
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into non-covalent interactions in dicopper(II,II) complexes bearing a naphthyridine scaffold: anion-dictated electrochemistry†
Jonathan
De Tovar
 *,
Christian
Philouze
,
Aurore
Thibon-Pourret
and
Catherine
Belle
*,
Christian
Philouze
,
Aurore
Thibon-Pourret
and
Catherine
Belle
Université Grenoble Alpes, CNRS, DCM, UMR 5250, 38000 Grenoble, France. E-mail: Jonathan.De-Tovar@univ-grenoble-alpes.fr
First published on 30th January 2024
Abstract
A family of bis(μ-hydroxido)dicopper(II,II) complexes bearing a naphthyridine-based scaffold has been synthesized and characterized. Cyclic voltammetry reveals that the nature of the anions present in the complexes plays a pivotal role in their electrochemical properties. X-ray diffraction, spectroscopic and electrochemical analysis data support the formation of intimate ion pairs by non-covalent interactions driving to a ca. 270 mV difference for the potential required to monooxidize the CuIICuII species.
Non-covalent interactions1,2 have been shown to play a pivotal role in the science of intermolecular relationships in chemistry3 and biology,4 such as protein folding2,5 and chiral recognition.6 Beyond classic hydrogen bond and van der Waals forces, non-covalent π interactions and C–F⋯H–C interactions7,8 have attracted the attention of the research community, since these forces are key control parameters involved in a variety of chemical transformations such as asymmetric transfer hydrogenations.6 Additionally, many of these chemical transformations are not only assisted by intra-molecular non-covalent interactions but also inter-molecular, which promotes the formation of ion pairs with interesting properties. Recently, it has been evidenced that the redox process associated with the ferrocenium/ferrocene couple (Fc+/0) is dependent on the nature of the anion present in the electrolyte due to the Fc+–anion ion pair interaction and steric constraints, which modulate the interfacial properties.9 Within this context, Kim and Yokota and co-workers observed that the potential associated for such a redox event can vary between 30 and up to 170 mV depending on the nature of the anion present in the medium. However, although these interactions offer a tuning possibility for building structure–activity relationships in catalysts, direct incorporation of specific π interactions into the design of molecular systems with precise control and reactivity remains a pivotal challenge.
We have initiated studies aimed at building and identifying catalytic systems in which these weak, yet important non-covalent interactions, could open new routes to enable challenging chemical transformations like selective C–H bond activation. Within this context and inspired by nature, Belle et al. found that dicopper(II,II) complexes containing a dipyridylethane naphthyridine hexadentate ligand not only act as a good compartmental ligand while allowing the isolation of dinuclear copper(II,II) complexes keeping the metallic centers in close proximity,10 but also have been demonstrated to activate toluene under electrochemically and environmentally friendly conditions.11
We envisioned, thus, that non-covalent interactions could purposely be used as dicopper(II,II) design tools for tuning both their oxidation potential and 2nd sphere effects, particularly when using tighter cages than the dipyridylethane naphthyridine ligand. For this purpose, a di((3,5-dimethyl)pyrazolyl)methane naphthyridine ligand (L) has been synthesized based on the Skraup–Doebner–von Miller quinoline synthesis12 starting with 1 and crotonaldehyde (2) yielding 3 followed by a subsequent oxidation of the methyl moieties into carboxaldehyde groups (compound 4) mediated by SeO2 (Scheme S1, ESI†).12 After synthesizing the dicarboxaldehyde–naphthyridine scaffold, bis(3,5-dimethylpyrazol-1-yl)methanone (5)13 was condensed with the former yielding ligand L (Scheme S1, ESI†). Then, three dicopper(II,II) molecular complexes named C1·(ClO4)2, C1·(BF4)2 and C1·(OTf)2 (where OTf− = CF3SO3−) have been synthesized (Scheme 1) allowing the evaluation of (i) the coordination properties of this ligand, (ii) the oxidative potential of the as-synthesized complexes for the couple E1/2(CuIIICuII/CuIICuII) and (iii) their stabilities under ambient conditions.
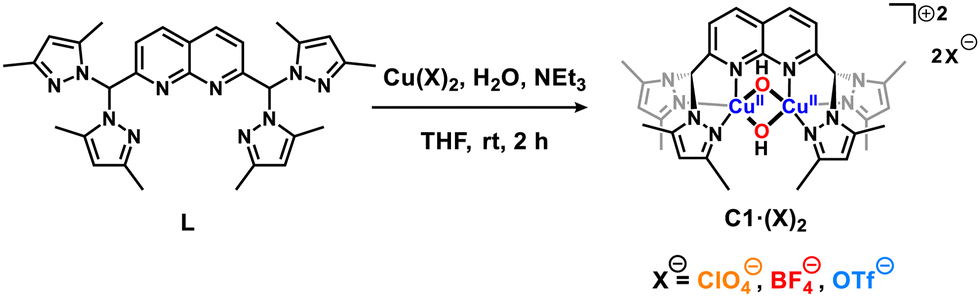 | ||
| Scheme 1 Synthesis of C1·(X)2 complexes prepared in this work. | ||
Suitable crystals for C1·(ClO4)2, C1·(BF4)2 and C1·(OTf)2 were obtained, crystallizing in the triclinic and orthorhombic space groups for the first two and the latter, respectively (see Table S1, ESI†). On the one hand, the molecular plots obtained (Fig. 1) evidence that the anions stay in the 2nd coordination sphere contrary to the previously reported dipyridylethane naphthyridine-based dicopper(II,II) complex, in which one triflate is coordinated to one copper(II) center in the solid-state.10 Instead, the ClO4−, BF4− and OTf− anions establish non-covalent interactions (hydrogen bonding, anion–π, C–H⋯π, B–F⋯H–C and C–F⋯H–C) with the C12+ scaffold, where the position of the anions nearby C12+ is governed by the nature of the former and the structure of the latter. Additionally, ligand L bears methyl-enriched pyrazole moieties, which lead to a dissimilar 2nd coordination sphere shell compared to the previously reported complex,11 making the presence of non-covalent interactions a pivotal factor to stabilize such complexes. On the other hand, although all complexes exhibit a dicopper bis(μ-hydroxido) core, the Cu⋯Cu and Cu⋯Nnaph (where naph = naphthyridine) bond lengths are rather similar for C1·(ClO4)2 and C1·(OTf)2 but longer for C1·(BF4)2. These variations can be attributed to the distinct hydrophobic and interactive nature of the anions. While ClO4− establishes not only anion–π interactions but also hydrogen bonds, as observed for OTf−, the latter, due to its perfluorinated methyl chain, locates itself in the less polar region of the C12+ scaffold. In contrast, BF4− anions, lacking an oxygen-enriched structure, steer clear of the relatively acidic protons associated with both bis(μ-hydroxido) and pyrazolo bridges, as well as the naphthyridine moieties, primarily interacting with the methyl groups. It is noteworthy that, even though the distance differs only by a few Å, the electronic and steric hindrance for complex C1·(BF4)2 differs significantly from those of C1·(ClO4)2 and C1·(OTf)2, allowing the coordination of solvent molecules to the copper(II) centers (Table S3, ESI†). Additionally, solvent molecules were found in the crystal structures of C1·(ClO4)2, C1·(BF4)2 and C1·(OTf)2, showing the presence of acetone, water and tetrahydrofuran molecules, respectively. All these differences are reflected in the naphthyridine scaffold, which is shown to be bent out of the NnaphCuCuNnaph plane by ca. 4.24°, 3.72° and 8.13° for C1·(ClO4)2, C1·(BF4)2 and C1·(OTf)2, respectively. Within this context, it is noteworthy to mention that the nature of the anion plays a pivotal role not only with the C12+ scaffold but also with these solvent molecules by allowing their proximity to the complex and trapping them into C12+ – anion pockets.
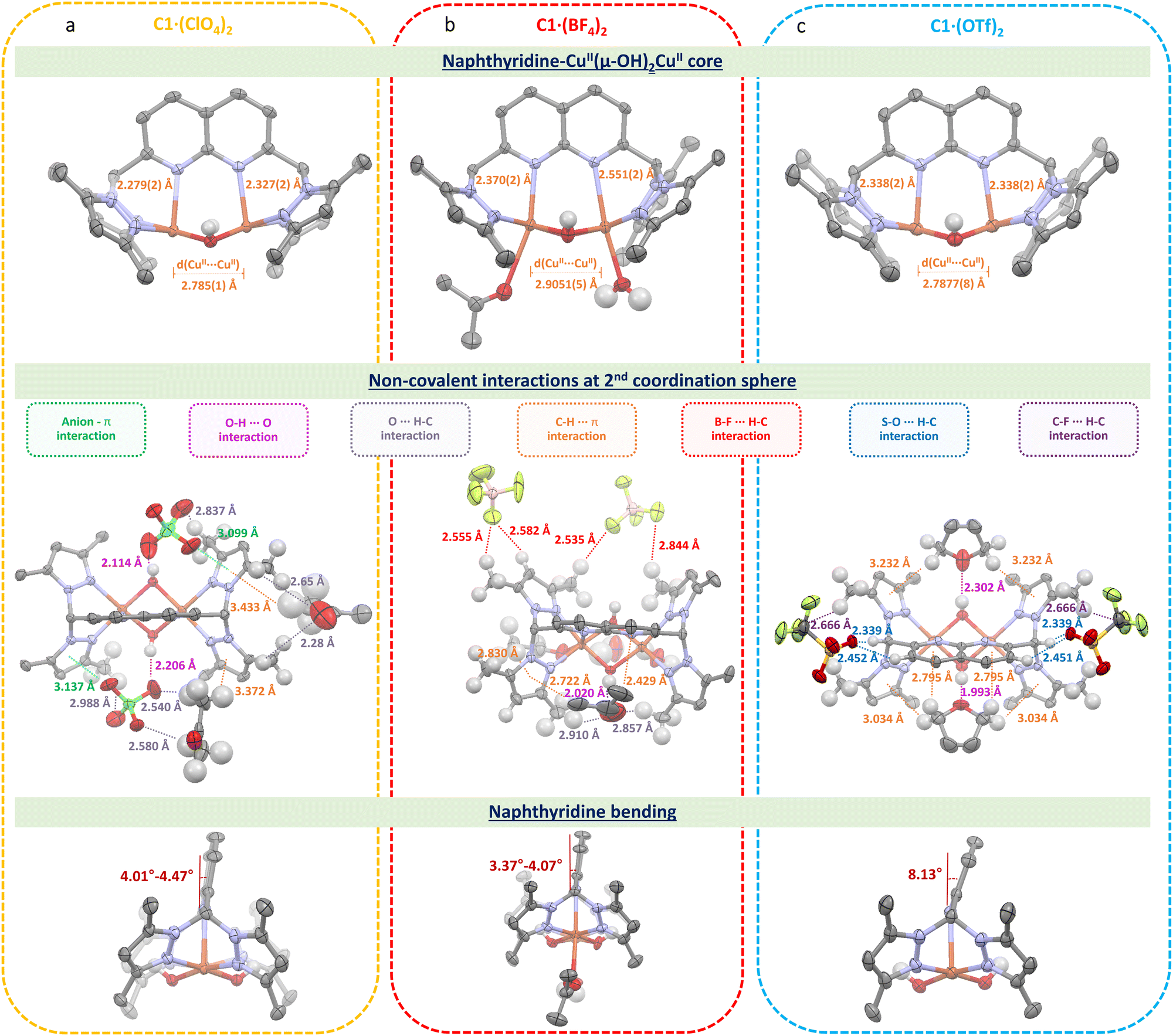 | ||
| Fig. 1 Displacement ellipsoid plots of (a) C1·(ClO4)2, (b) C1·(BF4)2 and (c) C1·(OTf)2 complexes at the 50% probability level. Cu–Nnaph and Cu⋯Cu distances (top), non-covalent interactions (middle) and naphthyridine bending angles (bottom). Solvent molecules, free anions and hydrogen atoms (except for the O–H bridges or when discussed) are omitted for clarity. | ||
Interestingly, green crystals were also obtained by the slow evaporation of the acetonitrile solution recovered from the washings of C1·(OTf)2. The single-crystal X-ray diffraction analysis revealed the unexpected formation of neutral mononuclear copper(II) complex C2 as a side product (see Scheme S2 and proposed mechanism of formation for C2 in the ESI†).
The previously reported naphthyridine-based dicopper(II,II) complex10 in CH3CN solution with 100 mM [nBu4N]ClO4 shows an anion exchange of OTf− by ClO4−. Then, in order to get further insights into these non-covalent interactions in solution, we analyzed the nature of the complex species under electrochemical conditions by determining their oxidative potentials, diffusion coefficients and hydrodynamic radii (Table S4, ESI†). One can expect an anion exchange process by perchlorates for both C1·(BF4)2 and C1·(OTf)2 forming complex C1·(ClO4)2. Interestingly and contrary to our expectations, when analyzing the oxidation peak potentials for this family of complexes (Fig. 2(a)), we observed that they appeared shifted to more anodic potentials following the trend Epa([C1·(ClO4)2]) < Epa([C1·(BF4)2]) < Epa([C1·(OTf)2]) and indicating that at least a few of these non-covalent interactions were maintained in solution, contrary to the reported behavior of the parent complex.9,11 From this, it can be inferred that (i) the inner C12+ nature for each of these complexes is kept under electrochemical conditions and must be studied individually and (ii) higher oxidation potentials are observed for anions with higher hydrophobic character. Additionally, the significant differences in the current intensities exhibited by the different complexes show that they diffuse differently towards the working electrode (Table S4, ESI†). We hypothesize that this behavior could be attributed to the anions present in the complexes, whereby they could keep and/or establish ion-pair interactions affecting the 1st and 2nd coordination spheres of the complexes in solution. As already evidenced in the solid-state, the nature of the anion is responsible for bending the naphthyridine scaffold out of the NnaphCuCuNnaph plane, diminishing both σ-donating and π-accepting electronic characters, which are translated towards higher oxidation potentials. More to this point, when such cyclic voltammetry was performed in CH3CN solution with 100 mM [nBu4N]BF4 instead of [nBu4N]ClO4, Epa for both C1·(BF4)2 and C1·(OTf)2 decreased ca. 140 mV and 190 mV, respectively, whereas that of C1·(ClO4)2 remained invariable (Fig. S20 and S21, ESI†). Under this scenario, CuIICuIII electro-generated species are favored and better stabilized by BF4−(electrolyte) anions for both C1·(BF4)2 and C1·(OTf)2 complexes. These phenomena appear by the fact that changing a ClO4− environment by BF4− intrinsically shifts the nature of the non-covalent interactions from anion–π, O–H⋯O and C–H⋯ to mainly B–F⋯H–C interactions, which are more prompted to be established by C1·(BF4)2 and C1·(OTf)2 rather than C1·(ClO4)2.
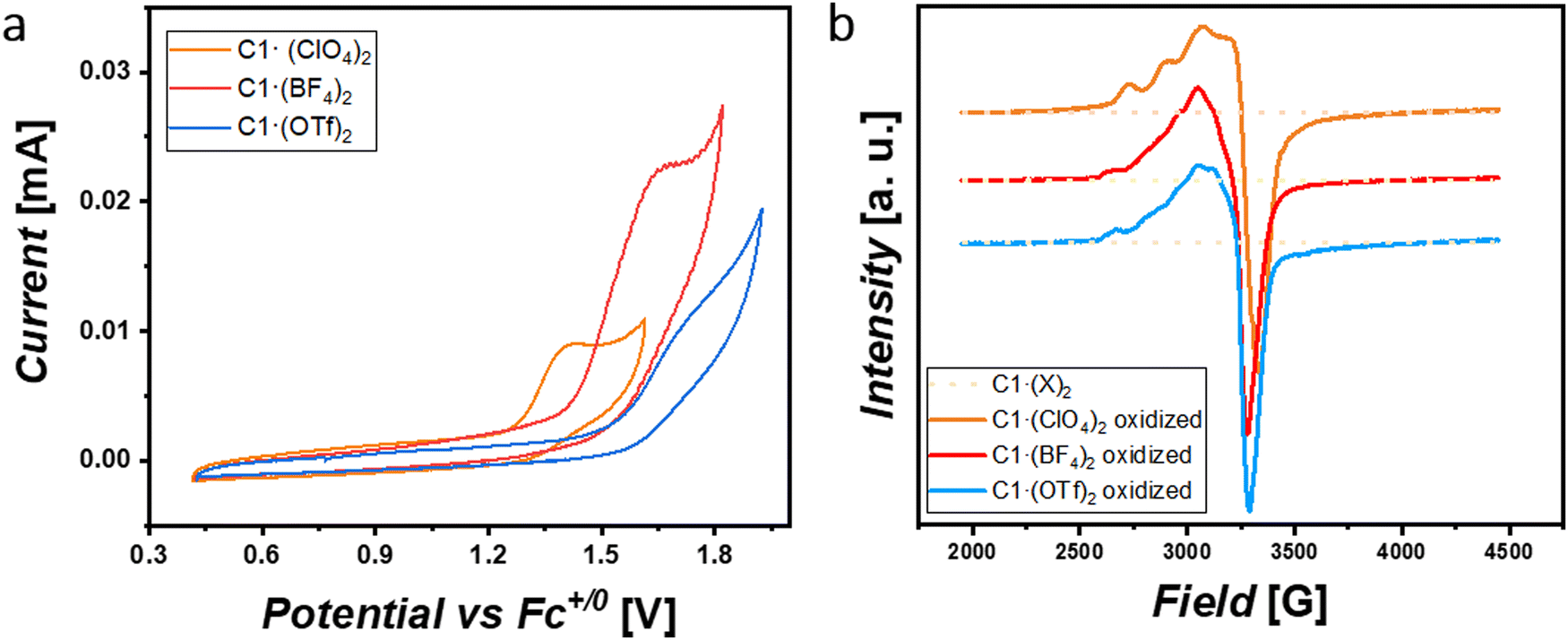 | ||
| Fig. 2 (a) Cyclic voltammograms for 1 mM solutions of C1·(ClO4)2 (orange), C1·(BF4)2 (red) and C1·(OTf)2 (blue) complexes in Ar-saturated anhydrous acetonitrile containing 0.1 M [n-Bu4N]ClO4. Scan rate = 50 mV s−1. (b) EPR spectra at 100 K for 1 mM solutions of C1·(ClO4)2 (orange), C1·(BF4)2 (red) and C1·(OTf)2 (blue) before and after electrochemical mono-oxidation in Ar-saturated anhydrous acetonitrile containing 0.1 M [n-Bu4N]ClO4. | ||
In addition to the electrochemical studies, the located spin density for the complexes prepared here was studied by electron paramagnetic resonance (EPR). Their spectra were recorded in anhydrous acetonitrile containing 0.1 M [n-Bu4N]ClO4 at 100 K (Table S4, ESI†). As shown in Fig. 2(b), all complexes except C2 (Fig. S23d, ESI†) exhibited a silent signal before oxidation pointing to the preservation of an antiferromagnetic behavior for the CuIICuII species. However, the C1·(X)2 family showed X-band signals after electrochemically passing a charge of ca. 0.16 C (Fig. S22, ESI†) corresponding to mono-oxidation from CuIICuII to CuIICuIII (Fig. 2(b)) for half of the complexes in solution. Interestingly, after simulating their monooxidized EPR spectra (Fig. S23, ESI†), we inferred that the three oxidized complexes exhibited a localized mixed-valent CuIICuIII behaviour at 100 K. Furthermore, the small variations observed in such EPR spectra for the three monooxidized complexes suggest an influence related to the presence of solvent and anion molecules through non-covalent interactions (2nd sphere effects).
In summary, we have demonstrated that the anions present in dicopper(II,II) complexes bearing a naphthyridine scaffold are able to bend the latter decreasing both σ-donating and π-accepting electronic properties, while establishing non-covalent interactions (hydrogen bonding, anion–π, O⋯C–H, C–H⋯π, B–F⋯H–C, S–O⋯C–H and C–F⋯H–C). Additionally, the presence of molecular pockets of dissimilar nature allows the accommodation of substrates like solvent molecules, a key step towards the development of a catalyst. Under this scenario, the anion nature dictates the availability of a pocket for a selected substrate. This was reflected in CuIII/CuII oxidation potentials ranging from 1.36, 1.57 and 1.63 V vs. Fc+/0 and matched with the increasing hydrophobic characters for the ClO4−, BF4− and OTf− anions, respectively, in [nBu4N]ClO4/CH3CN. Moreover, OTf− led to the activation and promotion of Csp3–Npyrazole bond cleavage through non-covalent interactions (Scheme S2, ESI†).
To the best of our knowledge, this work represents a unique scenario where non-covalent interactions have been successfully unraveled. This evidence provides a fresh set of guidelines for the design and applicability of novel catalysts, showcasing the significance of these subtle forces, which should not be overlooked. These forces can persist and actively participate in solution reactions. Further investigations in the near future are expected to delve into the role of the anion in C–H oxidation/oxygenation reactions.
This work was supported by the French Agence Nationale de la Recherche (ANR-22-CE07-0032). The authors are grateful to ICMG UAR 2607 for the analytical facilities (NMR, ESI-MS, EPR and X-Ray). This work has been partially supported by the CBH-EUR-GS (ANR-17-EURE-0003) and Labex ARCANE (ANR-11-LABX-0003-01) programs, in the framework of which this work has been carried out. Dr Florian Molton and Rodolphe Gueret are acknowledged for EPR and HR-ESI-MS analyses, respectively. We kindly acknowledge Dr Nicolas Le Poul, Dr Marius Réglier and Dr Jalila Simaan for fruitful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Y. Jin, Q. Zhen, D. Xiao, G. Tao, X. Xing, P. Yu and C. Xu, Nat. Commun., 2022, 13, 3276 CrossRef CAS PubMed.
- X. Lucas, A. Bauzá, A. Frontera and D. Quiñonero, Chem. Sci., 2016, 7, 1038 RSC.
- A. Gehlhaar, C. Wölper, F. van der Vight, G. Jansen and S. Schultz, Eur. J. Inorg. Chem., 2022, e202100883 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS PubMed.
- K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef PubMed.
- E. H. Krenske and K. N. Houk, Acc. Chem. Res., 2013, 46, 979 CrossRef CAS PubMed.
- V. Mzozoyana, F. R. van Heerden and C. Grimmer, Beilstein J. Org. Chem., 2020, 16, 190 CrossRef CAS PubMed.
- M. Morisue, M. Kawanishi, I. Ueno, T. Nakamura, T. Nabeshima, K. Imamura and K. Nozaki, J. Phys. Chem. B, 2021, 125, 9286 CrossRef CAS PubMed.
- R. A. Wong, Y. Yokota, M. Wakisaka, J. Inukai and Y. Kim, Nat. Commun., 2020, 11, 4194 CrossRef PubMed.
- J. A. Isaac, F. Gennarini, I. López, A. Thibon-Pourret, R. David, G. Gellon, B. Gennaro, C. Philouze, F. Meyer, S. Demeshko, Y. Le Mest, M. Réglier, H. Jamet, N. Le Poul and C. Belle, Inorg. Chem., 2016, 55, 8263 CrossRef CAS PubMed.
- J. A. Isaac, A. Thibon-Pourret, A. Durand, C. Philouze, N. Le Poul and C. Belle, Chem. Commun., 2019, 55, 12711 RSC.
- C. Vu, D. Walker, J. Wells and S. Fox, J. Heterocycl. Chem., 2002, 39, 829 CrossRef CAS.
- M. Tansky, Z. Gu and R. J. Comito, J. Org. Chem., 2021, 86, 1601 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2312255–2312259. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc06264c |
| This journal is © The Royal Society of Chemistry 2024 |