
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalysed intramolecular C(sp3)–H amination of alkyl azides
Kai
Wu
 a and
Chi-Ming
Che
*ab
a and
Chi-Ming
Che
*ab
aState Key Laboratory of Synthetic Chemistry, Department of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong, P. R. China. E-mail: cmche@hku.hk
bLaboratory for Synthetic Chemistry and Chemical Biology Limited Units 1503-1511, 15/F., Building 17W, Hong Kong Science Park, New Territories, Hong Kong, P. R. China
First published on 12th November 2024
Abstract
Iron-catalysed intramolecular C(sp3)–H amination of alkyl azides (N3R, R = alkyl) via the iron–alkylnitrene/alkylimido (Fe(NR)) intermediate, is an appealing synthetic approach for the synthesis of various N-heterocycles. This approach provides a direct atom-economy strategy for constructing C(sp3)–N bonds, with nitrogen gas as the only by-product and iron is a biocompatible, cheap, and earth-abundant metal. However, C(sp3)–H amination with alkyl azides is challenging because alkyl nitrenes readily undergo 1,2-hydride migration to imines. This article summarizes recent major advances in this field in terms of catalyst design, substrate scope expansion, stereoselectivity control, understanding of key reaction intermediates, and applications in the synthesis of complex natural products and pharmaceuticals.
 Kai Wu | Dr Kai Wu received his PhD degree in 2021 from The University of Hong Kong (HKU) in the field of organometallic chemistry under the supervision of Professor Chi-Ming Che. From 2021 to 2023, he worked as a postdoctoral fellow in State Key Laboratory of Synthetic Chemistry at HKU in Prof. Che's group. Currently, he is a Research Assistant Professor in the Chemistry Department of HKU. His research interest is in the development of novel synthetic approaches, typically centralizing on C–H activation via reactive metal–carbene, -imido/nitrene, and -oxo species, enabling challenging transformations to proceed with catalyst-controlled reactivity and selectivity. |
 Chi-Ming Che | Prof. Chi-Ming Che received his PhD degree in 1982 from The University of Hong Kong (HKU) under the supervision of Professor Chung-Kwong Poon. From 1980 to 1983, he studied at the California Institute of Technology under the supervision of Professor Harry B. Gray. Thereafter, he returned to HKU and was promoted to Chair Professor in Chemistry in 1992. He is the Zhou Guangzhao Professor in Natural Sciences (2016–present). He was elected as a member of the Chinese Academy of Sciences in 1995 and Foreign Associate of National Academy of Sciences, USA in 2013. He received First-Class Prize of State Natural Science Award of China (2007), Ryoji Noyori ACES Award (2016), Luigi Sacconi Medal (2020) and Silver Bauhinia Star of the HKSAR (2021). He has published over 1100 papers with a h-index of 131. |
1. Introduction
N-Heterocycles are ubiquitous in pharmaceuticals and natural products,1,2 and their synthesis is one of the most important and common synthetic tasks. Although methods to obtain these compounds are well studied,3–6 the most straightforward is C–H amination.7–19 The formation of C(sp3)–N bonds through metal–alkylnitrene/alkylimido (M = NR, R = alkyl) intermediates from alkyl azides is an ideal method for the synthesis of N-heterocycles because N2 is the sole by-product,20 and a variety of alkyl azide substrates are readily available. Various metal catalysts such as cobalt,21–25 ruthenium,26,27 nickel,28,29 and palladium complexes30,31 have been reported for C–H amination reactions. Among them, iron catalysts are the most attractive due to iron's biocompatibility and high earth abundance.Azides are simple and readily available Fe–nitrene precursors, with nitrogen being the only by-product, although elevated temperatures are often required to achieve decomposition of alkyl azides (Fig. 1). Another gas-releasing nitrene precursor is the dioxazolone,32 which is also readily prepared from abundant carboxylic acids, albeit with a slight penalty in atom economy compared to azides. On the other hand, hypervalent iodine reagents and hydroxylamine derivatives are believed to produce organic wastes (iodobenzene and hydroxyl compounds, respectively) that are not easy to remove from the reaction system. In contrast, the only by-product produced by the stable and cheap Chloramine T is inorganic waste (sodium chloride).33,34 Isoxazole is a less explored Fe–nitrene precursor that usually requires multi-step synthesis.35
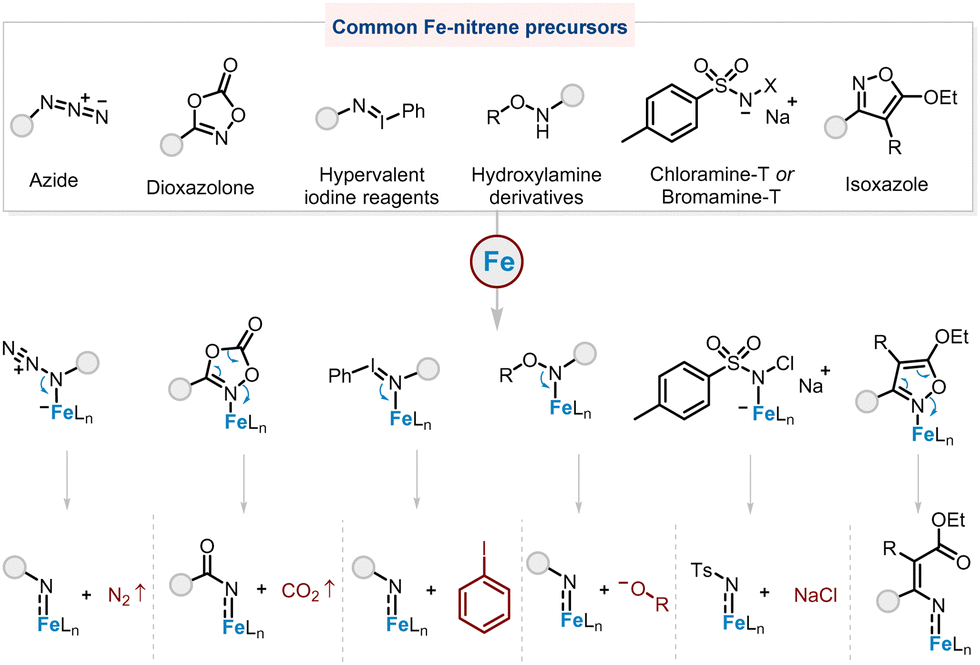 | ||
| Fig. 1 Common Fe–nitrene precursors. | ||
Nature provides blueprint for design and development of Fe catalysts for amination of inert C–H bonds. In biological systems, the FeIV–oxo porphyrin π-radical cation species, generated by the reaction of dioxygen with heme iron in cytochrome P450, is capable of hydroxylating inert C–H bonds.36,37 As such, the search for iron–nitrene/imido ligand multiple bond species that replicate the electronic structure of cytochrome P450 reactive iron–oxo intermediate38 would in principle constitute an effective strategy for the design of novel biomimetic iron catalysts for C–H bond amination reactions. In the literature, iron alkylimido/nitrene species are generally regarded as key reaction intermediates in Fe-catalysed intramolecular C(sp3)–H bond amination reactions of alkyl azides. They are usually formed through a series of reactions, involving coordination of organic azides with Fe-catalysts39 and nitrogen extrusion. Two possible reaction pathways for Fe–alkylimido/nitrene amination of C–H bonds are generally proposed, including (i) stepwise H-atom abstraction (HAA) and radical recombination mechanisms, and (ii) concerted mechanisms (the orchestration of C–N bond formation and C–H bond cleavage), to afford the desired N-heterocycles (Fig. 2(A)). Notably, in the Fe catalysed intramolecular amination of aliphatic, benzylic, O- and N-adjacent C(sp3)–H bonds, the FeIII radical imido, FeII and FeIII nitrenes, and FeV imido species have been suggested and in some cases identified as reaction intermediates.
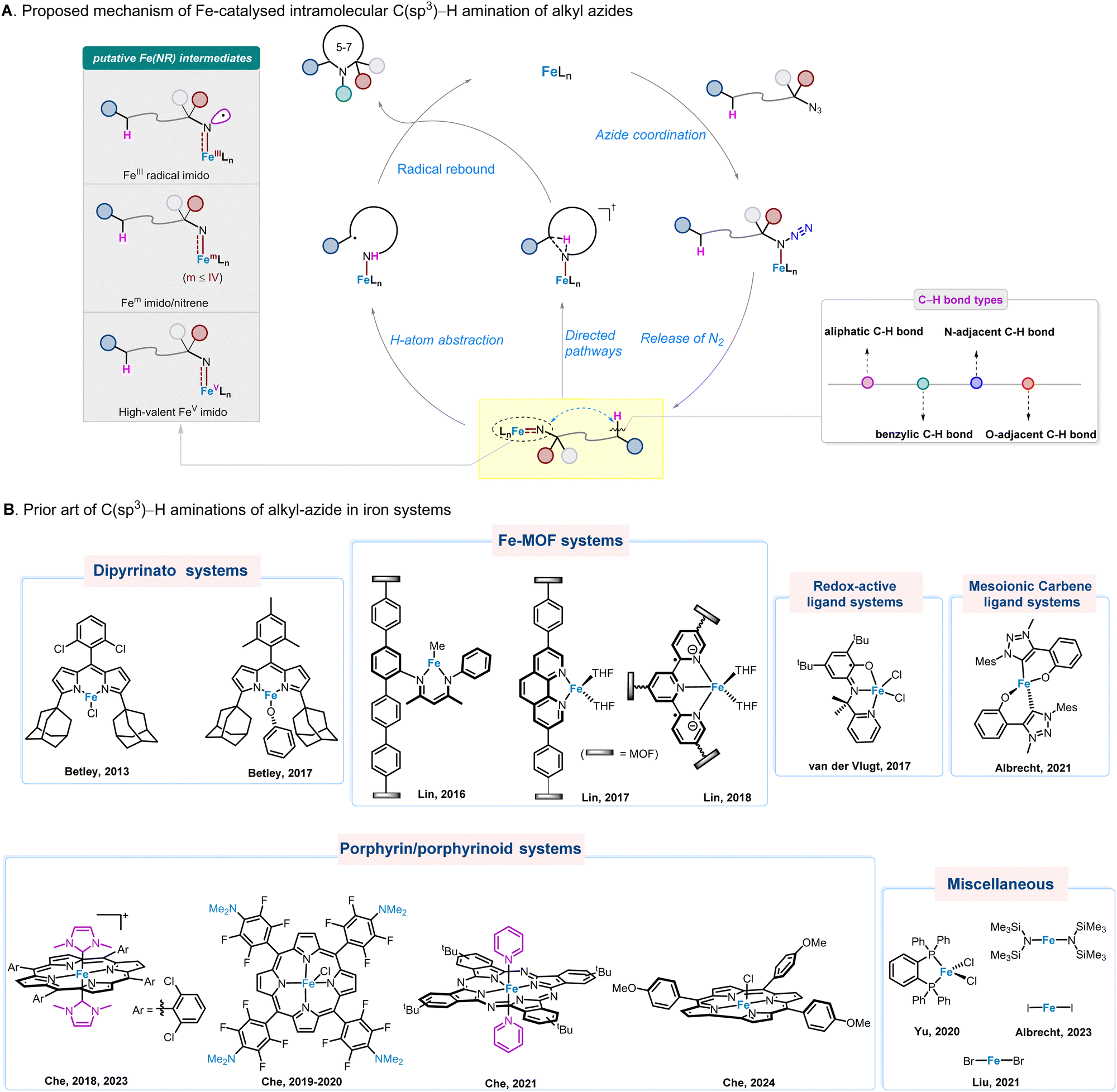 | ||
| Fig. 2 (A) Proposed mechanism of Fe-catalysed intramolecular C(sp3)–H amination of alkyl azides. (B) Prior art of C(sp3)–H activation in iron systems. | ||
Within this mechanistic scenario, various types of iron catalyst systems have been developed in recent years (Fig. 2(B)), with seminal work using the Fe–dipyrrinato catalyst reported by Betley and co-workers in year 2013.40 Here, we bring together the latest major advances in the field, providing insights into catalyst design and development, substrate scope expansion, key reaction intermediates, and applications in the synthesis and modification of complex heterocyclic natural products and bioactive compounds.
2. Fe-catalysed intramolecular C(sp3)–H amination of alkyl azides
2.1 Fe(III)–dipyrrinato ligand systems
In 2011, Betley and co-workers reported that the Fe(III)–dipyrrinato imido radical species generated by the decomposition of aryl and alkyl azides can react with the benzylic C–H bond of toluene through HAA reaction, followed by a radical recombination process.41 Thereafter, in 2013, these workers extended their studies to linear aliphatic azide substrates in an intramolecular fashion, which was the first example of Fe-catalysed intramolecular C(sp3)–H amination of alkyl azides (Scheme 1).40 Intriguingly, initial attempts found that only stoichiometric reaction could occur, possibly due to catalyst inhibition by the resulting N-heterocycle being coordinated with iron dipyrrinato species to form complex 1.16. The structure of 1.16 has been confirmed by X-ray crystal analysis. To overcome this issue, di-tert-butyl dicarbonate (Boc2O) was disclosed as the best protection reagent, which can reduce the nucleophilicity of pyrrolidine while maintaining catalyst efficiency, producing the product 1.1 in 93% yield under catalytic conditions. | ||
| Scheme 1 Fe(III)–dipyrrinato-catalysed intramolecular C(sp3)–H amination of alkyl azides. | ||
As depicted in Scheme 1, various C–H bonds, including O-adjacent, benzylic, allylic, aliphatic secondary and tertiary positions, are well-adapted to the catalyst, providing the corresponded products (1.1–1.12) in moderate to high yields, although the diastereoselectivity remains low. In particular, a tetra-substituted pyrrolidine 1.10 was also obtained in 58% yield. In the mechanistic study, radical clock experiments were performed and no ring-opening products were observed, suggesting that if radical intermediate 1.17 is indeed involved, its lifetime is short due to a fast recombination rate >1011 s−1. Alternatively, a direct C–H amination pathway cannot be excluded at this time.
2.2 MOF-supported iron complexes
In order to recover and reuse valuable catalysts, immobilization of homogeneous metal catalysts on MOFs has been extensively explored. A reliable synthetic approach to MOF modulation is the incorporation of orthogonal functional group on organic linkers. β-Diketiminate ligands (called NacNac) are a common class of bidentate ligands that coordinate with iron.42 Lin and co-workers recently reported the installation of this ligand in a UiO-type MOF to provide a Fe(II)–NacNac functionalized MOF catalyst (catalyst 1) that can effectively catalyse intramolecular C(sp3)–H amination of alkyl azides, and pyrrolidine 2.1 was obtained in 90% yield (Scheme 2).43 In comparison, when the homogeneous catalyst NacNac·Fe(Me) was used under identical reaction conditions, the product yield was only 30%. For product 2.2, TON goes up to 25. The sterically congested substrate was also well tolerated, yielding gem-dimethyl N-heterocycle 2.3 in quantitative yield, in sharp contrast to homogeneous catalysts where no desired product was detected. In addition, products (2.4–2.5) obtained via amination of allylic and aliphatic C–H bonds also show comparable efficiency, with yields of 40–90%. Notably, the recycled catalyst showed similar performance a second time, indicating that it is a highly active and reusable single-site solid catalyst.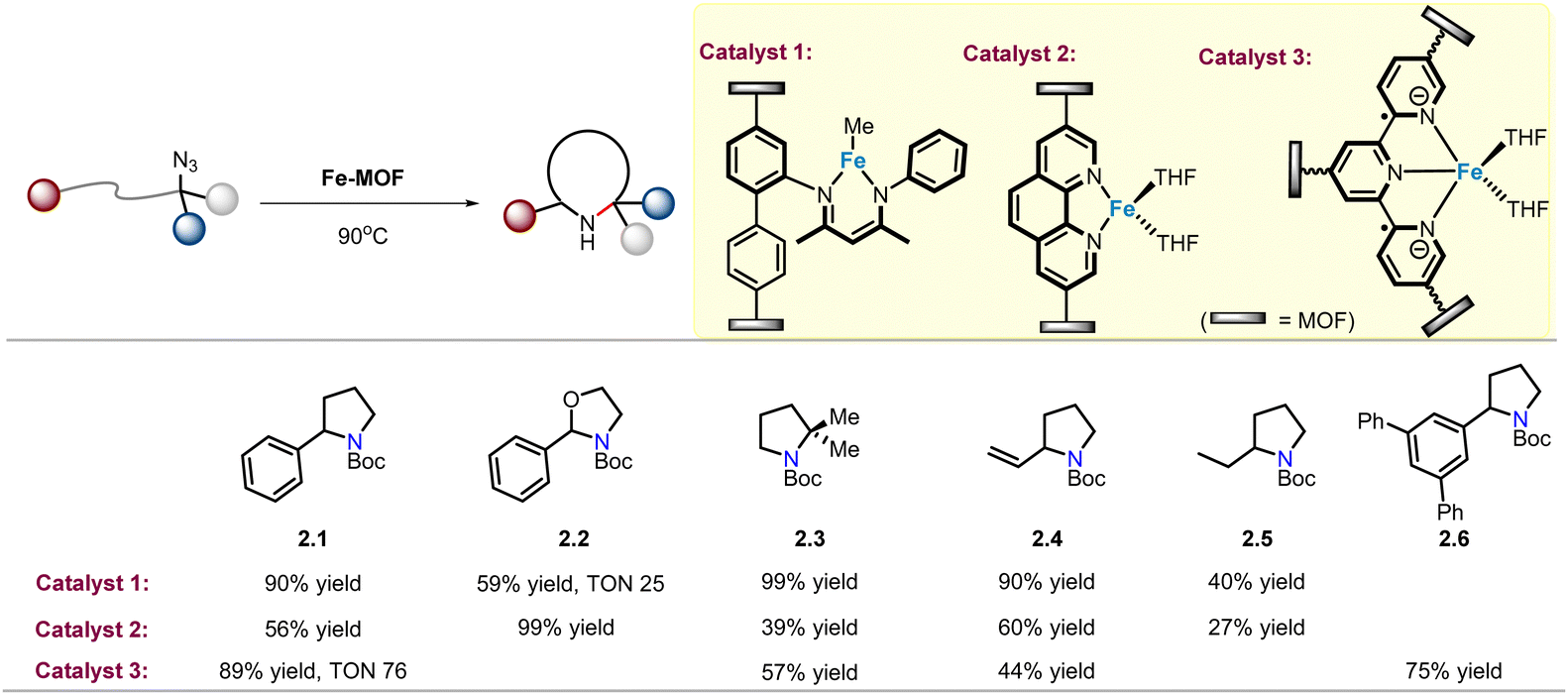 | ||
| Scheme 2 MOF supported iron catalysts for intramolecular C(sp3)–H amination of alkyl azides. | ||
Thereafter, Lin and co-workers developed a Fe-phenanthroline-based metal–organic framework (MOF) catalyst (catalyst 2) for the intramolecular C(sp3)–H amination of alkyl azides (Scheme 2).44 Although the yields of the same products (2.1, 2.3–2.5) were lower than those of the NacNac·Fe(Me) MOF catalyst, the formation of 2.2 (99% yield) was observed with excellent activity. In 2018, in order to eliminate the diffusion barrier of the Fe–MOF catalyst, the research group reported a FeBr2–terpyridine-based metal–organic layer (FeBr2·TPY-MOL) catalyst (catalyst 3), which represents a new 2D metal–organic layer (MOL) material category. This 2D material has easily accessible active sites that alleviate diffusion constraints compared to the smaller channels and pores of 3D MOFs. Using catalyst 3, product 2.1 was obtained, and the TON was 4 times higher than that using NacNac·Fe(Me) catalyst.45
2.3 Iron-catalysed diastereoselective intramolecular C–H amination of secondary aliphatic azides
Following their seminal work on the catalytic C–H amination of aliphatic azides to N-heterocycles in the presence of high-spin iron dipyrrin complexes, Betley and co-workers continued to investigate the enhancement of diastereoselectivity through catalyst modification.46 In their previous mechanistic studies, the identified fast radical rebound process or concerted mechanism had established HAA as the stereoselectivity-determining step.40 Therefore, the transition state of HAA is crucial for distinguishing two diastereotopic (diastereomeric) hydrogen atoms, which may be affected by ancillary anionic ligands.After screening a series of catalysts with different ancillary anionic ligands, a trigonal monopyramidal geometry with a phenoxide group in the Fe atom (complex 3.9) showed good performance in terms of product yield and diastereoselectivity. This contrasts with the inferior results for the distorted tetrahedral geometry of the bulkier alkoxide variants. As shown in Scheme 3, a series of 2,5-disubstituted pyrrolidines (3.1–3.5) were found to yield products with >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 syn/anti diastereoselectivity in moderate to good yields. The exception for naphthyl-containing substrates (e.g.3.6) remains unclear. Furthermore, for substrates that do not possess a sterically demanding α-azido group (3.8), the d.r. is still low under current conditions.
:1 syn/anti diastereoselectivity in moderate to good yields. The exception for naphthyl-containing substrates (e.g.3.6) remains unclear. Furthermore, for substrates that do not possess a sterically demanding α-azido group (3.8), the d.r. is still low under current conditions.
 | ||
| Scheme 3 Fe(III)–dipyrrinato-catalysed diastereoselective intramolecular C(sp3)–H amination of alkyl azides. | ||
2.4 FeIII catalysts with redox-active ligands
In 2017, van der Vlugt and co-workers developed a novel high-spin FeIII catalyst with redox-active pyridine–aminophenol ligand,30 which could be easily prepared under aerobic conditions in the presence of triethylamine.47 When the catalyst is used for the intramolecular benzylic C(sp3)–H amination of alkyl azides, the desired pyrrolidine product is obtained, accompanied by linear amine as major by-products. In particular, the TON of product 4.1 is as high as 620 despite a long reaction time (7 days). Next, the substrate scope was further investigated (Scheme 4). Notably, benzylic, allylic, and O-adjacent C–H aminations all occurred cleanly, yielding 5-membered N-heterocycles (4.1–4.4) in high yields. In the case of 4.4, a linear amine by-product was also detected (8% yield). For substrates containing a secondary benzylic C–H bond, the resulting product 4.5 was found to have considerable amounts of amine by-product (47% yield). Furthermore, considering substrates with stronger C–H bonds, such as primary C–H bonds and α-carbonyl C–H bonds, they are expected to be less effective for amination reactions. In fact, the amount of linear amine is usually greater than the desired C–H amination product (4.6–4.9).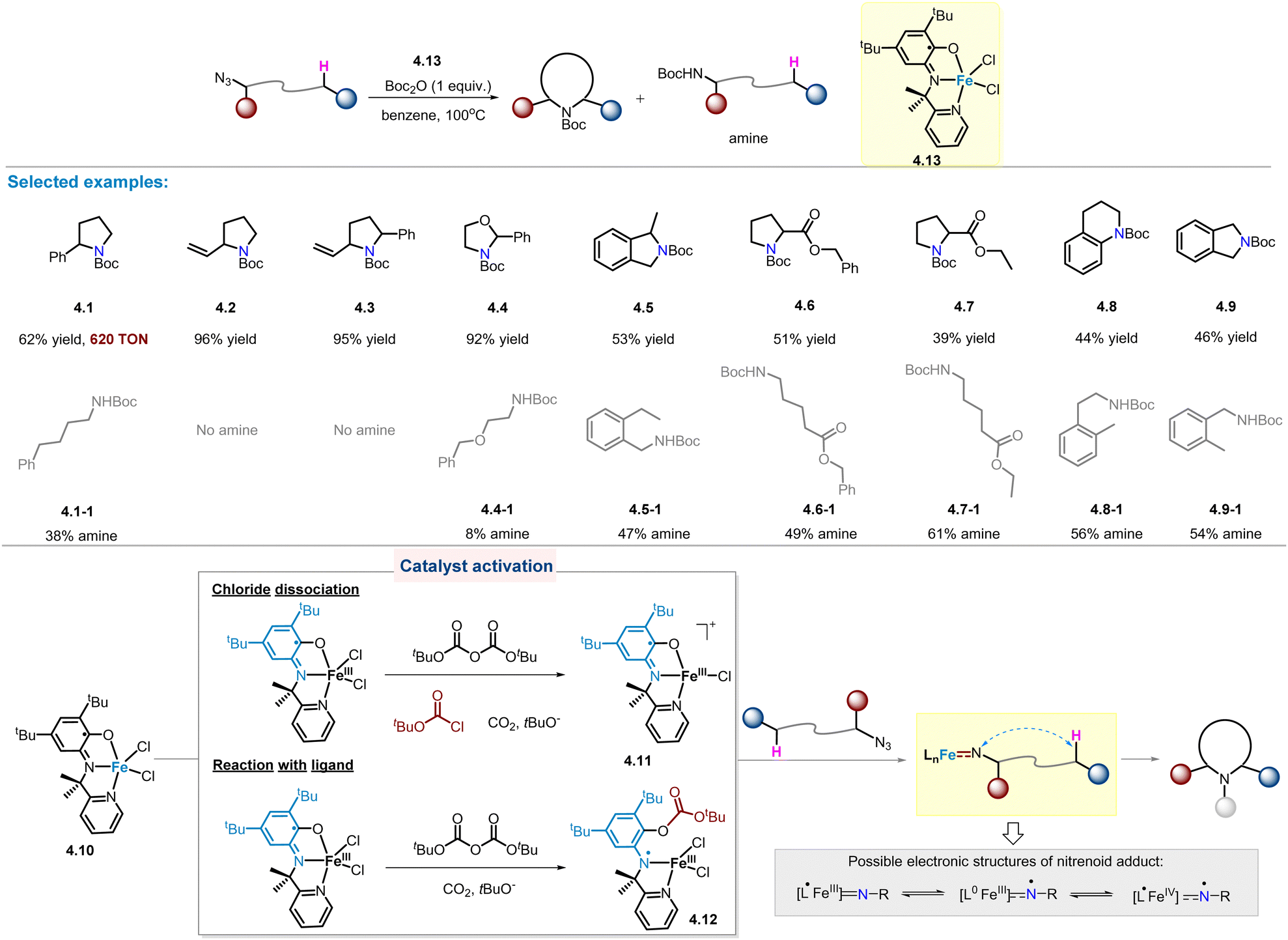 | ||
| Scheme 4 FeIII catalyst featuring with redox-active pyridine–aminophenol ligand for intramolecular C(sp3)–H amination of alkyl azides. | ||
Kinetic studies were performed to determine the rate-limiting step for this reaction (Scheme 4). Surprisingly, first-order kinetics of Boc2O were observed in rate-dependent experiments, implying that the reaction with Boc2O is the rate determining step in the overall process. Two possible ways are proposed for Boc2O to interact with 4.10, resulting in activated FeIII catalysts with higher affinity for alkyl azides: (1) chloride abstraction giving the four-coordinated cationic FeIII species 4.11, and (2) The N-centred radical FeIII species 4.12 is formed by the reaction of Boc2O with the phenolate fragment of the ligand.
2.5 Iron porphyrin catalysts
In 2014, Che and co-workers exploited the trans effect of NHC axial ligands in Ru porphyrins to improve the catalytic efficiency of C–H insertion reactions of carbene and aryl-nitrenes.48 Subsequently, Che and co-workers in 2018 reported an iron(III) porphyrin catalyst with an axial N-heterocyclic carbene ligand, FeIII(TDCPP)(IMe)2, which was found to catalyse the intramolecular C(sp3)–H amination of alkyl azides under microwave-assisted and thermal conditions (Scheme 5).49 This protocol has a broad substrate scope and can be used to efficiently aminate benzylic, allylic, and tertiary C–H bonds to the corresponding pyrrolidines (5.1–5.5) in up to 95% yield. For the amination of aliphatic and α-carbonyl C–H bonds, which were considered inefficient in previous reported systems, this method also gave good performance, affording α-alkyl (5.6–5.7), α-ester (5.9), and α-spiro (5.8) pyrrolidines (65–88% yields). Notably, two bridged pyrrolidines (5.10–5.11) were also obtained in 70–87% yield, which has not been reported in previous metal-catalysed intramolecular C(sp3)–H amination. To demonstrate the synthetic usefulness of this method, the reaction has been successfully applied to the syntheses of tropane (5.11), nicotine (5.12), leelamine (5.14) and cis-octahydroindole derivatives (5.13).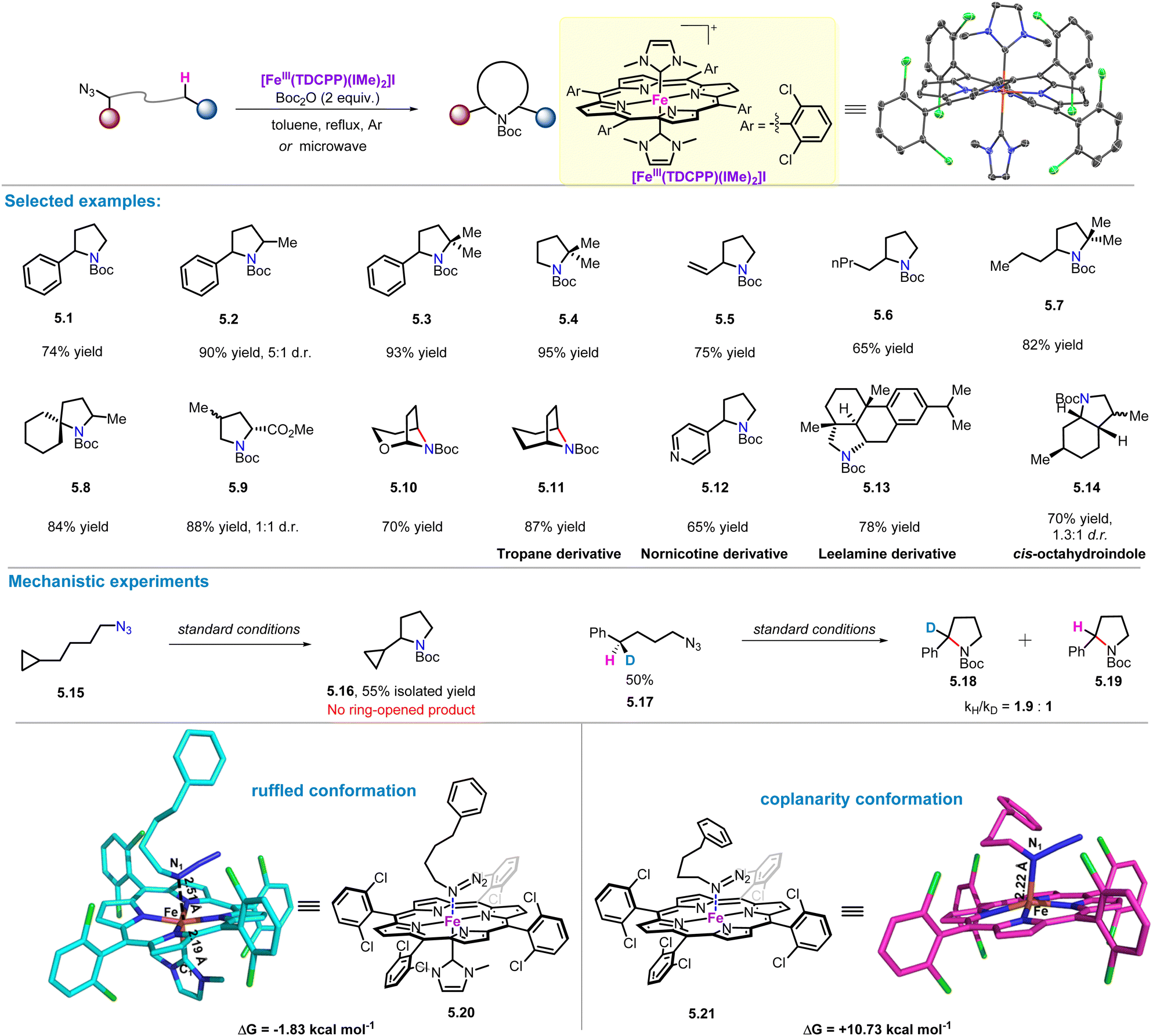 | ||
| Scheme 5 FeIII(TDCPP)(IMe)2-catalysed intramolecular C(sp3)–H amination of alkyl azides. | ||
In the radical clock experiment, the designed substrate 5.15 gave C–H amination product 5.16 in an isolated yield of 55%, and no ring-opened product was detected. Furthermore, in kinetic isotope effect (KIE) experiments, a kH/kD ratio of 1.9:1 was observed using 50% deuterated substrate 5.17. These mechanistic studies revealed a concerted or a fast rebound mechanism for the amination reaction. In addition, DFT calculations show that the introduction of the NHC axial ligand causes the porphyrin ring to adopt a ruffled conformation, resulting in adduct 5.20, whose energy is 12.56 kcal mol−1 higher than that of the adduct 5.21 without axial ligands.
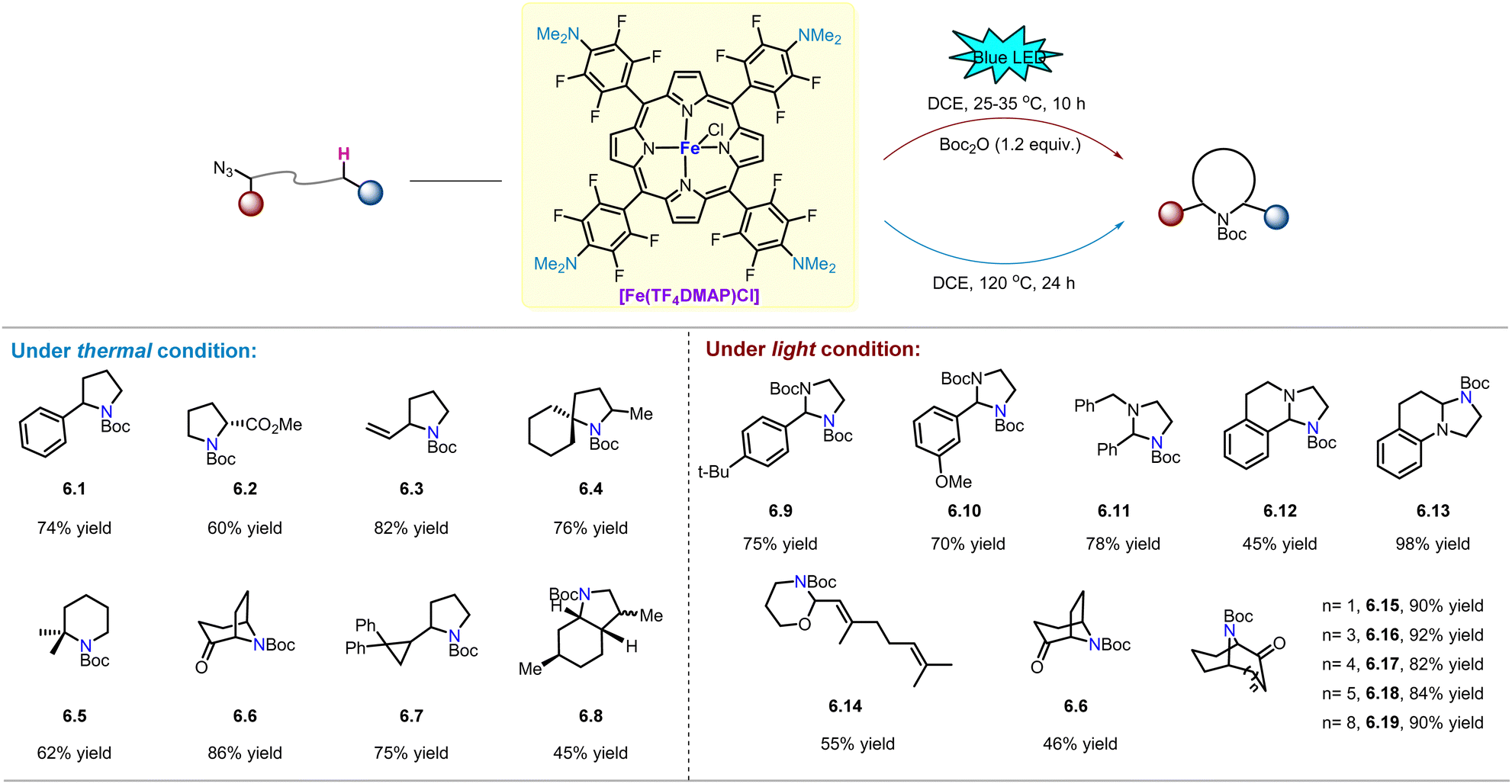
In 2014, Che and co-workers reported that FeIII(TF4DMAP)OTf (TF4DMAP = meso-tetrakis(o,o,m,m-tetrafluoro-p-(dimethylamino)phenyl)porphyrinato dianion, Scheme 6) effectively catalyses anti-Markovnikov oxidation of terminal aryl alkenes to aldehydes using H2O2 as the oxidant via a reactive iron–oxo porphyrin intermediate supported by TF4DMAP ligand.50 As nitrogen analogues of iron–oxo species, iron nitrene species with porphyrin ligands are expected to be highly reactive in Fe-catalysed intramolecular C(sp3)–H amination of alkyl azides. Subsequently, Che and co-workers reported the reactivity of FeIII(TF4DMAP)Cl for intramolecular C(sp3)–H amination of alkyl azides under light and thermal conditions (Scheme 6).
 | ||
| Scheme 6 [FeIII(TF4DMAP)OTf]-catalysed intramolecular C(sp3)–H amination of alkyl azides under light and thermal conditions. | ||
Under conventional thermal conditions,51 various C–H bonds, including benzylic, allylic, α-ester, and aliphatic C–H bonds, can be smoothly aminated to obtain the desired pyrrolidine (6.1–6.8) in 45–86% yield. In particular, product 6.7 with an intact cyclopropane ring was obtained in 75% yield, suggesting the existence of a fast radical rebound process or a concerted mechanism during C–H bond activation. Nonetheless, the iron–nitrene transfer reaction of organic azides still requires elevated temperature. Therefore, the development of organic azide photo-activation method provides a potential solution to this problem. In 2020, Che and co-workers reported that [FeIII(TF4DMAP)OTf] can play the dual role of photo-sensitizer and nitrene catalyst, catalysing intramolecular C(sp3)–H amination of alkyl azides under blue LED light (469 nm) irradiation at 25–35 °C.52 As shown in Scheme 6, various N- and O-containing alkyl azides undergo C–H amination to afford the corresponding imidazolidines (6.9–6.13) and 1,3-oxazinane derivative (6.14) in good yields, respectively. Furthermore, considering that nitrogen bridged bicyclic moieties are widely present in natural alkaloids,53 Che and co-workers applied iron-catalysed light-driven C–H amination to a series of medium-sized cyclic α-azidoketones to rapidly obtain the corresponding nitrogen bridged bicyclic compounds (6.6, 6.15–6.19). The yields of the compounds were 46–92%, although the yield of product 6.6 was lower than that obtained under thermal conditions.
2.6 Iron-diketiminate and -phosphine complexes
As described in Scheme 2, MOF-supported NacNac–Fe(II) complex with coordinating methyl ligand (catalyst 1, Scheme 2) is an efficient catalyst for C(sp3)–H amination of alkyl azides. In 2019, Yu and co-workers reported a simple catalytic system (catalyst 4) that can be readily obtained by mixing FeCl2 and NacNac ligands in a reaction vessel, and used this catalytic system to prepare a series of poly-substituted imidazolinones in good yields (Scheme 7).54 The substituents on the phenyl ring have little impact on the reaction efficiency, and the corresponding products (7.1–7.3) were obtained in excellent yields. Changes to cyclic 4- and 5-membered rings at the α-position were also well compatible, affording spiro-imidazolinones (7.6–7.7) in 79–94% yields. However, the diastereoselective-control of product 7.8 was still low. Although this catalytic system has the advantage of simplicity, high reaction temperatures (100 °C) and high catalyst loadings (20 mol%) are required to ensure complete conversion and high product yields. To address this problem, the same group demonstrated a more efficient iron–phosphine complex (catalyst 5, Scheme 7), which can be used at lower temperatures (40 °C) and lower amount of catalyst loading (5 mol%).55 The newly developed catalytic system provided the corresponding imidazolinone products (7.1–7.9) in yields comparable to those of the FeCl2–NacNac catalyst. Notably, two new products bearing furan (7.4) and ester (7.9) functional groups were obtained in high yields.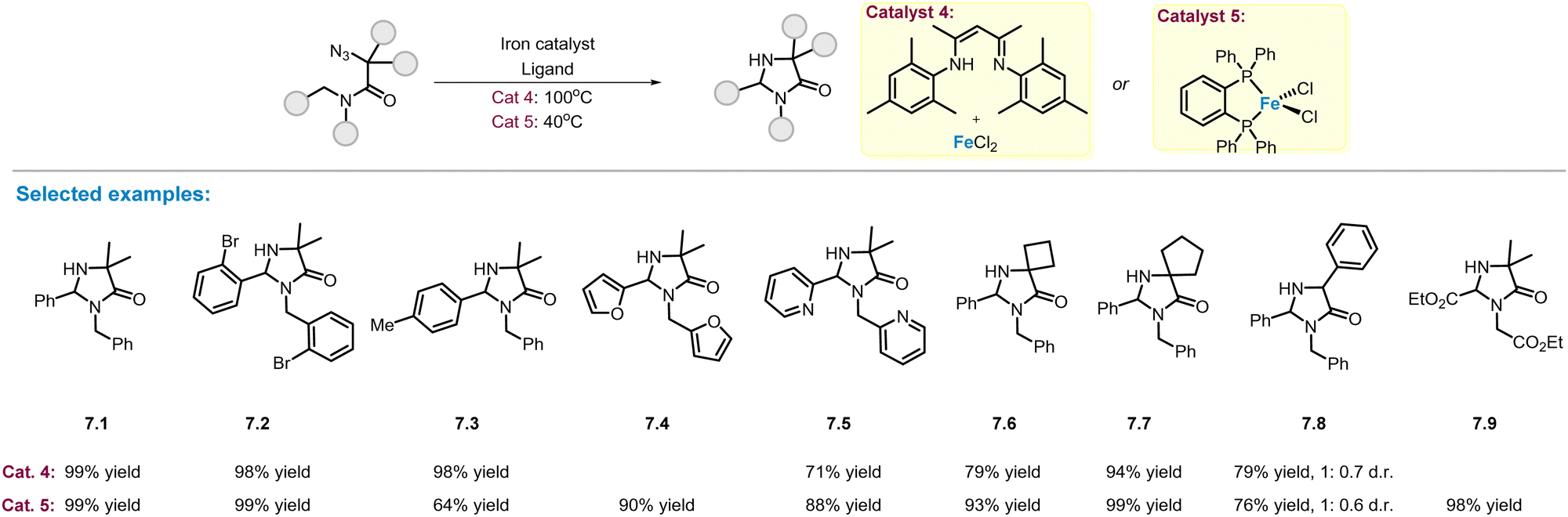 | ||
| Scheme 7 NacNac–Fe(II) and Fe(II)–phosphine complex-catalysed intramolecular C(sp3)–H amination of alkyl azides. | ||
2.7 FeBr2-catalysed intramolecular C(sp3)–H amination of alkyl azides for the synthesis of imidazolinones
As depicted in Scheme 7, the Yu group reports two novel iron catalysts for the synthesis of complex imidazolinones via C–H amination of tertiary alkyl azides. Surprisingly, Liu and co-workers found that the ligand-free iron catalyst FeBr2 can catalyse the C(sp3)–H amination of primary alkyl azides to generate a series of imidazolinones (8.1–8.5) in moderate yields (Scheme 8).56 Substrates with para-substituted aryl groups with different electronic properties (8.1–8.4) were well-tolerated by the catalyst, affording the desired products in 47–60% yields. Notably, the heteroaromatic substrate was found to have better performance (8.5, 75% yield). In comparison, no product of the tertiary alkyl aizde (8.6) was observed, possibly because this reaction is sensitive to steric hindrance.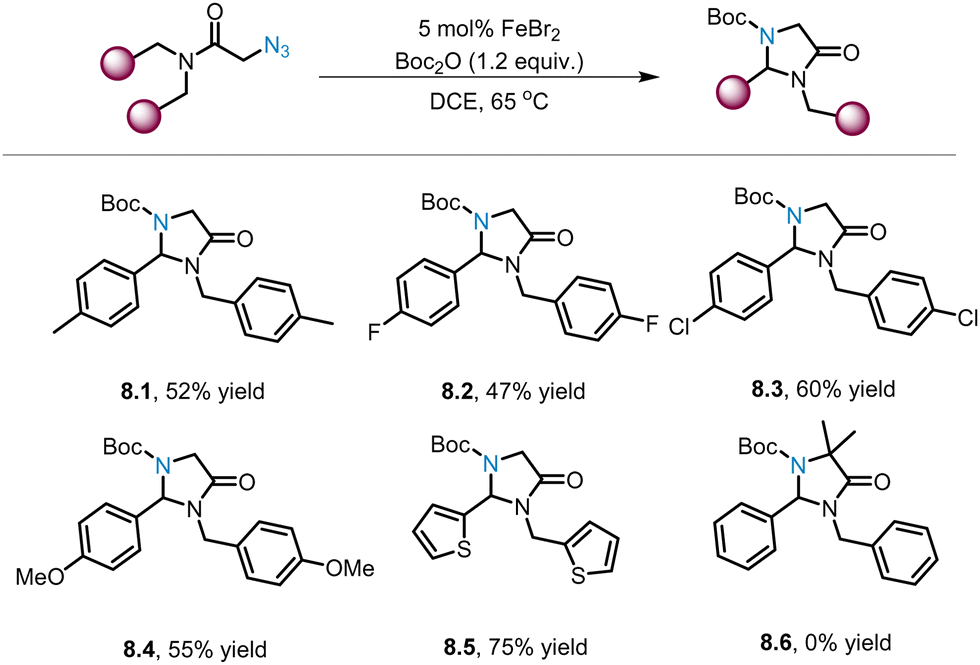 | ||
| Scheme 8 FeBr2-catalysed intramolecular C(sp3)–H amination of alkyl azides for the synthesis of imidazolinones. | ||
2.8 Iron(II)–phthalocyanine catalysts
Although significant progress has been made in catalyst design and mechanistic studies of Fe-catalysed intramolecular C(sp3)–H amination of alkyl azides, the application of this method in the preparation of structurally complex and pharmaceutically relevant molecules or natural products has not been as widespread. In 2021, Che and co-workers reported a soluble, air-stable and easily prepared iron(II)–phthalocyanine catalyst with pyridine as the axial ligand that can catalyse such reactions with a broad substrate scope. This reported iron(II)–phthalocyanine catalyst is particularly suitable for the late-stage functionalization of active pharmaceutical ingredients and the preparation of natural product derivatives/analogues (Scheme 9).57 For example, aliphatic azide substrates bearing heteroaromatic groups including indole (9.1) and furan (9.2) were well-tolerated, and the corresponding products were obtained in yields as high as 81%. As for extending the scope of C–H bonds to N- and O-adjacent C–H bonds, the corresponding amination products (9.2–9.3) were obtained in moderate yields. Importantly, unlike traditional routes that selectively produce five-membered ring products, this iron(II)–phthalocyanine catalyst is applicable to the synthesis of a series of uncommon 6-membered N-heterocycles (9.6–9.10, 9.12), and even an example of 7-membered heterocyclic compound (9.5) can be prepared.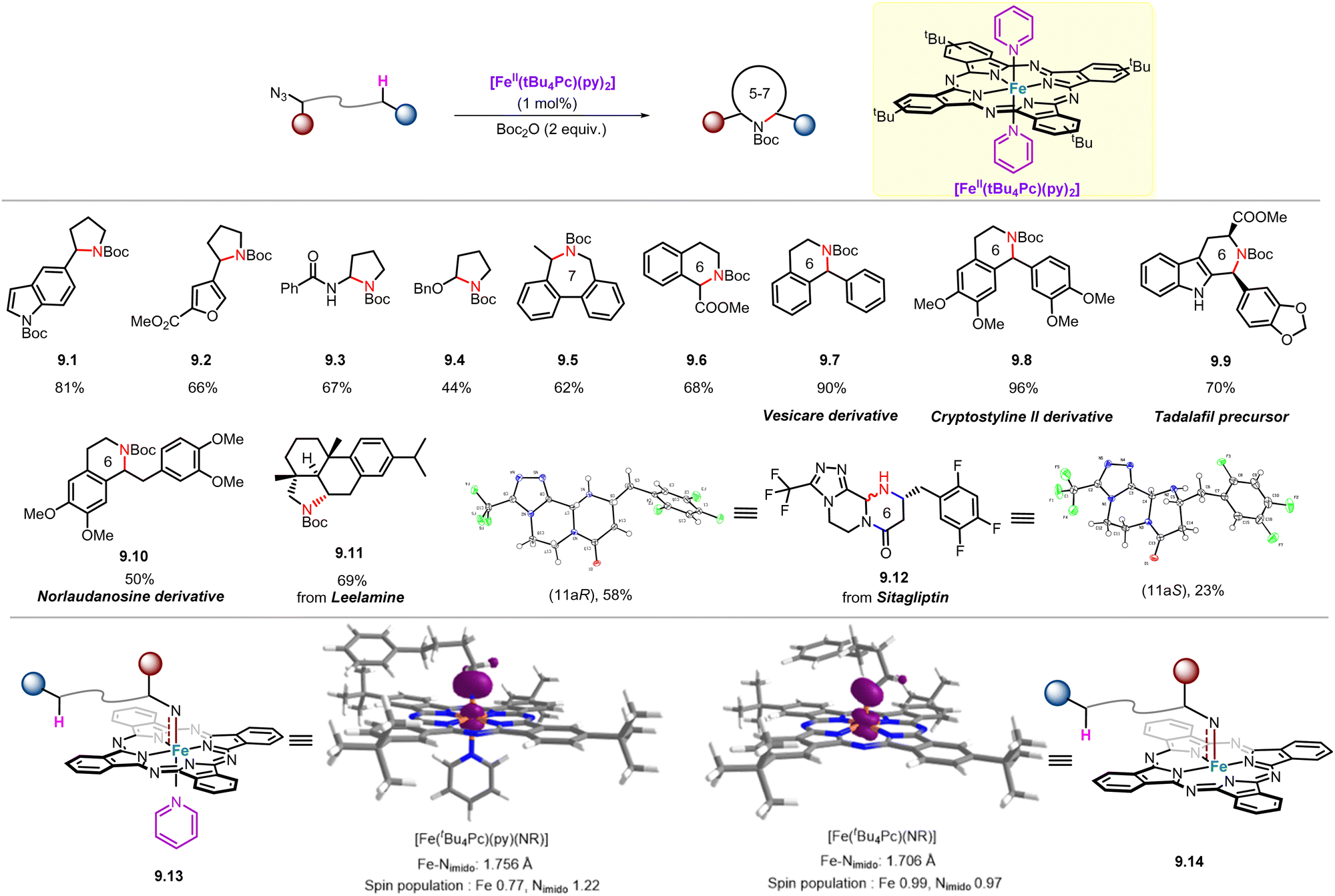 | ||
| Scheme 9 [Fe(tBu4Pc)(py)2]-catalysed intramolecular C(sp3)–H amination of alkyl azides. | ||
To better understand the reaction mechanism, we used DFT calculations to optimize the structures of two Fe(Pc)-imido species, [Fe(tBu4Pc)(py)(NR)] (9.13) (R = 4-phenylbutyl) and [Fe(tBu4Pc)(NR)] (9.14) (Scheme 9). The calculated Fe–N distance of the former (1.756 Å) is slightly longer than that of the latter (1.706 Å), which is attributed to the trans-effect of the axial pyridine ligand. The longer Fe–N distance of 9.13 may lead to its higher N-transfer reactivity in Fe(tBu4Pc)(py)2-catalyzed intramolecular C–H amination of alkyl azides.
2.9 Iron catalyst for C(sp3)–H amination of tertiary alkyl azides
Boc2O is often used as in situ protection reagent to suppress the nucleophilicity of N-heterocycle products known to poison catalysts. Recently, Albrecht and co-workers reported that for gem-dimethyl type or tertiary alkyl azide substrates, the reaction does not require Boc2O as an additive,58 although this was also observed in the synthesis of poly-substituted imidazolinones reported by the Yu group.54 Therefore, this improvement may be attributed to the resulting product having sterically crowded N-atoms, thereby reducing its ability to poison the catalyst, consistent with the inefficient process observed in obtaining products 10.7 and 10.8. In any case, a homoleptic C,O-chelated mesoionic carbene–iron complex was prepared, which is highly active in catalysing a variety of C–H bonds, including (hetero)benzylic (10.1–10.5), primary (10.8), secondary (10.6, 10.9–10.10), and tertiary (10.6, 10.11) C–H bonds, achieving an unprecedented high product TON (TON = 7600) (Scheme 10). Kinetic studies show a half-order dependence on catalyst concentration, suggesting a unique pathway involving the dimeric iron complex as the catalyst resting state.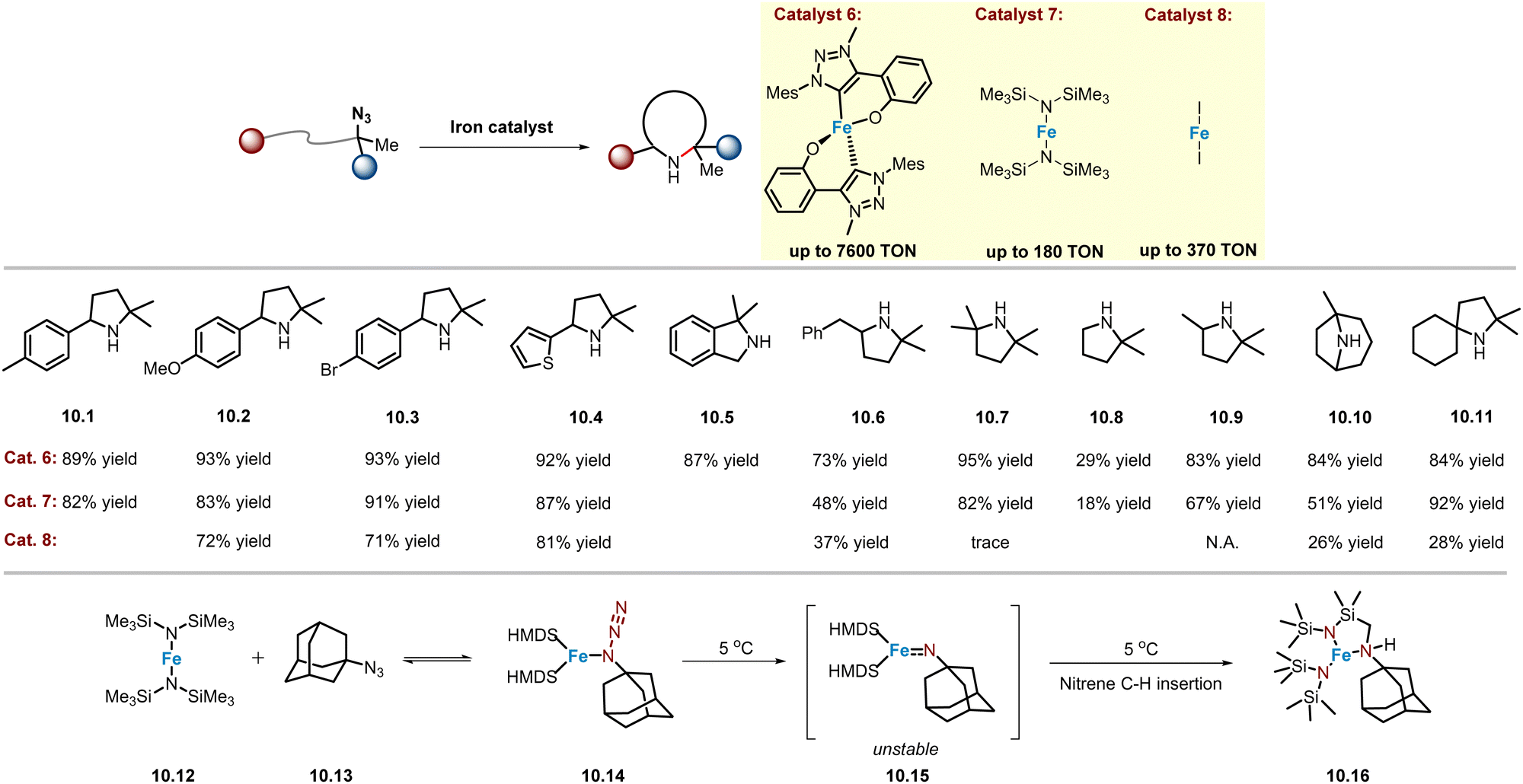 | ||
| Scheme 10 Mesoionic carbene–iron complex, Fe(HMDS)2- and FeI2-catalysed intramolecular C(sp3)–H amination of alkyl azides. | ||
An important finding was that Boc2O did not need to be used as an additive. Nonetheless, Albrecht and co-workers discovered simpler iron catalysts, which could significantly reduce the multistep synthesis required to obtain the catalyst precursors. In 2023, an air-sensitive Fe(HMDS)2 complex (catalyst 7, Scheme 10) was reported as a good catalyst, prepared in one step from commercially available starting materials.59 At the level of catalytic performance, in most cases Fe(HMDS)2 can provide product yields comparable to those obtained with catalyst 6 (Scheme 10), although a considerable decrease in efficiency was observed when obtaining products 10.6 and 10.10. Since catalyst 7 is air-sensitive, the same group disclosed an alternative and commercially available iron salt FeI2 (catalyst 8, Scheme 10) as a catalyst,60 which could rapidly and selectively catalyse the amination of various C–H bonds, achieving up to 370 product TON at 0.1 mol% catalyst loading.
Mechanistically, Fe catalysts are widely proposed to quickly reach equilibrium with the α-N coordination of alkyl azides, making it difficult to obtain stable iron α-N coordinated organoazide complexes. Albrecht and co-workers found that mixing of catalyst 7 (10.12) with 1-azidoadamantane (AdN3, 10.13) resulted in complex 10.14,61 whose structure was confirmed by single crystal X-ray diffraction analysis. Complex 10.14 could undergo nitrene C–H insertion at 5 °C to give 10.16, but attempts to obtain Fe(NAd) complex 10.15 were unsuccessful. However, in most cases, the catalytic efficiency is significantly lower than that of catalysts 6 and 7, especially for substrates with steric effect (10.7) or containing stronger C–H bonds (10.9), almost no reactivity is observed (Scheme 10).
2.10 Application of C(sp3)–H amination of alkyl azides in the synthesis of complex heterocyclic compounds
In our previously developed approach, the iron(III) porphyrin catalyst with the axial N-heterocyclic carbene ligand FeIII(TDCPP)(IMe)2 exhibited a broad substrate scope in the amination of various C–H bonds. The high reactivity of this catalyst allows us to use it in the preparation of complex nitrogen-containing compounds. It is gratifying that this catalyst has indeed been used to achieve the construction of the tetrahydroisoquinoline skeletons and the synthesis of polycyclic compounds (Scheme 11).62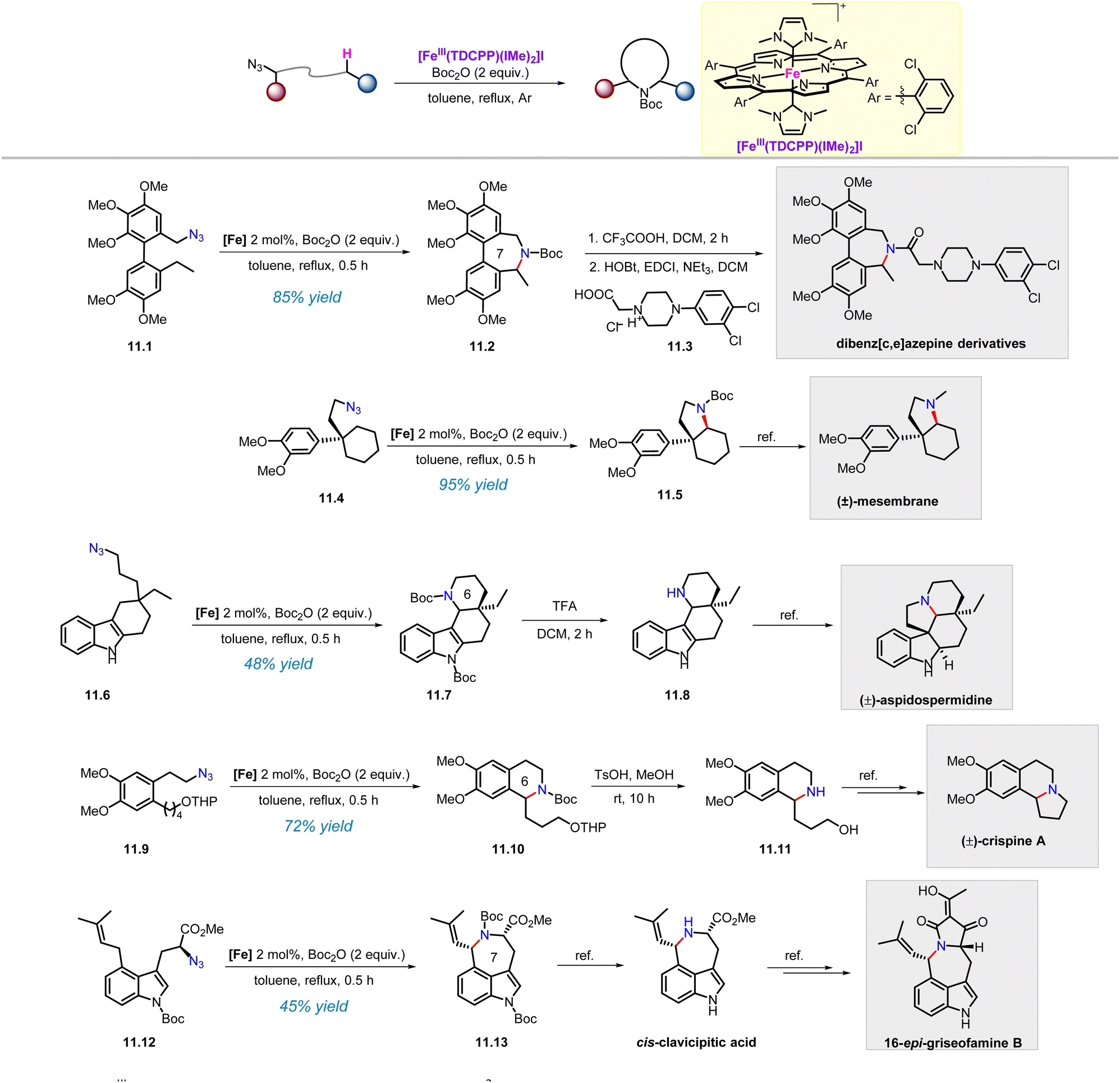 | ||
| Scheme 11 FeIII(TDCPP)(IMe)2-catalysed intramolecular C(sp3)–H amination of alkyl azides applied in complex settings. | ||
Dibenz[c,e]azepine is core structure in anti-obesity drugs and analogues of the alkaloid colchicine.63 Azide 11.1 underwent C(sp3)–H amination to afford the uncommon 7-membered ring product 11.2 in 85% yield under standard conditions. After removing the Boc group, the generated free amine intermediate was condensed with 11.3 to give the dibenz[c,e]azepines derivative, which showed potent cytotoxicity (6.5 μM) against the non-small lung cancer cell line NCI-H460. Next, intramolecular aliphatic C(sp3)–H amination of azide 11.4 afforded the fused ring product 11.5 in 95% yield, which could be further converted into (±)-mesembrane according to the reported procedure.64 Furthermore, starting from 11.6, the intramolecular C(sp3)–H amination product 11.7, which is a key intermediate to prepare (±)-aspidospermidine, was obtained in 46% yield with excellent diastereoselectivity.65 In addition, the tetrahydroisoquinoline skeleton 11.10 was also constructed via benzylic C–H amination of alkyl azide 11.9, and according to the literature, its double-deprotection in the presence of TsOH would allow the synthesis of (±)-crispine A.66 Seven-membered nitrogen heterocycles are attractive targets in the fields of medicinal chemistry and organic synthesis. So far, there have been no reports of their acquisition through C(sp3)–H activation. In this context, C(sp3)–H amination of alkyl azide 11.12 afforded the 7-membered ring product 11.13 in 45% yield, providing access to cis-clavicipitic acid and 16-epi-griseofamine B.67
2.11 FeV-alkylimido species for C(sp3)–H amination
Given the crucial role of high-valent iron–oxo (Fe(O)) species in enzymatic and/or synthetic C–H functionalization, it is attractive to explore high-valent Fe(NR) species for C–H functionalization. Very recently, Che and co-workers reported iron corrole-complex catalysed C(sp3)–H amination (Scheme 12).68 In this case, a series of di-substituted pyrrolidines (12.1–12.5) were obtained in 95–99% yields, with excellent diastereoselectivities (>99:1 d.r.) in all cases. And using a catalyst loading of 0.2 mol%, gram-scale product 12.1 was obtained. Notably, the TON of product 12.6 can reach 3880. This is a rare example of this type of reaction, which can produce up to several thousand TON of C–H amination product. Iron corrole complexes are also used in the synthesis or late-stage modification of a series of complex natural products and drugs, including cryptostyline II (12.7), geraniol (12.8), (S)-ibuprofen (12.9), (S)-(−)-citronellal (12.10), estrone (12.11) and nopol (12.12) to obtain the corresponding product derivatives. Compound 12.7 can be prepared with TON up to 1230. Interestingly, product 12.13 was produced from a 7-membered ring amination process, distinct from the previously reported 6-membered ring amination process,69 albeit in lower yield (18%).
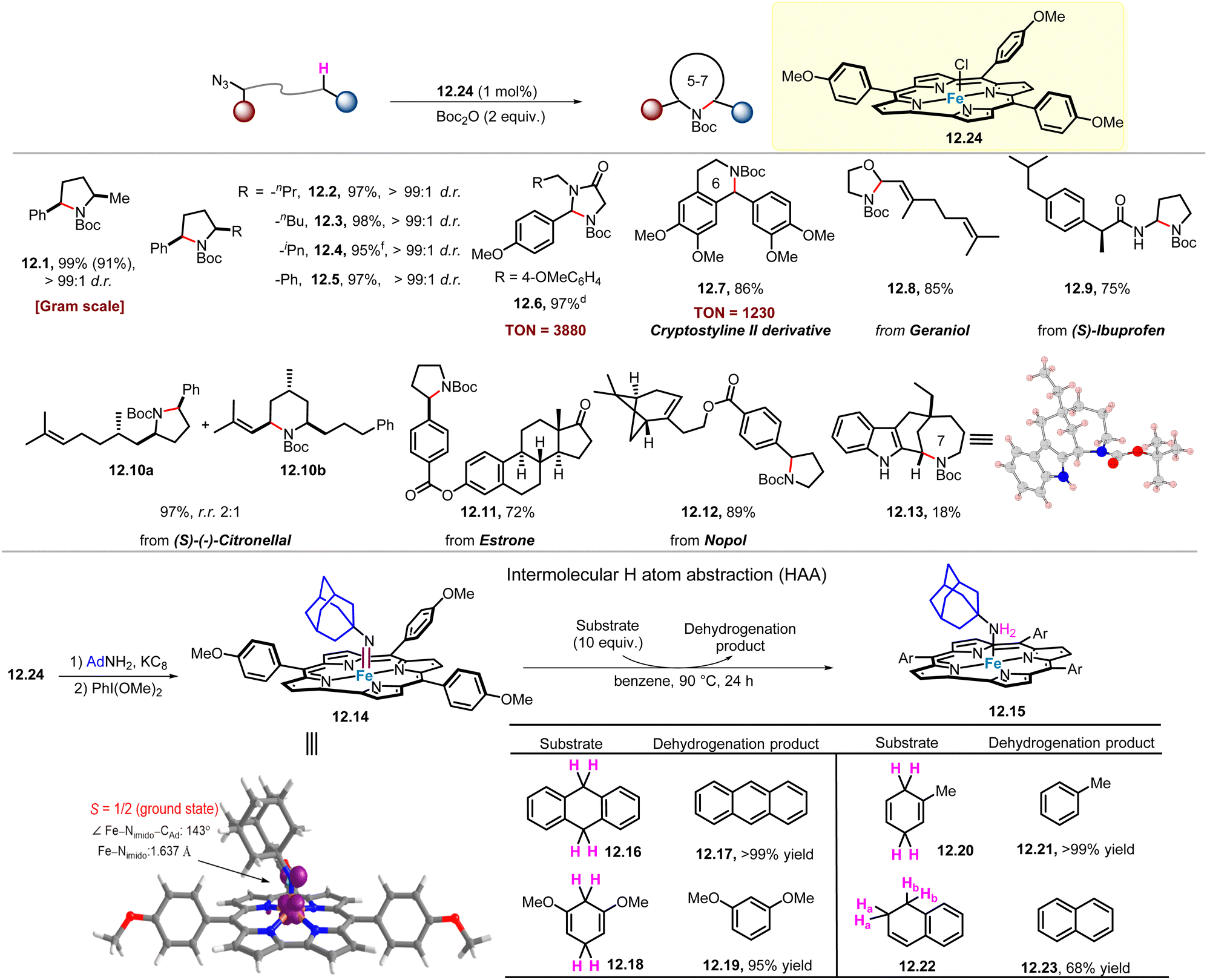 | ||
| Scheme 12 FeV-alkylimido for C(sp3)–H amination. | ||
Treatment of 12.24 with KC8/AdNH2 in benzene yields [FeIII(Tp-OMePC)(NH2Ad)] (12.15), which can be oxidized by PhI(OMe)2 to give [FeV(Tp-OMePC)(NAd)] (12.14). The prepared and well-characterized complex 12.14 represents the first example of FeV–alkylimido complexes that react with the C–H bonds of substrates (12.16, 12.18, 12.20, 12.22) to afford the corresponding dehydrogenation products (12.17, 12.19, 12.21, 12.23; yield 68–99%), through HAA mechanism (Scheme 12). These results support the critical role of FeV-alkylimido reaction intermediate in Fe(Cor)-catalysed intramolecular C(sp3)–H amination of alkyl azides. In addition, DFT calculations show that the structure of [FeV(Tp-OMePC)(NAd)] exhibits an energetically favoured doublet state, with a Fe–Nimido distance of 1.637 Å and a Fe–Nimido–Ad angle of 143°.
2.12 Enzyme-catalysed asymmetric alkyl–nitrene C(sp3)–H amination
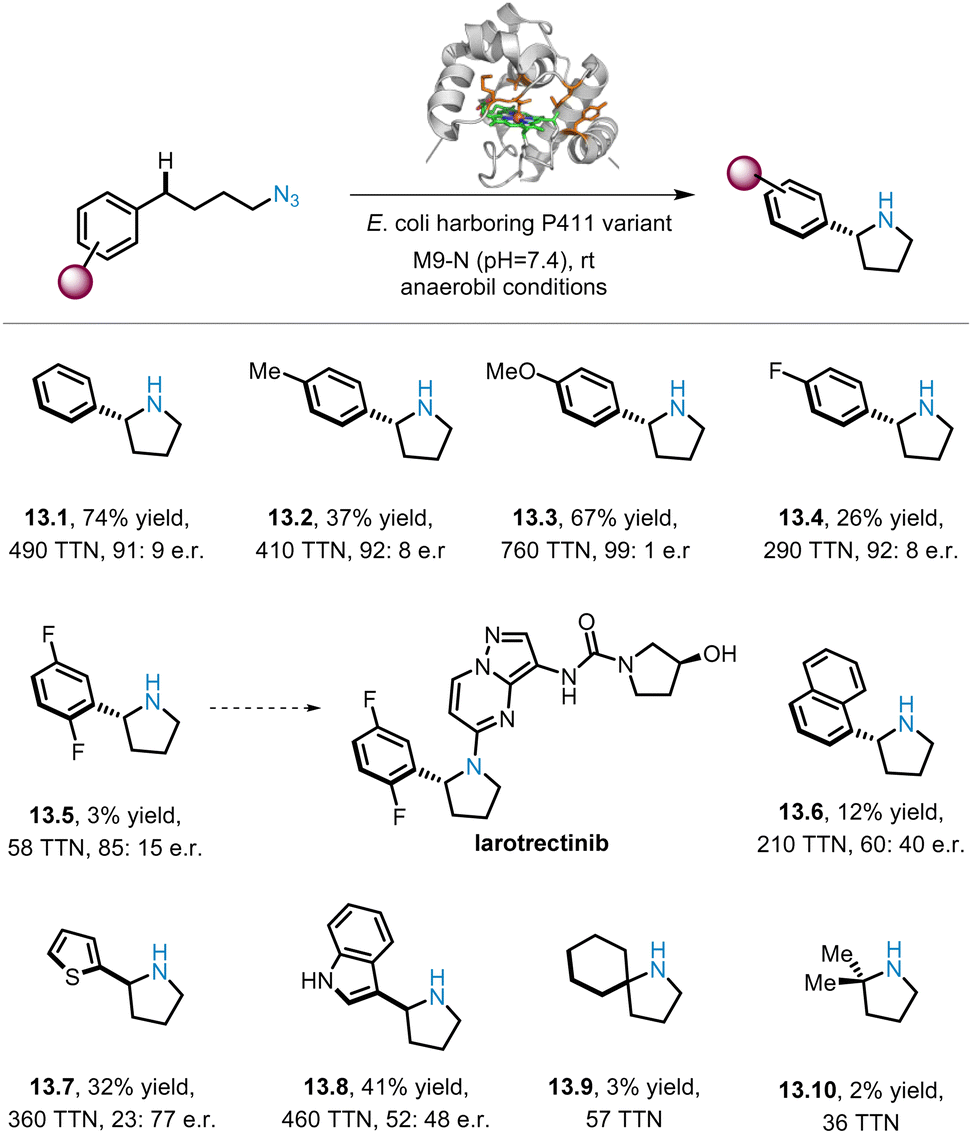
Despite the widespread success of iron-catalysed intramolecular C(sp3)–H aminations of alkyl azides, the construction of important chiral N-heterocyclic products such as pyrrolidines using this strategy remains a challenge. In 2023, Arnold and co-workers reported a variant of cytochrome P411 that can catalyse the insertion of alkyl nitrenes into C(sp3)–H bonds, thereby affording a series of pyrrolidine derivatives with good enantioselectivity (13.1–13.10).70 As shown in Scheme 13, substrates bearing various para-substituted Ph groups were subjected to benzylic C(sp3)–H amination to afford the corresponding pyrrolidines (13.1–13.4) with general high enantioselectivities (up to 99:1 e.r.), but lower yields were observed for the substrates with para-fluoro groups (e.g.13.4 26% yield). However, for the 2,5-difluoro substrate, trace amounts of the corresponding product (13.5) were obtained in 85:15 e.r., and compound 13.5 could be used as a building block for the preparation of the drug molecule Larotrectinib. Other substrates with different (hetero)aromatic rings, such as naphthalene (13.6), thiophene (13.7), and indole (13.8), are also compatible, producing products with moderate to low yields and e.r. Since unactivated aliphatic C–H bonds are challenging substrates, only small amounts of the products of 13.9 and 13.10 were obtained.
 | ||
| Scheme 13 Enzyme-catalysed asymmetric alkyl–nitrene C(sp3)–H amination. | ||
3. Conclusion and outlook
In summary, iron-catalysts supported by various ligands can be used for the intramolecular amination of C(sp3)–H bonds of alkyl azides to afford valuable N-heterocycles. Despite the higher catalyst loading, the Fe–dipyrrinatos system is attractive because it is a highly reactive catalyst, as revealed by the broader substrate scope and low reaction temperatures. On the other hand, MOF-supported Fe complexes with diketiminate, -phenanthroline and -terpyridine ligands systems can serve as recyclable and reusable catalysts with higher efficiency than homogeneous catalysts. Intriguingly, air-stable iron catalysts with redox-active ligands gave good product TON for primary aliphatic azide substrates, while iron-catalysts supported by mesoionic carbene ligands were found to be very efficient for tertiary azides, producing a high product TON (7600). For iron-porphyrin/porphyrinoid catalysts, the trans effect of axial ligands plays a critical role in their catalytic performance. These catalysts can be used in C(sp3)–H aminations of a wide scope of substrates with overall good performance. At the same time, this type of catalyst has a stable structure and can be used for the synthesis and late-stage modification of complex natural products and pharmaceuticals, with good performance. In particular, FeIII(Tp-OMePC)Cl is very efficient for the intramolecular C–H amination of common primary alkyl azides, delivering a high product TON of 3800.Mechanistic studies show that in most cases Fe–alkylimido/nitrene species undergo HAA and radical recombination processes during C–H bond amination reactions. However, to date, only a few iron imido/nitrene species have been isolated and fully characterized, showing the capability to react with C–H bonds through HAA in stochiometric reactions. Limited knowledge of reactive Fe–alkylimido/nitrene species prevents us from identifying the step(s) directly responsible for C–N bond formation, factors controlling reactivity and selectivity, and metal–ligand effects.
Given the potential explosive nature of aliphatic azides at high reaction temperature, it is important to develop robust, highly reactive iron-based catalyst systems that are capable of lowering the decomposition temperature of alkyl azides while maintaining high product turnover numbers. On the other hand, the use of energy transfer reactions to decompose alkyl azides in the presence of efficient photosensitizers provides an alternative to generate iron–nitrene species under mild conditions. These will facilitate stereo-controlled C(sp3)–H aminations of alkyl azides, including diastereoselective access to poly-substituted N-heterocycles and enantioselective aminations of various C–H bonds. Furthermore, the development of novel iron catalyst systems capable of intermolecular C–H amination of alkyl azides is highly appealing, as this would open up avenues to access non-cyclic N-containing building blocks, although a great challenge lies in the easily occurring intramolecular 1,2-hydride migration.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Hong Kong Research Grants Council General Research Fund (17315122, 17306824), the grants from the Innovation and Technology Commission (HKSAR, P. R. China) to the State Key Laboratory of Synthetic Chemistry and from the Laboratory for Synthetic Chemistry and Chemical Biology under the Health@InnoHK Program launched by the Innovation and Technology Commission, the Government of HKSAR.Notes and references
- R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef CAS PubMed.
- A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441–4557 CrossRef CAS PubMed.
- B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615–1637 CrossRef CAS.
- D. O'Hagan, Nat. Prod. Rep., 1997, 14, 637–651 RSC.
- G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484–4517 CrossRef CAS PubMed.
- D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC.
- J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092–4106 RSC.
- K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed.
- Y. Liu, K.-P. Shing, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2023, 13, 1103–1124 CrossRef CAS.
- Y. Liu, T. You, H.-X. Wang, Z. Tang, C.-Y. Zhou and C.-M. Che, Chem. Soc. Rev., 2020, 49, 5310–5358 RSC.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- F. Collet, C. Lescot and P. Dauban, Chem. Soc. Rev., 2011, 40, 1926–1936 RSC.
- R. Singh and A. Mukherjee, ACS Catal., 2019, 9, 3604–3617 CrossRef CAS.
- H. Hayashi and T. Uchida, Eur. J. Org. Chem., 2020, 909–916 CrossRef CAS.
- A. Fanourakis and R. J. Phipps, Chem. Sci., 2023, 14, 12447–12476 RSC.
- H. Khatua, S. Das, S. Patra, S. Nandi and B. Chattopadhyay, Tetrahedron Lett., 2024, 144, 155136 CrossRef CAS.
- Y. Liu, T. You, T.-T. Wang and C.-M. Che, Tetrahedron, 2019, 75, 130607 CrossRef.
- D. Possenti and G. Olivo, ChemCatChem, 2024, 16 DOI:10.1002/cctc.202400353.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- B. Plietker and A. Roeske, Catal. Sci. Technol., 2019, 9, 4188–4197 RSC.
- Y. Baek and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 7797–7806 CrossRef CAS PubMed.
- Y. Baek, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2019, 141, 16944–16953 CrossRef CAS PubMed.
- Y. Baek, A. Das, S.-L. Zheng, J. H. Reibenspies, D. C. Powers and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 11232–11243 CrossRef CAS PubMed.
- P. F. Kuijpers, M. J. Tiekink, W. B. Breukelaar, D. L. J. Broere, N. P. van Leest, J. I. van der Vlugt, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2017, 23, 7945–7952 CrossRef CAS PubMed.
- M. Goswami, P. Geuijen, J. N. H. Reek and B. de Bruin, Eur. J. Inorg. Chem., 2018, 617–626 CrossRef CAS.
- J. Qin, Z. Zhou, T. Cui, M. Hemming and E. Meggers, Chem. Sci., 2019, 10, 3202–3207 RSC.
- Z. Zhou, S. Chen, J. Qin, X. Nie, X. Zheng, K. Harms, R. Riedel, K. N. Houk and E. Meggers, Angew. Chem., Int. Ed., 2019, 58, 1088–1093 CrossRef CAS PubMed.
- Y. Dong, R. M. Clarke, G. J. Porter and T. A. Betley, J. Am. Chem. Soc., 2020, 142, 10996–11005 CrossRef CAS PubMed.
- Y. Dong, C. J. Lund, G. J. Porter, R. M. Clarke, S.-L. Zheng, T. R. Cundari and T. A. Betley, J. Am. Chem. Soc., 2021, 143, 817–829 CrossRef CAS PubMed.
- D. L. J. Broere, B. de Bruin, J. N. H. Reek, M. Lutz, S. Dechert and J. I. van der Vlugt, J. Am. Chem. Soc., 2014, 136, 11574–11577 CrossRef CAS PubMed.
- D. L. J. Broere, N. P. van Leest, B. de Bruin, M. A. Siegler and J. I. van der Vlugt, Inorg. Chem., 2016, 55, 8603–8611 CrossRef CAS PubMed.
- J. Kweon and S. Chang, Angew. Chem., Int. Ed., 2021, 60, 2909–2914 CrossRef CAS PubMed.
- R. Vyas, G.-Y. Gao, J. D. Harden and X. P. Zhang, Org. Lett., 2004, 6, 1907–1910 CrossRef CAS PubMed.
- L. Simkhovich and Z. Gross, Tetrahedron Lett., 2001, 42, 8089–8092 CrossRef CAS.
- P. S. Steinlandt, X. Xie, S. Ivlev and E. Meggers, ACS Catal., 2021, 11, 7467–7476 CrossRef CAS.
- P. R. O. De Montellano, Cytochrome P450: structure, mechanism, and biochemistry, Springer, 2005 Search PubMed.
- F. P. Guengerich, ACS Catal., 2018, 8, 10964–10976 CrossRef CAS PubMed.
- W. A. Nugent and J. M. Mayer, Metal-ligand multiple bonds: the chemistry of transition metal complexes containing oxo, nitrido, imido, alkylidene, or alkylidyne ligands, Wiley, New York, 1988 Search PubMed.
- W. Stroek, M. Keilwerth, L. A. Malaspina, S. Grabowsky, K. Meyer and M. Albrecht, Chem. – Eur. J., 2024, 30, e202303410 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591–595 CrossRef CAS PubMed.
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed.
- R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC.
- N. C. Thacker, Z. Lin, T. Zhang, J. C. Gilhula, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2016, 138, 3501–3509 CrossRef CAS PubMed.
- N. C. Thacker, P. Ji, Z. Lin, A. Urban and W. Lin, Faraday Discuss., 2017, 201, 315–327 RSC.
- Z. Lin, N. C. Thacker, T. Sawano, T. Drake, P. Ji, G. Lan, L. Cao, S. Liu, C. Wang and W. Lin, Chem. Sci., 2018, 9, 143–151 RSC.
- D. A. Iovan, M. J. T. Wilding, Y. Baek, E. T. Hennessy and T. A. Betley, Angew. Chem., Int. Ed., 2017, 56, 15599–15602 CrossRef CAS PubMed.
- B. Bagh, D. L. J. Broere, V. Sinha, P. F. Kuijpers, N. P. van Leest, B. de Bruin, S. Demeshko, M. A. Siegler and J. I. van der Vlugt, J. Am. Chem. Soc., 2017, 139, 5117–5124 CrossRef CAS PubMed.
- K.-H. Chan, X. Guan, V. K.-Y. Lo and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 2982–2987 CrossRef CAS PubMed.
- K.-P. Shing, Y. Liu, B. Cao, X.-Y. Chang, T. You and C.-M. Che, Angew. Chem., Int. Ed., 2018, 57, 11947–11951 CrossRef CAS PubMed.
- Y.-D. Du, C.-W. Tse, Z.-J. Xu, Y. Liu and C.-M. Che, Chem. Commun., 2014, 50, 12669–12672 RSC.
- Y.-D. Du, Z.-J. Xu, C.-Y. Zhou and C.-M. Che, Org. Lett., 2019, 21, 895–899 CrossRef CAS PubMed.
- Y.-D. Du, C.-Y. Zhou, W.-P. To, H.-X. Wang and C.-M. Che, Chem. Sci., 2020, 11, 4680–4686 RSC.
- S. Wonnacott and T. Gallagher, Mar. Drugs, 2006, 4, 228–254 CrossRef CAS.
- X. Zhao, S. Liang, X. Fan, T. Yang and W. Yu, Org. Lett., 2019, 21, 1559–1563 CrossRef CAS PubMed.
- S. Liang, X. Zhao, T. Yang and W. Yu, Org. Lett., 2020, 22, 1961–1965 CrossRef CAS PubMed.
- L. Wu, D. Zhong and W. Liu, Youji Huaxue, 2021, 41, 4083–4087 CrossRef CAS.
- T. You, S.-H. Zeng, J. Fan, L. Wu, F. Kang, Y. Liu and C.-M. Che, Chem. Commun., 2021, 57, 10711–10714 RSC.
- W. Stroek, M. Keilwerth, D. M. Pividori, K. Meyer and M. Albrecht, J. Am. Chem. Soc., 2021, 143, 20157–20165 CrossRef CAS PubMed.
- W. Stroek and M. Albrecht, Chem. Sci., 2023, 14, 2849–2859 RSC.
- W. Stroek, L. Hoareau and M. Albrecht, Catal. Sci. Technol., 2023, 13, 958–962 RSC.
- W. Stroek, M. Keilwerth, L. A. Malaspina, S. Grabowsky, K. Meyer and M. Albrecht, Chem. – Eur. J., 2024, 30, e202303410 CrossRef CAS PubMed.
- J. Fan, Y. Wang, X. Hu, Y. Liu and C.-M. Che, Org. Chem. Front., 2023, 10, 1368–1374 RSC.
- I. H. Hall, A. R. K. Murthy and S. D. Wyrick, J. Pharm. Sci., 1986, 75, 622 CrossRef CAS PubMed.
- P. Verma, A. Chandra and G. Pandey, J. Org. Chem., 2018, 83, 9968–9977 CrossRef CAS PubMed.
- J. Berges, B. Garcia and K. Muniz, Angew. Chem., Int. Ed., 2018, 57, 15891–15895 CrossRef CAS PubMed.
- T. R. Wu and J. M. Chong, J. Am. Chem. Soc., 2006, 128, 9646–9647 CrossRef CAS PubMed.
- T. Sheng, C. Ma, G. Zhang, X. Pan and Z. Liu, J. Nat. Prod., 2022, 85, 1128–1133 CrossRef CAS PubMed.
- T. You, K.-P. Shing, L. Wu, K. Wu, H.-H. Wang, Y. Liu, L. Du, R. Liang, D.-L. Phillips, X.-Y. Chang, J.-S. Huang and C.-M. Che, Adv. Sci., 2024 DOI:10.1002/advs.202401420.
- J. Fan, Y. Wang, X. Hu, Y. Liu and C.-M. Che, Org. Chem. Front., 2023, 10, 1368–1374 RSC.
- Z.-Y. Qin, S. Gao, Y. Zou, Z. Liu, J. B. Wang, K. N. Houk and F. H. Arnold, ACS Cent. Sci., 2023, 9, 2333–2338 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |