
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unlocking novel therapies: cyclic peptide design for amyloidogenic targets through synergies of experiments, simulations, and machine learning
Daria
de Raffele
ab and
Ioana M.
Ilie
 *ab
*ab
aUniversity of Amsterdam, van 't Hoff Institute for Molecular Sciences, Science Park 904, P.O. Box 94157, 1090 GD Amsterdam, The Netherlands. E-mail: i.m.ilie@uva.nl
bAmsterdam Center for Multiscale Modeling (ACMM), University of Amsterdam, P.O. Box 94157, 1090 GD Amsterdam, The Netherlands
First published on 7th December 2023
Abstract
Existing therapies for neurodegenerative diseases like Parkinson's and Alzheimer's address only their symptoms and do not prevent disease onset. Common therapeutic agents, such as small molecules and antibodies struggle with insufficient selectivity, stability and bioavailability, leading to poor performance in clinical trials. Peptide-based therapeutics are emerging as promising candidates, with successful applications for cardiovascular diseases and cancers due to their high bioavailability, good efficacy and specificity. In particular, cyclic peptides have a long in vivo stability, while maintaining a robust antibody-like binding affinity. However, the de novo design of cyclic peptides is challenging due to the lack of long-lived druggable pockets of the target polypeptide, absence of exhaustive conformational distributions of the target and/or the binder, unknown binding site, methodological limitations, associated constraints (failed trials, time, money) and the vast combinatorial sequence space. Hence, efficient alignment and cooperation between disciplines, and synergies between experiments and simulations complemented by popular techniques like machine-learning can significantly speed up the therapeutic cyclic-peptide development for neurodegenerative diseases. We review the latest advancements in cyclic peptide design against amyloidogenic targets from a computational perspective in light of recent advancements and potential of machine learning to optimize the design process. We discuss the difficulties encountered when designing novel peptide-based inhibitors and we propose new strategies incorporating experiments, simulations and machine learning to design cyclic peptides to inhibit the toxic propagation of amyloidogenic polypeptides. Importantly, these strategies extend beyond the mere design of cyclic peptides and serve as template for the de novo generation of (bio)materials with programmable properties.
 Daria de Raffele | Daria De Raffele is a postdoctoral researcher in the group of Dr Ioana M. Ilie. She obtained her PhD from Universitat Jaume I where she focused on the QM/MM theoretical study of enzyme promistuity. Her research focuses on the design of cyclic peptides to inhibit the toxic propagation of the prion protein. |
 Ioana M. Ilie | Dr Ioana M. ILIE is an Assistant Professor in the Computational Chemistry group at the University of Amsterdam. The research in her group focusses on multiscale simulations of biomolecular systems. In particular, her team relies on rational design and multiscale simulations to (1) understand and control the aggregation mechanisms of polypeptides and their response to the biological environment, (2) to design of peptide-based therapeutics for neurodegenerative diseases and (3) to develop smart (bio)materials with responsive properties. |
1 Introduction
Neurodegenerative disorders, such as Alzheimer's, Parkinson's and Creutzfeld-Jakob disease, affect over 50 million people world-wide, with over 10 million new cases a year. The world health organization projects that by 2040, neurodegeneration will become the second leading cause of death after cardiovascular disease.1 The associated polypeptides are intrinsically disordered (IDP) or rich in intrinsically disordered regions (IDRs), characterized by the lack of stable secondary structures in the native state.2 What they all share is the ability to accumulate in liquid-like membraneless organelles and/or form insoluble solid-like aggregates (e.g. oligomers, amyloid fibrils), which can alter protein functionality.3–7Various approaches have been developed to interfere with the accumulation processes by stabilizing or eliminating specific monomeric or aggregated forms of the responsible polypeptides.8,9 They rely primarily on the design of small molecules or antibodies that bind to monomeric or aggregated protein species, thereby making the substrate unavailable for conversion10 and/or sterically interfering with the aggregation process.9,11–13 Traditional small molecule drugs and protein-based therapeutics have made good contributions, yet their limitations in terms of selectivity, stability, and bioavailability,14,15 as well as their repeated failure in clinical trials16,17 have inspired the search for alternative therapeutic approaches. Among these, peptides and particularly cyclic peptides are attracting considerable attention due to their unique structural properties and diverse biological activities.18,19 Their cyclic nature confers enhanced stability and resistance to proteolytic degradation, while maintaining a robust binding to the target.20 Cyclic peptides have proven to be excellent candidates for cancer therapy,21 organ transplantation22 and inhibition of amyloid aggregation.23,24 Their size and functional properties ensure that the contact area is large enough to provide high selectivity,25 their ability to form salt-bridges and hydrogen bonds can lead to strong binding affinities,26 and cyclization increases their proteolytic stability.27
Amyloid-forming polypeptides, such as amyloid-β (Aβ42), α-synuclein (α-syn) and amylin (hIAPP), share the intrinsic disorder independently of their size or residue sequence. The cellular prion protein (PrPC) consists of a membrane-anchored ordered globular domain composed of three α-helices and a two stranded anti-parallel β-sheet preceded by a 100 residue unstructured flexible tail. Despite the well-defined secondary structure in its monomeric form, the cellular prion protein lacks a specific binding site accessible to potential small molecule inhibitors.8,28,29 Due to their properties, cyclic peptides can selectively intervene in the folding and aggregation process, bind even to targets lacking an easily accesible druggable pocket30 or heterogeneous and dynamic species,31 regulate the conformational stability of the target polypeptide and potentially halt or slow down disease onset or progression. Furthermore, the stability and permeability of cyclic peptides enable them to cross the blood–brain barrier,32 a crucial requirement for effective neurodegenerative disease therapies.
Advances in peptide synthesis techniques, combinatorial chemistry, and computational tools allow the de novo design and tuning of the structural elements, target specificity, binding affinities, solubility, cell permeability and proteolytic stability of natural and synthetic cyclic peptides. De novo design of cyclic peptides often rely on protein engineering strategies, such as rational design and directed evolution, which aid in the discovery and/or improvement of peptides for drug-related applications.33 Over the past years, computer simulations and machine learning enabled the exploration of a vast chemical space, accelerating the design and optimization of lead peptidomimetic candidates.34,35 Combined with directed evolution, they are versatile tools that enable an initial in silico screening step to scan the full combinatorial libraries and proposed mainly small molecules to be tested in vitro.36 While most of these models are trained on experimental data, more recently machine learning combined with molecular dynamics simulations successfully proposed, optimized and reduced the number of chemical compounds to be tested experimentally at a later stage.37 In contrast to small molecules and protein optimization, the use of machine learning for de novo peptide design is still in its early stages38–40 and its potential has been demonstrated mainly in non-therapeutic applications.39
In this paper, we provide an overview of the recent advancements of the utilization of cyclic peptides as therapeutic or imaging agents for neurodegenerative diseases, particularly focusing on the amyloid-β peptide, α-synuclein, amylin and the cellular prion protein. We emphasize on the importance of the synergy between computer simulations and experiments in light of the latest developments in machine learning for cyclic peptide design and optimization. Additionally, we provide a recipe for a potential approach to capitalize on the predictive power and results from computer simulations and AI in the development of cyclic peptide-based therapeutics.
2 Anti-amyloid therapeutic agents
2.1 Small molecules and antibodies
To date, extensive research efforts have been dedicated to the development and advancement of small molecules and antibodies targeting neurodegenerative targets into clinical trials.16,17,43,44 Small molecules have a low molecular weight (<900 Da) and hydrophobicity, and can therefore more readily traverse cell membranes and distribute throughout the body.45 They offer advantages such as oral administration and scalability for mass production.46 Nevertheless, they often bind to rigid targets with accessible druggable pockets, i.e. active sites or cavities on the surface of a protein with well defined structure that can accommodate small molecules. Proteins and peptides associated with neurodegenerative disorders are often intrinsically disordered and do not possess a well defined structure in their native state,2,47,48 which prevents the existence of druggable pockets and implicitly access to long-lived cavities for small-molecule binding. Despite their shortcomings, small molecules have been at the forefront of drug development against amyloid-β42, α-synuclein, amylin and prion protein condensation. Particularly, natural products and their degradation products were shown to alter the aggregation of the target polypeptide or modulate its toxic behavior49–64 (Fig. 1(a)). One example comes from curcumin, which is a natural product, that has various benefits such as preventing amyloid formation and promoting the formation of “off-pathway” soluble oligomers and prefibrillar aggregates;65,66 it can also disrupt Aβ40 fibrils and break down tau tangles.67,68 Additionally, the degradation products of curcumin – ferulic acid and vanillin – have a better solubility than curcumin. Ferulic acid can destabilize Aβ40 and Aβ42 fibrils69 and vanillin can prevent amyloid aggregation in living organisms.70 Likewise cholic acid, a primary bile acid, presents an anti-amyloidogenic behavior, inhibiting amyloid formation and preventing secondary nucleation.59 Vitamin k3, known for its anti-inflammatory and anti-cancer properties, inhibits the Aβ42 aggregation process and has a positive impact on reducing cytotoxicity in human neuronal cell line.62 We refer the interested reader to a comprehensive review of small molecule inhibitors for amyloid-β71 for amyloidogenic polypeptides in general.72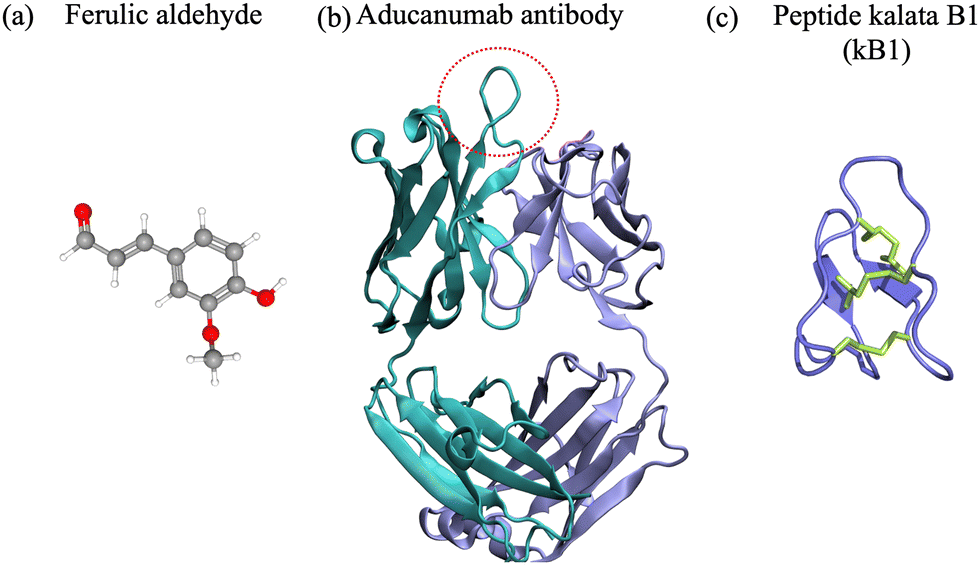 | ||
| Fig. 1 (a) Ferulic aldehyde (MW 194 Da) inhibits the Aβ42 multimerization. (b) Cartoon representation of the Aducanumab antibody (PDB ID: 6CO341) (MW 146 kDa). Highlighted are the heavy chain (cyan), light chain (light blue). The area enclosed by the red circle represents the binding interface between the antibody and the N-terminus of the Aβ42 peptide. (c) Naturally-occurring cyclotide kalata B1 (MW 2.92 kDa), derived from residues 306–311 of tau.42 | ||
Antibodies are Y-shaped proteins of larger molecular weight than small molecules (>150 kDa), which can recognize and bind to protein targets with high specificity and modulate their toxic behavior.73 In particular, monoclonal antibodies have been designed both for therapeutic and diagnostic applications. They bind to amyloidogenic polypeptides and/or their aggregates to stabilize a desired conformations and make the substrate unavailable for conversion10,43 and/or sterically interfere with the aggregation process.11–13 For instance, the DesAbs single-domain antibodies targeting Aβ42 epitopes74 interact with the monomeric peptide, bind with high affinity to the oligomeric species, but not the fibrillar structures, can inhibit secondary nucleation12 and suppress Aβ42-mediated toxicity in C. elegans.74 Aducanumab and lecanemab, approved anti-Aβ42 agents, are monoclonal antibodies effective for patients in the early stages of AD due to their ability to reduce amyloid deposits in the brain.43 Aducanumab (Fig. 1(b)) binds at the N-terminal residues 3 to 7 and can discriminate between monomers and aggregated species,44 while lecanemab binds to soluble Aβ42 protofibrils.75 Cinpanemab and prasinezumab, two monoclonal antibodies directed against α-synuclein aggregates failed in clinical trials due to the lack of positive effects in disease progression.16,17 The POM-family of antibodies (POMologues) has been developed to recognize a variety of epitopes along the sequence of the cellular prion protein10 and modulate its toxic effects.76 Notwithstanding, no drug against prion diseases is currently in clinical trials. Despite the recent success in the Alzheimer's field with aducanumab43 and the potential of gantenerumab,77 antibodies have limitations as therapeutics, including stability and immunogenicity,14,15 which can impact clinical efficiency.
2.2 Cyclic peptides
Cyclic peptides are naturally occurring or chemically synthesized macrocycles consisting of circular sequences of amino acids,78–81Fig. 1(c). Compared to small molecules, they can bind larger, more polar and solvent exposed protein surfaces.20 To mimic naturally occurring cyclic peptides, the macrocycles can be experimentally synthesized from linear precursors by connecting their N- and C-terminal residues via covalent bonds (head-to-tail cyclization).82 Chemically, cyclization can be achieved through lactamization or via disulfide bond formation ensuring the link between the two termini of the linear precursor.83 Head-to-tail cyclization restricts the dynamics of a peptide and can stabilize the formation of β-hairpins. Alternative approaches to favor other conformations such as α-helices can be achieved via stapling, i.e. via cross-linking of two or more side-chains. We refer the interested reader to detailed reviews on the synthesis of cyclic peptides84 and stapled peptides,85,86 and focus in the following paragraphs on head-to-tail cyclized peptides.Because of their physicochemical properties, cyclic peptides present a series of advantages as compared to their linear precursors, small molecules and biological therapeutic agents such as antibodies. First, the rigidity obtained through cyclization provides increased stability, higher resistance to proteolysis27 and enhanced cell permeability as compared to linear peptides.20,87,88 Second, their size and functional properties ensure that the contact area is large enough to provide high selectivity, and their ability to form salt-bridges and hydrogen bonds can lead to strong binding affinities.26 Hence they can maintain a robust antibody-like binding to (undruggable) interfaces with high affinity,20,79,89 due to their larger surface and implicitly the higher number of hydrogen bonding partners. Third, cyclic peptides have good in vivo stability, which contributes to enhanced retention and circulation, particularly if they are rich in non-canonical amino acids.20
Cyclic peptide-based therapeutics also face a series of challenges. Orally administered cyclic peptide-rich drugs struggle with poor oral bioavailability,78 because of the susceptibility of cyclic peptides to resist proteolytic degradation in the gastrointestinal tract.27 Nevertheless, different routes of administration, such as subcutaneous or via intravenous injections, overcome these difficulties and aid in the efficient delivery of the peptide-drug to the target.84 Another obstacle involves preventing off-target interactions, a challenge often tackled by selectively modifying natural amino acids in the sequence to non-natural ones.84
In the amyloid world, the cyclic peptide development has been growing over the past decade.30,78,80 Typical methods involve designing peptides rich in aromatic moieties, hydrophobic amino acids, or D-amino acids (due to the stereoselectivity for L-amino acids of proteases) that disrupt the aggregation process, i.e. β-breakers or agents that bind to monomeric and oligomeric species, competing with the responsible polypeptide to hinder its aggregation and/or toxic transformation.90 For instance, the RD2D3 D-peptide (H-ptlhthnrrrrrrprtrlhthrnr-NH2†), designed to modulate the binding of PrPC to Aβ42 oligomers, interferes with Aβ42-PrPC heteroassembly in a concentration-dependent manner.91 Its cyclic successor presents better in vitro potency and pharmakinetic properties92 and could potentially alter Aβ42 aggregation. The bicyclic DesBP peptide (RAACKLGIKACTSVYHACGGKRR) was rationally designed to bind monomeric Aβ42 at residues 31–36 and 38–4224,93 and was shown to alter the morphology of Aβ42 aggregates in a dose dependent manner. In particular, higher peptide concentrations lead to increased aggregate disorder and reduced cytotoxicity.93 Similarly, the BD1 cyclic peptide (O-ySGLIKWTTALLRTYC-NH2) was shown to inhibit α-synuclein fibril formation in vitro.94 The D,L-α-peptide CP-2 cyclic peptide (IJwHsK‡) prevents α-syn aggregation into toxic oligomers by an “off-pathway” mechanism.95 Particularly, it interacts with the N-terminus and the non-amyloidogenic region, altering the protein's membrane interaction properties and fibril morphologies, thereby preventing the toxic membrane disruption. The macrocyclic inhibitory peptides (MCIPs), were designed to bind to amyloids by mimicking human IAPP (hIAPP) interaction surfaces while maintaining only minimal hIAPP-derived self-/cross-recognition elements.96 Inhibitor selectivity was tuned by chirality, which lead to nanomolar binding affinities to hIAPP, to both amyloid-β40 and amyloid-β42 peptides, high proteolytic stability in human plasma and human brain–blood-barrier crossing ability.96 Also, disulfide-rich macrocyclic peptides are versatile scaffolds for stable biochemical tool development. Two examples are SFTI-1 (GRCTKSIPPICFPD, disulfide connectivity: Cys3–Cys11), a cyclic peptide that inhibits trypsin, and the kB1 cyclotide (GLPVCGETCVGGTCNTPGCTCSWPVCTRN, disulfide connectivity: Cys5–Cys19, Cys9–Cys21 and Cys14–Cys26), which have an inherent ability to inhibit the fibril growth of the tau-derived hexapeptide Ac-VQIVYK-NH2 (AcPHF6).42 Particularly, kB1 is a stronger inhibitor of tau fibrillizatiom than SFTI-1, enabling better binding and/or disruption of AcPHF6 fibrils. Recently, tau mimetic peptides (β-bracelets) have been designed starting from the high-resolution structure of the tau fibril fold by extracting β-strand sequences linked by β-arcs.97 The newly generated peptides self-assemble into parallel β-sheet fibrils and can serve as templates for the design of soluble inhibitors of tau seeding.
In terms of the cellular prion protein, no progress has been made on the therapeutic cyclic peptide market, despite its well defined secondary structure in the soluble form. Potential causes are the lack of druggable pockets or a stable unique binding region in the globular domain, and the intrinsically disordered nature of the tail. Though, the existence of monoclonal antibodies that bind in the nanomolar regime to PrPC indicate that putative interaction sites are available.10 We hypothesise that the rational design of cyclic peptides starting from available high resolution structures of PrPC in complex with monoclonal antibodies may serve as starting points for the design of cyclic peptides that can potentially stabilize the soluble isoform of the protein and therefore prevent its toxic transformation. Alternatively, by tweaking the environmental conditions through mild solvent alteration, e.g. by replacing water with D2O98 or by adding organic compounds,99,100 one can delicately alter the conformational landscape of the protein to reveal new (allosteric) druggable pockets without disturbing the protein's secondary structure. We refer the interested reader to a series of reviews on peptide-based strategies to interfere with protein misfolding and aggregation,101,102 a review on the therapeutic potential of cyclic90 and bicyclic peptides.103 Studies older than 10 years focusing on anti-amylin cyclic peptides and peptide-based inhibitors have been reviewed elsewhere.104
3 Design methods and pitfalls
3.1 Conventional peptide design approaches
To design a soluble peptide-based binder with simultaneously high target specificity, binding affinity, cell permeability and proteolitic stability requires prior knowledge of the molecular target and its environmental conditions. Experimentally, genetically encoded methods such as phage display105 or mRNA display106,107 allow the generation of libraries of cyclic peptides that bind with high affinity to the target.81,108 While these libraries offer the generation of a vast array of molecules, the chemical synthesis step as well as the numerous experimental trials are time and resource consuming. Furthermore, translating cyclic-peptide hits obtained through display technologies into clinical applications has proven challenging due to potential shortcomings in their pharmacological properties, including limited oral bioavailability, cell permeability, and solubility.108 Other approaches, such as directed evolution mimic the natural evolution process of a peptide by creating a diverse library to screen for mutants with improved characteristics.109 Directed evolution does not require information on the structure–function relationship of the substrate, and relies on an iterative procedure of random mutations and artificial selection to discover new and useful proteins, but is limited by the exhaustive pool of possibilities to be tested.Over the past years, rational design approaches for de novo peptide design have gained momentum. Rational design relies on a detailed understanding of the amino acid sequence, protein high resolution structure, function and interaction mechanisms.33 It involves the identification and mutation of key residues associated with protein stability to improve targeted physical and catalytic properties.33,110–112 Rational design relies on human intervention, which often offers an informed and efficient means to narrow down the search space for amino acids, resulting in a smaller and more manageable pool of effective peptides. De novo rational cyclic peptide design requires (a) high resolution three dimensional structures and biochemical/biophysical information of the target protein, and/or (b) detailed information of the ligand properties (e.g. hydrogen bonding abilities to the target, hydrophobicity, cyclization chemistry, existence of natural and non-natural residues) and conformations (i.e. the designed peptide may assume different conformations in the bound and unbound states).113 Recent advancements in cryo-electron microscopy (cryo-EM) have enabled the determination of the three-dimensional (3D) high resolution structures of new amyloidogenic aggregates and their monomeric precursors.114 These 3D structures corroborated with a comprehensive understanding of molecular interactions and structure–function relationships could enable the rational design of (cyclic) peptides tailored for amyloidogenic targets. As a matter a fact, rational design has been successfully used to generate the DesAbs antibodies targeting amyloid-β12 or specific α-synuclein and hIAPP epitopes.115
Starting from the high resolution structures of amyloid fibrils of tau, α-synuclein, and amyloid-β, miniproteins, ranging from 35 to 48 residues, were successfully designed to bind to the fibrillar tips of the targets and inhibit aggregation in in vitro and in vivo.116 First a library of peptide-based inhibitors was created using Rosetta. Subsequently, Rosetta's MotifGraft protocol117 was used to dock the inhibitors onto the fibrils and energy minimized. The top-ranking inhibitors, i.e. the best binders, were subjected to molecular dynamics simulations to assess the stability of the complexes. Lastly, Rosetta's ab initio structure prediction algorithm118 was employed for the final screening of inhibitors. Inhibitors with the most favorable energy predictions and the smallest root mean squared deviations from the original design were selected for experimental validation.
From a computational perspective, virtual screening allows fast screening of millions of compounds prior to experimental testing, thereby reducing cost and saving time. Virtual screening using cyclic peptides is limited by the availability of three-dimensional structures of the targets, by the absence of druggable pockets and by the lack of information on the structure of the designed cyclic peptide. To overcome some of the limitations, different computational techniques have been combined with machine learning to predict protein structures and complexes thereof. Notable examples include HADDOCK (High Ambiguity Driven protein–protein Docking),119–121 RosettaFold122 and AlphaFold2.123,124 HADDOCK uses biochemical and biophysical interaction data, such as nuclear magnetic resonance titration experiments or mutagenesis data, to facilitate the protein–protein docking process.119 Recent developments include the generation of cyclic peptide conformations and docking to the protein target using knowledge of the binding site on the protein side to drive the modeling.125 AlphaFold2 is a deep-learning algorithm that incorporates neural network architectures inspired by the physical and geometric aspects of protein structures.126 It employs insights from evolutionary conservation through the analysis of multiple sequence alignments. These alignments are generated by considering information from evolutionary related proteins, along with the 3D coordinates of a few homologous structures known as templates. Similarly, RoseTTAFold also utilizes multiple sequence alignments and a set of initial templates to accurately predict folded structures122 and protein–protein complexes.40,122 These technological advancements contribute significantly to the prediction protein structures through computational means. The intrinsic disorder associated with amyloidogenic polypeptides implies that the target protein lacks a stable structure and that its native state is better described by a diverse conformational ensemble rich in disordered structures.2,127,128 In this context, AlphaFold2 fails to predict such conformations, which often gives rise to unrealistic structures that do not accurately capture the states in the ensemble127–129 (Fig. 2). The lack of realistic and physically accurate ensembles of structures hampers the design of any type of inhibitor, which represents a limitation of these novel deep learning techniques.
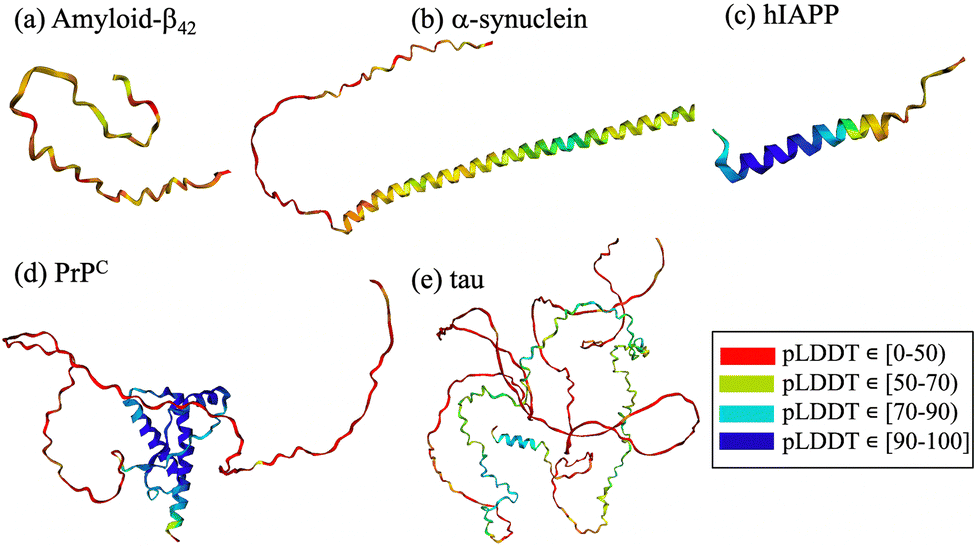 | ||
| Fig. 2 AlphaFold2 predictions for (a) amyloid-β42, (b) α-synuclein, (c) hIAPP, (d) PrPC and (e) tau. The structural elements are color-coded according to the confidence level of their AlphaFold2 prediction, red to blue for low to high confidence intervals, respectively. | ||
3.2 Molecular dynamics simulations in peptide design
Computer simulations are powerful tools that enable the generation of quantitative conformational ensembles for the target (intrinsically disordered) protein with properties comparable to experimental results.127,128,130 Moreover, molecular dynamics simulations can go beyond experimental resolution to provide valuable insights into the stability and dynamics of cyclic peptides,131 structural detail on their membrane permeability,132 as well as quantitative distributions of their target-bound and target-unbound states.128Recently extensive molecular dynamics simulations at full atomistic resolution (Table 1) have been used to successfully identify transient monomeric Aβ42 conformations that have characteristics of fibrillar structures.133 States of the monomeric, dimeric, oligomeric and fibrillar amyloidogenic polypeptides have been thoroughly characterized and have been reviewed elsewhere.2,128,130 The identified pool of structures could be potentially used for small molecule or cyclic peptide docking and design. Ideally, access to a well organized, reliable, and consistently maintained database of molecular dynamics trajectories of amyloidogenic polypeptides would avoid the repeated generation of similar trajectories and enable more rapid and consistent advancement in amyloid-related drug discovery. Example of such a publicly available database is the Molecular Dynamics Data Bank. The European Repository for Biosimulation Data.
For small molecule docking, snapshots from molecular dynamics simulations of the Aβ42 monomer,134 dimer50 or multimers52,135 have been clustered to generate representative ensembles to be prepared for docking, which can be experimentally validated.62 Briefly, curcumin and a set of curcumin derivatives were docked onto Aβ42 multimeric conformations generated with molecular dynamics simulations.50,52 Results revealed that the small molecules interact with high probability with the amyloidogenic driving domains 16KLVFF20 and 29GAIIG33 of Aβ42 and disrupt their secondary structure in the hexameric52 and dimeric arrangements.50 Interestingly, Silybin A (Sil A) and Silybin B (Sil B), two diasteroisomers of silibinin were shown to have different interaction preferences to Aβ40 and distinct biological response.51 Sil A binding the aromatic residues F19 and F20 slowed down aggregation, while Sil B interacting primarily with the C-terminus of the polypeptide fully abolished amyloid aggregation. Compelling evidence suggests that Silybin B is a powerful inhibitor also against the toxic self-assembly of hIAPP.53 Simulation and experimental work, revealed that the frequent interactions of Sil B with the S20–S29 sequence induces disorder in the amyloidogenic core and attenuates hIAPP toxicity and aggregation propensity.53 Myricetin, another polyphenolic flavonoid was shown to bind hIAPP at the amyloidogenic core and its C-terminus preventing aggregation and distorting the fibrils.136 The differential binding score (DIBS) was introduced to determine the binding preferences of ligands to an ensemble of IDP conformations by comparison against random coil ensembles of the same protein extracted from MD simulations.137 The validation was performed on epigallocatechin-3-gallate (EGCG) binding to the unstructured N-terminus of the tumor suppressor p53 protein, which compared favorably to experimental results. The predictive ability of simulations has been demonstrated in a translational study, in which atomistic simulations were used to design new polythiophene derivatives against prion aggregation, prior to in vivo testing.54 The compounds subsequently showed substantial prophylactic and therapeutic potency in prion-infected mice. Hence, simulations are powerful tools to generate conformational ensembles of the target polypeptide, which can act as scaffolds for the docking and design of molecules to target specific amyloid-forming regions.
The effects of antibodies on the structural and dynamic properties of amyloidogenic polypeptides have also revealed valuable insight into their modulating properties. Specifically, molecular dynamics simulations of Aducanumab in complex with Aβ42 revealed that the antibody sterically binds to monomeric, oligomeric and fibrillar species, with the binding site at the N-terminus (residues 2–7) preserved across all systems.41 Additionally, the results showed that the monomer unfolds and hydrophobically collapses on the antibody's surface, while in the complexes with aggregated species, the β-sheet structure of the peptide remains conserved.41 All-atom simulations of PrPC in complex with the neurotoxic POM1 and the innocuous POM6 antibodies revealed that the two antibodies, despite targeting similar epitopes, modulate differently the intrinsic flexibility of the protein28 and its orientation with respect to the cellular membrane.29 The information extracted from the simulations of amyloidogenic polypeptides in complex with antibodies could serve as starting points for the optimization and design of agents (e.g. antibodies, peptides) to bind with higher affinity towards selected species or for the rational design of cyclic peptides to modulate the target's conformational landscape enabling access to new binding sites.
Aside from the structure and the conformational landscape of the target, the conformations of the designed cyclic peptide in the target-bound and target-unbound states play an important role. Essentially, the designed cyclic peptides often adopt different conformations in solution as compared to the target-bound state. To design an efficient peptide-based inhibitor one needs to understand the conformational transitions of the cyclic peptides between the different states. While some peptide–protein complex structures are available, obtaining high resolution structures of cyclic peptides in solution is hampered by their low core-to-surface ratio, absence of specific couplings (e.g. NH-Hα) and diverse conformations in solution.131 Hence, molecular dynamics simulations have been successfully used to predict the energetically relevant conformational ensembles of cyclic peptides in solution, which compare favorably to available experimental data (e.g. NMR chemical shifts).138 We refer the interested reader to a comprehensive review of computational methods to characterize the behavior of cyclic peptides in solution131 and underline the synergistic effects of experimental and computational works.
Regarding the implications to the cyclic peptide design aspect, molecular dynamics simulations exceed experimental resolutions and can provide insight into the structural interactions between the peptide and the target at atomistic level of detail. For instance, macrocyclic peptides found in plants (cyclotides) have been experimentally shown to inhibit the aggregation of tau and amyloid-β42 fibrils.139 The peptide was subsequently docked onto 3D structures of Aβ42 fibrils and subjected to molecular dynamics simulations.140 The results explained experimental observations to reveal that the Cter-M cyclotide from C. ternatea (GLPTCGETCTLGTCYVPDCSCSWPICMKN) binds the Aβ42 fibril via hydrogen bonding, hydrophobic, electrostatic and π–π interactions, thereby inhibiting aggregation.140 Particularly, the peptide disrupts intermolecular hydrogen bonds and salt bridges in the Aβ42 fibril, which are crucial for its structural integrity. The effects occur within the first 50 ns of the simulations with disruptions in the fibril secondary structure at residues 2–7 and 38–41, resulting in the loss of extended β-sheet conformations. Importantly, the Aβ42 fibril in absence of the peptide maintains stable extended β-sheet conformations throughout the simulation trajectory.
Other approaches rely on available high resolution structures of protein complexes to identify linear interface motifs with an appropriate distances between residues to facilitate subsequent cyclization.141 In particular, backbone motifs of epitopes within protein–protein interfaces were identified and compared against available cyclic peptide databases to pinpoint promising candidates with desired structural features.141 Subsequently, the generated cyclic peptide–protein complexes underwent refinement through molecular dynamics simulations in explicit solvent to determine the binder with the highest target affinity. To validate the efficacy of this method, initial tests were conducted on a complex formed by the bovine trypsin inhibitor (BPTI) protein and the trypsin protease. The method identified a cyclic peptide that resembled the BPTI protein backbone at the interface, which is in agreement with experimentally known structures.
Despite extensive simulations, challenges remain when exploring the conformational space of IDPs both in the presence and in absence of modulators.142 Convergence is an issue due to the rugged free energy landscapes of the polypeptides, their size (which at times imposes the use of large simulation boxes) and/or kinetic traps. Some of these difficulties are overcome by using enhanced sampling techniques, implicit solvents and/or coarse-grained models, which together with advances in computing power enable the access to longer time- and length-scales. Alternative approaches, include reducing the size of the system by simulating fragments of the polypeptide of interest and using statistical mechanics tools to derive the conformational free energies of the full IDP.143 Current force fields struggle with over- or underestimating the properties of an IDP as compared to experimental quantities. Here, the IDP-tailored choices are the all-atom additive Charmm36m144 and Amber ff14IDPSFF,145 which have been fine tuned to reach experimental agreement and improve the conformational sampling of intrinsically disordered proteins.146 More recently, machine learning has been integrated into the development and improvement of force fields147,148 and novel techniques are emerging for IDP-specific force fields. An example is Charmm-NN, which uses atom-type based neural networks to calculate energies and forces149 and is subject to further improvements. A detailed overview of the challenges associated with IDP simulations and their reconciliation with experimental data have been reviewed in ref. 2 and 142. On the methodology side, the determination of the binding free energies of the cyclic peptide to the target also require special attention. For instance, using perturbation free energy calculations, a popular method with small molecules, one can determine the relative binding free energies and mechanistic detail, while preserving the flexibility of the complex.150 Nevertheless, the convergence still remains an issue. Alternatively, umbrella sampling, a technique that provided valuable insight into the themodynamics of monomer attachment to amyloid fibrils,143,151,152 would be a suitable choice for the determination of the binding free energies of a peptide to the target.
| Ref. | Peptide | Agent | Model | Solvent | Method | Samplinga |
|---|---|---|---|---|---|---|
| a Cumulative sampling over all replicas. Abbreviations. MD, molecular dynamics; H-REMD, Hamiltonian replica exchange molecular dynamics; MC/MD, hybrid Monte Carlo/molecular dynamics. RESPA, reversible multiple time scale molecular dynamics. b Two sets of simulations at different concentrations. | ||||||
| Barz et al.133 | Aβ42 monomer | — | Charmm36m | TIP3P | H-REMD | 40.8 μs |
| Jakubowski et al.52 | Aβ42 fibril | 94 small molecules | Charmm36m | TIP3P | MD | 10.4 μs |
| Dehabadi et al.50 | Aβ42 dimer | Ferulic aldehyde | Charmm36m | TIP3P | MD | 2.6 μs |
| Vanillin | 2.6 μs | |||||
| Sciacca et al.51 | Aβ40 monomer | SilA, SilB | Charmm36 | TIP3P | MD | 3 μs |
| Garcia-Vinuales et al.53 | hIAPP monomer | — | Charmm36m | TIP3P | MD | 6 μs |
| SilA | 6.5 μsb | |||||
| SilB | 6.5 μsb | |||||
| Dubey et al.136 | hIAPP fibril | Myricetin | Amber99sb | TIP3P | MD | 1.05 μs |
| Chen & Krishnan137 | p53-NTD | EGCG | OPLS-AA 2005 | TIP4D | MD | 500 ns |
| Frost & Zacharias41 | Aβ2–7 | AduFab | Charmm22* | TIP3P | MD | 500 ns |
| Aβ42 | AduFab | 1 μs | ||||
| Aβ42 dimer | 781 ns | |||||
| Aβ42 hexamer | 1 μs | |||||
| Aβ42 fubril | 254 ns | |||||
| Ilie & Caflisch28 | PrPC | — | Charmm36m | TIP3P | MD | 5 μs |
| POM1 | MD | 5 μs | ||||
| PrPC | POM6 | MD | 5 μs | |||
| Ilie et al.29 | PrPC | — | Charmm36 | ABSINTH | MC/MD | 4.8 μs MD + 240M MC |
| PrPC | POM1 | MC/MD | 4.8 μs MD + 240M MC | |||
| PrPC | POM6 | MC/MD | 4.8 μs MD + 240M MC | |||
| Kalmankar et al.140 | Aβ42 monomer | Cter-M cyclotide | OPLS3e | TIP4P | RESPA | 900 ns |
| Aβ17–42 pentamer | 900 ns | |||||
| Aβ11–42 fibril | 900 ns | |||||
| Aβ42 double fibril | 900 ns | |||||
3.3 Computational methods for cyclic peptide design
Various computational methods have proven essential in the design of cyclic peptides for amyloidogenic targets. TANGO,153 an algorithm developed to identify amyloidogenic sequences in proteins, was used to guide the search for cyclic peptides with improved binding affinity to Aβ40 oligomers.154 Residues 102–117 (PRRYTIAALLSPYSWS) from the G strand of protein transthyretin (TTR) were used as starting point to generate a peptide that binds Aβ40 and redirects it towards protease-sensitive, nonfibrillar aggregates. The peptide was head-to-tail cyclyzed and TANGO was used to select specific mutations that would retain or stabilize the antiparallel two-stranded β-sheet, resulting in a series of cyclic G (CG) peptides. Out of the five newly synthesized peptides, CG8 (TKVVTpPRYTIAKLSSPYSYSQ)§) had the most pronounced affinity towards Aβ40, results confirmed by ThT fluorescence analyses.154 Cyclization of CG8 via a disulfide bond using the simple cyclic peptide application (SCPA)155 within ROSETTA and the addition of a second D-proline (TKVVTpPRYTIAKLSSpPSYSQ) lead to increased peptide stability, enhanced conformational uniformity, and a higher β-sheet content.156 These findings highlight the added value of of cyclization and conformational homogeneity as design strategies.Des3PI (design of peptides targeting protein–protein interactions) is a novel computational fragment-based approach for designing cyclic peptides with high target specificity.157 This algorithm performs docking calculations of an amino acid library onto the targeted protein surface and subsequently connects residues with favorable target binding affinities to generate novel cyclic peptide sequences and structures. We envision that the potential of this method can be exploited to the maximum when combined with quantitative representations from molecular dynamics simulations to generate novel amyloid-binding cyclic peptides.
Among the computational methods employed in designing peptides, FoldX emerges as a powerful tool due to its ability to determine the free energy contributions of each atom at protein interfaces based on its own position relative to neighbours in the complex.158 It can thereby predict the impact of mutations on protein stability and optimize protein sequences for improved stability and desired functional properties. Relying on FoldX to perform an exhaustive thermodynamic profiling, the tandem peptide CAP1 was designed to inhibit tau aggregation.159 Both in vitro and in vivo experiments confirmed computational predictions by showing that CAP1 binds with high specificity and affinity (EC50 = 145 ± 49 nM) to tau aggregates, impeding their spread within cells. Additionally, CAP1 proved effective in hindering the ability of tau polymorphs obtained from the brains of Alzheimer's disease patients to initiate aggregation.
4 Machine learning for cyclic peptide design, property and activity prediction
Machine learning enables the rapid in silico screening and development of small molecules with therapeutic applications.36 On the peptide engineering side, machine learning has found recent applications in antibody optimization160,161 and enzyme evolution.34 The potential for cyclic peptide design is still in its initial stages and requires accurate and reliable training data. For instance, using conformational ensembles from molecular dynamics, data can be generated and incorporated in training sets to create machine learning models able to accurately predict structural ensembles of peptides and their complexes or to generate peptide sequences with improved physico-chemical properties. For improved performance and increased accuracy, experimental data (e.g. binding, toxicity) can be incorporated.In fact, by using data from molecular dynamics simulations of cyclic pentapeptides with diverse sequences and structural attributes as training datasets, machine learning models have been trained to predict structural ensembles for novel cyclic-peptide sequences, a method known as structural ensembles achieved by molecular dynamics and machine learning (StrEAMM).162 Alternative methods rely on generating comprehensive training datasets comprising sequences of blood–brain barrier penetrating linear peptides (BBPs) sourced from established databases and scientific literature, alongside non-BBPs peptides from UniProt to predict and explore novel BBPs with improved properties.163 AbDiffuser introduced a diffusion model tailored for the generation of three-dimensional antibody structures and corresponding sequences for biotechnological applications.164 Large protein families can be reliably mapped to a sequence ordinate using sequence alignment. AbDiffuser is an equivariant diffusion model designed to take advantage of these properties. The model adheres to physics-based constraints (e.g. bonds, torsional angles) and can accommodate different sequence lengths, thereby reducing the memory complexity. AbDiffuser relies on the Aligned Protein Mixer (APMixer), a neural network operating within the SE(3) equivariance framework to ensure consistent behavior, when subjected to rotations and translations in the three-dimensional space. Validation of the predictions through in silico and in vitro work underlines the importance of computational and experimental synergies when designing molecules with tailored properties.
Within the landscape of neurodegenerative diseases, MobiDB emerges as a resource that provides a comprehensive view of polypeptide disorder.165 This repository compiles an array of comprehensive data related to intrinsically disordered proteins (IDPs) and regions (IDRs) encompassing both experimental and computational information on protein disorder, (e.g. sequences, structures, and functional annotations). Its utility is extended to experimental scientists seeking detailed information about individual protein systems, as well as bioinformaticians who require substantial, unified protein datasets for building statistical classifiers. More recently, MobiDB integrates AlphaFold predictions sourced from AlphaFoldDB.166
A recent study highlighting the synergy between modern computational techniques and experiments, focused on developing a versatile method for designing proteins capable of targeting specific peptide sequences derived from armadillo proteins.167 Using no known structure, Monte Carlo simulations were employed to construct a hash table for bidentate side-chain-backbone interactions, to ensure the stability of the desired protein–peptide interface. Identified key residues were optimized using Rosetta to construct both the protein and peptide sequences while keeping the identified residues unchanged. To enhance the binding affinity and specificity, alanine scanning was performed and the binding free energies were determined to select the most favorable binders, validated by experimental techniques (e.g. X-ray crystallography, circular dichroism and biolayer interferometry). For IDPs, a similar approach may aid in the initial generation of polypeptide–cyclic peptide complexes than can then be investigated and optimized via molecular dynamics simulations.
An alternative approach known as hallucination relies on reversing deep neural networks trained to predict native protein structures, to design novel protein sequences and structures.168 Briefly, information encoded in several parameters of protein structure prediction networks containing learned representations and patterns that enable the networks to capture and predict various aspects of protein structures, including amino acid interactions and statistical relationships, is used to create realistic protein backbones and their corresponding amino acid sequences. First, random amino acid sequences are input into the trRosetta structure prediction network169 to predict distance maps. Then Monte Carlo sampling is employed in residue space to refine the sequences and improve the predicted structures. This process generates a diverse array of proteins with varied sequences and structures. To validate the physical manifestation of these hallucinations, synthetic genes for 129 hallucinated proteins were expressed and purified. Among these, 27 proteins exhibited circular dichroism spectra consistent with the target structures and the resolved three-dimensional structures of three selected proteins matched the hallucinated models, underlying the potential of the method in de novo protein design.
Chroma introduced a generative approach to design peptides with customized structures and functions.170 It employs a diffusion process, which incorporates conformational statistics of polymer ensembles (e.g. dihedral angles, bonds) and a neural architecture for molecular systems based on random graph neural networks for molecular systems. The model can be conditioned via external constraints (e.g. symmetries, substructures, and natural language prompts) to generate proteins with specific properties, including inter-residue distances, distinct structural domains, and semantic properties guided by classifiers.
A recent investigation explored the synergistic potential of integrating advanced deep learning methods with a Rosetta-based approach to enhance the accuracy and efficacy of designed protein sequences binding to specific target molecules.171 The success rate is defined by the Cα root mean squared deviations of the binder between structures generated with AlphaFold2126 or RoseTTAFold172 and Rosetta-designed structures. Large differences between them, i.e. deviations larger than 2.0 Å, indicate potential design pitfalls for protein binders. Complemented by confidence metrics from pairwise atomic environment predictions, successful binders are separated from those that do not perform well. The results show that AlphaFold2 or RoseTTAFold as evaluation filters in the protein design process increases the design success rate by 10-fold as compared to Rosetta.
Other strategies integrate RoseTTAFold,172 into denoising diffusion probabilistic models (DDPMs) to design novel proteins with specific structural or functional attributes.173 This effort gives rise to RFdiffusion,174 which incorporates RoseTTAFold as a denoising network within a generative diffusion model. Briefly, protein backbones are created from scratch by initializing frames of random residues and RFdiffusion is used to produce a refined and denoised prediction. Subsequently, sequences for these structures are generated employing the ProteinMPNN network.175 RFdiffusion predictions can be optimized by incorporating additional information (e.g. partial sequence and fold data) and enhanced through pre-trained weights and the application of loss functions.
Novel methods for cyclic peptide generation and design are rapidly emerging and might prove to be useful in the amyloidogenic polypeptide landscape. For instance, RINGER is a novel macrocycle conformer generator, which is a diffusion-based transformer model tailored to generate novel peptide macrocycles with specific sequences.176 Alphafold has been recently modified to predict the structure of macrocyclic peptides (AfCycDesign), which have been then experimentally validated.177 On the coarse-grained side, CycloPep emerges as a powerful tool to generate cyclic peptides compatible with the MA(R/S)TINI force field.178
5 A powerful trio: simulations, experiments and machine learning
The integration of computer simulations and experiments into machine learning powered engines enables the design, optimization and validation of custom protein-binding agents in an informed, fast and robust way. Taken together, these techniques have the necessary ingredients to generate novel, amyloid-specific and effective cyclic peptide binders, and hence make the next substantial step in the design of cyclic peptides as therapeutic agents or biomarkers against neurodegenerative diseases.Following the recipe introduced throughout this paper, there are at least four ingredients required for the successful de novo peptide design binding amyloidogenic targets (Fig. 3). First, the target scaffold and, in particular, quantitative distributions of conformations of this scaffold are necessary elements.127 Available three dimensional high resolution structures are excellent candidates, however in absence thereof, deep learning based methods such as AlphaFold2,126 RosettaFold122 or Chroma170 can accurately predict 3D models of protein structures even under user-specified environmental conditions.179 For IDPs, structure prediction is more challenging because of their native disorder characterised by a rugged free energy landscape.127 Fortunately, existing or predicted structures can be investigated to obtain quantitative conformational distributions using molecular dynamics simulations at full atomistic resolution (if the system size allows) or at coarse-grained level (when dealing with bigger targets or aggregates).2 For the latter, different methods can be employed to reinstate atomistic detail,180,181 which would enable to extract the statistically relevant states of the target, to be used in subsequent steps.
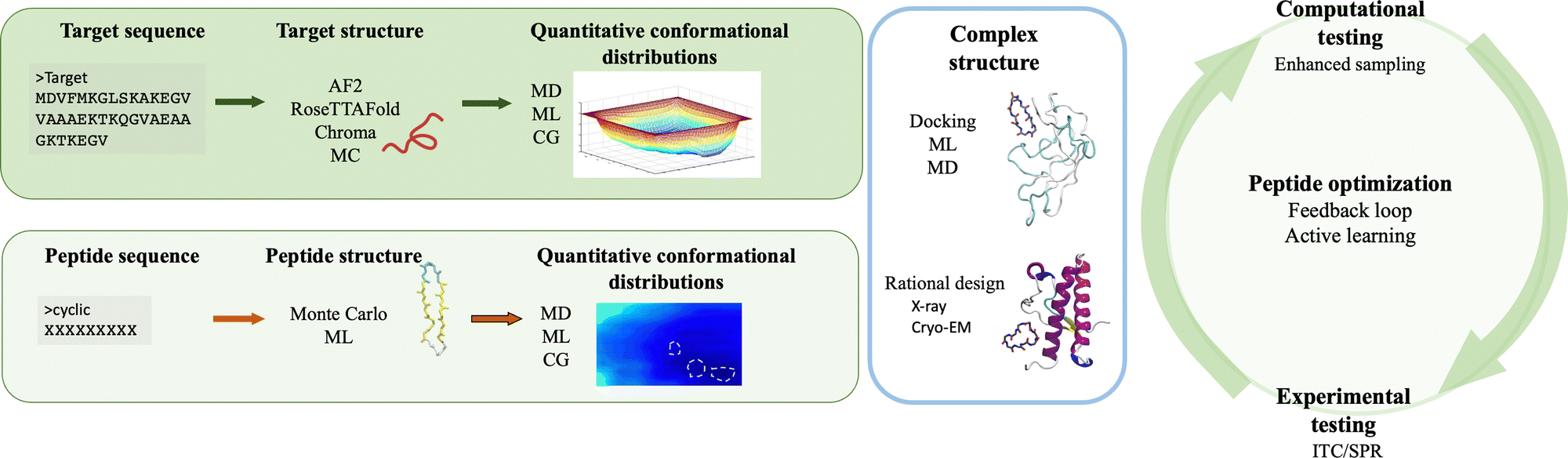 | ||
| Fig. 3 Schematic overview of the proposed de novo peptide design strategy against amyloidogenic targets. The first steps involve the preparation of the structures, the identification ofquantitative conformational distributions of the target (top left panel) and the binder (bottom left panel). After preparing the two structures, the peptide can be docked onto the monomeric or multimeric target to determine the structure of the complex. Alternatively, the complex can be rationally designed starting from high resolution structures (middle panel). Next, the binding of the peptides is computationally and experimentally tested. Importantly, the extracted data (e.g. binding free energies, kinetics) can be incorporated in feedback loops powered by machine learning engines (e.g. active learning cycle) to improve the peptide sequences and/or properties. After several cycles, the best binders are advanced into pre-clinical validation. Created with BioRender.com. | ||
Second, quantitative characterization of the conformations populated by the cyclic peptides in the target-bound versus target-unbound states are factors to be accounted for. The size of the peptides (below 20 residues) and their cyclic nature often limits their structure generation or even the complex prediction via deep learning approaches such as AlphaFold-Multimer182 or via experimental techniques.131 Assuming that the initial peptide–protein complex is known, e.g. from crystal structures183 or from de novo design,184 one can isolate the peptide from the complex and explore its conformational space in the unbound state via (enhanced sampling) molecular dynamics simulations at full atomistic resolution.131 For a cyclic peptide with unknown bound and unbound conformations, a convenient approach to obtain statistically meaningful conformations in solution is to generate its sequence by building its residues in an excluded volume-obeying manner,185 and sampling its conformational space via Monte Carlo simulations and/or relaxing it using (enhanced sampling) molecular dynamics simulations. Nevertheless, the latter contains no information on the conformations sampled by the peptide when bound to the target, which may represent a bottleneck when trying to dock to the target.
Third, a key aspect is the structure of the complex, which aids in understanding what type of interactions drive the assembly and which residues contribute the most to peptide binding and complex stability. Experimentally, a series of crystal structures of cyclic peptide–protein complexes have been resolved183 but none in complex with amyloidogenic targets. The thermodynamics and kinetics of peptide binding can be tested using methods such as surface plasmon resonance or isothermal titration calorimetry but none provides specific information on the binding epitope. Computationally, if the representative 3D structures are known, the peptide can be rationally designed and/or docked onto the target and enhanced sampling or deep learning techniques are employed to extract its binding free energy.186 Alternatively, in absence of known structures and/or unstably bound complexes, long molecular dynamics simulations could potentially reveal new binding sites. This approach may be efficient if the amyloidogenic target has a well defined secondary structure as is the case for 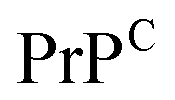 , or has druggable pockets. However, for polypeptides with a high degree of plasticity this is a resource intensive and potentially ineffective strategy, which would only slow down peptide design. Machine learning can facilitate the design of peptides, and corroborated with simulations and/or experiments, can aid in the estimation of binding affinities,187 and improve the peptide sequence for optimal binding to the target.171,188 Hence, if combined in an effective manner, computer simulations and machine learning can considerably increase peptide design and optimization efficiency, and can therefore speed up drug development.
, or has druggable pockets. However, for polypeptides with a high degree of plasticity this is a resource intensive and potentially ineffective strategy, which would only slow down peptide design. Machine learning can facilitate the design of peptides, and corroborated with simulations and/or experiments, can aid in the estimation of binding affinities,187 and improve the peptide sequence for optimal binding to the target.171,188 Hence, if combined in an effective manner, computer simulations and machine learning can considerably increase peptide design and optimization efficiency, and can therefore speed up drug development.
The fourth ingredient prior to clinical advancement is the experimental in vitro and in vivo validation. Given the complementarity of computational and experimental work, an attractive approach would be to integrate the trio, i.e. simulations, machine learning and experiments, into a dynamic and iterative engine. For instance, molecular dynamics simulations and deep learning, could be first used to predict and optimize protein and peptide conformations, stability, binding affinities, aiding in the selection of lead candidates prior to experimental validation. Then results from the trio can be incorporated into feedback loops37,189,190 that would allow the design of novel and improved peptide sequences, prediction of cyclic peptide bioactivity, better target selectivity, and off-target effects, thus aiding in the faster identification of potent and safe candidates. Hence, the unique integration of such methodologies can aid the design and optimization of novel experiments and computational work. Furthermore, an approach as such can significantly reduce the number of experiments that are required for validation and can increase the homogeneity across the experimental data sets (e.g. environmental conditions).39
6 Perspectives and outlook
In the last 20 years more than 16 cyclic peptide based therapeutics have been FDA and EMA-approved, mainly as antibiotics, anticancer therapeutics, antifungals and immunomodulators.80,191 Despite extensive research, no cyclic peptide-based drug for neurodegenerative diseases has passed clinical trials. The challenges arise due to the intrinsic disorder of the targets lacking traditional long-lived druggable pockets,90 the limited understanding of the associated mechanisms and the often ineffective integration and active feedback between disciplines. Here, we reviewed the design strategies for cyclic peptide design against amyloid-forming targets from a computational perspective and emphasized on the potential of the interconnection between computer simulations, experiments and machine learning in anti-amyloid cyclic peptide design for therapeutic, imaging and diagnostic applications. As such, we proposed a recipe, which can function like a digital twin i.e. creating scenarios relying on available information to improve performance and prevent design flaws, allowing for rapid analysis and accurate predictions.192 Essentially in cyclic peptide design, the digital twin would rely on information from computational and experimental findings to simulate the effect of a cyclic peptide-based drug on an amyloidogenic target, while enhancing the design and optimization of future peptide-based drugs, with better targeting abilities, reduced risk and lower cost. Clearly, the integration is not effortless and requires the efficient incorporation of extensive data from both experiments and simulations (e.g. binding constants, toxicity assays, morphological effects etc.) to be constantly exchanged between the physical and virtual machine. Importantly, while simulations essentially act as digital twins by themselves, the incorporation of homogeneous experimental data via machine learning powered engines can improve predictions, making the next substantial step in (peptide-based) drug design.The concepts and proposed strategies extend beyond drug design for therapeutic applications and hold the potential to aid in adjacent fields such as (bio)material design or controlled drug delivery.193 In essence, it all boils down to the gathering and the smart processing information from diverse sources to create a digital correspondent of a material capturing its composition, structure, responsiveness to external stimuli etc.194 to generate design rules for programmable and adaptable materials.195
Author contributions
Daria de Raffelle – data curation, formal analysis, investigation, visualization, writing – original draft, writing – review & editing. Ioana M. Ilie – conceptualization, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, writing – original draft, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I. M. I. acknowledges support from the Sectorplan Bèta & Techniek of the Dutch Government and the Dementia Research – Synapsis Foundation Switzerland.Notes and references
- K. Gammon, Nature, 2014, 515, 299–300 CrossRef PubMed
.
- I. M. Ilie and A. Caflisch, Chem. Rev., 2019, 119, 6956–6993 CrossRef CAS PubMed
- C. Hoffmann, R. Sansevrino, G. Morabito, C. Logan, R. M. Vabulas, A. Ulusoy, M. Ganzella and D. Milovanovic, J. Mol. Biol., 2021, 433, 166961 CrossRef CAS PubMed
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed
- C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS PubMed
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27–68 CrossRef CAS PubMed
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed
- A. Aguzzi, Nature, 2009, 459, 924–925 CrossRef CAS PubMed
- O. V. Galzitskaya, S. Y. Grishin, A. V. Glyakina, N. V. Dovidchenko, A. V. Konstantinova, S. V. Kravchenko and A. K. Surin, Int. J. Mol. Sci., 2023, 24, 3781 CrossRef CAS PubMed
- M. Polymenidou, R. Moos, M. Scott, C. Sigurdson, Y.-Z. Shi, B. Yajima, I. Hafner-Bratkovic, R. Jerala, S. Hornemann, K. Wuthrich, A. Bellon, M. Vey, G. Garen, M. N. G. James, N. Kav and A. Aguzzi, PLoS One, 2008, 3, 1–17 CrossRef PubMed
- R. H. Scannevin, Curr. Opin. Chem. Biol., 2018, 44, 66–74 CrossRef CAS PubMed
- F. A. Aprile, P. Sormanni, M. Podpolny, S. Chhangur, L.-M. Needham, F. S. Ruggeri, M. Perni, R. Limbocker, G. T. Heller, T. Sneideris, T. Scheidt, B. Mannini, J. Habchi, S. F. Lee, P. C. Salinas, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 13509–13518 CrossRef CAS PubMed
- E. Gibbs, J. Silverman, B. Zhao, X. Peng, J. Wang, C. Wellington, I. Mackenzie, S. Plotkin, J. Kaplan and N. Cashman, Sci. Rep., 2019, 9, 9870 CrossRef PubMed
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed
- H. Samaranayake, T. Wirth, D. Schenkwein, J. K. Räty and S. Ylä-Herttuala, Ann. Med., 2009, 41, 322–331 CrossRef CAS PubMed
- A. E. Lang, A. D. Siderowf, E. A. Macklin, W. Poewe, D. J. Brooks, H. H. Fernandez, O. Rascol, N. Giladi, F. Stocchi, C. M. Tanner, R. B. Postuma, D. K. Simon, E. Tolosa, B. Mollenhauer, J. M. Cedarbaum, K. Fraser, J. Xiao, K. C. Evans, D. L. Graham, I. Sapir, J. Inra, R. M. Hutchison, M. Yang, T. Fox, S. B. Haeberlein and T. Dam, N. Engl. J. Med., 2022, 387, 408–420 CrossRef CAS PubMed
- G. Pagano, K. I. Taylor, J. Anzures-Cabrera, M. Marchesi, T. Simuni, K. Marek, R. B. Postuma, N. Pavese, F. Stocchi, J.-P. Azulay, B. Mollenhauer, L. López-Manzanares, D. S. Russell, J. T. Boyd, A. P. Nicholas, M. R. Luquin, R. A. Hauser, T. Gasser, W. Poewe, B. Ricci, A. Boulay, A. Vogt, F. G. Boess, J. Dukart, G. D'Urso, R. Finch, S. Zanigni, A. Monnet, N. Pross, A. Hahn, H. Svoboda, M. Britschgi, F. Lipsmeier, E. Volkova-Volkmar, M. Lindemann, S. Dziadek, Š. Holiga, D. Rukina, T. Kustermann, G. A. Kerchner, P. Fontoura, D. Umbricht, R. Doody, T. Nikolcheva and A. Bonni, N. Engl. J. Med., 2022, 387, 421–432 CrossRef CAS PubMed
- L. Wang, N. Wang, W. Zhang, X. Cheng, Z. Yan, G. Shao, X. Wang, R. Wang and F. Caiyun, Signal Transduction Targeted Ther., 2022, 7, 1–48 CrossRef PubMed
- K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed
- P. Dougherty, A. Sahni and D. Pei, Chem. Rev., 2019, 119, 10241–10287 CrossRef CAS PubMed
- K. Kurrikoff, D. Aphkhazava and U. Langel, Curr. Opin. Pharmacology, 2019, 47, 27–32 CrossRef CAS PubMed
- C. Flores, G. Fouquet, I. C. Moura, T. T. Maciel and O. Hermine, Front. immunol., 2019, 10, 1–7 CrossRef PubMed
- V. Armiento, K. Hille, D. Naltsas, J. Lin, A. Barron and A. Kapurniotu, Angew. Chem., Int. Ed., 2020, 59, 3372–3384 CrossRef CAS
- T. Ikenoue, F. Aprile, P. Sormanni and M. Vendruscolo, Front. Neurosci., 2021, 15, 1–9 Search PubMed
- D. S. Nielsen, N. E. Shepherd, W. Xu, A. J. Lucke, M. J. Stoermer and D. P. Fairlie, Chem. Rev., 2017, 117, 8094–8128 CrossRef CAS PubMed
- K. Patel, L. J. Walport, J. L. Walshe, P. D. Solomon, J. K. K. Low, D. H. Tran, K. S. Mouradian, A. P. G. Silva, L. Wilkinson-White, A. Norman, C. Franck, J. M. Matthews, J. M. Guss, R. J. Payne, T. Passioura, H. Suga and J. P. Mackay, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26728–26738 CrossRef CAS PubMed
- H. Etayash, D. Pletzer, P. Kumar, S. K. Straus and R. E. W. Hancock, J. Med. Chem., 2020, 63, 9228–9236 CrossRef CAS PubMed
- I. M. Ilie and A. Caflisch, Biochim. Biophys. Acta, Proteins Proteomics, 2022, 1870, 11–12 CrossRef PubMed
- I. M. Ilie, M. Bacci, A. Vitalis and A. Caflisch, Biophys. J., 2022, 121, 2813–2825 CrossRef CAS PubMed
- X. Jing and K. Jin, Med. Res. Rev., 2020, 40, 753–810 CrossRef CAS PubMed
- J. Santos, P. Gracia, S. Navarro, S. Peña-Díaz, J. Pujols, N. Cremades, I. Pallarès and S. Ventura, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed
- S. Yamaguchi, S. Ito, T. Masuda, P.-O. Couraud and S. Ohtsuki, J. Controlled Release, 2020, 321, 744–755 CrossRef CAS
- Q. Liu, G. Xun and Y. Feng, Biotechnol. Adv., 2019, 37, 530–537 CrossRef CAS PubMed
- Z. Wu, S. B. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8852–8858 CrossRef CAS
- B. J. Wittmann, Y. Yue and F. H. Arnold, Cell Syst., 2021, 12, P1026–1045 CrossRef PubMed
- K. K. Yang, Z. Wu and F. H. Arnold, Nat. Met., 2019, 16, 687–694 CrossRef CAS PubMed
- B. Mohr, K. Shmilovich, I. S. Kleinwächter, D. Schneider, A. L. Ferguson and T. Bereau, Chem. Sci., 2022, 13, 4498–4511 RSC
- S. Ovchinnikov and P.-S. Huang, Curr. Opin. Struct. Biol., 2021, 65, 136–144 CrossRef CAS PubMed
- H. Narayanan, F. Dingfelder, A. Butte, N. Lorenzen, M. Sokolov and P. Arosio, Trends Pharmacol. Sci., 2021, 42, 151–165 CrossRef CAS PubMed
- M. Baek and D. Baker, Nat. Med., 2022, 19, 13–14 CAS
- C. V. Frost and M. Zacharias, Proteins: Struct., Funct., Bioinf., 2020, 88, 1592–1606 CrossRef CAS PubMed
- C. K. Wang, S. E. Northfield, Y.-H. Huang, M. C. Ramos and D. J. Craik, Eur. J. Med. Chem., 2016, 109, 342–349 CrossRef CAS PubMed
- J. Sevigny, P. Chiao, T. Bussière, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50–56 CrossRef CAS PubMed
- J. Arndt, F. Qian, B. Smith, C. Quan, K. Kilambi, M. Bush, T. Walz, R. Pepinsky, T. Bussière, S. Hamann, T. Cameron and P. Weinreb, Sci. Rep., 2018, 8, 6412 CrossRef PubMed
-
N. J. Yang and M. J. Hinner, Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins, Springer, New York, 2014, pp. 29–53 Search PubMed
- S. Stegemann, C. Moreton, S. Svanbäck, K. Box, G. Motte and A. Paudel, Drug Discovery Today, 2022, 28, 103344 CrossRef PubMed
- P. Kulkarni, S. Bhattacharya, S. Achuthan, A. Behal, M. K. Jolly, S. Kotnala, A. Mohanty, G. Rangarajan, R. Salgia and V. Uversky, Chem. Rev., 2022, 122, 6614–6633 CrossRef CAS PubMed
- C. J. Oldfield and A. K. Dunker, Annu. Rev. Biochem., 2014, 83, 553–584 CrossRef CAS PubMed
- L. L. N. Ngoc, S. G. Itoh, P. Sompornpisut and H. Okumura, Chem. Phys. Lett., 2020, 758, 137913 CrossRef CAS
- M. H. Dehabadi, A. Caflisch, I. M. Ilie and R. Firouzi, J. Phys. Chem. B, 2022, 126, 7627–7637 CrossRef CAS PubMed
- M. F. M. Sciacca, V. Romanucci, A. Zarrelli, I. Monaco, F. Lolicato, N. Spinella, C. Galati, G. Grasso, L. D'Urso, M. Romeo, L. Diomede, M. Salmona, C. Bongiorno, G. D. Fabio, C. L. Rosa and D. Milardi, ACS Chem. Neurosci., 2017, 8, 1767–1778 CrossRef CAS PubMed
- J. M. Jakubowski, A. A. Orr, D. A. Le and P. Tamamis, J. Chem. Inf. Model., 2019, 60, 289–305 CrossRef
- S. García-Viñuales, I. M. Ilie, A. M. Santoro, V. Romanucci, A. Zarrelli, G. D. Fabio, A. Caflisch and D. Milardi, Biochim. Biophys. Acta, Proteins Proteomics, 2022, 1870, 140772 CrossRef PubMed
- U. S. Herrmann, A. K. Schütz, H. Shirani, D. Huang, D. Saban, M. Nuvolone, B. Li, B. Ballmer, A. K. O. Åslund, J. J. Mason, E. Rushing, H. Budka, S. Nyström, P. Hammarström, A. Böckmann, A. Caflisch, B. H. Meier, K. P. R. Nilsson, S. Hornemann and A. Aguzzi, Sci. Transl. Med., 2015, 7, 299ra123 Search PubMed
- J. Zhao, A. Blayney, X. Liu, L. Gandy, W. Jin, L. Yan, J.-H. Ha, A. J. Canning, M. Connelly and C. Yang,
et al.
, Nat. Commun., 2021, 12, 986 CrossRef CAS PubMed
- P. Velander, L. Wu, F. Henderson, S. Zhang, D. R. Bevan and B. Xu, Biochem. Pharmacol., 2017, 139, 40–55 CrossRef CAS PubMed
- H. Ashrafian, E. H. Zadeh and R. H. Khan, Int. J. Biol. Macromol., 2021, 167, 382–394 CrossRef CAS PubMed
- M. Perni, C. Galvagnion, A. Maltsev, G. Meisl, M. B. Müller, P. K. Challa, J. B. Kirkegaard, P. Flagmeier, S. I. Cohen and R. Cascella,
et al.
, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1009–E1017 CrossRef CAS PubMed
- N. Majid, M. K. Siddiqi, A. N. Khan, S. Shabnam, S. Malik, A. Alam, V. N. Uversky and R. H. Khan, ACS Chem. Neurosci., 2019, 10, 4704–4715 CrossRef CAS PubMed
- S. K. Chaturvedi, N. Zaidi, P. Alam, J. M. Khan, A. Qadeer, I. A. Siddique, S. Asmat, Y. Zaidi and R. H. Khan, PLoS One, 2015, 10, e0136528 CrossRef PubMed
- M. K. Siddiqi, P. Alam, S. K. Chaturvedi, M. V. Khan, S. Nusrat, S. Malik and R. H. Khan, Biochim. Biophys. Acta, Proteins Proteomics, 2018, 1866, 549–557 CrossRef CAS PubMed
- P. Alam, S. K. Chaturvedi, M. K. Siddiqi, R. K. Rajpoot, M. R. Ajmal, M. Zaman and R. H. Khan, Sci. Rep., 2016, 6, 26759 CrossRef CAS PubMed
- P. Alam, M. K. Siddiqi, S. K. Chaturvedi, M. Zaman and R. H. Khan, Int. J. Biol. Macromol., 2017, 99, 477–482 CrossRef CAS PubMed
- Z. Gazova, K. Siposova, E. Kurin, P. Mucaji and M. Nagy, Proteins: Struct., Funct., Bioinf., 2013, 81, 994–1004 CrossRef CAS PubMed
- I. Morales, C. Cerda-Troncoso, V. Andrade and R. B. Maccioni, J. Alzheimers Dis., 2017, 60, 451–460 CAS
- R. Banerjee, Spectrochim. Acta, Part A, 2014, 117, 798–800 CrossRef CAS PubMed
- J. S. Rane, P. Bhaumik and D. Panda, J. Alzheimers Dis., 2017, 60, 999–1014 CAS
- A. Thapa, S. D. Jett and E. Y. Chi, ACS Chem. Neurosci., 2016, 7, 56–68 CrossRef CAS PubMed
- K. Ono, M. Hirohata and M. Yamada, Biochem. Biophys. Res. Commun., 2005, 336, 444–449 CrossRef CAS PubMed
- A. Kundu and A. Mitra, Plant Foods Hum. Nutr., 2013, 68, 247–253 CrossRef CAS PubMed
- S. Jokar, S. Khazaei, H. Behnammanesh, A. Shamloo, M. Erfani, D. Beiki and O. Bavi, Biophys. Rev., 2019, 11, 901–925 CrossRef CAS PubMed
- R. Roy and S. Paul, J. Phys. Chem. B, 2023, 127, 600–615 CrossRef CAS PubMed
- G. J. Weiner, Nat. Rev. Cancer, 2015, 15, 361–370 CrossRef CAS PubMed
- F. A. Aprile, P. Sormanni, M. Perni, P. Arosio, S. Linse, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, Sci. Adv., 2017, 3, e1700488 CrossRef PubMed
- L. Söderberg, M. Johannesson, P. Nygren, H. Laudon, F. Eriksson, G. Osswald, C. Möller and L. Lannfelt, Neurotherapeutics, 2022, 20, 195–206 CrossRef PubMed
- T. Sonati, R. R. Reimann, J. Falsig, P. K. Baral, T. O'Connor, S. Hornemann, S. Yaganoglu, B. Li, U. S. Herrmann, B. Wieland, M. Swayampakula, M. H. Rahman, D. Das, N. Kav, R. Riek, P. P. Liberski, M. N. G. James and A. Aguzzi, Nature, 2013, 501, 102–106 CrossRef CAS PubMed
- R. J. Bateman, J. Cummings, S. Schobel, S. Salloway, B. Vellas, M. Boada, S. E. Black, K. Blennow, P. Fontoura, G. Klein, S. S. Assunção, J. Smith and R. S. Doody, Alzheimer’s Res. Therapy, 2022, 14, 178 CrossRef CAS PubMed
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed
- E. Mihara, S. Watanabe, N. K. Bashiruddin, N. Nakamura, K. Matoba, Y. Sano, R. Maini, Y. Yin, K. Sakai, T. Arimori, K. Matsumoto, H. Suga and J. Takagi, Nat. Commun., 2021, 12, 1543 CrossRef CAS PubMed
- H. Zhang and S. Chen, RSC Chem. Biol., 2022, 3, 18–31 RSC
- T. Passioura, T. Katoh, Y. Goto and H. Suga, Annu. Rev. Biochem., 2014, 83, 727–752 CrossRef CAS PubMed
- H. Y. Chow, Y. Zhang, E. Matheson and X. Li, Chem. Rev., 2019, 119, 9971–10001 CrossRef CAS PubMed
- C. White and A. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS PubMed
- M. Erak, K. Bellmann-Sickert, S. Els-Heindl and A. G. Beck-Sickinger, Bioorg. Med. Chem., 2018, 26, 2759–2765 CrossRef CAS PubMed
- M. T. J. Bluntzer, J. OConnell, T. S. Baker, J. Michel and A. N. Hulme, Pept. Sci., 2020, 113, e24191 CrossRef
- P. M. Cromm, J. Spiegel and T. N. Grossmann, ACS Chem. Biol., 2015, 10, 1362–1375 CrossRef CAS PubMed
- V. Digiesi, V. de la Oliva Roque, M. Vallaro, G. Caron and G. Ermondi, Eur. J. Pharm. Biopharm., 2021, 165, 259–270 CrossRef CAS PubMed
- P. Matsson, B. C. Doak, B. Over and J. Kihlberg, Adv. Drug Delivery Rev., 2016, 101, 42–61 CrossRef CAS PubMed
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed
- B. D. Khairnar, A. Jha and J. M. Rajwade, J. Mater. Sci., 2023, 58, 9834–9860 CrossRef CAS
- N. S. Rösener, L. Gremer, E. Reinartz, A. König, O. Brener, H. Heise, W. Hoyer, P. Neudecker and D. Willbold, J. Biol. Chem., 2018, 293, 15748–15764 CrossRef PubMed
- S. Schemmert, L. C. Camargo, D. Honold, I. Gering, J. Kutzsche, A. Willuweit and D. Willbold, Int. J. Mol. Sci., 2021, 22, 6553 CrossRef CAS PubMed
- T. Ikenoue, F. A. Aprile, P. Sormanni, F. S. Ruggeri, M. Perni, G. T. Heller, C. P. Haas, C. Middel, R. Limbocker, B. Mannini, T. C. T. Michaels, T. P. J. Knowles, C. M. Dobson and M. Vendruscolo, Sci. Rep., 2020, 10, 1–15 CrossRef PubMed
- T. Ikenoue, M. Oono, M. So, H. Yamakado, T. Arata, R. Takahashi, Y. Kawata and H. Suga, ChemBioChem, 2023, 24, 1–7 CrossRef PubMed
- M. Chemerovski-Glikman, E. Rozentur-Shkop, M. Richman, A. Grupi, A. Getler, H. Y. Cohen, H. Shaked, C. Wallin, S. K. T. S. Wärmländer, E. Haas, A. Gräslund, J. H. Chill and S. Rahimipour, Chem. – Eur. J., 2016, 22, 14236–14246 CrossRef CAS PubMed
- A. Spanopoulou, L. Heidrich, H.-R. Chen, C. Frost, D. Hrle, E. Malideli, K. Hille, A. Grammatikopoulos, J. Bernhagen, M. Zacharias, G. Rammes and A. Kapurniotu, Angew. Chem., Int. Ed., 2018, 57, 14503–14508 CrossRef CAS PubMed
- B. H. Rajewski, K. M. Makwana, I. J. Angera, D. K. Geremia, A. R. Zepeda, G. I. Hallinan, R. Vidal, B. Ghetti, A. L. Serrano and J. R. Del Valle, J. Am. Chem. Soc., 2023, 145, 23131–23142 CrossRef CAS PubMed
- G. Giubertoni, L. Feng, K. Klein, G. Giannetti, Y. Choi, A. van der Net, G. Castro-Linares, F. Caporaletti, D. Micha, J. Hunger, A. Deblais, D. Bonn, A. Šaric, I. M. Ilie, G. H. Koenderink and S. Woutersen, bioRxiv, 2023.
- I. M. Ilie, C. Ehrhardt, A. Caflisch and G. Weitz-Schmidt, J. Chem. Inf. Model., 2023, 63, 3878–3891 CrossRef CAS PubMed
- A. Borsatto, O. Akkad, I. Galdadas, S. Ma, S. Damfo, S. Haider, F. Kozielski, C. Estarellas and F. L. Gervasio, eLife, 2022, 11, 1–25 CrossRef PubMed
- V. Armiento, A. Spanopoulou and A. Kapurniotu, Angew. Chem., Int. Ed., 2019, 59, 3372–3384 CrossRef PubMed
- J. L. Arenas, J. Kaffy and S. Ongeri, Curr. Opin. Chem. Biol., 2019, 52, 157–167 CrossRef PubMed
- S. Ullrich and C. Nitsche, Protein Sci., 2023 DOI:10.1002/pep2.24326
- R. K. Saini, D. Goyal and B. Goyal, Chem. Res. Tox., 2020, 33, 2719–2738 Search PubMed
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed
- N. Nemoto, E. Miyamoto-Sato, Y. Husimi and H. Yanagawa, FEBS Lett., 1997, 414, 405–408 CrossRef CAS PubMed
- R. W. Roberts and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12297–12302 CrossRef CAS PubMed
- C. Sohrabi, A. Foster and A. Tavassoli, Nat. Rev. Chem., 2020, 4, 90–101 CrossRef CAS PubMed
- J. D. Bloom and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9995–10000 CrossRef CAS PubMed
- Y.-W. Lin, Coord. Chem. Rev., 2017, 336, 1–27 CrossRef CAS
- J. Franceus, S. Dhaene, H. Decadt, J. Vandepitte, J. Caroen, J. Van der Eycken, K. Beerens and T. Desmet, Chem. Commun., 2019, 55, 4531–4533 RSC
- T. M. Caradonna and A. G. Schmidt, npj Vaccines, 2021, 6, 154 CrossRef CAS PubMed
-
V. Ramakrishnan, K. Patel and R. Goyal, De Novo Peptide Design: Principles and Applications, 2022 Search PubMed
- P. Ragonis-Bachar and M. Landau, Curr. Opin. Struc. Biol., 2021, 68, 184–193 CrossRef CAS PubMed
- P. Sormanni, F. A. Aprile and M. Vendruscolo, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 9902–9907 CrossRef CAS PubMed
- K. A. Murray, C. J. Hu, S. L. Griner, H. Pan, J. T. Bowler, R. Abskharon, G. M. Rosenberg, X. Cheng, P. M. Seidler and D. S. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2206240119 CrossRef CAS PubMed
-
D.-A. Silva, B. E. Correia and E. Procko, in Motif-Driven Design of Protein-Protein Interfaces, ed. B. L. Stoddard, 2016, vol. 1414, pp. 285–304 Search PubMed
- P. Bradley, K. M. Misura and D. Baker, Science, 2005, 309, 1868–1871 CrossRef CAS PubMed
- G. Van Zundert, J. Rodrigues, M. Trellet, C. Schmitz, P. Kastritis, E. Karaca, A. Melquiond, M. van Dijk, S. De Vries and A. Bonvin, J. Mol. Biol., 2016, 428, 720–725 CrossRef CAS PubMed
- G. C. van Zundert and A. M. Bonvin, Protein Struct. Predict., 2014, 1137, 163–179 CAS
- A. Vangone, J. Rodrigues, L. Xue, G. van Zundert, C. Geng, Z. Kurkcuoglu, M. Nellen, S. Narasimhan, E. Karaca and M. van Dijk,
et al.
, Proteins: Struct.,
Funct., Bioinf., 2017, 85, 417–423 CrossRef CAS PubMed
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, G. R. Lee, J. Wang, Q. Cong, L. N. Kinch and R. D. Schaeffer,
et al.
, Science, 2021, 373, 871–876 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Ždek and A. Potapenko,
et al.
, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- G. Casadevall, C. Duran and S. Osuna, JACS Au, 2023, 3, 1554–1562 CrossRef CAS PubMed
- V. Charitou, S. C. Van Keulen and A. M. Bonvin, J. Chem. Theory Comput., 2022, 18, 4027–4040 CrossRef CAS PubMed
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Ždek and A. Potapenko,
et al.
, Nature, 2021, 596, 583–589 CrossRef CAS PubMed
- K. M. Ruff and R. V. Pappu, J. Mol. Biol., 2021, 433, 167208 CrossRef CAS PubMed
- L. M. Pietrek, L. S. Stelzl and G. Hummer, Curr. Opin. Struc. Biol., 2023, 78, 102501 CrossRef CAS PubMed
- F. Pinheiro, J. Santos and S. Ventura, J. Mol. Biol., 2021, 433, 167059 CrossRef CAS PubMed
- W. Wang, Phys. Chem. Chem. Phys., 2021, 23, 777–784 RSC
- J. Damjanovic, J. Miao, H. Huang and Y.-S. Lin, Chem. Rev., 2021, 121, 2292–2324 CrossRef CAS PubMed
- M. Sugita, S. Sugiyama, T. Fujie, Y. Yoshikawa, K. Yanagisawa, M. Ohue and Y. Akiyama, J. Chem. Inf. Model., 2021, 61, 3681–3695 CrossRef CAS PubMed
- B. Barz, A. K. Buell and S. Nath, Chem. Commun., 2021, 57, 947–950 RSC
- M. Zhu, A. D. Simone, D. Schenk, G. Toth, C. M. Dobson and M. Vendruscolo, J. Chem. Phys., 2013, 139, 0351011–03510110 Search PubMed
- Y. Chebaro, P. Jiang, T. Zang, Y. Mu, P. H. Nguyen, N. Mousseau and P. Derreumaux, J. Phys. Chem. B, 2012, 116, 8412–8422 CrossRef CAS PubMed
- R. Dubey, S. H. Kulkarni, S. C. Dantu, R. Panigrahi, D. M. Sardesai, N. Malik, J. D. Acharya, J. Chugh, S. Sharma and A. Kumar, Biol. Chem., 2021, 402, 179–194 CAS
- Q.-H. Chen and V. Krishnan, Sci. Rep., 2021, 11, 22583 CrossRef CAS PubMed
- Q. N. N. Nguyen, J. Schwochert, D. J. Tantillo and R. S. Lokey, Phys. Chem. Chem. Phys., 2018, 20, 14003–14012 RSC
- N. V. Kalmankar, H. Hari, R. Sowdhamini and R. Venkatesan, J. Med. Chem., 2021, 64, 7422–7433 CrossRef CAS PubMed
- N. V. Kalmankar, B. R. Gehi and R. Sowdhamini, Front. Mol. Biosci., 2022, 9, 986704 CrossRef CAS PubMed
- B. L. Santini and M. Zacharias, Front. Chem., 2020, 8, 573259 CrossRef CAS PubMed
- K. Kasahara, H. Terazawa, T. Takahashi and J. Higo, Comput. Struct. Biotechnol. J., 2019, 17, 712–720 CrossRef CAS PubMed
- I. M. Ilie, D. Nayar, W. K. den Otter, N. F. A. van der Vegt and W. J. Briels, J. Chem. Theory Comput., 2018, 14, 3298–3310 CrossRef CAS PubMed
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmueller and A. D. MacKerell Jr, Nat. Met., 2016, 14, 71–73 CrossRef PubMed
- D. Song, R. Luo and H.-F. Chen, J. Chem. Inf. Model., 2017, 57, 1166–1178 CrossRef CAS PubMed
- J. Mu, H. Liu, J. Zhang, R. Luo and H.-F. Chen, J. Chem. Inf. Model., 2021, 61, 1037–1047 CrossRef CAS PubMed
- I. Poltavsky and A. Tkatchenko, J. Phys. Chem. Lett., 2021, 12, 6551–6564 CrossRef CAS PubMed
- T. Mueller, A. Hernandez and C. Wang, J. Chem. Phys., 2020, 152(5), 050902 CrossRef CAS PubMed
- P. Zhang and W. Yang, J. Chem. Phys., 2023, 159, 024118 CrossRef CAS PubMed
- K. Wallraven, F. L. Holmelin, A. Glas, S. Hennig, A. I. Frolov and T. N. Grossmann, Chem. Sci., 2020, 11, 2269–2276 RSC
- T. Gurry and C. M. Stultz, Biochem. J., 2014, 53, 6981–6991 CrossRef CAS PubMed
- N. Schwierz, C. V. Frost, P. L. Geissler and M. Zacharias, J. Phys. Chem. B, 2017, 121, 671–682 CrossRef CAS PubMed
- A.-M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed
- X. Lu, C. R. Brickson and R. M. Murphy, ACS Chem. Neurosci., 2016, 7, 1264–1274 CrossRef CAS PubMed
- G. Bhardwaj, V. K. Mulligan, C. D. Bahl, J. M. Gilmore, P. J. Harvey, O. Cheneval, G. W. Buchko, S. V. Pulavarti, Q. Kaas and A. Eletsky,
et al.
, Nature, 2016, 538, 329–335 CrossRef CAS PubMed
- C. B. Est, P. Mangrolia and R. M. Murphy, Prot. Eng., Design and Sel., 2019, 32, 47–57 CrossRef PubMed
- M. Delaunay and T. Ha-Duong, J. Comp.-Aided Mol. Des., 2022, 36, 605–621 CrossRef CAS PubMed
- J. Schymkowitz, J. Borg, F. Stricher, R. Nys, F. Rousseau and L. Serrano, Nucleic Acids Res., 2005, 33, W382–W388 CrossRef CAS PubMed
- N. Louros, M. Ramakers, E. Michiels, K. Konstantoulea, C. Morelli, T. Garcia, N. Moonen, S. DHaeyer, V. Goossens and D. R. Thal,
et al.
, Nat. Commun., 2022, 13, 1351 CrossRef CAS PubMed
- D. M. Mason, S. Friedensohn, C. R. Weber, C. Jordi, B. Wagner, S. Meng, P. Gainza, B. E. Correia and S. T. Reddy, bioRxiv, 2019.
- D. Mason, S. Friedensohn, C. Weber, C. Jordi, B. Wagner, S. Meng, R. Ehling, L. Bonati, J. Dahinden, P. Gainza, B. Correia and S. Reddy, Nat. Biomed. Eng., 2021, 5, 1–13 CrossRef PubMed
- J. Miao, M. L. Descoteaux and Y.-S. Lin, Chem. Sci., 2021, 12, 14927–14936 RSC
- X. Chen, Q. Zhang, B. Li, C. Lu, S. Yang, J. Long, B. He, H. Chen and J. Huang, Front. Genet., 2022, 13, 845747 CrossRef PubMed
- K. Martinkus, J. Ludwiczak, K. Cho, W.-C. Lian, J. Lafrance-Vanasse, I. Hotzel, A. Rajpal, Y. Wu, R. Bonneau and V. Gligorijevicet al., arXiv, 2023, preprint, arXiv:2308.05027.
- D. Piovesan, A. Del Conte, D. Clementel, A. M. Monzon, M. Bevilacqua, M. C. Aspromonte, J. A. Iserte, F. E. Orti, C. Marino-Buslje and S. C. Tosatto, Nucleic Acids Res., 2023, 51, D438–D444 CrossRef CAS PubMed
- M. Varadi and S. Velankar, Proteomics, 2022, 1–10 Search PubMed
- K. Wu, H. Bai, Y.-T. Chang, R. Redler, K. E. McNally, W. Sheffler, T. Brunette, D. R. Hicks, T. E. Morgan and T. J. Stevens,
et al.
, Nature, 2023, 616, 581–589 CrossRef CAS PubMed
- I. Anishchenko, S. J. Pellock, T. M. Chidyausiku, T. A. Ramelot, S. Ovchinnikov, J. Hao, K. Bafna, C. Norn, A. Kang and A. K. Bera,
et al.
, Nature, 2021, 600, 547–552 CrossRef CAS PubMed
- Z. Du, H. Su, W. Wang, L. Ye, H. Wei, Z. Peng, I. Anishchenko, D. Baker and J. Yang, Nat. Protoc., 2021, 16, 5634–5651 CrossRef CAS PubMed
- J. Ingraham, M. Baranov, Z. Costello, V. Frappier, A. Ismail, S. Tie, W. Wang, V. Xue, F. Obermeyer and A. Beam,
et al.
, BioRxiv, 2022, 1–12 Search PubMed
- N. R. Bennett, B. Coventry, I. Goreshnik, B. Huang, A. Allen, D. Vafeados, Y. P. Peng, J. Dauparas, M. Baek and L. Stewart,
et al.
, Nat. Commun., 2023, 14, 2625 CrossRef CAS PubMed
- M. Baek, F. DiMaio, I. Anishchenko, J. Dauparas, S. Ovchinnikov, J. Wang and D. Baker, RoseTTAFold: The first release of RoseTTAFold, https://zenodo.org/record/5068265, 2021 Search PubMed.
- J. Ho, A. Jain and P. Abbeel, Adv. Neural Inf. Process, 2020, 33, 6840–6851 Search PubMed
- J. L. Watson, D. Juergens, N. R. Bennett, B. L. Trippe, J. Yim, H. E. Eisenach, W. Ahern, A. J. Borst, R. J. Ragotte and L. F. Milles,
et al.
, Nature, 2023, 1–3 CAS
- J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. Wicky, A. Courbet, R. J. de Haas and N. Bethel,
et al.
, Science, 2022, 378, 49–56 CrossRef CAS PubMed
-
C. A. Grambow, H. Weir, N. L. Diamant, A. M. Tseng, T. Biancalani, G. Scalia and K. V. Chuang, RINGER: Rapid Conformer Generation for Macrocycles with Sequence-Conditioned Internal Coordinate Diffusion, 2023 Search PubMed
- S. A. Rettie, K. V. Campbell, A. K. Bera, A. Kang, S. Kozlov, J. De La Cruz, V. Adebomi, G. Zhou, F. DiMaio, S. Ovchinnikov and G. Bhardwaj, bioRxiv, 2023.
- A. Cabezón, M. Calvelo, J. R. Granja, N. Piñeiro and R. Garcia-Fandino, J. Colloid Interface Sci., 2023, 642, 84–99 CrossRef PubMed
- J. Ingraham, M. Baranov, Z. Costello, V. Frappier, A. Ismail, S. Tie, W. Wang, V. Xue, F. Obermeyer, A. Beam and G. Grigoryan, 2022.
- J. Roel-Touris and A. M. Bonvin, Comput. Struct. Biotechnol. J., 2020, 18, 1182–1190 CrossRef CAS PubMed
- W. G. Noid, J. Phys. Chem. B, 2023, 127, 4174–4207 CrossRef CAS PubMed
- R. Evans, M. O'Neill, A. Pritzel, N. Antropova, A. Senior, T. Green, A. Žídek, R. Bates, S. Blackwell, J. Yim, O. Ronneberger, S. Bodenstein, M. Zielinski, A. Bridgland, A. Potapenko, A. Cowie, K. Tunyasuvunakool, R. Jain, E. Clancy, P. Kohli, J. Jumper and D. Hassabis, bioRxiv, 2021, preprint DOI:10.1101/2021.10.04.463034.
- A. K. Malde, T. A. Hill, A. Iyer and D. P. Fairlie, Chem. Rev., 2019, 119, 9861–9914 CrossRef CAS PubMed
- M. Wendt, R. Bellavita, A. Gerber, N.-L. Efrém, T. Ramshorst, N. M. Pearce, P. R. J. Davey, I. Everard, M. Vazquez-Chantada, E. Chiarparin, P. Grieco, S. Hennig and T. N. Grossmann, Angew. Chem., Int. Ed., 2021, 60, 13937–13944 CrossRef CAS PubMed
- I. M. Ilie and A. Caflisch, J. Phys. Chem. B, 2018, 122, 11072–11082 CrossRef CAS PubMed
- M. Ciemny, M. Kurcinski, K. Kamel, A. Kolinski, N. Alam, O. Schueler-Furman and S. Kmiecik, Drug Discovery Today, 2018, 23, 1530–1537 CrossRef CAS PubMed
- S. Romero-Molina, Y. B. Ruiz-Blanco, J. Mieres-Perez, M. Harms, J. Münch, M. Ehrmann and E. Sanchez-Garcia, J. Prot. Res., 2022, 21, 1829–1841 CrossRef CAS PubMed
- F. Wan, D. Kontogiorgos-Heintz and C. de la Fuente-Nunez, Drug Discovery Today, 2022, 1, 195–208 CAS
- K. Shmilovich, S. S. Panda, A. Stouffer, J. D. Tovar and A. L. Ferguson, Digital Discovery, 2022, 1, 448–462 RSC
-
A. F. Rama Ranganathan, Method and apparatus using machine learning for evolutionary data-driven design of proteins and other sequence defined biomolecules, US20220348903A1, 2022 Search PubMed
- J. Li, K. Yanagisawa, M. Sugita, T. Fujie, M. Ohue and Y. Akiyama, J. Chem. Inf. Model., 2023, 63, 2240–2250 CrossRef CAS PubMed
- A. Fuller, Z. Fan, C. Day and C. Barlow, IEEE Access, 2020, 8, 108952–108971 Search PubMed
- R. Merindol and A. Walther, Chem. Soc. Rev., 2017, 46, 5588–5619 RSC
- D. Topolsky, A. Belyakov, V. Pochinskaia, I. Topolskaya, N. Yumagulov and D. Fedorov, 2022 IEEE International Multi-Conference on Engineering, Computer and Information Sciences (SIBIRCON), 2022.
- C. Kaspar, B. J. Ravoo, W. G. van der Wiel, S. V. Wegner and W. H. P. Pernice, Nature, 2021, 594, 345–355 CrossRef CAS PubMed
Footnotes |
| † Small letters indicate D-enantioneric amino acids. |
| ‡ J is the norleucine amino acid. |
| § Small letters indicate D-enantioneric amino acids. |
| This journal is © The Royal Society of Chemistry 2024 |