
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCsp2–H/F bond activation and borylation with iron†
Ethan
Zars
 a,
Lisa
Pick
b,
Achala
Kankanamge
a,
Michael R.
Gau
a,
Karsten
Meyer
b and
Daniel J.
Mindiola
*a
a,
Lisa
Pick
b,
Achala
Kankanamge
a,
Michael R.
Gau
a,
Karsten
Meyer
b and
Daniel J.
Mindiola
*a
aDepartment of Chemistry, University of Pennsylvania, 231 S 34th St, Philadelphia, PA 19104, USA. E-mail: mindiola@sas.upenn.edu
bDepartment of Chemistry & Pharmacy, Inorganic Chemistry, Friedrich-Alexander-Universität Erlangen – Nürnberg (FAU), Erlangen 91058, Germany. E-mail: karsten.meyer@fau.de
First published on 10th October 2024
Abstract
Reduction of [K2{(tBupyrr2pyr)Fe}2(μ-N2)] (1) with two equiv. of KC8 in the presence of crown-ether 18-C-6 yields the N2 adduct [{K(18-C-6)}2(tBupyrr2pyr)Fe(N2)] (2). Complex 2 heterolytically splits the Csp2–H bond of benzene to form [{K(18-C-6)}(tBupyrr2pyr)Fe(C6H5)] (3), whereby usage of a diboron B2pin2 promotes hydride elimination to form the salt [K(18-C-6)HB2Pin2] (4). Similarly, 3 can also be formed by cleavage of the C–F bond of fluorobenzene. Reaction of 3 with ClBcat yields [K(18-C-6)(thf)2][(tBupyrr2pyr)FeCl] (5) and PhBcat and the former can be reduced to 2 to complete a synthetic cycle for heterolytic benzene C–H activation and borylation.
The activation and functionalization of C–H bonds is a crucial step toward converting an unreactive and abundant substrate into more reactive or synthetically versatile functionalities.1 Much of the early literature on C–H activation focuses on the oxidative addition of C–H bonds at low valent precious metals.2 This research was performed in the context of cross-coupling reactions which typically go through catalytic cycles consisting of oxidative addition, transmetallation, and reductive elimination.3 A particularly useful target for C–H functionalization would be the generation of C–B bonds due to their prevalence in Suzuki–Miyaura cross coupling reactions.4 While C–H activation is usually accomplished with precious metals, there is precedent for this reaction at iron,5 which is the most abundant metal in Earth's crust.6 To this end, borylation of aryl Csp2–H bonds by photolysis of an iron boryl species has been reported,7 with more recent studies expanding this work to catalytic processes for C–H borylation.8 While mechanistic studies for arene C–H borylation reactions have been studied in detail with first row transition metals such as Co,9 similar studies clearly showing the bond forming and breaking processes at the Fe center have been exceptionally rare.5e,7b,7c,8a,8j,10
In this contribution, we show how a formally Fe0 and mononuclear Fe dinitrogen complex [{K(18-C-6)}2(tBupyrr2pyr)Fe(N2)] (2) (tBupyrr2pyr2− = 3,5-tBu2-bis(pyrrolyl)pyridine; 18-C-6 = 18-crown-6) can activate the Csp2–H bond of benzene (and the C–F bond of fluorobenzene) at room temperature to yield the ferrous phenyl complex [{K(18-C-6)}(tBupyrr2pyr)Fe(C6H5)] (3). We propose the C–H activation process to be heterolytic in nature by trapping KH with B2pin2 (pin = pinacolato) to form the adduct [K(18-C-6)HB2pin2] (4). The aryl ligand from 3 can be transmetallated using ClBcat (cat = catecholato) to yield the discrete salt [{K(18-C-6)(thf)2}(tBupyrr2pyr)FeCl] (5) along with the borane PhBcat (6). Finally, we show how 5 can be reduced to 2 to close a synthetic cycle for room temperature Csp2–X bond activation and borylation (Scheme 1).
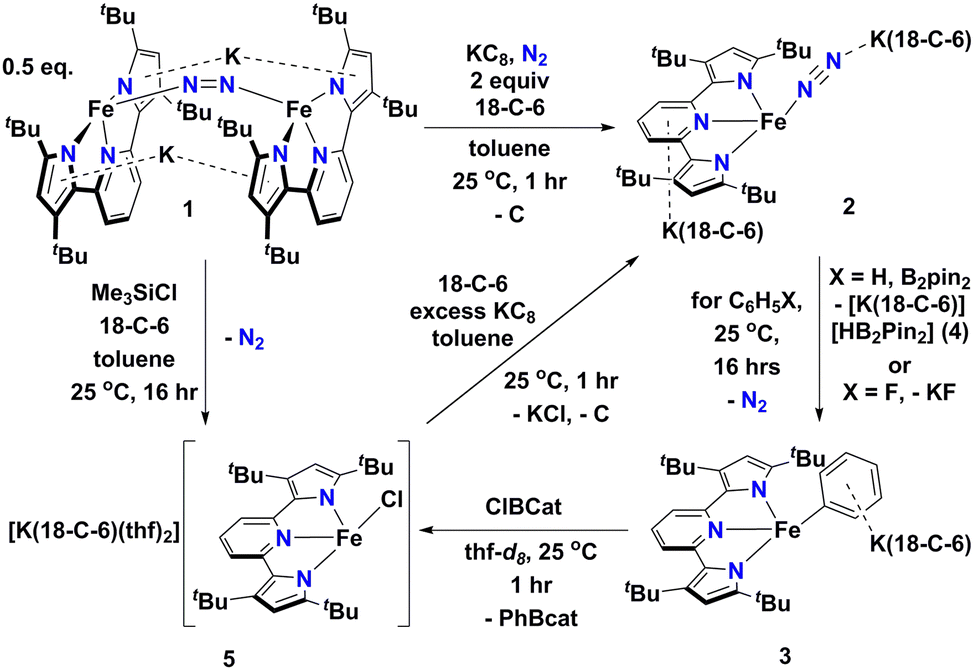 | ||
| Scheme 1 Reaction scheme outlining formation and reactions of [{K(18-C-6)}2(tBupyrr2pyr)Fe(N2)] (2). | ||
We have previously shown that reduction of the ferrous precursor [(tBupyrr2pyr)Fe(OEt2)]11 with one equiv. of KC8 yields the formally FeI end-on and bridging N2 complex [K2{(tBupyrr2pyr)Fe}2(μ-N2)] (1) in which the N2 ligand bridging the two Fe centers is topologically nonlinear.12 Such a geometry is retained upon further reduction to the formally Fe0 complex [{K2(18-C-6)}{(tBupyrr2pyr)Fe}2(μ-N2)] or via oxidative substitution of N2 with chalcogenides.13 The full extent of low oxidation state iron chemistry using the tBupyrr2pyr ligand platform, especially as it pertains to mononuclear complexes, has so far been unexplored. Treatment of 1 equiv. of 1 with four equivalents of 18-C-6 and two equivalents of KC8 in toluene at room temperature resulted in the initial formation of a brown residue. Allowing the reaction to stir for an additional hour at room temperature led to the conversion of the brown residue to a dark purple solution. After filtration and concentration in vacuo, the solution was cooled to −35 °C overnight to afford [{K(18-C-6)}2(tBupyrr2pyr)Fe(N2)] (2), which could be isolated in 79% yield as dark crystals (Scheme 1 and Fig. 1). The 1H NMR spectrum of 2 shows seven paramagnetically broadened and shifted resonances indicative of a Cs symmetric system (Fig. S1, ESI†). A single-crystal X-ray diffraction study (sc-XRD) of 2 shows a (tBupyrr2pyr)Fe scaffold with a terminal N2 ligand occupying the fourth coordination site and capped by a {K(18-C-6)}+ unit. The second {K(18-C-6)}+ is coordinated to the pyridyl portion of the meridional ligand. The geometry at the formally Fe(0) center in 2 can best be described as seesaw with a ∠Npyr–Fe–N2 angle of 140.7(2)° and a τ4 value of 0.51. The N2 ligand has an N–N bond length of 1.147(6) Å and a signature stretch at 1851 cm−1 in the IR spectrum (Fig. S21, ESI†) implying significant activation of the N2 ligand when compared to its free form (1.0975 Å, 2331 cm−1).14 The low energy vibration and elongated N–N bond for the N2 ligand implies not only the metal ion but the N2 ligand to be the likely locus of reduction.15 While we cannot determine the Fe oxidation state of 2 with complete certainty, a significantly shortened Fe–N2 bond length of 1.775(4) Å and solution state magnetic moment of 4.1 μB, measured by Evans Method in C6D6, which is above the expectation value for an S = 1 system, and implies contribution of a FeI atom and N2-centered radical to the complicated electronic structure. In contrast to the bridging nature of [{K2(18-C-6)(tBupyrr2pyr)Fe}2(μ-N2)], we propose the mononuclear structure of 2 to derive from the addition of more equivalents of 18-crown-6.
 | ||
| Fig. 1 ORTEP sc-XRD structures of complexes 2, 3, 4, and 5 at 50% probability level. Residual solvent molecules and H atoms (with the exception of the hydride in compound 4) have been omitted for clarity. Coordinated thf molecules coordinated to K+ in compound 5 have been also omitted for clarity. | ||
Interested in the oxidative chemistry of 2, we treated this species with one equivalent of bispinacolato diboron (B2pin2) in benzene expecting to observe reductive cleavage of the B–B bond. However, upon addition of the B2pin2, the reaction mixture gradually changed color from dark purple to red, and over 16 hours there was deposition of red crystals. A sc-XRD study on a single-crystal isolated from the reaction mixture confirms the structure to be instead the discrete phenyl salt [{K(18-C-6)}(tBupyrr2pyr)Fe(C6H5)] (3) which was isolated in 61% yield. The solid-state structure of 3 shows a formally FeII ion with an Fe–C bond length of 2.068(1) Å resulting from benzene C–H activation. The ∠Npyr–Fe–CAr angle is 123.88(5)° and the geometric index value of τ4 = 0.74 is consistent with a distorted cis-divacant octahedral coordination geometry. A 1H NMR spectroscopic study of these crystals in thf-d8 shows 9 paramagnetically shifted and broadened resonances showing preservation of a Cs symmetric system in solution (Fig. S2, ESI†). A room temperature solution state magnetic moment of 4.8 μB, determined by Evans method, is close to the spin only value for an S = 2 system in accord with a high spin FeII ion in 3. Examination of the supernatant from the reaction between 2 and B2Pin2 by 11B NMR spectroscopy in thf-d8 revealed 2 resonances at 30.97 ppm and 5.91 ppm (Fig. S9, ESI†). Resonances in these positions can be attributed to one trigonal planar and one pyramidal B atom as demonstrated by Marder and coworkers in a series of [{K(18-crown-6)}XB2pin2] molecules, where X = OtBu, OMe, and F.16 In our case, the B-containing molecule could be crystallized out of a concentrated thf solution at −35 °C and its structure determined to be [{K(18-crown-6)}HB2pin2] (4). A sc-XRD structure of 4 clearly shows a HB2pin2 core with two distinctly different B centers as well as a {K(18-C-6)}+ counter ion interacting with the hydride and nearby pinacolato oxygen (Fig. 1). 11B NMR spectroscopy of the crystals also showed, as expected, 2 resonances, albeit at slightly different chemical shifts (Fig. S8, ESI†) when compared to the crude material. We attribute this discrepancy to the lack of paramagnetic impurities in the crystalline material contributing to a noncontact paramagnetic shift.17 Transfer of a hydrogen atom to a borane has been reported before in the context of C–H activation at low valent iron centers, but diboron reagents have not been used to sponge out the hydride most likely stemming from heterolytic C–H bond cleavage.5e,5g Using other hydride acceptors such as imines and nitriles or other Lewis acids such as B(C6F5)3 did not result in the formation of 3 (Fig. S13–S17, ESI†). We found that complex 3 could also be generated by reaction of 2 with fluorobenzene at room temperature (Scheme 1) in a manner similar to Holland's work using low valent Co complexes.18 Unfortunately, our reaction was not quantitative thus resulting in formation of other intractable materials (Fig. S10, ESI†). Selective C–F bond activation is rare19 and C–F bonds are known to be stable toward catalytic C–H borylation protocols.8a,8c,8h,9c
Having demonstrated that 2 could undergo C–H activation of benzene we next turned to delivering or functionalizing the phenyl moiety of 3. Accordingly, treatment of 3 with one equivalent of ClBcat in thf-d8 resulted in a new paramagnetic molecule when judged by 1H NMR spectroscopy. 1H and 11B NMR spectroscopy confirmed that a second, diamagnetic species was formed, namely PhBCat (Fig. S19, ESI,†11B NMR δ = 32.6).20 The paramagnetic product was identified to be [{K(18-C-6)(thf)2}(tBupyrr2pyr)FeCl] (5) using a combination of 1H NMR, sc-XRD, solution magnetic susceptibility studies, as well as by an independent synthesis in 92% isolated yield via addition of 2 equiv. of Me3SiCl to the N2 precursor 1 (Scheme 1). A 1H NMR spectrum in thf-d8 is consistent with a plane of symmetry bisecting 5 given the observation of seven paramagnetically shifted and broadened resonances (Fig. S3, ESI†). A sc-XRD study on 5 shows the (tBupyrr2pyr)Fe core with a chloride ligand occupying the fourth coordination site and {K(18-crown-6)(thf)2}+ in the unit cell but not interacting with the iron complex. The Fe–Cl bond length is 2.2914(5) Å, which is longer than the Fe–Cl bond length of 2.1883(9) Å in the neutral ferric congener [(tBupyrr2pyr)FeCl].11 In the structure of 5 the ∠Npyr–Fe–Cl angle is 135.34(3)° and the geometric index τ4 value is 0.60, which makes this geometry best described as seesaw. A solution state magnetic moment of 5, determined by Evans method, is 5.4 μB and is consistent with an S = 2 system.
In the interest of investigating the electronic structures of these mononuclear formally FeII ate complexes (3 and 5), 57Fe Mössbauer spectroscopy and DC SQUID magnetometry measurements were performed on complex 5 as a representative example of this new class of compounds (Fig. 2). 57Fe Mössbauer spectroscopy of 5 at 77 K shows a quadrupole doublet with an isomer shift of δ = 0.82 mm s−1, a quadrupole splitting of ΔEQ = 1.02 mm s−1, and a linewidth of ΓFWHM = 0.32 mm s−1. These values are consistent with an S = 2 FeII ion on the tBupyrr2pyr ligand platform.11,13,21 This spin state was further confirmed by a DC SQUID magnetometry study which showed a field-independent magnetic moment of 5.28 μB (averaged between two independently measured samples) at 300 K. Unfortunately, due to difficulties in scaling up the synthesis of 3, we were unable to collect reliable 57Fe Mössbauer spectroscopy and DC SQUID magnetometry measurements on this complex in order to directly compare with 5. We can, however, be reasonably confident in our assignment of its oxidation and spin state as S = 2 FeII due to its solution state magnetic moment of 4.8 μB and similarities with 5 in its electronic absorption spectrum (Fig. S22 and S23, ESI†).
 | ||
| Fig. 2 (Top) Zero-field 57Fe-Mössbauer spectrum of 5 (solid state, 77 K). Measured data were fit with parameters δ = 0.82 mm s−1, ΔEQ = 1.02 mm s−1, and ΓFWHM = 0.32 mm s−1. (bottom) Temperature-dependent SQUID magnetization measurement of a powdered sample of 5 recorded from 2 to 300 K with an applied magnetic field of 1 T. Measured data were fit with parameters S = 2, TIP = 1023 × 10−6 emu, |D| = 12 cm−1, E/D = 0.14, and gav = 2.14. | ||
When complex 5 is reduced with an excess of KC8 and one equivalent of 18-C-6 under an N2 atmosphere, a gradual color change from red to dark purple is observed. Filtering the solution after one hour of reaction time and crystallization out of a concentrated toluene solution yields small black crystals that were identified as complex 2 by 1H NMR spectroscopy (Fig. S20, ESI†), thus completing a synthetic cycle of benzene activation and borylation involving the Fe0/FeII couple. Future work will involve attempts at rendering this system catalytic as well as expanding the substrate scope with the aim of site-selective C–H bond activation.
EZ synthesized the complexes, and wrote part of the manuscript. LP collected and analysed the Mössbauer and SQUID data. AK assisted with the synthesis of complexes. MRG solved and curated the crystal structures. KM and DJM provided funding and equipment for the project and helped compose the manuscript.
This work was supported by the U.S. Department of Energy, Office of Basic Energy Sciences (DOE-BES-DESC0023340) and the University of Pennsylvania (UPenn, D. J. M.), the Friedrich-Alexander-Universität Erlangen – Nürnberg, and the Alexander von Humboldt Foundation (K. M. and D. J. M.). A. K. thanks the Vagelos Integrated Program in Energy Research at UPenn for support.
Data availability
Data for this article including full synthetic procedures and characterization are available in the ESI.†Conflicts of interest
There are no conflicts to declare.References
- (a) S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. L. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed; (b) I. F. Yu, J. W. Wilson and J. F. Hartwig, Chem. Rev., 2023, 123, 11619–11663 CrossRef CAS PubMed; (c) R. Arevalo and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 9106–9123 CrossRef CAS PubMed.
- (a) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471–2526 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (c) B. A. Arndtsen, R. G. Bergman, T. A. Mobley and T. H. Peterson, Acc. Chem. Res., 1995, 28, 154–162 CrossRef CAS; (d) A. E. Shilov and G. B. Shul’pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed; (e) R. H. Crabtree, J. Chem. Soc., Dalton Trans., 2001, 2437–2450 RSC.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed; (b) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (c) X. Chen, K. M. Engle, D. H. Wang and Y. Jin-Quan, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- (a) S. Camadanli, R. Beck, U. Flörke and H.-F. Klein, Organometallics, 2009, 28, 2300–2310 CrossRef CAS; (b) M. V. Baker and L. D. Field, J. Am. Chem. Soc., 1987, 109, 2825–2826 CrossRef CAS; (c) L. D. Field, R. W. Guest and P. Turner, Inorg. Chem., 2010, 49, 9086–9093 CrossRef CAS PubMed; (d) S. Camadanli, R. Beck, U. Flörke and H.-F. Klein, Organometallics, 2009, 28, 2300–2310 CrossRef CAS; (e) A. Casitas, H. Krause, S. Lutz, R. Goddard, E. Bill and A. Fürstner, Organometallics, 2018, 37, 729–739 CrossRef CAS; (f) S. D. Ittel, C. A. Tolman, A. D. English and J. P. Jesson, J. Am. Chem. Soc., 1976, 98, 6073–6075 CrossRef CAS; (g) S. F. McWilliams, D. L. J. Broere, C. J. V. Halliday, S. M. Bhutto, B. Q. Mercado and P. L. Holland, Nature, 2020, 584, 221–226 CrossRef CAS PubMed; (h) W. D. Jones, G. P. Foster and J. M. Putinas, J. Am. Chem. Soc., 1987, 109, 5047–5048 CrossRef CAS; (i) M. K. Whittlesey, R. J. Mawby, R. Osman, R. N. Perutz, L. D. Field, M. P. Wilkinson and M. W. George, J. Am. Chem. Soc., 1993, 115, 8627–8637 CrossRef CAS; (j) C. A. Tolman, S. D. Ittel, A. D. English and J. P. Jesson, J. Am. Chem. Soc., 1979, 101, 1742–1751 CrossRef CAS; (k) A. K. Hickey, S. A. Lutz, C.-H. Chen and J. M. Smith, Chem. Commun., 2017, 53, 1245–1248 RSC.
- ed. W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Press, Boca Raton, FL, 97th edn, 2016, p. 19.
- (a) K. M. Waltz, X. He, C. Muhoro and J. F. Hartwig, J. Am. Chem. Soc., 1995, 117, 11357–11358 CrossRef CAS; (b) T. J. Mazzacano and N. P. Mankad, Chem. Commun., 2015, 51, 5379–5382 RSC; (c) K. M. Waltz, C. N. Muhoro and J. F. Hartwig, Organometallics, 1999, 18, 3383–3393 CrossRef CAS.
- (a) L. Britton, J. H. Docherty, G. S. Nichol, A. P. Dominey and S. P. Thomas, Chin. J. Chem., 2022, 40, 2875–2881 CrossRef CAS; (b) T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J.-B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062–4065 CrossRef CAS PubMed; (c) M. Kamitani, H. Kusaka and H. Yuge, Chem. Lett., 2019, 48, 898–901 CrossRef CAS; (d) J.-L. Tu, A.-M. Hu, L. Guo and W. Xia, J. Am. Chem. Soc., 2023, 145, 7600–7611 CrossRef CAS PubMed; (e) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258–17261 CrossRef CAS PubMed; (f) G. Yan, Y. Jiang, C. Kuang, S. Wang, H. Liu, Y. Zhang and J. Wang, Chem. Commun., 2010, 46, 3170–3172 RSC; (g) T. Kato, S. Kuriyama, K. Nakajima and Y. Nishibayashi, Chem. – Asian J., 2019, 14, 2097–2101 CrossRef CAS PubMed; (h) H. Lee, T. He and S. P. Cook, Org. Lett., 2023, 25, 1–4 CrossRef CAS PubMed; (i) M. Kamitani, Chem. Commun., 2021, 57, 13246–13258 RSC; (j) T. Hatanaka, Y. Ohki and K. Tatsumi, Chem. – Asian J., 2010, 5, 1657–1666 CrossRef CAS PubMed.
- (a) J. V. Obligacion, S. P. Semproni, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 10645–10653 CrossRef CAS PubMed; (b) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133–4136 CrossRef CAS PubMed; (c) T. P. Pabst and P. J. Chirik, J. Am. Chem. Soc., 2022, 144, 6465–6474 CrossRef CAS PubMed.
- S. R. Parmelee, T. J. Mazzacano, Y. Zhu, N. P. Mankad and J. A. Keith, ACS Catal., 2015, 5, 3689–3699 CrossRef CAS.
- K. Searles, S. Fortier, M. M. Khusniyarov, P. J. Carroll, J. Sutter, K. Meyer, D. J. Mindiola and K. G. Caulton, Angew. Chem., Int. Ed., 2014, 53, 14139–14143 CrossRef CAS PubMed.
- D. Sorsche, M. E. Miehlich, K. Searles, G. Gouget, E. M. Zolnhofer, S. Fortier, C.-H. Chen, M. Gau, P. J. Carroll, C. B. Murray, K. G. Caulton, M. M. Khusniyarov, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2020, 142, 8147–8159 CrossRef CAS PubMed.
- E. Zars, L. Gravogl, M. R. Gau, P. J. Carroll, K. Meyer and D. J. Mindiola, Chem. Sci., 2023, 14, 6770–6779 RSC.
- (a) N. Hazari, Chem. Soc. Rev., 2010, 39, 4044–4056 RSC; (b) P. L. Holland, Dalton Trans., 2010, 39, 5415–5425 RSC.
- J. B. Geri, J. P. Shanahan and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 5952–5956 CrossRef CAS PubMed.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed.
- J. D. Satterlee, Concepts Magn. Reson., 1990, 2, 69–79 CrossRef CAS.
- (a) T. R. Dugan, X. Sun, E. V. Rybak-Akimova, O. Olatunji-Ojo, T. R. Cundari and P. L. Holland, J. Am. Chem. Soc., 2011, 133, 12418–12421 CrossRef CAS PubMed; (b) T. R. Dugan, J. M. Goldberg, W. W. Brennessel and P. L. Holland, Organometallics, 2012, 31, 1349–1360 CrossRef CAS.
- (a) N. Toriumi, K. Yamashita and N. Iwasawa, Chem. – Eur. J., 2021, 27, 12635–12641 CrossRef CAS PubMed; (b) Q. K. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Angew. Chem., Int. Ed., 2021, 60, 20391–20399 CrossRef CAS PubMed; (c) Y. Nakamura, N. Yoshikai, L. Ilies and E. Nakamura, Org. Lett., 2012, 14, 3316–3319 CrossRef CAS PubMed; (d) Q.-K. Kang, Y. Lin, Y. Li, L. Xu, K. Li and H. Shi, Angew. Chem., Int. Ed., 2021, 60, 20391–20399 CrossRef CAS PubMed; (e) D. Guijarro and M. Yus, Tetrahedron, 2000, 56, 1135–1138 CrossRef CAS.
- D.-G. Yu and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 7097–7100 CrossRef CAS PubMed.
- (a) E. Zars, L. Gravogl, M. Gau, P. J. Carroll, K. Meyer and D. J. Mindiola, Inorg. Chem., 2022, 61, 1079–1090 CrossRef CAS PubMed; (b) D. Sorsche, M. E. Miehlich, E. M. Zolnhofer, P. J. Carroll, K. Meyer and D. J. Mindiola, Inorg. Chem., 2018, 57, 11552–11559 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI contains complete experimental details and spectral data. CCDC 2377358–2377361. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc04127e |
| This journal is © The Royal Society of Chemistry 2024 |