
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe thermal instability of hydrogen-substituted graphdiyne and its role in lithium–sulfur batteries†
Karam
Eeso
 a,
Bryan R.
Wygant
b,
Zhitao
Chen
a,
Akriti
Sarswat
a,
Timothy N.
Lambert
*bc and
Nian
Liu
*a
a,
Bryan R.
Wygant
b,
Zhitao
Chen
a,
Akriti
Sarswat
a,
Timothy N.
Lambert
*bc and
Nian
Liu
*a
aSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA 30332, USA. E-mail: nian.liu@chbe.gatech.edu
bSandia National Laboratories, Albuquerque, New Mexico 87185, USA. E-mail: tnlambe@sandia.gov
cCenter for Integrated Nanotechnologies, Albuquerque, New Mexico 87185, USA
First published on 5th November 2024
Abstract
This study reveals the role of thermal instability in hydrogen-substituted graphdiyne (HsGDY) and its impact on lithium–sulfur (Li–S) battery performance. HsGDY undergoes significant chemical and physical transformations when subjected to thermal heating, both in the presence and absence of sulfur. Our findings suggest that the structural transformation of HsGDY into a graphene-like structure is primarily responsible for enhancing sulfur trapping and reducing the polysulfide shuttle effect, rather than the previously hypothesized alkyne–sulfur chemical interactions.
To address the growing demand for energy storage, particularly in portable electronics and electric vehicles, lithium–sulfur (Li–S) batteries have garnered significant interest due to their superior theoretical energy density compared to conventional lithium-ion batteries. However, challenges such as the corrosion of the lithium anode through the “polysulfide shuttle” effect and the poor electronic conductivity of sulfur have limited their commercial viability due to poor cycle life and discharge rates. Efforts to mitigate these issues have led to the exploration of other materials to improve the performance and cycle life of Li–S batteries.1
To enhance the sulfur utilization and capacity retention of Li–S batteries, researchers have experimented with combining sulfur with various carbon materials,2–9 such as polyacrylonitrile (PAN)10 and graphene (G),3 aiming to leverage their conductive properties and structural benefits such as high surface area and tortuosity. These studies, along with our related work on FeSx/C cathode materials, where electrochemically reversible carbyne-polysulfide derivatives were proposed as the active species,9 led us to further consider graphdiyne (GDY) as a promising carbon material, given its unique two-dimensional structure composed of sp and sp2 hybridized carbon atoms. This structure forms an extended conjugated network, which facilitates superior electron mobility, high surface area, and chemical stability, making GDY an attractive material for energy storage applications. Additionally, GDY's reactive alkyne groups allow for versatile chemical functionalization, further enhancing its ability to interact with other species and improve performance.11–16 Upon more detailed inspection, it became clear that the lithium/carbon–sulfur GDY literature commonly invokes a mechanism involving electrochemically reversible alkyne–sulfur bonds for GDY, yet lacks sufficient physical and spectroscopic data to support this chemical conversion.12–14 As a result, we chose to evaluate the physicochemical changes of GDY following thermal and chemical (sulfur) exposure (i.e., the method used to prepare the electrodes) to understand the changes upon electrochemical cycling.
Central to this investigation was our hypothesis that the interaction between hydrogen-substituted graphdiyne (HsGDY) and sulfur during thermal treatment plays a pivotal role in enhancing the performance of Li–S batteries. For our studies, we utilized HsGDY, Fig. 2a, and propose that, as the mixture of sulfur and HsGDY is heated, the HsGDY ‘framework’ undergoes a constriction process that effectively traps molten sulfur within its structure, Fig. 1. This unique interaction is theorized to create a constrained environment for the sulfur, significantly affecting the dynamics of lithium polysulfide formation and retention during battery operation. Specifically, we hypothesize that the constricted space limits the escape of generated polysulfides from the cathode, addressing one of the primary challenges in Li–S battery technology—the notorious polysulfide shuttle effect. By impeding the migration of polysulfides out of the cathode due to increased tortuosity, which slows diffusion, this mechanism could substantially improve the battery's efficiency, cycle life, and overall performance. This study aims to explore this hypothesis through a series of experimental investigations, shedding light on the potential of thermal treatment in improving HsGDY–sulfur interactions for battery functionality while simultaneously probing for evidence of the previously proposed alkyne–sulfur interconversion reaction.
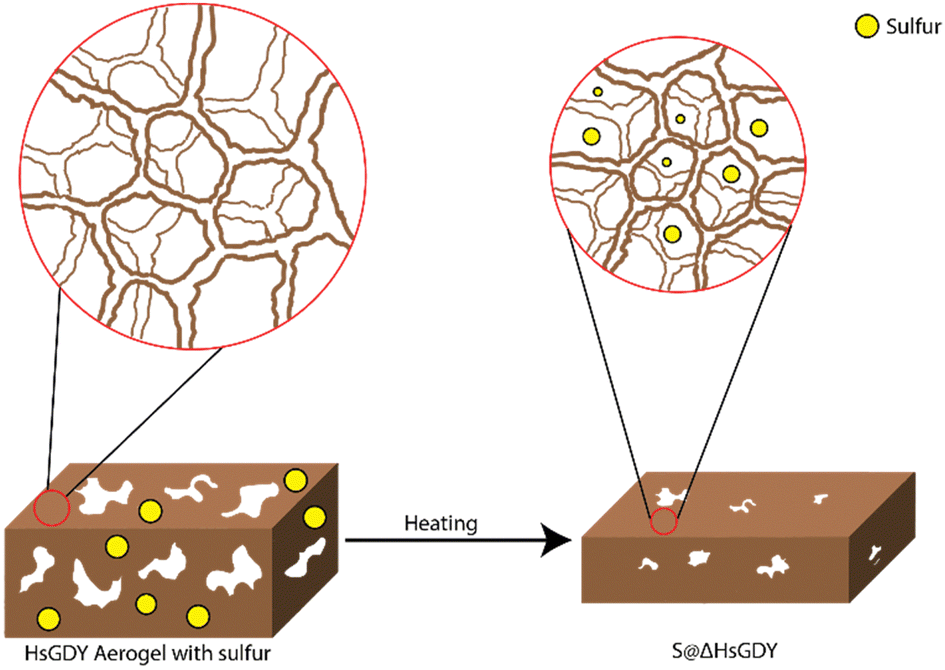 | ||
| Fig. 1 A graphical representation of how the structure of HsGDY changes to encapsulate sulfur. Initially the aerogel is porous with a larger surface area. However, the mesopores disappear and the sulfur is trapped deep inside the structure as HsGDY is heated with sulfur. | ||
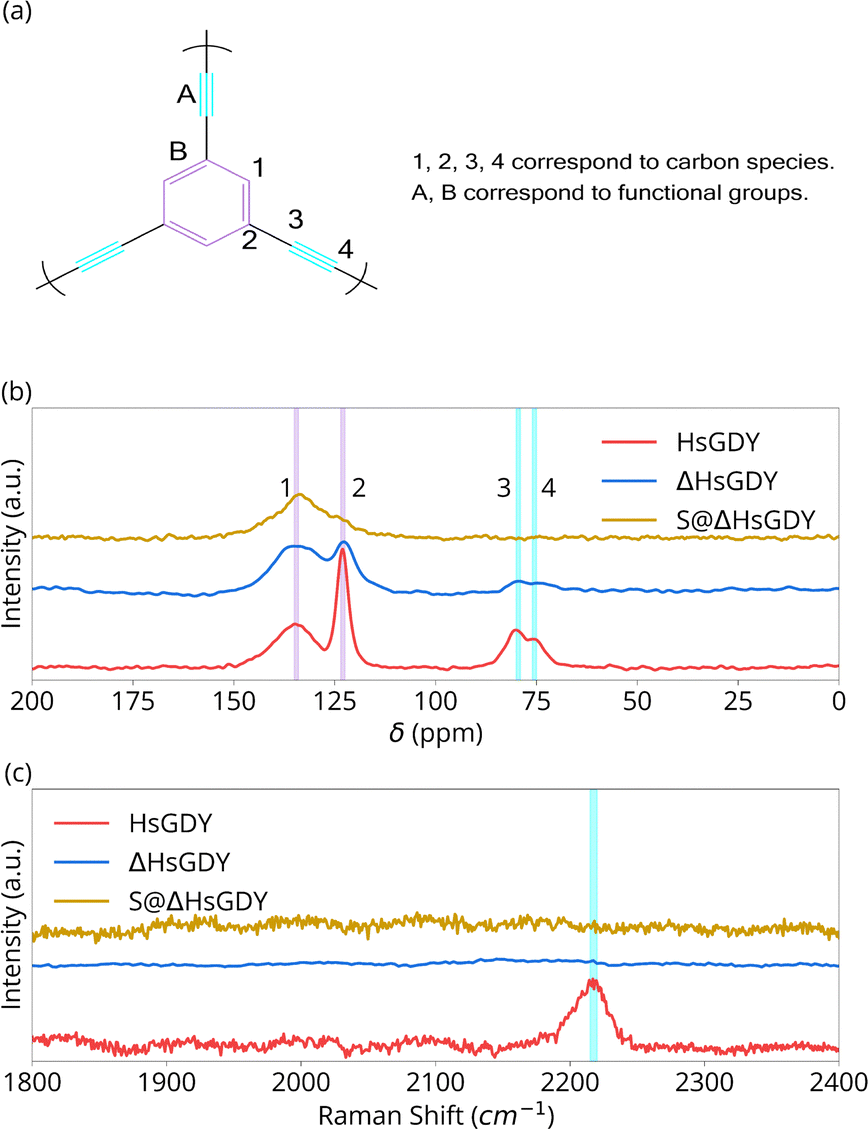 | ||
| Fig. 2 Chemical characterization of HsGDY, Heated HsGDY and S@HsGDY. (a) Carbon species and functional groups in (idealized) HsGDY; (b) solid-state 13C NMR; (c) Raman spectra. | ||
We began by preparing HsGDY according to a modified Glaser-type reaction (see ESI†).17 Briefly, triethynylbenzene (30 mg, 0.2 mmol) and copper chloride (6 mg, 0.06 mmol) were combined in pyridine (2 mL) in a glass vial. The vial was placed in a 40 °C water bath for 72 h, and then the solid was isolated, washed, and dried. The prepared HsGDY was then heated at 155 °C for 12 h in a tube furnace under argon to obtain ΔHsGDY. Similarly, S@ΔHsGDY was obtained when sulfur was heated together with HsGDY as a mixture in a mass ratio of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
To study the impact of the thermal conditions under which ΔHsGDY and S@ΔHsGDY were prepared, we conducted thermogravimetric analysis (TGA). Fig. S1 (ESI†) shows the full TGA results. The full TGA shows that there is mass loss even after 110 °C (loss below 110 °C can be attributed to water),13 likely from the evolution of hydrogen, methane, or carbon monoxide upon heating.18 Furthermore, the isothermal portion of the TGA has a mass loss of 7% over the 12 h period, further indicating that the HsGDY is not thermally stable. In response to this observed thermal instability, we performed a detailed chemical analysis.
Fig. 2 provides chemical characterization using solid state 13C NMR and Raman spectroscopy. 13C NMR peaks at 123.1 and 135.7 ppm correspond to the aromatic C–C and C–H sites, (assignments 2 and 1, respectively), Fig. 2b. Peaks at 75.5 and 81.8 ppm correspond to the alkyne carbons, C(sp)–C(sp) (assignment 4) and C(sp)–C(sp2) (assignment 3) sites.15 Pristine HsGDY shows a C(sp2):C(sp) of 3:1 (integration shown in Fig. S2, ESI†), while the ideal material should have a ratio of 1:1. Thus, the synthesized material has defects, likely from incomplete alkyne coupling.17,19 However, the major result of the NMR is the difference in chemical structure between the HsGDY and ΔHsGDY. The peaks at 75.5 and 81.8 ppm for ΔHsGDY decrease significantly, indicating that the triple bonds have reacted and that there is a major change in chemical structure—a change not previously reported in the GDY literature.7,13,14 Furthermore, this suggests that sulfur does not necessarily react with the triple bonds when HsGDY and sulfur are heated together to form the active material of the cathodes, as the triple bonds are partially lost even in the absence of S.
Additional analysis shows that the spectra exhibit the expected ratio of 1:1 in the pristine HsGDY for the C–H and C–C phenyl carbons (peak 1 at 135.7 and peak 2 at 123.1 ppm), while the ratio changes to 2.24:1 in ΔHsGDY. This implies that either (1) the bond between carbon 2 and 3 is breaking (possibly favored for pendant alkyne ‘arms’ that were not successfully coupled during the Glaser coupling reaction, i.e. ‘the defects’) resulting in a C–H bond, or (2) the alkyne bond is reacting (proposed to be crosslinking). More research will be required to better understand exactly what the ΔHsGDY structure is, but the most important aspect from 13C NMR analysis is significant reduction in the alkyne carbon peaks. 13C NMR data in Fig. 2b further reveals the differences between HsGDY and S@ΔHsGDY. Upon heating in the presence of sulfur, most of the aromatic C–C bonds (123.1 ppm) have shifted downfield, as demonstrated in Fig. 2b, and the peak previously assigned to the C–H aromatic bonds (135.7 ppm) broadens. This suggests that the S@ΔHsGDY sample is converted to a type of graphitic material,20,21 which exhibits a broad peak centered at 140 ppm.
Fig. 2c provides the Raman spectra for HsGDY and ΔHsGDY. In HsGDY, thepeak at 2230 cm−1 corresponds to the alkyne linkages;7,17 this peak is notably absent in ΔHsGDY, exemplifying the thermal instability of the alkyne functionality in HsGDY. To the best of our knowledge, this characterization has not been reported before in the literature. Unless the alkyne can be verified later upon charging an electrode, this would call into question electrochemical mechanism claims invoking reversible alkyne–S interactions for GDY.12–14
While 13C NMR clearly shows that the HsGDY material chemically changes with heating, such changes are not observed in the XPS measurements of these samples. The C 1s spectra for as-prepared and heated HSGDY are nearly indistinguishable (Fig. S3, ESI†), and even fitting of individual bonds does little to show differences between the two materials (Fig. S4 and S5, ESI†). The S XPS data indicates the formation of C–S bonds in the S@ΔHsGDY (Fig. S6, 165 eV, ESI†), suggestive of alkyne–sulfur or alkene–sulfur binding. As a result, although XPS has been used previously to support the thermal stability of HsGDY-like materials,13 the poor agreement between our C 1s and S 2p results show that 13C NMR is a more reliable means of probing thermal (in)stability in HsGDY.
Next, we explored what physical changes accompanied the material upon heating. Adsorption measurements in Fig. 3a show that the parent material adsorbs more than twice as much gas as the ΔHsGDY, leading to calculated surface areas of 1215 m2 g−1 (HsGDY) and 501 m2 g−1 (ΔHsGDY). Fig. 3b shows that the large pores (23–25 nm) decrease in size to 18 nm with heating, while the smaller pores (15 nm) either completely collapse or decrease below the minimum size of the fitting algorithm. These results suggest a collapse in structure, whereby heating the HsGDY causes a constriction of the material. Pores either collapse or close off, and the sheet structure tightens. This tightening of the interlayer spacing is confirmed by XRD, as shown in Fig. 3d. The amorphous peak shifts from 2-theta of 19° to 20.2° indicative of a 6% decrease in spacing (from 0.467 nm to 0.439 nm) upon heating. TEM also confirms the interlayer spacing decrease, Fig. S7 (ESI†), although SEM images of both compounds, Fig. S8 (ESI†), show little difference.
 | ||
| Fig. 3 Physical characterization of HsGDY and heated HsGDY (ΔHsGDY). (a) Shows the nitrogen adsorption results. (b) Shows the difference in calculated pore width and their frequency. (c) Shows the UV-Vis of the adsorption experiments. (d) Shows the XRD results. | ||
Fig. S9 (ESI†) shows the results of an adsorption test of HsGDY and ΔHsGDY when treated with a blue solution of Li2S8. After the 3-day exposure, the HsGDY solution is colorless, while the ΔHsGDY remains slightly colored, indicative of soluble sulfides remaining in solution. These results are consistent with the relatively higher surface area of the HsGDY vs. ΔHsGDY, as well as the ability of soluble sulfides to chemically react with the alkyne functional groups that are (more) present in the pristine HsGDY.13,14 These changes in solution Li2S8 concentration are further quantified using UV-Vis. The HsGDY solution exhibits a transmittance value of 98%, the ΔHsGDY shows 62%, and the control is at 18%, Fig. 3c.
S@ΔHsGDY, S+HsGDY (physically mixed) and S@Δgraphene were then further evaluated for electrochemical performance. S@Δgraphene was added for a reference since heating HsGDY with sulfur produces a graphitic compound, as discussed previously. Galvanostatic cycling data for CR2032 Li/(cathode) coin cells is provided in Fig. 4. Both S@Δgraphene and S@ΔHsGDY show a statistically significant (p < 0.001) better performance than the physically mixed S+HsGDY. The performance of S+HsGDY suggests that the existing alkyne bonds and high surface area (in HsGDY) do not contribute to an increase in performance, as suggested by others,12–14 but rather, it is the thermal treatment of S@HsGDY that leads to the performance gains. It can be argued that S+HsGDY was only physically mixed, so the sulfur would not be able to fully be trapped by the HsGDY. However, if the alkyne bonds were truly reversible as stated in the literature, then the performance should increase after the first charge, when much of the dissolved polysulfide would be reacted back to sulfur on the cathode. Furthermore, Fig. 3c shows that HsGDY demonstrated an increased adsorption of polysulfides, suggesting the S+HsGDY material should act similarly, increasing performance. However, the cycling results show the contrary, and this implies it is necessary to heat sulfur and HsGDY for increased performance. Furthermore, the cycling data shows a major decrease in specific capacity in the first 10 cycles for the S+HsGDY when compared to S@HsGDY, where the capacity fade follows a gentler exponential decay; this further suggests that HsGDY is not efficient at sulfur trapping, when all the other data suggests that it should. Neither ex situ Raman spectroscopy nor XPS was able to identify evidence of electrochemical cycling between alkyne and sulfur species (Fig. S10 and S11, ESI†).
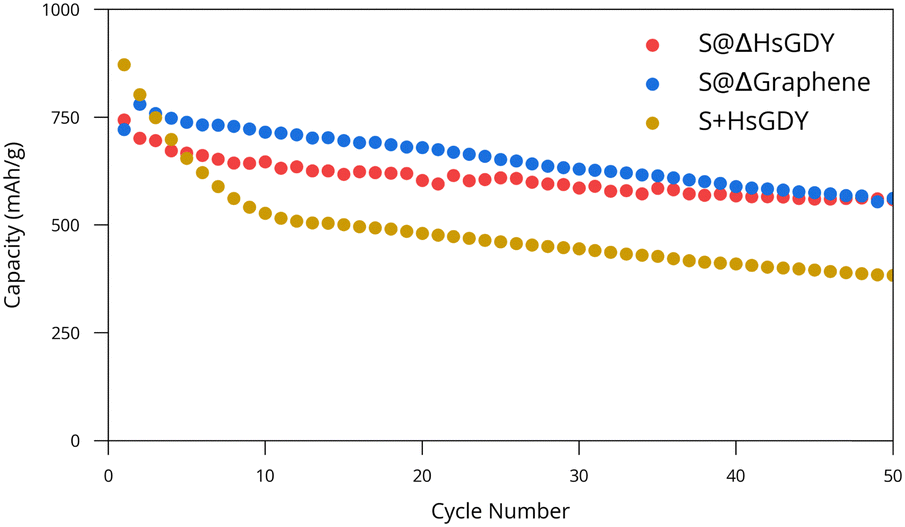 | ||
| Fig. 4 Cycling performance of S@HsGDY, S@graphene, and S+HsGDY. Best results are shown for each cathode material while triplicates are shown in Fig. S12 (ESI†). Capacity is in terms of mass of sulfur. | ||
Based on these observations, our hypothesis is that sulfur melts into the pores as the heating process occurs, and the HsGDY begins to simultaneously collapse in on itself, trapping the sulfur deep into the pores. This in situ trapping of sulfur provides the capacity increase relative to physically mixed materials. Although the performance is statistically lower than S@Δgraphene (p < 0.001), this is not practically significant since the means of the capacities are different by only 48 mA h g−1. However, there are some differences in the discharge capacity plots shown in Fig. 4. While both cathodes eventually decay in an exponential fashion, but the S@Δgraphene batteries initially increase in capacity before then decreasing like the S@ΔHsGDY batteries, suggesting an activation process occurs in the S@Δgraphene cathodes. Despite this, the charge/discharge curves in Fig. S13 (ESI†) do not show a noticeable difference between the materials, indicating the chemical processes themselves do not change. For broader context, we have included a comparison of our battery performance with other reported GDY-based and carbon-based Li–S batteries in Table S1 (ESI†). The performance reported in the GDY papers is comparable to our results, but some other carbon-based materials22 demonstrate significantly better performance. However, direct comparisons between GDY and other carbon-based materials are not straightforward because of differences in experimental conditions, such as heating procedures, electrolyte volumes, types of separators used, and other variables that can optimize the battery performance.
This study reveals the role of thermal instability in hydrogen-substituted graphdiyne (HsGDY) and its impact on lithium–sulfur (Li–S) battery performance. We found no spectroscopic evidence that alkynes participate in the sulfur electrochemistry; instead, we observed that they are mostly lost upon heating. HsGDY performance does improve after heating; however, this is likely due to the change in the interfaces between ΔHsGDY, sulfur, and the electrolyte. The thermal instability causes the HsGDY structure to collapse onto the sulfur, leading to mechanical trapping rather than chemical alkyne–sulfur trapping. This changes the interfaces since sulfur has a harder time escaping the collapsed structure, decreasing the shuttling effect and further improving performance. Further work will be needed to unequivocally establish the cycling mechanism; we suggest using cathodes harvested from cycled cells for additional characterization (e.g., NMR).
This work was supported by the Laboratory Directed Research and Development program at Sandia National Laboratories. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science. Sandia National Laboratories is a multi-program laboratory managed and operated by National Technology and Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International, Inc., for the U.S. Department of Energy's National Nuclear Security Administration under contract DE-NA-0003525. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. This work was performed in part at the Georgia Tech Institute for Electronics and Nanotechnology, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (ECCS-2025462).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Barghamadi, A. Kapoor and C. Wen, J. Electrochem. Soc., 2013, 160, A1256–A1263 CrossRef CAS
.
- C. Liang, N. J. Dudney and J. Y. Howe, Chem. Mater., 2009, 21, 4724–4730 CrossRef CAS
- Y. Feng, H. Zhang, Y. Zhang and X. Qu, ACS Omega, 2019, 4, 16352–16359 CrossRef CAS PubMed
- G. Tian,
et al.
, J. Mater. Chem. A, 2019, 7, 15640–15653 RSC
- S. S. Zhang, Energies, 2014, 7, 4588–4600 CrossRef
- K. Liu,
et al.
, ACS Appl. Mater. Interfaces, 2023, 15, 31478–31490 CrossRef CAS PubMed
- J. Li,
et al.
, Small, 2019, 15, 1805344 CrossRef PubMed
- J. Wang,
et al.
, Carbon, 2008, 46, 229–235 CrossRef CAS
- B. R. Wygant,
et al.
, J. Phys. Chem. C, 2022, 126, 9000–9008 CrossRef CAS
- S. Wei,
et al.
, J. Am. Chem. Soc., 2015, 137, 12143–12152 CrossRef CAS PubMed
- Q. Pan,
et al.
, ACS Appl. Mater. Interfaces, 2019, 11, 46070–46076 CrossRef CAS PubMed
- J. Gao,
et al.
, Chem. Eng. J., 2019, 373, 660–667 CrossRef CAS
- H. Du,
et al.
, Small, 2017, 13, 1702277 CrossRef PubMed
- Y. Yi,
et al.
, ACS Appl. Mater. Interfaces, 2021, 13, 59983–59992 CrossRef CAS PubMed
- J. He,
et al.
, Nat. Commun., 2017, 8, 1172 CrossRef PubMed
- L. C. Greenburg,
et al.
, Nano Lett., 2023, 23, 5967–5974 CrossRef CAS PubMed
- R. Du,
et al.
, Adv. Mater., 2014, 26, 8053–8058 CrossRef CAS PubMed
- I. Abed,
et al.
, BioResources, 2012, 7, 1200–1220 Search PubMed
- Y. Hu,
et al.
, Nat. Synth., 2022, 1, 449–454 CrossRef
- F. A. L. de Souza,
et al.
, J. Phys. Chem. C, 2016, 120, 27707–27716 CrossRef CAS
- Y. Wen,
et al.
, Nat. Commun., 2014, 5, 4033 CrossRef CAS PubMed
- P. Wang,
et al.
, eScience, 2023, 3, 100088 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04459b |
| This journal is © The Royal Society of Chemistry 2024 |