
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Compartmentalised single-chain nanoparticles and their function
Justus F.
Thümmler
 and
Wolfgang H.
Binder
*
and
Wolfgang H.
Binder
*
Institute of Chemistry, Faculty of Natural Science II, Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, D-06120, Halle (Saale), Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
First published on 15th November 2024
Abstract
Single-chain nanoparticles (SCNPs) are generated by intramolecular collapse and crosslinking of single polymer chains, thus conceptually resembling the structures of folded proteins. Their chemical flexibility and ability to form compartmentalised nanostructures sized ∼1 nm make them perfect candidates for numerous applications, such as in catalysis and drug delivery. In this review we discuss principles for the design, synthesis and analysis of SCNPs, with a focus on their compartmentalised structures, highlighting our own previous work. As such compartments offer the potential to generate a specific nanoenvironment e.g. for the covalent and non-covalent encapsulation of catalysts or drugs, they represent a novel, exciting, and expanding research area. Starting from the architectural and chemical design of the starting copolymers by controlling their amphiphilic profile, the embedding of blocks-, or secondary-structure-mimetic arrangements, we discuss design principles to form internal compartments inside the SCNPs. While the generation of compartments inside SCNPs is straightforward, their analysis is still challenging and often demands special techniques. We finally discuss applications of SCNPs, also linked to the compartment formation, predicting a bright future for these special nanoobjects.
 Justus F. Thümmler | Justus F. Thümmler studied chemistry at the Martin-Luther-University in Halle. After his specialization in physical chemistry of polymers and his master thesis at the Fraunhofer IMWS, he worked on his PhD thesis on near-infrared fluorescent single-chain nanoparticles and their nanostructural and photophysical characterization in the group of Professor Wolfgang H. Binder which he finished in 2024. In the Binder group he is currently working as researcher, continuing his work on single-chain nanoparticles. |
 Wolfgang H. Binder | Wolfgang H. Binder is currently a full professor of macromolecular chemistry at MLU since 2007. Born in Vienna, Austria (1969), he studied chemistry at the University of Vienna, obtaining his PhD in organic chemistry in 1995, followed by postdoctoral stays (1995–1997) with Prof. F. M. Menger (Emory University, Atlanta, GA) and Prof. J. Mulzer (University of Vienna), then completing his habilitation (2004), subsequently as associate professor from 2004 to 2007. His research interests are directed toward polymer synthesis, supramolecular chemistry, self-healing polymers, artificial membranes, and nanotechnology. |
1. Introduction
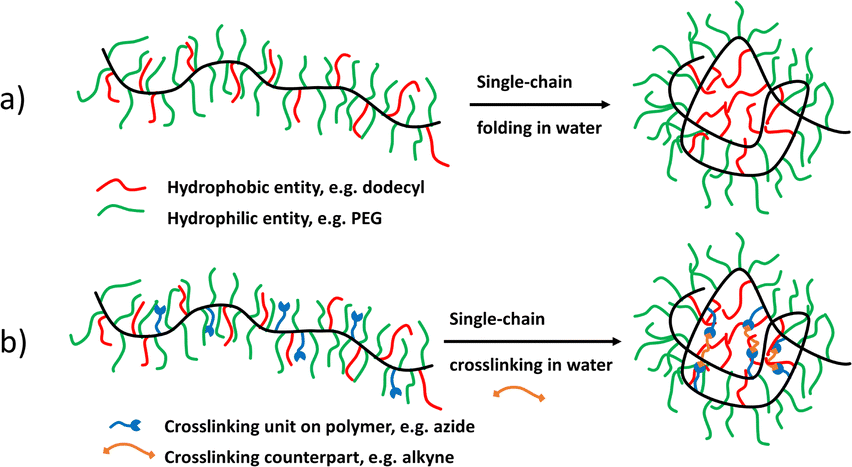
When components in a chemical or biological system require a specific localised environment, Nature has developed sophisticated separation entities on different length scales to spatially and temporally separate processes.1 Thus compartmentalisation is present in, e.g. each cellular compartment, like cell-membranes or organelles (mitochondria,2 cell-nuclei3); in viral capsids segregating and protecting the embedded nucleic-acids from the environment and mediating specific entry into cells;4 and most importantly in chemical reaction “organelles”, separating reactants in a chemical reaction and further allowing separation of the reactants after workup. Especially compartments on the nanoscale (below 100 nm) have emerged as a fundamental principle to direct chemical and biological reaction pathways, such as in enzymes in their hydrophobic/hydrophilic pockets5 allowing the specific positioning of reactants towards catalytic sites,6 or in (biomimetic) catalysis, then generating a specific reaction environment. Such compartments can be selective in hosting a specific sort of molecules in pockets, clefts, or porous structures, where a specific reactivity can be reached, given that dynamic processes are embedded to allow adaption and release of products. Organic (nano)particles like micelles, liposomes, lipid nanodiscs or proteins use organic molecules as building blocks often via self-assembly into higher organized systems. In the last decades many such compartmentalised systems have been developed, such as block-copolymer-assemblies,7 dendrimers,8 or nanogels,9 forming compartments in the nanometre range (e.g. biomembranes,10 micelles,11 dendritic boxes12) up to the micron range (such as liposomes13 or emulsion droplets14).Single-chain nanoparticles (SCNPs) have recently emerged as a special class of nanosystems displaying sizes in the range of 5–20 nm, depending on the polymer chain they are formed from.15 Based on their size, the chemical versatility of their generation and the multifaceted embedding of functional entities during their synthesis, they have been proven to be most versatile and adaptable in view of catalysis,16 drug delivery,17 or as enzyme-mimetics.18 SCNPs are crosslinked polymeric nanostructures that are formed from intramolecularly collapsed single polymer chains in a process reminiscent of protein folding.18 Conceptually, they are a special, extremely small type of nanogel, but due to their unique architecture, versatility of synthesis and the final properties they create a completely new class of polymeric nanoparticles.
It recently was discovered that even those small-sized SCNPs can display an internal structure equipping them with specialised functions beyond a pure encapsulation (see Fig. 1),19 usually significantly smaller than the already small particles themselves. The formation of these compartments inside those SCNPs is driven by molecular assembly (supramolecular/liquid crystalline assembly);20 polymer/polymer phase segregation;19b,19c or by amphiphilic segregation based on solvent/polymer segregation so finally guided by a sequential folding process.21 They are comparable to pockets normally found in, e.g. enzymes, where highly specialised reaction environments are generated to spatiotemporally catalyse specific reactions.
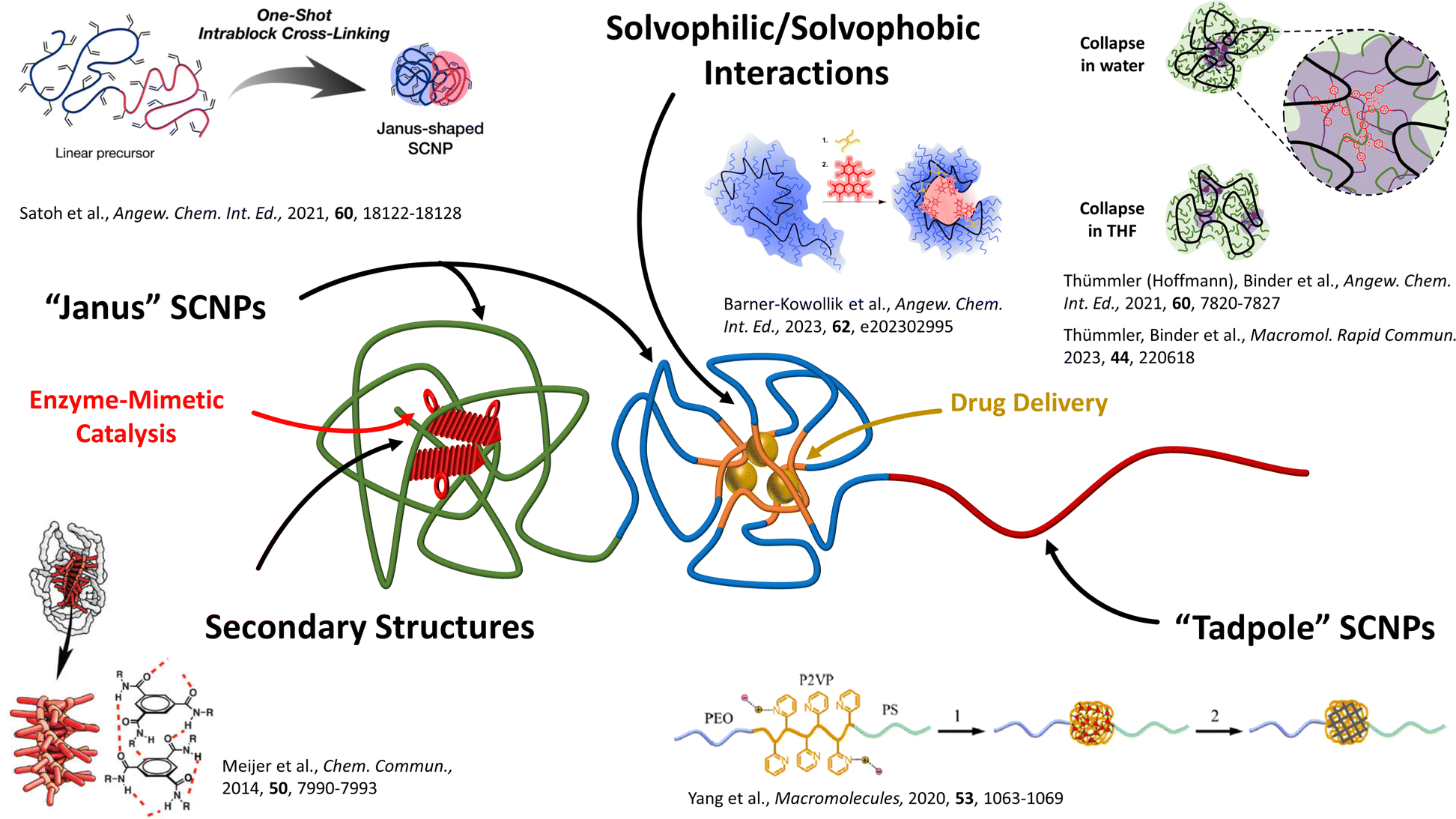 | ||
| Fig. 1 Illustration of compartmentalised structures in single-chain nanoparticles (SCNPs) and their applications in enzyme-mimetic catalysis and drug delivery. Block-copolymers can be collapsed into Janus-SCNPs by crosslinking the respective individual block. One or more non-functionalised blocks can create tadpole-shaped SCNPs, which structurally resemble surfactants or lipids. Secondary-structure-like compartments can be achieved, e.g. via H-bonding BTA-stacks. Solvophilic/solvophobic interactions are the main driving force of compartmentalised structures, since the solvophobic parts of the precursor polymer can phase-segregate, resulting in a core–shell structure. Reproduced with permission from: Wiley VCH, copyright © 2021,19b 2023 (open access CC BY-NC 4.0);19a,19e the Royal Society of Chemistry, copyright © 2014 (open access CC BY 3.0);22 and the American Chemical Society, copyright © 2020.23 | ||
They can be fundamental to tune optical effects with embedded dyes,24 induce special photothermal swelling effects19e,19f and conformational changes by photophysical crowding effects.25
Since Pomposo et al. have reviewed on internally and externally confined SCNPs in 2020,15a the development of confined/compartmentalised SCNPs has become a major interest among polymer researchers. It is in this article that we describe the latest developments in the generation of such structures also based on own contributions during the past years, with a focus on principles, analytical methods, as well as applications wherein the compartments are exploited. We focus on the principles guiding SCNP formation, together with the physicochemical effects guiding the formation of the internal compartments inside the SCNPs. With this perspective we take our starting point at previously published papers on SCNPs, moving beyond the chemical and physical principles described therein.
2. Design of compartmentalised single-chain nanoparticles (SCNPs)
Similar to proteins, which are formed by collapse of a single peptide-chain, SCNPs are generated by single-chain collapse at high dilutions dominated by solvent-assisted intramolecular vs. intermolecular-assembly (Fig. 2).26 Subsequent internal crosslinking interactions, e.g. via H-bonds, click-reactions, or metal complexation, can preserve the collapsed chains in these conformations.27 Experimentally, single-chain collapse is conducted at high dilutions (<10−5–10−6 M).26 The internal crosslinking interactions then preserve the collapsed chains in the so reached conformations.27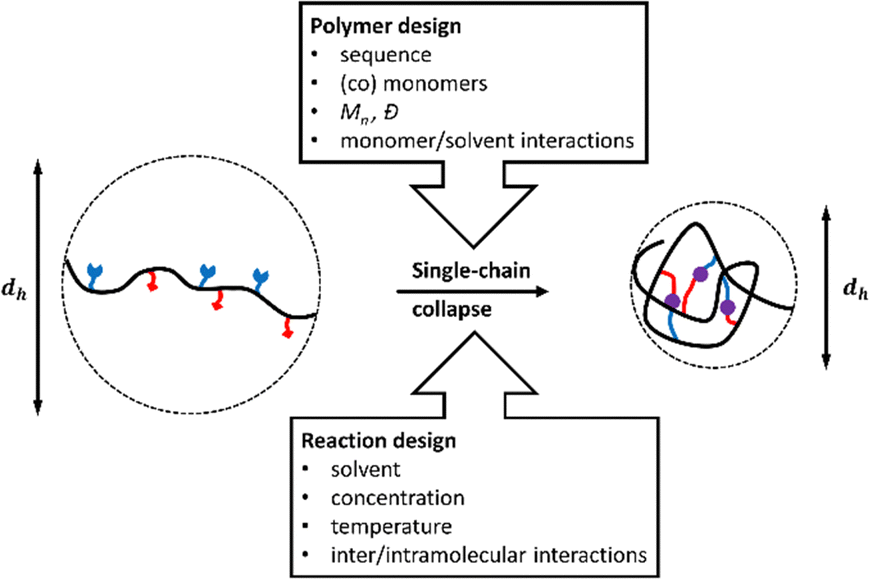 | ||
| Fig. 2 Factors influencing the structure of compartimentalized single-chain nanoparticles (SCNPs) by collapse of a single polymer chain, often with a reduction of the hydrodynamic diameter dh. | ||
Critical aspects to control formation of compartments inside the SCNPs are therefore (a) the design of the polymer that is folded into a SCNP, controlling its shape, internal structure, solubility, biocompatibility, or stimuli-responsive behaviour; (b) the conditions of chain-collapse (such as solvens and temperature), and the subsequent crosslinking methods to “freeze-in” the previously generated structures. As the processes involved during SCNP formation are comparable to the folding of peptides into secondary structures with the additional quest to prevent a subsequent aggregation of the folded macromolecules, the final shape of the SCNP as well as the internal compartments and functionality are strongly influenced by the architecture of the individual polymer chains, the solvent that is used during the collapse reaction, and the solvent/monomer-interactions of the individual monomer units during chain-collapse and -crosslinking. It is evident that, e.g. random copolymers without specific solvent/monomer interactions are expected to result in more random, sparse structures in the final SCNP, whereas the use of sequential, diblock-/triblock- or sequence-specific copolymers, can easily yield compartmentalised SCNPs, as shown in Fig. 3.28
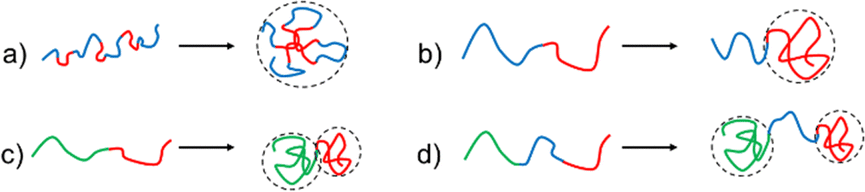 | ||
| Fig. 3 Examples of compartment-formation achieved by different types of copolymers, leading to single-chain nanoparticles. (a) A random copolymer collapsing into a globular or random shaped SCNP; block copolymers collapsing into (b) a tadpole-shaped SCNP, (c) a Janus SCNP, and (d) a dumbbell-shaped SCNP. Blue: non-crosslinking polymer segments. Red and green: potentially crosslinking polymer segments. | ||
Control over the copolymers’ architecture during synthesis is crucial to reach the desired SCNP with a specific compartmentalised structure. Often living polymerisation techniques are used to this purpose, such as controlled radical polymerisations like atom transfer radical polymerisation (ATRP),29 nitroxide mediated polymerisation (NMP),30 reversible-addition–fragmentation chain-transfer (RAFT) polymerization,24,31 or Ru-catalysed living radical polymerisation;32 as well as other methods like living anionic and cationic polymerisation,19c,33 group transfer polymerisation (GTP)34 or ring opening metathesis polymerisation (ROMP).35 Together with the placement of the individual monomers, these polymerisation techniques reach low polydispersities and high molecular weights (>20 kDa up to 100 kDa or more), all resulting in precisely engineered sizes (5–20 nm) and chemical compositions of the SCNPS. Bioinspired SCNPs are especially attractive for biomedical application because of their biocompatibility when embedding polypeptides and carbohydrates as backbone materials, optionally generated by biosynthesis,36 which can further be modified then deviating from their natural composition and structure to yield highly functional SCNPs.37 Polypeptides can also be produced synthetically, by performing ring opening polymerisations of N-carboxyanhydrides.38
2.1. Collapsing methods
The condition upon single-chain collapse can engineer a chemical functionality inside/around the SCNP to form different types of particles from one and the same polymer chain. In this chapter we are highlighting some basic principles of the analysis and formation of SCNPs.Firstly, preventing intermolecular crosslinking and aggregation is central and must be ensured during the collapse and crosslink procedure of SCNPs, since only then the intramolecular interactions will result in (stable & useful) single-chain nanoparticles. To this end, various methods have been developed, all having in common that they require high dilutions in the range of <1 mg mL−1 for polymers, equal to ∼10−6 M for reactive groups along the polymer chains.26,39
Generating SCNPs on a reasonable scale with high reproducibility requires (a) fast and efficient reactions under high dilutions and (b) defined experimental conditions during single-chain collapse. Often SCNP syntheses are conducted using a continuous-addition protocol as developed by Harth et al.,17,30a,39b,40 where a concentrated polymer solution is slowly dropped into a solution with the reactants under vigorous stirring, see Fig. 4b. The droplet entering the reaction solution thus should react efficiently resulting in intramolecular crosslinks only. Since all reactive groups along the polymer chains of this droplet are so completely consumed, the reactive groups of the next droplet will again react only intramolecularly under these diluted conditions allowing syntheses under much higher final concentrations and yields in the gram-scale.41
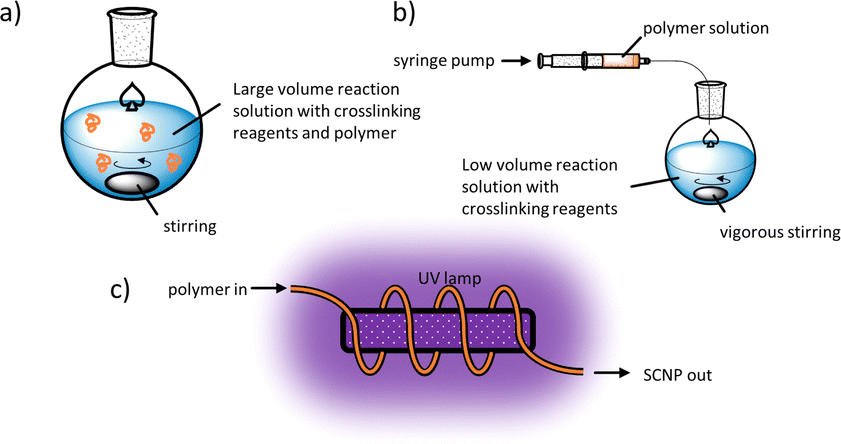 | ||
| Fig. 4 Illustration of experimental procedures for the synthesis of SCNPs using (a) a large volume reaction solution with a low concentration of polymer, (b) the continuous-addition protocol, and (c) a photochemical flow reactor. | ||
Upscaling of SCNP-synthesis is possible with flow reactors, wherein the reaction is taking place along a tube through which a solution is flowing, addressing larger scale syntheses. As reported by Barner-Kowollik and Diesendruck et al.,42 a polymer that contains photodimerizing groups can flow through a tube that is wrapped around an ultraviolet (UV) lamp, as depicted in Fig. 4c, to effect photo crosslinking after single-chain collapse. This way SCNPs can be synthesised with shorter irradiation times and higher conversions than in a comparable reaction in a batch reactor, making this method quite efficient.
2.2. Crosslinking methodologies to stabilise formed SCNPs and their compartments
The crosslinking strategies used to stabilize the initially collapsed SCNP into the final, irreversible structure are crucial in maintaining the initially, collapsed polymer conformation. Both, covalent and non-covalent crosslinks can be applied. In addition, dynamic crosslinks, usually supramolecular or dynamic covalent bonds, allow for an additional responsive element inside the SCNPs, potentially allowing a later chain reorganization or “error” corrections of eventually misfolded compartments.Thus supramolecular interactions such as hydrogen bonds are effective to form SCNPs, using ureidopyrimidinone29a,35a,43 or Hamilton wedges with cyanuric acid derivatives.44 Chiral benzene-1,3,5-tricarboxamides (BTA) have emerged as a noncovalent crosslinking method that do not only provides hydrogen bonds as linking units, but embeds a chiral compartment inside the SCNP core, similar to α-helices in proteins.20b,22,29d,35d,45 β-Hairpin-like single-chain structures can be formed using supramolecular quadrupole π-stacking interactions between electron-rich benzene moieties and electron deficient pentafluorobenzene moieties in an ABC-block copolymer with a polystyrene, and a polypentafluorostyrene block, separated by a spacer block.46 Other supramolecular interactions, such as metal complexes can be used for the crosslinking of a single polymer chain with the ligands covalently bound to the polymer chain, thereby utilising the complexation of a metal ion.23,35f,47 This interaction can also be used to incorporate a metal catalyst inside the SCNP to synthesize an enzyme analogue.35e,35f,47f,47g,48
Less dynamic but more direct is the use of covalent crosslinks, which fix the internal nanostructures even if external properties like solvent quality are changed. Besides simple organic reactions, like nucleophilic substitutions,30c ester/amide formations,21b,25,36b,37b,49 or polymerisations of vinyl groups33,50 and cyclic esters,51 “click” reactions52 have been proven useful due to their high efficiency under mild (even aqueous) reaction conditions. The copper catalysed azide–alkyne click reaction (CuAAC) became one of the most important methods used by us for covalently crosslinking SCNPs, based on its large free reaction enthalpies (>50 kJ mol−1), resulting in high yields, and the insensitivity to many other functional groups and solvents used therein.53 We19d–g,52,54 and others40,55 have used the CuAAC extensively to stabilize the collapsed polymers and transform them into the final SCNPs under mild conditions. The wide variety of possible covalent reactions then allows the subsequent incorporation of functionalities even after the completed single-chain collapse reaction, like the incorporation of a specific number of dye molecules,19e or the embedding of responsivities to external stimuli19f to change the structure of the SCNPs. This further allows synthesis of covalently crosslinked SCNPs that can be dynamically opened and closed by photothermal-effects, transferring energy to the outside of the SCNP-shell.56 Further mild crosslinking chemistries are photodimerizations29c,35g,42a,57 and disulfide bridges35c,58 reaching dynamic covalent SCNPs that are especially interesting for biomedical applications since their responsivities can increase their drug delivery efficiency and biodegradability.50
3. Compartmentalised SCNPs
To generate compartments located at the outside of an SCNP block copolymers can be used. Depending on the type, order, and functionality of the respective blocks differently shaped SCNP systems can be achieved, resulting in phase segregating properties of the different parts of the SCNPs. Examples for compartmentalised SCNPs are shown in Fig. 1, including their chemical properties, collapse methodologies, and special features. As depicted in Fig. 3b, collapsing a block copolymer with one non-reactive, solvophilic block and s solvophobic and reactive block, tadpole-shaped SCNPs can be synthesised19c resembling the structure of surfactants. Because of their exposed blocks such SCNPs can further aggregate into micelle- or vesicle-like superparticles via their exposed blocks, relying on Flory–Huggins principles.29c,59 Block copolymers with two reactive blocks can collapse into two-sided “Janus” SCNPs (see Fig. 3c).35h,57i Satoh et al. showed that also blocks with the same crosslinking functionality will collapse into Janus SCNPs because of the higher reactivity of proximal sites.19b Adding a non-reactive spacer block in between the crosslinkable blocks then generates dumbbell-shaped SCNPs as depicted in Fig. 3d.49a,60Forming compartments in the inner core of the SCNPs usually starts with the formation of “unimolecular micelles”, similar to single-chain nanoparticles, but formed without subsequent crosslinking chemistry. They can form core–shell-like structures in water with hydrophilic sidechains in the shell and hydrophobic sidechains in the core, as depicted in Fig. 5a. Their hydrophilicity in the shell can be achieved by using poly ethylene glycol (PEG)32,61 or ionic sidegroups.62 The core-building, hydrophobic sidechains can be introduced by using either aliphatic32,61c,62 or fluorinated sidechains,61a,61b which in turn forms compartments by hydrophilic/hydrophobic solvent interactions. Those assemblies have been used as antimicrobial compounds61c and are interesting as drug delivery systems, covalently binding or encapsulating drugs inside their hydrophobic core,62 as controlled drug-release systems, similar to classical micellar structures (Table 1).61a
 | ||
| Fig. 5 (a) An amphiphilic random copolymer folding into a unimolecular micelle in water. (b) An amphiphilic random copolymer with crosslinking units collapsing and crosslinking into a single-chain nanoparticle. | ||
| Chemical Structure | M n /kDa | Collapse Methodology | D h /nm | Special Features/Applications | Ref. |
|---|---|---|---|---|---|

|
10–40 | Hydrophilic–hydrophobic interactions in water (unimolecular micelles) | 12–15 | Two hydrophilic units, single-fluorophore encapsulation via endgroup functionalisation | 62 |
|
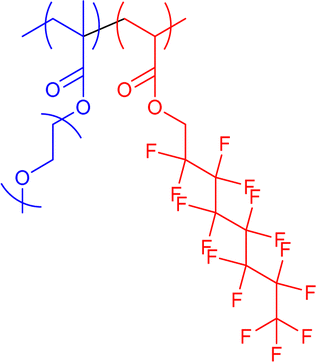
13–44 | Hydrophilic–hydrophobic interactions in water (unimolecular micelles) | 5–8 | Encapsulation of drug cargos, thermoresponsive behaviour, invertible micelle upon solvent exchange to hexafluorobenzene | 61a,61b,63 |
|
320–460 | Anthracene photodimerization in THF/hexane mixtures | 10 | Core–shell structure formation in non-aqueous system | 35g |
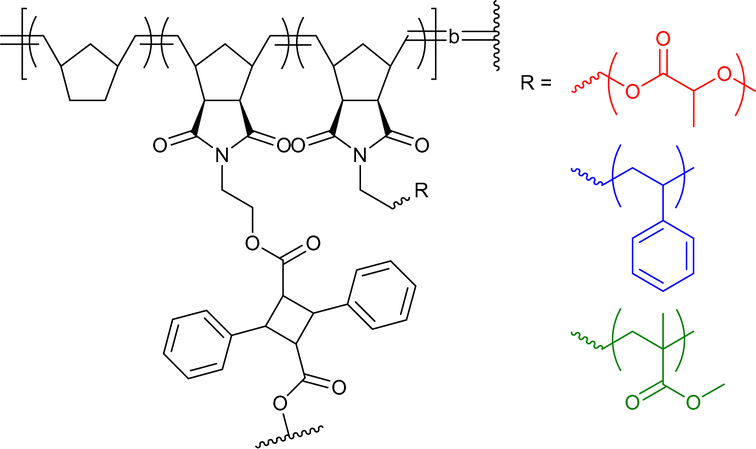
|
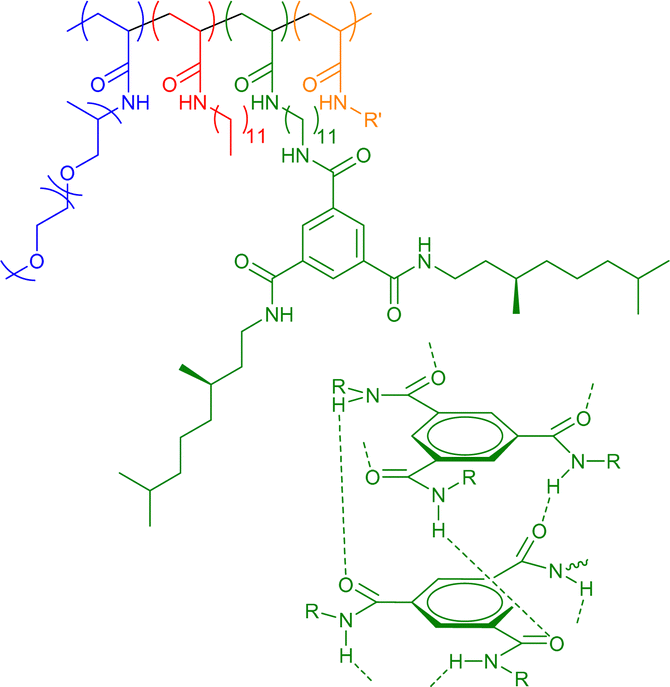
8–93 | Chiral H-bonding of BTA in water | 10–14 | Helical structure in SCNP core, incorporation of dyes, catalysts, and other functionalities (R’ in structure) | 45e,45f,64 |
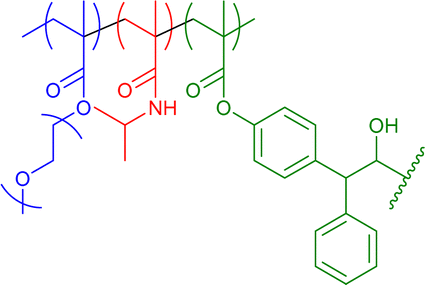
|
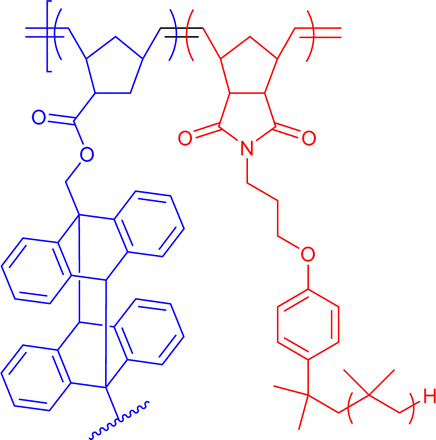
300–430 | Cinnamic acid photodimerization in THF | 6.6 | Differently miscible blocks, block-order-dependent nanostructure | 35h |
|
18–29 | Benzophenone photocrosslink in water at different temperatures | 12–16 | SCNP size dependent on crosslinking temperature, thermoresponsive swelling-deswelling of core | 65 |
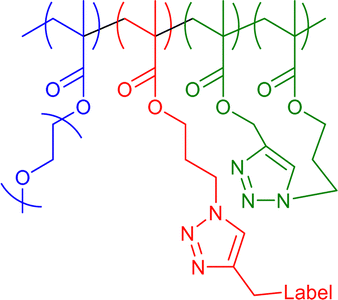
|
36–44 | Internal CuAAC in water | 5–10 | Covalent encapsulation of multiple fluorescent and radical labels, thermoresponsive swelling-deswelling of core | 19f,19g |
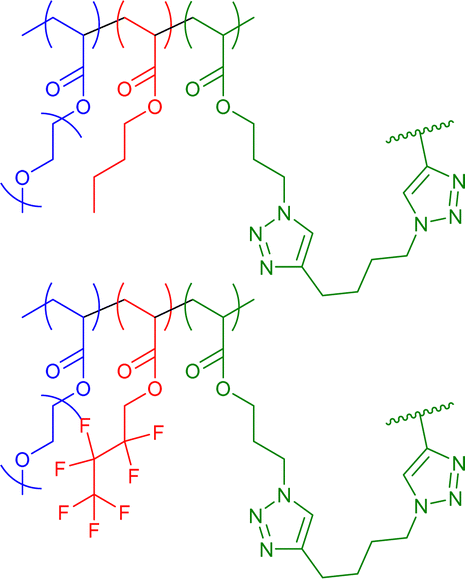
|
13–19 | CuAAC in water with external bivalent crosslinker | 6–12 | Differently hydrophobic environments for either aliphatic or fluorinated SCNP core, specific encapsulation of aliphatic/fluorinated cargos | 54 |
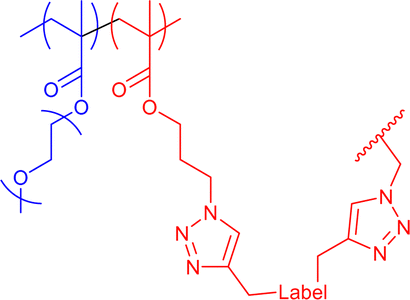
|
35 | CuAAC in water or THF with external bivalent and functional crosslinker | 6–12 | Internal structure dependent on solvent medium and crosslinker bulkiness, covalent encapsulation of multiple crosslinking fluorescent and radical labels | 19e |
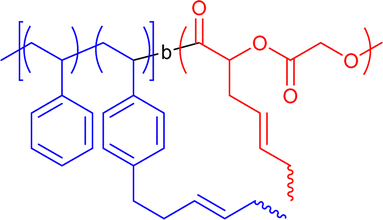
|
20–23 | Intrablock crosslinking via olefin methathesis in DCM | 6–8 | Janus-shaped SCNP, one-shot crosslinking of spatially close reactive sites, lamellar phase separation | 19b |
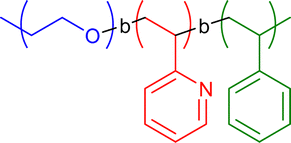
|
n.a. | Co complexation in o-dichlorobenzene | 12 | Tadpole structure with two tails, magnetic and catalytically active cobalt generation upon thermolysis | 23 |
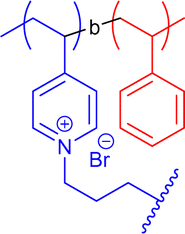
|
475 | Amine quaternisation in DMF | 34 | Tadpole SCNP, head can actively graft on inert surfaces | 66 |
The synthesis of crosslinked, compartmentalised SCNPs is based on the same mechanism as the formation of unimolecular micelles but with an additional crosslinking reaction to retain the compartmentalised structure even after concentration or solvent changes. As depicted in Fig. 5b, the different interacting individual segments can lead to the formation of core–shell structured SCNPs. Ideally, the crosslinking moieties are solvophobic so that the inner core of the SCNP is stabilised, with the crosslinking reactions now taking place only in the core, resulting in stronger compartmentalised structures.35d,35g,67
As mentioned above the solvents in which the collapse and crosslinking reactions take place have a major influence on the internal compartmentalised structures of the SCNP. A solvent or solvent mixture that shows good quality for all segments of the precursor polymer, including the crosslinking units, will result in non-controlled, sparse SCNPs,35d,35g while a solvent with a poor quality for all segments can result in multi-chain aggregation instead of the collapse of single chains.35g Exemplary, Simon et al. showed that the single-chain collapse of a comb-copolymer with poly-isobutylene (PIB) sidechains (Mn = 3.5 kDa) and anthracene crosslinking units resulted in different compartimentations inside the formed particles, depending on the solvent quality.35g The photodimerization of the anthracene units in pure tetrahydrofuran (THF), a good solvent for both monomer units, resulted in low conversions and sparse SCNPs, while the same reaction in pure hexane, which is only a good solvent for the PIB units, resulted in multi-chain particles. Only a mixture of both solvents was able to yield high conversions and well-defined core–shell structured SCNPs.
We19e used amphiphilic polymers to prepare core–shell structured SCNPs, reaching different compartments depending on the solvent and crosslinker used during single-chain collapse. The precursor polymer, a random copolymer with hydrophilic PEGMA monomers and hydrophobic azide-functionalised monomers, allows crosslinks via CuAAC in mixture with hydrophobic, bivalent alkynes with and without functionality (radical TEMPO label, fluorescent aBOD-label, octadiyne) for a subsequent analysis of the SCNPs (see Fig. 6).
 | ||
| Fig. 6 Concept to create differently structured SCNPs by adjusting the interactions of comonomers during solvent-induced collapse. Reproduced with permission from Wiley VCH, copyright © 2023 (open access CC BY-NC-ND 4.0).19e | ||
Chain collapse was performed by CuAAC in water and THF as selective and non-selective solvent, respectively. It was hypothesised that the reaction in water would lead to core–shell structured SCNPs because of the amphiphilic nature of the precursor polymer and the hydrophobicity of the crosslinkers, while the same reaction in THF, a solvent which is almost equally well solubilizes all monomers of the polymer and crosslinkers, would lead to SCNPs without defined internal structures.
Extensive analyses via CW-EPR and time resolved fluorescence spectroscopy revealed that the internal structure is strongly guided by the polarity of the solvent, resulting in formation of dense hydrophobic cores, when the collapse reactions are performed in water. Using solely the labelled crosslinkers the formation of a core–shell structure was reduced, whereas mixtures of the rigid label-crosslinkers with the flexible octadiyne resulted in the desired core–shell structures as depicted in Fig. 7.
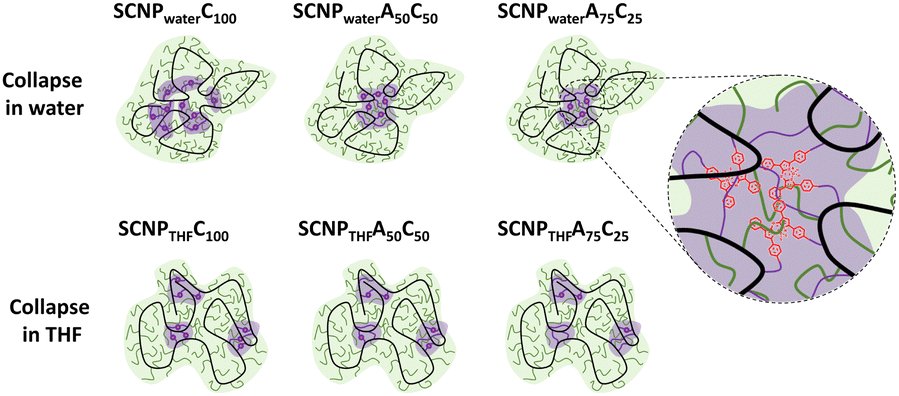 | ||
| Fig. 7 Schematic illustration of the collapse-solvent and crosslinker-composition-dependence of the internal nanostructure of amphiphilic SCNPs (green: hydrophilic regions, violet: hydrophobic regions). Only those SCNPs formed a core–shell structure which were crosslinked in water with flexible crosslinkers. Reproduced with permission from Wiley VCH, copyright © 2023 (open access CC BY-NC-ND 4.0).19e | ||
The relation between the flexibility and length of a crosslinker and the resulting nanostructure of the SCNP is known in literature,68 caused by the formation of long range interactions for longer and more flexible crosslinkers, resulting in more compact and dense SCNPs directly linked to the desired applications of SCNPs. We embedded fluorescent dyes for pump–probe photoacoustic (PA) imaging as crosslinkers, so exponentially increasing PA signals, while the unstructured SCNPs showed only weak and non-exponential PA signals.
We further tuned the hydrophobicity of the SCNP core by changing the fraction of hydrophobic units in a similarly designed SCNP system (see Fig. 8) to reach a closer mimicry of proteins and enzymes, modifying with (i) purely hydrophobic, aliphatic sidechains and (ii) hydrophobic, fluorinated sidechains.54
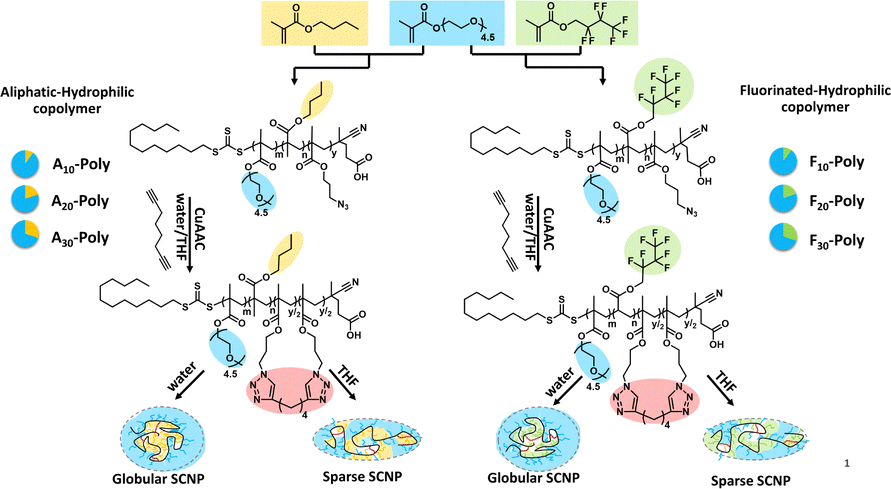 | ||
| Fig. 8 Synthesis of random copolymers from different methacrylate monomers, displaying different monomer/solvent interactions. Crosslinking after single-chain collapse generates single-chain nanoparticles with different compartments. Reproduced and adapted with permission from the Royal Society of Chemistry, copyright © 2024 (open access CC BY 3.0). | ||
By tuning the composition of the copolymer (variation of the hydrophobic (aliphatic/fluorinated) and hydrophilic monomers) not only core–shell structured SCNPs but different compartments were formed inside the aliphatic and fluorinated cores. Solvochromic dyes were added as probes, hydrophobicity-selective via an aliphatic or a fluorinated sidechain, proving by time resolved fluorescence spectroscopy a stronger affinity of the fluorinated dye to the fluorinated core. Similar, an aliphatic EPR label was found to preferably enter the aliphatic core compared to the fluorophilic core. Hence, the different types of cores effected their affinity to bind external hydrophobic probes, mimicking the selectivity of enzymatic structures.
Response of the compartment formation to temperature can be reached via thermoresponsive segments in the precursor polymer chain, e.g. poly(oligo ethylene glycol) methacrylates (PEGMA) or poly N-isopropyl acrylamides (PNIPAM). These polymers show lower critical solution temperature (LCST) behavior in water, becoming insoluble in water above the cloud point temperature Tcp. As demonstrated by Lu et al.,65 at temperatures well below Tcp of those segments, the SCNP synthesis will result in sparse non-compartmentalised SCNPs, whereas at temperatures above Tcp, core–shell structured SCNPs with a dense core will be obtained as depicted in Fig. 9. Conducting the SCNP synthesis at medium temperatures, a core–shell structure is generated, but in a less dense state. Swelling of the core with water leads to a thermoresponsivity of the core itself.
 | ||
| Fig. 9 Simulated chain conformations of a thermoresponsive precursor polymer chain at the indicated temperatures, resulting in so structured SCNPs upon single-chain crosslinking. This figure has been published in CCS Chemistry (2020, open access); “Controlling the Chain Folding for the Synthesis of Single-Chain Polymer Nanoparticles Using Thermoresponsive Polymers” is available under DOI: https://doi.org/10.31635/ccschem.020.202000190.65 | ||
Using CW-EPR19d and time resolved fluorescence spectroscopy19f we demonstrated that the cloud point temperature of the outer shell of the SCNPs can differ from the that of the internal compartments, which leads to contraction of the SCNP system at increased temperatures, still below the macroscopically observed Tcp. As SCNPs are monomolecular confined nanogels, they swell with the dispersing solvent.69 Due to the higher hydrophobicity inside the core of the structured SCNPs compared to their outer shells, the core shows lower and less defined cloud point temperatures, in turn leading to deswelling of the cores by increasing the temperature, still keeping the SCNPs dispersed. This leads to the previously mentioned contraction of the SCNPs and decreases of dh, as followed by increased of diffusions, e.g. by DOSY-NMR spectroscopy, as well as decreased rotation correlation times in CW-EPR-spectra. Polarity and density changes inside the SCNP cores which follow this contraction play an important role in PA imaging, wherein absorbed NIR light generates heat by pulsed laser excitations of fluorescent dyes. Each laser pulse then changes the core of the SCNP, as depicted in Fig. 10, in turn modulating the photophysical properties of the fluorescent dye, resulting in fluctuating signals. So far, this behavior, an interesting contrast mechanism for PA imaging, has only been observed in our SCNPs, caused by the SCNPs’ internal nanostructure.
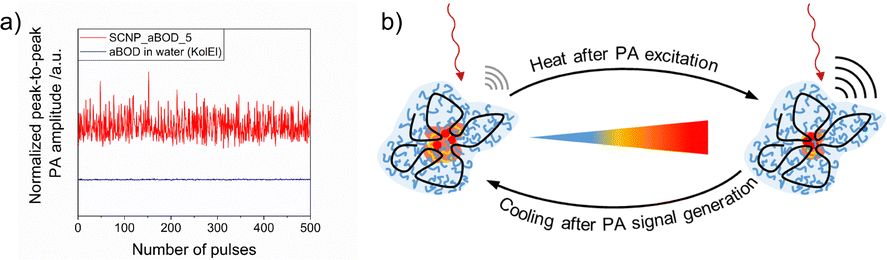 | ||
| Fig. 10 (a) Fluctuating PA signal of the thermoresponsive SCNPs. (b) Schematic interpretation of the thermoresponsive behavior. Reproduced and adapted with permission from the Royal Society of Chemistry, copyright © 2023.19f | ||
Compactions of SCNPs as described rely on different thermo-responsivity of core and shell of the SCNPs. A different approach to switch between compacted and expanded SCNPs was followed by Barner-Kowollik et al. by introducing cis/trans-switchable azo-dyes as crosslinkers.25 Those dyes are known to reversibly switch between their cis- and trans-conformation by irradiating with different wavelengths of light. By irradiation with 620 nm light, compactions of the SCNPs were observed by forcing the azo-dye into the cis-conformation, while subsequent irradiation with 415 nm light switched the dyes back into the trans-conformation, in turn expanding the SCNPs.
Other principles to generate segregated regions inside SCNPs are liquid crystals,45f,64ae.g. by introducing stilbenes or azobenzenes as side chains to the precursor polymer, resulting in compartmentalised SCNPs upon crosslinking. Inside the relatively unordered SCNPs, those groups will still be able to self-assemble and form (now dynamic) ordered structures which segregate from the polymer backbone.57f,59c Thus supramolecular motifs generate internal compartments inside SCNPs via the benzene-1,3,5-tricarboxamide (BTA) group introduced by Meijer and Palmans.45f,64a BTA molecules form supramolecular assemblies that build helical stacks with threefold intermolecular hydrogen bonds, resembling α-helices of proteins. When BTA molecules are part of a precursor polymer that is dissolved in water, they will self-assemble inside the SCNP's interior, forming intramolecular crosslinks of the polymer chain, segregated from other parts of the polymer to modulate the overall shape of the resulting SCNP.20b,22,29d,35d,45a,70 These helical stacked structures can be used as platforms for catalytic sites, enabling enzyme-like internal structures which significantly increases catalytic activities.45b–d,71
Stepwise folding of individual chains segments of a precursor polymer is the main approach to reach externally compartmentalised SCNPs, like Janus- or dumbbell-shaped SCNPs from block-copolymers after single-chain collapse and crosslink, as described above.31b,43c,49a,57i This process tunes the nanostructure and shape of SCNPs originating from random copolymers, e.g. by first preorganising a precursor chain into single-chain ring structure via a hetero Diels–Alder reaction of the endgroups, and then crosslinking the main chain reactive groups via click-chemistry, as shown by Pomposo et al.55f This process resulted in significantly stronger compacted SCNPs, compared to the SCNPs, which were obtained by the conventional, non-preorganised, precursor polymer.
4. Characterization of SCNPs and their internal compartments
The characterization of the internal compartmentalisation of SCNPs is challenging due to the usually rather non-defined nature of the SCNPs interior, the inherent dynamics of the SCNP, and polydispersity-effects of the precursor polymer together with the formation of mixed phases inside the SCNPs, or chemical changes of the crosslinking moieties. Traditional methods focus on analysis methods to describe their size and shape, including chromatography, microscopy, and scattering techniques. Collapsing linear precursor polymer chains into SCNPs is usually followed by size reductions that can be measured as hydrodynamic diameters dh.39b,72 While a non-crosslinked precursor chain is potentially able to reach its full length, the (covalently) crosslinked SCNP is normally unable to change its conformation and is trapped in its collapsed position, resulting in reduced hydrodynamic diameters compared to the precursor chain, as depicted in Fig. 2. This reduction in dh, usually measured by gel permeation chromatography (GPC) dynamic light scattering (DLS) or diffusion-ordered NMR spectroscopy (DOSY-NMR), can be a proof of a successful single-chain collapse with solely intramolecular crosslinks and the absence of intermolecular interactions, that would lead to increased values of dh.Water-based GPC measurements have been used to prove the localization of hydrophobic parts of the system inside the SCNP-core.19e,19g After single-chain collapse, elution times should be shifted to higher retention times in SEC, indicative of lower apparent molecular weights, because of the reduced hydrodynamic diameter of the SCNP after crosslinking. We in some cases observed an opposite trend with a shift to lower retention times or seemingly larger hydrodynamic diameters. Contrary to results from DLS and DOSY-NMR measurements, indicative of enthalpic contributions in addition to the expected entropic contribution during GPC separation.72 This in turn hints to the formation of compartments, when comparing a precursor polymer and the resulting SCNP, as there is a significant, unexpected polarity change after the collapse reaction by hiding the non-polar parts of an amphiphilic polymer in its hydrophobic core.
Another indirect method to prove the location of the hydrophobic parts of an amphiphilic copolymer in the core of the resulting SCNPs investigates changes in the LCST behavior.19e,19g The cloud point temperatures Tcp of LCST systems are sensitive to changes of the polarity of the sample, resulting in lower Tcp values when hydrophobic units are contributing to the solubility of the sample. Comparing Tcp values of the precursor polymers to those of the resulting SCNPs shows increased Tcp values for the SCNPs. Hence, solubilities increase after single-chain collapses, indicating, that the hydrophobic parts of the polymers are hidden in a hydrophobic core.
Although methods like SEC, DLS, and DOSY-NMR can give good information about the hydrodynamic diameter of a SCNP and its relative change after single-chain collapse, these methods usually lack information about the SCNP's shape, since dh is defined as the diameter of a perfect hard sphere with the same diffusion properties as the SCNP, ignoring its actual shape. Scattering techniques, such as small angle X-ray or neutron scattering (SAXS, SANS), and static light scattering (SLS), can be helpful as powerful characterization methods, providing information about the dimensions of the SCNP via its radius of gyration rG, but additionally about its shape, e.g. if the SCNP is sparse, globular, stick-like, or compartmentalised.20b,47a,61b,67,73
Besides size and shape characterization in solution via chromatographic and scattering techniques, methods like atomic force microscopy (AFM) and transmission electron microscopy (TEM) have been used to visualize and analyze SCNPs in the dry state. AFM is a universal method that is theoretically able to visualize any type of SCNP. Nevertheless, its exact interpretation and comparison to other techniques like DLS or DOSY-NMR can be challenging, since the SCNPs tend to lose their shape in the dry state. This, in addition to poor resolutions with SCNP's sizes often being below the widths of the AFM cantilever tips, can lead to unreliable results.29a,35a,74 TEM results in more accurate results for SCNPs when compared to other techniques.59b However, TEM is not universally applicable for all SCNPs, limited by heavy atoms required for achieving good contrasts. Although direct visualizations of the internal compartments using microscopic techniques are barely possible due to insufficient resolutions, Pomposo et al. and Meijer et al. were able to use TEM to visualize the formation of metal-based local domains inside an SCNP (see Fig. 11a);48a,75 and high-resolution AFM to visualize differently dense parts of the SCNPs (Fig. 11b).29a
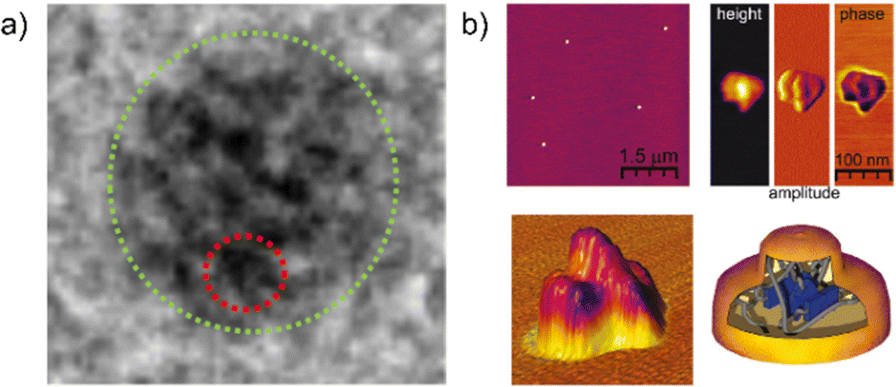 | ||
| Fig. 11 (a) TEM image of local domains of a Cu-folded SCNP. Reprinted with permission from the American Chemical Society, copyright © 2018 (open access).75 (b) High resolution AFM image of a SCNP showing a dense core and a less dense shell. Reprinted with permission from the American Chemical Society, copyright © 2010.29a | ||
NMR-spectroscopy has been widely used to indirectly address compartment formations in SCNPs by analysis of differences in mobility between the inner/outer parts via half-widths of resonances in 1H-NMR spectra,32 as a qualitative measure for the mobility of the respective chemical entities. Usually freely moving groups will show a sharp half-width, while hindered movements will result in broad signals. Hence, the chemical groups that are in the core of a compartmentalised SCNP will appear broadened in comparison to those in the less confined shell. More complex (2D)-NMR techniques, e.g. nuclear Overhauser effect spectroscopy (NOESY)19b,46,62a or T2-relaxation68 can give detailed information about the formation of compartments with specific chemical groups and their density inside the SCNP, in addition to STD-NMR.76
CW-EPR-spectroscopy on internalised radical (TEMPO) and time resolved fluorescence spectroscopy on fluorescent labels19d,e,g were used by us to directly reveal internal compartments inside SCNPs. Thus CW-EPR spectroscopy proved the formation of confined cores inside the SCNPs in which the TEMPO-labels were embedded, by probing spin-exchange between clustered TEMPO-molecules inside the compartment. As depicted in Fig. 12a and b Heisenberg exchange coupling had to be included in the simulations of the spectra, an effect occurring when the distance between two or more radical labels is below 1 nm, indicating high local concentrations of the labels. This proved the agglomeration of the labels in a confined area of the SCNP, sized smaller than 1 nm. Further concentration-dependent measurements revealed that the labels were located inside the SCNP's core, and not on its surface, as shown in Fig. 12c and d. If the labels were located in the outer shell, collisions of individual SCNPs at high concentrations would have led to at least minor changes of the shapes of the spectra, which were found to be concentration independent. (Time resolved) decay associated fluorescence spectroscopy (DAS) also represents a powerful method to understand compartmentalization in SCNPs, and was used to further confirm the results of the CW-EPR spectra by showing comparable effects, as shown in Fig. 13. Whereas a non-SCNP-bound dye (aBOD) in water fluoresced in a single exponential decay with a short fluorescence lifetime, covalently binding the same dye to a linear polymer chain created two additional decays with fluorescence lifetimes in the ns-range now embedded in polymer regions, that could form hydrophobic environments with less contact of aBOD to the fluorescence quenching water. In addition, the SCNP-bound aBOD molecules now showed the formation of excitonic couples. Similar to the Heisenberg exchange in the CW-EPR spectra, those couples only occur when two or more dye molecules are in close proximity, again proving the formation of confined structures inside the SCNPs with high local label concentrations. Other fluorescence techniques follow the formation of SCNPs and their confined structures, via Förster resonance energy transfer (FRET) systems inside SCNPs.77 With this FRET system, the spatial proximity of the functionalities inside the SCNP can be proven.
 | ||
| Fig. 12 CW-EPR spectroscopy of SCNPs. (a) Spectra revealed Heisenberg exchange coupling of individual TEMPO labels, included to the spectral simulation for a perfect fit. (b) Schematic interpretation of the Heisenberg exchange coupling. (c) Concentration independent shape of the spectra proving the location of the labels inside the SCNPs, and not on their surface, as depicted in (d). Reproduced and adapted with permission from Wiley VCH, copyright © 2020 (open access CC BY 4.0).19g | ||
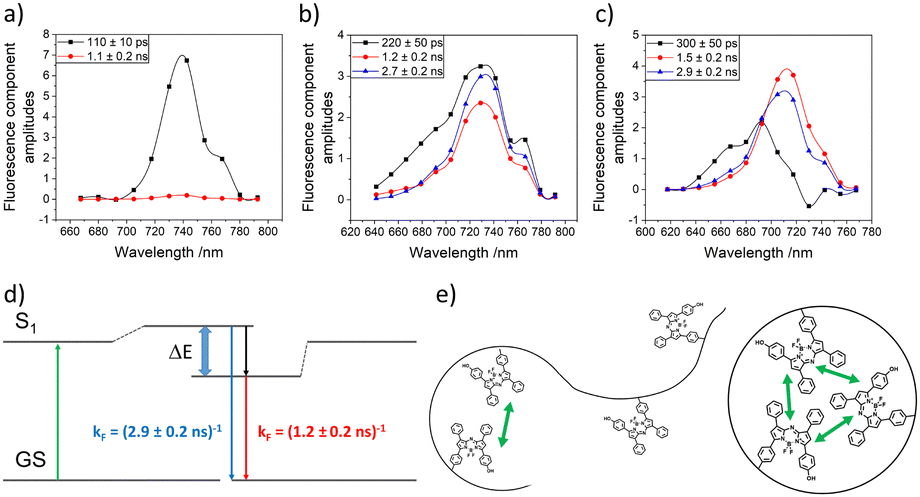 | ||
| Fig. 13 Decay associated fluorescence spectra of (a) aBOD in water (KolEl), (b) the aBOD labelled linear polymer, and (c) the aBOD labelled SCNP. (d) Jablonsky diagram of the excitonic coupling of multiple aBOD molecules. (e) Schematic interpretation of the nanostructure of the aBOD labelled linear polymer and SCNP. Reproduced and adapted with permission from Wiley VCH, copyright © 2020 (open access CC BY 4.0).19g | ||
5. Applications of SCNPs
Single-chain nanoparticles have various applications that arise from their extremely small size, internal and external compartments, and specific functionalities, reminiscent of catalytic sites. Thus, just like enzymes, SCNPs can be designed to act as catalysts with high catalytic activity and specificity.16 Their enzyme-like structure is also an important factor in their application for biomedical applications.17 Next to these two main applications, SCNPs can also be used for detecting specific chemicals,35b,78 antifouling,38,61c catching of carbon nanotubes,57g or as surfactants.59a5.1. SCNPs as enzyme mimetic catalysts
Enzymatic reactions are usually superior in reaction speed, specificity, and activity under ambient conditions.79 Inspired by those reactions, artificial receptors and catalysts, such as macrocyclic compounds, star or helical polymers, dendrimers, micelles, vesicles, or polymerosomes have been designed trying to copy enzymes,16 imitating the unique internal structure of enzymes. Enzymes provide specific local environments, characterised by, e.g. hydrophobic, compartmentalised cavities which are stabilised by H-bonds, hydrophobic assemblies or disulfide bonds. Often also so-called cofactors are localised in those compartments, which additionally effect the catalytic activity. SCNPs are conceptually synthetic analoga to enzymes, based on collapsed/folded single polymer chains providing compartments with specific local properties, e.g. hydrophobic, confined environments, or the opportunity to bind catalytic sites.15a Meijer et al. demonstrated via CW-EPR measurements that especially the hydrophobic environment – or rather the retardation of water from those compartments – are crucial for their catalytic activity.45d Similar to enzymes, catalytically active SCNPs are able to provide high selectivities for reactions by providing hydrophobic compartments and confined spaces to which specific compounds can bind and react with each other, even with the possibility to bind cofactors.47g,48a,71c,80 Both, organic as well as metal-complex catalysts can be incorporated into the hydrophobic compartments inside SCNPs that can catalyse, e.g. the reduction of ketones,71a,73a oxidation of alcohols,71c aldol reactions,71b,71d,81 Glaser couplings,48a,82 Sonogashira and Suzuki couplings,47f,81,83 CuAAC,35e,35f,80 phenol hydroxylations,47g photocatalytic reactions,19a,71f,84 benzoin condensations,47h or intramolecular hydroaminations,85 even in aqueous environments, in which some of those reactions are normally poor in yield or conversion. One advantage that SCNP-based, enzyme-mimetic catalytic systems have over their natural analoga is their stimuli-responsive behaviour, allowing to recycle the catalytic system while maintaining the catalytic activity.71b,86 Biocompatibility allows their introduction into living cells, where they can perform their catalytic reactions either alone or together with natural enzymes.35f,55g,71f Hence, it is possible to specifically generate harmful molecules or other components inside of specific cells like tumour tissues, i.e., those cells can be killed specifically without harming healthy tissue.5.2. SCNPs for biomedical applications
Next to their use as carriers of catalytic sites, the main field of interest in the research of compartmentalised SCNPs is in their application for biomedical purposes, providing perfect environments for the encapsulation of drugs or to covalently bind labels as imaging contrast agents. However, the design of SCNPs for biomedical applications is complex due to several basic requirements:87 (i) water solubility; (ii) ability to carry and protect hydrophobic drugs; (iii) non-toxicity; (iv) biocompatibility/stability in biological media; (v) biodegradability; and (vi) tissue specificity. To overcome the limitations in water solubility, the precursor polymers need to be designed to result in SCNPs with a polar shell, compatible with water. Often PEG sidechains are used to provide a proper water solubility,43e,45f,50,61a,61b,88e.g. using (oligo ethylene glycol) methacrylate (OEGMA) monomers as part of the precursor polymer, extended to quarternized amines,89 alcohols,90,91 acrylic acid,92 or sugar carbohydrate backbones.36a,36b,93 PEG based shells not only enable good water solubilities, but provide the SCNP with biocompatibility and low toxicity.43e,45f,51a,61c,71f Next to PEG, also other precursor polymer designs which are based e.g. on PNIPAM,78b acrylic acid,92 or carbohydrates36,93 can provide good biocompatibilities with cell viabilities of up to 10 mg mL−1, hiding toxic entities in the cores without harming any living tissue.Biocompatibility together with the stability of SCNPs in biological media are major issues controlling interactions within biological media, which are complex aqueous mixtures containing salts, glucose, amino acids, vitamins, and natural macromolecular assemblies like enzymes, all able to interact with, and alter the SCNP structure.45f Recent approaches towards biocompatible SCNPs use natural surfaces via carbohydrates, but also proteins and polypeptides have already been modified to be the basis of SCNPs with less harmful interactions with biological media.37,94 To achieve the same biocompatibility and stability on synthetic SCNPs, the “stealth effect” can be used, mostly found for PEG based systems,95 moving through biological media with reduced clearance.
Tissue specificity, e.g. targeting tumour tissue, can so be achieved via the enhanced permeation and retention effect.96 This effect uses the leaky vasculature of tumours, accumulating nanoparticles only there, since the nanoparticles can easily leave the blood vessels through the leaks and enter this tissue. The chemical flexibility of SCNPs can be used to incorporate different functionalities that enable targeting of specific tissues or even specific compartments of a living cell, such as nitric oxide sensors,78b antigens,36b hormones,92 or proteins89 that only bind to specific tissue types. Targeting of specific cell types or cell compartments has been achieved in SCNPs by adding cell compartment specific moieties like Concanavalin A binding glucose,93 by the formation of polyplexes,91a or by adjusting the surface charge of the SCNPs.90b,91b
6. Conclusions
Compartimentized single-chain nanoparticles (SCNPs) are novel and unique polymeric structures with a wide range of applications, especially in enzyme-mimetic catalysis and biomedical methods. There are countless possibilities to create nanosized compartments inside SCNPs for specific applications, precisely designed in view of the material (natural or synthetic), the nature of crosslinking (covalent/non-covalent/dynamic-covalent), their shape (sparse, globular, tethered), and the nature of the formed compartments. The nanosized compartments, coupled to the size of the SCNPs & their dynamics are what makes them unique candidates as catalysts, drugs, or nanooptical entities. Not only can those “subunits” be tuned via their secondary structure, the polymer architecture, and, most prominent, the amphiphilic behaviour, but also aspects of responsivity, such as temperature-induced volume transition, be easily transferred. However, the techniques to catch the dynamic and often ambiguous nature of the internal compartments remains challenging, requiring special and combined microscopic and spectroscopic techniques, a field expanding rapidly, considering 2D-NMR-spectroscopy&EPR-methods, both probing spatially close subunits of the SCNPs, but also classical fluorescence allows to collect information about the dynamics and the binding specificity of compartments inside SCNPs. It is this detailed knowledge which pushes forward the development of SCNPs, their design and synthesis, coupled to the intricate knowledge of physics inside small confinements, as the compartments inside present.Author contributions
Both authors have contributed to the planning, writing, and reviewing process of this article.Data availability
All data are stored in our internal data management system and in the originally, already published papers cited in this Feature Article. Original data (all data files) are available on request, but can be collected and stored (if desired) before acceptance of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the DFG project BI1337/17-1, the project BI1337/18-1 and the “PoliFaces” initiative for financial support.Notes and references
- L. Bar-Peled and N. Kory, Nat. Metab., 2022, 4, 1232–1244 CrossRef PubMed.
- M. H. Schuler, A. M. English, T. Xiao, T. J. Campbell, J. M. Shaw and A. L. Hughes, Mol. Cell, 2021, 81, 3786–3802 CrossRef CAS PubMed.
- P. Bhat, D. Honson and M. Guttman, Nat. Rev. Mol. Cell Biol., 2021, 22, 653–670 CrossRef CAS PubMed.
- J. T. Trinh, Q. Shao, J. Guan and L. Zeng, Nat. Commun., 2020, 11, 3813 CrossRef CAS PubMed.
- Y. Wu, Q. Hu, Y. Che and Z. Niu, Chem. Sci., 2024, 15, 6200–6217 RSC.
- M.-C. Wu and T. Yow Tsong, J. Phys. Soc. Jpn., 2013, 82, 114801 CrossRef.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- J. Wang, B. Li, L. Qiu, X. Qiao and H. Yang, J. Biol. Eng., 2022, 16, 18 CrossRef PubMed.
- A. V. Kabanov and S. V. Vinogradov, Angew. Chem., Int. Ed., 2009, 48, 5418–5429 CrossRef CAS PubMed.
- (a) M. Hoffmann, J. Eisermann, F. A. Schoffmann, M. Das, C. Vargas, S. Keller and D. Hinderberger, Biochim. Biophys. Acta, Biomembr., 2021, 1863, 183681 CrossRef CAS PubMed; (b) M. Hoffmann, D. Haselberger, T. Hofmann, L. Muller, K. Janson, A. Meister, M. Das, C. Vargas, S. Keller, P. L. Kastritis, C. Schmidt and D. Hinderberger, Biomacromolecules, 2021, 22, 3901–3912 CrossRef CAS PubMed.
- R. C. Oliver, J. Lipfert, D. A. Fox, R. H. Lo, S. Doniach and L. Columbus, PLoS One, 2013, 8, e62488 CrossRef CAS PubMed.
- M. Nikzamir, Y. Hanifehpour, A. Akbarzadeh and Y. Panahi, J. Inorg. Organomet. Polym., 2021, 31, 2246–2261 CrossRef CAS.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- V. K. B. Adjedje, E. Schell, Y. L. Wolf, A. Laub, M. J. Weissenborn and W. H. Binder, Green Chem., 2021, 23, 9433–9438 RSC.
- (a) E. Verde-Sesto, A. Arbe, A. J. Moreno, D. Cangialosi, A. Alegría, J. Colmenero and J. A. Pomposo, Mater. Horiz., 2020, 7, 2292–2313 RSC; (b) Y. Shao and Z. Z. Yang, Prog. Polym. Sci., 2022, 133 Search PubMed.
- J. Rubio-Cervilla, E. Gonzalez and J. A. Pomposo, Nanomaterials, 2017, 7, 341–360 CrossRef PubMed.
- A. P. P. Kröger and J. M. J. Paulusse, J. Controlled Release, 2018, 286, 326–347 CrossRef PubMed.
- S. Wijker and A. R. A. Palmans, ChemPlusChem, 2023, 88, e202300260 CrossRef CAS PubMed.
- (a) K. Mundsinger, B. T. Tuten, L. Wang, K. Neubauer, C. Kropf, M. L. O'Mara and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2023, 62, e202302995 CrossRef CAS PubMed; (b) K. Watanabe, N. Kaizawa, B. J. Ree, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Angew. Chem., Int. Ed., 2021, 60, 18122–18128 CrossRef CAS PubMed; (c) K. Watanabe, S. Katsuhara, H. Mamiya, T. Yamamoto, K. Tajima, T. Isono and T. Satoh, Chem. Sci., 2019, 10, 3330–3339 RSC; (d) A. H. Roos, J. F. Hoffmann, W. H. Binder and D. Hinderberger, Soft Matter, 2021, 17, 7032–7037 RSC; (e) J. F. Thümmler, A. H. Roos, J. Krüger, D. Hinderberger, F. J. Schmitt, G. Tang, F. G. Golmohamadi, J. Laufer and W. H. Binder, Macromol. Rapid Commun., 2023, 44, e2200618 CrossRef PubMed; (f) J. F. Thümmler, R. Maragani, F. J. Schmitt, G. Tang, S. M. Rahmanlou, J. Laufer, H. Lucas, K. Mäder and W. H. Binder, Chem. Commun., 2023, 59, 11373–11376 RSC; (g) J. F. Hoffmann, A. H. Roos, F. J. Schmitt, D. Hinderberger and W. H. Binder, Angew. Chem., Int. Ed., 2021, 60, 7820–7827 CrossRef CAS PubMed.
- (a) G. M. T. Huurne, A. R. A. Palmans and E. W. Meijer, CCS Chem., 2019, 64–82 CrossRef; (b) P. J. M. Stals, M. A. J. Gillissen, T. F. E. Paffen, T. F. A. Greef, P. Lindner, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2014, 47, 2947–2954 CrossRef CAS.
- (a) P. H. Maag, F. Feist, V. X. Truong, H. Frisch, P. W. Roesky and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2023, 62, e202309259 CrossRef CAS PubMed; (b) I. Mohamed Irshadeen, V. X. Truong, H. Frisch and C. Barner-Kowollik, Chem. Commun., 2022, 58, 12975–12978 RSC.
- N. Hosono, A. R. Palmans and E. W. Meijer, Chem. Commun., 2014, 50, 7990–7993 RSC.
- D. Xiang, B. Y. Jiang, F. X. Liang, L. T. Yan and Z. Z. Yang, Macromolecules, 2020, 53, 1063–1069 CrossRef CAS.
- S. Wijker, R. Monnink, L. Rijnders, L. Deng and A. R. A. Palmans, Chem. Commun., 2023, 59, 5407–5410 RSC.
- A. E. Izuagbe, V. X. Truong, B. T. Tuten, P. W. Roesky and C. Barner-Kowollik, Macromolecules, 2022, 55, 9242–9248 CrossRef CAS.
- C. K. Lyon, A. Prasher, A. M. Hanlon, B. T. Tuten, C. A. Tooley, P. G. Frank and E. B. Berda, Polym. Chem., 2015, 6, 181–197 RSC.
- S. Mavila, O. Eivgi, I. Berkovich and N. G. Lemcoff, Chem. Rev., 2016, 116, 878–961 CrossRef CAS PubMed.
- J. A. Pomposo, I. Perez-Baena, F. Lo Verso, A. J. Moreno, A. Arbe and J. Colmenero, ACS Macro Lett., 2014, 3, 767–772 CrossRef CAS PubMed.
- (a) E. B. Berda, E. J. Foster and E. W. Meijer, Macromolecules, 2010, 43, 1430–1437 CrossRef CAS; (b) N. Hosono, A. M. Kushner, J. Chung, A. R. Palmans, Z. Guan and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 6880–6888 CrossRef CAS PubMed; (c) F. Zhou, M. Xie and D. Chen, Macromolecules, 2013, 47, 365–372 CrossRef; (d) N. Hosono, M. A. Gillissen, Y. Li, S. S. Sheiko, A. R. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed; (e) P. T. Dirlam, H. J. Kim, K. J. Arrington, W. J. Chung, R. Sahoo, L. J. Hill, P. J. Costanzo, P. Theato, K. Char and J. Pyun, Polym. Chem., 2013, 4, 3765–3773 RSC; (f) E. A. Appel, J. Dyson, J. del Barrio, Z. Walsh and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 4185–4189 CrossRef CAS PubMed.
- (a) E. Harth, B. Van Horn, V. Y. Lee, D. S. Germack, C. P. Gonzales, R. D. Miller and C. J. Hawker, J. Am. Chem. Soc., 2002, 124, 8653–8660 CrossRef CAS PubMed; (b) J. Willenbacher, K. N. R. Wuest, J. O. Mueller, M. Kaupp, H. A. Wagenknecht and C. Barner-Kowollik, ACS Macro Lett., 2014, 3, 574–579 CrossRef CAS PubMed; (c) S. Gillhuber, J. O. Holloway, H. Frisch, F. Feist, F. Weigend, C. Barner-Kowollik and P. W. Roesky, Chem. Commun., 2023, 59, 4672–4675 RSC.
- (a) B. S. Murray and D. A. Fulton, Macromolecules, 2011, 44, 7242–7252 CrossRef CAS; (b) T. K. Claus, J. Zhang, L. Martin, M. Hartlieb, H. Mutlu, S. Perrier, G. Delaittre and C. Barner-Kowollik, Macromol. Rapid Commun., 2017, 38 Search PubMed.
- T. Terashima, T. Sugita, K. Fukae and M. Sawamoto, Macromolecules, 2014, 47, 589–600 CrossRef CAS.
- Y. Shao, Y. L. Wang, Z. Tang, Z. Wen, C. Chang, C. Wang, D. Sun, Y. Ye, D. Qiu, Y. Ke, F. Liu and Z. Yang, Angew. Chem., Int. Ed., 2022, 61, e202205183 CrossRef CAS PubMed.
- K. Watanabe, R. Tanaka, K. Takada, M. J. Kim, J. S. Lee, K. Tajima, T. Isono and T. Satoh, Polym. Chem., 2016, 7, 4782–4792 RSC.
- (a) E. J. Foster, E. B. Berda and E. W. Meijer, J. Polym. Sci., Polym. Chem., 2011, 49, 118–126 CrossRef CAS; (b) M. A. J. Gillissen, I. K. Voets, E. W. Meijer and A. R. A. Palmans, Polym. Chem., 2012, 3, 3166–3174 RSC; (c) B. T. Tuten, D. M. Chao, C. K. Lyon and E. B. Berda, Polym. Chem., 2012, 3, 3068–3071 RSC; (d) G. M. ter Huurne, M. A. J. Gillissen, A. R. A. Palmans, I. K. Voets and E. W. Meijer, Macromolecules, 2015, 48, 3949–3956 CrossRef CAS; (e) J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS PubMed; (f) Y. Bai, X. Feng, H. Xing, Y. Xu, B. K. Kim, N. Baig, T. Zhou, A. A. Gewirth, Y. Lu, E. Oldfield and S. C. Zimmerman, J. Am. Chem. Soc., 2016, 138, 11077–11080 CrossRef CAS PubMed; (g) C. H. Liu, L. D. Dugas, J. I. Bowman, T. Chidanguro, R. F. Storey and Y. C. Simon, Polym. Chem., 2020, 11, 292–297 RSC; (h) J. Nam, S. Kwon, Y. G. Yu, H. B. Seo, J. S. Lee, W. B. Lee, Y. Kim and M. Seo, Macromolecules, 2021, 54, 8829–8838 CrossRef CAS.
- (a) R. Gracia, M. Marradi, U. Cossio, A. Benito, A. Perez-San Vicente, V. Gomez-Vallejo, H. J. Grande, J. Llop and I. Loinaz, J. Mater. Chem. B, 2017, 5, 1143–1147 RSC; (b) R. Gracia, M. Marradi, G. Salerno, R. Perez-Nicado, A. Perez-San Vicente, D. Dupin, J. Rodriguez, I. Loinaz, F. Chiodo and C. Nativi, ACS Macro Lett., 2018, 7, 196–200 CrossRef CAS PubMed; (c) N. Blanco-Cabra, J. Movellan, M. Marradi, R. Gracia, C. Salvador, D. Dupin, I. Loinaz and E. Torrents, npj Biofilms Microbiomes, 2022, 8, 52 CrossRef CAS PubMed.
- (a) M. Hebel, J. Gacanin, T. Luckerath, D. Y. W. Ng and T. Weil, Macromol. Rapid Commun., 2022, 43, e2100413 CrossRef PubMed; (b) P. Malo de Molina, T. P. Le, A. Iturrospe, U. Gasser, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Omega, 2022, 7, 42163–42169 CrossRef CAS PubMed.
- X. Tian, R. Xue, F. Yang, L. Yin, S. Luan and H. Tang, Biomacromolecules, 2021, 22, 4306–4315 CrossRef CAS PubMed.
- (a) A. Nitti, R. Carfora, G. Assanelli, M. Notari and D. Pasini, ACS Appl. Nano Mater., 2022, 5, 13985–13997 CrossRef CAS; (b) M. K. Aiertza, I. Odriozola, G. Cabanero, H. J. Grande and I. Loinaz, Cell. Mol. Life Sci., 2012, 69, 337–346 CrossRef CAS PubMed.
- A. Sanchez-Sanchez, I. Perez-Baena and J. A. Pomposo, Molecules, 2013, 18, 3339–3355 CrossRef CAS PubMed.
- A. M. Hanlon, R. W. Chen, K. J. Rodriguez, C. Willis, J. G. Dickinson, M. Cashman and E. B. Berda, Macromolecules, 2017, 50, 2996–3003 CrossRef CAS.
- (a) O. Galant, H. B. Donmez, C. Barner-Kowollik and C. E. Diesendruck, Angew. Chem., Int. Ed., 2021, 60, 2042–2046 CrossRef CAS PubMed; (b) O. Galant, C. E. Diesendruck and S. Spatari, Org. Process Res. Dev., 2023, 28, 1607–1617 CrossRef.
- (a) P. J. M. Stals, M. A. J. Gillissen, R. Nicolaÿ, A. R. A. Palmans and E. W. Meijer, Polym. Chem., 2013, 4, 2584–2597 RSC; (b) C. C. Cheng, F. C. Chang, H. C. Yen, D. J. Lee, C. W. Chiu and Z. Xin, ACS Macro Lett., 2015, 4, 1184–1188 CrossRef CAS PubMed; (c) J. L. Zhang, G. Gody, M. Hartlieb, S. Catrouillet, J. Moffat and S. Perrier, Macromolecules, 2016, 49, 8933–8942 CrossRef CAS; (d) H. W. van Roekel, P. J. Stals, M. A. Gillissen, P. A. Hilbers, A. J. Markvoort and T. F. de Greef, Chem. Commun., 2013, 49, 3122–3124 RSC; (e) C. C. Cheng, D. J. Lee, Z. S. Liao and J. J. Huang, Polym. Chem., 2016, 7, 6164–6169 RSC.
- T. S. Fischer, D. Schulze-Sunninghausen, B. Luy, O. Altintas and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2016, 55, 11276–11280 CrossRef CAS PubMed.
- (a) T. Mes, R. van der Weegen, A. R. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 5085–5089 CrossRef CAS PubMed; (b) M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Polym. Chem., 2014, 52, 12–20 CrossRef CAS; (c) Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13105 CrossRef CAS PubMed; (d) P. J. M. Stals, C. Y. Cheng, L. van Beek, A. C. Wauters, A. R. A. Palmans, S. Han and E. W. Meijer, Chem. Sci., 2016, 7, 2011–2015 RSC; (e) E. Archontakis, L. Deng, P. Zijlstra, A. R. A. Palmans and L. Albertazzi, J. Am. Chem. Soc., 2022, 144, 23698–23707 CrossRef CAS PubMed; (f) L. Deng, L. Albertazzi and A. R. A. Palmans, Biomacromolecules, 2022, 23, 326–338 CrossRef CAS PubMed.
- J. Lu, N. Ten Brummelhuis and M. Weck, Chem. Commun., 2014, 50, 6225–6227 RSC.
- (a) A. Sanchez-Sanchez, A. Arbe, J. Kohlbrecher, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2015, 36, 1592–1597 CrossRef CAS PubMed; (b) Z. Zhu, N. Xu, Q. Yu, L. Guo, H. Cao, X. Lu and Y. Cai, Macromol. Rapid Commun., 2015, 36, 1521–1527 CrossRef CAS PubMed; (c) I. Berkovich, S. Mavila, O. Iliashevsky, S. Kozuch and N. G. Lemcoff, Chem. Sci., 2016, 7, 1773–1778 RSC; (d) F. Wang, H. Pu, M. Jin and D. Wan, Macromol. Rapid Commun., 2016, 37, 330–336 CrossRef CAS PubMed; (e) L. N. Neumann, D. A. Urban, P. Lemal, S. Ramani, A. Petri-Fink, S. Balog, C. Weder and S. Schrettl, Polym. Chem., 2020, 11, 586–592 RSC; (f) J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC; (g) S. Thanneeru, S. S. Duay, L. Jin, Y. Fu, A. M. Angeles-Boza and J. He, ACS Macro Lett., 2017, 6, 652–656 CrossRef CAS PubMed; (h) S. Garmendia, S. B. Lawrenson, M. C. Arno, R. K. O'Reilly, D. Taton and A. P. Dove, Macromol. Rapid Commun., 2019, 40, e1900071 CrossRef PubMed.
- (a) A. Sanchez-Sanchez, A. Arbe, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2014, 3, 439–443 CrossRef CAS PubMed; (b) H. Rothfuss, N. D. Knofel, P. W. Roesky and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 5875–5881 CrossRef CAS PubMed.
- (a) R. K. Roy and J. F. Lutz, J. Am. Chem. Soc., 2014, 136, 12888–12891 CrossRef CAS PubMed; (b) A. Sanchez-Sanchez and J. A. Pomposo, J. Nanomater., 2015, 2015, 1–7 Search PubMed; (c) J. Liang, J. J. Struckhoff, P. D. Hamilton and N. Ravi, Langmuir, 2017, 33, 7660–7668 CrossRef CAS PubMed.
- Y. Gao, V. I. Bohmer, D. Zhou, T. Zhao, W. Wang and J. M. Paulusse, J. Controlled Release, 2016, 244, 375–383 CrossRef CAS PubMed.
- (a) E. H. H. Wong, S. J. Lam, E. Nam and G. G. Qiao, ACS Macro Lett., 2014, 3, 524–528 CrossRef CAS PubMed; (b) E. H. H. Wong and G. G. Qiao, Macromolecules, 2015, 48, 1371–1379 CrossRef CAS.
- (a) W. H. Binder and C. Kluger, Curr. Org. Chem., 2007, 10, 1791–1815 CrossRef; (b) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS; (c) W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS; (d) N. Li and W. H. Binder, J. Mater. Chem., 2011, 21, 16717–16734 RSC; (e) D. Döhler, P. Michael and W. H. Binder, Acc. Chem. Res., 2017, 50, 2610–2620 CrossRef PubMed; (f) S. Neumann, M. Biewend, S. Rana and W. H. Binder, Macromol. Rapid Commun., 2020, 41, e1900359 CrossRef PubMed.
- (a) M. Meldal and F. Diness, Trends Chem., 2020, 2, 569–584 CrossRef CAS; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- M. Alqaisi, J. F. Thümmler, F. Lehmann, F.-J. Schmitt, L. Lentz, F. Rieder, D. Hinderberger and W. H. Binder, Polym. Chem., 2024, 15, 2949–2958 RSC.
- (a) A. R. de Luzuriaga, N. Ormategui, H. J. Grande, I. Odriozola, J. A. Pomposo and I. Loinaz, Macromol. Rapid Commun., 2008, 29, 1156–1160 CrossRef; (b) A. R. de Luzuriaga, I. Perez-Baena, S. Montes, I. Loinaz, I. Odriozola, I. García and J. A. Pomposo, Macromol. Symp., 2010, 296, 303–310 CrossRef; (c) L. Oria, R. Aguado, J. A. Pomposo and J. Colmenero, Adv. Mater., 2010, 22, 3038–3041 CrossRef CAS PubMed; (d) B. V. Schmidt, N. Fechler, J. Falkenhagen and J. F. Lutz, Nat. Chem., 2011, 3, 234–238 CrossRef CAS PubMed; (e) N. Ormategui, I. García, D. Padro, G. Cabañero, H. J. Grande and I. Loinaz, Soft Matter, 2012, 8, 734–740 RSC; (f) J. Rubio-Cervilla, H. Frisch, C. Barner-Kowollik and J. A. Pomposo, Macromol. Rapid Commun., 2019, 40, e1800491 CrossRef PubMed; (g) J. Chen, K. Li, J. S. L. Shon and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 4565–4569 CrossRef CAS PubMed; (h) I. Perez-Baena, I. Loinaz, D. Padro, I. García, H. J. Grande and I. Odriozola, J. Mater. Chem., 2010, 20, 6916–6922 RSC.
- J. F. Thümmler, R. Maragani, F.-J. Schmitt, G. Tang, S. M. Rahmanlou, J. Laufer, H. Lucas, K. Mäder and W. H. Binder, Chem. Commun., 2023, 59, 11373–11376 RSC.
- (a) J. He, L. Tremblay, S. Lacelle and Y. Zhao, Soft Matter, 2011, 7, 2380–2386 RSC; (b) W. Fan, X. Tong, Q. Yan, S. Fu and Y. Zhao, Chem. Commun., 2014, 50, 13492–13494 RSC; (c) P. G. Frank, B. T. Tuten, A. Prasher, D. Chao and E. B. Berda, Macromol. Rapid Commun., 2014, 35, 249–253 CrossRef CAS PubMed; (d) C. A. Tooley, S. Pazicni and E. B. Berda, Polym. Chem., 2015, 6, 7646–7651 RSC; (e) W. Z. Fan, X. Tong, F. Farnia, B. Yu and Y. Zhao, Chem. Mater., 2017, 29, 5693–5701 CrossRef CAS; (f) W. Z. Fan, X. Tong, G. Li and Y. Zhao, Polym. Chem., 2017, 8, 3523–3529 RSC; (g) M. Bilgi, D. K. Balta, B. A. Temel and G. Temel, J. Polym. Sci., Polym. Chem., 2018, 56, 2709–2714 CrossRef CAS; (h) H. Frisch, J. P. Menzel, F. R. Bloesser, D. E. Marschner, K. Mundsinger and C. Barner-Kowollik, J. Am. Chem. Soc., 2018, 140, 9551–9557 CrossRef CAS PubMed; (i) X. Ji, Y. Zhang and H. Zhao, Chemistry, 2018, 24, 3005–3012 CrossRef CAS PubMed; (j) H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2019, 58, 3604–3609 CrossRef CAS PubMed; (k) H. Frisch, D. Kodura, F. R. Bloesser, L. Michalek and C. Barner-Kowollik, Macromol. Rapid Commun., 2020, 41, e1900414 CrossRef PubMed.
- C. F. Song, L. Y. Li, L. Z. Dai and S. Thayumanavan, Polym. Chem., 2015, 6, 4828–4834 RSC.
- (a) F. Xu, Z. Fang, D. Yang, Y. Gao, H. Li and D. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 6717–6723 CrossRef CAS PubMed; (b) J. Wen, L. Yuan, Y. Yang, L. Liu and H. Zhao, ACS Macro Lett., 2013, 2, 100–106 CrossRef CAS PubMed; (c) W. Wen, T. Y. Huang, S. Guan, Y. B. Zhao and A. H. Chen, Macromolecules, 2019, 52, 2956–2964 CrossRef CAS.
- (a) Z. Cui, L. Huang, Y. Ding, X. Zhu, X. Lu and Y. Cai, ACS Macro Lett., 2018, 7, 572–575 CrossRef CAS PubMed; (b) Z. G. Cui, H. Cao, Y. Ding, P. Gao, X. H. Lu and Y. L. Cai, Polym. Chem., 2017, 8, 3755–3763 RSC.
- (a) M. Calosi, E. Guazzelli, S. Braccini, M. Lessi, F. Bellina, G. Galli and E. Martinelli, Polymers, 2022, 14, 774 CrossRef CAS PubMed; (b) E. Guazzelli, E. Masotti, M. Kriechbaum, F. Uhlig, G. Galli and E. Martinelli, Macromol. Chem. Phys., 2023, 224, 2200360 CrossRef CAS; (c) T. K. Nguyen, S. J. Lam, K. K. Ho, N. Kumar, G. G. Qiao, S. Egan, C. Boyer and E. H. Wong, ACS Infect. Dis., 2017, 3, 237–248 CrossRef CAS PubMed.
- (a) R. Liu and J. S. Lindsey, ACS Macro Lett., 2019, 8, 79–83 CrossRef CAS PubMed; (b) R. Liu, S. J. Liu, G. F. Hu and J. S. Lindsey, New J. Chem., 2020, 44, 21293–21308 RSC; (c) S. J. Liu, J. Rong, R. Liu and J. S. Lindsey, ACS Appl. Polym. Mater., 2021, 3, 1767–1776 CrossRef CAS.
- A. Delledonne, E. Guazzelli, S. Pescina, A. Bianchera, G. Galli, E. Martinelli and C. Sissa, ACS Appl. Nano Mater., 2023, 6, 15551–15562 CrossRef CAS PubMed.
- (a) S. Wijker, L. Deng, F. Eisenreich, I. K. Voets and A. R. A. Palmans, Macromolecules, 2022, 55, 6220–6230 CrossRef CAS PubMed; (b) L. Deng, A. Sathyan, C. Adam, A. Unciti-Broceta, V. Sebastian and A. R. A. Palmans, Nano Lett., 2024, 24, 2242–2249 CrossRef CAS PubMed; (c) L. Deng, A. R. Olea, A. Ortiz-Perez, B. Sun, J. Wang, S. Pujals, A. R. A. Palmans and L. Albertazzi, Small Methods, 2024, e2301072 CrossRef PubMed.
- H. Zhang, L. Zhang, J. C. You, N. B. Q. Zhang, L. X. Z. Yu, H. Y. Zhao, H. J. Qian and Z. Y. Lu, CCS Chem., 2021, 3, 2143–2154 CrossRef CAS.
- X. Zhao, D. Li, J. Zhu, Y. Fan, J. Xu, X. Huang, Z. Nie and D. Chen, ACS Macro Lett., 2024, 13, 882–888 CrossRef CAS PubMed.
- S. Basasoro, M. Gonzalez-Burgos, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2016, 37, 1060–1065 CrossRef CAS PubMed.
- S. Y. Liao, L. X. Wei, L. A. Abriata and F. Stellacci, Macromolecules, 2021, 54, 11459–11467 CrossRef CAS.
- J. Jelken, S. H. Jung, N. Lomadze, Y. D. Gordievskaya, E. Y. Kramarenko, A. Pich and S. Santer, Adv. Funct. Mater., 2022, 32, 2107946 CrossRef CAS.
- M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2013, 46, 4120–4125 CrossRef CAS.
- (a) T. Terashima, T. Mes, T. F. De Greef, M. A. Gillissen, P. Besenius, A. R. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed; (b) E. Huerta, P. J. Stals, E. W. Meijer and A. R. Palmans, Angew. Chem., Int. Ed., 2013, 52, 2906–2910 CrossRef CAS PubMed; (c) M. Artar, E. R. J. Souren, T. Terashima, E. W. Meijer and A. R. A. Palmans, ACS Macro Lett., 2015, 4, 1099–1103 CrossRef CAS PubMed; (d) E. Huerta, B. van Genabeek, P. J. Stals, E. W. Meijer and A. R. Palmans, Macromol. Rapid Commun., 2014, 35, 1320–1325 CrossRef CAS PubMed; (e) Y. L. Liu, P. Turunen, B. F. M. de Waal, K. G. Blank, A. E. Rowan, A. R. A. Palmans and E. W. Meijer, Mol. Syst. Des. Eng., 2018, 3, 609–618 RSC; (f) Y. Liu, S. Pujals, P. J. M. Stals, T. Paulohrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- J. Engelke, J. Brandt, C. Barner-Kowollik and A. Lederer, Polym. Chem., 2019, 10, 3410–3425 RSC.
- (a) I. Perez-Baena, F. Barroso-Bujans, U. Gasser, A. Arbe, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 775–779 CrossRef CAS PubMed; (b) A. Sanchez-Sanchez, S. Akbari, A. Etxeberria, A. Arbe, U. Gasser, A. J. Moreno, J. Colmenero and J. A. Pomposo, ACS Macro Lett., 2013, 2, 491–495 CrossRef CAS PubMed; (c) A. Sanchez-Sanchez, S. Akbari, A. J. Moreno, F. L. Verso, A. Arbe, J. Colmenero and J. A. Pomposo, Macromol. Rapid Commun., 2013, 34, 1681–1686 CrossRef CAS PubMed; (d) B. Mei, A. J. Moreno and K. S. Schweizer, ACS Nano, 2024, 18, 15529–15544 CrossRef CAS PubMed.
- A. P. P. Kröger, R. J. E. A. Boonen and J. M. J. Paulusse, Polymer, 2017, 120, 119–128 CrossRef.
- J. A. Pomposo, A. J. Moreno, A. Arbe and J. Colmenero, ACS Omega, 2018, 3, 8648–8654 CrossRef CAS PubMed.
- S. Walpole, S. Monaco, R. Nepravishta and J. Angulo, Methods Enzymol., 2019, 615, 423–451 CAS.
- P. H. Maag, F. Feist, H. Frisch, P. W. Roesky and C. Barner-Kowollik, Chem. Sci., 2024, 15, 5218–5224 RSC.
- (a) Z. Lu, J. Zhang, W. Yin, C. Guo and M. Lang, Macromol. Rapid Commun., 2022, 43, e2200156 CrossRef PubMed; (b) J. Hu, M. R. Whittaker, H. Duong, Y. Li, C. Boyer and T. P. Davis, Angew. Chem., Int. Ed., 2014, 53, 7779–7784 CrossRef CAS PubMed; (c) J. Pinacho-Olaciregui, E. Verde-Sesto, D. Taton and J. A. Pomposo, Macromol. Rapid Commun., 2024, 45, e2400116 CrossRef PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., 2010, 123, 118–142 CrossRef.
- T. M. Xiong, E. S. Garcia, J. Chen, L. Zhu, A. J. Alzona and S. C. Zimmerman, Chem. Commun., 2022, 58, 985–988 RSC.
- M. Meir, A. Abuaf, D. Subhomoy and M. Yithak, RSC Adv., 2023, 13, 1580–1586 RSC.
- A. Blazquez-Martin, E. Verde-Sesto, A. Arbe and J. A. Pomposo, Angew. Chem., Int. Ed., 2023, e202313502 CAS.
- R. Lambert, A. L. Wirotius, S. Garmendia, P. Berto, J. Vignolle and D. Taton, Polym. Chem., 2018, 9, 3199–3204 RSC.
- D. Arena, E. Verde-Sesto, I. Rivilla and J. A. Pomposo, J. Am. Chem. Soc., 2024, 146, 14397–14403 CrossRef CAS PubMed.
- P. H. Maag, F. Feist, H. Frisch, P. W. Roesky and C. Barner-Kowollik, Macromolecules, 2022, 55, 9918–9924 CrossRef CAS.
- (a) S. Liu, M. Q. Tang, J. Pang, J. T. Hu, W. J. Chen, J. Cheng, Z. W. Liu, H. H. Zhao and R. Tan, ACS Sustainable Chem. Eng., 2022, 10, 11760–11772 CrossRef CAS; (b) N. D. Knofel, H. Rothfuss, J. Willenbacher, C. Barner-Kowollik and P. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 4950–4954 CrossRef PubMed.
- Y. Li, D. Maciel, J. Rodrigues, X. Shi and H. Tomas, Chem. Rev., 2015, 115, 8564–8608 CrossRef CAS PubMed.
- C. T. Adkins, J. N. Dobish, S. Brown and E. Harth, ACS Macro Lett., 2013, 2, 710–714 CrossRef CAS PubMed.
- J. Liu, G. Feng, R. Liu, N. Tomczak, L. Ma, G. G. Gurzadyan and B. Liu, Small, 2014, 10, 3110–3118 CrossRef CAS PubMed.
- (a) A. P. P. Kroger, N. M. Hamelmann, A. Juan, S. Lindhoud and J. M. J. Paulusse, ACS Appl. Mater. Interfaces, 2018, 10, 30946–30951 CrossRef PubMed; (b) N. M. Hamelmann, J. D. Paats and J. M. J. Paulusse, ACS Macro Lett., 2021, 10, 1443–1449 CrossRef CAS PubMed.
- (a) N. M. Hamelmann, S. Uijttewaal, S. D. Hujaya and J. M. J. Paulusse, Biomacromolecules, 2022, 23, 5036–5042 CrossRef CAS PubMed; (b) N. M. Hamelmann, J. D. Paats, Y. Avalos-Padilla, E. Lantero, L. Spanos, I. Siden-Kiamos, X. Fernandez-Busquets and J. M. J. Paulusse, ACS Infect. Dis., 2023, 9, 56–64 CrossRef CAS PubMed.
- A. B. Benito, M. K. Aiertza, M. Marradi, L. Gil-Iceta, T. Shekhter Zahavi, B. Szczupak, M. Jimenez-Gonzalez, T. Reese, E. Scanziani, L. Passoni, M. Matteoli, M. De Maglie, A. Orenstein, M. Oron-Herman, G. Kostenich, L. Buzhansky, E. Gazit, H. J. Grande, V. Gomez-Vallejo, J. Llop and I. Loinaz, Biomacromolecules, 2016, 17, 3213–3221 CrossRef CAS PubMed.
- A. P. P. Kröger, M. I. Komil, N. M. Hamelmann, A. Juan, M. H. Stenzel and J. M. J. Paulusse, ACS Macro Lett., 2019, 8, 95–101 CrossRef PubMed.
- J. E. F. Radu, L. Novak, J. F. Hartmann, N. Beheshti, A.-L. Kjøniksen, B. Nyström and J. Borbély, Colloid Polym. Sci., 2007, 286, 365–376 CrossRef.
- N. J. Butcher, G. M. Mortimer and R. F. Minchin, Nat. Nanotechnol., 2016, 11, 310–311 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |