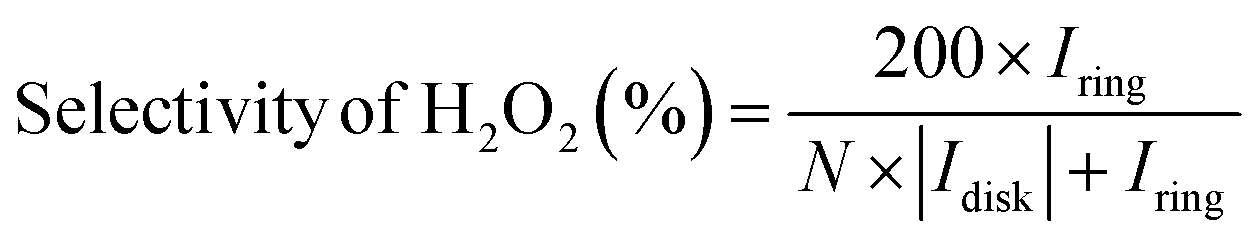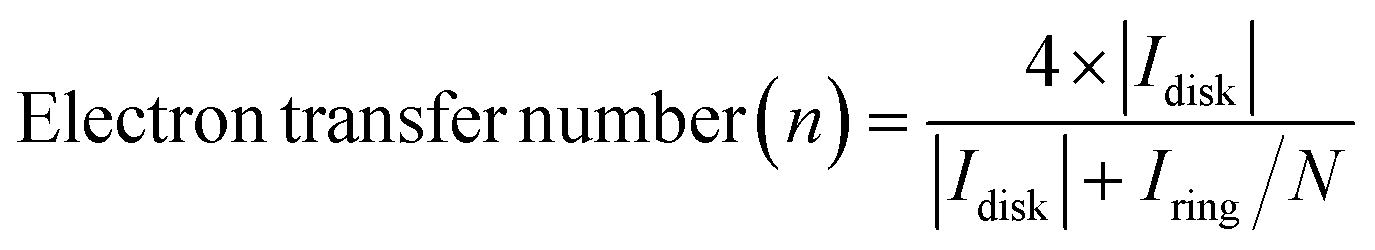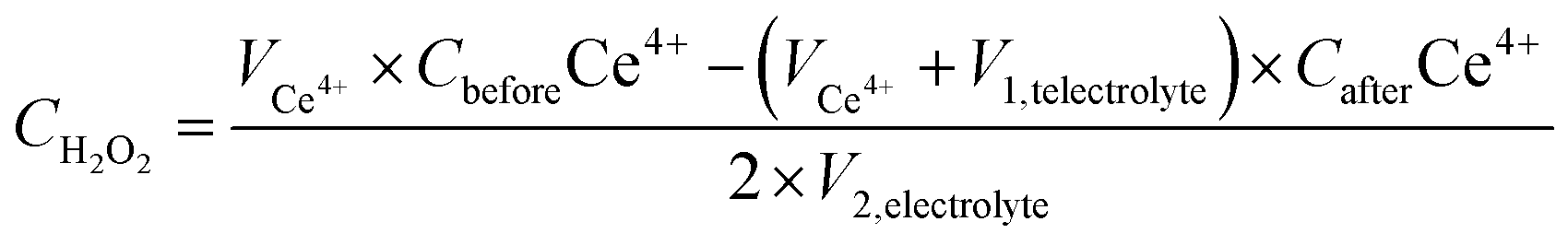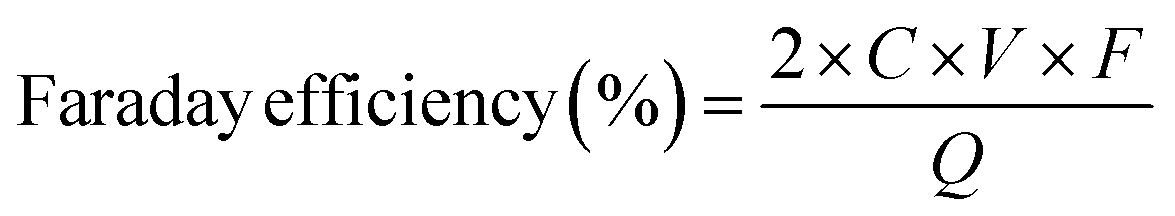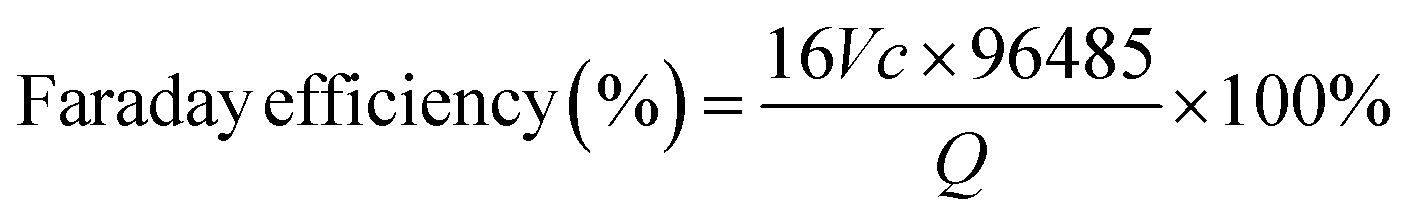DOI:
10.1039/D4CC01476F
(Highlight)
Chem. Commun., 2024,
60, 5232-5244
Recent advances in electrosynthesis of H2O2via two-electron oxygen reduction reaction
Received
31st March 2024
, Accepted 17th April 2024
First published on 18th April 2024
Abstract
The electrosynthesis of hydrogen peroxide (H2O2) via a selective two-electron oxygen reduction reaction (2e− ORR) presents a green and low-energy-consumption alternative to the traditional, energy-intensive anthraquinone process. This review encapsulates the principles of designing relational electrocatalysts for 2e− ORR and explores remaining setups for large-scale H2O2 production. Initially, the review delineates the fundamental reaction mechanisms of H2O2 production via 2e− ORR and assesses performance. Subsequently, it methodically explores the pivotal influence of microstructures, heteroatom doping, and metal hybridization along with setup configurations in achieving a high-performance catalyst and efficient reactor for H2O2 production. Thereafter, the review introduces a forward-looking methodology that leverages the synergistic integration of catalysts and reactors, aiming to harmonize the complementary characteristics of both components. Finally, it outlines the extant challenges and the promising avenues for the efficient electrochemical production of H2O2, setting the stage for future research endeavors.
1. Introduction
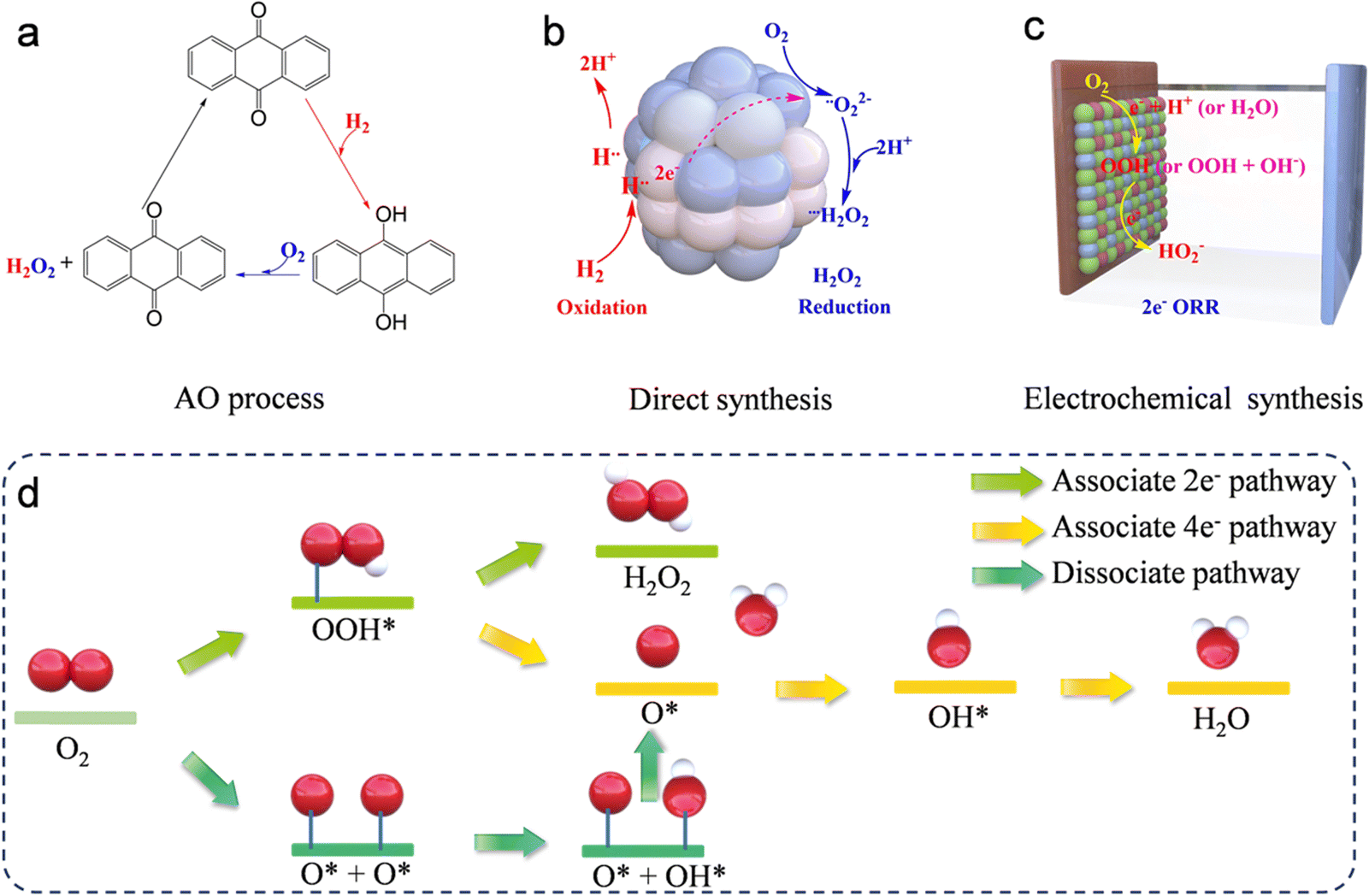
Hydrogen peroxide (H2O2) is one of the most significant industrial chemicals all over the world, with a wide range of applications that extend from paper and textile manufacturing to wastewater treatment systems, polymer and pharmaceutical syntheses, and organic degradation due to its remarkable oxidative and environmentally benign properties.1–5 However, 95% of the H2O2 product is produced by the anthraquinone oxidation (AO) process, which requires the presence of a noble palladium catalyst, hydrogenation, and oxidation steps (Fig. 1a), along with the recycling of excessive organic solvent and distillation of H2O2 from the reaction mixture.6,7 These disadvantages result in high costs, excessive energy consumption, and multiple operations. These limitations have prompted researchers to investigate alternative, environmentally friendly technologies for synthesizing H2O2. A notable alternative to the conventional AO approach for manufacturing H2O2 is the direct synthesis process, which utilizes hydrogen (H2) and oxygen (O2) as the initial reactants (Fig. 1b). This method employs a catalytic reaction, typically mediated by palladium/platinum (Pd/Pt) or similar catalysts, to combine these gases directly into H2O2 under carefully managed conditions. However, the direct synthesis of H2O2 (DS-H2O2) utilizing H2 and O2 faces several challenges, including the inherent safety risks due to the explosive nature of the H2 and O2 reaction, lower yields which reduce efficiency, the necessity for costly noble metal catalysts, operational complexities requiring precise control over reaction conditions to maintain selectivity, and generally lower production rates that may not meet industrial-scale demands.8–10 Therefore, to facilitate widespread adoption, it is essential to develop other efficient and cost-effective methods for the synthesis of H2O2.
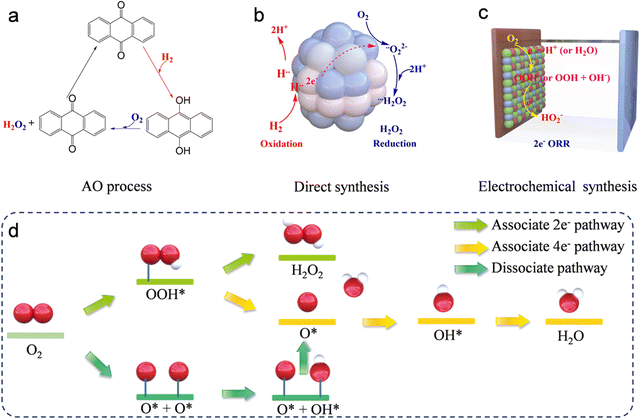 |
| | Fig. 1 Illustration of hydrogen peroxide production via (a) traditional anthraquinone process, (b) the direct synthesis, and (c) electrochemical two-electron oxygen reduction reaction. (d) Illustration of the proposed ORR process. | |
Notably, the electrochemical approach, utilizing the two-electron oxygen reduction reaction (2e− ORR) pathway, presents a promising method for generating H2O2. This technique is advantageous due to its portability and enhanced safety profile (Fig. 1c).6 Additionally, the electrocatalytic 2-electron oxygen reduction reaction (2e− ORR) occurs in an aqueous electrolyte (for example, aqueous KOH, phosphate-buffered saline (PBS), H2SO4, HClO4, and Na2SO4) with O2 as the starting material.11–15 This process avoids the use of excessive organic solvents required in the AO process and eliminates the direct contact between H2 and O2 in the DS-H2O2 process, indicating that it is more environmentally friendly and safer compared to the other two methods. Furthermore, the 2e− ORR can be driven at low voltage, suggesting that it can be integrated with renewable energy sources. These advantages position it as a sustainable alternative to supersede both the AO and DS-H2O2 processes. In the 2e− ORR, H2O2 is produced directly through the reduction of O2 at the cathode. Notwithstanding the advantages, the sluggish reaction kinetics coupled with side reactions significantly impede the overall energy efficiencies. Consequently, the essential conditions for the optimization of H2O2 electrochemical synthesis involve the deliberate design of specialized catalysts. These catalysts must favor the selective 2e− ORR pathway, while inhibiting the competing 4e− ORR pathway, and should exhibit high activity, superior selectivity, and strong stability. To date, optimal practical outcomes in 2e− ORR have been achieved through rational engineering of catalysts that offer affordability, abundance, and durability.16–20 The engineering strategies involve microstructural engineering, heteroatom doping engineering, and metal hybridization engineering. Many recent studies have confirmed that the dimensions and types of pore structures significantly affect the 2e− ORR performance.19,21–24 For example, three-dimensional (3D) materials can effectively avoid stacked structures, thereby favoring the exposure of more catalytically active sites. The mesopore structure in the materials can reduce the retention time of H2O2 on the catalyst surface, thus enhancing the productivity of H2O2.22,23 In addition to the influence of structure on the 2e− ORR performance, the composition of the catalysts significantly impacts the performance. The doping of heteroatoms and metallic hybridization are subjects of extensive study because of the ability of heteroatom incorporation into substrates to activate otherwise inert atoms and the inherent high activity of metals in catalyzing the oxygen reduction reaction.25 Moreover, given that the objective of the 2e− ORR is to supplant the conventional AO process, the setups for large-scale H2O2 production assume critical importance. It is essential to design a logical setup that can maximize catalyst utilization and facilitate continuous production.
The exploration of electrochemical synthesis of H2O2 through a 2e− ORR represents a burgeoning research domain. Despite the abundance of comprehensive reviews on the 2e− ORR, the focus on integrating catalyst engineering involving basic design principles with existing large-scale setups for H2O2 production remains limited. This review discusses the reaction mechanisms, performance assessment, design principles of catalysts, and setups for large-scale H2O2 production.
2. Reaction mechanisms of H2O2 production via 2e− ORR
In the cathode of an electrochemical cell, the electrochemical oxygen reduction reaction (ORR) can follow two distinct pathways in aqueous environments: 2e− ORR and 4e− ORR.25–28 In the 2e− ORR, H2O2 is the primary product, whereas in the 4e− ORR, the final product is H2O. These reactions are influenced by distinct catalysts and environmental conditions. Thus, the selection of the catalyst and reaction media play critical roles in determining the predominant reduction pathway. Typically, the ORR unfolds through either a dissociative or an associative pathway. The dissociative pathway involves breaking the O–O bond upon oxygen adsorption, leading to the formation of adsorbed oxygen (O), which is sequentially reduced to adsorbed hydroxide (OHads) and adsorbed water (H2Oads). Conversely, the associative mechanism retains the O–O bond, with the end products being either water (H2O) or H2O2, contingent upon the capacity of the catalysts to split the O–O bond within the *OOH intermediate. Regarding H2O2 production via the 2e− ORR, as illustrated in Fig. 1d, the O2 molecule initially attaches to the active sites, undergoing hydrogenation to form *OOH, which is finally reduced to H2O2. Conversely, if the O–O bond is broken, the resulting *O undergoes further reduction, leading to the unwanted formation of H2O. Thus, preserving the O–O bond and fine-tuning the adsorption energy of *OOH are essential steps to maximize H2O2 yield. For an ideal 2e− ORR catalyst, it is critical to strongly adsorb O2 molecules to initiate *OOH formation. Moreover, the adsorption at active sites should be sufficiently weak to enable straightforward desorption of *OOH, leading to H2O2 production. In summary, the production of H2O2 through the 2e− ORR is a dual-step hydrogenation process involving the generation of *OOH intermediates and H2O2 at active sites, see eqn (1) and (2).| | | * + O2 + H+ + e− = *OOH | (1) |
| | | *OOH + H+ + e− = H2O2 + * | (2) |
where * represents the active sites.
According to the discussion above, the adsorption energy of *OOH determines the 2e− ORR pathway. The limiting potential (UL) is defined as the minimal potential at which all steps of the reaction proceed in a thermodynamically favorable direction. Consequently, the theoretical overpotential can be determined by calculating the difference between the limiting potential and equilibrium potential.29 The graphical representation of ULversus (vs.) ΔG*OOH for the 2e− ORR demonstrates a volcanic shape. At the extreme left of the volcano plot, the *OOH intermediate exhibits overly strong adsorption to the active sites, steering the ORR mechanism towards a 4e− process. This indicates a reduced selectivity for the 2e− ORR pathway, albeit with elevated activity. Conversely, at the extreme right of the volcano plot, the *OOH intermediate on the surface of active sites exhibits insufficient surface binding, resulting in heightened selectivity for the 2e− ORR pathway, albeit at the cost of diminished activity. Thus, to achieve optimal activity and selectivity for the 2e− ORR, active sites should ideally be positioned near the peak of the volcano plot.
In addition, it should also be noted that the catalytic process of 2e− ORR varies in different electrolytes. The half-cell reactions can be expressed as follows (eqn (3a) and (4a)).30 However, studies on the H2O2 production mechanism in neutral electrolytes are scarce. In such electrolytes, both hydroxyl species and protons are sparse. Therefore, further investigation into the ORR pathway is necessary to uncover the impact of pH on H2O2 production. The occurrence of 2e− ORR inevitably accompanies the 4e− ORR, as shown in eqn (3b) and (4b).
| | | Acid media: O2 + 2H+ + 2e− = H2O2, E0 = 0.70 V vs. RHE at PH < 11.6 | (3a) |
| | | O2 + 4H+ + 4e− = 2H2O, E0 = 1.23 V vs. RHE | (3b) |
| | | Basic media: O2 +2H2O + 2e− = H2O2 +2OH−, E0 = 0.74 V vs. RHE at PH = 13 | (4a) |
| | | O2 + 2H2O + 4e− → 4OH−, E0 = 1.23 V vs. RHE | (4b) |
where
E0 represents the standard equilibrium potential, while RHE refers to the reversible hydrogen electrode.
3. Performance assessment: rotating-ring disk electrode and applicable devices
The utilization of the rotating ring-disk electrode (RRDE) technique constitutes a pivotal methodology in the comprehensive evaluation of H2O2 electrosynthesis and the elucidation of oxygen reduction reaction (ORR) mechanisms. This advanced approach enables the accurate determination of H2O2 yields resulting from the ORR by initially enabling the reduction of molecular O2 at the disk electrode, thereby generating both H2O and H2O2. Subsequently, the rotation of the disk electrode expedites the transfer of produced H2O2 to a platinum ring electrode, where H2O2 is oxidized back to O2. This process facilitates the quantification of H2O2 through the generation of a measurable ring current. Elevated currents observed at the disk electrode are indicative of robust ORR activity, whereas augmented ring currents indicate significant H2O2 production, thereby evidencing the efficacy of H2O2 production. By assessing the currents measured at both the disk and ring electrodes, the number of electrons transferred (n) and the selectivity towards H2O2 production (%) can be meticulously calculated, as shown in eqn (5)–(7).| | 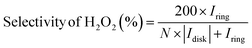 | (5) |
| | 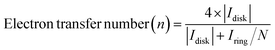 | (6) |
| |  | (7) |
where Iring, Idisk, and N represent the ring current, disk current, and the collection efficiency of the RRDE, respectively. The n values approaching 2 indicate reactions predominantly following a two-electron pathway, resulting in H2O2 production, while n values nearing 4 imply the predominance of a four-electron pathway leading to the formation of H2O. The applied potential on the ring electrode is set at 1.2 V (vs. RHE) to quantify the H2O2, where the H2O2 is oxidized back to O2.31
The true efficiency of catalysts for H2O2 production can be assessed through electrolyzer setups. Such configurations provide a practical measure of H2O2 electrosynthesis performance, offering a more applicable comparison to the RRDE method. This approach allows for the evaluation of catalyst stability and the potential for large-scale production over prolonged periods. The concentration of H2O2 can be measured using conventional titration methods, such as Ce(SO4)2 titration or iodometric titration. The H2O2 concentration obtained in applicable devices can be ascertained through the transformation of yellow Ce4+ ions to colorless Ce3+ ions, facilitated by H2O2, allowing for the determination of H2O2 concentration based on the change in Ce4+ ion concentration before and after the reaction (see eqn (8)–(10)). In iodometric titration, the process involves the oxidation of I- ions into I2 within an acidic environment. Following this, the introduction of a starch aqueous solution to the solution containing I2 results in the formation of a dark blue complex. To conclude the titration and evaluate the H2O2 concentration, Na2S2O3 is gradually added to the dark blue mixture until the color vanishes (see eqn (11)–(13)).
| | | 2Ce4+ + H2O2 = 2Ce3+ + 2H+ + O2 | (8) |
| | 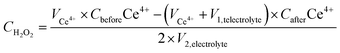 | (9) |
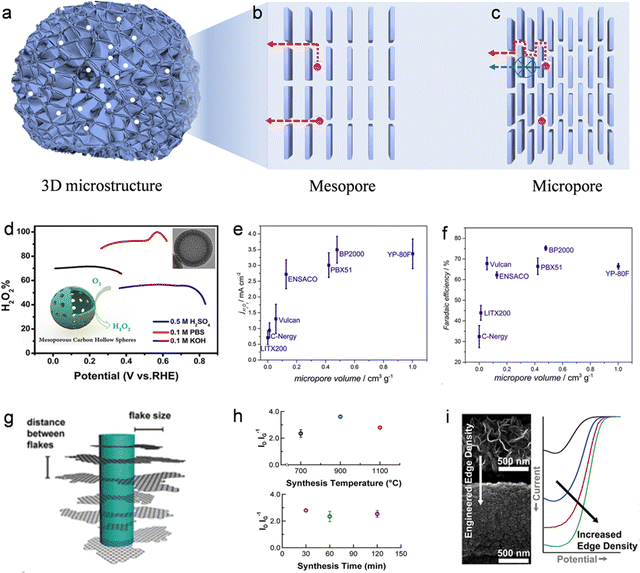
| | 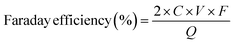 | (10) |
where
CH2O2 is the real H
2O
2 concentration in an applicable device,
VCe4+,
V1,telectrolyte,
CbeforeCe
4+CafterCe
4+, and
V2,electrolyte represent the volume of the used Ce(SO
4)
2 solution, the volume of electrolyte taken from the electrolyzer, the initial concentration of Ce(SO
4)
2, the final concentration of Ce(SO
4)
2 after H
2O
2 is added, and the total electrolyte volume, respectively. In
eqn (10),
C represents the concentration of H
2O
2,
V denotes the volume of the electrolyte,
F stands for the Faraday constant (96
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
485 C mol
−1), and
Q refers to the charge consumed.
| | | H2O2 +2KI + H2SO4 → I2 + K2SO4 + 2H2O | (11) |
| | | I2 + 2Na2S2O3 → Na2S4O6 + 2NaI | (12) |
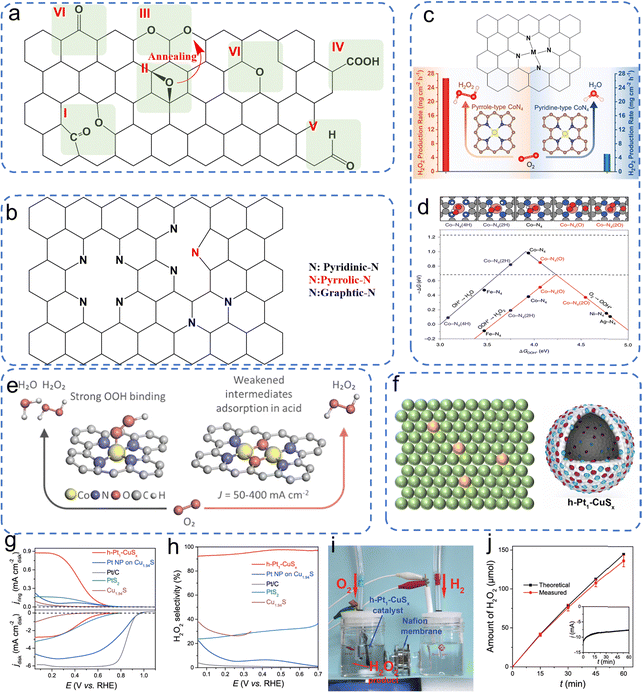
| | 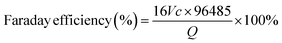 | (13) |
where the
V,
c, and
Q represent the consumed volume of Na
2S
2O
3, the concentration of Na
2S
2O
3, and the applied charge during the electrolysis, respectively.
4. Strategies to design catalysts for the 2e− ORR
4.1 Microstructure engineering
The engineering of microstructures of materials plays a vital role in boosting catalytic performance due to their influence on the specific surface area (SSA), exposure of catalytic active sites, mass diffusion, and conductivities. The microstructures are influenced by both their dimensions and porosities. Materials processing similar chemical compositions with different dimensions and porosities usually display completely different features in terms of catalytic performance. Therefore, understanding the effects of microstructures can lead to an effective material design. For example, three-dimensional (3D) materials predominantly contain porous structures including macropores, mesopores and micropores. Such controllable pore structures can purposely regulate the 2e− ORR coordination of reaction intermediates by changing the exposure of active sites and the retention time of H2O2 in the specialized pore size, thus determining the overall performance of a catalyst.22,23 Regarding materials with two-dimensional (and one-dimensional) structures, such as graphene (and typical carbon nanotubes (CNTs)), they are distinguished by distinctive flake-like and linear morphologies, wherein the active sites are constituted by surface functional groups and/or structural defects. Furthermore, the two-dimensional (2D) and one-dimensional (1D) substrates serve as efficacious carriers for electron transfer processes. In this section, the effects of microstructures of materials regarding the dimensions and porosities on the 2e− ORR performance will be comprehensively discussed.
Regarding the materials with 3D structure and sufficient pore structure, the 2e− ORR performance of a material is affected by the SSA and types of pore structure in 3D materials, as shown in Fig. 2a. Chai et al. reported the synthesis of mesoporous carbon hollow spheres exhibiting an exceptionally high specific surface area (SSA) of 1362.4 m2 g−1. These spheres displayed a remarkably high selectivity for H2O2 production, reaching 99.9% in a neutral phosphate-buffered saline (PBS) electrolyte,19 as shown in Fig. 2d. The unique hollow morphology and the structural features of the radial channels contribute to an increased SSA, which in turn facilitates mass transfer during the oxygen reduction reaction process. In an analogous research, a hierarchically porous carbon material was developed, boasting an ultrahigh SSA exceeding 2500 m2 g−1. It is claimed that the SSA plays a pivotal role in enhancing the performance of H2O2 production.21 The optimal porous architecture enhances the exposure of electroactive sites, thereby augmenting the utilization efficiency of these sites for H2O2 production. Therefore, the 3D structure with high SSA is beneficial for both exploring the actives and accelerating the mass transfer for H2O2 production. The inherent relation between the structural features and H2O2 production performance is deeply studied by analyzing the states of H2O2 in the pore structure because materials processing high SSA include macropores, mesopores and micropores. In the realm of catalysis, the presence of mesopore structures within catalysts significantly accelerates the migration route of H2O2 due to enhanced mass transport capabilities, in contrast to the restricted mobility inherent in micropore structures.22,23 This phenomenon is illustrated in Fig. 2b and c: the H2O2 generated in the mesopore structure can easily diffuse into the bulk electrolyte, reducing the decomposition of H2O2 and thus enhancing the production. However, in micropore structures, H2O2 is either transiently held within micropores or compelled to traverse extended distances to reach the catalyst surface. This distinct discrepancy between mesopore and micropore configurations markedly influences the residency duration of H2O2 within the catalyst structure. For example, nitrogen-doped carbons with a mesoporous architecture reveal a notable H2O2 selectivity exceeding 90% in acidic electrolytes. Such remarkable H2O2 selectivity can be attributed to the fast mass transfer and minimized contact time of H2O2 within these mesoporous structures.22 In another research, Chuanxin He et al. highlighted that micro/mesoporous carbon architectures can elevate the local pH and expedite the generation of intermediates (*O2 and *OOH) via inducing a localized pseudo-alkaline environment, thereby enhancing the efficiency of H2O2 production.32 Furthermore, the pore volume also plays a crucial role in determining the selectivity towards H2O2 production in the 2e− ORR. As documented in Fig. 2e and f, Chorkendorff and colleagues elucidated that carbon materials endowed with substantial microporous volumes manifest a pronounced selectivity for H2O2 production.24 Specifically, Fig. 2e and f demonstrate an increase in H2O2 current density concomitant with the augmentation of micropore volume, underscoring the beneficial impact on 2e− ORR efficiency. The production volume of H2O2 is positively correlated with the extent of microporous volume, implying that these micropores act as auxiliary active sites, enhancing the catalytic process.
 |
| | Fig. 2 Schematic of the (a) 3D microstructure, (b) mesopore and (c) micropore. (d) H2O2 selectivity of mesoporous carbon hollow spheres. Reproduced from ref. 22. Copyright (2014) American Chemical Society. (e) Current densities of H2O2 and (f) faradaic efficiency as a function of micropore volume. Reproduced from ref. 24. Copyright (2018) Elsevier. (g) Ordered structure of graphene flakes on carbon nanowires. (h) Representative Raman spectra of NT-3DFGs. (i) SEM image and representative rotating disk electrode linear sweep voltammetry (LSV) curves (n = 3) for glassy carbon (black) and NT-3DFGs. Reproduced from ref. 34.Copyright (2020) American Chemical Society. | |
In addition to the SSA and pore structure, microstructural engineering also involves the design of geometrical structures, which is mainly related to the rational utilization of active sites. As shown in Fig. 2g, both commonly used graphene and CNTs, when utilized as 2e− ORR catalysts, are functionalized with active epoxy, hydroxyl, carbonyl groups, and the defects.2,11–13,33 The rational design of ordered microstructures has been regarded as an effective strategy to improve the H2O2 production in 2e− ORR by taking full advantage of the edge sites with functional groups. A graphene-based hybrid nanomaterial, known as nanowire-templated out-of-plane three-dimensional fuzzy graphene (NT-3DFG), has been developed.34 This material is characterized by its unique arrangement of independent graphene layers on nanowires, as depicted in Fig. 2g. The innovative composite structure not only prevents the graphene layers from stacking but also significantly enhances the accessibility of the edge sites on graphene flakes. Adjusting the density of these out-of-plane graphene flakes by simply changing the annealing temperature and time (Fig. 2h) allows for the modulation of both the density of active sites for ORR and the overall ORR activity of NT-3DFG. The finely regulated NT-3DFG sample exhibits exceptional 2e− ORR performance, including an onset potential of 0.79 ± 0.01 V vs. RHE and a high selectivity for hydrogen peroxide production (94 ± 2% H2O2). Thanks to the optimized arrangement of vertically aligned graphene on nanowires, the limiting current density for the oxygen reduction reaction experienced an enhancement, concurrent with the increase in edge density (Fig. 2i).
4.2 Heteroatom doping engineering
Heteroatom doping engineering involves introducing alloplastic atoms into an inert substrate, which can regulate the local electronic structure to activate the inert atoms, thus enhancing the catalytic activity.25 In addition, heteroatoms can also introduce active functional groups, which function as active sites for 2e− ORR, into the substrate to achieve a high-performance catalyst, such as the oxygen-bearing functional groups on the carbon-based materials. Generally, heteroatom doping engineering involves doping non-metallic/metallic heteroatoms into carbonaceous/non-carbonaceous materials. Regarding the heteroatom-doped carbonaceous materials, elements such as boron (B), nitrogen (N), oxygen (O), fluorine (F), phosphorous (P), and sulfur (S) are doped into carbon substrates to activate the electrochemically inactive atoms by regulating the electronic conductivity or introducing defect sites.7,35,36
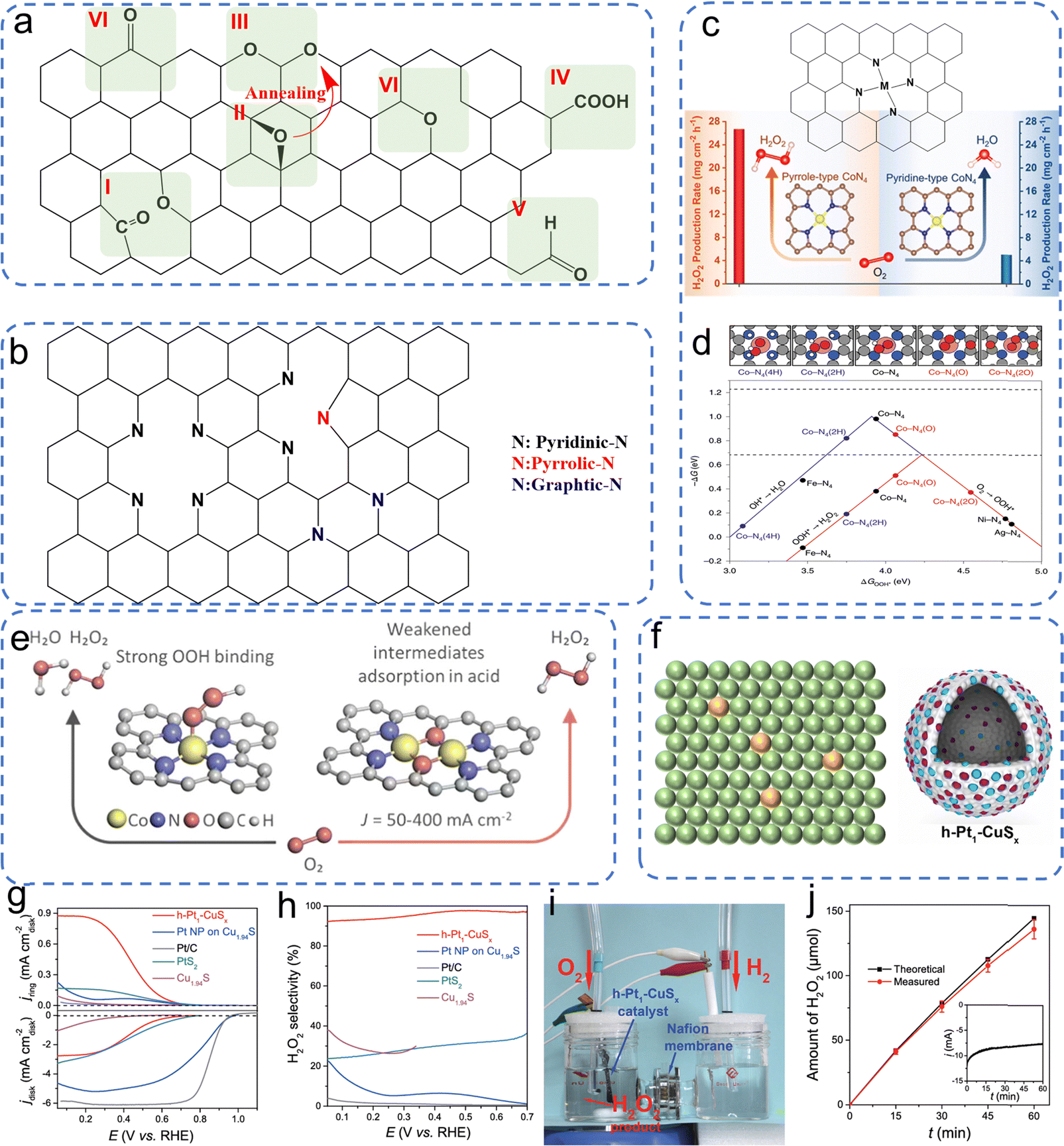
Great attention has been paid to the oxygen-doped carbonaceous materials (O–C). The oxygen incorporation into carbons can greatly localize the π electron distribution, achieving high H2O2 selectivity towards the 2e− ORR, while the activity of O–C is limited because of the high resistance of surface charge transfer. Studies on the effects of oxygen dopants on the 2e− ORR mainly focus on identifying the specific oxygen-bearing functional groups. As reported in numerous works, as shown in Fig. 3a, the quinone functions, carbonyl, and ether/epoxy can function as active sites for the 2e− ORR.13,17,28,37–39 Typically, the catalytic activities of these oxygen-bearing groups are different in various chemical coordination environments. For example, when oxygen atoms were introduced into carbon to prepare quinone-rich, carbonyl-rich, and etheric-rich carbon,38 the quinone functional groups, as shown in Fig. 3a (area I), were confirmed as the most active site compared to the other oxygen groups. More importantly, the catalytic activity of an oxygen group is highly dependent on the geometric position. For example, the C–O groups located at the base of few-layered mildly reduced graphene oxide (mrGO) gradually transform into ordered arrangements of ring ether groups at the edges of the mrGO when annealed at 600 °C (see Fig. 3a, area II and III). Such rebuilt ether groups from the basal plane position to the edge position resulted in elevated peroxide formation activity.37 Besides, the other oxygen group, –COOH, was experimentally confirmed as an active site for 2e− ORR. Chemical titration experiments using PH, BA, and 2-BrPE as titrants to specifically react with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH, and COOH groups, respectively, were performed to identify the 2e− ORR active sites.17 The results indicated that the COOH groups on the carbon sheet contributed most to the 2e− ORR performance, and DFT calculations demonstrated that the COOH on the edge (see Fig. 3a, area IV) has the highest catalytic activity towards 2e− ORR. These studies have thoroughly investigated the types of oxygen groups and their geometric positions on the graphene base, which implies that the precise design of catalysts significantly affects the 2e− ORR performance.
O, C–OH, and COOH groups, respectively, were performed to identify the 2e− ORR active sites.17 The results indicated that the COOH groups on the carbon sheet contributed most to the 2e− ORR performance, and DFT calculations demonstrated that the COOH on the edge (see Fig. 3a, area IV) has the highest catalytic activity towards 2e− ORR. These studies have thoroughly investigated the types of oxygen groups and their geometric positions on the graphene base, which implies that the precise design of catalysts significantly affects the 2e− ORR performance.
 |
| | Fig. 3 Illustration of (a) oxygen dopant, (b) nitrogen dopant and (c) metal–Nx site on a graphene substrate. Reproduced from ref. 52. Copyright (2022) American Chemical Society. (d) DFT calculations of different metal–Nx sites on the volcano plot. Reproduced from ref. 51. Copyright (2020) Springer Nature. (e) Illustration of diatomic cobalt sites on a carbon substrate. Reproduced from ref. 57. Copyright (2024) American Chemical Society. (f) Illustration of atomically dispersed metallic atoms on non-carbonaceous substrates and schematic representation of the structural evolution of h-Pt1-CuSx. (g) Linear sweep voltammetry (LSV) curves were obtained utilizing a rotating ring-disk electrode (RRDE) in an electrolyte solution of 0.1 M O2-saturated HClO4. (h) The selectivity towards H2O2. (i) The electrochemical device employed for the electrosynthesis of H2O2. (j) The quantification of H2O2 produced over time. Reproduced from ref. 20. Copyright (2019) Elsevier. | |
In addition to the O dopant, the nitrogen (N) dopant is also widely studied for 2e− ORR due to the big difference in electronegativity between the nitrogen atom and carbon atom. Regarding nitrogen-doped carbonaceous materials, the main active sites contain pyridinic, pyrrolic, and graphitic N (see Fig. 3b). The pyridinic N has a lone pair of electrons, which implies it can be easily reduced or protonated, especially in acidic electrolytes.40 Such a characteristic hinders the capacity to attenuate the O–O bond, resulting in a predisposition towards the formation of H2O2. However, in neutral or alkaline pH environments, there was a notable reduction in selectivity for H2O2, which can be attributed to the enhanced feasibility of water (H2O) generation via the cleavage of the O–O bond. Consequently, carbons dominated by pyridinic sites demonstrated enhanced performance in the 2e− ORR within acidic electrolytes.41 Nevertheless, both pyrrolic N and graphitic N also demonstrated their catalytic activity towards the 2e− ORR. Specifically, the catalytic activity in pyrrolic N-dominated carbons arises from the positive charge on the adjacent carbon, induced by the high electronegativity of the pyrrolic N within the carbon network.7,42 In the case of graphitic nitrogen, it donates electrons to the π-conjugated system of carbon–carbon bonds, thereby facilitating the adsorption of molecular oxygen. This electron donation subsequently leads to a proton-coupled electron transfer, facilitating the formation of the *OOH intermediate species.43 In addition to the most widely explored O and N dopants, other non-metallic elements including boron (B), fluorine (F), phosphorus (P), and sulfur (S) were introduced into the carbon base to enhance the 2e− ORR performance.35,40,44–48
Moreover, the incorporation of metallic dopants has significantly attracted the interest of researchers. This heightened focus is largely attributed to the comparatively limited catalytic activity of non-metallic dopants within carbon-based frameworks, despite their pronounced selectivity for H2O2 production. Recent research has delved into highly active single-atom catalysts, which are composed of atomically dispersed metals and substrates. This category encompasses non-precious metals like Fe, Co, Ni, and Mn,49–53 as well as precious metals such as Pt, Os, Ir, Rh, Ru, and Pd.20,54–56 Generally, the active metallic atoms (M) are combined with nitrogen atoms, as shown in Fig. 3c, to construct M–Nx sites on the carbon substrates. According to the DFT calculations shown in Fig. 3d, many metals, for example, nickel (Ni) and silver (Ag) exhibit a preference for producing H2O2 rather than H2O compared to metals like ruthenium (Ru), iron (Fe), and cobalt (Co), which is attributed to their enhanced protophilic properties for H2O2 production. While for the Ru, Fe, and Co active metal centers, their preference lies in breaking the *OOH intermediates to *O, which leads to H2O formation. Based on the calculated volcano plot, almost no active M–N4 sites are close to the ideal volcano plot for H2O2 production (ΔGOOH* = 4.22 eV). It is beneficial to subtly adjust the M–N4 site to retain most of its significant catalytic efficiency in the generation of H2O2. Optimization of the synergy between the metal center and the surrounding atomic configuration, coupled with alterations to the metal center of the M–N4 moiety, is necessary to achieve the desired ΔGOOH* value and enhance the production of H2O2. For example, the Co–N4 moiety can be tailored by fine-tuning its surrounding oxygen atomic configuration; as shown in Fig. 3d, the new site Co–N4(O), incorporated in nitrogen-doped graphene (NG), demonstrated optimized H2O2 production.51 Besides, atomic Co–Nx–C sites on oxygen group-functionalized carbons, denoted as Co–POC–O, were synthesized through the processes of pyrolysis and oxidation of a cobalt-coordinated porphyrin framework precursor, demonstrating remarkable performance in the electrochemical synthesis of H2O2.49 Interestingly, Yijin Kang et al. found that the pyrrole-type Co–N4 primarily facilitates the 2e− ORR, whereas the pyridine-type Co–N4 is instrumental in the 4e− ORR (Fig. 3c). Significantly, the pyrrole-type Co–N4 exhibited a remarkable H2O2 selectivity of 94% under acidic conditions. The observed enhancement in catalytic performance is ascribed to a diminished interaction between O2 and the reactive intermediate *OOH, thereby augmenting H2O2 production.52 Additionally, subsequent research has demonstrated that incorporating a second cobalt atom into the Co–N4 coordination to create novel diatomic cobalt sites, as illustrated in Fig. 3e, attenuates the binding affinity of *OOH and *HOOH.57 These newly engineered diatomic cobalt sites thus inhibit the cleavage of the O–O bond while concurrently mitigating the parasitic hydrogen peroxide reduction reaction (H2O2RR). Although atomically dispersed metals on carbon substrates are mostly reported, for example, CuSx and titanium nitride, they can be used as substrates to load active atoms for H2O2 production, in which the active atoms are surrounded by other metallic atoms or nonmetallic atoms (Fig. 3f).20,55 Fabrication of hollow nanospheres by atomically dispersing platinum (Pt) on an amorphous CuSx support (h-Pt1-CuSx), featuring a substantial concentration of single atomic Pt sites (24.8 at%), was reported (Fig. 3f).20 The active Pt atoms facilitate the activation of the inert CuSx substrate, while CuSx concurrently prevents the aggregation of Pt atoms. This novel catalyst demonstrates a high ability to consistently convert O2 into H2O2 with selectivity ranging between 92% and 96% across a broad potential window of 0.05 to 0.7 V vs. RHE in HClO4 electrolyte (Fig. 3g and h). Moreover, an electrochemical device capable of directly synthesizing H2O2 from H2 and O2 was developed, achieving H2O2 production rates as high as 546 ± 30 mol kgcat−1 h−1 (Fig. 3i and j).
4.3 Metal hybridization engineering
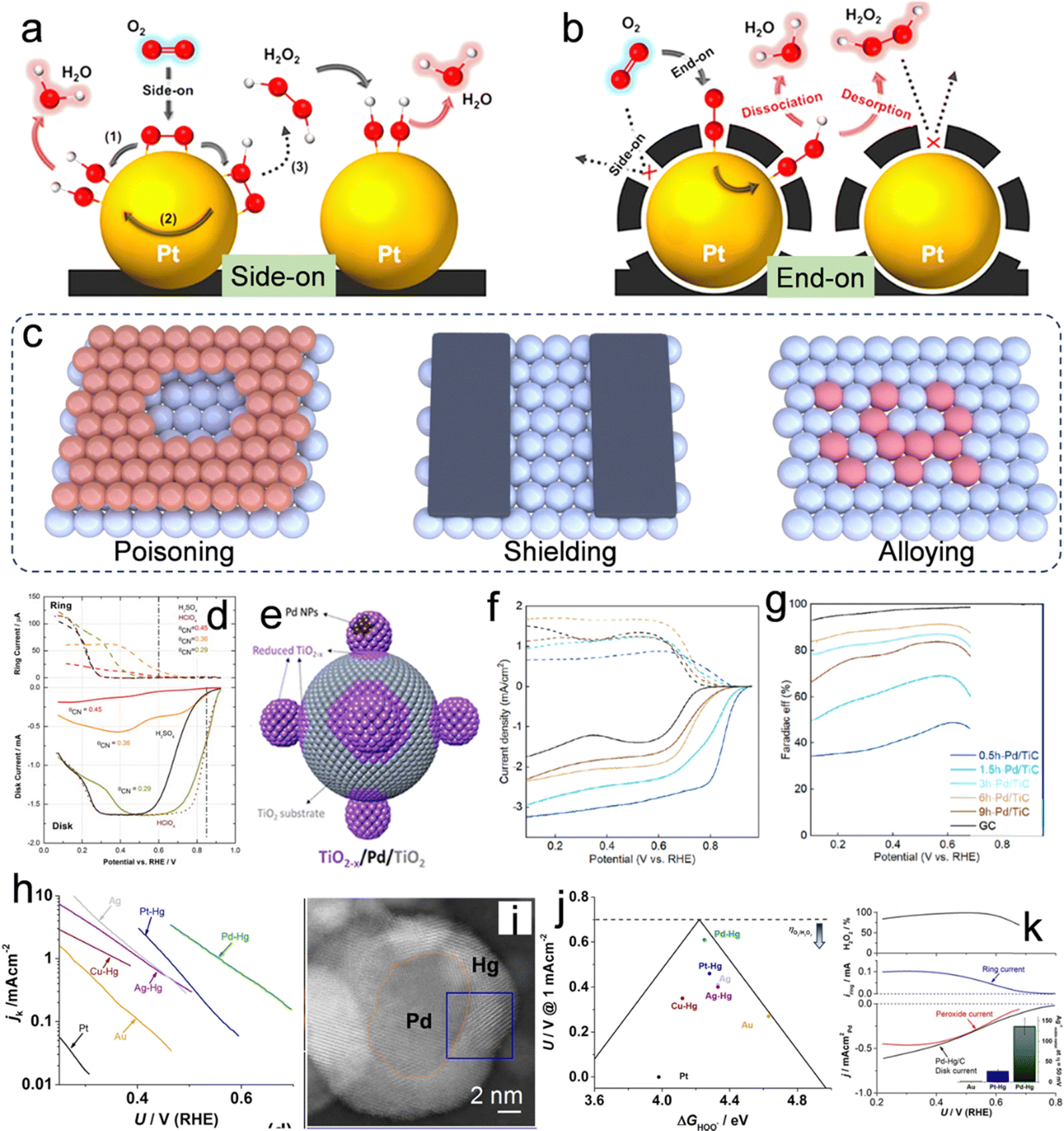
Metallic catalysts, such as Pt and Pd, demonstrate notable catalytic efficiency in ORR, while predominantly facilitating the four-electron (4e−) pathway.6 The strategic design of an active metallic catalyst capable of suppressing the 4e− pathway while promoting the 2e− pathway holds substantial importance. Hence, gaining an in-depth understanding of the interactions between molecular oxygen (O2) and the catalytic surface is crucial for the development of effective catalysts. As elucidated in “Section 2: Reaction mechanisms of H2O2 production via 2e− ORR,” the protonation of O2 to form *OOH intermediates represents a significant hurdle in the 2e− ORR process. Therefore, the initial step for the 2e− ORR is when the adsorbed O2 on the catalytic surface captures a proton to form *OOH. The adsorption of O2 manifests in two distinct configurations: dissociative side-on adsorption and associative end-on adsorption.58Fig. 4a and b illustrate that the side-on adsorption mechanism extends the bond length and attenuates the double bond in O2, yielding H2O as the product. However, the end-on adsorption mechanism leads to the adsorption of oxygen in the form of *OOH, which is capable of producing both H2O2 and H2O. Therefore, the principal challenge in the design of 2e− ORR catalysts lies in altering the O2 adsorption mode from a “side-on” to an “end-on” configuration. To effectuate the transition from a “side-on” to an “end-on” adsorption configuration, two strategic approaches are proposed: (i) surface poisoning/shielding, which involves the deliberate obstruction of exposed ensemble sites with inert species (Fig. 4c), effectively altering the surface chemistry to favor “end-on” adsorption; and (ii) the alloying of the active metal with an inert metal, which facilitates the isolation of active metal atoms (Fig. 4c), thereby providing a distinct geometric arrangement conducive to the desired adsorption mode.
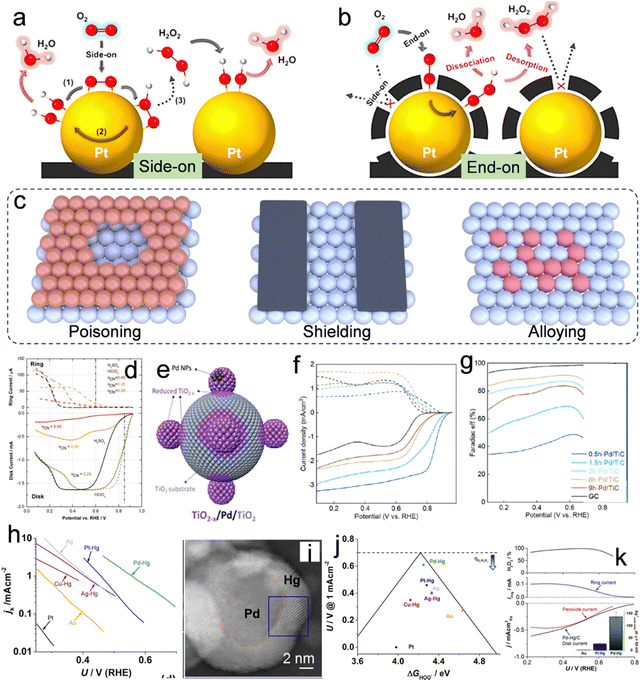 |
| | Fig. 4 O2 adsorption on Pt/C and C(Pt)/C in the form of (a) side-on configuration and (b) end-on configuration. Reproduced from ref. 58. Copyright (2014) American Chemical Society. (c) Illustration of surface poisoning and shielding on the surface of the metal and metallic alloying. (d) Polarization curves obtained from RRDE experiments for the ORR on a Pt (111)–CNad electrode with varying CNad coverages, conducted in a 0.05 M H2SO4 solution. Reproduced from ref. 59. Copyright (2015) Elsevier. (e) Illustrative schematic demonstrating the encapsulation process of a TiO2−x overlayer on Pd NPs via ball milling. (f) LSV curves obtained using an RRDE and corresponding (g) Faraday efficiency for the production of H2O2. Reproduced from ref. 64. Copyright (2022) American Chemical Society. (h) Kinetic current density for H2O2 as a function of the applied potential. (i) TEM images of the prepared Pd-Hg alloy. (j) The potential needed to achieve a kinetic current density of 1 mA cm−2 for H2O2 on polycrystalline catalysts as a function of the calculated *OOH binding energy. (k) LSV curves obtained using an RRDE and corresponding H2O2 selectivity. Reproduced from ref. 68. Copyright (2014) American Chemical Society. | |
Partial occlusion of the active metal surface can be achieved through the employment of specific poisonous ions, including Cl−, SCN−, CN−, Br− and S2−, as well as other entities such as amorphous carbon, TiO2, etc., to enhance H2O2 selectivity.1,6 Introducing cyanide ions (CN−) into the H2SO4 electrolyte modifies the ORR selectivity towards the 2e− pathway while reducing the overall ORR activity (Fig. 4d).59 This modification can be attributed to changes in the configuration of species adsorbed on the Pt surfaces of the Pt(111) catalyst. Besides, other ions, for example, Cl−, Br−, and S2− were also found to poison the Pt site in acidic electrolytes due to their strong adsorption ability.60–62 Although ion poisoning can effectively facilitate the 2e− ORR pathway, the introduction of heteroatom ions into electrolytes and reduced overall activity of the catalysts remain the challenge. Therefore, the direct design of active materials with “self-shielding” characteristics for enhanced 2e− ORR performance is another effective strategy. These “self-shielding” materials are designed based on the principle of changing the “side-on” type to the “end-on” type. For example, when Pt nanoparticles were coated with an amorphous carbon layer (ACL), as shown in Fig. 4b, it was observed that the composite of Pt@ACL facilitated the adsorption of O2 in an end-on configuration, significantly improving the performance of H2O2 electrosynthesis.58 Importantly, the catalyst demonstrated enhanced catalytic stability, which is likely attributed to the shielding of Pt catalysts by the carbon layers enveloping them. This protective mechanism effectively mitigates the risks of leaching and ripening under conditions of high corrosivity during operation.58 Similarly, Markovic et al. reported a Pt(111) catalyst enveloped by a calix[4]arene. Because of the strong inhibition of O2 adsorption and reduced exposure of Pt sites, the ORR activity on such catalysts is reduced with the concomitant formation of H2O2.63 Furthermore, an intriguing category of support effect, which leverages the advantages of encapsulation, is the strong metal–support interaction.64 As shown in Fig. 4e, the TiO2−x/Pd/TiO2 sample was prepared by using an unprecedented time-dependent mechanical milling method. During this process, the TiO2 substrate was reduced to TiO2−x overlayers and coated with Pd nanoparticles (Pd NPs). This process facilitated a valence band restructuring and a reduction in the d-band center of the surface Pd atoms, significantly influencing the electronic and geometric properties of the catalyst. These alterations led to optimized adsorption energies of OOH* intermediates. A progressive decrease in the limiting current density of the disk current, accompanied by an augmentation in the ring current, was discernible from the 0.5 h-Pd/TiC to the 6 h-Pd/TiC samples. This trend suggests a transition from the 4e− ORR pathway to the 2e− ORR pathway (Fig. 4f). The augmentation in the faradaic efficiency of H2O2 production, escalating from approximately 35% to exceed 90%, substantiates this observation (Fig. 4g).
Analogous to the surface poisoning/shielding strategy, which modulates the performance by altering the direct contact interface between the catalytic sites and O2, metallic alloying, which involves combining an active metal with an inert metal that exhibits weak interaction with O2, can isolate the highly active metals (For example, Pt and Pd dominate the 4e− ORR pathway), and alter the adsorption geometry of the O2 molecule. For example, noble metals and their alloys, such as Pd–Au,65,66 and Pd–Hg,67,68 are currently state-of-the-art catalysts that demonstrate near-zero overpotential for oxygen reduction along with an extremely high H2O2 selectivity of up to 98%. For example, Au NPs embellished with Pd atoms have exhibited an augmented production of H2O2. Through an investigation into the influence of Pd content on the Au surface, it was discerned that Pd does not segregate on the Au NP surface until its concentration surpasses 8%; at this point the selectivity for H2O2 production can reach 95% in 0.1 M HClO4.69 Nevertheless, an excessive concentration of Pd leads to the formation of Pd ensemble sites, which preferentially dissociate the oxygen bond (O–O), thereby diminishing the selectivity for H2O2 production. Subsequent research has revealed that isolating Rh, Pd, Pt, or Rh sites on Au (111) surfaces enhances the performance of the 2e− ORR relative to pure gold.70 Furthermore, E. L. Stephens systematically studied the non-noble metallic Hg alloys for 2e− ORR.68 As shown in Fig. 4h, the curves delineating the partial current densities associated with hydrogen peroxide production, relative to the applied potential, were plotted. The findings underscore the critical impact of catalyst composition on the requisite overpotential. Remarkably, the Pd–Hg alloy demonstrated the highest current at minimal overpotential. However, Pt–Hg, Ag–Hg, and Cu–Hg demonstrated a sequential escalation in overpotential. The Pd–Hg alloy exhibits a core–shell configuration, wherein Pd constitutes the core and Hg forms the shell, as illustrated in Fig. 4i. According to density functional theory (DFT) calculations presented in Fig. 4j, the Pd–Hg alloy is positioned near the apex of the volcano plot, demonstrating the minimum overpotential or heightened catalytic activity. The LSV curves shown in Fig. 4k further confirm the high H2O2 selectivity, surpassing 95% within the voltage window of 0.35 to 0.55 V vs. RHE in 0.1 M HClO4.
5. Designing setups for H2O2 production
To realize industrial production of H2O2via the electrochemical synthetic strategy, rationally designing setups is another crucial engineering aspect that aims to achieve an efficient device for stable H2O2 products. Therefore, the designed setup should maintain the original active sites for oxygen adsorption, ensure effective mass migration, facilitate excellent H2O2 desorption and exhibit good stability.71
Although the commonly used RRDE method can maximize the advantages of a catalyst, the H2O2 production is so limited that it greatly hinders the industrial production. The one-pot configuration, depicted in Fig. 5a, employs the catalyst-coated carbon cloth directly as the working electrode, alongside a graphite rod serving as the counter electrode and an additional reference electrode.11 The operational conditions for this setup closely mimic those of the RRED system. Li et al. studied H2O2 production using this one-pot setup reactor. In this system, the production rate of H2O2 reached 115.32mmol (gcat h)−1 at 0.5V vs. RHE, and the Faraday efficiency (FE) reached 89.0%.11 However, the generated H2O2 is present throughout the electrolyte, which can be decomposed on the anode surface.72 Nevertheless, an effective approach to prevent the decomposition of H2O2 at the anode involves coupling it with an anode that possesses a hydrogen oxidation reaction (HOR) functionality, capable of oxidizing H2O into H2O2. In a recent study, Xia et al.73 elucidated the elevated efficiency of a membrane-free flow cell designed for the electrocatalytic production of H2O2. They observed a significant increase in FE to 200%. This remarkable enhancement is attributed to the application of hydrophilic carbon fiber paper, which functions dually as both the cathode and the anode, facilitating the concurrent generation of H2O2. An alternative strategy to mitigate the decomposition of H2O2 at the anode involves employing a membrane to segregate the cathode and anode chambers. In a similar study, Yamanaka et al. conducted a study on H2O2 production using an H-cell in a one-pot batch reactor (Fig. 5b), where they noted a pronounced degradation in H2O2 electrosynthesis efficiency in comparison to experiments conducted in an H-cell equipped with a membrane.72 The integration of a membrane within the H-cell effectively precludes the migration of H2O2 to the anode; however, the increased separation between the cathode and the anode results in an elongated ion diffusion path, thereby escalating solution resistance. Additionally, the limited solubility of O2 in liquid electrolytes further constrains the maximum rate of H2O2 production achievable. Furthermore, the discontinuous nature of H-cells and the accumulation of H2O2 on the electrode surface leads to further reduction of H2O2.
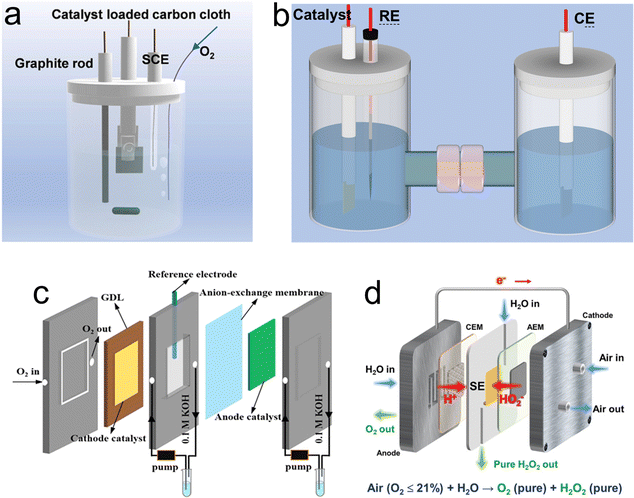 |
| | Fig. 5 Different types of setups for H2O2 production. (a) One-pot setup. Reproduced from ref. 11. Copyright (2022) Elsevier. (b) H-cell. (c) Illustration of the flow cell setup. Reproduced from ref. 17. Copyright (2021) Wiley-VCH GmbH. (d) solid–electrolyte (SE) cell system. Reproduced from ref. 78. Copyright (2023) Wiley-VCH GmbH. | |
To address the drawbacks of the one-pot setup and H-cell, gas-diffusion electrodes (GDEs) were developed by integrating a hydrophobic layer (gas-diffusion layer, GDL) and catalysts, as shown in Fig. 5c.17 The GDEs are meticulously engineered by coating the catalyst onto the gas diffusion layer, establishing three-phase interfaces (TPIs) when immersed in the electrolyte. The TPIs play a crucial role in ensuring the efficient transport of O2, thereby maintaining a consistent flow of O2 directly to the catalyst layer.74–77 This configuration is instrumental in optimizing the efficacy of the electrode by ensuring that the catalytic sites are uniformly accessible to O2. Through this strategic layering and interface design, the GDEs achieve a delicate balance between oxygen permeability and catalytic activity, thereby maximizing the overall performance of catalytic electrodes in H2O2 production. Utilizing this apparatus, the oxygen-doped carbon nanosheet (OCNS900) was affixed onto the gas diffusion layer (GDL) to fabricate the cathode for the oxygen reduction reaction (ORR), while an iridium dioxide (IrO2)-coated titanium sheet was employed as the anode for the oxygen evolution reaction (OER). The rate of H2O2 production was quantified to be 770 mmol g(catalyst h)−1 following an operational period of 1 hour.17 Interestingly, for the electrosynthesis of pure H2O2 solution, a solid–electrolyte (SE) cells system was constructed (Fig. 5d), avoiding the need for expensive and intricate post-treatment processes necessary for purifying H2O2 solutions in conventional setups illustrated in Fig. 5a–c. For example, both an anion exchange membrane (AEM) and a proton exchange membrane (PEM), specifically Nafion 117, were employed to facilitate anion and cation exchange, respectively. The poly(styrene-co-divinylbenzene) ion-exchange resin sulfonate (HEOWNS, P-72091, 75-200 mesh) was utilized as the solid electrolyte (SE). The cathode and anode sides were supplied with airflow and deionized water, respectively.78 Chuangang Hu and colleagues utilized N and S co-doped TCNTs (N,S-TCNTs) as cathode catalysts to achieve a high FE of 95.1% at a current density of 150 mA cm−2. Concurrently, at a current density of 300 mA cm−2, the highest production rate of 4.35 mmol cm−2 h−1 can be achieved.78
6. Conclusions and perspectives
In this review, an exhaustive summary of various engineering strategies aimed at identifying efficient 2e− ORR catalysts has been provided. This summary encapsulates discussions on microstructure engineering, heteroatom doping, and metal hybridization techniques. A 3D ordered mesopore structure is regarded as an efficient configuration for a good 2e− ORR catalyst as it increases the exposure of active sites and facilitates mass transport of H2O2. Besides, heteroatom doping is commonly used to guide the design of active sites, particularly in the field of carbonaceous materials. The inert atoms on the substrates are activated after the incorporation of heteroatoms (For example, B, N, O, F, and P) via regulating the local electronic structure and introducing active functional groups, thus inducing an enhanced 2e− ORR performance. Metal hybrid engineering, particularly involving noble metals such as Pt and Pd that are recognized for yielding highly active components for the 4e− ORR, is also used to induce a 2e− pathway via the utilization of poisoning/shielding and alloying. Transforming the O2 adsorption configuration from “side-on” to “end-on” on the metallic surface can leverage the geometric effects of the catalyst for H2O2 production. In addition, the setups including the one-pot setup, H-cell, flow cell setup and solid–electrolyte (SE) cell for large-scale H2O2 production are also discussed. Considerable attention has been dedicated to the engineering of catalysts and electrochemical reactors. Despite noteworthy progress, the scaling up of H2O2 electrochemical production remains nascent compared to the traditional anthraquinone process. Consequently, this area presents both challenges and opportunities for the industrial production of H2O2via 2e− ORR.
The development of efficient 2e− ORR catalysts that exhibit both high activity and selectivity remains a formidable challenge. Among the most extensively studied heteroatom-doped carbonaceous materials, an increase in selectivity typically coincides with a reduction in activity. Although the incorporation of metallic atoms into carbonaceous substrates has been shown to enhance the activity of H2O2 production up to a certain extent, it invariably leads to a compromise in H2O2 selectivity. More critically, a catalyst recognized for its superior 2e− ORR performance often exhibits a lower half-wave potential compared to its counterpart favored for 4e− ORR performance, suggesting that greater energy input is required to facilitate the electrosynthesis process. Additionally, although noble metallic alloys have shown high activity and selectivity, their high cost significantly limits their application on a large scale. Therefore, a profound understanding of the mechanisms governing the 2e− ORR is urgently required. Recent investigations have leveraged advanced in situ techniques, such as operando ATR-IR spectroscopy, in situ X-ray absorption near edge structure (XANES), in situ Raman spectroscopy, and density functional theory (DFT) calculations. These methodologies offer novel perspectives on pinpointing active sites and elucidating the structural evolution of catalysts. Such insights are paramount for the rational design of efficacious 2e− ORR catalysts.
Looking ahead, there is anticipation for the exploration of single-atom catalysts on low-cost substrates combined with simple preparation processes that ensure high activity, high selectivity, and affordability. Given that these catalysts will ultimately be used for large-scale H2O2 production, the integration of catalysts and reactors is also crucial. The methods for depositing these catalysts onto substrates need to be refined through techniques such as air spraying, ultrasonic spraying, and in situ growth, to ensure optimal ion/electron/mass transfer and gas flow. A reaction chamber that is resistant to the electrolyte and has a rational flow channel configuration, maximizing the conversion rate of O2, is vital for the electrosynthesis process. Therefore, there is a pressing need for focused efforts towards the development of reactors that not only offer superior catalytic performance but also possess chambers with excellent corrosion resistance.
In addition to the catalysts discussed above for H2O2 production, the electrolyte should also be focused on. Electrolytes provide the environments for the oxygen reduction reaction, which may affect the stability of the intermediate *OOH and finally regulate the H2O2 production. For example, Wu et al. found that cetyltrimethylammonium bromide (CTAB) can accelerate the 2e− ORR performance of carbon black (CB) in 0.1 M KOH electrolyte, while sodium dodecylsulfate (SDS) weakened the 2e− ORR performance due to the “pull-off” effect of the cationic surfactant.79 Furthermore, P. Strasser et al. observed notable cation (K+)-induced electrocatalytic enhancement effects on the 2e− ORR in acidic environments.80 In basic electrolytes, cations such as Cs+, K+, and Li+ enhance the 2e− ORR performance of oxygen-functionalized carbon in accordance with the ionic size of the cation: Cs+> K+ > Li+.81 Therefore, rationally exploring the additives for the electrolyte is of great importance.
Author contributions
Ao Yu: writing, review and editing – original draft. Shengwen Liu – review and editing. Yang Yang: supervision, funding acquisition, project administration.
Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, and there is no professional or other personal interest of any nature or kind in any product, service, and/or company that could be construed as influencing the position presented in, or the review of, the manuscript entitled.
Acknowledgements
Y. Y. acknowledges the financial support from the National Science Foundation under grant no. CBET-1949840 and ACS PRF (65481-ND10). A. Y. and S. L. acknowledge the financial support from the Preeminent Postdoctoral Program (P3) at UCF.
References
- J. H. Kim, Y.-T. Kim and S. H. Joo, Curr. Opin. Electrochem., 2020, 21, 109–116 CrossRef CAS
 .
.
- K. Dong, Y. Lei, H. Zhao, J. Liang, P. Ding, Q. Liu, Z. Xu, S. Lu, Q. Li and X. Sun, J. Mater. Chem. A, 2020, 8, 23123–23141 RSC .
- T. Lian, C. Huang, F. Liang, X. Li and J. Xi, ACS Appl. Mater. Interfaces, 2019, 11, 45692–45701 CrossRef CAS PubMed .
- N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed .
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed .
- X. Huang, M. Song, J. Zhang, T. Shen, G. Luo and D. Wang, Nano-Micro Lett., 2023, 15, 86 CrossRef CAS PubMed .
- A. Byeon, W. C. Yun, J. M. Kim and J. W. Lee, Chem. Eng. J., 2023, 456, 141042 CrossRef CAS .
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS .
- Q. Liu, J. C. Bauer, R. E. Schaak and J. H. Lunsford, Appl. Catal., A, 2008, 339, 130–136 CrossRef CAS .
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef CAS .
- A. Yu, G. Ma, L. Zhu, R. Zhang, Y. Li, S. Yang, H.-Y. Hsu, P. Peng and F.-F. Li, Appl. Catal. B, 2022, 307, 121161 CrossRef CAS .
- A. Yu, G. Ma, L. Zhu, Y. Hu, R. Zhang, H.-Y. Hsu, P. Peng and F.-F. Li, Nanoscale, 2021, 13, 15973–15980 RSC .
- Z. Y. Lu, G. X. Chen, S. Siahrostami, Z. H. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. C. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS .
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z.-Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 CrossRef CAS PubMed .
- Q. Zhang, X. Tan, N. M. Bedford, Z. Han, L. Thomsen, S. Smith, R. Amal and X. Lu, Nat. Commun., 2020, 11, 4181 CrossRef PubMed .
- S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund, D. Sokaras, T. R. Kim, Y. Liu, X. Yan, E. Nilsson, R. Sinclair, J. K. Norskov, T. F. Jaramillo and Z. Bao, J. Am. Chem. Soc., 2018, 140, 7851–7859 CrossRef CAS PubMed .
- S. Chen, T. Luo, K. Chen, Y. Lin, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, X. Li, J. Hu, H. Li, M. Zhu and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 16607–16614 CrossRef CAS PubMed .
- G. Chen, J. Liu, Q. Li, P. Guan, X. Yu, L. Xing, J. Zhang and R. Che, Nano Res., 2019, 12, 2614–2622 CrossRef CAS .
- Y. Pang, K. Wang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, ACS Catal., 2020, 10, 7434–7442 CrossRef CAS .
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS .
- S. Wang, H. Liu, D. Ye, Q. Lan, X. Zhu, Y. Yang, R. Chen and Q. Liao, Sep. Purif. Technol., 2022, 289, 120687 CrossRef CAS .
- J. Park, Y. Nabae, T. Hayakawa and M.-A. Kakimoto, ACS Catal., 2014, 4, 3749–3754 CrossRef CAS .
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, ACS Sustainable Chem. Eng., 2018, 6, 311–317 CrossRef CAS .
- V. Čolić, S. Yang, Z. Révay, I. E. L. Stephens and I. Chorkendorff, Electrochim. Acta, 2018, 272, 192–202 CrossRef .
- H. He, S. Liu, Y. Liu, L. Zhou, H. Wen, R. Shen, H. Zhang, X. Guo, J. Jiang and B. Li, Green Chem., 2023, 25, 9501–9542 RSC .
- D. Zhang, E. Mitchell, X. Lu, D. Chu, L. Shang, T. Zhang, R. Amal and Z. Han, Mater. Today, 2023, 63, 339–359 CrossRef CAS .
- X. Guo, S. Liu, X. Wan, J. Zhang, Y. Liu, X. Zheng, Q. Kong and Z. Jin, Nano Lett., 2022, 22, 4879–4887 CrossRef CAS PubMed .
- G. Zhu, T. Chen, Y. Hu, L. Ma, R. Chen, H. Lv, Y. Wang, J. Liang, X. Li, C. Yan, H. Zhu, H. Liu, Z. Tie, Z. Jin and J. Liu, Nano Energy, 2017, 33, 229–237 CrossRef CAS .
- D. H. Mok, S. Back and S. Siahrostami, Angew. Chem., Int. Ed., 2024, e202404677 Search PubMed .
- H. Yang, N. An, Z. Kang, P. W. Menezes and Z. Chen, Adv. Mater., 2024, 2400140 CrossRef PubMed .
- Y. Pang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, J. Mater. Chem. A, 2020, 8, 24996–25016 RSC .
- L. Jing, Q. Tian, W. Wang, X. Li, Q. Hu, H. Yang and C. He, Adv. Energy Mater., 2024, 2304418 CrossRef .
- L. Han, Y. Y. Sun, S. Li, C. Cheng, C. E. Halbig, P. Feicht, J. L. Hübner, P. Strasser and S. Eigler, ACS Catal., 2019, 9, 1283–1288 CrossRef CAS .
- D. San Roman, D. Krishnamurthy, R. Garg, H. Hafiz, M. Lamparski, N. T. Nuhfer, V. Meunier, V. Viswanathan and T. Cohen-Karni, ACS Catal., 2020, 10, 1993–2008 CrossRef CAS .
- Y. Xia, X. Zhao, C. Xia, Z.-Y. Wu, P. Zhu, J. Y. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed .
- N. Yang, L. Li, J. Li, W. Ding and Z. Wei, Chem. Sci., 2018, 9, 5795–5804 RSC .
- H. W. Kim, M. B. Ross, N. Kornienko, L. Zhang, J. Guo, P. Yang and B. D. McCloskey, Nat. Catal., 2018, 1, 282–290 CrossRef .
- G.-F. Han, F. Li, W. Zou, M. Karamad, J.-P. Jeon, S.-W. Kim, S.-J. Kim, Y. Bu, Z. Fu, Y. Lu, S. Siahrostami and J.-B. Baek, Nat. Commun., 2020, 11, 2209 CrossRef CAS PubMed .
- X. Lu, D. Wang, K.-H. Wu, X. Guo and W. Qi, J. Colloid Interface Sci., 2020, 573, 376–383 CrossRef CAS PubMed .
- V. Perazzolo, C. Durante, R. Pilot, A. Paduano, J. Zheng, G. A. Rizzi, A. Martucci, G. Granozzi and A. Gennaro, Carbon, 2015, 95, 949–963 CrossRef CAS .
- D. Iglesias, A. Giuliani, M. Melchionna, S. Marchesan, A. Criado, L. Nasi, M. Bevilacqua, C. Tavagnacco, F. Vizza, M. Prato and P. Fornasiero, Chem, 2018, 4, 106–123 CAS .
- L. Li, C. Tang, Y. Zheng, B. Xia, X. Zhou, H. Xu and S.-Z. Qiao, Adv. Energy Mater., 2020, 10, 2000789 CrossRef CAS .
- J. Zhang, G. Zhang, S. Jin, Y. Zhou, Q. Ji, H. Lan, H. Liu and J. Qu, Carbon, 2020, 163, 154–161 CrossRef CAS .
- K. Zhao, Y. Su, X. Quan, Y. Liu, S. Chen and H. Yu, J. Catal., 2018, 357, 118–126 CrossRef .
- W. Wang, X. Lu, P. Su, Y. Li, J. Cai, Q. Zhang, M. Zhou and O. Arotiba, Chemosphere, 2020, 259, 127423 CrossRef CAS PubMed .
- N. Jia, T. Yang, S. Shi, X. Chen, Z. An, Y. Chen, S. Yin and P. Chen, ACS Sustainable Chem. Eng., 2020, 8, 2883–2891 CrossRef CAS .
- H. Kim, H. Yang, J. Kang and N. Takeuchi, Carbon, 2021, 182, 242–253 CrossRef CAS .
- Y. Sun, S. Li, B. Paul, L. Han and P. Strasser, J. Electroanal. Chem., 2021, 896, 115197 CrossRef CAS .
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2019, 31, e1808173 CrossRef PubMed .
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed .
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K.-S. Lee, S.-P. Cho, J. S. Yoo, Y.-E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed .
- S. Chen, T. Luo, X. Li, K. Chen, J. Fu, K. Liu, C. Cai, Q. Wang, H. Li, Y. Chen, C. Ma, L. Zhu, Y.-R. Lu, T.-S. Chan, M. Zhu, E. Cortés and M. Liu, J. Am. Chem. Soc., 2022, 144, 14505–14516 CrossRef CAS PubMed .
- Y. Tian, M. Li, Z. Wu, Q. Sun, D. Yuan, B. Johannessen, L. Xu, Y. Wang, Y. Dou, H. Zhao and S. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202213296 CrossRef CAS PubMed .
- C. H. Choi, M. Kim, H. C. Kwon, S. J. Cho, S. Yun, H.-T. Kim, K. J. J. Mayrhofer, H. Kim and M. Choi, Nat. Commun., 2016, 7, 10922 CrossRef CAS PubMed .
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed .
- J. H. Kim, D. Shin, J. Lee, D. S. Baek, T. J. Shin, Y.-T. Kim, H. Y. Jeong, J. H. Kwak, H. Kim and S. H. Joo, ACS Nano, 2020, 14, 1990–2001 CrossRef CAS PubMed .
- H. Huang, M. Sun, S. Li, S. Zhang, Y. Lee, Z. Li, J. Fang, C. Chen, Y.-X. Zhang, Y. Wu, Y. Che, S. Qian, W. Zhu, C. Tang, Z. Zhuang, L. Zhang and Z. Niu, J. Am. Chem. Soc., 2024, 146(13), 9434–9443 CrossRef CAS PubMed .
- C. H. Choi, H. C. Kwon, S. Yook, H. Shin, H. Kim and M. Choi, J. Phys. Chem. C, 2014, 118, 30063–30070 CrossRef CAS .
- E. G. Ciapina, P. P. Lopes, R. Subbaraman, E. A. Ticianelli, V. Stamenkovic, D. Strmcnik and N. M. Markovic, Electrochem. Commun., 2015, 60, 30–33 CrossRef CAS .
- V. Stamenkovic, N. M. Markovic and P. N. Ross, J. Electroanal. Chem., 2001, 500, 44–51 CrossRef CAS .
- N. M. Marković, H. A. Gasteiger, B. N. Grgur and P. N. Ross, J. Electroanal. Chem., 1999, 467, 157–163 CrossRef .
- D. He, L. Zhong, S. Gan, J. Xie, W. Wang, Z. Liu, W. Guo, X. Yang and L. Niu, Electrochim. Acta, 2021, 371, 137721 CrossRef CAS .
- B. Genorio, D. Strmcnik, R. Subbaraman, D. Tripkovic, G. Karapetrov, V. R. Stamenkovic, S. Pejovnik and N. M. Marković, Nat. Mater., 2010, 9, 998–1003 CrossRef CAS PubMed .
- J. Zhang, J. Ma, T. S. Choksi, D. Zhou, S. Han, Y.-F. Liao, H. B. Yang, D. Liu, Z. Zeng, W. Liu, X. Sun, T. Zhang and B. Liu, J. Am. Chem. Soc., 2022, 144, 2255–2263 CrossRef CAS PubMed .
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS .
- E. Pizzutilo, S. J. Freakley, S. Cherevko, S. Venkatesan, G. J. Hutchings, C. H. Liebscher, G. Dehm and K. J. J. Mayrhofer, ACS Catal., 2017, 7, 5699–5705 CrossRef CAS .
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed .
- A. Verdaguer-Casadevall, D. Deiana, M. Karamad, S. Siahrostami, P. Malacrida, T. W. Hansen, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, Nano Lett., 2014, 14, 1603–1608 CrossRef CAS PubMed .
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. Schiffrin, J. Am. Chem. Soc., 2011, 133, 19432–19441 CrossRef PubMed .
- J. S. Jirkovský, I. Panas, S. Romani, E. Ahlberg and D. J. Schiffrin, J. Phys. Chem. Lett., 2012, 3, 315–321 CrossRef .
- Y. Tian, D. Deng, L. Xu, M. Li, H. Chen, Z. Wu and S. Zhang, Nano-Micro Lett., 2023, 15, 122 CrossRef CAS PubMed .
- I. Yamanaka and T. Murayama, Angew. Chem., Int. Ed., 2008, 120, 1926–1928 CrossRef .
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS .
- W. Sun, B. A. Peppley and K. Karan, J. Power Sources, 2005, 144, 42–53 CrossRef CAS .
- P. J. Hamilton and B. G. Pollet, Fuel Cells, 2010, 10, 489–509 CrossRef CAS .
- R. E. Rosli, A. B. Sulong, W. R. W. Daud, M. A. Zulkifley, T. Husaini, M. I. Rosli, E. H. Majlan and M. A. Haque, Int. J. Hydrogen Energy, 2017, 42, 9293–9314 CrossRef CAS .
- N. Aukland, A. Boudina, D. S. Eddy, J. V. Mantese, M. P. Thompson and S. S. Wang, J. Mater. Res., 2004, 19, 1723–1729 CrossRef CAS .
- Y. Long, J. Lin, F. Ye, W. Liu, D. Wang, Q. Cheng, R. Paul, D. Cheng, B. Mao, R. Yan, L. Zhao, D. Liu, F. Liu and C. Hu, Adv. Mater., 2023, 35, 2303905 CrossRef CAS PubMed .
- K.-H. Wu, D. Wang, X. Lu, X. Zhang, Z. Xie, Y. Liu, B.-J. Su, J.-M. Chen, D.-S. Su, W. Qi and S. Guo, Chem, 2020, 6, 1443–1458 CAS .
- J. L. Hübner, L. E. B. Lucchetti, H. N. Nong, D. I. Sharapa, B. Paul, M. Kroschel, J. Kang, D. Teschner, S. Behrens, F. Studt, A. Knop-Gericke, S. Siahrostami and P. Strasser, ACS Energy Lett., 2024, 1331–1338, DOI:10.1021/acsenergylett.3c02743 .
- J. Lee, J. S. Lim, G. Yim, H. Jang, S. H. Joo and Y. J. Sa, ACS Appl. Mater. Interfaces, 2021, 13, 59904–59914 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abcde
*abcde