
Upcycling of waste polyesters for the development of a circular economy
Huaiyuan
Zhao†
ab,
Yingdan
Ye†
a,
Yibin
Zhang
a,
Lei
Yang
b,
Weichen
Du
b,
Songlin
Wang
*b and
Zhaoyin
Hou
 *ab
*ab
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, Department of Chemistry, Zhejiang University, Hangzhou 310028, China. E-mail: zyhou@zju.edu.cn; Fax: 86-571-88273283; Tel: 86-571-88273283
bZhejiang Hengyi Petrochemical Research Institute Co., Ltd, Hangzhou 311200, China
First published on 30th October 2024
Abstract
The rapidly increasing production and widespread application of plastics have brought convenience to our lives, but they have consumed a huge amount of nonrenewable fossil energy, leading to additional CO2 emissions and generation of an enormous amount of plastic waste (also called white pollution). Chemical recycling and upcycling of waste plastic products (also called waste plastic refineries) into recycled monomers and/or valuable chemicals can decrease the dependence on fossil energy and/or reduce the emission of CO2, enabling the full utilization of carbon resources for the development of a circular economy. Polyesters, a vital class of plastics, are ideal feedstocks for chemical recycling due to the easily depolymerizable ester bonds compared to polyolefins. Among them, polyethylene terephthalate (PET) is the most widely used product, making its chemical recycling to a circular carbon resource a hot topic with significant concerns. In this feature article, recent progress in depolymerization of waste polyesters (PET and/or PET-containing materials) and the subsequent upgrading of depolymerized monomers (or intermediates) to valuable chemicals was reviewed and prospected. Newly reported technologies, such as thermal catalysis, photocatalysis, electrocatalysis, and biocatalysis, were discussed. The achievements, challenges, and potential of industrial applications of chemical recycling of polyesters were addressed.
1. Introduction
Plastics have various advantages, including light weight, outstanding chemical stability, excellent impact resistance and high abrasion resistance. Additionally, the convenience of molding, coloring and processing of plastics has led to their rapidly increased production and application across a wide range of fields, making them indispensable materials in our life (Fig. 1A).1–4 Since 1950, annual production of plastics has surged from just 2 million tons to nearly 390 million tons in 2021.5 More importantly, plastic production continues to rise and is expected to reach 1.2 billion tons per year by 2060.6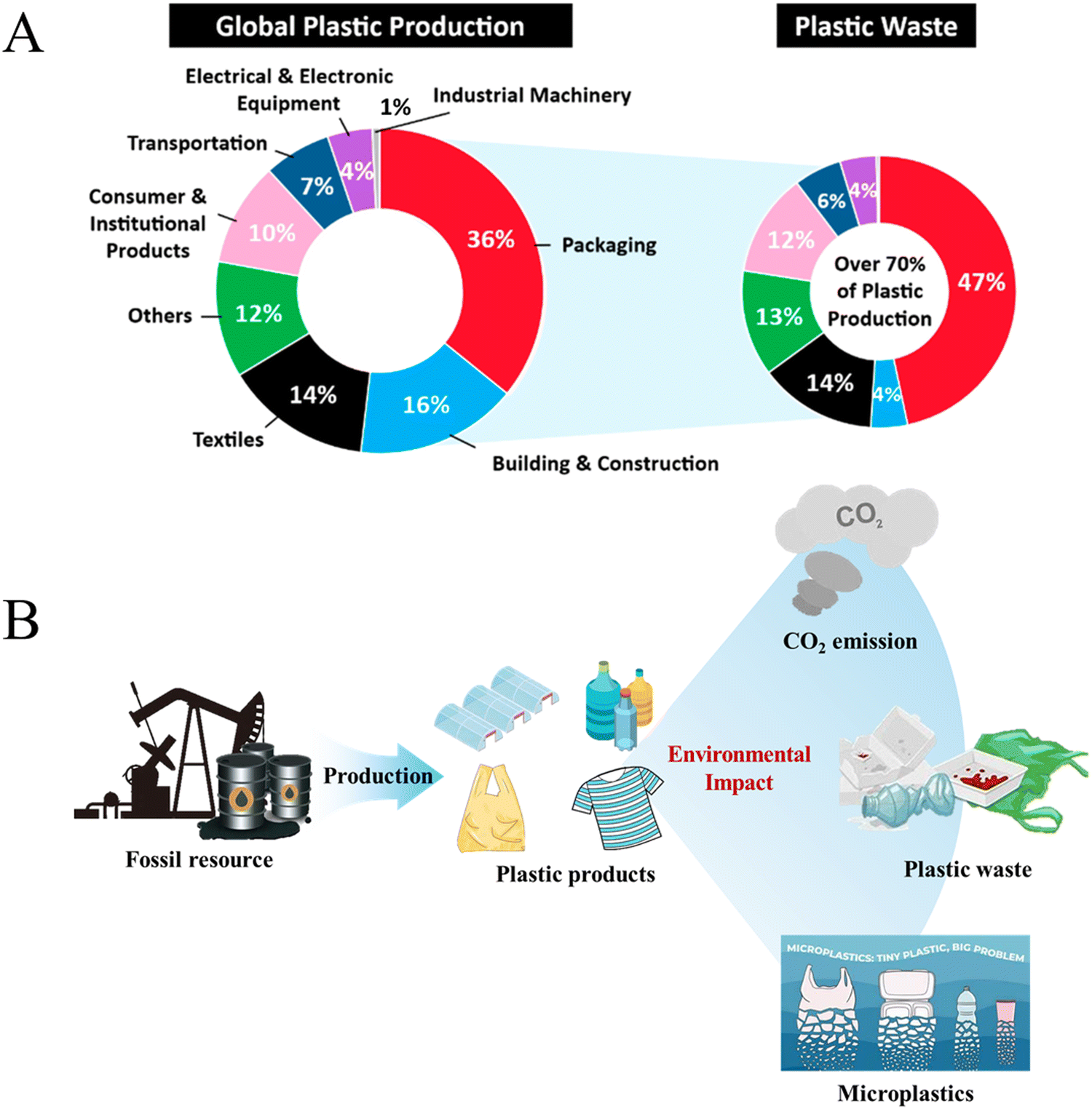 | ||
| Fig. 1 (A) Applications of plastics. Reproduced from ref. 4 with permission from Elsevier, copyright [2023]. (B) The present life cycle of plastics. | ||
Along with the increasing production and demand for plastics, associated energy and environmental challenges are becoming pronounced.7,8 Currently, the vast majority of plastic feedstocks are nonrenewable fossil resources, and the share of plastic production in global oil consumption is expected to rise from 6% in 2014 to 20% by 2050.9 At the same time, the plastic production process also releases significant amounts of greenhouse gases, and it was projected that global greenhouse gas (GHG) emissions from the plastic life cycle will increase from 1.7 billion tons of CO2 equivalent in 2015 to 6.5 billion tons of CO2 equivalent by 2050, contributing substantially to climate change (Fig. 1B).10 Therefore, more sustainable feedstocks, such as biomass and recycled waste plastics, are being considered as the ideal substitutes for the next generation of plastics.
A significant proportion of plastic products have a short service life, with about 40% being discarded after a single use or within one month, and this short lifespan leads to severe pollution and resource wastage caused by plastic waste. Under natural conditions, plastics can break down gradually into fragments of varying sizes due to factors such as weathering, ultraviolet radiation and human activities (Fig. 1B).11 Among these fragments, microplastics (MPs, 1 μm–5 mm) constitute 92.4% of plastic debris.12 These microplastics can enter organisms and accumulate through the food chain over time, leading to a series of harmful diseases due to bioaccumulation.13,14 Recycling of waste plastics has become an urgent issue concerning resource consumption and environmental remediation all over the world.15–18
Polyolefins, such as polyethylene (PE), polypropylene (PP), and polystyrene (PS), are the most widely used petroleum-based polymers, accounting for over 75% of global polymer production (Fig. 2A).19 Polyolefin monomers are typically linked by stable C–C bonds, making the depolymerization of polyolefins into monomers or chemicals difficult under mild reaction conditions due to the stability and similarity of C–C bonds.20,21 Polyesters, including polyethylene terephthalate (PET), poly(lactic acid) (PLA), poly(3-hydroxybutyrate) (PHB), polycaprolactone (PCL), and poly(ethylene-2,5-furandicarboxylate) (PEF), are the second largest class of plastic materials after polyolefins, valued for their durability, processability, and low cost.22–24 Among these polyesters, PET, the primary representative of polyester plastics, accounts for about 8% of global plastic production (Fig. 2A).19 PET is widely used in the packaging industry because of its excellent barrier properties for oxygen, carbon dioxide and moisture.25,26 At the same time, PET fiber has the characteristics of lightness, durability and wear resistance, possessing a broad application space in textile and automobile manufacturing industries (Fig. 2B).27–29 The market share of PET fiber was 54% of the global fiber production in 2021.30 Unfortunately, the service life of the PET-based products is short; in this case, using waste PET-based products as raw materials and/or the source of carbon instead of nonrenewable fossil energy is of great importance for the development of a circular economy. PLA, one of the most commercially promising degradable plastics, has excellent physical properties and processing properties and is extensively used in packaging, fiber and 3D printing fields.31,32 Unlike polyolefins, polyesters can be more easily depolymerized into their monomers or other valuable chemicals through highly selective C–O bond breakage.33,34 However, approximately 85% of polyester waste is currently incinerated, sent to landfills, or discarded, with only 15% being recycled, most of which is downcycled into lower-value applications.30
 | ||
| Fig. 2 (A) Worldwide plastic production classified by the plastic type. Reproduced from ref. 19 with permission from Royal Society of Chemistry, copyright [2021]. (B) Market segments of PET-based products. Reproduced from ref. 29 with permission from Royal Society of Chemistry, copyright [2024]. | ||
Improving the recovery and recycle efficiency of waste polyesters is undoubtedly crucial for reducing plastic pollution and decreasing over-dependence on fossil resources. Chemical recycling and upcycling of waste polyesters have garnered increasing attention because they can promote the sustainable development of the plastics industry and enhance economic efficiency.35–40 In this feature article, the current recycling technologies of waste PET-based products were introduced in short, recent progress in chemical recycling routines and catalysts was reviewed in detail, and direct depolymerization of a wide range of PET-based materials (including waste clothes, bottles, packing materials, films and mixed plastics) and the subsequent upgrading of depolymerized products were emphasized. The newly reported one-pot conversion of waste polyesters into high-valued chemicals via thermal catalysis, photocatalysis, electrocatalysis, and biocatalysis was also introduced. Additionally, outlook and perspectives on the need for improvement and the industrial applications of chemical upcycling of waste polyesters were proposed, and we hope this review can inspire the development of more advanced strategies to address the challenges posed by waste polyesters.
2. Current recycling technologies of waste PET-based products
Until 1980s, most PET-based waste products were directly disposed via landfills and incineration, which brought a great risk to the environment and ecosystem as toxic substances, incineration flue gases and microplastic particles were released into the earth, air and oceans. Then, the concept of recycling waste PET began to sprout and gained ground as the production of PET-based products increased rapidly. In the past few decades, primary recycling, mechanical recycling and energetic recycling were developed and industrialized in many countries. More recently, chemical recycling and/or upcycling have attracted more and more attention. These achievements and progress in the above technologies were well reviewed in recently published works.41–43Primary recycling, also known as closed-loop recycling, is mainly adopted for uncontaminated polyester waste, using melting to make it a raw material for the manufacture of new polyesters. This type of recycling mainly takes place inside the production plant to ensure that the recycling process is pure and efficient, which is not suitable for the recycling of polyester waste produced in daily life.44
Mechanical recycling is the most popular method for recycling plastics because of its simplicity and low cost, and it involves collection, sorting, shredding, melting, and further production of secondary raw materials. It was reported that up to 83% of the plastic waste of the plastic-producing industry in Germany could be recycled via mechanical recycling in 2021.41 But this technology alone was only suitable for thermoplastic polymers, as the purity and cleanliness of raw feedstock are required. In this case, mainly postindustrial polyester waste is recycled, and most recycled products are added to another manufacturing process and partly replace raw materials. On the other hand, mechanical recycling of postconsumer polyester waste is difficult as it is often heavily contaminated with dust, oil or biological substances. Besides, the recycled polyester is impure due to mixing with other kinds of wastes together and the unreliable separation process. Ensuring the separation of each type of plastic is a key step during the process of mechanical recycling. At the same time, the melting process also damages the integrity of primary polyesters, which decreases their quality to lower value products.45
Energetic recycling, also known as incineration, can convert plastic waste into heat and debris and emit gases.46 Energy balance closure and sensitivity analyses indicated that the heat released from the combustion of PE and PP reached 42.98 and 44.00 MJ kg−1, respectively.47 However, this process is not suitable for polyesters with low energy density and halogen containing polymers, such as PVC, as the combustion process will emit toxic halogen gases and large amounts of GHG.48
Chemical recycling can depolymerize waste polyesters into monomers through chemical reactions, which can then be resynthesized to new polyesters or other types of materials.42 This method of recycling is highly flexible and efficient in dealing with different sources and various types of waste polyesters via the corresponding chemical path. According to the reaction medium, it can be divided into hydrolysis, glycolysis, ammonolysis and alcoholysis.29 Subsequent chemical upgrading can turn these depolymerized products into high-value chemicals.
It can be concluded that the above treatment and/or the recycling path of waste polyesters have their own characteristics and applicable scenarios. Life cycle assessment (LCA), technical and economic analysis (TEA), or other relevant sustainability metric determination can be used to critically evaluate the suitability of various recycling methods under certain conditions. Present primary recycling, mechanical recycling and energetic recycling of postindustrial polyester waste are simple and economically viable, but insufficient for achieving a sustainable circular economy. Chemical recycling of polyester waste can realize the recycling and upgrading of resources, which is more in line with the requirements of a circular economy.
3. Recent progress in chemical recycling routines and catalysts
Polyesters have been extensively studied as potential feedstocks for depolymerization into monomers, oligomers, or valuable molecules, primarily due to the chemical susceptibility of their ester groups. Various solvents and/or catalysts have been successfully developed for the depolymerization of polyesters, including water, alcohols, and amines.193.1. Hydrolysis of polyesters
Hydrolysis of PET with water (H2O) can produce terephthalic acid (PTA) and ethylene glycol.49 PTA, a commodity chemical, is currently produced from petroleum based p-xylene via oxidation and refinement.50 In this case, hydrolyzing waste PET to produce PTA can reduce the dependence on fossil resources and enable the recycling of PET.51 Thirty years ago, Campanelli et al. carried out the hydrolysis of PET with a large volume of water and found that PET could be hydrolyzed completely to PTA within 2 h at 265 °C.52 Recently, Onwucha et al. reported that the yield of PTA increased from ∼86 to ∼98% with the hydrolysis time prolonged from 6 to 24 h at 200 °C in pure water (the PET/water ratio was 0.35).53 Additionally, the hydrolysis of PLA, PHB, and PCL under neutral conditions can also yield the corresponding monomers with over 80% efficiency at high temperature (>200 °C).54–56 However, most polyesters have high crystallinity and strong hydrophobicity, making it difficult to dissolve them in water; at the same time, the hydrolysis of ester bonds in water requires a high activation energy, leading to the need for high reaction temperature.Unlike the above catalyst-free hydrolysis process, catalysis can reduce the activation energy of PET hydrolysis and accelerate the reaction rate effectively; especially, acidic hydrolysis of PET can produce high-purity PTA and EG. Besides inorganic acids (such as sulfuric acid and phosphoric acid),57 organic acids (like p-toluenesulfonic acid monohydrate (pTSA), 2-naphthalenesulfonic acid (2-NSA), and 1,5-naphthalenedisulfonic acid tetrahydrate (1,5-NDSA)) have also been proven to be effective for the hydrolysis of PET.58 Yang et al. found that using PTA, the basic unit of PET, as an acid catalyst can also promote the hydrolysis of PET, and it can avoid generation of acidic waste.59 This approach achieved a PTA yield of 95.5% with a high purity at 220 °C in 3 h, eliminating the need for complex purification steps.
Another promising process involves using heterogeneous catalysts. Kang et al. conducted microwave-assisted PET hydrolysis to recover PTA using ZSM-5 catalysts with different Si/Al ratios and ionic types.60 It was found that H@ZSM-5-25, which has the highest total acid content and primarily Brønsted acidic sites, achieved nearly 100% PTA yield when PET was hydrolyzed at 230 °C for 30 min. The activation energy over H@ZSM-5-25 was only 1.2 kJ mol−1, which is much lower than that required without a catalyst (19.64 kJ mol−1, Fig. 3A). The mechanism of PET depolymerization via hydrolysis in the presence of Brønsted acid catalysts, such as H@ZSM-5-25, is illustrated in Fig. 3B, that is, the generated hydroxonium ions reacted with the carbonyl groups of PET to form hydroxyl groups at first, resulting in the formation of a carbo-cation. Next, water launched a nucleophilic attack through proton transfer, conforming to thermodynamic stability. Finally, further steps followed the typical hydrolysis process of an ester, forming PTA and EG.
 | ||
| Fig. 3 (A) Schematic of the hydrolysis of PET over ZSM-5 assisted by microwaves and 1/T − ln(k) curves of H+@ZSM-5-25 and under catalyst-free conditions. (B) PET hydrolysis mechanism with an acid catalyst. Reproduced from ref. 60 with permission from Elsevier, copyright [2020]. (C) Schematic of the acid hydrolysis of PET over PSSA and 0.5 M, 1 M, 2 M solutions of PSSA on the PET surface (a)–(c) and 0.5 M, 1 M, and 2 M solutions of H2SO4 on the PET surface (d)–(f). Reproduced from ref. 61 with permission from Elsevier, copyright [2022]. | ||
Lately, Abedsoltan et al. disclosed that poly(4-styrenesulfonic acid) (PSSA) as a catalyst exhibited a short induction time and high catalytic activity for the acidic hydrolysis of PET, attributed to the hydrophobic nature of PSSA, which enhanced wettability and adsorption on PET surfaces (Fig. 3C).61
Besides the popularly reported hydrolysis of PET, Yu et al. discovered that PHB (an expensive bio-based polyester) could be hydrolyzed to its monomer (3-hydroxybutyric acid) with concentrated sulfuric acid (80–98 wt%), and the yield of 3-hydroxybutyric acid reached 50% at 70 °C within 0.5 h. However, 3-hydroxybutyric acid gradually dehydrated to crotonic acid (with up to a 90% yield) as the reaction progressed, due to the carbonizing and dehydrating effects of the concentrated sulfuric acid.62
Simultaneously, many research studies disclosed that hydrolysis of polyesters with alkalies (such as NaOH, KOH, Na2CO3 and K2CO3) can be conducted at low temperature, but additional acidification and separation steps are indispensable to get free PTA and EG (Fig. 4).63,64 Mishra et al. carried out the alkaline hydrolysis of PET in an aqueous NaOH solution, where EG was recovered using salting-out technology and PTA was obtained via the acidification of disodium terephthalate. The yield of PTA and EG was almost equivalent to the conversion of PET at 90–150 °C for 90 min (and reached 98.46% at 150 °C).65 More recently, several works found that tetrahydrofuran (THF), dimethyl isosorbide (DMI), dimethylsulfoxide (DMSO), 1,3-dimethyl-2-imidazolidinone, and 1,4-dioxane can be used as co-solvents to enhance the depolymerization of PET.64,66,67 At the same time, it was found that NaOH can also accelerate the hydrolysis of PLA68 and PHB62 to their monomer (L-lactic acid and 3-hydroxybutyric acid). These results confirmed that alkaline hydrolysis can reduce the reaction temperature obviously, but this process requires subsequent acidification, resulting in the formation of large amounts of salt.
 | ||
| Fig. 4 Experimental procedure of PET alkaline hydrolysis. Reproduced from ref. 64 with permission from American Chemical Society, copyright [2023]. | ||
Subcritical and supercritical fluids, as effective reaction media, were also used to enhance the performance of polyester hydrolysis.69 Colnik et al. found that the yield of PTA reached 90% when the hydrolysis of PET was carried out in the subcritical region (300 °C) for 30 min, but isophthalic acid, benzoic acid, acetaldehyde, 1,4-dioxane and CO2 formed easily in supercritical water when the reaction temperature and time exceeded 350 °C and 30 min. The energy consumption of subcritical hydrothermal degradation of PET needs high costs due to the heating of large amounts of water. Calculated electricity consumption for hydrothermal degradation of 1 kg of PET waste was estimated to be 36.2 kW h on the laboratory scale and 4.3 kW h on the industrial scale.70 When tungsten-facilitated TiO2 solid acid was added in the hydrolysis of waste PET bottles in supercritical CO2, they can be hydrolyzed to PTA, EG, and diethylene glycol (DEG) completely within 15 h at 160 °C and 15 MPa.71 The subcritical mixture of CO2–H2O is also efficient for the hydrolysis of PET, achieving 85% yield of PTA at 200 °C for 100 min. More interestingly, the performance of this process remained the same even when polyolefins and/or colorants appeared in the discarded PET.72
The achievements of the above works in the hydrolysis of PET are summarized in Table 1, and it can be found that monomers can be obtained via hydrolysis of polyesters, but the reaction conditions are harsh. The addition of acid/base catalysts and the use of supercritical/subcritical methods and other types of treatments or co-solvents can effectively accelerate the hydrolysis of polyesters under mild conditions, and more efforts are essential in order to pave the way for economical utilization.
| Catalyst | Temp. (°C) | Time (min) | Conv. (%) | YieldPTA (%) | Methods or co-solvents used | Ref. |
|---|---|---|---|---|---|---|
| — | 200 | 1440 | — | 98.0 | — | 53 |
| HNO3 | 120 | 140 | 87.4 | 87.2 | — | 73 |
| pTSA | 150 | 180 | — | >90 | — | 58 |
| 2-NSA | 150 | 180 | — | >90 | — | 58 |
| 1,5-NDSA | 150 | 480 | — | >90 | — | 58 |
| Terephthalic acid | 220 | 180 | 100.0 | 95.5 | — | 59 |
| H@ZSM-5-25 | 230 | 30 | 100.0 | 100.0 | Micro-wave | 60 |
| PSSA | 150 | 1440 | 100.0 | 92.0 | — | 61 |
| NaOH | 80 | 20 | 95.2 | ∼95 | — | 63 |
| NaOH | 150 | 90 | 98.5 | 98.5 | — | 65 |
| NaOH | 95 | 60 | — | 84.0 | Trioctylmethylammonium bromide | 74 |
| NaOH | 110 | 120 | ∼100 | ∼100 | [C16H33N(CH3)3]3PW12O40 | 75 |
| NaOH | 90 | 45 | 100.0 | 99.0 | Ultrasound, tetrabutyl ammonium iodide | 76 |
| NaOH | 60 | 60 | 98.0 | 98.0 | EG/DMSO | 64 |
| NaOH | 60 | 60 | 91.0 | 91.0 | EG/1,3-dimethyl-2-imidazolidinone | 64 |
| NaOH | 60 | 60 | 58.0 | 58.0 | EG/1,4-dioxane | 64 |
| KOH | 60 | 60 | 100.0 | 97.5 | EG/THF | 66 |
| KOH | 100 | 30 | 100.0 | 99.6 | EG/DMI | 67 |
| — | 300 | 30 | — | 90.0 | Sub- and supercritical water | 70 |
| WOx/TiO2 | 160 | 900 | 100.0 | — | Supercritical CO2 | 71 |
| — | 200 | 100 | 99.0 | 85.0 | Subcritical CO2–H2O | 72 |
3.2. Glycolysis of polyesters
Glycolysis refers to the reaction of polyesters in a diol solution (such as EG, DEG, propylene glycol, etc.) to produce monomers that contain terminal hydroxyl groups. The glycolysis reaction routes of PET, PEF and PLA in EG or DEG are shown in Fig. 5.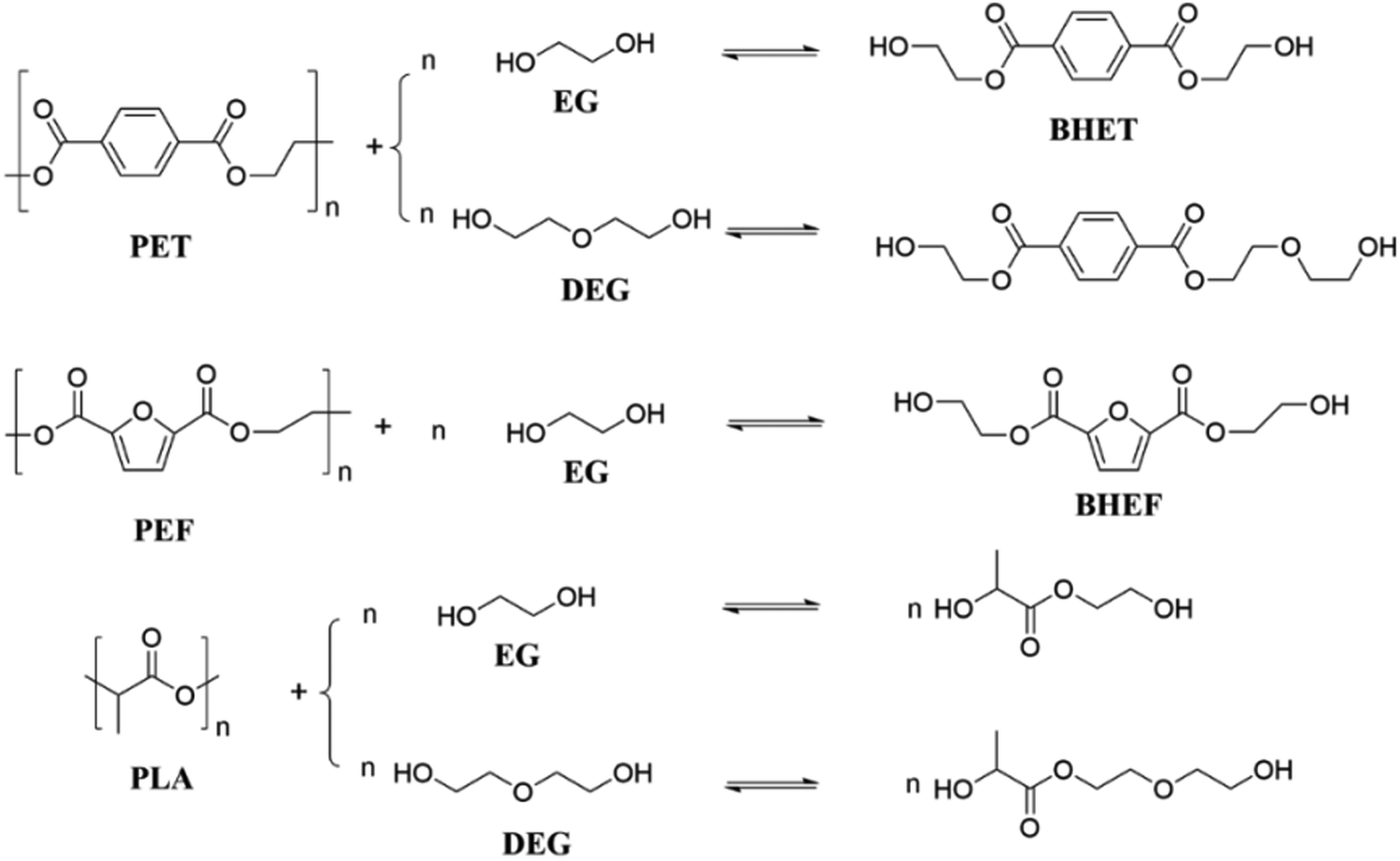 | ||
| Fig. 5 Glycolysis of PET, PEF and PLA with EG or DEG. | ||
Glycolysis of PET with EG is one of the most straightforward methods for commercializing PET recycling. The recovered bis(2-hydroxyethyl) terephthalate (BHET) can be repolymerized into new polyesters or upgraded into other high-value chemicals. Several international chemical companies, such as DuPont, Dow Chemical, and Shell, have established facilities dedicated to this process. PET and excess EG are transformed into BHET via transesterification reaction at temperatures ranging from 170 to 300 °C. Both homogeneous and heterogeneous catalysts have been found to promote the glycolysis of PET.
Metal acetates, such as Zn(OAc)2, Mn(OAc)2, and Co(OAc)2, are the most commonly reported homogeneous catalysts for the glycolysis of PET.37 Mohammadi et al. found that the combined use of Zn(OAc)2 and microwave irradiation reduced the reaction time to 5 min at 240 °C and 400 W, achieving a BHET yield of 96.3%. When Sb2O3 was added as the catalyst, the yield of oligomers reached 96.7% (Fig. 6A).77 Additionally, metal chlorides, metal carbonates, and metal sulfates have also been employed as catalysts for the glycolysis of PET.42,78,79
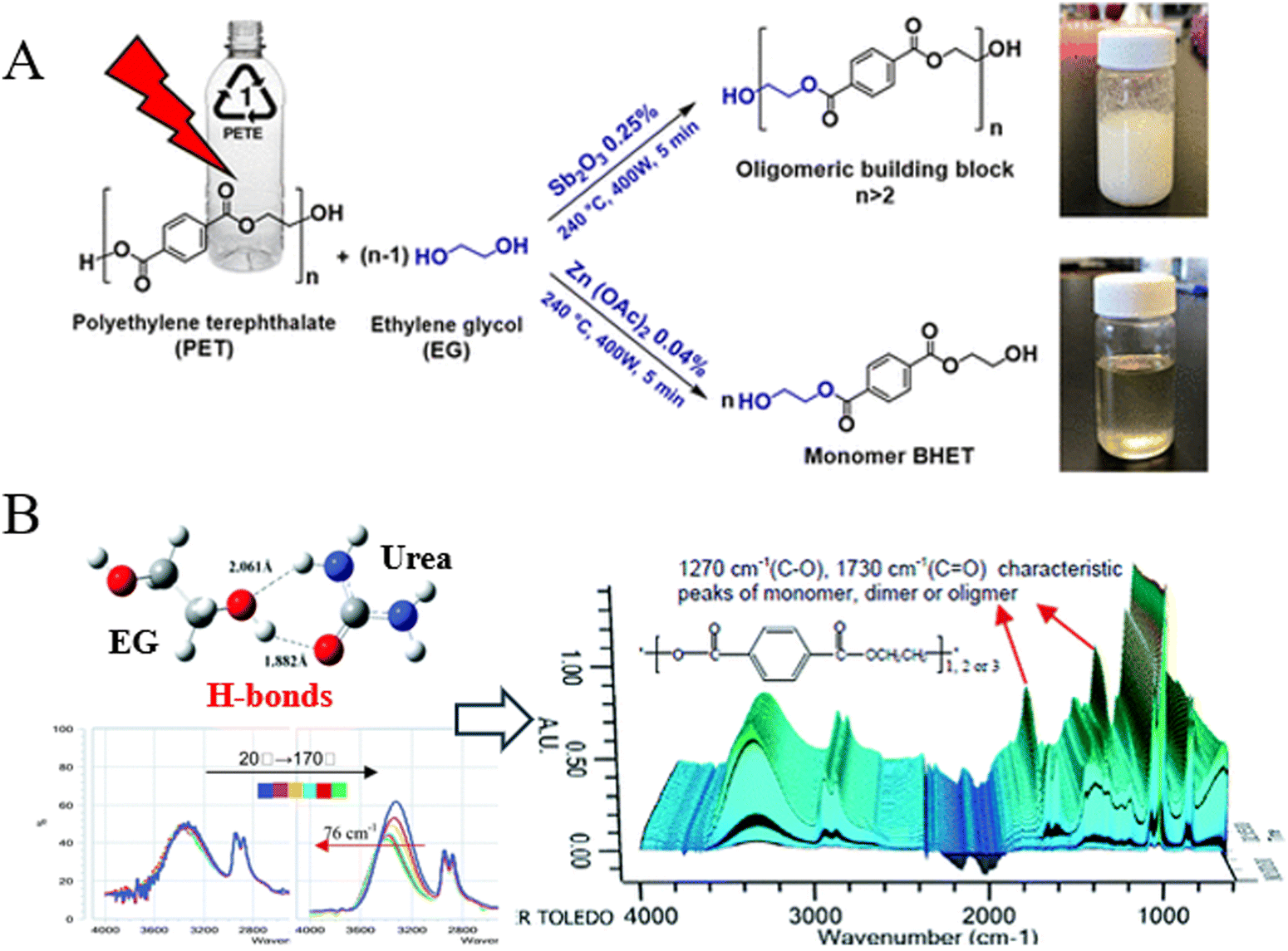 | ||
| Fig. 6 (A) Microwave-assisted glycolysis of PET with EG in the presence of Sb2O3 and Zn(OAc)2. Reproduced from ref. 77 with permission from American Chemical Society, copyright [2023]. (B) Schematic of the hydrogen bond formed between EG and urea promoting the glycolysis of PET. Reproduced from ref. 80 with permission from Royal Society of Chemistry, copyright [2012]. | ||
Besides the above salts and oxides, Wang et al. found that urea was an efficient green catalyst for the glycolysis of PET, achieving 100% conversion of PET with 77.7% selectivity toward BHET when the ratio of mPET![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mEG:murea in the feed was 1:4:0.1 at 180 °C for 3 h, attributed to the hydrogen bond formed between EG and urea, which promotes the glycolysis of PET (Fig. 6B). Moreover, urea remained effective even after ten recycles.80 At the same time, it was also reported that cyanamide served as an effective catalyst for the glycolysis of PET, achieving a BHET yield of 95.21% at 190 °C within 2.5 h.81 Ionic liquids (ILs), as a new class of solvents, have been shown to be promising for the glycolysis of PET. In 2010, it was reported that 1-butyl-3-methylimidazolium tetrachloroferrate [bmim][FeCl4] exhibited a higher activity than FeCl3 and [bmim]Cl for the glycolysis of PET due to the synergistic effect of its cation and anion.82 The cation of this ionic liquid, [bmim]+, interacts with the carbonyl oxygen (C
:mEG:murea in the feed was 1:4:0.1 at 180 °C for 3 h, attributed to the hydrogen bond formed between EG and urea, which promotes the glycolysis of PET (Fig. 6B). Moreover, urea remained effective even after ten recycles.80 At the same time, it was also reported that cyanamide served as an effective catalyst for the glycolysis of PET, achieving a BHET yield of 95.21% at 190 °C within 2.5 h.81 Ionic liquids (ILs), as a new class of solvents, have been shown to be promising for the glycolysis of PET. In 2010, it was reported that 1-butyl-3-methylimidazolium tetrachloroferrate [bmim][FeCl4] exhibited a higher activity than FeCl3 and [bmim]Cl for the glycolysis of PET due to the synergistic effect of its cation and anion.82 The cation of this ionic liquid, [bmim]+, interacts with the carbonyl oxygen (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the ester of PET; meanwhile, the anion of the catalyst, [FeCl4]−, interacts with the hydrogen in the hydroxyl group of ethylene glycol, making the oxygen of ethylene glycol more negative and the attack on the carbon cation of the ester group easier. This synergic effect made the depolymerization of PET over the [bmim][FeCl4] catalyst much easier. In 2015, the same group further found that [bmim]2[CoCl4] exhibited superior catalytic activity for the glycolysis of PET under mild conditions (175 °C, 1.5 h), achieving a complete conversion of PET with 81.1% yield of BHET.83 Recently, Liu et al. developed a series of metal-free cholinyl ILs, which are cheaper and environmentally friendly compared to traditional imidazole-based ILs, and they found that the yield of BHET was as high as 85.2% at 180 °C for 4 h with 5 wt% choline acetate IL ([Ch][OAc]).84
O) in the ester of PET; meanwhile, the anion of the catalyst, [FeCl4]−, interacts with the hydrogen in the hydroxyl group of ethylene glycol, making the oxygen of ethylene glycol more negative and the attack on the carbon cation of the ester group easier. This synergic effect made the depolymerization of PET over the [bmim][FeCl4] catalyst much easier. In 2015, the same group further found that [bmim]2[CoCl4] exhibited superior catalytic activity for the glycolysis of PET under mild conditions (175 °C, 1.5 h), achieving a complete conversion of PET with 81.1% yield of BHET.83 Recently, Liu et al. developed a series of metal-free cholinyl ILs, which are cheaper and environmentally friendly compared to traditional imidazole-based ILs, and they found that the yield of BHET was as high as 85.2% at 180 °C for 4 h with 5 wt% choline acetate IL ([Ch][OAc]).84
As heterogeneous catalysts, transition metal-substituted polyoxometalates (POMs) K6SiW11MO39(H2O) (M = Zn2+, Mn2+, Co2+, Cu2+, Ni2+) have also shown excellent activity for the glycolysis of PET under mild conditions.85 Among them, a small amount of K6SiW11ZnO39(H2O) (with a 0.13% catalyst-to-PET molar ratio) can achieve complete depolymerization of PET in 0.5 h at 185 °C, with BHET yield exceeding 84%. Additionally, this catalyst could be recycled up to 8 times without significant loss of activity. Fang et al. also developed a class of POMs (Na12[WZnM2(H2O)2(ZnW9O34)2]) with a sandwich structure and multiple transition metal active sites (M = Zn2+, Mn2+, Co2+, Cu2+, Ni2+) for the glycolysis of PET, and it was found that these catalysts demonstrated a better activity than traditional heteropolyacid catalysts.86
Zeolites and metal oxides also play important roles in the glycolysis of PET. In 2008, Shukla et al. found that the yield of BHET reached 65% over a Y-type zeolite (Si/Al = 0.8) at 196 °C after 8 h.87 Recently, Mo et al. reported that Ti doped SBA-15 (Ti/SBA-15) can increase the yield of BHET to 87.2% at 190 °C within 45 min (Fig. 7A).88 Yun et al. demonstrated that CeO2 NPs with a size of 2.7 nm exhibited excellent catalytic performance, achieving a PET conversion rate of 98.6% and a BHET yield of 90.3% at 196 °C within 15 min.89 It is proposed that the oxygen-enriched defects in CeO2 NPs induced the production of Ce3+, providing activation sites that significantly accelerated the glycolysis reaction (Fig. 7B). In detail, the oxygen of the carbonyl group (CO) in PET was activated over the Ce3+ Lewis acid site of defective CeO2 to make the carbon of the carbonyl group electropositive. Then, free lone pairs of electrons on the oxygen of ethylene glycol attacked the carbon cation in ester linkages. Finally, the C–O bond in PET was broken and a new C–O bond with EG formed. As a result, PET is converted to oligomers and then intermediate products are further transformed into the BHET monomer. The oxygen-rich defects in CeO2 NPs induced the production of Ce3+, providing activation sites that significantly accelerated the glycolysis reaction. Cao et al. disclosed that over O2-assisted defect-rich ZnO the conversion rate of PET (100%) and the yield of BHET (92.4%) in air were 3.5 and 10.6 times higher, respectively, than in nitrogen, which was attributed to the abundance of reactive oxygen species and defects on the (100) crystal surface of ZnO in air.90 They further synthesized Mo/ZnO nanosheets for high-efficiency glycolysis of PET, where the doped Mo replaced Zn atoms at defect sites, forming Mo–Zn bonds. This increased the polarity of the adsorbed O2 molecules, which can facilitate electron transfer and accelerate the fracture of C–O bonds in PET (Fig. 7C).91
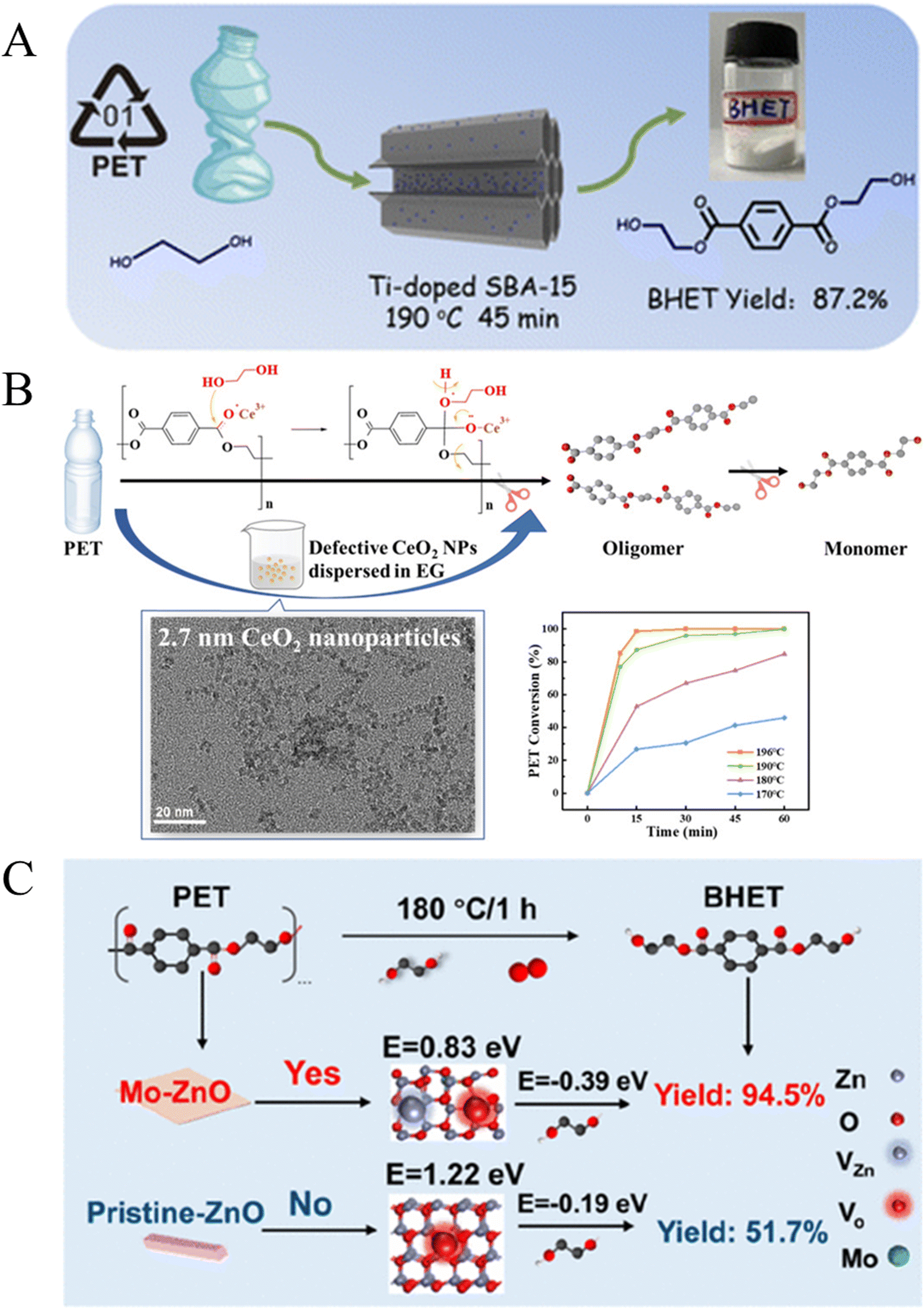 | ||
| Fig. 7 (A) Schematic illustration of the glycolysis of PET over Ti/SBA-15. Reproduced from ref. 88 with permission from Royal Society of Chemistry, copyright [2023]. (B) Ultrasmall CeO2 nanoparticles with oxygen-rich defects for the glycolysis of PET. Reproduced from ref. 89 with permission from American Chemical Society, copyright [2022]. (C) Acceleration of PET glycolysis by ultrathin ZnO nanosheets with Mn doping. Reproduced from ref. 91 with permission from American Chemical Society, copyright [2022]. | ||
In addition, Gabirondo et al. performed the glycolysis of PEF using a mixed catalyst of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and benzoic acid (BA); it was found that the yield of bis(2-hydroxyethyl) furanoate (BHEF) was 92% using a 7.5 wt% catalyst at 180 °C for 1.5 h. The recovered BHEF could be re-polymerized into PEF via polycondensation, enabling the closed recycling of PEF.92 The hydroxy-terminated compounds obtained from the glycolysis of PLA can be used as macromolecular crosslinkers for epoxy natural rubber (ENR) or as curing precursors for the production of thermosetting polyesters.93 More recently, Wu et al. reported that by premixing zinc acetate and cheap alkalis in ethylene glycol, the yield of BHET could reach 86.4% with a complete glycolysis of PET at 180 °C within 2 h, and this performance was attributed to the free hydroxide ions that could enhance the nucleophilicity of oxygen in ethylene glycol and make it easy to attack carbonyl groups, and also accelerate the swelling and dissolution of PET. At the same time, the in situ generated Zn-glycolate and zinc oxide nanoparticles would activate the oxygen in the carbonyl group, making the carbon cations more electropositive. This process can significantly reduce industrial costs and has been found to be promising for achieving large-scale industrial applications.94
In summary, various catalysts, such as metal acetates, organic catalysts, ionic liquids, polyoxometalates, zeolites and metal oxides, have been tested to accelerate the glycolysis of PET and the results are summarized in Table 2. It can be found that glycolysis is one of the most promising methods for industrial-level chemical recycling of end-of-life PET. This process can be carried out at atmospheric pressure and low temperature, and the BHET product is easily separated via crystallization, presenting unique advantages. We think that more efforts are needed in the future to enhance the yield of BHET with low EG/PET in the feed and find efficient technologies to treat real polyester wastes produced in our daily life.
| Catalyst | Cat:PET (wt%) |
Mass ratio EG/PET | Temp. (°C) | Time (min) | Conv. (%) | YieldBHET (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Zn(OAc)2 | — | 2.5 | 240 | 5 | 100.0 | 96.3 | 77 |
| Urea | 10.0 | 4.0 | 180 | 180 | 100.0 | 77.7 | 80 |
| Cyanamide | 5.0 | 11.0 | 190 | 150 | — | 95.2 | 81 |
| [bmim][FeCl4] | 20.0 | 4.0 | 178 | 240 | 100.0 | 59.2 | 82 |
| [bmim]2[CoCl4] | 16.7 | 7.0 | 175 | 90 | 100.0 | 81.1 | 83 |
| [Ch][OAc]) | 5.0 | 4.0 | 180 | 240 | 98.2 | 85.2 | 84 |
| NaOH/Zn(OAc)2 | 2.0 | 1.9 | 180 | 120 | 100.0 | 86.4 | 94 |
| Urea/ZnCl2 | 5.0 | 4.0 | 170 | 30 | 100.0 | 83.0 | 95 |
| 1,3-Dimethylurea/Zn(OAc)2 | 5.0 | 4.0 | 190 | 20 | 100.0 | 82.0 | 96 |
| K6SiW11ZnO39(H2O) | 2.0 | 4.0 | 185 | 30 | 100.0 | 84.1 | 85 |
| Na12WZn3(H2O)2(ZnW9O34)2 | 0.5 | 4.0 | 190 | 40 | 100.0 | 84.5 | 86 |
| Y-Zeolite | 1.0 | 6.0 | 196 | 480 | — | 65.0 | 87 |
| Ti/SBA-15 | 12.5 | 7.0 | 190 | 45 | 99.9 | 87.2 | 88 |
| CeO2-2.7 nm | 1.0 | 7.0 | 196 | 15 | 98.6 | 90.3 | 89 |
| O2-assisted defect-rich ZnO | 1.0 | 4.0 | 180 | 60 | 100.0 | 92.4 | 90 |
| Mo/ZnO | 1.0 | 4.0 | 180 | 60 | 100.0 | 94.5 | 91 |
| Fe3O4 | 2.0 | 13.0 | 210 | 30 | 100.0 | >93 | 97 |
| ZnO–Fe3O4 | 1.0 | 6.0 | 190 | 30 | 100.0 | 92.3 | 98 |
| ZIF-8 | 1.0 | 5.0 | 197 | 90 | 100.0 | 77.0 | 99 |
| MAF-6 | 1.0 | 6.0 | 180 | 240 | 92.4 | 81.7 | 100 |
| MOF-74 Mg/Mn | 5.0 | — | 190 | 240 | 100.0 | 91.8 | 101 |
| MgZnAl LDH | 1.0 | 10.0 | 196 | 180 | 100.0 | 75.0 | 102 |
| Co-Al-CO3 LDH | 1.0 | 11.0 | 180 | 120 | 100.0 | 96.0 | 103 |
3.3. Ammonolysis of polyesters
Ammonolysis of PET can produce various fatty amines with high added value, which can be used as chain extenders in the synthesis of polyamide resins and hydrogels due to their strong nucleophilic reactivity. The reaction pathway is shown in Fig. 8.104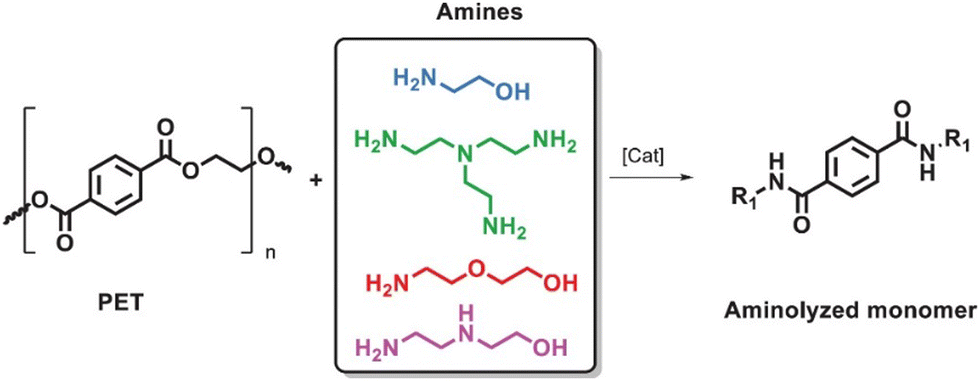 | ||
| Fig. 8 Aminolysis of PET with amines. Reproduced from ref. 104 with permission from Wiley, copyright [2022]. | ||
Shukla et al. developed an ammonolysis process of PET using sodium acetate (1% by weight of PET) and ethanolamine (a PET molar ratio of 1:6), and they found that the yield of N,N′-bis(2-hydroxyethyl) terephthalamide (BHETA) from PET fibers reached 91.1% under reflux for 8 h, which was higher than the yield from PET plastic bottles (83.2%).105 This difference was attributed to the molecular weight of PET and its chain distribution. The oligomers recovered from the hydroxyethyl ethylenediamine ammonolysis of PET could be used to synthesize polyester amide resins, which exhibited better properties than conventional polyester amide resins.106
Palekar et al. found that the ammonia depolymerization of PET with ethanolamine in ILs (1-hexyl-3-methylimidazolium trifluoromethanesulfonate) proceeded quickly, and the yield of BHETA reached 89% within 1 h at 196 °C, using a PET-to-ethanolamine mass ratio of 1:6 and 0.2 g of ILs.107 Achilias et al. used microwave heating to further reduce the ammonolysis time, finding that PET could be completely depolymerized with ethanolamine in 3 min, achieving a 100% BHETA yield when the irradiation power was set to 100 W.108
Ammonolysis of PLA can produce nitrogen-containing compounds, such as pyridine, acetonitrile, and alanine, which can be directly used as solvents or intermediates in the chemical industry.109–111 Xu et al. found that waste PLA could be effectively converted to pyridine via thermal catalytic ammonification processes (Fig. 9A). Under the atmosphere of NH3 and at a temperature of 500 °C, the yield of pyridine from PLA reached 24.8% over HZSM-5 (Si/Al = 25).109 The same group further reported that the Co/HZSM-5 (Si/Al = 25) catalyst can achieve the rapid pyrolysis of waste PLA to acetonitrile in an ammonia atmosphere, and the yield of acetonitrile reached 50.2% at 650 °C with a residence time of 2.5 seconds under NH3 flow.110 Additionally, Shao et al. developed a process of conversion of PLA waste into a new 3D printable material, finding the PLA waste could be completely ammonolyzed by ethanolamine within 60 min at 100 °C, producing N-lactoylethanolamine (N-LEA) with a PLA-to-EA mass ratio of 1:4. N-LEA could further be converted into a 3D printing photocured resin through reactions shown in Fig. 9B.112
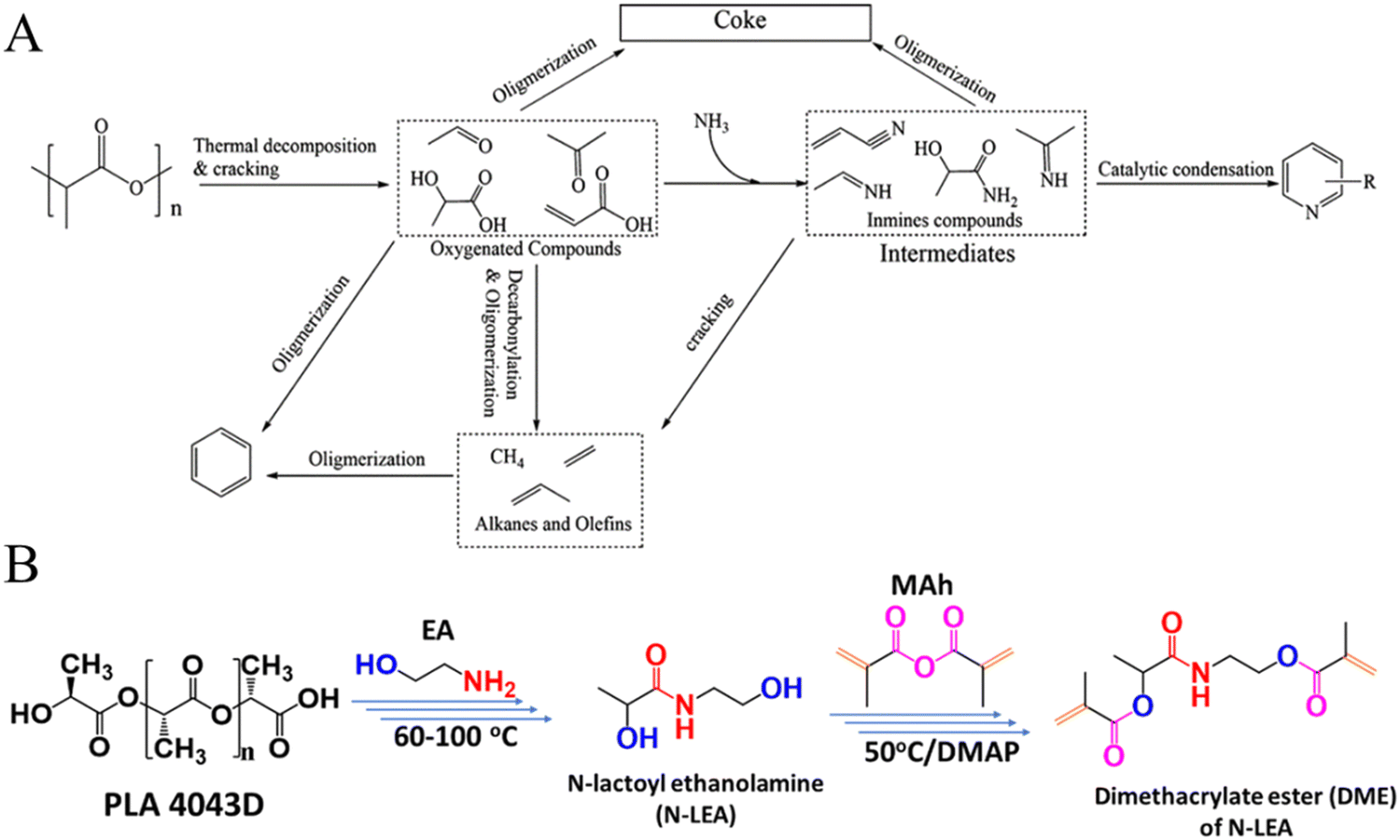 | ||
| Fig. 9 (A) Proposed reaction pathway from PLA to pyridines. Reproduced from ref. 109 with permission from American Chemical Society, copyright [2015]. (B) General procedure of synthesis starting from PLA to form diacrylate ester DME. Reproduced from ref. 112 with permission from Royal Society of Chemistry, copyright [2022]. | ||
Ammonolysis is a promising method of recycling of waste polyesters to high valued chemicals, but the number of reported works is limited, and hence more efficient routines and/or catalysts need to be developed for this upgrading process.
3.4. Methanolysis of polyesters
Alcoholysis processes of polyesters involve the depolymerization of polyesters by alcohols, where alcohols attack the carbonyl group of the ester bond, breaking the polymer chain. Among them, methanolysis of PET with methanol can produce dimethyl terephthalate (DMT) and EG. In particular, methanolysis exhibits high reactivity due to the small steric hindrance and strong nucleophilic ability of methanol.113Metal salts were reported often as the homogenous catalysts for this reaction. Mishra et al. found that a mixture of lead acetate and zinc acetate in equal amounts can promote the methanolysis of PET, achieving quantitative conversion of PET to DMT and EG at 140 °C within 2 h.114 Pham et al. also reported that waste PET could be depolymerized completely at 25 °C in 14 h, with a DMT yield of 93.1% after 24 h at the molar ratios of K2CO3, water, methanol, and dichloromethane to PET repeating units of 0.2, 0.4, 50 and 50, respectively.115
Apart from the above homogenous catalysts, solid bases and solid acids were also tested for methanolysis of polyesters in recent works. Tang et al. disclosed that MgO/NaY catalysts with varying MgO contents were active for the methanolysis of PET, and their activity enhanced with the increased basic sites. The best conversion of PET and the yield of DMT reached 99 and 91%, respectively, when 21% MgO/NaY was utilized (at a concentration of 4 wt% and a mixed methanol-to-PET mass ratio of 6) at 200 °C for 30 min.116 Additionally, the same group also developed a low-cost calcined sodium silicate solid base catalyst, which can completely alcoholyze PET and achieve a 95% DMT yield at 200 °C in 30 min.117 Du et al. found that ZnO nanoparticles with a uniform size of 4 nm can be used as a pseudo-homogeneous catalyst for PET methanolysis, and the degradation rate of PET and the yield of DMT reached approximately 97 and 95%, respectively, at 170 °C in 15 min.118 Recently, Ye et al. synthesized an environmentally friendly and stable TixSi1−xO2 solid acid catalyst for the catalytic upcycling of waste PET to DMT; it was found that the yield of DMT over Ti0.5Si0.5O2 (2.5 wt% PET) reached 98.2%, with complete degradation of PET in 2 h at 160 °C. SEM images of PET residual at different reaction times and the possible reaction routine for the alcoholysis of PET with methanol over Ti0.5Si0.5O2 are shown in Fig. 10A.119 It was concluded that the compact PET particles might be etched with these adsorbed Ti0.5Si0.5O2 NPs to form a porous structure, along with the formation of oligomers with [OOC–C6H4–COO–C2H4]17–18 units (around the interface between the catalyst and PET). Then, these oligomers in the reaction mixture were further decomposed to intermediates A and B, and DMT formed from these intermediates. More recently, Zhang et al. found that Cu/SiO2-IM also exhibited excellent activity, with the conversion of PET and the selectivity of DMT reaching 92.35 and 99.0% at 200 °C without any initial gas pressure. The high activity can be attributed to the small metal particles, the balance between Cu+ and Cu0 active sites, and a high ratio of Brønsted acid sites to Lewis acid sites, and the proposed reaction mechanism of PET methanolysis over the Cu/SiO2 catalyst is shown in Fig. 10B.120
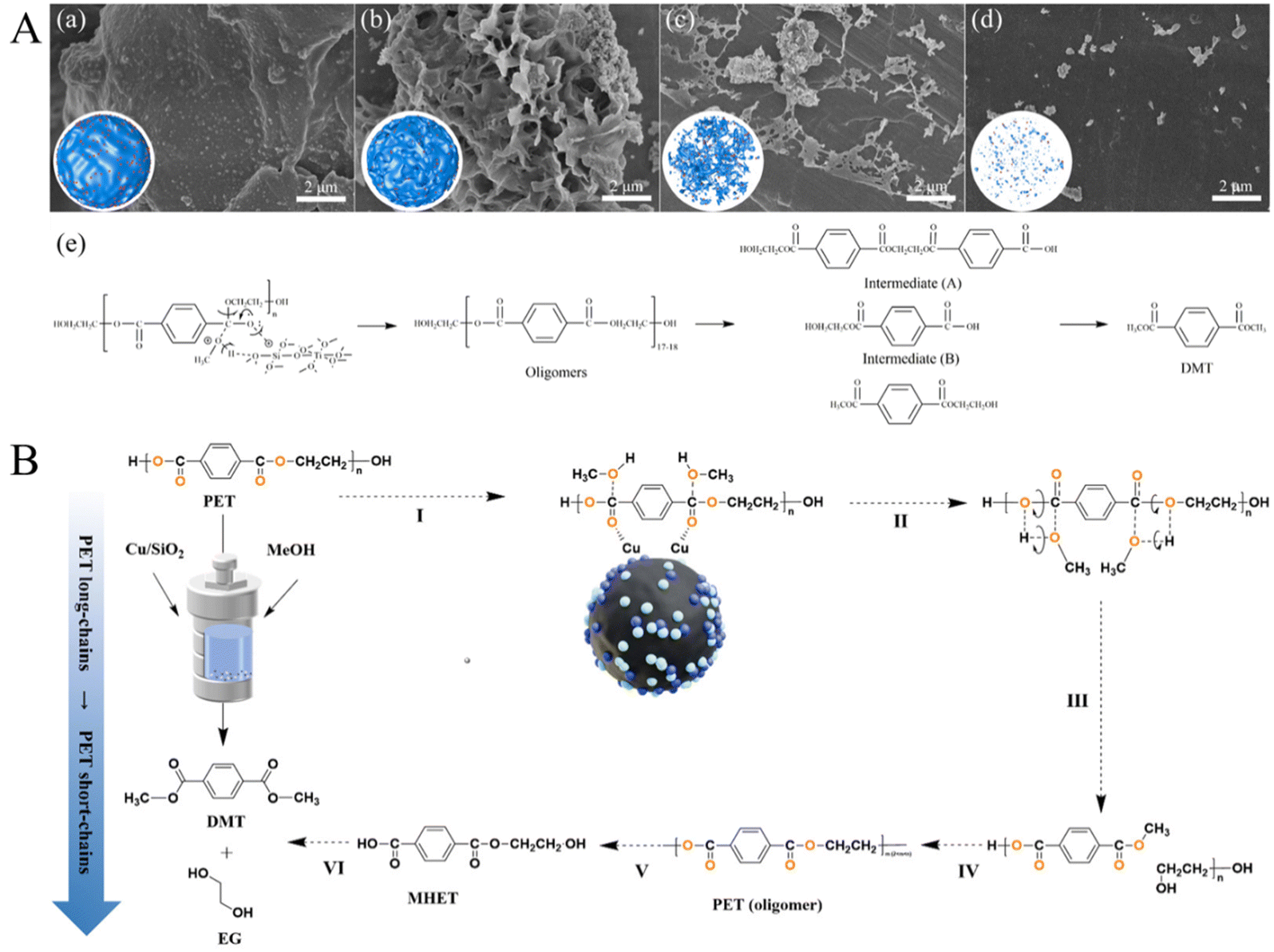 | ||
| Fig. 10 (A) SEM images of remained PET at different reaction times ((a): 0 h, (b): 0.5, (c): 2 h, and (d): 4 h). (e) Possible reaction routine for the alcoholysis of PET with methanol over Ti0.5Si0.5O2. Reproduced from ref. 119 with permission from Royal Society of Chemistry, copyright [2023]. (B) Proposed reaction mechanism for PET methanolysis over the Cu/SiO2 catalyst. Reproduced from ref. 120 with permission from Royal Society of Chemistry, copyright [2024]. | ||
As the reaction conditions of supercritical methanolysis of PET (in methanol) are relatively milder compared to that of supercritical hydrolysis of PET in water, Sako et al. tried to methanolize PET in supercritical methanol (at 300 °C and 9–23 MPa), achieving an 80% yield of DMT in just 0.5 h.121 Liu et al. further found that this reaction could be accelerated by co-addition of CO2 in supercritical methanol, and the yield of DMT increased to 95% at 270 °C in 40 min, with a methanol-to-PET mass ratio of 6:1.122
At the same time, a variety of ILs have been tested to catalyze the methanolysis of PET. Liu et al. found that an IL ([HDBU][Im]) derived from an imidazole anion can promote the methanolysis of PET. After a 3-h reaction at 140 °C with an n(PET):(CH3OH):([HDBU][Im]) molar ratio of 1:5:0.1, the degradation rate of PET reached 100%, and the yield of DMT was 75%.123 It was suggested that PET or PC could be partially dissolved or swelled in the reaction medium, and alcohol was activated via the formation of a hydrogen bond with [HDBU][Im], and simultaneously, the cation [HDBU]+ underwent an electrostatic interaction with the oxygen of carboxyl groups in PET or PC, which was inclined to form the carbonyl carbocation through electron transfer. Then, the activated alcohol (oxyanion) that stripped the hydrogen proton became more favorable to facilitate the nucleophilic attack at the carbonyl carbocation. This process resulted in fragmentation or disconnection of the long molecule chain of PET or PC into the smaller oligomers. Similarly, the oligomers were further attacked by the activated alcohol to afford the monomer and dialkyl carbonate. Additionally, Liu et al. also found that an IL with Brønsted–Lewis acid properties, [HO3S–(CH2)3–NEt3]Cl–ZnCl2, can catalyze the alcoholysis of PET with n-butanol with satisfactory performance.124
Lee et al. proposed a mechanochemical ball milling method for methanolysis of PET to recover DMT, utilizing physical processes, such as collision, shearing, and grinding to induce depolymerization reactions. PET powder (2.8 g) and methanol (70 equivalents) were ball milled for 6 h at 650 rpm, resulting in a mixture of DMT and EG. After treatment, pure DMT (2.4 g, 82%) was obtained, and this technology is also suitable for recycling PLA and polycarbonate (PC).113
Besides PET, waste PLA can also be recovered to form methyl lactate and ethyl lactate via methanolysis or ethanolysis, respectively. Various catalysts have been tested for the alcoholysis recovery of PLA, including ILs, solid acids, and metal complexes.125 In 2023, Ye et al. found that a SiO2@UiO-66-derived solid acid (SO42−/ZrO2/SiO2) is effective for the methanolysis of waste PLA to methyl lactate (MLA), achieving a yield of 92.7% at 140 °C in 5 h.126 More recently, our group disclosed that WOx/ZrP catalysts with highly dispersed WOx active sites anchored on the surface of zirconium phosphate (ZrP) were more stable for the methanolysis of PLA. The yield of MLA over the 10% WOx/ZrP catalyst reached 94.5% at 160 °C within 4 h due to the abundant WOx active sites and strong acidity.127
The above works confirmed that methanolysis of PET with methanol can form DMT and EG, and methanolysis can utilize colored PET, carpet fiber, blended plastics, and resins rejected from the mechanical recycling system, which has a much broader range of feedstocks. More interestingly, the boiling point of DMT (∼285 °C) is lower than that of BHET (∼375 °C); it is easy to achieve higher levels of purity for DMT by employing techniques such as crystallization and distillation.128 Since 2020, increasing attention has been paid to the methanolysis of PET with methanol, and the results are summarized in Table 3.
| Catalyst | Cat:PET (w/w) (%) |
Mass ratio of EG/PET | Temp. (°C) | Time (min) | Conv. (%) | YieldDMT (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Pt(OAc)2 and Zn(OAc)2 | 6.0 | 2.4 | 140 | 120 | 100.0 | 97.8 | 114 |
| K2CO3 | 14.4 | 8.3 | 25 | 1440 | 100.0 | 93.1 | 115 |
| Aluminium triisopropoxide | 10.0 | 15.8 | 200 | 120 | — | 64.0 | 129 |
| MgO/NaY | 4.0 | 6.0 | 200 | 30 | 99.0 | 91.0 | 116 |
| Calcined Na2SiO3 | 5.0 | 5.0 | 200 | 30 | 100.0 | 95.0 | 117 |
| Bamboo leaf ash | 20.8 | 8.2 | 200 | 60 | — | 78.0 | 130 |
| ZnO NPs | 3.5 | 6.0 | 170 | 15 | 97.0 | 95.0 | 118 |
| Ti0.5Si0.5O2 | 2.5 | 7.9 | 160 | 120 | 100.0 | 98.2 | 119 |
| Cu/SiO2-IM | 40.0 | 52.7 | 200 | 90 | 92.4 | 91.5 | 120 |
| Cu/ZrO2 | 24.0 | 23.7 | 220 | 360 | 100.0 | 98.0 | 131 |
| [HDBU][Im] | — | 0.8 | 140 | 180 | 100.0 | 75.0 | 123 |
| PIL-Zn2+ | 2.0 | 4.0 | 170 | 60 | 100.0 | 90.3 | 132 |
| Polymeric NC sheets | 100.0 | 30.0 | 185 | 360 | — | 85.0 | 133 |
| Zn(II)-complexes | 5.0 | 3.2 | 100 | 720 | 100.0 | 76.0 | 134 |
| Supercritical methanolysis | — | 3.2–23.7 | 300 | 30 | 100.0 | 80.0 | 121 |
| Supercritical methanolysis | — | 6.0 | 270 | 40 | 100.0 | 95.0 | 122 |
| Ball milling | — | 12.1 | r.t. | 360 | 99.0 | 82.0 | 113 |
Depolymerization of polyesters is the first step in the chemical recycling process. Various monomers can be obtained via hydrolysis, glycolysis, ammonolysis, and methanolysis of polyesters, which can then be used to resynthesize plastics or further converted into other value-added chemicals. The polymerization monomer can be obtained directly by the hydrolysis reaction. However, neutral hydrolysis requires extremely high reaction temperature, and hydrolysis under acidic or alkaline conditions needs complicated post-treatment. Glycolysis reaction can realize the continuous recovery of waste polyesters under normal pressure, but problems such as a long reaction time and low product yield need to be solved. Ammonolysis is a potential method to upgrade polyesters, while the use of toxic and expensive chemicals should be considered. As for methanolysis reaction, the compatibility of feedstock is strong and the depolymerized products are easy to purify; unfortunately, the reaction conditions of high temperature and high pressure cannot be avoided under supercritical conditions. Exploring novel and efficient catalysts for polyester depolymerization to avoid the harsh reaction conditions is of significant research importance for advancing polyester recycling. The depolymerization mechanism of polyesters can mainly involve two key steps. One is that the cation of catalysts (electron-withdrawing group) interacts with –CO in polyesters, enhancing the electro-positivity of carbonyl carbon. The other one is that the anion (electron-donating group) reacts with the H atom of the degradation medium (water, alcohols or amines) to make the O in –OH or N in –NH2 more electronegative. The two activated parts react with each other to break the ester bond and further complete the degradation of polyesters. Improving the electron-withdrawing and electron-donating abilities of catalysts can effectively increase the degradation rate of polyesters, but the too strong ability of these catalysts is prone to promote carbonization or dehydration reaction, resulting in a decreased yield of target products. Besides, polyester materials have strong hydrophobicity and rigid structures, increasing the hydrophobicity and reducing the size of catalysts can increase the contact between catalysts and polyester, enhancing the reaction activity.
4. Chemical recycling of a wide-range of waste PET-based products
As the PET-based products show excellent stability, processability and biological non-toxicity, PET is widely utilized in the production of drinking bottles, textile cloth, packaging films and so on. Direct utilization of these real waste PET-based products as raw feedstocks is of great significance to promote the large-scale applications of waste PET recycling.4.1. Chemical recycling of PET bottles
PET bottles are some of the most used commodity plastics in beverage and drinking water packaging and are often disposable items deserving special attention due to their mass-production and high daily consumption. Enayati et al. developed a model for the self-sufficient chemical recycling of PET bottles via glycolysis,135 in which the pyrolysis products of bottle labels (prepared at 800 °C) were selected as catalysts (containing CaCO3, CaO, and TiO2) for the glycolysis of PET bottles, and the conversion of PET reached 100% with a 95.8% yield of BHET over a 1.0 wt% catalyst at 200 °C in 1.5 h (Fig. 11A). Chan et al. proposed a method for waste PET bottle conversion to cross-linked hydrogels via aminolysis using tri- and tetraamines for plastic waste valorization, in which, reaction of PET with diethylenetriamine (DETA) or triethylenetetramine (TETA) at 190°C resulted in a complete dissolution of PET within 30min. The aminolysis products of PET rich in amino groups could be readily cross-linked by ethylene glycol diglycidyl ether (EGDE) to form a hydrogel (Fig. 11B).136
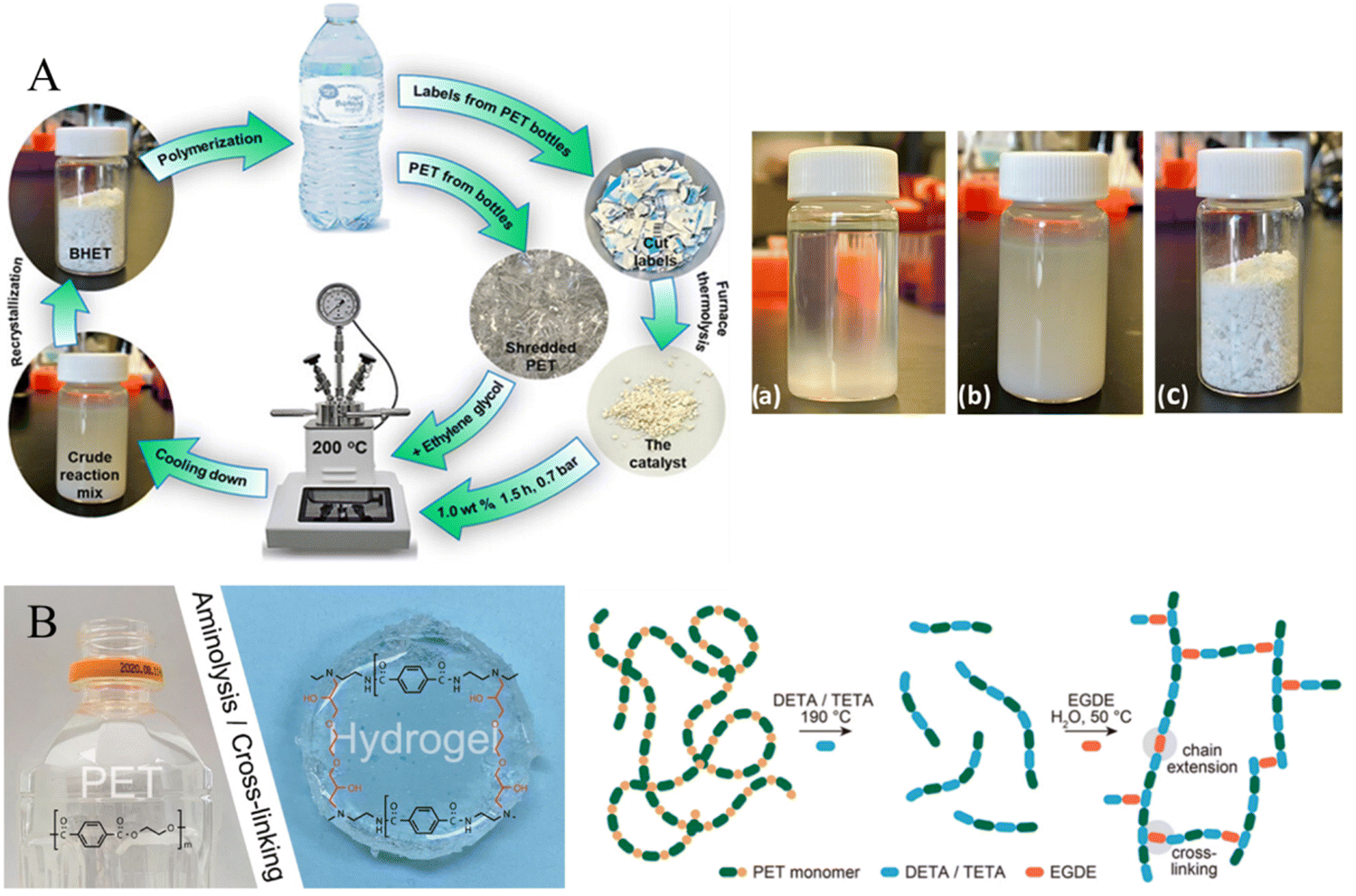 | ||
| Fig. 11 (A) Schematic illustration of the self-sufficient chemical recycling of PET bottles via glycolysis and the photo of the reaction mixture of PET glycolysis at 70 °C (a), room temperature (b), and BHET (c). Reproduced from ref. 135 with permission from American Chemical Society, copyright [2023]. (B) The photo of a hydrogel prepared from a PET bottle and schematic representation of PET-derived hydrogel formation by cross-linking of oligomeric products of PET aminolysis. Reproduced from ref. 136 with permission from Elsevier, copyright [2021]. | ||
4.2. Recycling of PET from mixed plastics
In daily life, recycled polyesters are often a mixture of multiple plastics, and how to deal with the plastic mixture is an urgent problem to be solved. Xie et al. presented a metal-free catalytic sorting method for targeted deconstruction of multilayer packaging materials. Commercial milk bags (PET/polyamide (PA)/PE) and vacuum seal storage bags (PET/PE) were added directly as feedstocks using N-methylpiperidine as a catalyst in a methanol solution, and they achieved a 90% yield of DMT and EG at 160 °C for 1 h (Fig. 12A).137 The solid residues isolated from the reaction mixture were essentially close to pure low density PE or PA, suggesting that it is feasible to recycle PET from packaging materials. Cao et al. further reported an oxygen-vacancy rich catalyst, Fe/ZnO nanosheets (Vo-rich Fe/ZnO NSs), for the methanolysis of different kinds of PET and mixed polyester wastes.138 It was found that PET particles, slices and bottles could be converted via methanolysis at 160 °C to obtain DMT with high yield. Under the same conditions, PET and polycarbonate (PC) were converted into DMT and bisphenol A completely (both yields reached 99% within 1 h). For the mixture of PET/PP packing boxes and PET/PE thin films, the yields of DMT were up to 99 and 96% at 160 °C in 1 h with a 1 wt% catalyst, while PP and PE hardly underwent any reaction (Fig. 12B). | ||
| Fig. 12 (A) Deconstruction of post-consumer multilayer packaging materials, which contained PET/PA/PE or PET/PE. Reproduced from ref. 137 with permission from Cell Press, copyright [2024]. (B) Catalytic depolymerization of mixed polyester wastes. Experiments were carried out under an air atmosphere. Reproduced from ref. 138 with permission from Nature, copyright [2024]. | ||
4.3. Recycling of PET from waste textile
Unlike waste pure PET-based materials, such as bottles, films and packing materials, chemical recycling of PET-containing textile waste is more difficult. This might be attributed to the fact that most textiles are mixed fibers and contain additives or colorants, in which the polyester is interlaced tightly with other fibers (such as cotton, nylon, spandex, dyes, and finishes). Chen et al. have proposed a strategy to use polyesters in textiles via decolorization of waste textiles, glycolysis of decolorized polyester textiles and purification of the BHET product (Fig. 13A).139 It was found that the yield of BHET reached 89.2% with 99.2% purity from a mixture of randomly picked waste polyester textile samples that contained a library of additives with different colors. More interestingly, the recovered BHET could be polycondensed to produce new polyester materials that have comparable properties to those of PET derived from petroleum-based BHET and the dyes also have the potential to be recycled. More recently, Andini et al. disclosed that several postconsumer mixed textile wastes could be glycolysed to form BHET over a ZnO catalyst under microwave irradiation followed by a simple solvent dissolution, which enables the separation of cotton and nylon (Fig. 13B).30 Compared with the depolymerization of individual components, it was found that the polyester and spandex contents in mixed textiles could be depolymerized completely to BHET and MDA, respectively, while cotton and nylon in the mixed textiles remained intact. Some of the recovered components can be integrated directly into the textile and clothing manufacturing for textile-to-textile recycling. This process holds the potential to achieve a global textile circularity rate of 88%.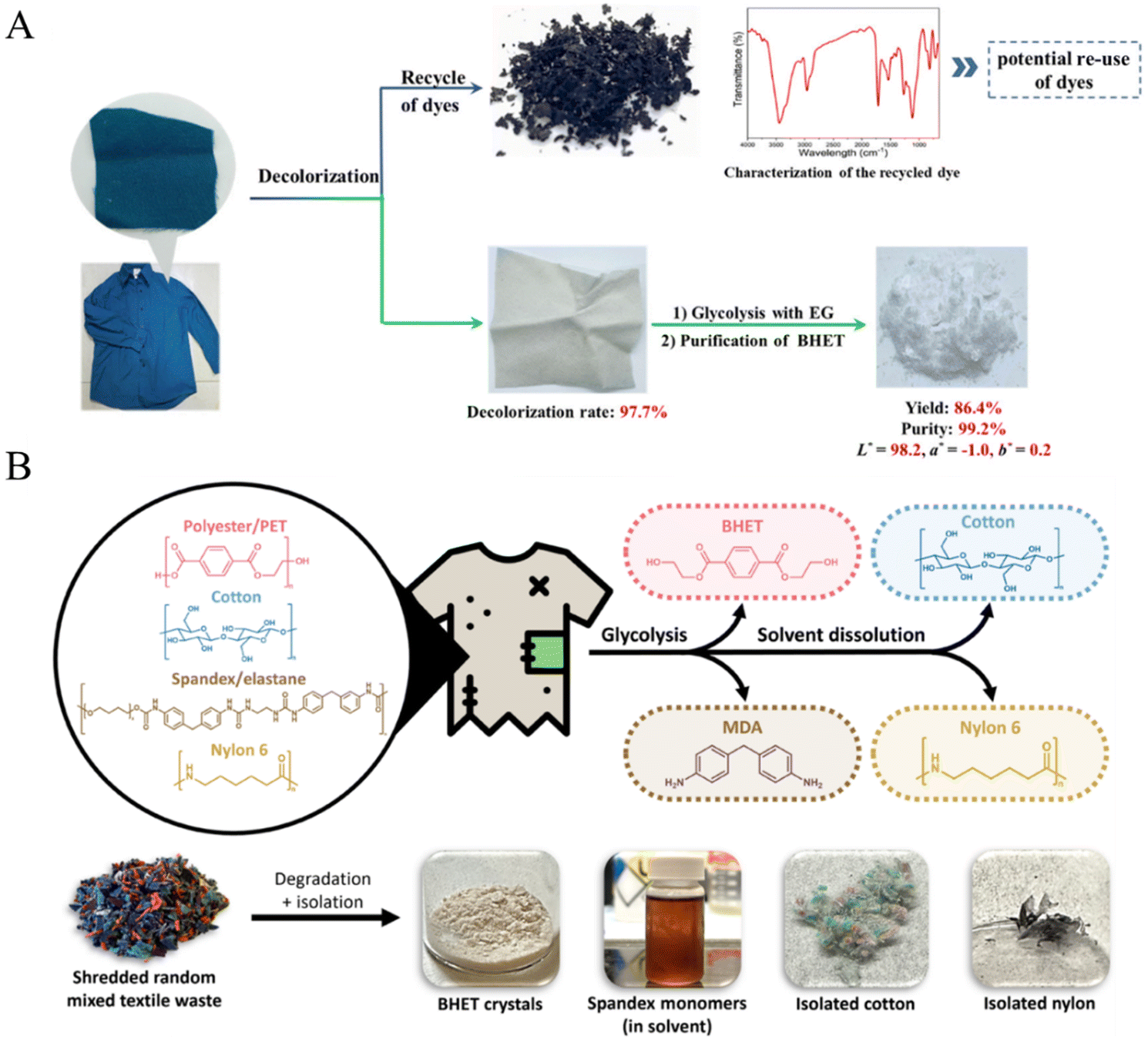 | ||
| Fig. 13 (A) Predecolorization of a waste polyester textile produces recycled dyes and decolorized textiles, which are then subjected to glycolysis and purification to afford white and purified BHET. Reproduced from ref. 139 with permission from Royal Society of Chemistry, copyright [2023]. (B) Overview of the full chemical recycling process of real mixed textile waste (polyester, cotton, spandex, and nylon) using MW-assisted glycolysis and solvent dissolution. The photo of isolated components obtained from real mixed textile waste with unknown compositions. MDA, 4,4′-methylenedianiline. Reproduced from ref. 30 with permission from Science, copyright [2024]. | ||
The recycling of real PET-based products has attracted worldwide attention, but there are still many problems and challenges. PET bottles are mainly used for beverage and drinking water packaging with high quality and less impurities, making them possess high recycling value. However, the transparency and viscosity of PET after recycling cannot meet the requirements of food grade, which can only be downgraded as chemical fiber. The optimal path for the recycling of PET-based bottles is to perform food-grade recycling first via developing bottle-to-bottle technology and equipment and then downgrade the materials after recycling several times. The PET film is often used in agricultural films, plastic bags and soft packaging, but its recycling rate is quite low. On the one hand, a soft film is mostly a composite material, including PET, PE, PP and PVC, which is necessary to carry out complicated classification. Meanwhile, the recycled film is usually broken and small with light weight, bringing great challenges to the recycling in the later stage. Also, the mechanical strength will decrease after regeneration, and the residue of additives will also affect the quality. In order to improve the recycling of the PET film, focusing on classified collection is the first step, and then developing a tiered recovery strategy suitable for the PET composite film to fully tap its value is essential (especially chlorine-containing polymers). Waste textiles are the largest source of waste PET, and the importance of waste textile recycling is beyond doubt, but the real recycling of waste textiles is more difficult than imagined. The biggest problem is that the most common type of waste textile is polyester-cotton blend, which requires separation of polyesters and cotton fibers before recycling them separately. However, the traditional mechanized loosening means find it difficult to separate different fibers, and the technical difficulty, environmental protection requirements and cost will become high via chemical means. In addition, the decolorization of waste textiles is another difficulty. For solving these urgent problems, it may be feasible to find suitable solvents for the dissolution of fibers and dyes.
In order to promote the industrial recycling of waste polyesters, it is recommended to conduct economic evaluation of advanced recycling technologies in the whole process of recycling (including reaction, process, integrated facility, waste treatment and supply chain level) to realize successful commercialization. Besides, the guidance and support policies are also essential.
5. Upgrading of depolymerized monomers and intermediates
Depolymerization of polyesters is the first step in the chemical recycling process. Various monomers and/or intermediates can be obtained via hydrolysis, glycolysis, ammonolysis, and methanolysis of polyesters, which can be utilized to resynthesize plastics or further convert into other valued chemicals. Recently, it was reported that these depolymerized products of PET obtained via hydrolysis (PTA), glycolysis (BHET), and methanolysis (DMT) can be converted to 1,4-cyclohexanedimethanol (CHDM) via consecutive hydrogenation (Fig. 14).140 CHDM is a versatile monomer than ethylene glycol for the synthesis of high-quality, non-toxic polyester fibers with low relative density, high melting point, and high transparency. It was confirmed that CHDM is a crucial raw material for the production of poly(ethylene terephthalate-co-1,4-cyclohexylenedimethylene terephthalate) (PETG).141 | ||
| Fig. 14 Reaction paths from PET to CHDM. | ||
5.1. Upgrading of depolymerized PTA to CHDM
Upgrading of PTA to CHDM involves two steps: consecutive hydrogenation of the benzene ring and further hydrogenation of the carboxyl group. In 2006, Zhang et al. demonstrated that CHDM could be synthesized via a two-step hydrogenation of PTA. In the first step, the conversion of PTA and the selectivity of 1,4-cyclohexanedicarboxylic acid (CHDA) reached 99 and 99%, respectively, using a Pd/C catalyst at 250 °C and 12 MPa H2 for 1 h. Then, CHDM formed in high selectivity (98.3%) at 250 °C and 10 MPa H2 in 6 h with nearly complete conversion of CHDA (99.4%) over an Ru–Sn/C catalyst.142 RuSn and RuSnPd catalysts were also tested and demonstrated to be efficient for the separated hydrogenation of CHDA to CHDM.143,144 However, this two-step technology was conducted in different reactors under high H2 pressure, which increased the complexity of operation and the costs of production.A one-step hydrogenation method can simplify this process and has attracted the interest of researchers gradually. In 2012, Zhao et al. found that the Ru–Sn–B/Al2O3 catalyst was suitable for the one-pot hydrogenation of PTA to CHDM in a stainless steel batch reactor, but under two reaction stages. The first stage was conducted at 150 °C and 6 MPa H2, followed by a second stage at 230 °C and 10 MPa H2, and the final conversion of PTA reached 100% with a 86.3% yield of CHDM.145 Zhang et al. developed a dual catalyst system by physically mixing a Ru–Sn/Al2O3 catalyst with a Pd/Al2O3 catalyst, achieving a PTA conversion of 96.3% and a CHDM yield of 72.2% at lower pressure. Initially, the reactor was pressurized with H2 to 4 MPa and heated to 180 °C. After reacting for 1.5 h, the pressure and temperature were increased to 8 MPa and 230 °C, and the reaction was continued for another 5 h.146 More recently, the same group further disclosed that doping a Pd/Al2O3 catalyst with Ce can raise the yield of CHDM under the same reaction conditions. They found that the addition of 2% Ce increased the yield of CHDM obviously from 72.2 to 85.5%, indicating that Ce facilitated the activation of H–H and CO bonds.147 Additionally, Choi et al. combined a Pd/Y-zeolite catalyst with an Ru–Sn–Pt/Y-zeolite catalyst to prepare CHDM in a fixed-bed reactor; it was found that the conversion of PTA reached 100%, with a CHDM selectivity of 93% at 230 °C and 8 MPa H2.148
The above studies indicated that CHDM, a versatile monomer, could be synthesized via the consecutive hydrogenation of PTA, but this routine requires a noble metal catalyst, high H2 pressure, and complex reaction procedures.
5.2. Upgrading of depolymerized BHET to CHDM
BHET, a product of the glycolysis of polyesters, is currently used as the monomer of recycled polyesters instead of the petroleum based PTA. Recently, it was reported that BHET can also be upgraded to the versatile CHDM monomer via a two-step hydrogenation process, with bis(2-hydroxyethyl) cyclohexane-1,4-dicarboxylate (BHCD) as the intermediate. In 2016, Hou et al. developed a one-pot technology for the conversion of BHET to CHDM over a trimetallic RuPtSn/Al2O3 catalyst via two-step reaction stages: the initial step was performed at 170 °C for 2 h followed by 260 °C for 6 h under a 5 MPa H2 atmosphere. The final yield of CHDM reached 87.1% with a complete conversion of BHET.149 However, the reusability of this catalyst was restricted by the elution of Sn.In batch autoclaves, Guo et al. also found that CHDM could be prepared via a two-step hydrogenation. In the first step (at 155 °C, 3 h, and 7 MPa H2), the conversion of BHET reached 100%, with a 95% yield of BHCD over a Pd/C catalyst, and then, CHDM formed in the second step via the further hydrogenation of BHCD over a Cu–Zn/Al2O3 catalyst at 270 °C, 10 h, and 10.5 MPa H2, and the final yield of CHDM reached 78%.150 The competition between hydroxyl and ester groups increases the difficulty in selectively hydrogenating ester groups, leading to the formation of byproducts (such as ethyl 4-methylbenzoate (EMB) and cyclohexanemethanol (CHM)), meanwhile, high reaction temperature and high H2 pressure also increase the risk of this process, making it important to reduce both for large-scale applications.
Previous works conducted in our laboratory found that a mesoporous HZSM-5 zeolite-supported Ru catalyst (Ru/m-HZSM-5) can catalyze the selective hydrogenation of the benzene ring in BHET toward BHCD under mild conditions (120 °C and 3 MPa), and the yield of BHCD reached 95.6% within 2 h (Fig. 15A).151 Then, the further hydrogenation of BHCD to CHDM could be carried out in a continuous fixed-bed reactor over a cheap Cu/MgAl2O4 catalyst; the yield of CHDM reached 98% with a complete conversion of BHCD at 240 °C and 4 MPa H2. The excellent performance was primarily due to the highly dispersed Cu particles embedded in the framework of spinel MgAl2O4, which provided strong metal–support interactions as well as high Cu+ content and strong basicity of the Cu/MgAl2O4 catalyst (Fig. 15B).152
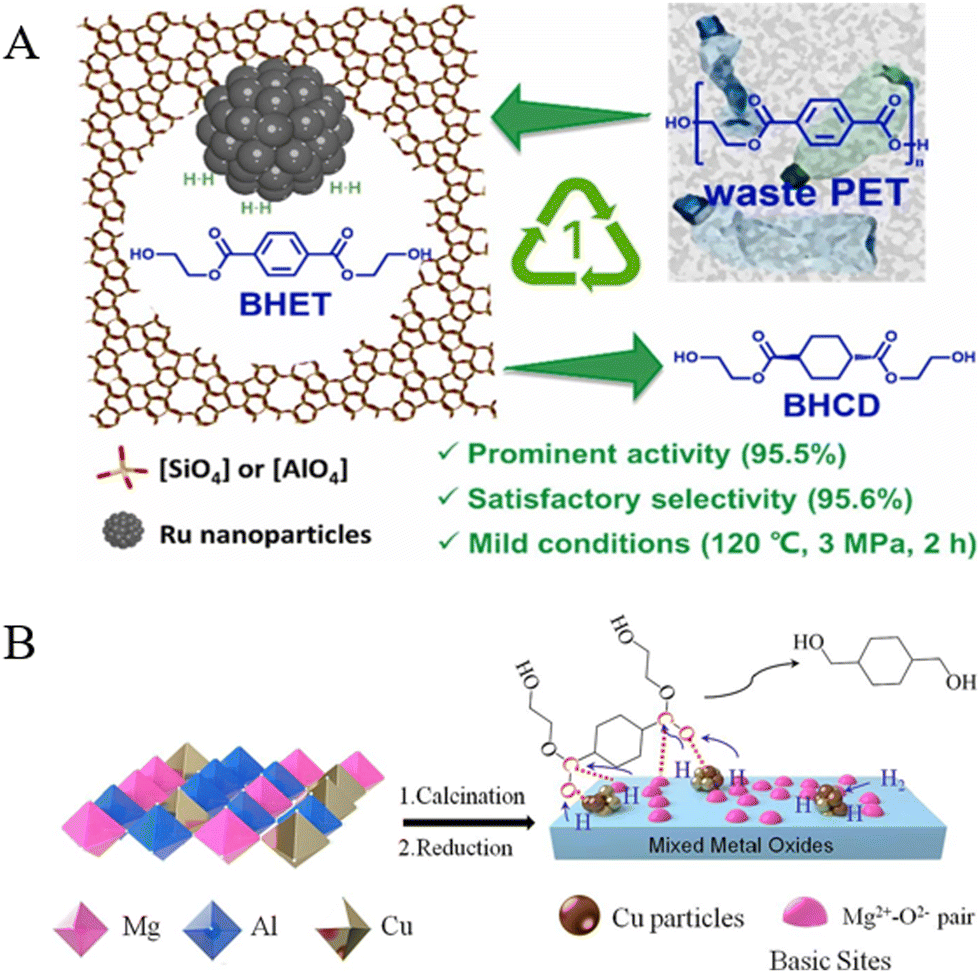 | ||
| Fig. 15 (A) Schematic illustration of the hydrogenation of BHET to BHCD over Ru/m-HZSM-5. Reproduced from ref. 151 with permission from Elsevier, copyright [2024]. (B) The preparation of Cu/MgAl2O4 and the hydrogenation of BHCD to CHDM. Reproduced from ref. 152 with permission from Elsevier, copyright [2024]. | ||
5.3. Upgrading of depolymerized DMT to CHDM
Currently, the industrialized CHDM is produced via a two-step process in two reactors from DMT. In the first step, DMT is transformed into dimethyl 1,4-cyclohexanedicarboxylate (DMCD) through the highly exothermic hydrogenation of the phenyl group over Pd-based catalysts at 160–180 °C and 30–48 MPa H2.153,154 In the second reactor, DMCD is converted into CHDM via the hydrogenation of the ester groups into hydroxyl groups over a conventional Cu–Cr catalyst at around 200 °C and 4 MPa H2.155 But this process is limited by the high H2 pressure in the first stage and the Cu–Cr catalyst used in the second hydrogenation step is toxic to the environment.Recently, it was reported that Ru-based catalysts were more active for the hydrogenation of aromatic rings in DMT due to their moderate adsorption and activation capacity for aromatic rings and H2, as well as their lower cost compared to Pd.156 Chen et al. discovered that heterogeneous bimetallic RuPd catalysts exhibited 88.5% conversion of DMT with a 96.2% selectivity of DMCD under 8.0 MPa H2 and at 180 °C for 6 h.157 Zhang et al. found that Ru supported on spherical Al2O3 with a high specific surface area and well-developed pores (Pd/HTC-Al2O3) exhibited excellent activity, achieving about 96.0% conversion of DMT and 95.0% yield of DMCD at 180 °C, 8 MPa H2, and a hydrogen/DMT molar ratio of 120.158 Yu et al. have prepared uniformly a highly dispersed Ru catalyst on Al20SBA-15 (Si/Al ratio = 20) by chemical fluid deposition, employing supercritical CO2 as the solvent. They achieved a DMCD yield of 93.4% at 100 °C and 4.14 MPa H2 in 0.5 h.159 Previous work conducted in our laboratory also found that a nitrogen-doped graphene-coated Ru-based catalyst (Ru@G-CS) was extremely active for the hydrogenation of DMT to DMCD at 160 °C and 2.5 MPa with a DMT/Ru molar ratio of 833; the yield of DMCD reached 98.5% within 4 h. This prominent performance was attributed to the high dispersion of Ru, promoted by nitrogen atoms in the N-doped graphene skeleton and a strong interaction between Ru and the support.160 Recently, Yan et al. reported a yolk–shell structured SiO2@Ru@NC catalyst, which is capable of catalyzing the conversion of DMT to DMCD at 90 °C, 2 MPa H2 and a DMT/catalyst weight ratio of 5; the conversion of DMT and the selectivity of DMCD reached 97.7 and 94.3%, respectively, within 2 h (Fig. 16A).161
 | ||
| Fig. 16 (A) Synthesis process and morphology of yolk–shell structured SiO2@Ru@NC and its performance in the hydrogenation of DMT to DMCD. Reproduced from ref. 161 with permission from Royal Society of Chemistry, copyright [2024]. (B) The hydrogen reduction of a NiAl-LDH precursor at various temperatures. Reproduced from ref. 162 with permission from American Chemical Society, copyright [2016]. (C) The effect of K doping of the Ni/SiO2 catalyst. Reproduced from ref. 163 with permission from Royal Society of Chemistry, copyright [2023]. | ||
Apart from the popularly reported precious metal catalysts, Ni catalysts were also tested for the hydrogenation of DMT to DMCD. Fan et al. fabricated a double-confined nickel catalyst, Ni/Ni(Al)Ox/AlOx, with Ni nanoparticles implanted in a weakly crystalline Ni(Al)Ox matrix and embedded in amorphous AlOx networks through the hydrogen reduction of a NiAl-LDH precursor. They achieved a 93.3% selectivity toward DMCD and almost complete conversion of DMT at 90 °C, 4 MPa H2 in 4 h in a batch reactor (Fig. 16B).162 A recent work showed that post-impregnated doping with potassium could significantly improve the performance of Ni/SiO2 catalysts; the selectivity of DMCD reached 97 with a 98% conversion of DMT at 100 °C and 2.5 MPa for 4 h, attributed to the increased presence of Ni(0) species and the reduction of moderate acidic sites modified by KF (Fig. 16C).163
In the second step, Cu catalysts have been verified to be highly effective for the hydrogenation of esters in DMCD. Zhang et al. reported that the gas-phase hydrogenation of DMCD to CHDM was successfully performed using a well-dispersed copper-based catalyst derived from a CuMgAl LDH precursor, benefiting from the synergistic effect between surface-active Cu0 sites and Lewis base sites, and the conversion of DMCD remained 100% with 99.8% selectivity for CHDM up to 200 h at 240 °C and 6 MPa.164 Previous work conducted in our laboratory disclosed that a Cu1/Mg3Sc2O6 catalyst derived from a new LDH-structured Cu1Mg3Sc2(OH)12CO3 material possesses enlarged surface area, enhanced basicity, and reduced acidity compared to Cu1/Mg3Al2O6. This Cu1/Mg3Sc2O6 catalyst exhibited prominent activity and selectivity for the conversion of DMCD to CHDM, achieving a 96.3% yield at 250 °C, 2 MPa, and a WHSV of 0.49 h−1 and retaining this performance for at least 300 h.165 Well-dispersed Cu supported on MgO (Cu/MgO)166 catalysts and Cu nanoparticles socketed in situ into copper phyllosilicate nanotube catalysts167 also exhibited high catalytic performance, with high conversion (>98%) and selectivity to CHDM (>96%) at 220 °C and 5–6 MPa H2. Additionally, Lou et al. found that 5 wt% Re supported on activated carbon showed catalytic performance with complete conversion of DMCD and a 66% yield of CHDM, and a specific rate reaching 950 mmol DMCD gRe−1 h−1 at 220 °C and 10 MPa H2 due to the medium strong acid sites and high dispersion of exposed Re sites.168
Apart from the two-step hydrogenation technology, several works have reported the exploration of one-pot conversion of DMT to CHDM. Hungria et al. found that a tri-metallic Ru5PtSn/SiO2 catalyst can achieve one-pot conversion of DMT to CHDM under mild conditions, with a conversion of 63.9% and selectivity of 71.2% at 120 °C and 4 MPa H2.169 On the surface of an Al2O3 supported tri-metallic RuPtSn catalyst, the conversion of DMT and the selectivity of CHDM increased to 98.2% and 75.1%, respectively, in programmed reaction steps (180 °C and 6 MPa H2 for 2 h at first, followed by 260 °C and 8.5 MPa H2 for 8 h).170 Xiao et al. also reported a one-pot tandem catalytic process for converting DMT to CHDM using a physical mixture of 5% Pd/CMK-3 and Cu–MgAlOx catalysts in a batch reactor. The final conversion of DMT reached 99.5%, and the selectivity for CHDM increased to 82.3% at 250 °C and 5 MPa H2 for 3 h.171 However, there is still room for improvement in the yield of CHDM in one-pot reactions.
5.4. Upgrading of depolymerized products from other polyesters
Lactic acid, the hydrolysis product of PLA, is an important platform compound and can be further converted into high-value chemicals.172 Acrylic acid, one of the fastest growing commodity chemicals, is currently produced from fossil propene. Holmen found that the direct conversion of lactic acid to acrylic acid (AA) occurred successfully over a CaSO4/Na2SO4 catalyst via dehydration, achieving a maximum yield of 68% at 400 °C.173 Another valuable fine chemical, 2,3-pentanedione (PD), could be obtained from two lactic acid molecules via dehydration and condensation. Miller et al. disclosed that different pore glass-supported Cs hydroxides can achieve a 49% PD yield (64% selectivity) by decreasing Lewis acidity at 280 °C.174 Acetaldehyde can also be synthesized via the decarboxylation or decarbonylation of lactic acid over an SiO2-supported silicotungstic acid catalyst at 275 °C.175 Moreover, lactic acid can also be used as a raw material for the production of 1,2-propanediol via hydrogenation176 and pyruvic acid via oxidation.177PHB, another popular biodegradable plastic, is used in the production of optical materials, magnetic powders and biological nanomaterials, and its depolymerized products were also upgraded to various chemicals. Li et al. utilized 3-hydroxybutyric acid (3-HBA), the hydrolysis product of PHB, as a raw material to prepare crotonic acid (CA) via dehydration and further produced propylene through decarboxylation.178 PHB could be converted into ethyl 3-hydroxybutyrate (HBEt), which was then cyclized with cyclohexene oxide to form the new bicyclic monomer 4-methyloctahydro-2H-benzo[b][1,4]dioxepin-2-one (4-MOHB).179
6. One-pot transformation of polyesters into valuable chemicals
Besides yielding monomers for the re-synthesis of new polyesters, the direct depolymerization of waste polyesters can also convert them into valuable chemicals in a one-pot technology. This approach maximizes the utilization of the carbon resources in waste polyesters, enabling effective resource utilization. In published works, thermal catalysis, electrocatalysis, photocatalysis, and biocatalysis have been demonstrated to show significant feasibility in the one-pot transformation of polyesters into valuable chemicals.180,181 Li et al. reviewed the recent progress in photocatalysis, electrocatalysis, and their integration in plastic conversion strategies. It was concluded that these approaches show high reactivity and selectivity under environmentally benign conditions and provide alternative reaction pathways for plastic conversion to value-added chemicals and fuels.182 More recently, Liang et al. also summarized the progress in clean catalytic technologies to convert plastics into fuels, materials and fine chemicals, including photo-, electro-, and photoelectrochemical catalysis and solar thermal electrochemical processes. The degradation mechanisms, various processes, optimized conditions and pretreatment methods for upcylcing of various plastics were discussed in detail. It was concluded that these techniques could be conducted under mild conditions using clean energy and are environmentally sustainable.1836.1 One-pot conversion of polyesters via thermocatalysis
Hu et al. reported a self-designed three-tooth clip-on ruthenium complex based on a quinaldine skeleton to upgrade PET into high-value-added chemicals via a transesterification/hydrogenation relay strategy. They found that the rate determining step of this conversion process is the esterification of polyesters into oligomer fragments, followed by hydrogenation using specially designed quinaldine Ru(II) complexes. This approach is effective for several commercially available or post-consumer PET products to be degraded to high-yield p-phenyldimethyl alcohol under mild conditions (80 °C, 5 bar H2) (Fig. 17A).184 In the direct depolymerization of poly(L-lactic acid) (PLA), McGuire et al. found that the yield of L-lactide (L-LA) reached 92% in a catalyst system composed of tin(II) bis(2-ethylhexanoate) (Sn(Oct)2) and glycerolethoxylate (GEO) at 160 °C (Fig. 17B), and this catalyst retained a high L-LA yield for four recycles.185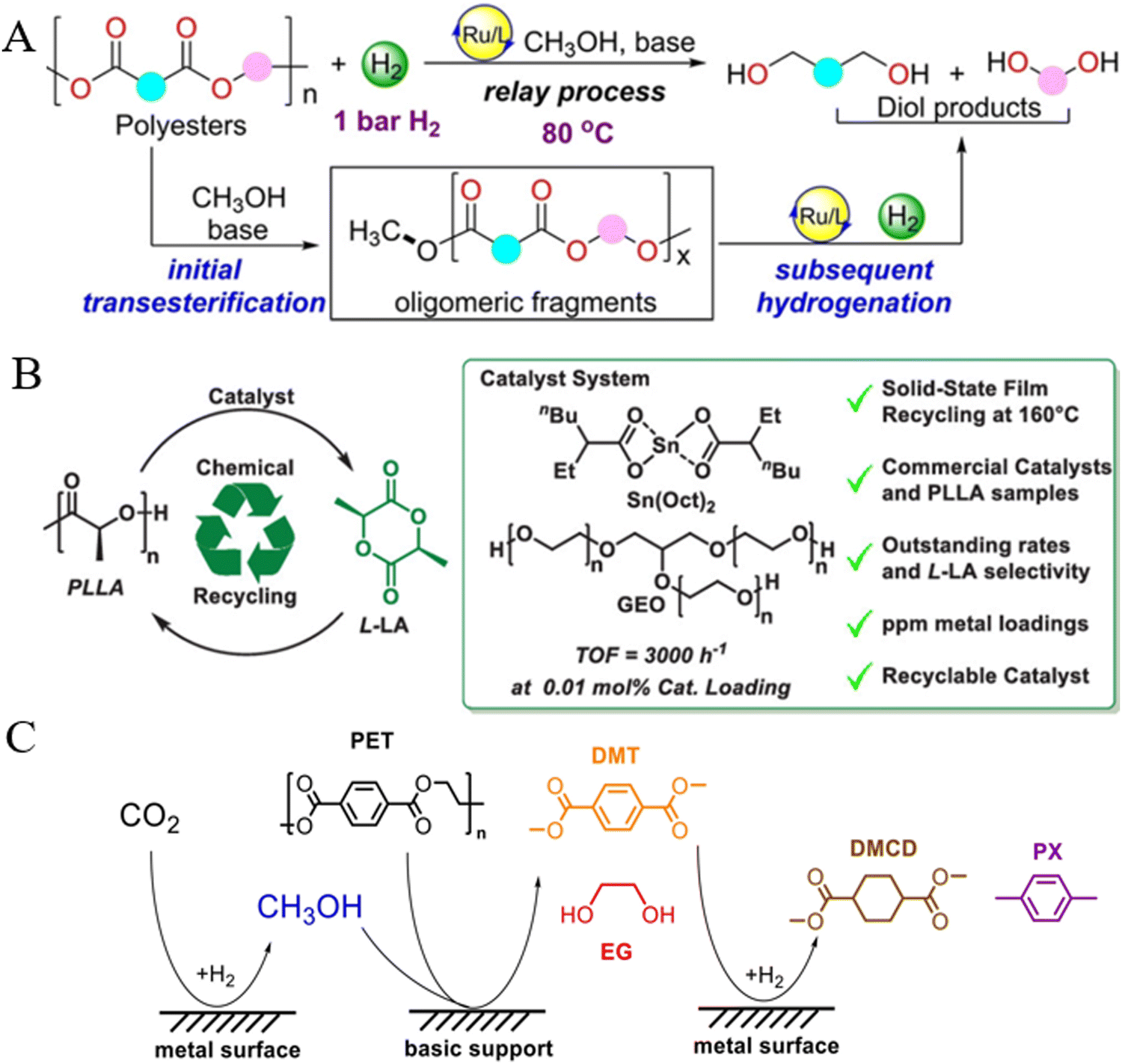 | ||
| Fig. 17 (A) Ester exchange/hydrogenation relay strategy for degradation of PET. Reproduced from ref. 184 with permission from Wiley, copyright [2023]. (B) The Sn(II)/alcohol catalyst system is used to recycle PLLA into L-LA. Reproduced from ref. 185 with permission from American Chemical Society, copyright [2023]. (C) The scheme of dual-promoted CO2 hydrogenation and PET degradation reaction systems. Reproduced from ref. 186 with permission from Wiley, copyright [2022]. | ||
Li et al. found that the synergic coupling of three reactions (CO2 hydrogenation, PET methanol alcoholysis and DMT hydrogenation) successfully took place over CuFeCr catalysts in the presence of in situ formed methanol (Fig. 17C). The coupling reaction enabled the hydrogenation of CO2 to methanol to overcome the thermodynamic equilibrium limit and the calculated methanol production rate (11 mmol per g-cat) was twice higher than that without PET.186 By using ionic liquids (ILs) containing chlorine as both catalysts and solvents, with ZnCl2 as a Lewis acid catalyst, Cao et al. achieved the upcycling of mixed PET and PVC plastics. They found that tetrabutylphosphonium chloride (Bu4PCl) ILs were able to extract and store HCl from PVC, and the in situ generated HCl promoted the depolymerization of PET with ZnCl2 at 230 °C, producing TPA and upgrading EG into 1,2-dichloroethane. This strategy not only simplifies the separation process in plastic recycling but also reduces the generation of chlorine-containing waste.187
Recently, Sun et al. presented an innovative process for directly transforming PET into CHDM over a dual-catalyst system (reduced graphene oxide supported Pd and oxalate-gel-derived copper–zinc oxide) in one pot. This method efficiently transformed PET into polyethylene-1,4-cyclohexanedicarboxylate (PECHD), which is then converted into CHDM with an overall yield of 95% and a notably higher trans/cis ratio of up to 4.09/1, far exceeding that of commercial CHDM. Various post-consumer PET plastics were suitable for this method and the yields of CHDM were between 78 and 89% across different substrates, showing promising industrial application potential.188
6.2. One-pot conversion of polyesters via electrocatalysis
Electrocatalysis is considered a promising technology for recycling and converting waste polyesters using renewable electric energies (such as solar and wind), as it can be performed at mild temperatures and pressures.181Zhou et al. designed a technique to recover PET into TPA, potassium dicarboxylate (KDF), and H2 in a KOH electrolyte using a bifunctional CoNi0.25P electrocatalyst. It was found that PET could be depolymerized into TPA and EG in an alkaline solution, and then, EG was oxidized to formate on a CoNi0.25P anode over a wide potential window (1.3–1.7 V vs. RHE), while H2 was produced at the cathode. TPA was obtained by using formic acid to acidify the electrolyte, and the electrolyte could be further converted into solid KDF after concentration and crystallization (Fig. 18A).189 Liu et al. have prepared a CuCo2O4 nanowire array/nickel foam electrocatalyst (Fig. 18B), over which EG obtained by the depolymerization of PET was oxidized to formate with 93% Faraday efficiency and 86% selectivity at a high current density of 100 mA cm−2.190
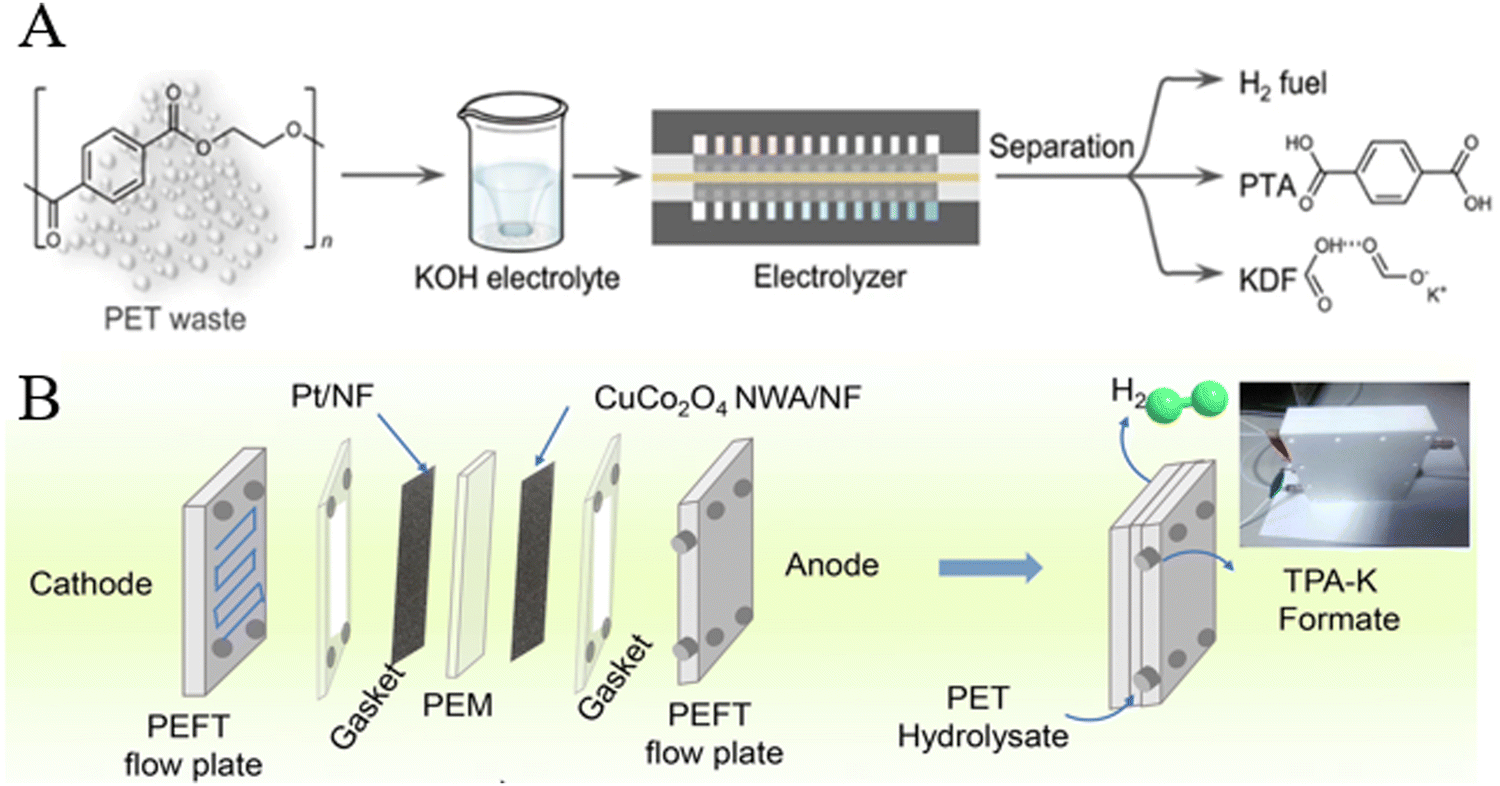 | ||
| Fig. 18 (A) Electrocatalytic PET upcycling to commodity chemicals and H2 using a CoNi0.25P electrocatalyst. Reproduced from ref. 189 with permission from Nature, copyright [2021]. (B) Schematic illustration of electro-reforming PET in a two-electrode MEA flow reactor. Reproduced from ref. 190 with permission from Royal Society of Chemistry, copyright [2022]. | ||
The electrocatalytic recovery of PLA was also reported by Li et al. with cobalt selenide nanosheets on nickel foam (0.1-CoSe2/NF) prepared by a simple hydrothermal and selenization treatment method. This material can be utilized as a bifunctional catalyst for the assisted hydrolysis of PLA. At a potential of 1.5 V, the yield of acetic acid reached 87% after a 30-h potentiostatic test, attributed to its high specific surface area, abundant active sites, and porous structure.191
6.3. One-pot conversion of polyesters via photocatalysis
Photocatalysis is a process that uses light energy to drive chemical reactions. In recent years, numerous studies have focused on the photocatalytic recovery of polyesters under mild conditions. When light irradiates on the photocatalyst, electrons in the valence band (VB) are excited to the conduction band (CB), generating pairs of photogenerated electrons (e−) and holes (h+). The photogenerated electrons in the CB reduce H2O to H2, while the holes in the VB migrate to the catalyst surface, where they oxidize and decompose polyesters into small molecule compounds.192,193In 2018, Uekert et al. reported the use of a CdS/CdOx quantum dot photocatalyst for the photocatalytic reforming of waste polyester (PLA and PET) into small molecules and H2 in an alkaline aqueous solution. They found that PLA could be reformed into pyruvate compounds via photocatalysis, while PET was converted into formate, acetate, glycolate, and lactate.194 The same group also found a carbon nitride/nickel phosphide (CNx|Ni2P) photocatalyst, which could photocatalytically convert polyesters into various carboxylate salts and H2.195 Recently, Zhang et al. synthesized a defect-rich nickel-based thiophosphate and cadmium sulfide composite photocatalyst (d-NiPS3/CdS), which could be used for the photocatalytic recycling of polyesters to produce high-value chemicals and hydrogen. At room temperature, d-NiPS3/CdS can convert waste PLA and PET plastics with hydrogen production efficiencies up to 39.76 mmol per g-cat per h (PLA) and 31.38 mmol per g-cat per h (PET), respectively (Fig. 19). The oxidation products of PLA were acetate and pyruvate, while the oxidation products of PET were formate, acetate, and glycolate. The efficiency of photocatalytic polyester recovery could be improved as d-NiPS3 enhanced the rapid separation of photogenerated electron–hole pairs generated on CdS, accelerating hydrogen production and polyester degradation.196
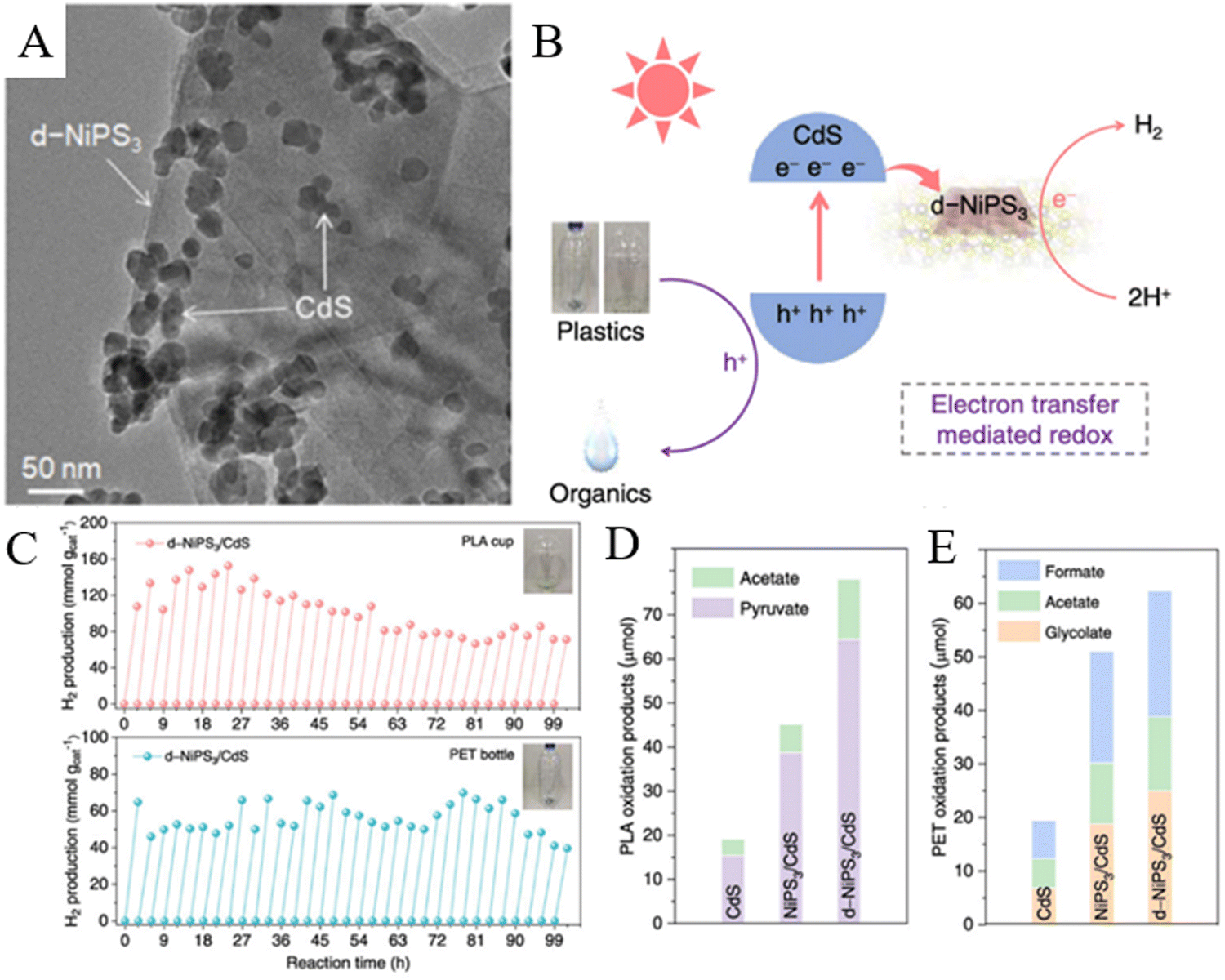 | ||
| Fig. 19 (A) HRTEM image of d-NiPS3/CdS, (B) mechanism of d-NiPS3/CdS photocatalytic reforming of plastics, (C) consecutive H2 production via long-term photoreforming of PLA cups and PET bottles using d-NiPS3/CdS, (D) and (E) organic products from PLA and PET for different photocatalysts following 9 h of photoreforming. Reproduced from ref. 196 with permission from American Chemical Society, copyright [2023]. | ||
Because photocatalytic reforming of polyesters requires pretreatment with a lye and faces challenges in achieving precise chemical conversion, the separation of these mixed degraded products is difficult. Additionally, the photocatalytic recycling of polyesters is restricted due to its limited efficiency and the generation of CO2.197 More efficient photocatalysts for polyester recycling remains essential.
6.4. One-pot conversion of polyesters via biocatalysis
Biocatalysis is also a commonly reported technique for recycling polyesters, typically carried out at mild temperature and pressure.198–200 Biocatalysis primarily relies on the action of enzymes, making it essential to screen and discover suitable enzymes for this purpose.201–203Yoshida et al. isolated a novel bacterium, Ideonella sakaiensis 201-F6, by screening natural microbial communities exposed to PET in the environment. This strain could produce two different enzymes to depolymerize PET. Specifically, PET hydrolase (PETase) hydrolyzes PET to 2-hydroxyethyl methyl terephthalate (MHET), and MHET hydrolase (MHETase) further hydrolyzes MHET to PTA and EG.204 To improve the efficiency of biocatalytic PET recovery, Tournier et al. further enhanced a PET hydrolase using computer-aided enzyme engineering, achieving 90% PET depolymerization within 10 h at 65 °C with a productivity of 16.7 g PTA per L per h (200 g of PET per kg of suspension, an enzyme concentration of 3 mg per g PET).205 More recently, Lu et al. invented a stable and efficient PET hydrolytic enzyme (FAST-PETase) based on a structure-predicting machine learning algorithm, which demonstrated excellent activity under harsh conditions (50–72 °C and pH 6.5–8). 51 types of PET-based products could be completely degraded by FAST-PETase within 1 week and the degradation liquid could be repolymerized to produce new polyesters (Fig. 20).206 Myburgh et al. also reported the recombinant expression of a fungal keratinase-like enzyme (CLE1) in Saccharomyces cerevisiae, producing a crude supernatant that effectively hydrolyzed different types of PLA materials. The codon-optimized Y294[CLEns] strain showed optimal enzyme production and hydrolysis capabilities, releasing up to 9.44 g L−1 lactic acid from 10 g L−1 PLA membrane.207
 | ||
| Fig. 20 (A) Complete depolymerization of 51 types of post-consumer PET products and (B) schematic of the closed-loop PET recycling process. Reproduced from ref. 206 with permission from Nature, copyright [2022]. | ||
More interestingly, Sadler and Wallace demonstrated that engineered Escherichia coli could direct upcycling of PTA to vanillin, and 79% conversion to vanillin was achieved under optimized conditions, and they also found that post-consumer PET from a plastic bottle could be directly used as the feedstock. This achievement paved the way for the biological upcycling of post-consumer plastic waste into vanillin using an engineered microorganism.208 Recently, Schubert et al. disclosed that the activity PET-hydrolase, LCCICCG, was limited by conformational constraints in the PET polymer; however, endo-type cuts could promote chain mobility and hence the density of attack sites on the surface, which gradually promoted the formation of soluble products.209 Li et al. further developed an efficient chemical-enzymatic approach to convert mixed plastic wastes into MHET using combined three discovered BHETases (KbEst, KbHyd, and BrevEst) from nature and quantum theoretical computations. They found that the yield of MHET reached 98.26% in 40 h, and BrevEst accomplished the highest BHET hydrolysis (∼87% efficiency in 12 h) for yielding analytical-grade MHET.210
Previous studies have confirmed that waste polyester plastics (such as PET and PLA) can be transformed into monomers or other valuable chemicals via thermal catalysis, electrocatalysis, photocatalysis, or biocatalysis. The design of catalysts with high activity and stability can not only significantly reduce energy consumption and reaction time, but also improve the yield of recovered products.42 It is worth mentioning that although many advanced technologies appear to be promising, assessment of stability and economical efficiency is often conducted under ideal conditions and ignores factors such as the pretreatment of waste polyesters, the purification of products and the difficulty in scaling up, which can lead to inaccurate forecasts.
7. Conclusions and outlook
Polyesters, important components of plastics, are widely used in production and daily life due to their excellent properties. The development of chemical recycling technology offers a viable solution for the efficient utilization of waste polyesters, reducing the dependence on fossil energy, mitigating environmental pollution and contributing to carbon neutrality. On the basis of recent publications, several depolymerization routines of waste polyesters (particularly PET), such as hydrolysis, glycolysis, ammonolysis, and methanolysis, were summarized, and the newly reported recycling of actual PET-based products (including bottles, films, clothes and plastic mixtures) was introduced. Subsequently, further upgrading of the depolymerized products from PET (including PTA, BHET and DMT) to CHDM via one-pot approaches and/or two-step hydrogenation processes was highlighted. Additionally, the one-pot conversion of polyesters to valuable compounds via thermal catalysis, photocatalysis, electrocatalysis, and biocatalysis was identified as a promising transformation pathway.Honestly, many achievements have been reported on recycling and upcycling of waste PET to recycled monomers and/or value-added products (such as CHDM). Among them, hydrolysis, glycolysis, ammonolysis, and methanolysis of waste PET to monomers have been verified on the industrial scale with promising economic benefits compared with the fresh fossil derived counterpart. The newly developed photocatalysis, electrocatalysis and biocatalysis technologies might make these processes more energy-conserving, and the explorations made using textile and mixed plastics directly as feedstocks could pave the way for elimination of plastic waste and decrease the dependence on nonrenewable fossil energy for a sustainable development.
However, significant efforts are still needed in terms of efficiency, environmental protection, versatility, and resource consumption. Hydrolysis uses water as a solvent and can directly produce recyclable PTA, but it requires high reaction temperature. Glycolysis and methanolysis can be carried out under milder conditions, but side reactions happened easily. Aminolysis requires special attention due to the use of toxic and expensive nitrogenous chemicals. Thermocatalytic technology can efficiently recycle and convert polyesters into specific fine chemicals via controlled pathways. However, this process has drawbacks such as high energy consumption and equipment requirements. Meanwhile, enhancing the performance of catalysts for desired reaction routes is challenging. The reaction conditions for electrocatalysis, photocatalysis, and biocatalysis are relatively mild, but these technologies face issues such as the small scale of recovery reactions, tedious pretreatment of raw materials, and complex post-processing steps. Furthermore, the efficiency and stability of catalysts need to be improved, and there is a desire to expand the high-value utilization of polyester depolymerized products.
In published works, assessment of economic and environmental sustainability was often ignored. Many limitations, such as the collection, transportation, sorting and pretreatment of plastic wastes, were not addressed in most academic research studies. These shortcomings can lead to inaccurate or misleading predictions, reduce opportunities for optimization and limit industrial relevance.211 To promote the real applications of waste polyesters via recycling and upgrading on a large scale, we think that the following aspects deserve greater attention. First, waste polyesters come from a wide range of sources and pre-treatment processes, such as sorting, decolorization, and impurity removal, before the depolymerization process and hence it must be carefully performed because many residues have a negative influence on the quality of the final product. Striving for a green and low-carbon cycle is the final objective. Reducing energy and chemical consumption as well as the circulation of materials in the recycling process is a vital initiative. Besides, a deep understanding of the depolymerization mechanism, especially in the presence of heterogenous catalysts, can enhance the effectiveness of depolymerization, which is the direction of long-term efforts. Additionally, conducting research on the deep processing of recycled products and expanding the range of polyester-derived products to generate valuable chemicals can increase the profitability of the industry. Finally, the separation and purification of recycled products must not be overlooked. From an industrial perspective, waste polyester refineries could be a viable option for the future, while special technologies, including pretreatment, catalysts, reactors, and separation systems must be studied in detail for the realization of waste polyester refineries The development of waste polyester refineries will play an increasingly important role in the sustainable development of society.
Author contributions
Huaiyuan Zhao: reference searching, formal analysis, writing – original draft; Yingdan Ye: reference searching, formal analysis, writing – original draft; Yibin Zhang: validation, formal analysis; Lei Yang: validation, formal analysis; Weichen Du: validation, formal analysis; Songlin Wang: conceptualization, validation, supervision; and Zhaoyin Hou: conceptualization, resources, writing – reviewing and editing, supervision, funding acquisition.Data availability
This submission is an invited review; it does not contain experiments or data. All data used in this submission are from published references.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024C01045), National Natural Science Foundation of China (Contract No. 21773206) and the Fundamental Research Funds for the Central Universities (No. 226-2022-00055).Notes and references
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- T. Muringayil Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Case Stud. Chem. Environ. Eng., 2024, 9, 100673 CrossRef CAS.
- Y. Wang, R.-J. van Putten, A. Tietema, J. R. Parsons and G.-J. M. Gruter, Green Chem., 2024, 26, 3698–3716 RSC.
- Y. Chen, L. Bai, D. Peng, X. Wang, M. Wu and Z. Bian, Environ. Sci.: Adv., 2023, 2, 1151–1166 CAS.
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, Chem. Soc. Rev., 2024, 53, 7309–7327 RSC.
- C. J. Rhodes, Sci. Prog., 2018, 101, 207–260 CrossRef PubMed.
- J. Zheng and S. Suh, Nat. Clim. Change, 2019, 9, 374–378 CrossRef.
- R. G. Santos, G. E. Machovsky-Capuska and R. Andrades, Science, 2021, 373, 56–60 CrossRef CAS PubMed.
- M. Kumar, X. Xiong, M. He, D. C. W. Tsang, J. Gupta, E. Khan, S. Harrad, D. Hou, Y. S. Ok and N. S. Bolan, Environ. Pollut., 2020, 265, 114980 CrossRef CAS PubMed.
- L. J. J. Meijer, T. van Emmerik, R. van der Ent, C. Schmidt and L. Lebreton, Sci. Adv., 2021, 7, eaaz5803 CrossRef PubMed.
- L.-Z. Yin, X.-Q. Luo, J.-L. Li, Z. Liu, L. Duan, Q.-Q. Deng, C. Chen, S. Tang, W.-J. Li and P. Wang, J. Hazard. Mater., 2024, 474, 134728 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- E. T. C. Vogt and B. M. Weckhuysen, Nature, 2024, 629, 295–306 CrossRef CAS PubMed.
- K. M. Van Geem, Science, 2023, 381, 607–608 CrossRef CAS PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS PubMed.
- C. Li, C. Guo, V. Fitzpatrick, A. Ibrahim, M. J. Zwierstra, P. Hanna, A. Lechtig, A. Nazarian, S. J. Lin and D. L. Kaplan, Nat. Rev. Mater., 2020, 5, 61–81 CrossRef.
- T. Iwata, Angew. Chem., Int. Ed., 2015, 54, 3210–3215 CrossRef CAS PubMed.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- R. Nisticò, Polym. Test., 2020, 90, 106707 CrossRef.
- T. Fernández-Menéndez, D. García-López, A. Argüelles, A. Fernández and J. Viña, Polym. Test., 2020, 90, 106729 CrossRef.
- A. F. Sousa, R. Patrício, Z. Terzopoulou, D. N. Bikiaris, T. Stern, J. Wenger, K. Loos, N. Lotti, V. Siracusa, A. Szymczyk, S. Paszkiewicz, K. S. Triantafyllidis, A. Zamboulis, M. S. Nikolic, P. Spasojevic, S. Thiyagarajan, D. S. van Es and N. Guigo, Green Chem., 2021, 23, 8795–8820 RSC.
- R. Atakan, S. Sezer and H. Karakas, J. Ind. Text., 2020, 49, 835–857 CrossRef CAS.
- T. El Darai, A. Ter-Halle, M. Blanzat, G. Despras, V. Sartor, G. Bordeau, A. Lattes, S. Franceschi, S. Cassel, N. Chouini-Lalanne, E. Perez, C. Déjugnat and J.-C. Garrigues, Green Chem., 2024, 26, 6857–6885 RSC.
- E. Andini, P. Bhalode, E. Gantert, S. Sadula and D. G. Vlachos, Sci. Adv., 2024, 10, eado6827 CrossRef PubMed.
- A. Z. Naser, I. Deiab and B. M. Darras, RSC Adv., 2021, 11, 17151–17196 RSC.
- Z. Liu, Y. Wang, B. Wu, C. Cui, Y. Guo and C. Yan, Int. J. Adv. Manuf. Technol., 2019, 102, 2877–2889 CrossRef.
- N. Malik, P. Kumar, S. Shrivastava and S. B. Ghosh, Int. J. Plast. Technol., 2017, 21, 1–24 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Mater., 2017, 1, 0046 Search PubMed.
- S. S. Borkar, R. Helmer, F. Mahnaz, W. Majzoub, W. Mahmoud, M. M. Al-Rawashdeh and M. Shetty, Chem. Catal., 2022, 2, 3320–3356 CrossRef CAS.
- Z. Jia, L. Gao, L. Qin and J. Yin, RSC Sustainability, 2023, 1, 2135–2147 RSC.
- O. Guselnikova, O. Semyonov, E. Sviridova, R. Gulyaev, A. Gorbunova, D. Kogolev, A. Trelin, Y. Yamauchi, R. Boukherroub and P. Postnikov, Chem. Soc. Rev., 2023, 52, 4755–4832 RSC.
- K. Wang, C. Guo, J. Li, K. Wang, X. Cao, S. Liang and J. Wang, J. Environ. Chem. Eng., 2024, 12, 113539 CrossRef CAS.
- C. Shi, E. C. Quinn, W. T. Diment and E. Y. X. Chen, Chem. Rev., 2024, 124, 4393–4478 CrossRef CAS PubMed.
- A. Schade, M. Melzer, S. Zimmermann, T. Schwarz, K. Stoewe and H. Kuhn, ACS Sustainable Chem. Eng., 2024, 12, 12270–12288 CrossRef CAS.
- Y. Weng, C.-B. Hong, Y. Zhang and H. Liu, Green Chem., 2024, 26, 571–592 RSC.
- M. Wang, Y. Li, L. Zheng, T. Hu, M. Yan and C. Wu, Polym. Chem., 2024, 15, 585–608 RSC.
- S. M. Al-Salem, P. Lettieri and J. Baeyens, Waste Manage., 2009, 29, 2625–2643 CrossRef CAS PubMed.
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer, S. R. Nicholson, G. T. Beckham and A. C. Carpenter, ACS Sustainable Chem. Eng., 2023, 11, 965–978 CrossRef CAS.
- O. Eriksson and G. Finnveden, Energy Environ. Sci., 2009, 2, 907–914 RSC.
- B. S. Adeboye, B. Z. Adewole, A. M. Adedoja, S. O. Obayopo, S. A. Mamuru, M. O. Idris, I. K. Okediran and A. A. Asere, Waste Disposal Sustainable Energy, 2021, 3, 289–298 CrossRef.
- P. Cai, J. Fu, M. Zhan, W. Jiao, T. Chen and X. Li, J. Cleaner Prod., 2022, 342, 130762 CrossRef CAS.
- E. Feghali, L. Tauk, P. Ortiz, K. Vanbroekhoven and W. Eevers, Polym. Degrad. Stab., 2020, 179, 109241 CrossRef CAS.
- Y. He, Y. Luo, M. Yang, Y. Zhang, L. Zhu, M. Fan and Q. Li, Appl. Catal., A, 2022, 630, 118440 CrossRef CAS.
- H. Sun, Z. Chen, J. Zhou, L. Chen and W. Zuo, J. Environ. Chem. Eng., 2024, 12, 112558 CrossRef CAS.
- J. R. Campanelli, M. R. Kamal and D. G. Cooper, J. Appl. Polym. Sci., 1993, 48, 443–451 CrossRef CAS.
- C. N. Onwucha, C. O. Ehi-Eromosele, S. O. Ajayi, M. Schaefer, S. Indris and H. Ehrenberg, Ind. Eng. Chem. Res., 2023, 62, 6378–6385 CrossRef CAS.
- H. Tsuji, H. Daimon and K. Fujie, Biomacromolecules, 2003, 4, 835–840 CrossRef CAS PubMed.
- T. Saeki, T. Tsukegi, H. Tsuji, H. Daimon and K. Fujie, Polymer, 2005, 46, 2157–2162 CrossRef CAS.
- H. Tsuji, T. Ono, T. Saeki, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2005, 89, 336–343 CrossRef CAS.
- S. Mishra, A. S. Goje and V. S. Zope, Polym.-Plast. Technol., 2003, 42, 581–603 CrossRef CAS.
- H. Abedsoltan and M. R. Coleman, J. Appl. Polym. Sci., 2022, 139, e52451 CrossRef CAS.
- W. Yang, R. Liu, C. Li, Y. Song and C. Hu, Waste Manage., 2021, 135, 267–274 CrossRef CAS PubMed.
- M. J. Kang, H. J. Yu, J. Jegal, H. S. Kim and H. G. Cha, Chem. Eng. J., 2020, 398, 125655 CrossRef CAS.
- H. Abedsoltan, I. S. Omodolor, A. C. Alba-Rubio and M. R. Coleman, Polymer, 2021, 222, 123620 CrossRef CAS.
- J. Yu, D. Plackett and L. X. L. Chen, Polym. Degrad. Stab., 2005, 89, 289–299 CrossRef CAS.
- S. Ügdüler, K. M. Van Geem, R. Denolf, M. Roosen, N. Mys, K. Ragaert and S. De Meester, Green Chem., 2020, 22, 5376–5394 RSC.
- H. Chen and H. Hu, Ind. Eng. Chem. Res., 2023, 62, 12925–12934 CrossRef CAS.
- S. Mishra and A. S. J. P. R. E. Goje, Polym. React. Eng., 2003, 11, 963–987 CrossRef CAS.
- S. Zhang, W. Xu, R. Du, X. Zhou, X. Liu, S. Xu and Y.-Z. Wang, Green Chem., 2022, 24, 3284–3292 RSC.
- H. Yu, Y. Wang, L. Chen, C. Wei, T. Mu and Z. Xue, Green Chem., 2023, 25, 7807–7816 RSC.
- M. Yagihashi and T. Funazukuri, Ind. Eng. Chem. Res., 2010, 49, 1247–1251 CrossRef CAS.
- Ž. Knez, M. K. Hrnčič, M. Čolnik and M. Škerget, J. Supercrit., 2018, 133, 591–602 CrossRef.
- M. Čolnik, Ž. Knez and M. Škerget, Chem. Eng. Sci., 2021, 233, 116389 CrossRef.
- W.-Z. Guo, H. Lu, X.-K. Li and G.-P. Cao, RSC Adv., 2016, 6, 43171–43184 RSC.
- D. Osei, L. Gurrala, A. Sheldon, J. Mayuga, C. Lincoln, N. A. Rorrer and A. R. C. Morais, Green Chem., 2024, 26, 6436–6445 RSC.
- S. Mishra, A. S. Goje and V. S. Zope, Polym. React. Eng., 2003, 11, 79–99 CrossRef CAS.
- V. A. Kosmidis, D. S. Achilias and G. P. Karayannidis, Macromol. Mater. Eng., 2001, 286, 640–647 CrossRef CAS.
- Y. Wang, H. Wang, H. Chen and H. Liu, Chin. J. Chem. Eng., 2022, 51, 53–60 CrossRef CAS.
- N. R. Paliwal and A. K. Mungray, Polym. Degrad. Stab., 2013, 98, 2094–2101 CrossRef CAS.
- S. Mohammadi, M. G. Bouldo and M. Enayati, ACS Appl. Polym. Mater., 2023, 5, 6574–6584 CrossRef CAS PubMed.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, S. Arnaiz and J. I. Gutiérrez-Ortiz, Polym. Degrad. Stab., 2010, 95, 1022–1028 CrossRef.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320 CrossRef.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566 RSC.
- Z. Wang, Y. Jin, Y. Wang, Z. Tang, S. Wang, G. Xiao and H. Su, ACS Sustainable Chem. Eng., 2022, 10, 7965–7973 CrossRef CAS.
- H. Wang, R. Yan, Z. Li, X. Zhang and S. Zhang, Catal. Commun., 2010, 11, 763–767 CrossRef CAS.
- Q. Wang, Y. Geng, X. Lu and S. Zhang, ACS Sustainable Chem. Eng., 2015, 3, 340–348 CrossRef CAS.
- Y. Liu, X. Yao, H. Yao, Q. Zhou, J. Xin, X. Lu and S. Zhang, Green Chem., 2020, 22, 3122–3131 RSC.
- Y. Geng, T. Dong, P. Fang, Q. Zhou, X. Lu and S. Zhang, Polym. Degrad. Stab., 2015, 117, 30–36 CrossRef CAS.
- P. Fang, B. Liu, J. Xu, Q. Zhou, S. Zhang, J. Ma and X. Lu, Polym. Degrad. Stab., 2018, 156, 22–31 CrossRef CAS.
- S. R. Shukla, V. Palekar and N. Pingale, J. Appl. Polym. Sci., 2008, 110, 501–506 CrossRef CAS.
- S. Mo, Y. Guo, X. Liu and Y. Wang, Catal. Sci. Technol., 2023, 13, 6561–6569 RSC.
- L.-X. Yun, H. Wu, Z.-G. Shen, J.-W. Fu and J.-X. Wang, ACS Sustainable Chem. Eng., 2022, 10, 5278–5287 CrossRef CAS.
- J. Cao, Y. Lin, T. Zhou, W. Wang, Q. X. Zhang, B. Pan and W. J. I. Jiang, iScience, 2023, 28, 107492 CrossRef PubMed.
- J. Cao, Y. Lin, W. Jiang, W. Wang, X. Li, T. Zhou, P. Sun, B. Pan, A. Li and Q. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 5476–5488 CrossRef CAS.
- E. Gabirondo, B. Melendez-Rodriguez, C. Arnal, J. M. Lagaron, A. Martínez de Ilarduya, H. Sardon and S. Torres-Giner, Polym. Chem., 2021, 12, 1571–1580 RSC.
- B. Nim, M. Opaprakasit, A. Petchuk and P. Opaprakasit, Polym. Degrad. Stab., 2020, 181, 109363 CrossRef CAS.
- J. Wu, F. Yang, D. Shi, Z. Miao, J. Wang, D. Wang and Y. Zhang, ChemSusChem, 2024, e202401922 CrossRef PubMed.
- Q. Wang, X. Yao, Y. Geng, Q. Zhou, X. Lu and S. J. G. C. Zhang, Green Chem., 2015, 17, 2473–2479 RSC.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300 CrossRef CAS.
- Q. Sun, Y.-Y. Zheng, L.-X. Yun, H. Wu, R.-K. Liu, J.-T. Du, Y.-H. Gu, Z.-G. Shen and J.-X. Wang, ACS Sustainable Chem. Eng., 2023, 11, 7586–7595 CrossRef CAS.
- L.-X. Yun, Y. Wei, Q. Sun, Y.-T. Li, B. Zhang, H.-T. Zhang, Z.-G. Shen and J.-X. Wang, Green Chem., 2023, 25, 6901–6913 RSC.
- Q. Suo, J. Zi, Z. Bai and S. Qi, Catal. Lett., 2017, 147, 240–252 CrossRef CAS.
- R.-X. Yang, Y.-T. Bieh, C. H. Chen, C.-Y. Hsu, Y. Kato, H. Yamamoto, C.-K. Tsung and K. C. W. Wu, ACS Sustainable Chem. Eng., 2021, 9, 6541–6550 CrossRef CAS.
- M. A. Baluk, P. J. Trzebiatowska, A. Pieczyńska, D. Makowski, M. Kroczewska, J. Łuczak and A. Zaleska-Medynska, J. Environ. Manage., 2024, 363, 121360 CrossRef CAS PubMed.
- G. Eshaq and A. E. ElMetwally, J. Mol. Liq., 2016, 214, 1–6 CrossRef CAS.
- D. Thomas, R. Ranjan and B. K. J. R. S. George, RSC Sustainability, 2023, 1, 2277–2286 RSC.
- M. D. de Dios Caputto, R. Navarro, J. L. Valentín and Á. Marcos-Fernández, J. Polym. Sci., 2022, 60, 3269–3283 CrossRef CAS.
- S. R. Shukla and A. M. Harad, Polym. Degrad. Stab., 2006, 91, 1850–1854 CrossRef CAS.
- V. Jamdar, M. Kathalewar and A. Sabnis, J. Polym. Environ., 2018, 26, 2601–2618 CrossRef CAS.
- V. S. Palekar, R. V. Shah and S. R. Shukla, J. Appl. Polym. Sci., 2012, 126, 1174–1181 CrossRef CAS.
- D. S. Achilias, G. P. Tsintzou, A. K. Nikolaidis, D. N. Bikiaris and G. P. Karayannidis, Polym. Int., 2011, 60, 500–506 CrossRef CAS.
- L. Xu, Q. Yao, Z. Han, Y. Zhang and Y. Fu, ACS Sustainable Chem. Eng., 2016, 4, 1115–1122 CrossRef CAS.
- Z. Yuan, X. Zhang, Q. Yao, Y. Zhang and Y. Fu, J. Anal. Appl. Pyrolysis, 2019, 140, 376–384 CrossRef CAS.
- S. Tian, Y. Jiao, Z. Gao, Y. Xu, L. Fu, H. Fu, W. Zhou, C. Hu, G. Liu, M. Wang and D. Ma, J. Am. Chem. Soc., 2021, 143, 16358–16363 CrossRef CAS PubMed.
- L. Shao, Y.-C. Chang, C. Hao, M.-E. Fei, B. Zhao, B. J. Bliss and J. Zhang, Green Chem., 2022, 24, 8716–8724 RSC.
- H. W. Lee, K. Yoo, L. Borchardt and J. G. Kim, Green Chem., 2024, 26, 2087–2093 RSC.
- S. Mishra and A. S. Goje, Polym. Int., 2003, 52, 337–342 CrossRef CAS.
- D. D. Pham and J. Cho, Green Chem., 2021, 23, 511–525 RSC.
- S. Tang, F. Li, J. Liu, B. Guo, Z. Tian and J. Lv, J. Environ. Chem. Eng., 2022, 10, 107927 CrossRef CAS.
- S. Tang, F. Li, J. Liu, B. Guo, Z. Tian and J. Lv, J. Chem. Technol. Biotechnol., 2022, 97, 1305–1314 CrossRef CAS.
- J.-T. Du, Q. Sun, X.-F. Zeng, D. Wang, J.-X. Wang and J.-F. Chen, Chem. Eng. Sci., 2020, 220, 115642 CrossRef CAS.
- B. Ye, R. Zhou, Z. Zhong, S. Wang, H. Wang and Z. Hou, Green Chem., 2023, 25, 7243–7252 RSC.
- Y. Zhang, J. Gao, C. Jiang, G. Luo, J. Fan, J. H. Clark and S. Zhang, Green Chem., 2024, 26, 6748–6759 RSC.
- T. Sako, T. Sugeta, K. Otake, N. Nakazawa, M. Sato, K. Namiki and M. Tsugumi, J. Chem. Eng. jpn, 2005, 30, 342–346 CrossRef.
- J. Liu and J. Yin, Ind. Eng. Chem. Res., 2022, 61, 6813–6819 CrossRef CAS.
- M. Liu, J. Guo, Y. Gu, J. Gao and F. Liu, ACS Sustainable Chem. Eng., 2018, 6, 15127–15134 CrossRef CAS.
- S. Liu, Z. Wang, L. Li, S. Yu, C. Xie and F. Liu, J. Appl. Polym. Sci., 2013, 130, 1840–1844 CrossRef CAS.
- R. Petrus, D. Bykowski and P. Sobota, ACS Catal., 2016, 6, 5222–5235 CrossRef CAS.
- B. Ye, R. Zhou, C. Wang, Z. Wang, Z. Zhong and Z. Hou, Appl. Catal., A, 2023, 649, 118936 CrossRef CAS.
- Y. Ye, H. Han, Y. Zhang, H. Zhao, W. Du, P. Chen and Z. Hou, Appl. Catal., A, 2024, 686, 119917 CrossRef CAS.
- A. Tullo, C&EN Global Enterp., 2023, 101, 20–26 Search PubMed.
- H. Kurokawa, M.-a Ohshima, K. Sugiyama and H. Miura, Polym. Degrad. Stab., 2003, 79, 529–533 CrossRef CAS.
- Z. T. Laldinpuii, V. Khiangte, S. Lalhmangaihzuala, C. Lalmuanpuia, Z. Pachuau, C. Lalhriatpuia and K. Vanlaldinpuia, J. Polym. Environ., 2022, 30, 1600–1614 CrossRef CAS.
- J. Cheng, J. Xie, Y. Xi, X. Wu, R. Zhang, Z. Mao, H. Yang, Z. Li and C. Li, Angew. Chem., Int. Ed., 2024, 63, e202319896 CrossRef CAS PubMed.
- Z. Jiang, D. Yan, J. Xin, F. Li, M. Guo, Q. Zhou, J. Xu, Y. Hu and X. Lu, Polym. Degrad. Stab., 2022, 199, 109905 CrossRef CAS.
- Y. Xu, Y. Ji, Y. Liu, W. Deng, F. Huang and F. Zhang, ChemCatChem, 2024, 16, e202301763 CrossRef CAS.
- J. M. Payne, G. Kociok-Köhn, E. A. C. Emanuelsson and M. D. Jones, Macromolecules, 2021, 54, 8453–8469 CrossRef CAS.
- M. Enayati, S. Mohammadi and M. G. Bouldo, ACS Sustainable Chem. Eng., 2023, 11, 16618–16626 CrossRef CAS PubMed.
- K. Chan and A. Zinchenko, J. Environ. Chem. Eng., 2021, 9, 106129 CrossRef CAS.
- S. Xie, C. Wang, W. Hu, J. Z. Hu, Y. Wang, Z. Dong, N. N. Intan, J. Pfaendtner and H. Lin, Cell Rep. Phys. Sci., 2024, 5, 102145 CrossRef CAS.
- J. Cao, H. Liang, J. Yang, Z. Zhu, J. Deng, X. Li, M. Elimelech and X. Lu, Nat. Commun., 2024, 15, 6266 CrossRef CAS PubMed.
- Z. Chen, H. Sun, W. Kong, L. Chen and W. Zuo, Green Chem., 2023, 25, 4429–4437 RSC.
- Z. Sun, K. Wang, Q. Lin, W. Guo, M. Chen, C. Chen, C. Zhang, J. Fei, Y. Zhu, J. Li, Y. Liu, H. He and Y. Cao, Angew. Chem., Int. Ed., 2024, 63, e202408561 CrossRef CAS PubMed.
- X. Wei, W. Zheng, X. Chen, J. Qiu, W. Sun, Z. Xi and L. Zhao, Chem. Eng. Sci., 2024, 294, 120103 CrossRef CAS.
- N. Z. Zhang, E. T. Jin and X. X. Li, CN Pat., 113056446A, 2021 Search PubMed.
- Z. Zhu, Z. Lu, B. Li and S. Guo, Appl. Catal., A, 2006, 302, 208–214 CrossRef CAS.
- Y. Hara and K. Endou, Appl. Catal., A, 2003, 239, 181–195 CrossRef CAS.
- G. X. Zhao, Study on the Catalytic Performance of Catalytic Hydrogenation of PTA for Preparation of Cyclohexane Dimethanol, Beijing University of Chemical Technology, 2012 Search PubMed.
- R. Zhang, H. Jin, L. Ma and S. Yang, RSC Adv., 2023, 13, 27036–27045 RSC.
- R. Zhang, H. Jin, L. Ma, S. Yang and G. He, Catal. Commun., 2024, 187, 106887 CrossRef CAS.
- Y. H. Choi, H. W. Park and S. J. Park, CN Pat., 106164028b, 2019 Search PubMed.
- D. Hou, J. Xin, X. Lu, X. Guo, H. Dong, B. Ren and S. Zhang, RSC Adv., 2016, 6, 48737–48744 RSC.
- X. Guo, J. Xin, X. Lu, B. Ren and S. Zhang, RSC Adv., 2015, 5, 485–492 RSC.
- W. Ou, H. Wang, Y. Ye, H. Zhao, Y. Zhang and Z. Hou, J. Hazard. Mater., 2024, 476, 134964 CrossRef CAS PubMed.
- X. Jiang, Z. Chang, L. Yang, W. Du and Z. Hou, Fuel, 2024, 363, 130944 CrossRef CAS.
- F. Zhang, J. Chen, P. Chen, Z. Sun and S. Xu, AIChE J., 2012, 58, 1853–1861 CrossRef CAS.
- Y. Huang, Y. Ma, Y. Cheng, L. Wang and X. Li, Ind. Eng. Chem. Res., 2014, 53, 4604–4613 CrossRef CAS.
- S. Zhang, Q. Hu, G. Fan and F. Li, Catal. Commun., 2013, 39, 96–101 CrossRef CAS.
- K. Zhang, Q. Meng, H. Wu, J. Yan, X. Mei, P. An, L. Zheng, J. Zhang, M. He and B. Han, J. Am. Chem. Soc., 2022, 144, 20834–20846 CrossRef CAS PubMed.
- J. Chen, X. Liu and F. Zhang, Chem. Eng. J., 2015, 259, 43–52 CrossRef CAS.
- F. Zhang, J. Chen, P. Chen, Z. Sun and S. Xu, AIChE J., 2012, 58, 1853–1861 CrossRef CAS.
- W. Yu, S. Bhattacharjee, W.-Y. Lu and C.-S. Tan, ACS Sustainable Chem. Eng., 2020, 8, 4058–4068 CrossRef CAS.
- C. Wang, B. Ye, H. Han, S. Wang, L. Yang and Z. Hou, Chin. J. Inorg. Chem., 2023, 39, 2091–2102 CAS.
- L. Yan, Y. Shen, Z. Zou, X. Zhang, Z. Yu, G. Wang and C. Chen, Inorg. Chem. Front., 2024, 11, 3285–3295 RSC.
- Q. Fan, X. Li, Z. Yang, J. Han, S. Xu and F. Zhang, Chem. Mater., 2016, 28, 6296–6304 CrossRef CAS.
- H. Xiao, C. Zhang, J. Zhao, Z. Zheng and Y. Li, RSC Adv., 2023, 13, 16363–16368 RSC.
- S. Zhang, G. Fan and F. Li, Green Chem., 2013, 15, 2389–2393 RSC.
- Z. Chang, B. Ye, Z. Zhong, S. Wang, H. Wang, W. Du and Z. Hou, J. Mater. Chem. A, 2024, 12, 1003–1011 RSC.
- H. Liu, Q. Hu, G. Fan, L. Yang and F. Li, Catal. Sci. Technol., 2015, 5, 3960–3969 RSC.
- X. Gong, M. Wang, H. Fang, X. Qian, L. Ye, X. Duan and Y. Yuan, Chem. Commun., 2017, 53, 6933–6936 RSC.
- J. Luo, E. Qu, Y. Zhou, Y. Dong and C. Liang, Appl. Catal., A, 2020, 602, 117669 CrossRef CAS.
- A. B. Hungria, R. Raja, R. D. Adams, B. Captain, J. M. Thomas, P. A. Midgley, V. Golovko and B. F. G. Johnson, Angew. Chem., Int. Ed., 2006, 45, 4782–4785 CrossRef CAS PubMed.
- X. Li, Z. Sun, J. Chen, Y. Zhu and F. Zhang, Ind. Eng. Chem. Res., 2014, 53, 619–625 CrossRef CAS.
- X. Xiao, H. Xin, Y. Qi, C. Zhao, P. Wu and X. Li, Appl. Catal., A, 2022, 632, 118510 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- R. Holmen, US Pat., 2859240, 1958 Search PubMed.
- M. S. Tam, R. Craciun, D. J. Miller and J. E. Jackson, Ind. Eng. Chem. Res., 1998, 37, 2360–2366 CrossRef CAS.
- B. Katryniok, S. Paul and F. Dumeignil, Green Chem., 2010, 12, 1910–1913 RSC.
- L. Huang, Y. Zhu, H. Zheng, M. Du and Y. Li, Appl. Catal., A, 2008, 349, 204–211 CrossRef CAS.
- M. Ai and K. Ohdan, Appl. Catal., A, 1997, 150, 13–20 CrossRef CAS.
- Y. Li and T. J. Strathmann, Green Chem., 2019, 21, 5586–5597 RSC.
- Z. Li, Y. Shen and Z. Li, ACS Sustainable Chem. Eng., 2022, 10, 8228–8238 CrossRef CAS.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- J. Li, H. Ma, G. Zhao, G. Huang, W. Sun and C. Peng, ChemSusChem, 2024, 17, e202301352 CrossRef CAS PubMed.
- X. Liang, X. Li, Q. Dong, T. Gao, M. Cao, K. Zhao, E. Lichtfouse, A. Patrocinio and C. Wang, Chem. Eng. J., 2024, 482, 148827 CrossRef CAS.
- Y. Hu, S. Zhang, J. Xu, Y. Liu, A. Yu, J. Qian and Y. Xie, Angew. Chem., Int. Ed., 2023, 62, e202312564 CrossRef CAS PubMed.
- T. M. McGuire, A. Buchard and C. Williams, J. Am. Chem. Soc., 2023, 145, 19840–19848 CrossRef CAS PubMed.
- Y. Li, M. Wang, X. Liu, C. Hu, D. Xiao and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202117205 CrossRef CAS PubMed.
- R. Cao, M.-Q. Zhang, Y. Jiao, Y. Li, B. Sun, D. Xiao, M. Wang and D. Ma, Nat. Sustainability, 2023, 6, 1685–1692 CrossRef.
- Z. Sun, K. Wang, Q. Lin, W. Guo, M. Chen, C. Chen, C. Zhang, J. Fei, Y. Zhu, J. Li, Y. Liu, H. He and Y. Cao, Angew. Chem., Int. Ed., 2024, 63, e202408561 CrossRef CAS PubMed.
- H. Zhou, Y. Ren, Z. Li, M. Xu, Y. Wang, R. Ge, X. Kong, L. Zheng and H. Duan, Nat. Commun., 2021, 12, 4679 CrossRef CAS PubMed.
- F. Liu, X. Gao, R. Shi, E. C. M. Tse and Y. Chen, Green Chem., 2022, 24, 6571–6577 RSC.
- Y. Li, Y. Zhao, H. Zhao, Z. Wang, H. Li and P. Gao, J. Mater. Chem. A, 2022, 10, 20646 RSC.
- S. Chu, B. Zhang, X. Zhao, H. S. Soo, F. Wang, R. Xiao and H. Zhang, Adv. Energy Mater., 2022, 12, 2200435 CrossRef CAS.
- J. Meng, Y. Zhou, D. Li and X. Jiang, Sci. Bull., 2023, 68, 1522–1530 CrossRef CAS PubMed.
- T. Uekert, M. F. Kuehnel, D. W. Wakerley and E. Reisner, Energy Environ. Sci., 2018, 11, 2853–2857 RSC.
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed.
- S. Zhang, H. Li, L. Wang, J. Liu, G. Liang, K. Davey, J. Ran and S.-Z. Qiao, J. Am. Chem. Soc., 2023, 145, 6410–6419 CrossRef CAS PubMed.
- A. Garratt, K. Nguyen, A. Brooke, M. J. Taylor and M. G. Francesconi, ACS Environ. Au, 2023, 3, 342–347 CrossRef CAS PubMed.
- R. Wei and W. Zimmermann, Microb. Biotechnol., 2017, 10, 1302–1307 CrossRef CAS PubMed.
- R. P. Magalhães, J. M. Cunha and S. F. Sousa, Int. J. Mol. Sci., 2021, 22, 11257 CrossRef PubMed.
- B. Zhu, D. Wang and N. Wei, Trends Biotechnol., 2022, 40, 22–37 CrossRef CAS PubMed.
- X. Han, W. Liu, J.-W. Huang, J. Ma, Y. Zheng, T.-P. Ko, L. Xu, Y.-S. Cheng, C.-C. Chen and R.-T. Guo, Nat. Commun., 2017, 8, 2106 CrossRef PubMed.
- C. DelRe, Y. Jiang, P. Kang, J. Kwon, A. Hall, I. Jayapurna, Z. Ruan, L. Ma, K. Zolkin, T. Li, C. D. Scown, R. O. Ritchie, T. P. Russell and T. Xu, Nature, 2021, 592, 558–563 CrossRef CAS PubMed.
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed.
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196–1199 CrossRef CAS PubMed.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CrossRef CAS PubMed.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS PubMed.
- M. W. Myburgh, L. Favaro, W. H. van Zyl and M. Viljoen-Bloom, Bioresour. Technol., 2023, 378, 129008 CrossRef CAS PubMed.
- J. Sadler and S. Wallace, Green Chem., 2021, 23, 4665–4672 RSC.
- S. W. Schubert, T. B. Thomsen, K. S. Clausen, A. Malmendal, C. J. Hunt, K. Borch, K. Jensen, J. Brask, A. S. Meyer and P. Westh, ChemSusChem, 2024, 17, e202301752 CrossRef CAS PubMed.
- A. Li, L. Wu, H. Cui, Y. Song, X. Zhang and X. Li, ChemSusChem, 2024, 17, e202301612 CrossRef CAS PubMed.
- K. D. Nixon, Z. O. G. Schyns, Y. Luo, M. G. Ierapetritou, D. G. Vlachos, L. T. J. Korley and T. H. Epps III, Nat. Chem. Eng., 2024, 1, 615–626 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |