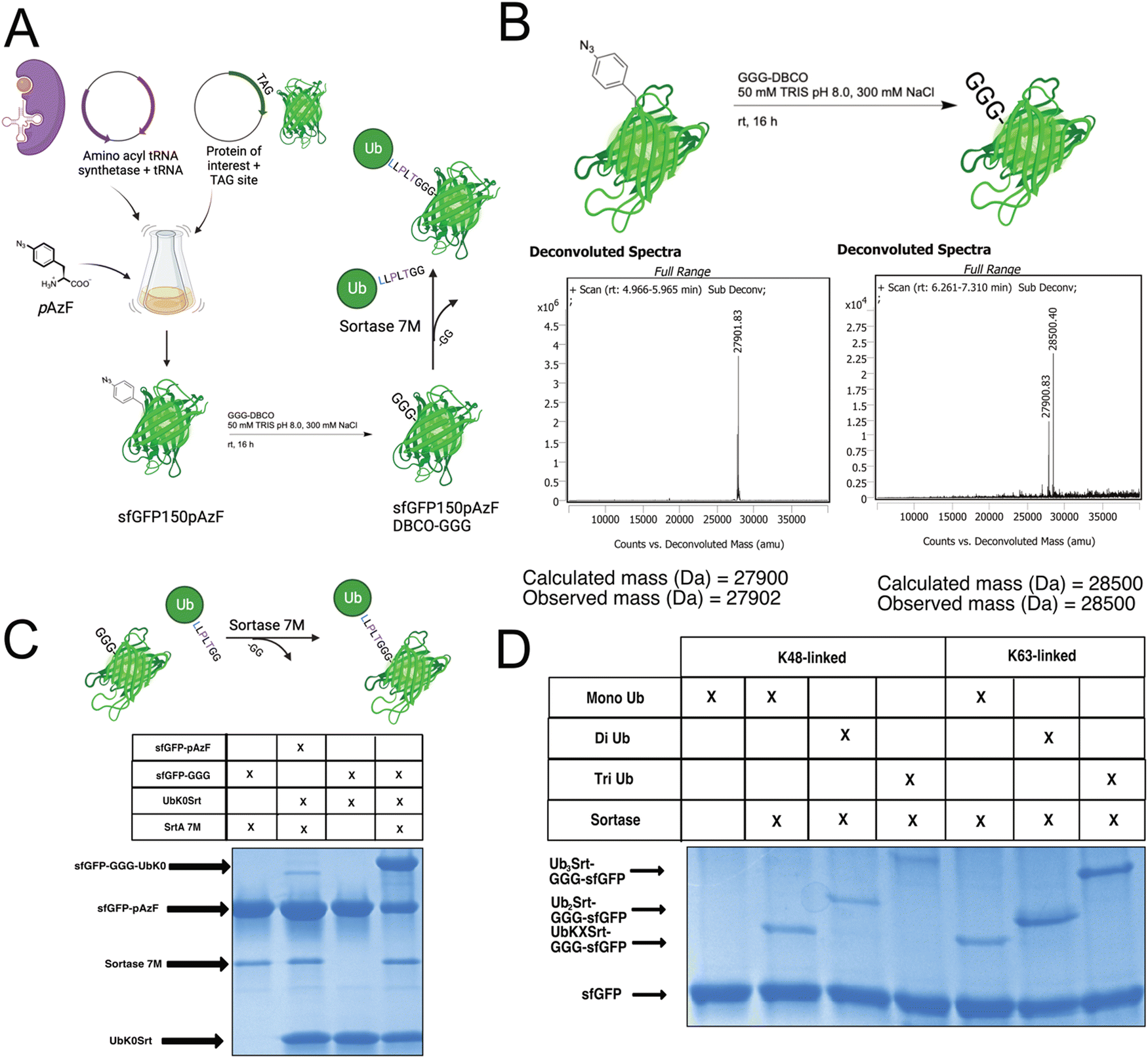 | ||
| Fig. 3 Generating internally mono and polyubiquitinated sfGFP via click chemistry and sortase. (A) Schematic of workflow for genetic code expansion, click chemistry, and ubiquitination via sortase. (B) Reaction conditions and deconvoluted intact protein mass spec showing successful GGG-DBCO attachment to sfGFP150pAzF. (C) SDS-PAGE gel stained with Coomassie showing site-specific monoubiquitination of sfGFPpAzF150-DBCO-GGG via sortylation (approx. 60% conversion based on densitometry). (D) SDS-PAGE gel stained with Coomassie Blue showing site specific attachment of K48 and K63-linked mono, di, and tri ubiquitin onto sfGFP-150pAzF-DBCO-GGG. | ||
Generating polyubiquitinated PCNA
To further exemplify the broader uses of this approach, we tested this workflow on DNA replication protein PCNA. PCNA is a protein which trimerizes to form a “clamp” around the DNA, and thus is a key protein in DNA replication as its classic function assists in recruiting and maintaining attachment of the DNA polymerase to the template DNA.32 PCNA serves critical roles in maintaining genomic integrity in cells; if the replication machinery encounters DNA damage lesions, PCNA is ubiquitinated in response and facilitates either “error-free” homologous template switching or error-prone translesion synthesis. The decision between template switching or translesion synthesis is dependent on the ubiquitination of PCNA; the former being enabled by K63-linked polyubiquitination, and the latter being enabled by mono-ubiquitination of PCNA.27 PCNA has been reported to have additional possible modes of ubiquitination,33,34 but these are understudied due to a lack of tools.To generate site-specifically (poly)ubiquitinated PCNA, we first performed GCE to incorporate the pAzF click handle into the protein (Fig. 4A and B, left). We selected K164 as the point of conjugation given its significance in DNA translesion synthesis. To do so, we mutated the K164 position to an Amber stop codon (TAG) for GCE and added a C-terminal His-tag for purification. pAzF incorporation was robust at this site, with the final PCNA product showing high fidelity incorporation of the pAzF (Fig. 4B, left and Fig. S5, ESI†). Following purification, the PCNA-164pAzF was subjected to SPAAC with GGG-DBCO to install the oligoglycine handle for sortylation (Fig. 4B). Following SPAAC, we utilized SrtA 7M to conjugate purified mono (Fig. 4C), di, and triubiquitin linked by either K48 or K63 to the PCNA trimers at site 164. Both K48-linked and K63-linked polyubiquitin were successfully conjugated onto this position and were visible by SDS-PAGE stained with Coomassie Blue (Fig. 4D) as well as Western blotting against ubiquitin, K48-linked polyubiquitin, or K63-linked polyubiquitin (Fig. 4E and Fig. S7, ESI†) thus demonstrating our approach.
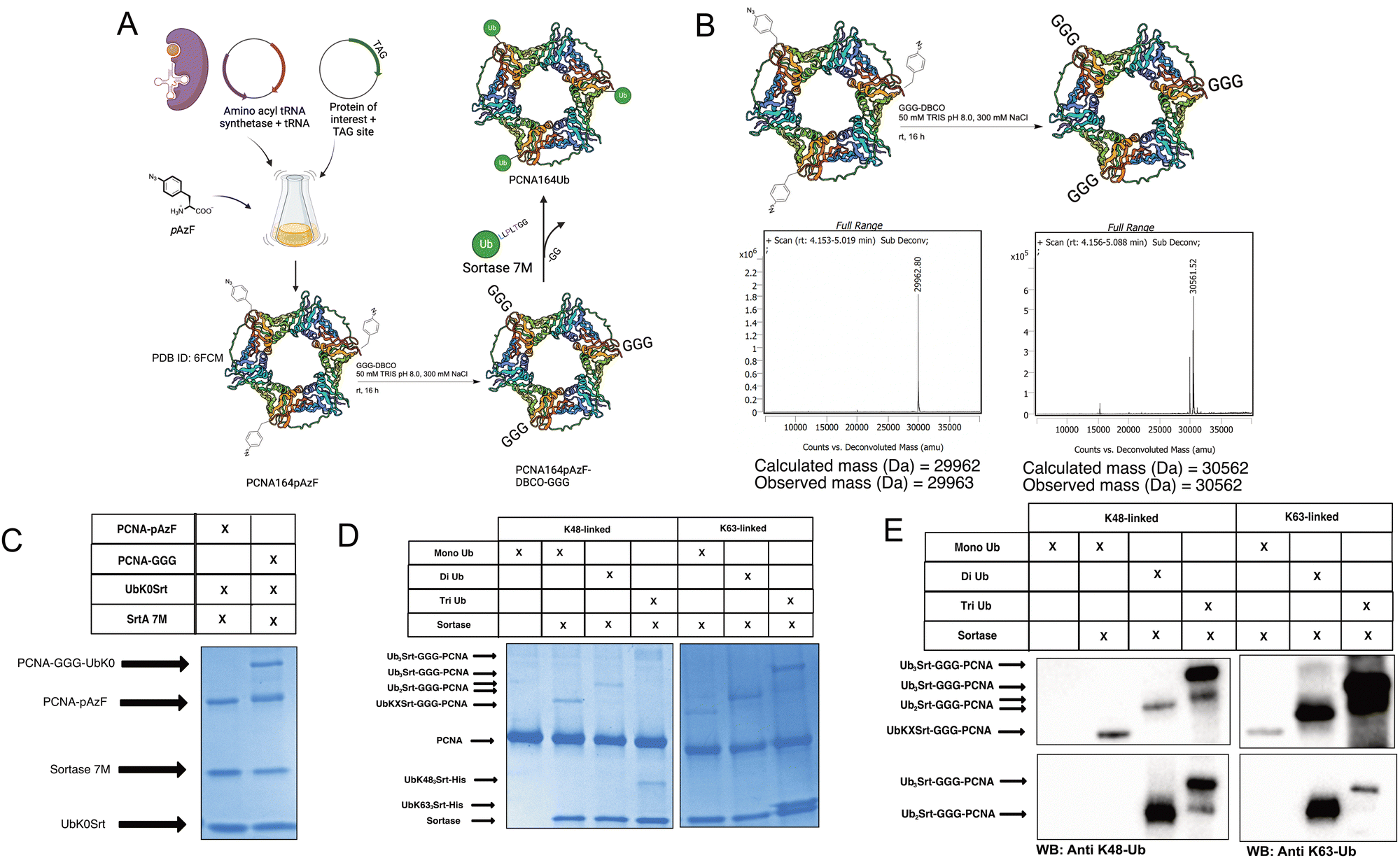 | ||
| Fig. 4 Generating site-specifically polyubiquitinated PCNA. (A) Schematic showing general workflow of generation of PCNA-164Ubn. [PDB ID: 6FCM]35 (B) Schematic and deconvoluted intact-protein mass spec showing pAzF incorporation and GGG-DBCO click chemistry. (C) SDS-PAGE gel stained with Coomassie showing click-dependent monoubiquitination of PCNA164pAzF. (approx. 60% conversion based on densitometry). (D) SDS-PAGE gel stained with Coomassie showing mono, di, and triubiquitination of PCNA with K48-linked and K63-linked polyubiquitin. (E) Western blots showing successful conjugation of polyubiquitins to PCNA164pAzF-DBCO-GGG. Immunoblot against ubiquitin (top), K48-linked polyubiquitin (bottom left), and K63-linked polyubiquitin (bottom right). | ||
Given pre-existing work in the field (outlined in ref. 16 and 19), we wanted to highlight some key features of our technology compared to what is currently published. The Lang group has previously developed and reported a system for GCE which incorporates a novel unnatural amino acid, azido-glycylglycyl lysine (AzGGK), which enables the use of sortase mediated ligation directly onto lysine residues at the position of interest on a protein. This generates an isopeptide linkage that is more akin to a native ubiquitination, which may be desirable in instances where a near-native linkage is critical to the experimental system it is being applied to. The incorporation of pAzF in our hands is very robust and while the click chemistry results in a less native linkage between the ubiquitin and protein of interest, the use of commercially available pAzF and GGG-DBCO abrogate the need for chemical synthesis of the AzGGK, making this strategy more accessible to biochemistry labs and to those who may want to avoid chemical synthesis of reagents. The Lang group has also used a derivation of this technology to generate polyubiquitin chains by generating ubiquitin dimers via and E1/E2 system and using the AzGGK/sortase approach to make longer ubiquitin chains by incorporating the AzGGK into the ubiquitin monomers themselves. While this system presents a robust approach to generate polyubiquitin chains, our strategy which utilizes wild-type ubiquitin leaves opportunity for GCE to be used in alternative ways (e.g. incorporating click or photoaffinity handles, post-translational modifications such as phosphorylation) throughout the ubiquitin chains without the complication of introducing multiple GCE sites into the ubiquitin monomers. Finally, through our blended approach using SrtA 7M for forward conjugation and SrtA 5M in the presence of diglycine for cleavage, we can recover unspent ubiquitin from the reactions by purification since the system disfavors hydrolysis of the sortase recognition motif in unconjugated ubiquitin monomers.
Conclusions
Here we present a method for the facile generation and conjugation of defined ubiquitin chains to proteins of interest, thus providing a new technology to biochemically test hypotheses surrounding ubiquitination events. In doing so, we demonstrate the utility of a transient C-terminal His-tag to facilitate enrichment and purification of sortase compatible ubiquitin chains and the application of GCE and SPAAC to conjugate bespoke chains to sites of interest. Additionally, we have demonstrated and expanded upon differing activities which have been previously noted36 between the SrtA 5M mutants and the 7M mutants and leveraged their differential activities to our benefit.Taken together, the work presented here expands on the current knowledge, use, and applications of sortase technology, as well as providing valuable methods for generating ubiquitinated protein substrates via sortase-mediated ligation. This suite of reagents and methodologies provides access to ubiquitinated proteins with greater control and homogeneity than previous approaches, which will enable new avenues for the exploration of the ubiquitin code and enable us, and others, to explore the ubiquitin code more extensively.
Author contributions
GMB: conceptualization, funding acquisition, investigation, methodology, resources, supervision, writing original draft, writing – review and editing. NRR: investigation, methodology, resources, writing original draft, writing – review and editing. JNB: investigation, resources, writing – review and editing. MIN: resources, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a research grant from the Basser Center for BRCA and the National Institutes of Health R35-GM142505 (to G. M. B.) and support for the NIH Chemistry Biology Interface Training Grant (T32 GM133398) to N. R. R. J. N. B. was supported by the NSF Graduate Research Fellowship. Figures were created using some features from Biorender.com. We are grateful to Maricela Guzman for her support of work, members of the Burslem lab for useful discussion and to Donita Brady, PhD for her critical evaluation and discussion of this manuscript.Notes and references
- V. Akimov, I. Barrio-Hernandez, S. V. F. Hansen, P. Hallenborg, A. K. Pedersen, D. B. Bekker-Jensen, M. Puglia, S. D. K. Christensen, J. T. Vanselow, M. M. Nielsen, I. Kratchmarova, C. D. Kelstrup, J. V. Olsen and B. Blagoev, Nat. Struct. Mol. Biol., 2018, 25, 631–640 CrossRef CAS PubMed.
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS PubMed.
- V. Chau, J. W. Tobias, A. Bachmair, D. Marriott, D. J. Ecker, D. K. Gonda and A. Varshavsky, Science, 1989, 243, 1576–1583 CrossRef CAS PubMed.
- R. M. Hofmann and C. M. Pickart, Cell, 1999, 96, 645–653 CrossRef CAS PubMed.
- Y. Kulathu and D. Komander, Nat. Rev. Mol. Cell Biol., 2012, 13, 508–523 CrossRef CAS.
- M. A. Michel, D. Komander and P. R. Elliott, in The Ubiquitin Proteasome System: Methods and Protocols, ed. T. Mayor and G. Kleiger, Springer, New York, New York, NY, 2018, pp. 73–84 DOI:10.1007/978-1-4939-8706-1_6.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS.
- A. Shanmugham, A. Fish, M. P. A. Luna-Vargas, A. C. Faesen, F. E. Oualid, T. K. Sixma and H. Ovaa, J. Am. Chem. Soc., 2010, 132, 8834–8835 CrossRef CAS.
- W. Gui, G. A. Davidson and Z. Zhuang, RSC Chem. Biol., 2021, 2, 450–467 RSC.
- G. M. Burslem, Biochim. Biophys. Acta, Gen. Subj., 2022, 1866, 130079 CrossRef CAS PubMed.
- K. A. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon and A. Brik, Angew. Chem., Int. Ed., 2011, 50, 6137–6141 CrossRef CAS PubMed.
- S. Tang, L.-J. Liang, Y.-Y. Si, S. Gao, J.-X. Wang, J. Liang, Z. Mei, J.-S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 13333–13337 CrossRef CAS PubMed.
- R. Meledin, S. M. Mali, S. K. Singh and A. Brik, Org. Biomol. Chem., 2016, 14, 4817–4823 RSC.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y.-C. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS PubMed.
- M. Fottner, J. Heimgärtner, M. Gantz, R. Mühlhofer, T. Nast-Kolb and K. Lang, J. Am. Chem. Soc., 2022, 144, 13118–13126 CrossRef CAS PubMed.
- M. Fottner, A. D. Brunner, V. Bittl, D. Horn-Ghetko, A. Jussupow, V. R. I. Kaila, A. Bremm and K. Lang, Nat. Chem. Biol., 2019, 15, 276–284 CrossRef CAS PubMed.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760–763 CrossRef CAS PubMed.
- H. Abujubara, J. C. J. Hintzen, S. Rahimi, I. Mijakovic, D. Tietze and A. A. Tietze, Chem. Sci., 2023, 14, 6975–6985 RSC.
- H. Mao, S. A. Hart, A. Schink and B. A. Pollok, J. Am. Chem. Soc., 2004, 126, 2670–2671 CrossRef CAS PubMed.
- H. H. Wang, B. Altun, K. Nwe and A. Tsourkas, Angew. Chem., Int. Ed., 2017, 56, 5349–5352 Search PubMed.
- W. Yu, K. P. Gillespie, B. Chhay, A.-S. Svensson, P.-Å. Nygren, I. A. Blair, F. Yu and A. Tsourkas, Bioconjugate Chem., 2021, 32, 1058–1066 CrossRef CAS PubMed.
- F. Tian, G. Li, B. Zheng, Y. Liu, S. Shi, Y. Deng and P. Zheng, Chem. Commun., 2020, 56, 3943–3946 RSC.
- H. Hirakawa, S. Ishikawa and T. Nagamune, Biotechnol. J., 2015, 10, 1487–1492 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11399–11404 CrossRef CAS PubMed.
- B. M. Dorr, H. O. Ham, C. An, E. L. Chaikof and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13343–13348 CrossRef CAS.
- Z. A. Wang, S. D. Whedon, M. Wu, S. Wang, E. A. Brown, A. Anmangandla, L. Regan, K. Lee, J. Du, J. Y. Hong, L. Fairall, T. Kay, H. Lin, Y. Zhao, J. W. R. Schwabe and P. A. Cole, J. Am. Chem. Soc., 2022, 144, 3360–3364 CrossRef CAS PubMed.
- L. Fan, T. Bi, L. Wang and W. Xiao, Biochem. J., 2020, 477, 2655–2677 CrossRef CAS PubMed.
- K. Martinez-Fonts and A. Matouschek, Biochemistry, 2016, 55, 1898–1908 CrossRef CAS PubMed.
- M. Fottner, M. Weyh, S. Gaussmann, D. Schwarz, M. Sattler and K. Lang, Nat. Commun., 2021, 12, 6515 CrossRef CAS PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- M. O’Donnell, L. Langston and B. Stillman, Cold Spring Harbor Perspect. Biol., 2013, 5, a010108 Search PubMed.
- K. N. Choe and G.-L. Moldovan, Mol. Cell, 2017, 65, 380–392 CrossRef CAS PubMed.
- S. Zhao and H. D. Ulrich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7704–7709 CrossRef CAS PubMed.
- D. Ohayon, A. De Chiara, P. M.-C. Dang, N. Thieblemont, S. Chatfield, V. Marzaioli, S. S. Burgener, J. Mocek, C. Candalh, C. Pintard, P. Tacnet-Delorme, G. Renault, I. Lagoutte, M. Favier, F. Walker, M. Hurtado-Nedelec, D. Desplancq, E. Weiss, C. Benarafa, D. Housset, J.-C. Marie, P. Frachet, J. El-Benna and V. Witko-Sarsat, J. Exp. Med., 2019, 216, 2669–2687 CrossRef CAS.
- N. Pishesha, J. R. Ingram and H. L. Ploegh, Annu. Rev. Cell Dev. Biol., 2018, 34, 163–188 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed methods, supplemental figures, and construct sequences. See DOI: https://doi.org/10.1039/d3cb00229b |
| This journal is © The Royal Society of Chemistry 2024 |