
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical deconstructive functionalization of arylcycloalkanes via fragmentation of anodically generated aromatic radical cations
Hussain A.
Maashi
ab,
Abdulrahman H.
Husayni
a,
James
Harnedy
a and
Louis C.
Morrill
 *ac
*ac
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: lcm71@bath.ac.uk
bDepartment of Chemistry, College of Science, University of Bisha, Bisha 61922, Saudi Arabia
cDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 13th September 2024
Abstract
This highlight summarises electrochemical approaches for the deconstructive functionalization of arylcycloalkanes via the fragmentation of anodically generated aromatic radical cations. A diverse range of deconstructive functionalization processes is described, including discussion on the electrochemical reaction conditions employed, scope and limitations, and reaction mechanisms, in addition to highlighting future opportunities in this burgeoning area of sustainable synthesis.
 Hussain A. Maashi | Hussain A. Maashi obtained a Bachelors in Chemistry at Jazan University (Saudi Arabia) and a Masters in Chemistry at Western Illinois University (United States). He is now pursuing a PhD at Cardiff University under the direction of Dr Louis Morrill investigating the development of new synthetic methodology enabled by electrochemistry. |
 Abdulrahman H. Husayni | Abdulrahman H. Husayni obtained a Bachelors in Chemistry at Jazan University (Saudi Arabia) and a Masters in Chemistry at Hampton University (United States). He is now pursuing a PhD at Cardiff University under the direction of Dr Louis Morrill investigating the development of new synthetic methodology enabled by electrochemistry. |
 James Harnedy | James Harnedy obtained a Masters in Chemistry at the University of Nottingham (United Kingdom) and a PhD at Cardiff University under the direction of Dr Louis Morrill. He is now a scientist at Domainex (Cambridge, United Kingdom). |
 Louis C. Morrill | Louis Morrill received his PhD from the University of St Andrews in 2014 under the direction of Prof. Andrew Smith and undertook postdoctoral research at UC Berkeley with Prof. Richmond Sarpong. In June 2015, he started his independent research career at Cardiff University, before relocating to the University of Bath in 2024. Research in the group is focused on inventing new reactions in organic chemistry and developing sustainable catalytic methodologies for synthesis. |
1. Introduction
Deconstructive functionalization can be described as the process of breaking chemical bonds, followed by utilization of the formed reactive intermediates to access functionalized products. This is a powerful synthetic strategy, which can provide expedient access to a diverse range of useful functionalized molecules that are challenging to access via other approaches.1 The deconstructive functionalization of cyclic systems, such as cycloalkanes, is particularly interesting, as it can provide direct access to difunctionalized products where the distance between the newly installed functional groups is determined by the size of the ring. However, for even the most strained cycloalkane, cyclopropane, these systems require activation before the cleavage of strong C–C σ-bonds can occur. A powerful strategy for the deconstructive functionalization of cycloalkanes is the single electron oxidation of the aromatic group within arylcycloalkanes,2 which can been achieved through the use of copper catalysts in combination with a strong oxidant,3 photoredox catalysts,4 direct photo-oxidation,5 or via electrochemical oxidation (vide infra). The formation of the corresponding aromatic radical cation significantly weakens benzylic C–C σ-bonds within the cycloalkane rings, which can facilitate bond cleavage to form radical and cation intermediates that can be utilized for subsequent functionalization processes.Organic electrochemistry has recently undergone a renaissance due to the increasing global challenge to develop more sustainable synthetic methods to produce chemicals for society,6 in addition to the increasing availability of standardized batch and flow electrochemical reactors.7 It is ideally suited to the selective generation of aromatic radical cation intermediates due to the inherent ability to precisely control the potential at the working electrode (anode), combined with the wide array of electrode materials available that can be employed to optimize substrate–electrode interactions.8 In recent years, there has been a significant increase in reports that describe the development of electrochemical methods for deconstructive functionalization of arylcycloalkanes, which proceed via the fragmentation of anodically generated aromatic radical cations. Electrochemical approaches can address drawbacks associated with complementary approaches, such as avoiding the use of strong oxidants, metal (photo)catalysts, high energy UV light irradiation, or strong acid solutions.3–5 This highlight will summarise these developments, providing a critical analysis of each reported method with respect to electrochemical reaction conditions employed, substrate scope and limitations, and reaction mechanism, whilst also providing a perspective on opportunities for further development and understanding in this burgeoning area of synthetic organic electrochemistry (Scheme 1).
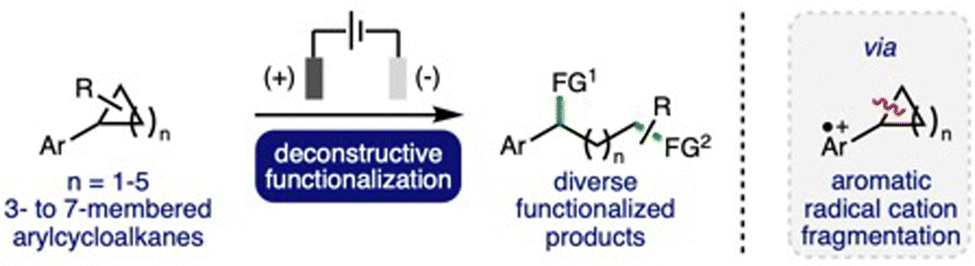 | ||
| Scheme 1 Electrochemical deconstructive functionalization of arylcycloalkanes via fragmentation of anodically generated aromatic radical cations. FG = functional group. | ||
2. Arylcyclopropanes
The inherent ring strain that exists within the cyclopropane scaffold provides a thermodynamic driving force for ring-opening reactions.9 As such, cyclopropanes have been established as a versatile C3 building block in organic synthesis.10 However, challenges exist with respect to controlling the selectivity of C–C bond cleavage and the subsequent functionalization. Existing strategies for the selective deconstructive functionalization of electronically unbiased arylcyclopropanes include transition metal-catalyzed C–C bond oxidative addition, and electrophilic activation using Lewis acids.11 Drawbacks to these approaches include the use of directing groups to facilitate regioselective C–C bond activation, and compatibility issues between Lewis acids and some classes of nucleophiles. An attractive alternative approach involves the single electron oxidation of the aromatic ring within the arylcyclopropane to form the corresponding aromatic radical cation (see Scheme 2 for oxidation potentials of representative arylcyclopropanes),12 which significantly weakens the β-C–C σ-bonds within the cyclopropane and promotes regioselective nucleophilic attack. This section provides an overview of developed electrochemical strategies for the deconstructive functionalization of arylcyclopropanes,13 which proceed via the fragmentation of anodically generated aromatic radical cations.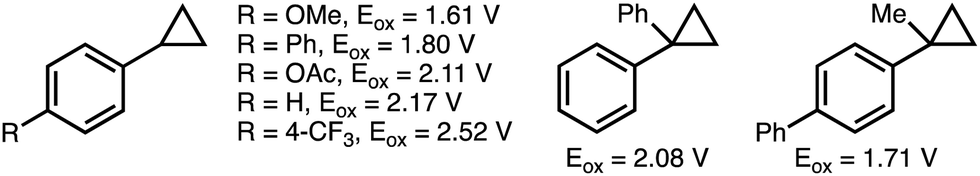 | ||
| Scheme 2 Reported oxidation potentials of representative arylcyclopropanes. All oxidation potentials (Eox) quoted vs. Ag/AgCl.12 | ||
In a seminal early report (1970), Shono reported the anodic oxidation of arylcyclopropanes in methanol (Scheme 3).14 Employing galvanostatic (constant current) electrolysis (j = 80 mA cm−2), a selection of arylcyclopropanes with different substituents, both on the aromatic ring and on the cyclopropane scaffold, underwent ring opening to form 1,3-dimethoxyether products in good yields and with high faradaic efficiencies. It was found that higher product yields could be obtained using potentiostatic (constant potential) electrolysis (Eanode = 1.7 V vs. SCE). The transformation was not stereospecific, with trans-1,2-disubstituted cyclopropanes resulting in the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. of diether products. Plotting the half-wave oxidation potentials of various substituted arylcyclopropanes against Hammett's σ+ constants, coupled with no observed formation of alternative products that may arise from cyclopropane oxidation, led the authors to propose a mechanism that involved the initial anodic oxidation of the aromatic ring to the corresponding aromatic radical cation, which triggers mesolytic cleavage of a β-C–C σ-bond in the adjacent strained cyclopropane ring. Methanol addition followed by further anodic oxidation and trapping of the resulting benzylic carbocation with methanol affords the 1,3-dimethoxy ether product. The cathodic reaction is the conversion of protons to hydrogen gas.
:1 d.r. of diether products. Plotting the half-wave oxidation potentials of various substituted arylcyclopropanes against Hammett's σ+ constants, coupled with no observed formation of alternative products that may arise from cyclopropane oxidation, led the authors to propose a mechanism that involved the initial anodic oxidation of the aromatic ring to the corresponding aromatic radical cation, which triggers mesolytic cleavage of a β-C–C σ-bond in the adjacent strained cyclopropane ring. Methanol addition followed by further anodic oxidation and trapping of the resulting benzylic carbocation with methanol affords the 1,3-dimethoxy ether product. The cathodic reaction is the conversion of protons to hydrogen gas.
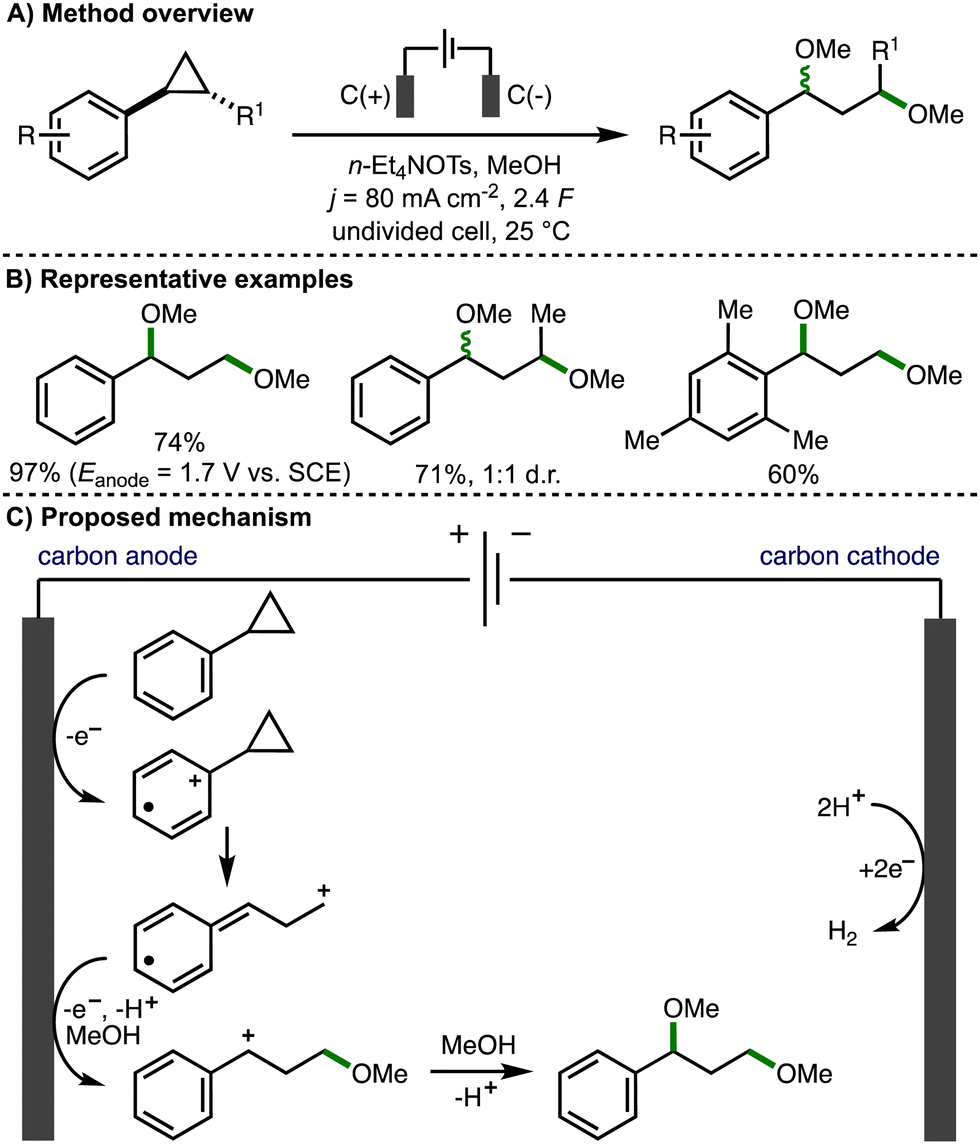 | ||
| Scheme 3 1,3-Dimethoxylation of arylcyclopropanes (Shono). | ||
Over 50 years later, in 2021, Zhang, Lei and co-workers revisited this strategy with the aim to expand the scope of the electrochemical process to access 1,3-difunctionalized molecules (Scheme 4).15 By employing Et3N·3HF as a nucleophilic fluoride source with Pt plate electrodes under galvanostatic electrolysis conditions, the authors reported the synthesis of a broad range of 1,3-difluorinated products in good yields. The protocol exhibits good functional group tolerance and can be performed on gram scale. Various electron-withdrawing groups (e.g., SO2Me, CO2Me, CF3) can be incorporated into the aromatic ring, however, arylcyclopropanes containing electron-releasing aromatic substituents (e.g., OMe, SMe) were not tolerated. Various di- and tri-substituted arylcyclopropane scaffolds underwent deconstructive difluorination in good yields, although a low diastereoselectivity was observed when trans-1,2-diphenylcyclopropane was employed as the substrate. The authors also demonstrated the electrosynthetic method on derivatives of naturally occurring compounds, including diacetonefructose and L-menthol. Cyclic voltammetry experiments confirmed that the aromatic ring within the arylcyclopropane is crucial for oxidation. density functional theory (DFT) analysis revealed that the distal C atom within the cyclopropane radical cation possesses a partial positive charge making it susceptible to nucleophilic attack. The authors also gained supporting evidence for benzylic radical and benzylic cation intermediates via trapping experiments. As such, a mechanism was presented that involves initial anodic oxidation of the arylcyclopropane to form a radical cation intermediate, which results in weakening of the Cα–Cβ bond within the cyclopropane ring. This intermediate is proposed to undergo a three-electron SN2 reaction with fluoride to generate a benzylic radical. Further anodic oxidation and trapping of the resulting benzylic carbocation with fluoride furnishes the observed 1,3-difluorinated product. Furthermore, through variation of the electrochemical reaction conditions, the authors were able to access alternative 1,3-difunctionalization products. This included 1,3-oxyfluorination via the addition of methanol as a co-solvent, lowering the reaction temperature to 0 °C, and employing carbon cloth as the anode material. The observed selectivity was rationalized by faster reaction of methanol with the arylcyclopropane radical cation due the lower relative nucleophilicity of Et3N·3HF, followed by intercepting the benzylic carbocation with fluoride due to the excess quantity of Et3N·3HF being present in the reaction system. Ethers could also be employed as nucleophiles in combination with Et3N·3HF to access 1,3-oxyfluorination products using slightly modified electrochemical reaction conditions.
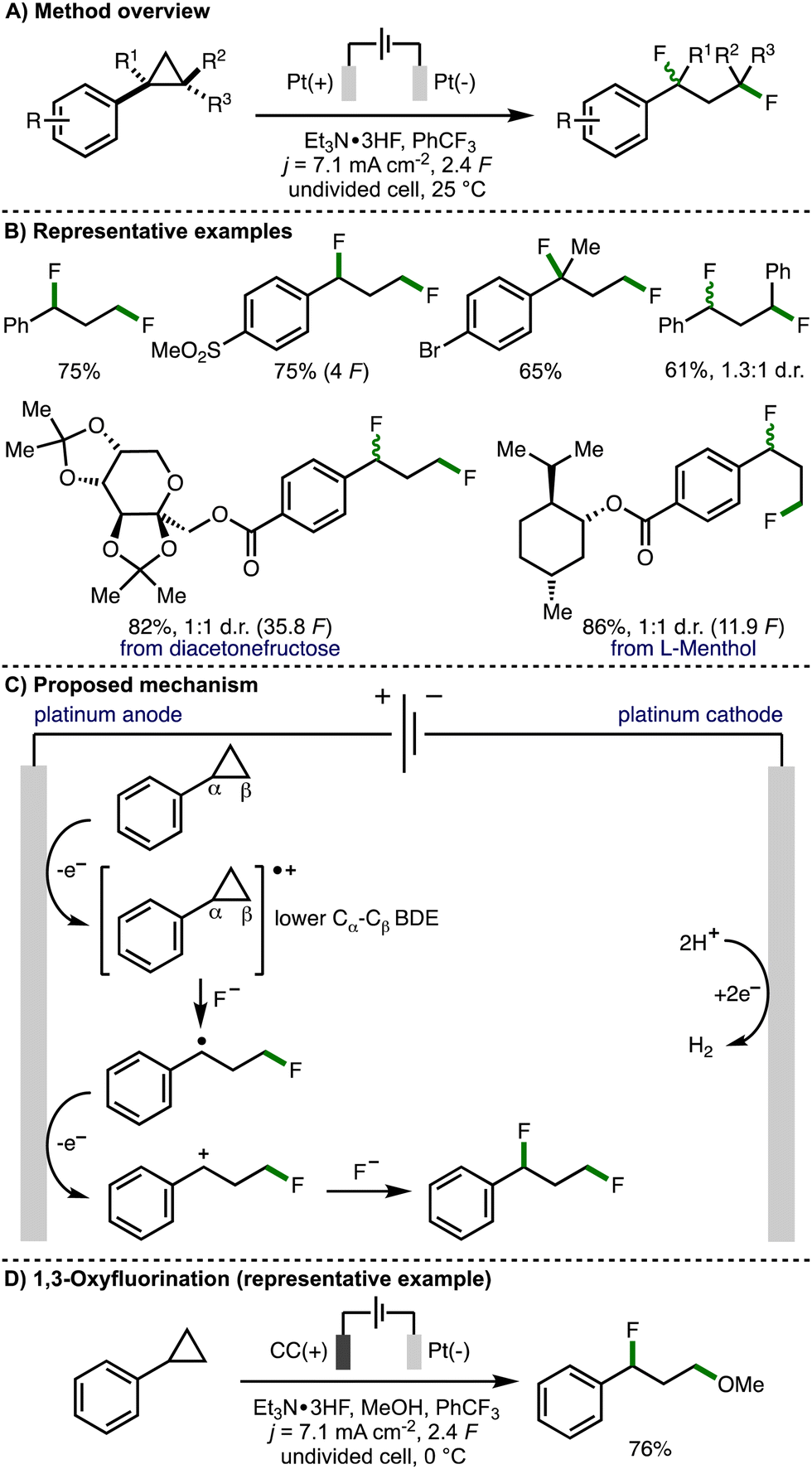 | ||
| Scheme 4 1,3-Difluorination and 1,3-oxyfluorination of arylcyclopropanes (Zhang and Lei). | ||
In 2023, Banerjee and co-workers reported the electrochemical 1,3-oxofluorination of gem-difluoroarylcyclopropanes to generate α-trifluoromethyl substituted acetophenone derivatives (Scheme 5).16 Optimized electrochemical reaction conditions consisted of an undivided cell equipped with a graphite anode and a nickel cathode, galvanostatic electrolysis (i = 4 mA), under an O2 atmosphere, employing Et3N·3HF a fluoride source in acetonitrile and using n-Bu4NOAc as a supporting electrolyte. Employing these conditions, a range of gem-difluoroarylcyclopropanes, bearing a selection of substituents on both the aromatic ring and the cyclopropane scaffold, could be converted to the corresponding α-trifluoromethyl substituted acetophenone derivatives in synthetically useful yields. This included derivatives of naturally occurring compounds such as Euginol. In general, the electrochemical protocol worked well for arylcyclopropane substrates that contained electron-rich aromatic rings. Poor conversion was observed for substrates containing electron-deficient aromatic rings, which is likely due to the increased oxidation potential of these molecules. Alkyl groups could be added to the methylene position of the gem-difluoroarylcyclopropanes to access the corresponding acetophenone derivatives in good yields. Mechanistic experiments were performed to exclude the possibility of a chain propagation mechanism and to provide supporting evidence for the formation of a benzylic radical intermediate. It was also found that benzyl alcohols undergo oxidation to the corresponding acetophenone derivative under the electrochemical reaction conditions, suggesting that these could be present as intermediates. Based on these observations, the authors proposed a reaction mechanism that initiates with anodic oxidation of the aromatic ring present within the gem-difluoroarylcyclopropane to the corresponding aromatic radical cation. Subsequent regioselective ring-opening is proposed to occur via a three-electron SN2 nucleophilic attack of fluoride at the difluoro-substituted carbon atom to form the trifluoromethyl group and a benzylic radical, which is intercepted by either oxygen or cathodically generated superoxide radical anion to form a hydroperoxide intermediate. Further elimination of water would generate the observed α-trifluoromethyl substituted acetophenone products. Alternatively, the authors also proposed that the hydroperoxide intermediate could be converted into a benzylic alcohol, with further anodic oxidation to give the acetophenone.
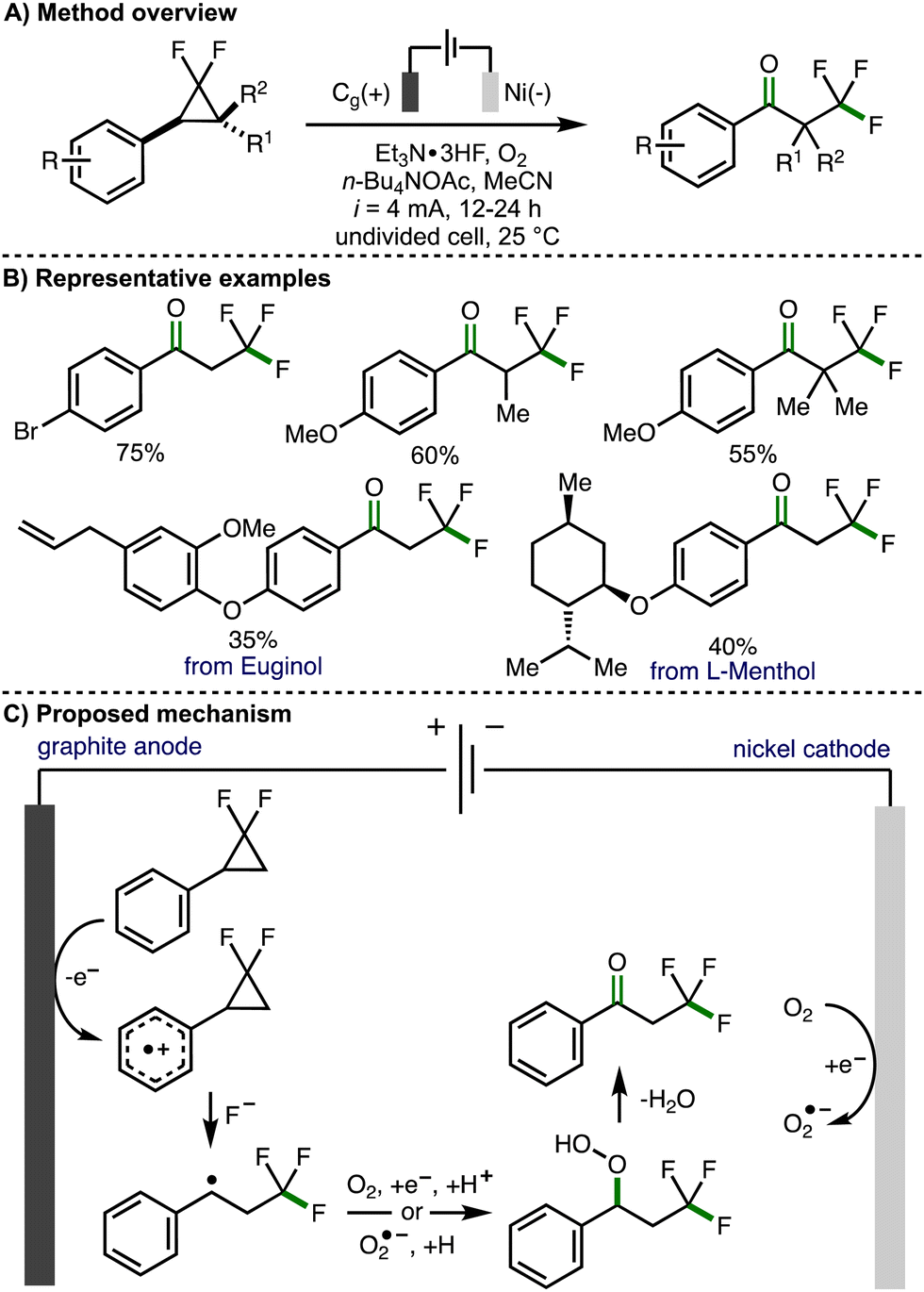 | ||
| Scheme 5 1,3-Oxofluorination of gem-difluoroarylcyclopropanes (Banerjee). | ||
Also in 2023, Feng and co-workers disclosed an electrochemical method for the 1,3-fluorofunctionalization of arylcyclopropanes (Scheme 6).17 In contrast to the previous report from Lei (cf., Scheme 4), which provided access to benzylic fluorides, here the opposite regioselectivity is observed, and a tertiary alkyl fluoride motif is introduced. This is explained by the existence of a tight ion pair between the fluorinating agent (tetrafluoroborate anion) and the anodically generated arylcyclopropane radical cation, which enabled a peripheral fluorine incorporation. Employing galvanostatic electrolysis (i = 5 mA), a graphite anode and nickel plate cathode, n-Et4NBF4 as both electrolyte and fluoride source in the presence of methanol, the 1,3-fluoromethoxylation of arylcyclopropanes was realised. Using optimized reaction conditions, a selection of 1,2,2-trisubstituted arylcyclopropanes participated in this transformation. In general, arylcyclopropanes that contained electron-rich aromatic rings (i.e., those bearing electron-releasing substituents) resulted in highest product yields. By varying the nucleophile used in conjunction with n-Et4NBF4, a variety of alternative 1,3-fluorofunctionalizations were developed, including 1,3-fluoroalkoxylation with various alcohols, 1,3-fluoroacyloxylation using alkyl acetic acids, and 1,3-fluoroamination using N-heterocycles (e.g., pyrazoles, 1H- and 2H-indazoles). A selection of reactions were performed to gain mechanistic insight, including a radical clock experiment that provided supporting evidence for the involvement of a benzylic radical intermediate. Based upon this, and related published literature, the authors proposed that the arylcyclopropane undergoes anodic oxidation to generate a radical cation, which participates in a three-electron nucleophilic attack by fluoride from the tetrafluoroborate anion via a tight ion pair to generate a benzylic radical. Further anodic oxidation would access a benzylic carbocation, which is intercepted by cathodically generated methoxide (when methanol is employed as the second nucleophile) to generate the observed 1,3-fluoromethoxylated product.
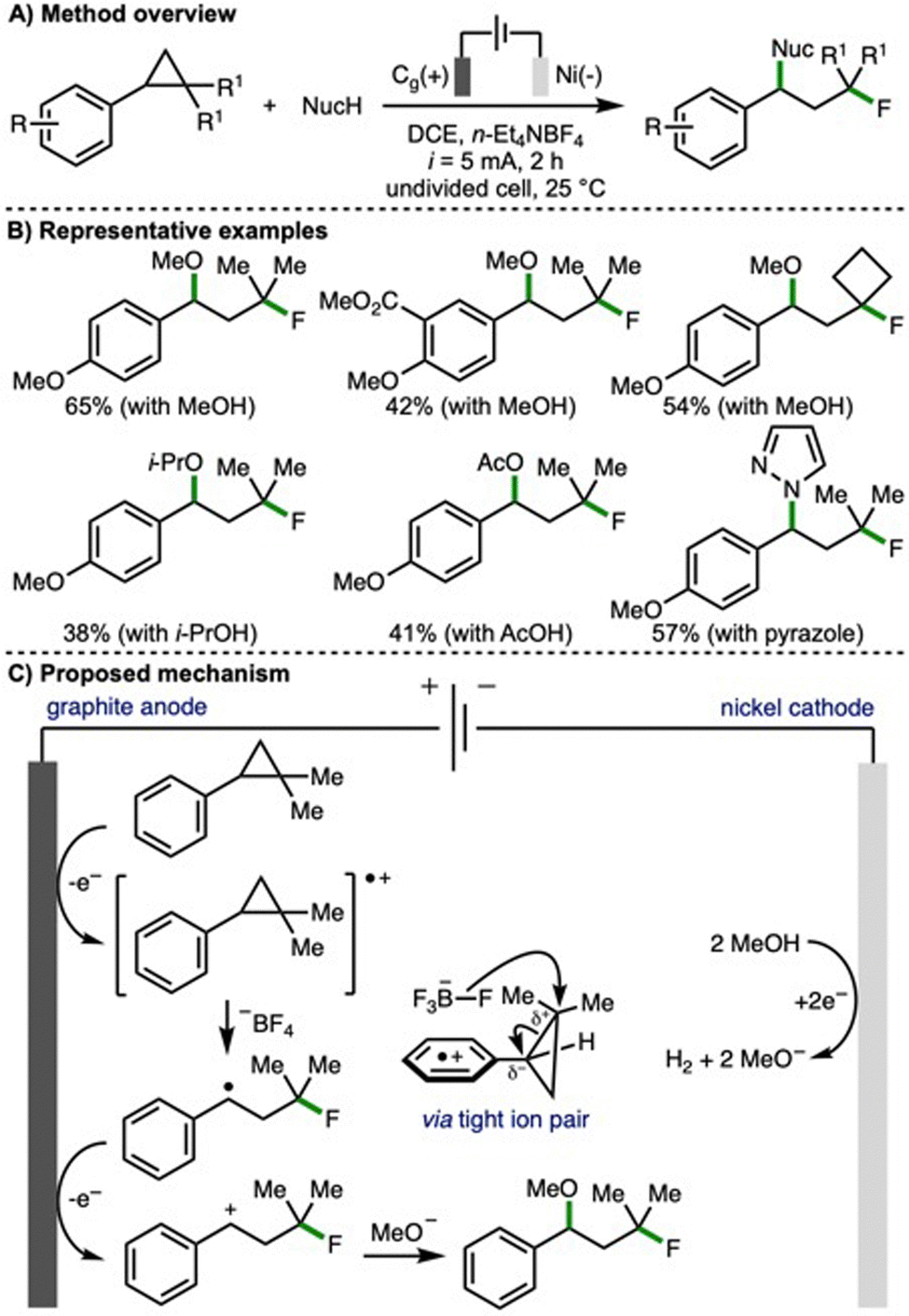 | ||
| Scheme 6 1,3-Fluorofunctionalization of arylcyclopropanes (Feng). | ||
In 2023, Song, Li and co-workers reported the direct electrochemical conversion of unactivated arylcyclopropanes to 1,3-diols – an important structural motif found across a broad range of biologically active compounds (Scheme 7).18 This transformation has several inherent challenges, including several potential undesired reactions such as further alcohol oxidation (due to the similar oxidation potentials of the arylcyclopropane reagents and the 1,3-diol products) and oxetane formation (via intramolecular nucleophilic attack). Regardless, the electrochemical deconstructive 1,3-dihydroxylation of arylcyclopropanes was achieved using galvanostatic electrolysis (i = 10 mA), carbon cloth electrodes, and a solvent system composed of water and acetone. The scope of arylcyclopropanes reported was broad, including a variety of substituents on both the aromatic ring (e.g., halogens, esters, amides etc.) and cyclopropane scaffold. For substrates bearing an electron-withdrawing substituent on the ring, resulting in a higher oxidation potential of the arylcyclopropane substrate, the addition of AcOH and elevated reaction temperatures (50 °C) were required to achieve satisfactory product yields. The electrochemical procedure could be used to produce 1,3-diols on gram scale and was also applied to the late-stage 1,3-dihydroxylation of derivatives of various biologically active compounds, including ciprofibrate and sulbactam. Labelling experiments with H2O18 confirmed that water was the source of the hydroxyl groups within the products. Cyclic voltammetry was used to investigate and compare the oxidation of alkylcyclopropanes vs. arylcyclopropanes, which provided supporting evidence that the reaction initiates via oxidation of the aromatic ring rather than oxidation of the cyclopropane unit. The ring opening is highly regioselective (e.g., for 1,2-disubstituted arylcyclopropanes), with cleavage of the most substituted cyclopropane C–C bond observed. This was attributed to the larger LUMO orbital coefficient at the substituted carbon, making it more susceptible to nucleophilic attack. Radical trapping experiments provided support for benzy conditions lic radical intermediates. As such, the authors suggested that the reaction proceeds via anodic oxidation of the aromatic ring within the arylcyclopropane, followed by regioselective nucleophilic attack of water to form a benzylic radical. Subsequent anodic oxidation and water trapping of the benzylic carbocation generates the observed 1,3-diol products.
 | ||
| Scheme 7 1,3-Dihydroxylation of arylcyclopropanes (Song and Li). | ||
Later in 2023, Song, Li and co-workers reported an electrochemical method for 1,3-oxohydroxylation of arylcyclopropanes (Scheme 8).19 Compared to their previous report describing 1,3-dihydroxylation of arylcyclopropanes (cf., Scheme 7), the electrochemical conditions were modified in several ways, including the use of a platinum cathode, increased current (i = 20 mA) and longer reaction times (130 minutes). Using optimized electrochemical reaction conditions, a wide variety of monosubstituted, 1,2-disubstituted, and 1,2,2-trisubstituted arylcyclopropanes were converted into the corresponding β-hydroxyketone products in synthetically useful yields. Substoichiometric quantities of DDQ (20 mol%) were added when using arylcyclopropane substrates that contained electron-withdrawing aromatic substituents, presumably acting as a redox mediator to facilitate anodic oxidation of the arylcyclopropane. 1,2-arylalkylcyclopropanes were selectively converted to β-hydroxyketone products without any observed formation of 1,3-diketones. The electrochemical method could be performed on gram scale and was also applied to the synthesis of derivatives of biologically active compounds, including flurbiprofen. For several substrates, 1,3-diols were observed as byproducts. The validity of 1,3-diols as reaction intermediates was supported through their presence during reaction-time profiles, alongside the observed conversion of 1,3-diols to β-hydroxyketones using the optimized electrochemical reaction conditions. As such, the authors proposed the same reaction mechanism as previously used to generate the 1,3-diol (cf., Scheme 7(C)), with further selective anodic oxidation of the benzylic hydroxyl group to the form the observed β-hydroxyketone products.
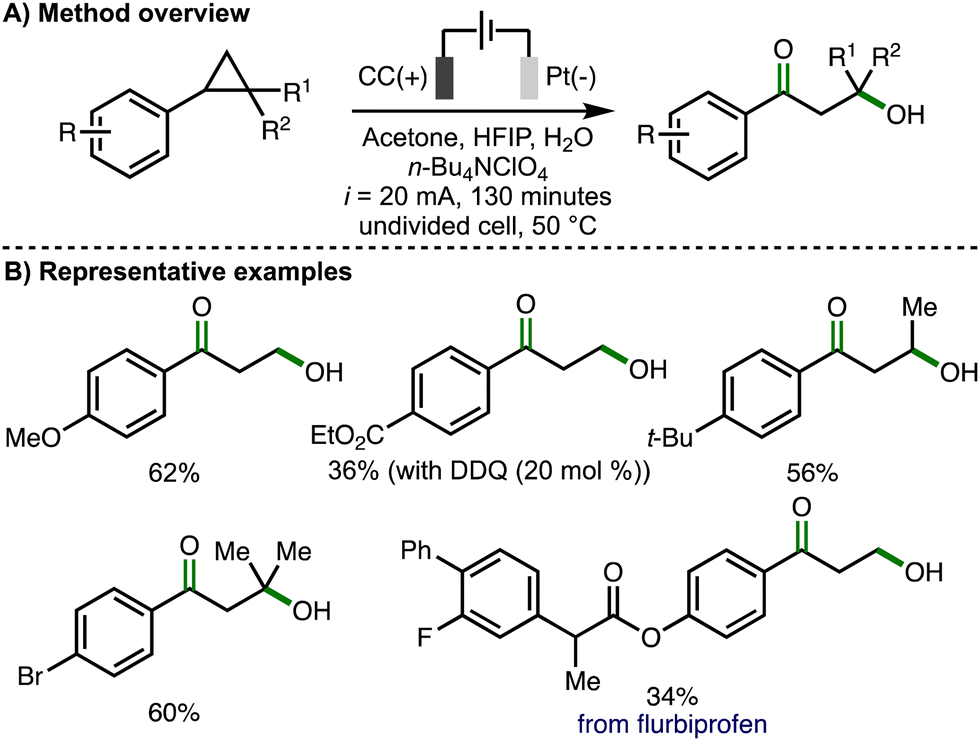 | ||
| Scheme 8 1,3-Oxohydroxylation of arylcyclopropanes (Song and Li). | ||
Also in 2023, Song, Li and co-workers published a separate study describing the electrochemical regioselective 1,3-hydroxyfunctionalization of arylcyclopropanes (Scheme 9).12 The strategy was to employ various nucleophiles to intercept the anodically generated arylcyclopropane radical cation in the presence of water. Challenges with this approach included the chemo- and regioselectivity considerations that arise from the presence of two competing nucleophiles, in addition to the determination of electrochemical reaction conditions suitable for various types of nucleophiles. Initially, the 1,3-hydroxyalkynylation of arylcyclopropanes was developed using galvanostatic electrolysis (i = 10 mA), a graphite rod anode and platinum cathode, employing alkynyltrifluoroborate salts as a C-based nucleophile in the presence of water. It was found that this procedure was applicable across a selection of monoarylcyclopropanes and 1,1-diarylcyclopropanes, forming the corresponding products in moderate yields, which was attributed towards the competitive formation of 1,3-diols and low substrate conversion in some cases. Impressively, various alternative nucleophiles could be employed to enable other 1,3-hydroxyfunctionalizations, including 1,3-hydroxyamidation (using diarylsulfonamides), 1,3-hydroxynitroesterification (using NaNO3), 1,3-hydroxysulfonylation (using sulfonic acids), 1,3-hydroxybromination (using NBS, which presumably generates bromide at the cathode), 1,3-hydroxychlorination (using CaCl2), and 1,3-fluorohydroxylation (using Et3N·3HF). For each class of nucleophile, modifications to the electrochemical reaction conditions (e.g., electrode material, solvent system, electrolyte, redox mediators etc.) were required to achieve satisfactory yields. The observed regioselectivity, with the addition of the hydroxyl group at the benzylic position in most cases, was rationalized by the arylcyclopropane radical cation being preferentially intercepted by the stronger nucleophiles (e.g., alkynyltrifluoroborates, nitrate anion etc.), and then trapping the benzylic carbocation with water due to it being present in excess quantities. The alternative regioselectivity observed for the 1,3-fluorohydroxylation process, which was also observed by Lei and co-workers (cf., Scheme 4(C)), was attributed to the lower nucleophilicity of Et3N·3HF in water. Supported by various experiments, the authors proposed an analogous reaction mechanism to their concurrently reported 1,3-dihydroxylation process (cf., Scheme 7(C)).
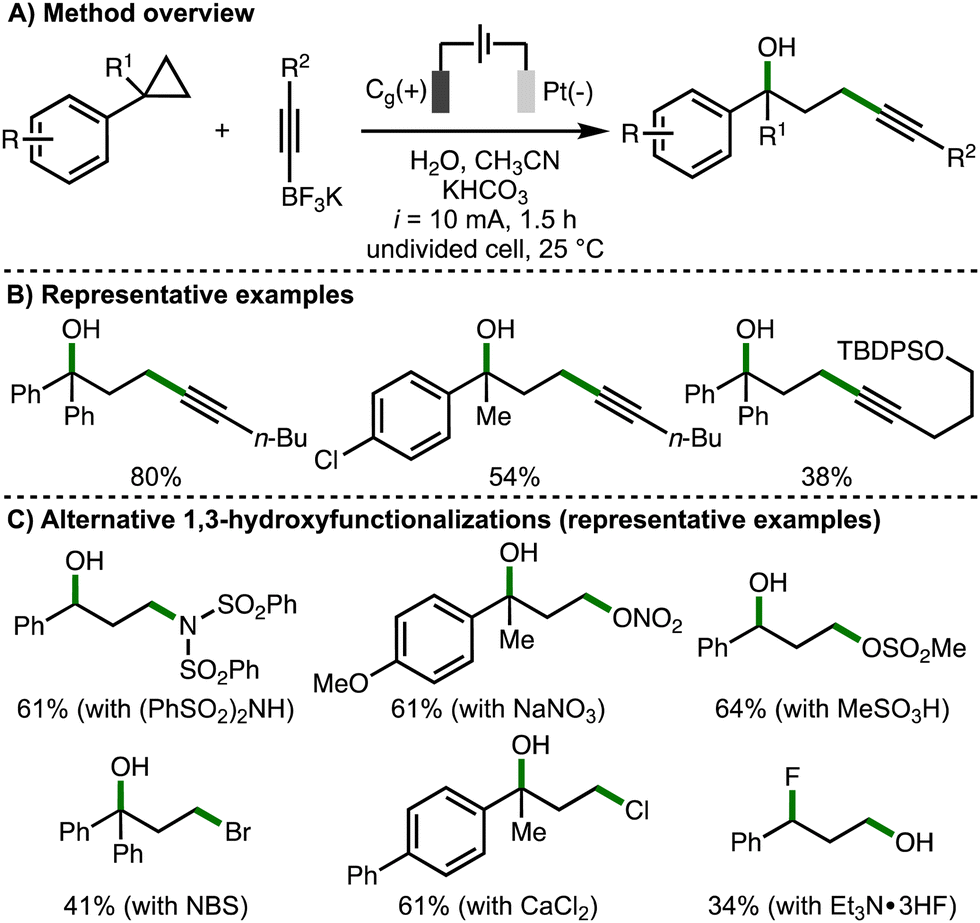 | ||
| Scheme 9 1,3-Hydroxyfunctionalization of arylcyclopropanes (Song and Li). | ||
In 2023, Luo, Xiao and co-workers reported an electrochemical 1,3-alkoxyimidation of unactivated arylcyclopropanes (Scheme 10).20 This four-component system was composed of the arylcyclopropane, acetonitrile, carboxylic acids, and alcohols. Again, a significant challenge associated with this approach is accurately controlling the reactivity, chemo- and regioselectivity of each component in order to achieve the desired transformation. Nevertheless, employing an undivided cell equipped with Pt electrodes, galvanostatic electrolysis conditions (i = 5 mA), and a mixed solvent system composed of MeCN and MeOH, the envisaged four-component coupling was achieved. It was found that the reaction must be performed under Ar, with significant quantities of undesired by-product 3-methoxy-1-phenylpropan-1-one formed when the reaction was performed open to air. Employing optimized electrochemical reaction conditions, a large range of aryl and alkyl carboxylic acids could be employed to access various 1,3-alkoxyimide products, including a derivative of the drug molecule probenecid. With respect to the arylcyclopropane component, those that contained electron-withdrawing aromatic substituents were well tolerated, but electron-donating aromatic substituents represented a limitation of the electrosynthetic method. The alcohol co-solvent could also be varied to access different ethers within the products. Cyclic voltammetry experiments revealed that cyclopropylbenzene had a lower oxidation potential than either benzoic acid or methanol, which both showed no obvious oxidation peaks in the region of 0–2.5 V. As such, the authors proposed a reaction mechanism that initiates with anodic oxidation of the arylcyclopropane to the corresponding radical cation, which is intercepted by cathodically generated methoxide (when methanol is employed as the co-solvent). The formed benzylic radical undergoes further anodic oxidation to give a benzylic carbocation, which is intercepted by acetonitrile to form a nitrilium ion. Subsequent trapping with benzoate followed by 1,3-acyl transfer (Mumm rearrangement) provides access to the observed 1,3-alkoxyimide products.
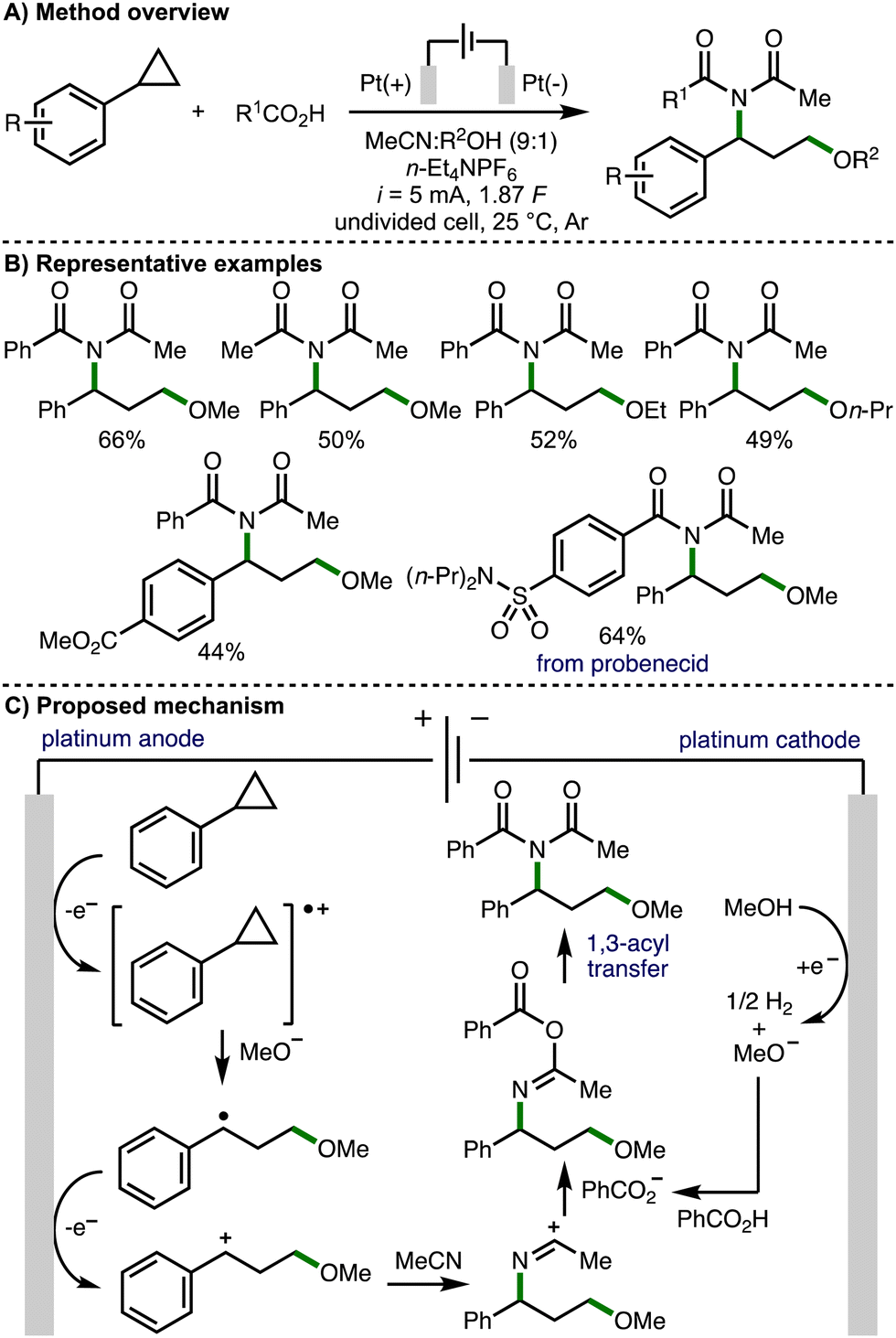 | ||
| Scheme 10 1,3-Alkoxyimidation of arylcyclopropanes (Luo and Xiao). | ||
3. Donor–acceptor cyclopropanes and cyclobutanes
The introduction of vicinal donor and acceptor substituents within cyclopropane and cyclobutane scaffolds polarizes the C(sp3)–C(sp3) bond and enables more facile bond cleavage.21 As such, donor–acceptor (D–A) cyclopropanes/cyclobutanes are valuable C3/C4 building blocks in organic synthesis, respectively.22 Most commonly, these molecules are activated by coordination of the acceptor moiety to Lewis acids, which further polarizes the vicinally substituted C–C bonds, facilitating heterolytic bond cleavage such that D–A cyclopropanes/cyclobutanes can engage in reactions via their formal zwitterionic form. These dipole species participate in a broad range of transformations, including cycloadditions, ring-opening reactions, and re-arrangement processes. An alternative mode of substrate activation involves the single electron oxidation of the aromatic ring, which weakens the vicinally substituted C–C bonds within D–A cyclopropanes/cyclobutanes, promoting the formation of synthetically useful radical cation intermediates. This section provides an overview of developed electrochemical strategies for the deconstructive functionalization of D–A cyclopropanes/cyclobutanes, which proceed via the fragmentation of anodically generated aromatic radical cations.In 2021, Jacob, Werz and co-workers reported the first example of the electrochemical activation of donor–acceptor cyclopropanes and cyclobutanes, which provided access to β- and γ-hydroxyketones, respectively (Scheme 11).23 Employing an undivided cell, potentiostatic electrolysis conditions (Ecell = 2.40 V), glassy carbon electrodes, n-Bu4NBF4 as supporting electrolyte, hexafluoroisopropanol (HFIP) as solvent, and an O2 balloon, a selection of donor–acceptor cyclopropanes were converted to the corresponding to β-hydroxyketones in high yields via direct anodic oxidation. Direct electrolysis of donor–acceptor cyclobutanes using the same conditions initially formed 1,2-dioxanes, where could be converted into the corresponding γ-hydroxyketones in up to quantitative yields by treatment with triethylamine via the Kornblum–DeLaMare rearrangement. For donor–acceptor cyclopropanes containing electron-rich aromatic rings (e.g., methoxy substituted aromatics), the solvent was switched from HFIP to MeCN, the cell potential was lowered to 1.30 V, and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) was employed in stoichiometric quantities as a redox mediator in order to access the desired products. A variety of synthetic and computations experiments were performed to gain mechanistic insight. Divided cell electrochemical studies revealed that the transformation initiates via oxidation of the donor–acceptor cyclopropane/cyclobutane substrates. Trapping experiments provided supporting evidence for a benzylic carbocation intermediate, whilst inhibition experiments suggested a radical mechanism is operative. Full conversion to products was observed after passing 0.3 F of charge, proving that a radical chain propagation is involved. As such, the authors proposed a reaction mechanism that starts with anodic oxidation of the aromatic ring within the substrate to generate the corresponding aromatic radical cation, which undergoes mesolytic cleavage of the cyclopropane moiety. Radical combination with triplet oxygen followed by intramolecular 5–exo–trig cyclization generates a 1,2-dioxolane radical cation that undergoes single electron transfer (SET) with the starting donor–acceptor cyclopropane (chain propagation) to form a 1,2-dioxolane, which converts to the observed β-hydroxyketones in situ via Kornblum–DeLaMare rearrangement. Quantum-chemical calculations were consistent with this proposed mechanism. In a separate follow-up study, Werz and co-workers reported the electrocatalytic synthesis of 1,2-dioxolanes from tetrasubstituted donor–acceptor cyclopropanes employing galvanostatic electrolysis conditions.24
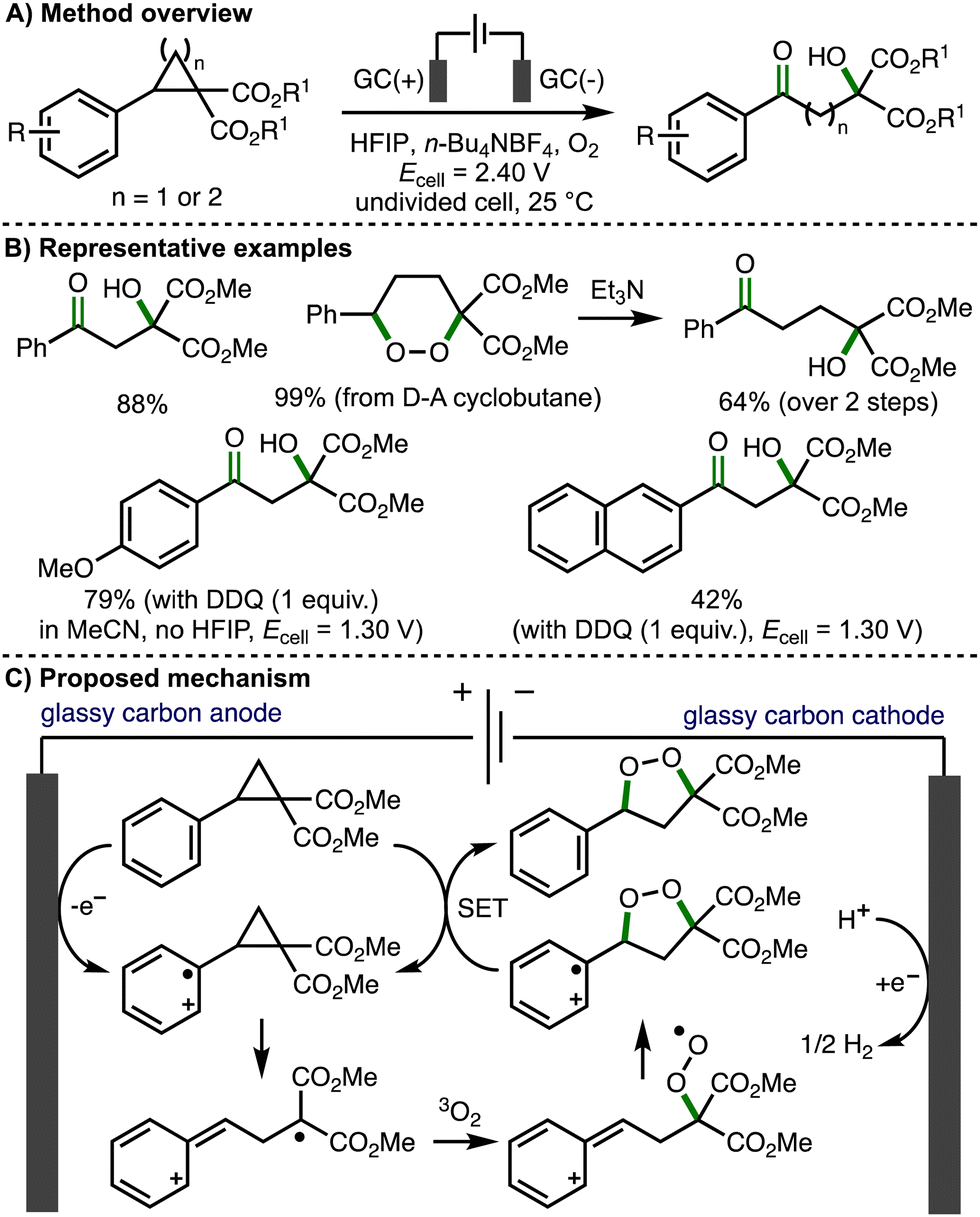 | ||
| Scheme 11 β- and γ-hydroxyketone formation via electrochemical activation of donor–acceptor cyclopropanes and cyclobutanes, respectively (Jacob and Werz). | ||
Concurrently, Banerjee and co-workers were investigating closely related electrosynthetic transformations. In 2021, they disclosed an electrochemical method for the 1,3-oxohydroxylation of donor–acceptor cyclopropanes (Scheme 12).25 In comparison to the electrochemical reaction conditions employed by Jacob, Werz and co-workers (cf, Scheme 11(A)), galvanostatic electrolysis (i = 1 mA) was employed and acetonitrile was used as the solvent instead of HFIP. The authors found that donor–acceptor cyclopropanes containing aromatic rings bearing electron-releasing substituents (e.g., Me, OMe) performed best, providing access to the corresponding β-hydroxyketone products in high yields, with those bearing electron-withdrawing substituents (e.g., NO2) being unreactive. This observation was attributed towards the higher oxidation potential of electron-deficient aromatic systems, which was confirmed via cyclic voltammetry experiments. A similar reaction mechanism was proposed to that described previously (cf., Scheme 11(C)). The authors performed the electrosynthetic method on gram scale and also demonstrated the synthetic utility of the products via derivatization.
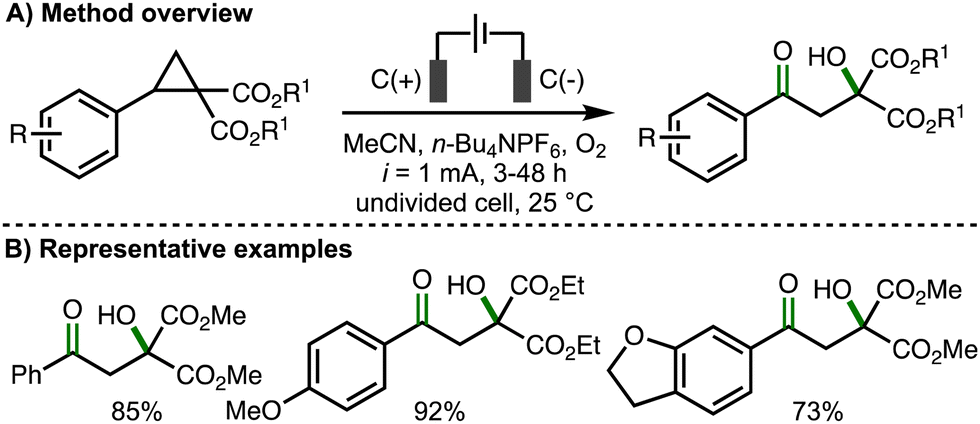 | ||
| Scheme 12 1,3-Oxohydroxylation of donor–acceptor cyclopropanes (Banerjee). | ||
Also in 2021, Werz and co-workers reported Friedel–Crafts-type reactions with electrochemically generated electrophiles from donor–acceptor cyclopropanes and cyclobutanes (Scheme 13).26 In comparison to their previous study, nucleophilic aromatics were added in excess (10 equiv.), an Ar atmosphere was used (instead of oxygen), and an increased cell potential (Ecell = 6–10 V) was employed. Interestingly, an alternating current mode was utilized in order to suppress electrode surface passivation during electrolysis. Using these modified conditions, the deconstructive arylation of various donor–acceptor cyclopropanes and cyclobutanes was achieved. The use of HFIP was crucial for reactivity, which was attributed towards its ability to stabilize radical cation intermediates. With respect to the substrate scope, donor–acceptor cyclopropanes and cyclobutanes that contained aromatic rings bearing halogen, methyl and aryl substitution were well tolerated and the electrochemical reaction could be performed using 1 mmol of cyclopropane substrate. Heteroaromatics, or aromatic rings containing substituents that were strongly electron-releasing (e.g., 4-OMe) or electron-withdrawing were either not successful. A selection of nucleophilic aromatic reaction partners were employed, including p-xylene, toluene, mesitylene, allyl benzene, indane, and tetralin, which all gave the corresponding products in good yields. Electron-rich aromatics such as anisole gave the products in lower yields, and as a mixture of regioisomers. Based upon their previous studies, the authors suggested that the reaction mechanism proceeds via initial anodic oxidation of the donor–acceptor cyclopropane to generate the aromatic radical cation, which undergoes mesolytic cleavage to release ring strain (SN1-type pathway). The resulting benzylic carbocation is intercepted by aromatic nucleophiles, which after rearomatization and hydrogen atom abstraction from solvent generates the observed products. However, when an enantioenriched donor–acceptor cyclopropane (94% e.e.) was employed, the corresponding arylated product was formed in 43% e.e., which suggested that a SN2-type pathway, where the aromatic nucleophile directly intercepts the cyclopropane radical cation, is competitive.
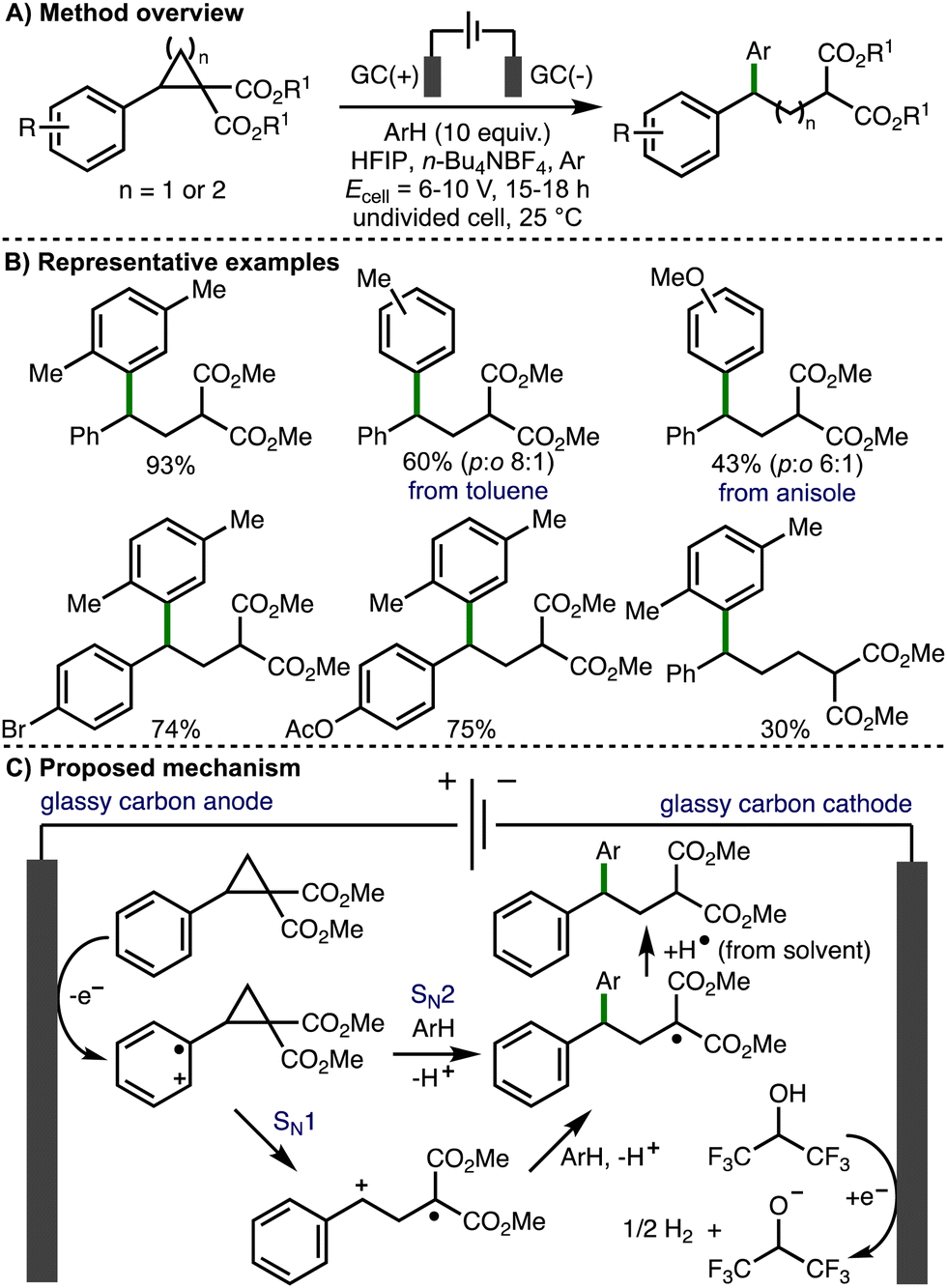 | ||
| Scheme 13 Friedel–Crafts-type reactions with electrochemically generated electrophiles from donor–acceptor cyclopropanes and cyclobutanes (Werz). | ||
4. 5-, 6-, and 7-Membered arylcycloalkenes and arylcycloalkanols
Cyclopropanes and cyclobutanes possess considerably more ring strain than their 5-, 6- or 7-membered counterparts. As such, the successful deconstructive functionalization of these larger ring systems typically requires the incorporation of additional substituents on the arylcycloalkane scaffold in order to stablise the key reactive intermediates formed upon C–C bond cleavage and to provide a sufficient driving force for reaction. This section provides an overview of developed electrochemical strategies for the deconstructive functionalization of 5-, 6-, and 7-membered arylcycloalkenes and arylcycloalkanols, which proceed via the fragmentation of anodically generated aromatic radical cations.In 1996, Ogibin and co-workers reported the electrochemical cleavage of double bonds in conjugated cycloalkenylbenzenes (Scheme 14).27 Employing galvanostatic electrolysis (j = 100 mA cm−2) in an undivided cell equipped with a graphite anode and stainless steel cathode, using MeOH as solvent and n-Bu4NBF4 as supporting electrolyte at 60 °C, it was found that 5-, 6-, and 7-membered phenyl-substituted cycloalkenes could be converted into their corresponding ring-opened ketal–acetal products in good yields. Upon passing 2 F of charge, 1,2-dimethoxycycloalkylbenzenes were formed (as a mixture of diastereomers), which confirmed these as reaction intermediates. The requirement to pass 6 F of charge in order to obtain ring-opened products in high yields (theoretically requiring only 4 F of charge) was attributed to the higher oxidation potential of the 1,2-dimethoxycycloalkylbenzene intermediates compared to the alkenylbenzene starting materials, which resulted in competitive anodic oxidation of methanol. It was subsequently demonstrated that the ketal–acetal products could be converted into dicarbonyl compounds and monoacetals via treatment with dilute sulfuric acid.28 The procedure was also applied successfully to the deconstructive methoxylation of indene, 1,2-dihydronaphthalane, and acenaphthene.29 The authors propose a reaction mechanism that starts with the anodic conversion of cycloalkenylbenzenes into the corresponding 1,2-dimethoxycycloalkylbenzenes.30 These intermediates undergo further single electron anodic oxidation to form an aromatic radical cation, which undergoes mesolytic cleavage to form a benzylic radical and oxocarbenium ion. Further anodic oxidation and trapping with methanol provides the observed ketal–acetal products.
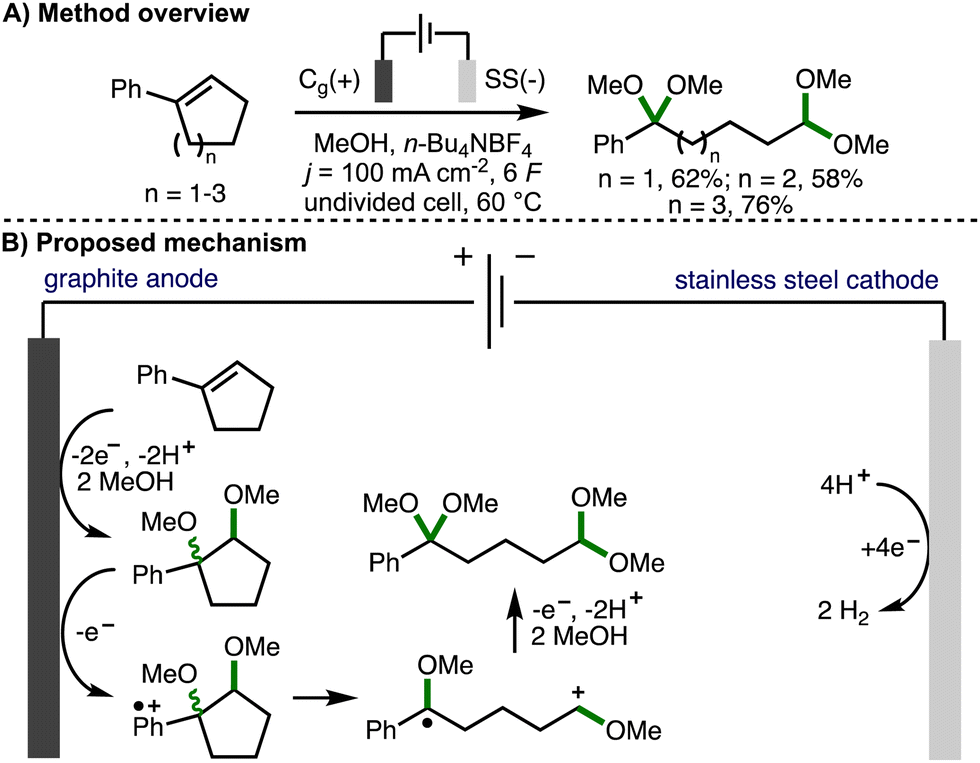 | ||
| Scheme 14 Electrochemical cleavage of double bonds in conjugated cycloalkenylbenzenes (Ogibin). | ||
In 2023, our group reported the deconstructive functionalization of unstrained cycloalkanols via electrochemically generated aromatic radical cations (Scheme 15).31 Employing galvanostatic conditions (i = 10 mA) and an undivided cell equipped with a graphite anode and platinum cathode, using a mixed solvent system of CH2Cl2:MeOH (3:1) and n-Bu4NBF4 as supporting electrolyte at room temperature, it was found that passing 3 F of charge enabled the conversion of a broad range or aryl-substituted cycloalkanols to the corresponding distally methoxylated ketone products. With respect to the substrate scope, the presence or an aromatic ring was required at either the 1- or 2-positions within the cycloalkanol scaffold. The electronics of the aromatic rings could be varied through the incorporation of electron-donating or electron-withdrawing functional groups. Heteroaryl substituents (e.g., 3-pyridyl, 2-benzothiazolyl) were tolerated and the method could be applied to ring expansion by employing bicyclic cycloalkanol substrates. 5-, 6- and 7- membered aryl-substituted cycloalkanols underwent deconstructive methoxylation to give the corresponding distally methoxylated ketones in good yields. The nucleophile could be varied, including different alcohols (for alkoxylation), n-Bu4NOAc (for acetoxylation), and N-heterocycles (e.g., pyrazole for amination). The electrochemical procedure was demonstrated on gram-scale in flow. In comparison to batch, the flow process could be performed with lower electrolyte concentration and with increased current density, which resulted in higher productivity (4.1 mmol h−1vs. 0.08 mmol h−1). Mechanistic experiments confirmed that the presence of an aromatic ring and 1,2-disubstitution were both required for deconstructive functionalization to occur. Furthermore, the same products were formed when the hydroxyl functional group was substituted with a methyl ether, which ruled out the possibility of alkoxy radical intermediates. As such, a mechanism was proposed that initiated with anodic oxidation of the aromatic ring, present at the 1- and/or 2-position of the cycloalkanol scaffold, to form the corresponding aromatic radical cation. This species undergoes hydroxyl-assisted mesolytic cleavage with further anodic oxidation and proton loss to generate a benzylic carbocation, which is intercepted by methanol to form the observed distally methoxylated ketone products.
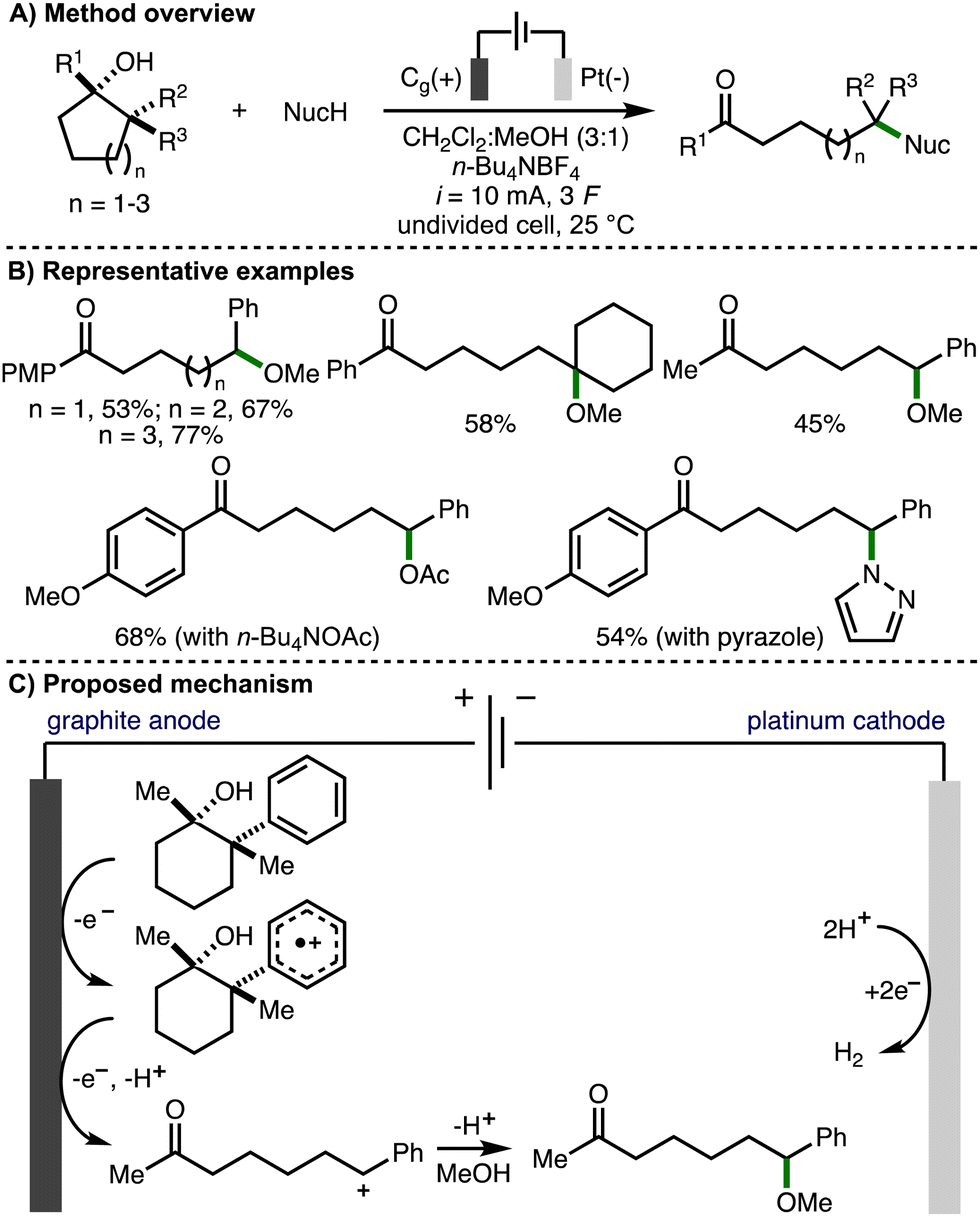 | ||
| Scheme 15 Deconstructive functionalization of unstrained cycloalkanols via electrochemically generated aromatic radical cations (Morrill). | ||
In 2024, Guo, Xia and co-workers reported the electrochemical ring-expansion/functionalization of unstrained secondary cycloalkanols, including those that contained a 2-arylalcohol structural motif (Scheme 16).32 The optimized reaction conditions employed galvanostatic electrolysis (i = 5 mA) and an undivided cell equipped with a graphite anode and platinum cathode, using acetonitrile as solvent, n-Bu4NClO4 as supporting electrolyte, and p-toluenesulfonic acid (TsOH) as an additive at room temperature under air. It was found that the addition of TsOH improved both conversion to the desired product and the diastereomeric ratio. After 5 h reaction time (3.1 F of charge passed), a range of secondary 2-arylcycloalkanols underwent ring expansion in the presence of external nucleophiles to form the corresponding cyclic ether products in synthetically useful yields and with a general preference for the syn diastereomer. It was found that 4-, 5- and 6-membered cycloalkanols underwent ring expansion and that various aromatic substituents (e.g., 4-F, 4-t-Bu, 4-OMe) were tolerated. The external nucleophile was also varied to include a selection of N-heterocycles (e.g., substituted pyrazoles, 1,2,3-triazoles, benzotriazoles, tetrazole) and p-hydroxybenzyl alcohol. Cyclic voltammetry was used to confirm that the 2-arylcycloalkanol substrates undergo preferential oxidation in the presence of N-heterocyclic nucleophiles. The authors proposed that the reaction mechanism initiates via anodic oxidation of the aromatic ring to form the corresponding aromatic radical cation. Subsequent mesolytic cleavage, single electron oxidation and deprotonation would generate a benzylic carbocation intermediate. Intramolecular nucleophilic addition of the carbonyl oxygen atom would form an oxocarbenium ion, which could be intercepted by the external nucleophile in a diastereoselective fashion to generate the observed cyclic ether products. An alternative mechanistic pathway involving β-scission of anodically generated alkoxy radicals was also suggested. The cathodic generation of hydrogen gas was facilitated by the addition of p-toluenesulfonic acid.
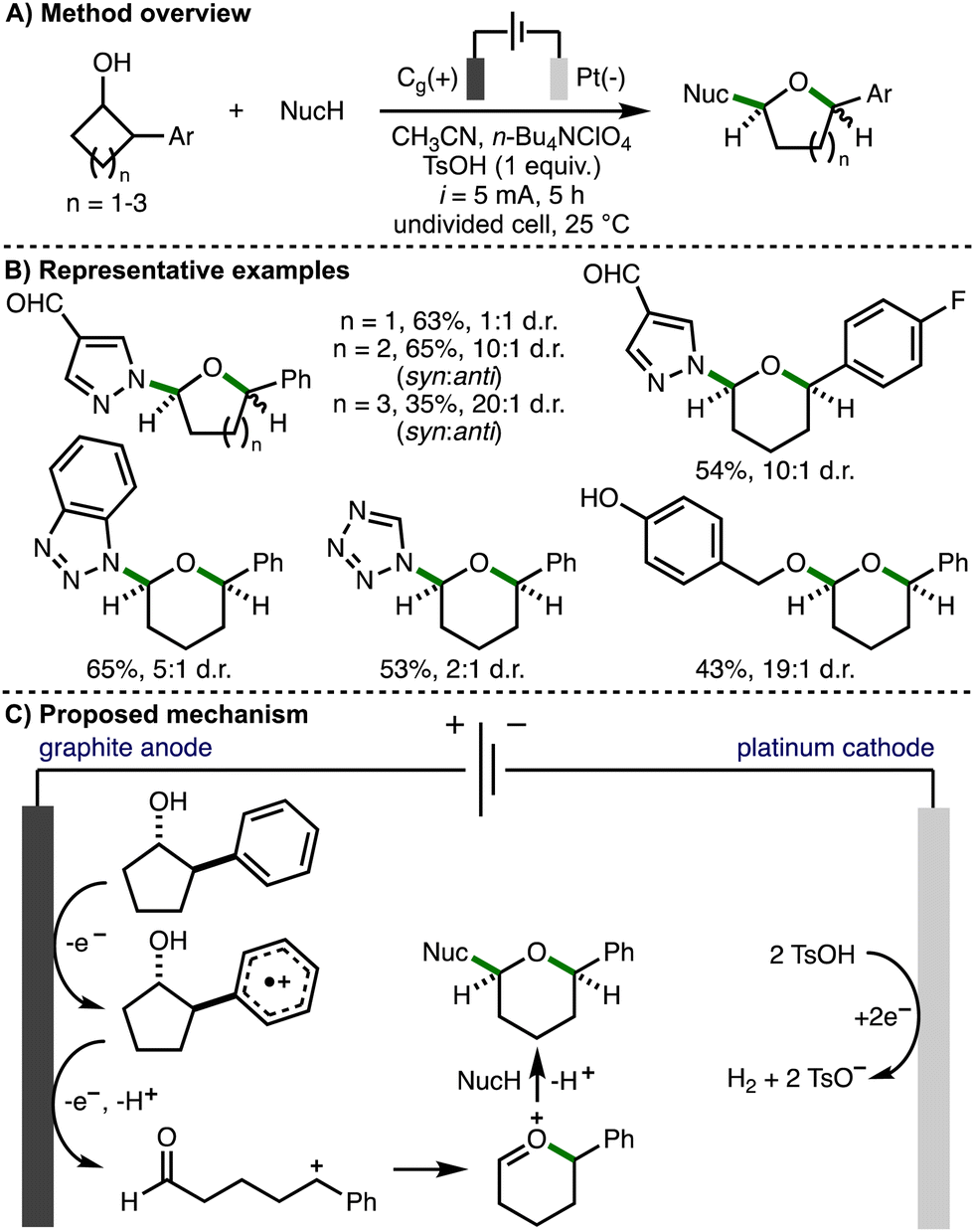 | ||
| Scheme 16 Electrochemical ring-expansion/functionalization of unstrained secondary cycloalkanols (Guo and Xia). | ||
5. Conclusion and outlook
This highlight summarises electrochemical approaches for the deconstructive functionalization of arylcycloalkanes via the fragmentation of anodically generated aromatic radical cations. The advances in this area have been highlighted, and categorized according to the type of cycloalkane substrate employed, namely arylcyclopropanes, donor–acceptor cyclopropanes/cyclobutanes, and 5-, 6- and 7-membered arylcycloalkenes and arylcycloalkanols. An array of electrochemical deconstructive functionalization processes have been covered, including the diverse 1,3-difunctionalization of arylcyclopropanes and donor–acceptor cyclopropanes, the 1,4-oxohydroxylation of donor–acceptor cyclobutanes, and the deconstructive alkoxylation, acetoxylation, and amination of 5-7-membered arylcycloalkanols. Despite a sharp increase in the number of publications in this area over the past few years, many opportunities remain. These include, but are not limited to: (1) the development of novel electrochemical strategies for the deconstructive functionalization of arylcycloalkanes, which address limitations (e.g., substrate scope) associated with existing approaches. For example, the majority of reported methods only work well for arylcycloalkane substrates bearing either electron-rich or electron-poor aromatic rings, rarely both. This observation may be due to multiple factors, including: variation in the oxidation potential of the substrate and/or nucleophile employed for functionalization; over-oxidation, or instability, of products under the employed electrochemical conditions; other undesired side reactions. The use of redox mediators (indirect anodic generation of aromatic radical cations) or potentiostatic (constant potential) electrolysis may help address such issues by improving selectivity for the desired products; (2) the use of various techniques (e.g., synthetic and computational experiments, electroanalytical tools) in order to gain deeper insight into the electrochemical reaction mechanisms. For example, the mechanistic pathway between the anodically generated aromatic radical cation and the observed functionalized products is reported to proceed either via mesolytic cleavage followed by nucleophilic attack or via a three-electron SN2 type reaction. It is likely that substituents on the arylcycloalkane scaffold will influence the predominant reaction mechanism; (3) diversifying the range functionalizations possible to include various bond formations by further diversifying the array of reagents that are compatible with the electrochemical reaction conditions and can be employed as nucleophiles; (4) the application of the developed electrosynthetic methodologies towards the synthesis and/or derivatization of biologically active molecules; (5) the development of enantioselective processes (many of the products formed contain new stereogenic centers); (6) the development of electrochemical deconstructive functionalization processes that proceed via the fragmentation of cathodically generated aromatic radical anions; (7) employment of flow electrochemistry to improve efficiency and further enhance sustainability metrics. It is anticipated that many new and exciting electrochemical transformations involving deconstructive functionalization will be discovered to address these and other challenges in the coming years.Data availability
No primary research results have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the School of Chemistry at Cardiff University and the Department of Chemistry at the University of Bath for generous support. We also thank the Saudi Arabia cultural mission in the UK and the Department of Chemistry at the University of Bisha (H. A. M.) and the Department of Chemistry at Jazan University (A. H. H.) for PhD studentships.References
- For selected reviews, see: (a) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed; (b) S. P. Morcillo, Angew. Chem., Int. Ed., 2019, 58, 14044–14054 CrossRef CAS PubMed.
- For pioneering early work, see: (a) V. R. Rao and S. S. Hixson, J. Am. Chem. Soc., 1979, 101, 6458–6459 CrossRef CAS; (b) K. Mizuno, J. Ogawa and Y. Otsuji, Chem. Lett., 1981, 741–744 CrossRef CAS.
- For selected examples, see: (a) S. Yang, L. Wang, H. Zhang, C. Liu, L. Zhang, X. Wang, G. Zhang, Y. Li and Q. Zhang, ACS Catal., 2019, 9, 716–721 CrossRef CAS; (b) L. H. Wang, X. M. Wang, G. Zhang, S. B. Yang, Y. Li and Q. Zhang, Org. Chem. Front., 2019, 6, 2934–2938 RSC; (c) H. Zhang, H. Xiao, F. Jiang, Y. Fang, L. Zhu and C. Li, Org. Lett., 2021, 23, 2268–2272 CrossRef CAS PubMed.
- For selected examples, see: (a) Z. Lu, J. D. Parrish and T. P. Yoon, Tetrahedron, 2014, 70, 4270–4278 CrossRef CAS PubMed; (b) L. Ge, D. X. Wang, R. Xing, D. Ma, P. J. Walsh and C. Feng, Nat. Commun., 2019, 10, 4367 CrossRef PubMed; (c) H. Liu, Y. Li, D. X. Wang, M. M. Sun and C. Feng, Org. Lett., 2020, 22, 8681–8686 CrossRef CAS PubMed; (d) Z. Zuo and A. Studer, Org. Lett., 2022, 24, 949–954 CrossRef CAS PubMed; (e) L. Ge, C. Zhang, C. Pan, D.-X. Wang, D.-Y. Liu, Z.-Q. Li, P. Shen, L. Tian and C. Feng, Nat. Commun., 2022, 13, 5938 CrossRef CAS PubMed.
- For selected examples, see: (a) R. Pitts, B. Ling, J. A. Snyder, A. E. Bragg and T. Lectka, J. Am. Chem. Soc., 2016, 138, 6598–6609 CrossRef PubMed; (b) D. Petzold, P. Singh, F. Almqvist and B. König, Angew. Chem., Int. Ed., 2019, 58, 8577–8580 CrossRef CAS PubMed; (c) T. V. T. Nguyen, M. D. Wodrich and J. Waser, Chem. Sci., 2022, 13, 12831–12839 RSC.
- For selected reviews, see: (a) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed; (b) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- (a) M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed; (b) C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef.
- For selected reviews, see: (a) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (b) D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- (a) K. B. Wiberg, Angew. Chem., Int. Ed. Engl., 1986, 25, 312–322 CrossRef; (b) E. V. Anslyn and D. A. Dougherty, Chapter 2: Strain and Stability, Modern Physical Organic Chemistry, University Science, Sausalito, CA, 2006 Search PubMed.
- For selected reviews, see: (a) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227–263 CrossRef CAS PubMed; (b) S. T. Sivanandan, B. Krishna, T. V. Baiju and C. Mohan, Eur. J. Org. Chem., 2021, 6781–6805 CrossRef CAS.
- For selected reviews, see: (a) I. M. Taily, D. Saha and P. Banerjee, Org. Biomol. Chem., 2021, 19, 8627–8645 RSC; (b) K. Chen, Z.-Q. Li, C. Zhu and C. Feng, Trends Chem., 2023, 5, 160–161 CrossRef CAS.
- W. Sheng, X. Huang, J. Cai, Y. Zheng, Y. Wen, C. Song and J. Li, Org. Lett., 2023, 25, 6178–6183 CrossRef CAS PubMed.
- R. Kumar, N. Banerjee, P. Kumar and P. Banerjee, Chem. – Eur. J., 2023, 29, e202301594 CrossRef CAS PubMed.
- T. Shono and Y. Matsumura, J. Org. Chem., 1970, 35, 4157–4160 CrossRef CAS.
- P. Peng, X. Yan, Z. Liu, L. Zeng, Y. Chen, H. Zhang and A. Lei, Nat. Commun., 2021, 12, 3075 CrossRef CAS PubMed.
- S. Dutt, R. Kumar, N. Banerjee, D. Saha and P. Banerjee, Adv. Synth. Catal., 2024, 366, 526–532 CrossRef CAS.
- Y. Yue, Y. Song, S. Zhao, C. Zhang, C. Zhu and C. Feng, Org. Lett., 2023, 25, 7385–7389 CrossRef CAS PubMed.
- J. Cai, Y. Wen, W. Sheng, X. Huang, Y. Zheng, C. Song and J. Li, Green Chem., 2023, 25, 6618–6622 RSC.
- X. Huang, J. Cai, Y. Zheng, C. Song and J. Li, Adv. Synth. Catal., 2024, 366, 201–206 CrossRef CAS.
- W. Zhou, P. Chen, Z.-Q. Li, L.-T. Xiao, J. Bai, X.-R. Song, M.-J. Luo and Q. Xiao, Org. Lett., 2023, 25, 6919–6924 CrossRef CAS PubMed.
- For selected reviews, see: (a) H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef CAS PubMed; (b) M. Yu and B. L. Pagenkopf, Tetrahedron, 2005, 61, 321–347 CrossRef CAS.
- For selected reviews, see: (a) T. F. Schneider, J. Kaschel and D. B. Werz, Angew. Chem., Int. Ed., 2014, 53, 5504–5523 CrossRef CAS PubMed; (b) H.-U. Reissig and R. Zimmer, Angew. Chem., Int. Ed., 2015, 54, 5009–5011 CrossRef CAS PubMed; (c) A. Ghosh, R. Dey and P. Banerjee, Chem. Commun., 2021, 57, 5359–5373 RSC; (d) K. Ghosh and S. Das, Org. Biomol. Chem., 2021, 19, 965–982 RSC.
- S. Kolb, M. Petzold, F. Brandt, P. G. Jones, C. R. Jacob and D. B. Werz, Angew. Chem., Int. Ed., 2021, 60, 15928–15934 CrossRef CAS PubMed.
- G. A. Oliver, S. Kolb and D. B. Werz, Synlett, 2024, 35, 963–966 CrossRef CAS.
- D. Saha, I. M. Taily and P. Banerjee, Eur. J. Org. Chem., 2021, 5053–5057 CrossRef CAS.
- S. Kolb, N. L. Ahlburg and D. B. Werz, Org. Lett., 2021, 23, 5549–5553 CrossRef CAS PubMed.
- Y. N. Ogibin, A. I. Ilovaisky and G. I. Nikishin, J. Org. Chem., 1996, 61, 3256–3258 CrossRef CAS.
- Y. N. Ogibin, A. I. Ilovaisky and G. I. Nikishin, Russ. Chem. Bull., 1996, 45, 1939–1941 CrossRef.
- Y. N. Ogibin, A. I. Ilovaisky and G. I. Nikishin, Electrochim. Acta, 1997, 42, 1933–1941 CrossRef CAS.
- Y. N. Ogibin, A. Ilovaisky and G. I. Nikishin, Chapter 2: Olefins and Aromatics, Novel Trends in Electroorganic Synthesis, Springer, Tokyo, Japan, 1998 Search PubMed.
- J. Harnedy, H. A. Maashi, A. A. M. A. El Gehani, M. Burns and L. C. Morrill, Org. Lett., 2023, 25, 1486–1490 CrossRef CAS PubMed.
- L. Zhao, P. Hu, J. Tian, X. Zhang, C. Yang, L. Guo and W. Xia, Org. Lett., 2024, 26, 4882–4886 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |