
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkenylation of unactivated alkanes: synthesis of Z-alkenes via dual Co-TBADT catalysis†
Vetrivelan
Murugesan
,
Anagha
Syam
,
Guru Vigknesh
Anantharaj
and
Ramesh
Rasappan
 *
*
School of Chemistry, Indian Institute of Science Education and Research Thiruvananthapuram, Vithura, Thiruvananthapuram, Kerala 695551, India. E-mail: rr@iisertvm.ac.in
First published on 4th November 2024
Abstract
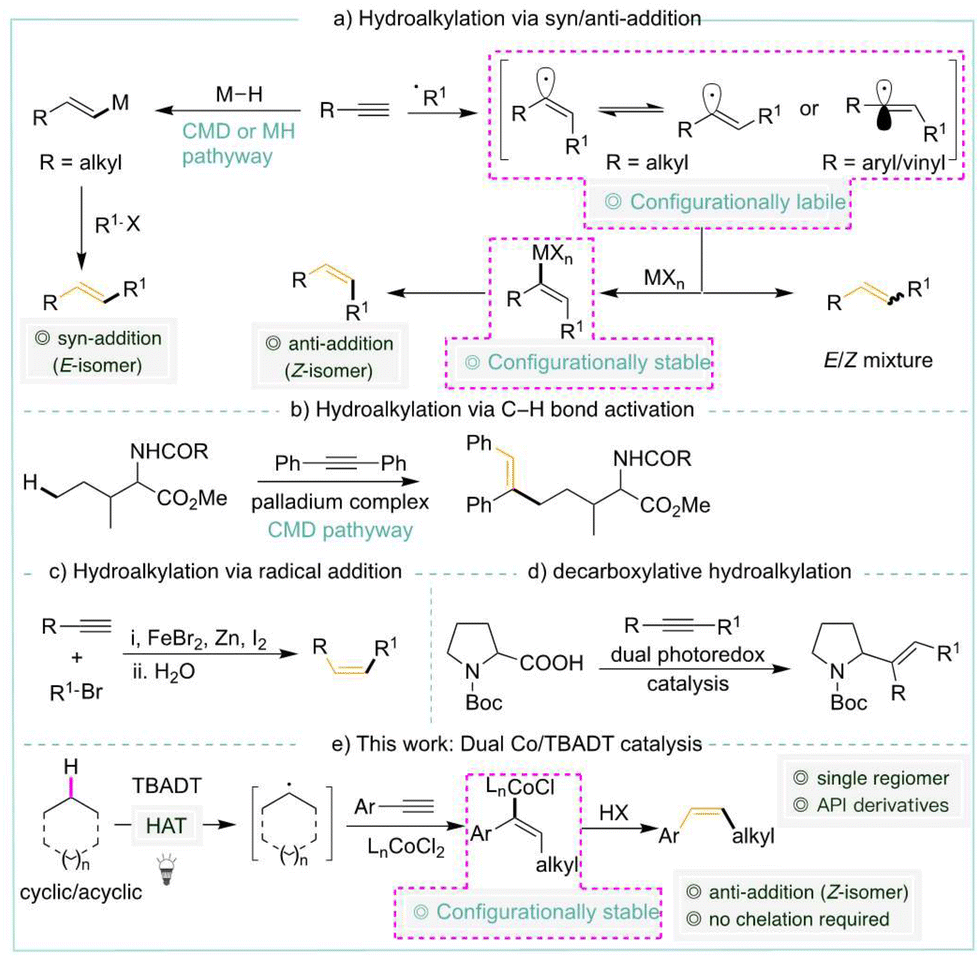
Hydroalkylation of terminal alkynes via C–H activation is the most atom-economical and straightforward method for synthesizing alkenes. They remain confined to using C(sp2)–H or activated C(sp3)–H bonds. A chelating group enabled the alkenylation of C(sp3)–H bonds, resulting in E alkenes. Protocols by which alkenylation of unactivated C(sp3)–H bonds occurs without a chelating group via metal-hydride or radical pathways remain unknown. Our cobalt-HAT catalysis achieves the desired Z alkene with excellent regio- and diastereoselectivity via C–H activation.
In organic synthesis, alkenes serve as versatile and unique building blocks.1 Among the numerous methods that exist, transition metal catalyzed hydroalkylation2 of readily available terminal alkynes is regarded as one of the most versatile methods for the stereo- and regio-controlled synthesis of alkenes. The direct addition of a C–H bond across an alkyne is notably the most atom- and step-economical method.3 Despite notable advancements in the utilization of C(sp2)–H and activated C(sp3)–H bonds for hydroalkylation,4 unactivated C(sp3)–H bonds are seldom encountered, necessitating the presence of a chelating group, and are predominantly employed with internal alkynes (Scheme 1b).5
 | ||
| Scheme 1 Hydroalkylation of alkynes. | ||
Transition metal-mediated hydroalkylation via C–H activation typically yields syn-adducts by concerted metalation–deprotonation (CMD) or metal-hydride routes (Scheme 1a). Conversely, the alternate approach of adding carbon-centred radicals to alkynes results in limited stereoselectivity6 owing to the in situ formation of vinyl radicals with either π (linear) or σ (bent and rapidly inverting) geometry.7 The vinyl radical, fortunately, generates configurationally stable vinyl-metal species in situ,8 which have been utilized successfully in stereospecific transformations.9
Although radical additions to alkenes have been extensively studied,10 additions to alkynes are less prevalent (Scheme 1c).6b,11 This can be attributed to the higher singlet–triplet energy gap and/or the generation of a highly reactive intermediate vinyl radical.7b,12 On the other hand, photocatalysis has been effectively employed to synthesize alkenes through HAT processes.13 Employing Ni/Ir dual catalysis, the group of Macmillan9b and Rueping9c recently revealed a decarboxylative hydroalkylation (Scheme 1d). There are currently no known methods for accommodating unactivated alkanes in hydroalkylation in the absence of chelating groups via radical additions, metal hydride, or CMD. Herein, we demonstrate the application of cobalt-HAT catalysis in generating the desired Z alkene with high regio- and diastereoselectivity.
We commenced our study with substrates 1-(tert-butyl)-4-ethynylbenzene 1a and cyclohexane 2a. Under the optimized conditions involving 20 mol% CoCl2 and 6 mol% TBADT, 3a was obtained in 83% isolated yield with a ratio of 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 (Z:E) (entry 18). A comprehensive selection of nickel complexes was evaluated in light of our prior expertise in dual TBADT-nickel catalysis (Table S1, ESI†).14 Nevertheless, when NiCl2·dtbpy and NiBr2·phen were utilized, the cross-coupled product 3a was obtained only in 37% and 40% yield (Table 1, entries 1 and 2), respectively, with a moderate Z:E ratio. Significantly, the majority of entries utilizing a nickel complex exhibited complete consumption of alkyne 1a (Table S1, ESI†). Pleasingly, a dramatic improvement in the yield was observed when CoCl2 was employed (Table S3, ESI†). Additionally, in order to augment the reaction, a variety of ligands (Table 1, entries 3–8) were utilized, comprising phen, tMephen, dMebpy, and dtbpy (Table 1, entries 3–5 and 7). Of the several ligands that were evaluated, those based on bipyridine demonstrated an enhanced yield for the synthesis of 3a. In particular, ligand dMeObpy produced 3a with a 74% yield (entry 6). The moderate yields were attributable to an incomplete consumption of alkyne 1a. Having identified the optimal ligand, reactions were conducted by altering the catalyst loadings. A significant improvement in yield was observed when the precomplexed cobalt catalyst was used with decreased loading of TBADT (Table 1, entry 9).
:7 (Z:E) (entry 18). A comprehensive selection of nickel complexes was evaluated in light of our prior expertise in dual TBADT-nickel catalysis (Table S1, ESI†).14 Nevertheless, when NiCl2·dtbpy and NiBr2·phen were utilized, the cross-coupled product 3a was obtained only in 37% and 40% yield (Table 1, entries 1 and 2), respectively, with a moderate Z:E ratio. Significantly, the majority of entries utilizing a nickel complex exhibited complete consumption of alkyne 1a (Table S1, ESI†). Pleasingly, a dramatic improvement in the yield was observed when CoCl2 was employed (Table S3, ESI†). Additionally, in order to augment the reaction, a variety of ligands (Table 1, entries 3–8) were utilized, comprising phen, tMephen, dMebpy, and dtbpy (Table 1, entries 3–5 and 7). Of the several ligands that were evaluated, those based on bipyridine demonstrated an enhanced yield for the synthesis of 3a. In particular, ligand dMeObpy produced 3a with a 74% yield (entry 6). The moderate yields were attributable to an incomplete consumption of alkyne 1a. Having identified the optimal ligand, reactions were conducted by altering the catalyst loadings. A significant improvement in yield was observed when the precomplexed cobalt catalyst was used with decreased loading of TBADT (Table 1, entry 9).
|
|
|||
|---|---|---|---|
| Entry | Deviation from above | 1a |
3a
(Z:E) |
|
a Optimized conditions: 0.15 mmol of 1a, 7.5 mmol of 2a, 6 mol% of PC – TBADT (portion wise), 20 mol% of Co-I (portion wise), CH3CN:PhH (4:1, 0.08 M), 390 nm, 27 °C, 48 h.
b Determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
c CH3CN [0.1 M].
d Isolated yields.
e 5 mol% of TBADT.
f 7.5 mol% of TBADT; bpy – 2,2′-bipyridine; phen – 1,10-phenanthroline; dMeObpy – 4,4′-dimethoxy-2,2′-bipyridine; dMebpy – 4,4′-dimethyl-2,2′-bipyridine; dtbpy – 4,4-di-tert-butyl-2,2-dipyridyl; tMephen – 3,4,7,8-tetramethyl-1,10-phenanthroline.
|
|||
| 1c | (7.5 mol%) NiCl2·dtbpy | n.d. | 37 (79:21) |
| 2c | (7.5 mol%) NiBr2·phen | n.d. | 40 (80:20) |
| 3 | (10 mol%) CoCl2, phen | 25 | 60 (80:20) |
| 4 | (10 mol%) CoCl2, tMephen | 29 | 48 (81:19) |
| 5 | (10 mol%) CoCl2, dMebpy | 23 | 61 (79:21) |
| 6 | No deviation | 15 | 74 (84:16) |
| 7 | (10 mol%) CoCl2, dtbpy | 18 | 68 (87:13) |
| 8 | (10 mol%) CoCl2, (20 mol%) dmap | 19 | 32 (82:18) |
| 9 | (10 mol%) PC, (10 mol%) Co-I | T | 89 (80:20) |
| 10 | (5 mol%) PC, (10 mol%) Co-I | 27 | 71 (90:10) |
| 11 | Same as entry 10, 60 h | 9 | 80 (91:9) |
| 12 | Same as entry 10, acetone (0.08 M) | 88 | 5 (75:25) |
| 13 | Same as entry 10, DMSO (0.08 M) | 91 | <5 |
| 14 | (5 mol%) PC, (5 mol%) Co-I | 22 | 58 (80:20) |
| 15 | (7.5 mol%) PC, (15 mol%) Co-I | 6 | 93 (86:14) |
| 16 | (7.5 mol%) PC, (23 mol%) Co-I | 6 | 87 (92:8) |
| 17 | (5 mol%) PC, (20 mol%) Co-I | 25 | 73 (95:5) |
| 18a | (4 + 2 mol%) PC, (16 + 4 mol%) Co-I | <5 |
83 (93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :7) :7)
|
| 19 | Same as entry 15, 25 eq. of CyH | 42 | 37 (90:10) |
| 20e | (30 mol%) An added as sensitizer | 32 | 55 (91:9) |
| 21e | (20 mol%) BP added as sensitizer | 38 | 43 (85:15) |
| 22 | Without TBADT or CoCl2·dMeObpyf | 99 | n.d. |
| 23a | Under air | 15 | 51 (89:11) |
| 24a | 20 eq. H2O added | 10 | 47 (85:15) |
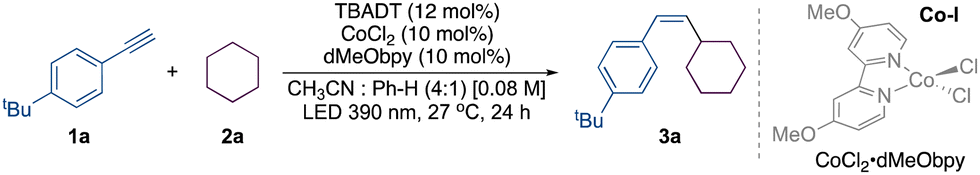
A further decrease in the loading of TBADT from 12 mol% to 5 mol% was shown to have a negative impact (Table 1, entry 10). No reaction was visually discernible in DMSO or acetone (Table 1, entries 12 and 13). The ideal reaction conditions necessitate the utilization of a 4:1 mixture of CH3CN:PhH. None of the alternative solvents that were evaluated (Table S2, ESI†) proved to be effective. Consequently, our study focused on altering the proportion between TBADT and CoCl2, as well as adjusting the loading of the catalyst (Table 1, entries 14–17). While 7.5% TBADT increased the yield of 3a, it also decreased the Z:E ratio of 3a to 86:14 (Table 1, entry 15). Control experiments conducted in the absence of TBADT or CoCl2·dMeObpy demonstrate the indispensability of these particular reagents (Table 1, entry 22). The yield of 3a was diminished when the quantity of cyclohexane was reduced (Table 1, entry 19). Photosensitizers are recognized for their ability to promote E to Z isomerization.15 However, the use of photosensitizers such as anthracene (An) and benzophenone (BP) did not prove beneficial in enhancing the E/Z ratio (entries 20 and 21). The reaction can be performed at 4 mmol scale; the complete consumption of 1a necessitated 5 days, resulting in the isolation of 3a with a yield of 70% (Z:E – 92:8) (SI-21, ESI†).
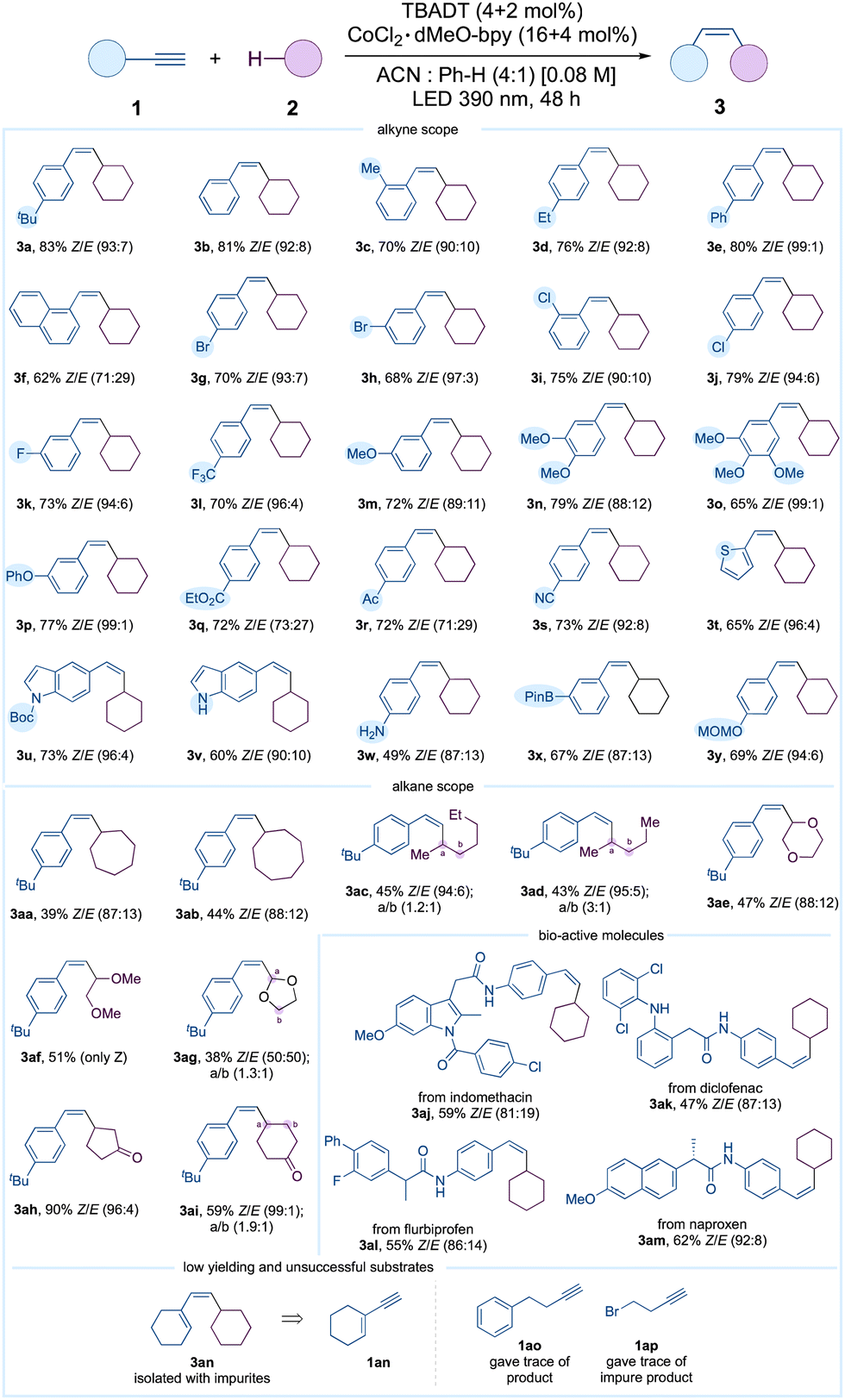
Table 2 depicts the scope of alkynes and alkanes. Phenyl acetylenes incorporating tert-butyl, methyl, ethyl, phenyl, and π-extended naphthyl groups exhibited efficient cross-coupling reactions. The aryl bromides (1g and 1h) and chlorides (1i and 1j) remained intact. The medicinally significant fluoro and trifluoromethyl derivatives 1k and 1l were synthesized with respective yields of 73% and 70%. The compatibility of the methyl and phenyl ethers 1m–1p was also demonstrated. Pleasingly, no competing reactions involving esters and ketones were observed. The nitrile group was likewise compatible. We employed heterocycles including thiophene and indole derivatives 1t–1v under the optimized conditions. Consequently, high yields of the corresponding coupled products 3t–3v were effectively obtained. Remarkably, the Boc protecting group, known for its high sensitivity, remained stable throughout the reaction. Equally striking was the compatibility of the unprotected amine; indole 1v and aniline 1w produced 3v and 3w in 60% and 49% isolated yields, respectively. Perhaps importantly, the compatibility of the boronic ester ensured that subsequent functionalization remained viable. In addition, the MOM group 3y, which is particularly sensitive to acid, remained stable under the optimized reaction conditions. Furthermore, the scope of alkanes 2 was expanded. Cycloheptane and cyclooctane were compatible, producing 3aa and 3ab in satisfactory yields, as predicted. Acyclic alkanes were also found to be compatible, yielding a mixture of isomers. We also illustrate the effectiveness of the approach in instances of polarity-matched and mismatched HATs. As anticipated, the polarity-matched α-heteromethylenes underwent successful functionalization to yield the corresponding alkenes 3ae–3ag. The polarity-mismatched α-methylenes in carbonyl compounds were intact; as demonstrated in Table 2, cyclopentanone and cyclohexanone exhibited compatibility and produced the cross-coupled products 3ah and 3ai with good yields. The API derivatives; indomethacin 3aj, diclofenac 3ak, flurbiprofen 3al, and naproxen 3am derivatives were obtained with satisfactory yields. Of note, when the enyne 1an was employed, a mixture of impure products was obtained. The alkyl alkynes including but-3-yn-1-ylbenzene 1ao and 4-bromobut-1-yne 1ap were recovered intact. Regrettably, N-methylmorpholine, N-methylpyrrolidine, and tetrahydrothiophene did not yield the coupled product. It is interesting that the formation of 3a was entirely suppressed when 2 eq. of N-methylmorpholine was added to the default reaction (SI-87, ESI†).
|
The introduction of deuterated methanol under the optimized reaction conditions led to a 70% incorporation of deuterium in the cross-coupled product 3a-D. Utilizing deuterated aryl acetylene 1m-D, we identified 100% deuterium incorporation in the product 3m-D. These results rule out the potential for aryl acetylide to serve as an intermediate. NMR analysis of the reaction crude in the presence of TEMPO revealed the presence of the TEMPO adduct 4, confirming cyclohexyl radical intermediacy. Additionally, radical clock substrate 1aq offered the cyclized product 3aq′ in conjunction with uncyclized product 3aq, which confirms the presence of a vinyl radical intermediate (Scheme 2).
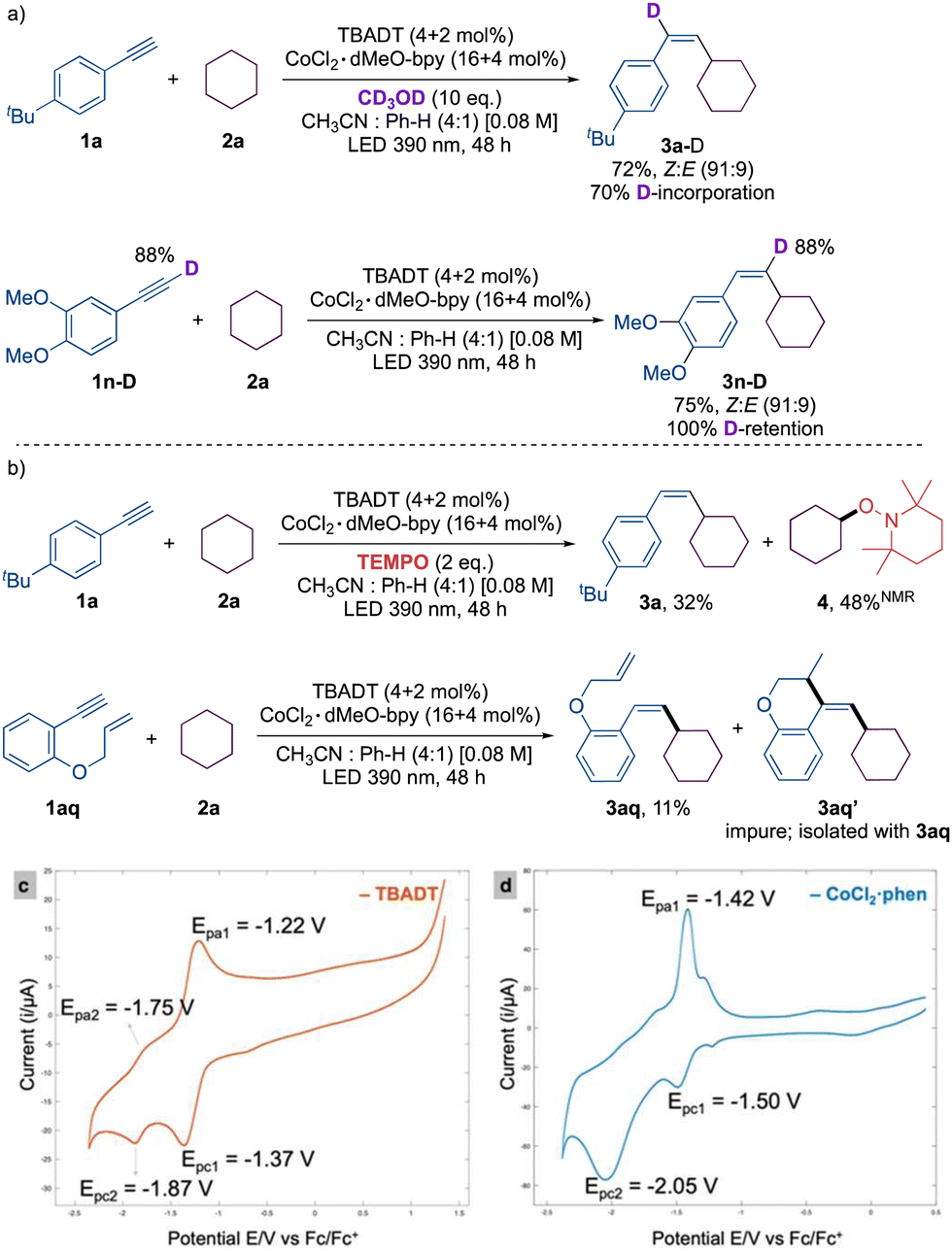 | ||
| Scheme 2 (a) Deuterium labelling experiments, and (b) radical trap experiments. Cyclic voltammogram of (c) TBADT and (d) CoCl2·1,10-phenanthroline. | ||
A cyclic voltammogram was conducted to gain a more comprehensive understanding of the mechanism, as we anticipated that the effective reducing agent would be [W10O32]6− rather than [W10O32]5−.16 The cyclic voltammetry (CV) of TBADT showed two chemically reversible reductions at potentials of −1.37 V and −1.87 V, which correspond to the reduction of [W10O32]4−/[W10O32]5− and [W10O32]5−/[W10O32]6−, respectively. In contrast to the irreversible reduction peak observed in the CV of the default complex, Co(II)Cl2·dMeObpy (Co-I), an equally effective complex, CoCl2·phen (entry 5, Table S4, ESI†), displayed two subsequent reductions. The reversible peak at −1.5 V is presumably assigned to Co(II)/Co(I) and the irreversible peak at −2.05 V is assigned to Co(I)/Co(0).17 Given that the reduction potential of [W10O32]4−/[W10O32]5− is more anodic than that of Co(II) to Co(I), it is probable that [W10O32]6− reduces the Co(II) complex. Furthermore, as the reduction of Co(I) to Co(0) happens at a more cathodic potential, the active low valence species is attributed to the Co(I) species.
Based on our prior14,16b and current studies, we postulated a hypothetical mechanism in Scheme 3. The catalytic cycle begins when the 390 nm LED excites TBADT (LMCT (ligand-to-metal charge transfer): oxygen → tungsten), resulting in the short-lived excited species [W10O32]4−*. Subsequent intersystem crossing generates the long-lived triplet wO species, which undertakes a HAT event to produce the alkyl radical IV and pentavalent decatungstate, [W10O32]5−. The pentavalent decatungstate undergoes disproportionation, resulting in the regeneration of [W10O32]4− and the emergence of the strong reductant [W10O32]6−. This reductant then reduces the Co(II) complex VII, generating the low-valent Co(I) complex VIII. In parallel, the alkyl radical that is in situ-generated is captured by an alkyne, resulting in the formation of a thermodynamically unstable transitory vinyl radical. Ultimately, the vinyl radical is captured by the low valent Co(I) complex VIII in a stereoselective manner or the generated species IX is more favourable than X because the cyclohexyl group and the ligands attached to the cobalt center exhibit steric hinderance in X. Following this, the vinyl cobalt intermediate is protodemetallated in order to generate the desired alkene product and restore the Co(II) complex VII. The alternate pathway wherein the alkyl radical (IV) combines with Co(I) and subsequently undergoes hydroalkylation is excluded as it would result in the formation of the E-isomer.
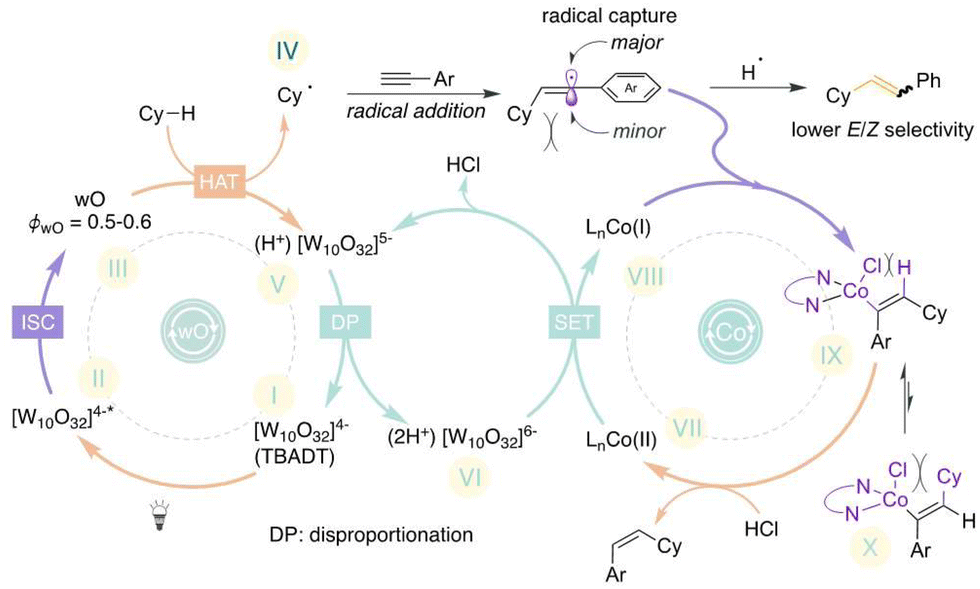 | ||
| Scheme 3 Hypothetical mechanism. | ||
In conclusion, a protocol was developed to facilitate the hydroalkylation of terminal alkynes through the C–H activation of unactivated cyclic/acyclic alkanes. The inclusion of sensitive functional groups like carbonyl, nitrile, and Bpin as well as acid-sensitive groups like MOM and Boc demonstrates the mitigation of the reaction conditions. The scope of alkanes was broadened to include acyclic alkanes and APIs. A mechanism has been disclosed by preliminary mechanistic investigations.
We thank IISER Trivandrum and Science and Engineering Research Board; CRG/2023/005485 and RSC Research Fund grant; R23-8225379124 for the financial support. A. S. acknowledges CSIR. V. M. acknowledges IISER Trivandrum for the fellowship.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Flynn and W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed; (b) C. Oger, L. Balas, T. Durand and J. Galano, Chem. Rev., 2013, 113, 1313 CrossRef CAS PubMed.
- (a) X. Lu, J. Liu, X. Lu, Z. Zhang, T. Gong, B. Xiao and Y. Fu, Chem. Commun., 2016, 52, 5324 RSC; (b) A. Suess, M. Uehling, W. Kaminsky and G. Lalic, J. Am. Chem. Soc., 2015, 137, 7747 CrossRef CAS.
- (a) Y. Minami and T. Hiyama, Acc. Chem. Res., 2016, 49, 67 CrossRef CAS PubMed; (b) L. Yang and H. Huang, Chem. Rev., 2015, 115, 3468 CrossRef CAS PubMed; (c) K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS; (b) B. Zhou, H. Chen and C. Wang, J. Am. Chem. Soc., 2013, 135, 1264 CrossRef CAS; (c) N. Chernyak, S. I. Gorelsky and V. Gevorgyan, Angew. Chem., Int. Ed., 2011, 50, 2342 CrossRef CAS.
- (a) Y. Nakao, E. Morita, H. Idei and T. Hiyama, J. Am. Chem. Soc., 2011, 133, 3264 CrossRef CAS; (b) C. Lin, Z. Chen, Z. Liu and Y. Zhang, Org. Lett., 2017, 19, 850 CrossRef CAS PubMed; (c) S. Maity, S. Agasti, A. Earsad, A. Hazra and D. Maiti, Chem. – Eur. J., 2015, 21, 11320 CrossRef CAS PubMed; (d) M. Li, Y. Yang, D. Zhou, D. Wan and J. You, Org. Lett., 2015, 17, 2546 CrossRef CAS; (e) J. Xu, Z. Zhang, W. Rao and B. Shi, J. Am. Chem. Soc., 2016, 138, 10750 CrossRef CAS PubMed.
- (a) Y. Zhang, L. Huang, K. Cheng, B. Yao and J. Zhao, Synthesis, 2009, 3504 CrossRef; (b) S. Feng, X. Xie, W. Zhang, L. Liu, Z. Zhong, D. Xu and X. She, Org. Lett., 2016, 18, 3846 CrossRef CAS PubMed; (c) W. Chen, M. Zhang and Y. Zhao, Synthesis, 2016, 1342 CrossRef; (d) J. Li, J. Zhang, H. Tan and D. Wang, Org. Lett., 2015, 17, 2522 CrossRef CAS.
- (a) T. P. M. Goumans, K. van Alem and G. Lodder, Eur. J. Org. Chem., 2008, 435 CrossRef CAS; (b) U. Wille, Chem. Rev., 2013, 113, 813 CrossRef CAS.
- J. M. Huggins and R. G. Bergman, J. Am. Chem. Soc., 1981, 103, 3002 CrossRef CAS.
- (a) C. Cheung, F. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932 CrossRef CAS PubMed; (b) N. Till, R. Smith and D. MacMillan, J. Am. Chem. Soc., 2018, 140, 5701 CrossRef CAS PubMed; (c) H. Yue, C. Zhu, R. Kancherla, F. Liu and M. Rueping, Angew. Chem., Int. Ed., 2020, 59, 5738 CrossRef CAS.
- A. Capacci, J. Malinowski, N. McAlpine, J. Kuhne and D. MacMillan, Nat. Chem., 2017, 9, 1073 CrossRef CAS.
- (a) M. Huang, W. Hao, G. Li, S. Tu and B. Jiang, Chem. Commun., 2018, 54, 10791 RSC; (b) H. Han, Y. Lee, Y. Jung and S. Han, Org. Lett., 2017, 19, 1962 CrossRef CAS; (c) H. Liang, Y. Ji, R. Wang, Z. Zhang and B. Zhang, Org. Lett., 2019, 21, 2750 CrossRef CAS PubMed; (d) N. Chalotra, A. Ahmed, M. Rizvi, Z. Hussain, Q. Ahmed and B. Shah, J. Org. Chem., 2018, 83, 14443 CrossRef CAS PubMed; (e) M. Alkan-Zambada and X. Hu, J. Org. Chem., 2019, 84, 4525 CrossRef CAS.
- (a) R. Gómez-Balderas, M. L. Coote, D. J. Henry, H. Fischer and L. Radom, J. Phys. Chem. A, 2003, 107, 6082 CrossRef; (b) B. Giese and S. Lachhein, Angew. Chem., Int. Ed., 1982, 21, 768 CrossRef.
- (a) K. Ohmatsu and T. Ooi, Nat. Synth., 2023, 2, 209 CrossRef; (b) Z. Ye, Y. Yu, Y.-M. Lin, Y. Chen, S. Song and L. Gong, Nat. Synth., 2023, 2, 766 CrossRef; (c) H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed; (d) M.-J. Zhou, L. Zhang, G. Liu, C. Xu and Z. Huang, J. Am. Chem. Soc., 2021, 143, 16470 CrossRef CAS.
- (a) V. Murugesan, A. Muralidharan, G. Anantharaj, T. Chinnusamy and R. Rasappan, Org. Lett., 2022, 24, 8435 CrossRef CAS PubMed; (b) V. Murugesan, A. Ganguly, A. Karthika and R. Rasappan, Org. Lett., 2021, 23, 5389 CrossRef CAS PubMed.
- T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2021, 122, 2650 CrossRef.
- (a) I. Perry, T. Brewer, P. Sarver, D. Schultz, D. DiRocco and D. MacMillan, Nature, 2018, 560, 70 CrossRef CAS; (b) R. Pilli, K. Selvam, B. S. S. Balamurugan, V. Jose and R. Rasappan, Org. Lett., 2024, 26, 2993 CrossRef CAS; (c) X. Hu, I. Cheng-Sánchez, W. Kong, G. A. Molander and C. Nevado, Nat. Catal., 2024, 7, 655–665 CrossRef CAS PubMed.
- D. P. Hickey, C. Sandford, Z. Rhodes, T. Gensch, L. R. Fries, M. S. Sigman and S. D. Minteer, J. Am. Chem. Soc., 2019, 141, 1382 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04651j |
| This journal is © The Royal Society of Chemistry 2024 |