
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interphase formation versus fluoride-ion insertion in tunnel-structured transition metal antimonites†
Alice R.
Giem
ab,
Jaime R.
Ayala
 ab,
Jingxiang
Cheng
ab,
Conan
Weiland
c,
Cherno
Jaye
c,
Daniel A.
Fischer
c and
Sarbajit
Banerjee
*ab
ab,
Jingxiang
Cheng
ab,
Conan
Weiland
c,
Cherno
Jaye
c,
Daniel A.
Fischer
c and
Sarbajit
Banerjee
*ab
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: banerjee@chem.tamu.edu
bDepartment of Material Science & Engineering, Texas A&M University, College Station, TX 77843, USA
cMaterial Measurement Laboratory, National Institute of Standards and Technology, Gaithersburg, MD 20899, USA
First published on 6th November 2024
Abstract
Fluoridating reagents are used to model interfacial reactions in fluoride-ion batteries. Topochemical F-ion insertion is seen for one-dimensional (1D) tunnel-structured FeSb2O4 but interphase formation comprising antimony (oxy)fluorides is observed for MnSb2O4.
Grid-level energy storage requires a diversity of battery chemistries so as to alleviate stresses on current mineral supply chains and to provide safer alternatives to Li-ion batteries.1–3 “Beyond Li” alternatives such as Na-ion batteries are yet to realize comparable energy densities, and often do little to diversify materials palettes.4,5 Fluoride-ion batteries, which implement fluoride anions as charge carriers have received considerable attention as an orthogonal alternative with the potential to alleviate supply chain challenges and preclude the need for metal electrodeposition.6–10 Fluoride conversion electrodes have shown promise but challenges remain with regards to high temperatures for successful operation, large volume changes, and sluggish fluoride-ion diffusion.6–8 Much recent attention has focused on the design of insertion-based fluoride electrode materials where the integrity of the structural framework of insertion electrodes is preserved upon F-ion insertion/deinsertion.11 However, a key consideration is the competition between ion insertion and interphase formation.12 Here, we use fluoridating reagents, XeF2 and NOBF4, as models for interfacial electrode reactions to contrast F-ion reactivity in 1D tunnel-structured FeSb2O4 and MnSb2O4, which both adopt the schafarzikite-type structure but with distinctive oxidation potentials (0.771 and 1.56 vs. standard hydrogen electrode (SHE) for Fe2+/Fe3+ and Mn2+/Mn3+, respectively).13 We utilize a broad suite of spectroscopic methods to probe chemical reactivity in model F-ion insertion hosts to differentiate between interphase formation and insertion reactions.
The schafarzikite-type MA2O4 structure sketched in Fig. 1 (M = transition metal, A = p-block element with stereochemically active lone pairs) forms 1D tunnels defined by corner-shared [MO6] octahedra and [AO4] tetrahedra.14–16 The stereochemical activity of 5s2 electron lone pairs of Sb3+ cations defines a large one-dimensional tunnel with an abundance of interstitial sites that can accommodate F-ions (Fig. 1B). In FeSb2O4, electron lone pair repulsions mediate frustrated coordination and ensure low site-to-site diffusion barriers whilst redox compensation is engendered through formal oxidation of Fe centers upon F-ion insertion.14 The greater oxidation potential of the d-block cation in MnSb2O4 holds promise for accessing a higher voltage but the more challenging redox also renders parasitic reactions more probable.17 While reversible room-temperature F-ion insertion with stoichiometries up to x ≈ 1 can be accessed for FeSb2O4Fx,14,15 complex interphasic products are instead stabilized upon reaction with MnSb2O4.
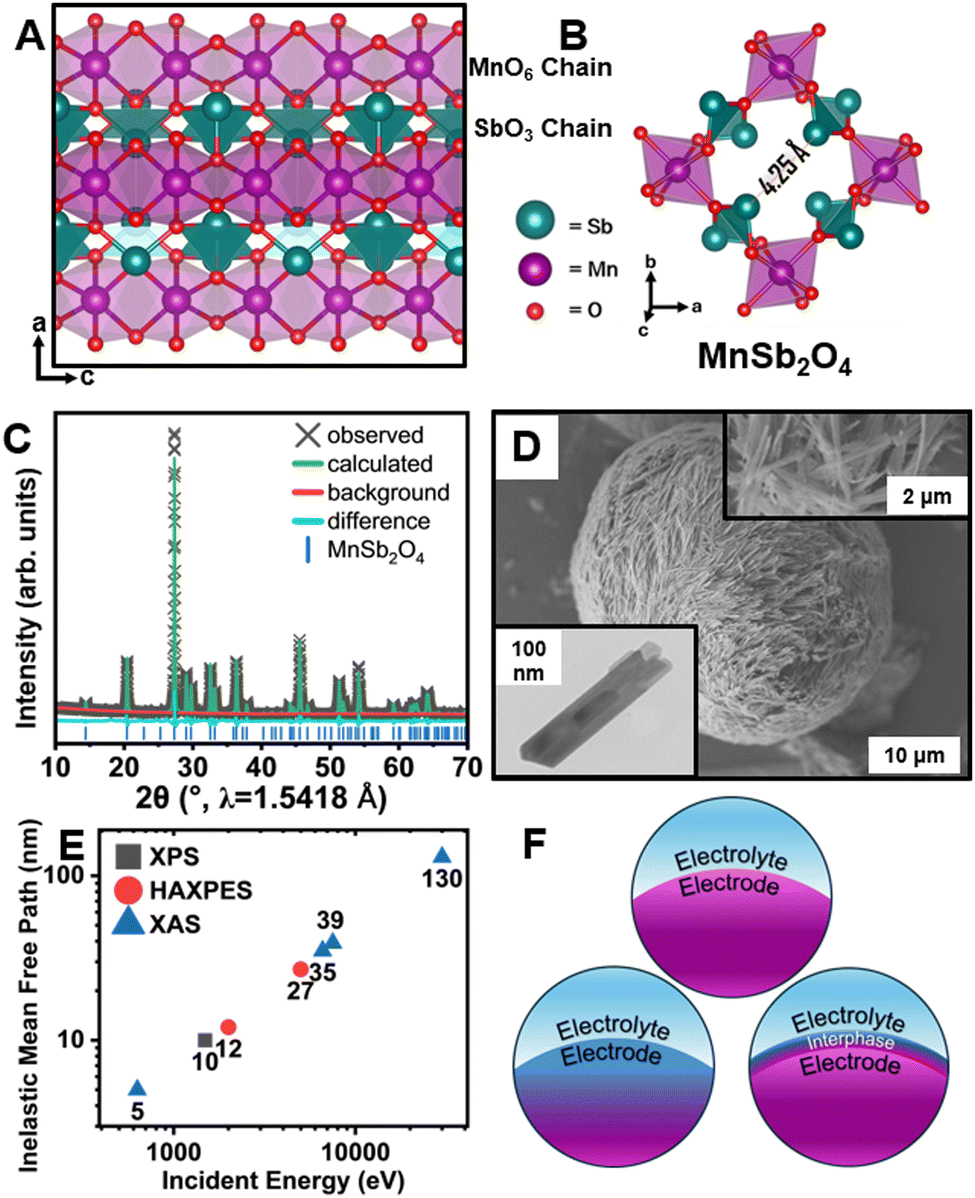 | ||
| Fig. 1 Structure and characterization of MnSb2O4. (A) Schafarzikite-type MnSb2O4 viewed along (010); (B) 1D tunnel of MnSb2O4 viewed along the c direction. (C) Rietveld-refinement to powder XRD pattern used to obtain the structures in (A) and (B). (D) SEM image of MnSb2O4 nanorods; top inset shows magnified SEM; bottom inset shows a TEM image. (E) IMFP of an electron calculated based upon the incident energy for XPS, HAXPES, and XAS measurements. (F) Schematic illustration of F-ion reactivity. | ||
Interphase formation at oxide surfaces has attracted much recent attention given the critical role of interphasic products in mediating ion transport, surface stresses, and preservation of electrolyte/electrode connectivity.18,19 Little is known about interphase formation in anion batteries. Here, we use diverse X-ray absorption and emission spectroscopy probes of varying penetration depth (Fig. 1E) to examine interphase formation in MnSb2O4 (Fig. 1F).
MnSb2O4 powders were obtained through a hydrothermal synthesis as described in the ESI.† A powder X-ray diffraction (XRD) pattern and Rietveld refinement is shown in Fig. 1C (see also Table S1, ESI†).20 MnSb2O4 is isostructural to FeSb2O4 and crystallized in the tetragonal P42/mbc space group. MnSb2O4 affords a larger tunnel width of 4.25 Å (Fig. 1B) as compared to 4.14 Å for FeSb2O4.21,22 The obtained powders adopt a single-crystalline needle-like morphology (Fig. 1D and Fig. S1, ESI†).
Fluoridation reactions were initially performed using XeF2 in acetonitrile under a diverse range of conditions as described in the Methods section.17 Under these conditions, FeSb2O4 can be fluoridated to insert unitary fluoride ions per Fe center.14,15 Powder XRD measurements and results from Rietveld refinement for fluoridation of MnSb2O4 are shown in Fig. S2 and Table S2 (ESI†). Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) characterization is shown in Fig. S3 with Fig. S4 (ESI†) showing transmission electron microscopy (TEM) imaging including lattice resolved high-resolution TEM and selected area electron diffraction (SAED). Minimal changes of lattice parameters are observed in powder X-ray diffraction even though EDS elemental maps demonstrate some reactivity and remanence of fluorine-containing species. Definitive identification of the reaction products is enabled by synchrotron X-ray spectroscopy measurements, which provide a means of distinguishing insertion and interphase formation (Fig. 1F).
X-ray absorption near-edge structure (XANES) spectroscopy measurements of the as-prepared and fluoridated MnSb2O4 are shown in Fig. 2. Soft X-ray (surface sensitive) Mn L-edge (2p–3d) spectra for XeF2-treated MnSb2O4 are plotted in Fig. 2A. Corresponding Fe L-edge data for XeF2-reacted FeSb2O4 are plotted in Fig. 2B. In contrast to FeSb2O4, where F-ion insertion brings about oxidation of the Fe center,14,15 the Mn L3-edge spectra with a pronounced absorption at 640 eV characteristic of divalent Mn do not evidence signatures of Mn oxidation upon treatment with XeF2.23 As such, both powder XRD and Mn L-edge XANES measurements do not support the notion of F-ion insertion. However, Fig. 2C shows that reaction of MnSb2O4 results in the emergence of the F K-edge (1s–2p), characteristic of fluoride moieties at the surface. This and the EDS measurements in Fig. S2 (ESI†) suggest that fluorine is incorporated at least on particle surfaces through alternative reaction pathways.
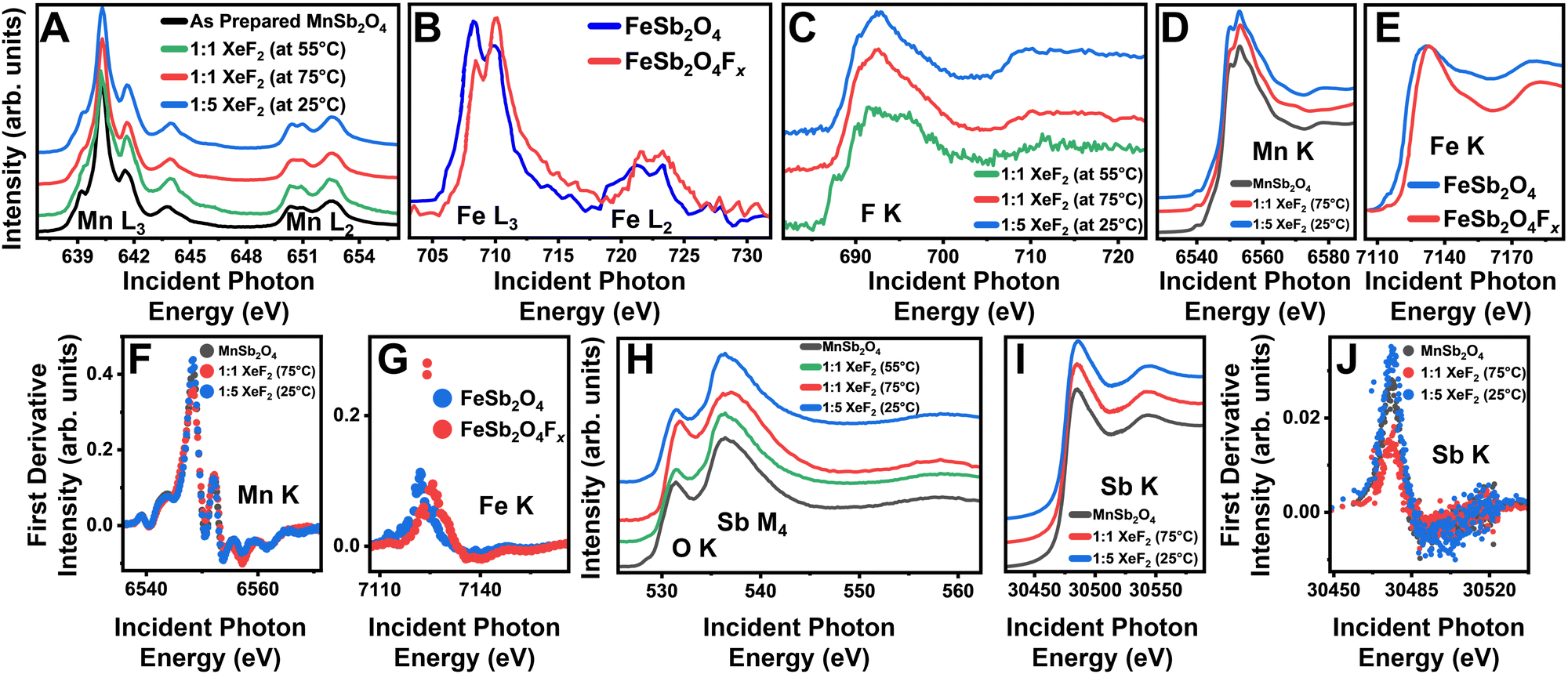 | ||
Fig. 2 XANES characterization of fluoridated products. XANES spectra collected for the as-prepared MnSb2O4 (black) and fluoridated products (green 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of XeF2/CH3CN at 55 °C, red 1:1 molar ratio of XeF2/CH3CN at 75 °C, and blue 1:5 molar ratio of XeF2 in CH3CN at 25 °C) at the (A) Mn L3-/L2-edges, (B) Fe L3/L2-edges, (C) F K-edge, (D) Mn K-edge, and (E) Fe K-edge. Additional spectra in (F) represent the first derivative of the Mn K-edge spectra, (G) first derivative of the Fe K-edge spectra, (H) Sb M4-, O K-edges, first derivative of the Mn K-edge spectra, (I) Sb K-edge, and (J) first derivative of the Sb K-edge spectra. Panels (B), (E), and (G) are adapted with permission from Zaheer et al.14 Copyright (2020) ACS Energy Lett. :1 molar ratio of XeF2/CH3CN at 55 °C, red 1:1 molar ratio of XeF2/CH3CN at 75 °C, and blue 1:5 molar ratio of XeF2 in CH3CN at 25 °C) at the (A) Mn L3-/L2-edges, (B) Fe L3/L2-edges, (C) F K-edge, (D) Mn K-edge, and (E) Fe K-edge. Additional spectra in (F) represent the first derivative of the Mn K-edge spectra, (G) first derivative of the Fe K-edge spectra, (H) Sb M4-, O K-edges, first derivative of the Mn K-edge spectra, (I) Sb K-edge, and (J) first derivative of the Sb K-edge spectra. Panels (B), (E), and (G) are adapted with permission from Zaheer et al.14 Copyright (2020) ACS Energy Lett. | ||
Hard X-ray (“bulk”-sensitive) XANES measurements have been collected for Mn, Sb, and Fe at their corresponding K-edges (1s–2p). Contrasting Fig. 2D and E respectively, the Mn and Fe K-edges measured for XeF2-reacted MnSb2O4 and FeSb2O4 show pronounced differences. The Fe K-edge spectrum shows a pronounced shift of ca. 3 eV of the white-line absorption, indicating bulk oxidation concomitant with F-ion insertion.14,15 In contrast, only a subtle change in the shape of the Mn K-edge is observed (a ca. 5 eV shift is anticipated for the oxidation of Mn2+ to Mn3+).24 The differential spectrum in Fig. 2F further does not show significant changes again arguing against bulk F-ion insertion in MnSb2O4 (contrast to Fig. 2G for FeSb2O4).
To examine the fluoride product evidenced from Fig. 2C and Fig. S2 (ESI†), we next explore surface and bulk probes of Sb bonding. Surface-sensitive Sb M4-edge (3d–4f) XANES spectra in Fig. 2H show modified relative intensities of pre-edge and white line absorption features at 531 eV and 536 eV, respectively. Subtle shifts of the absorption features indicate a change in the coordination environment of Sb whilst preserving a nominally trivalent antimony species. Sb K-edge (1s–2p) spectra in Fig. 2I do not show a pronounced shift of the white-line absorption; the differential spectra of the Sb K-edge plotted in Fig. 2J show an increase in the intensity of the peak at 30475 eV. The modification of the Sb local structure is more evident in surface-sensitive Sb M4-edge XANES as compared to the Sb K-edge, which probes deeper into the sample. As such, the XANES results suggest the stabilization of amorphous interphasic products that are predominantly a mixture of fluorides and oxyfluorides of trivalent antimony.25
We next probe XeF2-reacted MnSb2O4 using hard X-ray photoemission spectroscopy (HAXPES).26 The Mn 2p core level of the as-prepared MnSb2O4 is shown in Fig. 3A. The satellite peak at 650 eV binding energy is a distinct feature of Mn2+.27Fig. 3B and C contrast the Mn 2p core levels of MnSb2O4 after XeF2 treatment. Integrating the satellite peaks corroborates that the Mn centers do not undergo bulk oxidation as would be expected upon F-ion insertion. Fig. 3D shows the emergence of remnant F-containing species upon XeF2 treatment. Fig. 3E depicts the Sb 3d core level with the Sb 3d3/2 centred at approximately 540 eV, verifying the preservation of the Sb3+ formal oxidation state. The Sb 3d5/2 core level feature overlaps with the O 1s core level. The decrease in area observed in the Sb 3d5/2/O 1s peak indicates a less oxygen rich surface upon 1:3 XeF2 treatment (Fig. 3E), suggesting accumulation of fluoride/oxyfluoride interphasic products.
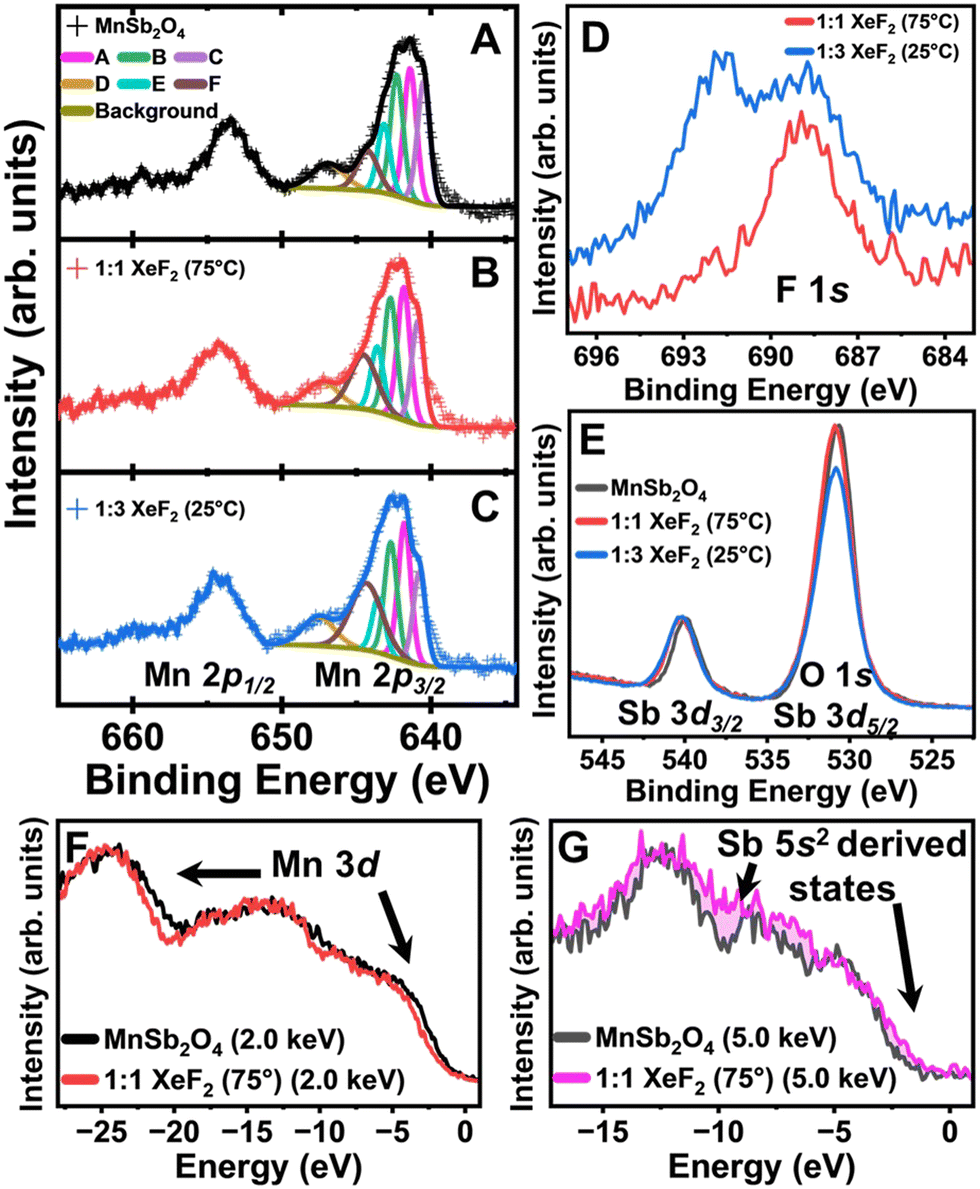 | ||
| Fig. 3 HAXPES spectra of MnSb2O4 and derivatives collected at 2 keV incident energy unless otherwise stated; (A) Mn 2p core level of the as-prepared MnSb2O4 shown with fits (A)–(F) for Mn 2p3/2; (B) Mn 2p core level after a 1 mol treatment with XeF2 in CH3CN; (C) Mn 2p core level after a 3 mol excess treatment with XeF2; (D) F 1s core level spectra after XeF2 treatments; (E) Sb 3d core level. VB spectra of MnSb2O4 before and after the reaction with 1 mol XeF2 collected at an incident energy of (F) 2 keV and (G) 5 keV. | ||
To probe the electronic structure of MnSb2O4 before and after XeF2 treatment, valence band (VB) spectra were acquired using energy-variant HAXPES (Fig. 3E and F). The photoionization cross-sections of orbitals have an incident energy-dependent decay that can be utilized to decipher orbital contributions.14,22 Orbital subshells with higher orbital angular momentum (e.g., d and f shells) contribute greater intensity to the VB spectrum at lower incident energies. In contrast, at higher energies, the s and p shells make a greater contribution to the VB.14 A subtle diminution in intensity is visible for fluoridated MnSb2O4 at 2 keV incident energy indicating less Mn 3d character upon fluoridation. At 5 keV incident energy (Fig. 3G), an increase in the states near the Fermi level suggests greater contributions from the Sb lone-pair states hybridized with F 2p states upon fluoridation. These observations are furthermore consistent with the accumulation of an Sb-rich (and Mn-poor) interphasic product. While increasing temperature accelerates F-ion insertion in FeSb2O4,16 the reactivity of MnSb2O4 and the speciation of the interphasic product is not substantially modified from room temperature as consistent with a surface passivation reaction.
Fig. S5 (ESI†) shows density of states (DOS) calculations for MnSb2O4 and FeSb2O4. Atom-projected partial DOS plots illustrate that Fe 3d–O 2p states are located at the valence band edge and above Sb-derived states. In contrast, Mn 3d–O 2p states are strongly overlapped with Sb states at the valence band edge of MnSb2O4. As such, Fe and Sb are preferentially oxidized by the fluoridating reagents in FeSb2O4 and MnSb2O4, respectively. Fig. S6 (ESI†) shows that F-ion insertion to form a putative MnSb2O4Fx is unlikely because of strong distortions needed to push Mn states above Sb states.
The most significant change in HAXPES spectra is observed in the F 1s region (Fig. 3D). Treatment with a 1:1 molar ratio of XeF2 yields a singular F 1s feature, but upon treatment with a 3-molar excess of XeF2, a second F 1s feature is observed. The features can be ascribed to a combination of surface insertion and interphase formation or fluoride/oxyfluoride interphases. An alternative fluoridation agent, NOBF4, has furthermore been examined because of its high oxidizing power.28 NOBF4 has an oxidation potential of 1.7 V vs. SHE, which is greater than the oxidation potential of Mn by 0.14 V. Fig. S7 (ESI†) shows a powder XRD pattern acquired for MnSb2O4 after reaction with NOBF4 in acetonitrile. The reaction does not bring about an evident change of lattice parameters (Table S3, ESI†). SEM-EDS in Fig. S8 (ESI†) provides confirmation of fluoride incorporation with Fig. S9 (ESI†) showing TEM, including lattice-resolved high-resolution TEM and SAED.
Fig. S10 (ESI†) plots a Mn L3-edge XANES spectrum acquired for MnSb2O4 after treatment with NOBF4. The spectrum shows a slight shift of the white line absorption and diminution of the pre-edge feature at 639 eV. Fig. S11 (ESI†) shows Sb M4-edge and O K-edge XANES spectra for the as-prepared MnSb2O4 and after treatment with a 3-molar excess of NOBF4. Similar to Fig. 2B, no shift of the white line absorption energy is observed, which indicates retention of a nominally trivalent oxidation state for Sb. However, a pronounced modification of relative intensities of features centred at 531 eV and 536 eV is suggestive of a change in the Sb local coordination environment. F K-edge XANES spectra in Fig. S11B (ESI†) indicate the presence of anionic fluoride species after NOBF4 treatment. Core-level HAXPES spectra (Fig. S11B–D, ESI†) show pronounced changes suggestive of the accumulation of an interphasic product. The O 1s core level is significantly decreased in intensity as compared to overlapping Sb 3d states, indicating that there is less oxygen on the surface (Fig. S11C, ESI†). The shift in binding energy for the Sb 3d3/2 core level further suggests an alteration of the surficial Sb local coordination environment. Based on these observations, the interphase layer is most-likely formed by amorphous antimony fluorides and oxyfluorides, such as SbF3 or a F-rich oxyfluoride such as Sb3O2F5 (Fig. S11E–G, ESI†). Table S4 (ESI†) provides balanced chemical equations for the formation of each of these species. While NOBF4 is a stronger oxidizing reagent, based on the electronic structure considerations depicted in Fig. S5 (ESI†), it promotes increased antimony oxidation and exacerbates the formation of the interphasic product. Fig. S12 (ESI†) depicts crystal structures of the candidate materials but based on X-ray scattering, the interphasic products do not appear to have long-range order.
Understanding the competitive nature of insertion versus interphase formation is key to the design and discovery of insertion electrodes of fluoride-ion batteries. We find a sharp contrast between low-dimensional FeSb2O4 and MnSb2O4, both of which crystallize in a schafarzikite-type structure with a 1D tunnel. While the former reversibly accommodates F-ions in interstitial sites along its tunnel, the latter is characterized by very limited bulk F-insertion. The insertion of fluoride-ions is instead constrained by the formation of an interphase layer on the surfaces of the MnSb2O4 particles. While Mn affords a higher oxidation potential, the overlap of Mn 3d-derived states with Sb states at the valence band edge promotes the preferential formation of amorphous antimony fluoride or antimony oxyfluoride interphasic products. Understanding the reactivity of model systems will assist in the future realization of viable F-ion insertion electrodes and full batteries.
The authors acknowledge support from the Welch Foundation under award A-1978-20190330. This research used resources of the National Synchrotron Light Source II under Contract DE-SC0012704.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Notes and references
- A. Zeng, et al. , Nat. Commun., 2022, 13, 1341 CrossRef CAS PubMed.
- C. Xu, et al. , Commun. Mater., 2020, 1, 1–10 CrossRef.
- P. Xu, et al. , Trends Chem., 2021, 3, 620–630 CrossRef CAS.
- B. Esser, et al. , Adv. Energy Mater., 2024, 2402824 CrossRef.
- H. S. Hirsh, et al. , Adv. Energy Mater., 2020, 10, 2001274 CrossRef CAS.
- M. A. Reddy and M. Fichtner, J. Mater. Chem., 2011, 21, 17059–17062 RSC.
- A. W. Xiao, et al. , Joule, 2021, 5, 2823–2844 CrossRef CAS.
- V. K. Davis, et al. , Science, 2018, 362, 1144–1148 CrossRef CAS PubMed.
- D. McTaggart, et al. , ChemSusChem, 2023, 16, e202300486 CrossRef CAS PubMed.
- V. Vanita, et al. , J. Mater. Chem. A, 2024, 12, 8769–8784 RSC.
- M. A. Nowroozi, et al. , J. Mater. Chem. A, 2018, 6, 4658–4669 RSC.
- J. L. Andrews, et al. , ACS Energy Lett., 2022, 7, 2340–2348 CrossRef CAS.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1–21 CrossRef CAS.
- W. Zaheer, et al. , Cell Rep. Phys. Sci., 2021, 2, 100592 CrossRef CAS.
- W. Zaheer, et al. , ACS Energy Lett., 2020, 5, 2520–2526 CrossRef CAS.
- B. P. de Laune, et al. , Inorg. Chem., 2017, 56, 10078–10089 CrossRef CAS PubMed.
- C. K. Blakely, et al. , J. Solid State Chem., 2020, 289, 121490 CrossRef CAS.
- W. Li, et al. , Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- C. D. Quilty, et al. , Chem. Rev., 2023, 123, 1327–1363 CrossRef CAS PubMed.
- M. Roelsgaard, et al. , Dalton Trans., 2016, 45, 18994–19001 RSC.
- K. Tolborg, et al. , IUCrJ, 2020, 7, 480–489 CrossRef CAS PubMed.
- J. V. Handy, et al. , Chem. Mater., 2022, 34, 1439–1458 CrossRef CAS.
- B. Gilbert, et al. , J. Phys. Chem. A, 2003, 107, 2839–2847 CrossRef CAS.
- T. S. Wu, et al. , Appl. Phys. Lett., 2012, 101, 022408 CrossRef.
- S. Imran Ali and M. Johnsson, Dalton Trans., 2016, 45, 12167–12173 RSC.
- Z. W. Lebens-Higgins, et al. , J. Phys. Chem. Lett., 2020, 11, 2106–2112 CrossRef CAS PubMed.
- E. S. Ilton, et al. , Appl. Surf. Sci., 2016, 366, 475–485 CrossRef CAS.
- A. R. Wizansky, et al. , J. Solid State Chem., 1989, 81, 203–207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04331f |
| This journal is © The Royal Society of Chemistry 2024 |