
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A liquid-infiltrated Al2O3 framework electrolyte enables aqueous zinc batteries†
Rongyu
Deng
ab,
Yi
Yuan
c,
Zixuan
Li
c,
Alex W.
Robertson
 *b and
Feixiang
Wu
*a
*b and
Feixiang
Wu
*a
aSchool of Metallurgy and Environment, Engineering Research Center of the Ministry of Education for Advanced Battery Materials, Central South University, Changsha, 410083, China. E-mail: feixiang.wu@csu.edu.cn
bDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK. E-mail: alex.w.robertson@warwick.ac.uk
cDepartment of Materials, University of Oxford, Oxford, OX1 3PH, UK
First published on 12th November 2024
Abstract
Aqueous zinc-ion battery anodes face the twin challenges of dendrite growth and severe side reactions. Here a liquid-infiltrated Al2O3 framework electrolyte (LIAFE) is developed to address these issues and enables stable long-life Zn anodes for over 4000 hours. The LIAFE shows a uniform morphology and exhibits enhanced performance.
Zinc metal is considered the most promising anode material for aqueous zinc ion batteries due to its natural abundance, low cost, and high theoretical volumetric energy density (5855 mA h cm−3) and gravimetric capacities (820 mA h g−1).1–3 However, several challenges associated with zinc metal anodes have hindered the development of aqueous zinc-ion batteries, such as the growth of dendrites leading to batteries failure, and severe side reactions leading to interface deterioration and performance degradation.4–7 To address these issues, various strategies have been employed, including surface coatings,8,9 separator modifications,10 additive engineering,11,12 and the development of new electrolytes.13,14
These newly developed electrolytes have spanned solid electrolytes,15 quasi-solid-state electrolytes,16 and solid–liquid hybrid electrolytes.17 Inorganic solid electrolytes include zinc-ion conductors, like ZnPS3 electrolyte, which exhibits an ionic conductivity of 2.0 × 10−3 S cm−1 thus enabling a stable cycle performance.15 Quasi-solid-state electrolytes, particularly hydrogel systems, offer higher ionic conductivity, but their low mechanical properties and poor stability result in reduced cycling stability.18 Solid–liquid hybrid electrolytes, such as ‘soggy-sand’ electrolytes,19,20 quasi-decoupled solid–liquid hybrid electrolytes,15 and densified electrolytes,21 exhibit good cycling stability under high voltage conditions. These hybrid electrolytes combine the mechanical strength of inorganic solid electrolytes with the high ionic conductivity of liquid electrolytes,22 effectively modifying the electrolyte-electrode interface to suppress dendrite formation and side reactions, thereby delivering improved electrochemical performance. The primary mechanism of hybrid electrolytes is based on the interaction between water molecules and solid components, in addition, the solids enhance mechanical strength and act to suppress zinc dendrites.23 However, despite their advantages, solid–liquid hybrid electrolytes still contain a significant amount of liquid. Further reduction of the liquid content in the electrolyte by increasing the proportion of solid components should enhance the electrochemical performance.
Herein, a liquid-infiltrated Al2O3 framework electrolyte (LIAFE) is developed, which exhibits stable long-life zinc anodes with a uniform morphology. By employing aluminum oxide (Al2O3) as a framework to bind water molecule activity, LIAFE electrolytes exhibit excellent long cycle stability. Zn/Zn symmetric cells using LIAFE electrolytes demonstrate a long cycle life of over 4000 hours, and the zinc anodes after cycling offer a flat surface morphology without any dendrite growth.
The LIAFE electrolytes are simple to prepare. The preparation begins with the formation of a solid Al2O3 tablet skeleton (Fig. 1a). Initially, the Al2O3 is evenly dispersed in the basis electrolyte (BE), which is a 2 M ZnSO4 solution for symmetric cells and half-cells, or 2 M ZnSO4 with additional 0.1 M MnSO4 for Zn/MnO2 full cells. Then, the mixture is rapidly frozen using liquid nitrogen and promptly transferred to a freeze-drying apparatus. After 48 hours of freeze-drying, the sample is removed and ground to obtain a powder of Al2O3 uniformly mixed with the solute. Finally, the powder is pressed into 13 mm diameter solid tablet under a pressure of 10 tons. The scanning electron microscope (SEM) image reveals that the solid-state tablet skeleton mainly consists of Al2O3 and solute powders, with uniformly distributed voids and gaps across the surface (Fig. 1b), allowing the basis electrolyte to evenly infiltrate through solid-state tablets. The inset in Fig. 1b provides a digital photograph showing the side view of a solid tablet. After the BE is applied to the surface of the tablet, it is allowed to infiltrate for ten minutes, which is long enough for it to fully infiltrate the voids within the solid skeleton, forming the LIAFE (Fig. S1, ESI†). The voids and gaps in the solid tablet act as channels for efficient transport of zinc ions after fully absorbing basis electrolytes (Fig. 1c). The cathodic linear sweep voltammetry (LSV) shows that LIAFE exhibits a higher onset potential compared to BE. Moreover, the response current of the cell using BE electrolyte is ten times greater than that of the cell using LIAFE electrolytes (Fig. S2, ESI†), indicating that LIAFE effectively inhibits the hydrogen evolution reaction (HER). These results demonstrate that LIAFE effectively mitigates side reactions at the electrode–electrolyte interface. This improvement is attributed to the presence of Al2O3, which reduces the number of water molecules in direct contact with zinc anodes, thereby minimizing undesirable reactions.
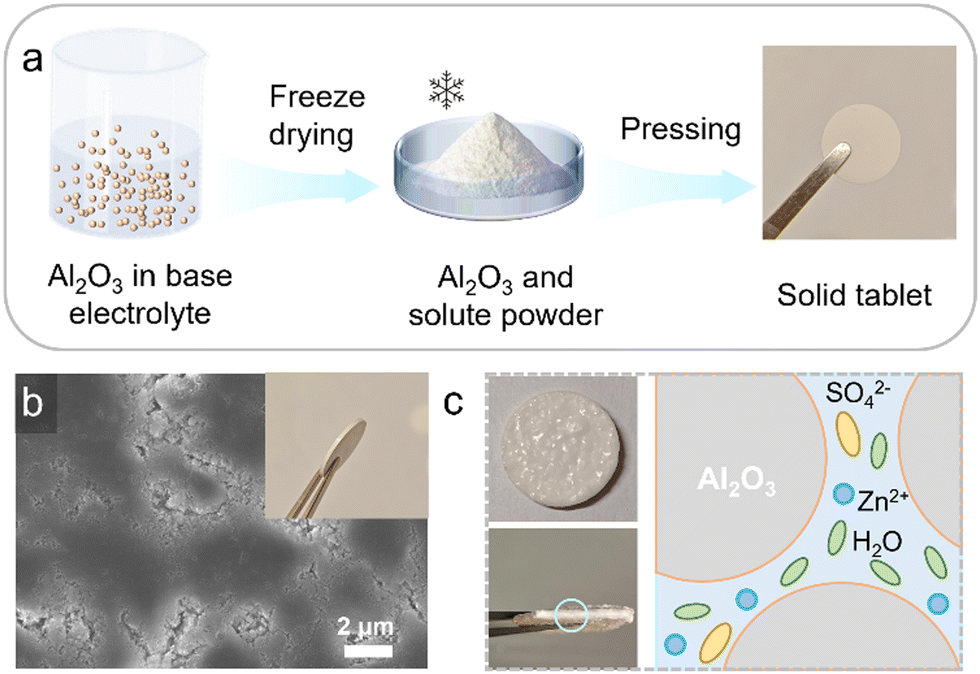 | ||
| Fig. 1 (a) Schematic illustration for the process of preparing solid tablets. (b) SEM images of solid tablets, the inset shows a photo of a tablet. (c) Photos of LIAFE and a scheme mechanism sketching the proposed structure. | ||
A symmetric Zn/Zn cell was used to evaluate the long-term cycling performance of the zinc anode with LIAFE. The rate performance was assessed by varying the applied current density, with it is sequentially increased from 0.1 to 0.2, 0.5 and 1 mA cm−2, and then decreased back to 0.5, 0.2, 0.1 mA cm−2. Zn/Zn cells with only the BE electrolyte exhibited significant changes in the overpotential, which indicates poor rate performance of BE electrolyte during Zn stripping/plating (Fig. 2a). Additionally, when current density was returned to 0.1 mA cm−2, the overpotential gradually increased with over the course of continued cycling, which is attributed to the gradual deterioration of the cell due to interfacial side reactions. In sharp contrast, Zn/Zn cells using LIAFE show a small overpotential change with current variation, demonstrating that LIAFE provides a fast zinc ion transport channel for electrochemical reactions. More importantly, when the current density returns to 0.1 mA cm−2, Zn/Zn cells with LIAFE cycle stably for more than 4000 hours without any overpotential increase, indicating that LIAFE effectively restrains the deterioration of the anode interfaces during long-term cycling (Fig. S3, ESI†). Zn/Zn cells with LIAFE exhibit exceptional cycling stability at a current density of 0.2 mA cm−2 with a fixed capacity of 0.1 mA h cm−2 (Fig. 2b). In comparison, cells using just the BE show a high overpotential and fail after 162 hours. Conversely, Zn/Zn cell using LIAFE exhibits an ultra-long lifespan of more than 4500 h with much smaller overpotential. The insets of Fig. 2b reveal the voltage hysteresis between BE and LIAFE in the first 10 h, suggesting an overpotential of 124 mV and 70 mV with BE and LIAFE, respectively. The cell shows a cycle life of more than 1000 hours at a higher fixed capacity of 0.2 mA h cm−2 (Fig. S4, ESI†). Furthermore, when the current density increases to 0.5 mA cm−2 with capacity of 0.25 mA h cm−2, cells using LIAFE cycle stably for 1000 hours, while the cell with BE shorted out after 141 hours of cycling (Fig. 2c). Insets in Fig. 2c show that the overpotential of the cell using just the BE is significantly higher than that of cells using LIAFE at 0.5 mA cm−2. More importantly, the overpotential of the cell with LIAFE remains stable over 1000 hours, indicating that LIAFE significantly enhances the long-term cycling stability of the zinc metal anode compared to BE. In addition, the cell shows good cycling stability at higher current densities. At 1 mA cm−2, 0.5 mA h cm−2, the cell with BE begins to exhibit short circuits after just 91 hours of cycling. By comparison, cells using LIAFE demonstrate significantly improved stability, cycling stably for over 600 hours (Fig. S5, ESI†). These results indicate that LIAFE provides fast Zn2+ channels for Zn stripping/plating and maintains the integrity of the electrochemical interface.
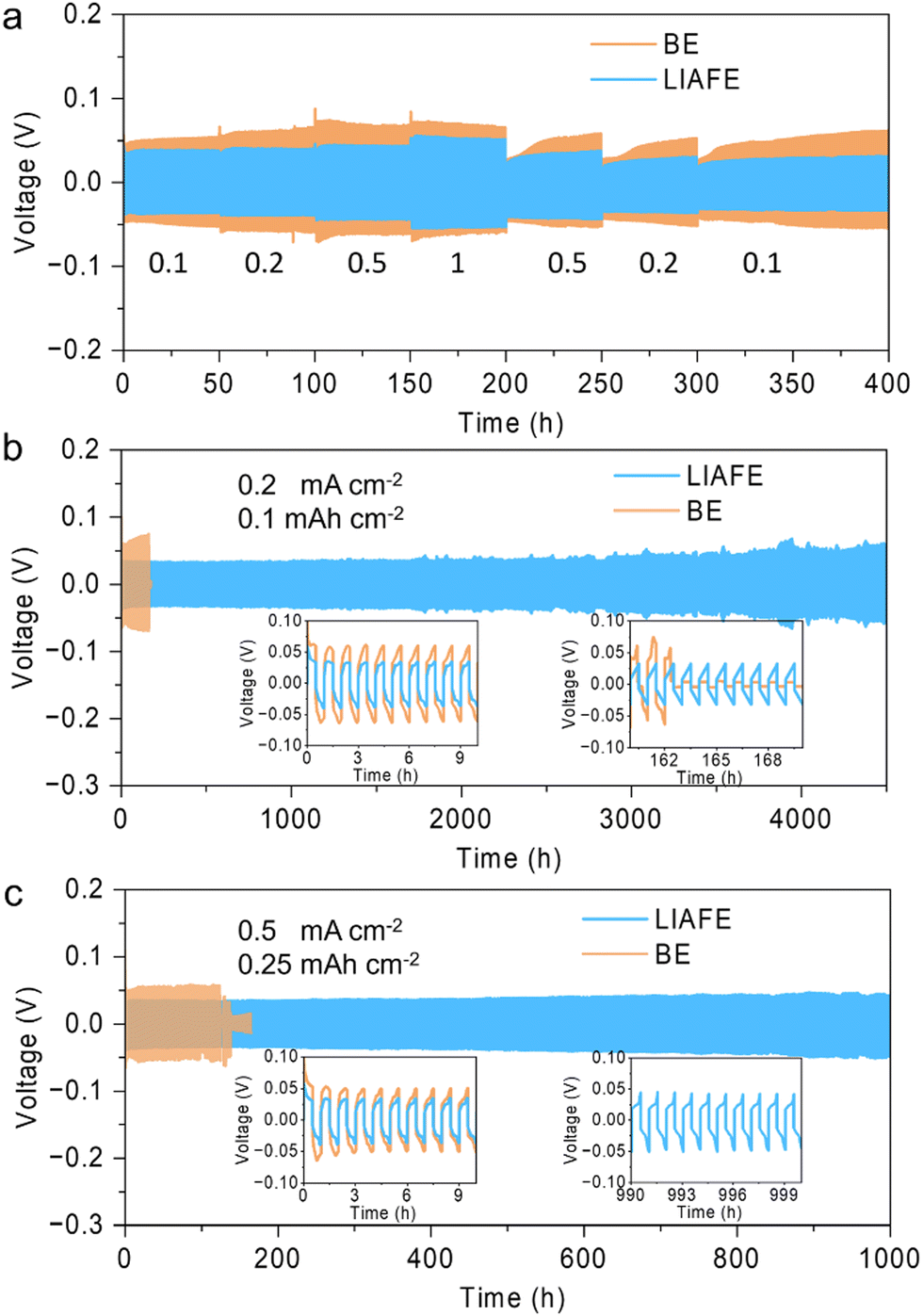 | ||
| Fig. 2 Electrochemical performance of symmetric Zn/Zn cells with LIAFE. (a) Rate performance at current density from 0.1 to 1 mA cm−2. (b) Cycling performance at 0.2 mA cm−2, 0.1 mA h cm−2. (c) Cycling performance at 0.5 mA cm−2, 0.25 mA h cm−2. | ||
To further analyze the interface, the Zn/Zn symmetric cell cycled at current density of 0.2 mA cm−2 was disassembled for characterization. The zinc anode of the cell using BE exhibits a significantly rough and disordered deposition morphology, with obvious zinc dendrites on the surface (Fig. S6, ESI†). The X-ray diffraction (XRD) patterns reveal the presence of substantial zinc hydroxide sulfate (ZHS) by-products, Zn4SO4(OH)6 4H2O, on the surface of the zinc anode (Fig. S7, ESI†). These by-products contribute to surface roughening, leading to disordered zinc deposition and poor cycling stability. In sharp contrast, the zinc anode cycled with LIAFE demonstrates an extremely uniform surface morphology, characterized by closely stacked flat hexagonal structures (Fig. 3a). The corresponding XRD patterns indicate the ZHS present on the surface of zinc anodes contains only one water molecule, forming Zn4SO4(OH)6 H2O (Fig. 3b), unlike the multi-hydrated ZHS observed with BE. The water-deficient ZHS has a denser structure, acting similarly to a solid electrolyte interphase (SEI), which promotes uniform and consistent Zn2+ deposition while preventing interface deterioration caused by continuous contact between zinc anodes and electrolytes. The formation of this water-deficient ZHS is attributed to the Al2O3 solid tablet skeleton in the LIAFE, which reduces the number of water molecules at the zinc anode–electrolyte interface, leading to the generation of ZHS with a single water molecule. Additionally, the strong mechanical hardness of the skeleton inhibits disordered deposition at the interface, controlling the growth of ZHS and resulting in a flat morphology. Higher magnification SEM images provide a more detailed view of the water-deficient ZHS on the surface of the zinc anode (Fig. S8, ESI†). The hexagonal ZHS sheets uniformly cover the zinc anode, with Zn stripping/plating occurring beneath this layer, leading to a more uniform and dense zinc deposition.
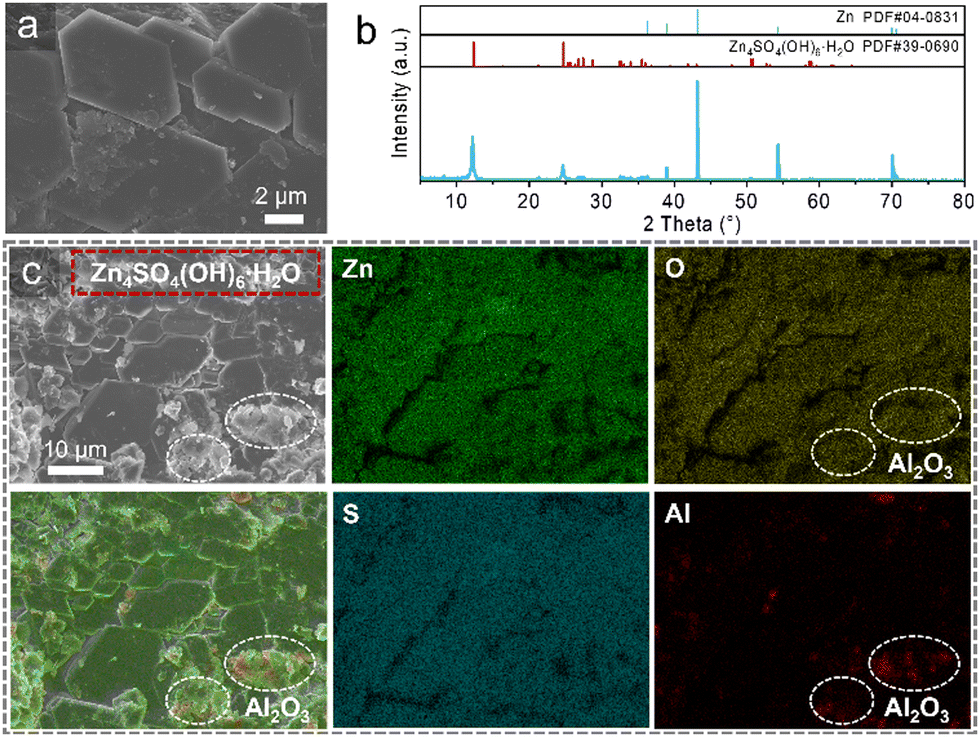 | ||
| Fig. 3 (a) SEM images of zinc anodes obtained from the Zn/Zn cell with LIAFE after 2500 hours cycle. (b) The XRD patterns of Zn, Zn4SO4(OH)6 H2O, and the cycled zinc anode with LIAFE. (c) The EDS mapping of the cycled zinc anode. | ||
Furthermore, the EDS mapping of the zinc anode after cycling 2500 hours at a current density of 0.2 mA cm−2 demonstrates the uniform and dense water-deficient ZHS layer (Fig. 3c). The elemental distribution maps indicate the distribution of the elements of Zn4SO4(OH)6 H2O on the surface of the zinc anode. This water-deficient ZHS forms a dense protective layer on the surface by close contact accumulation. In contrast to the water-rich ZHS, which has a loose structure on the zinc anode cycled with BE, the water-deficient ZHS provides superior protection to the interface and promotes more uniform zinc deposition. The distribution positions of the elements in Zn, O, S are consistent, and all of them are uniformly distributed across the surface of zinc anodes in the form of hexagonal dense stacking, which indicates that water-deficient ZHS is sufficiently dense to avoid zinc anodes from directly contacting with electrolytes. In addition, several small particles are observed on the surface of the zinc anodes with LIAFE, which are residual Al2O3 particles, resulting from the close contact of the Al2O3 skeleton with the zinc anode. The locations of these residual Al2O3 particles are identified by the elemental distribution of Al. These results indicate that LIAFE induces the formation of a dense water-deficient ZHS layer on the surface of zinc anodes, effectively promoting the long-term cycle stability of Zn anodes.
Zn/Cu half cells are tested at 0.2 mA cm−2 with a fixed capacity of 0.2 mA h cm−2, which showed that the half-cell using LIAFE works smoothly over 500 cycles and maintains a high coulombic efficiency (CE) of 98.7% at 500th cycle. In contrast, the cell with BE fails after 135 cycles with a CE of 97.9% (Fig. S9, ESI†). Furthermore, in the electrochemical impedance spectroscopy (EIS) of full Zn/MnO2 cells, charge transfer resistance (Rct) is the resistance of Zn2+ transferring from solvated ionic state crossing the interface and inserting into electrodes.24 The Rct of the cell using BE is 294 Ω while the Rct of the cell with LIAFE is only 98 Ω with a resting time of 0 h (Fig. 4a). After 17 hours of left standing, the Rct of cells with BE increase to 1006 Ω, whereas the Rct of cells with LIAFE decrease to 72 Ω (Fig. 4b), which indicates that BE passivates the interface during rest, while LIAFE is able to stabilize the interface. Moreover, the ionic conductivity of the LIAFE electrolyte is measured by conducting EIS on stainless-steel electrodes symmetric battery. The ionic conductivities of 40 mS cm−1 for BE and 11 mS cm−1 for LIAFE (Fig. S10, ESI†). Cyclic voltammetry (CV) shows that LIAFE exhibits stronger peak currents for redox reactions, suggesting that LIAFE enhances the capacity of Zn/MnO2 full cells (Fig. 4c).
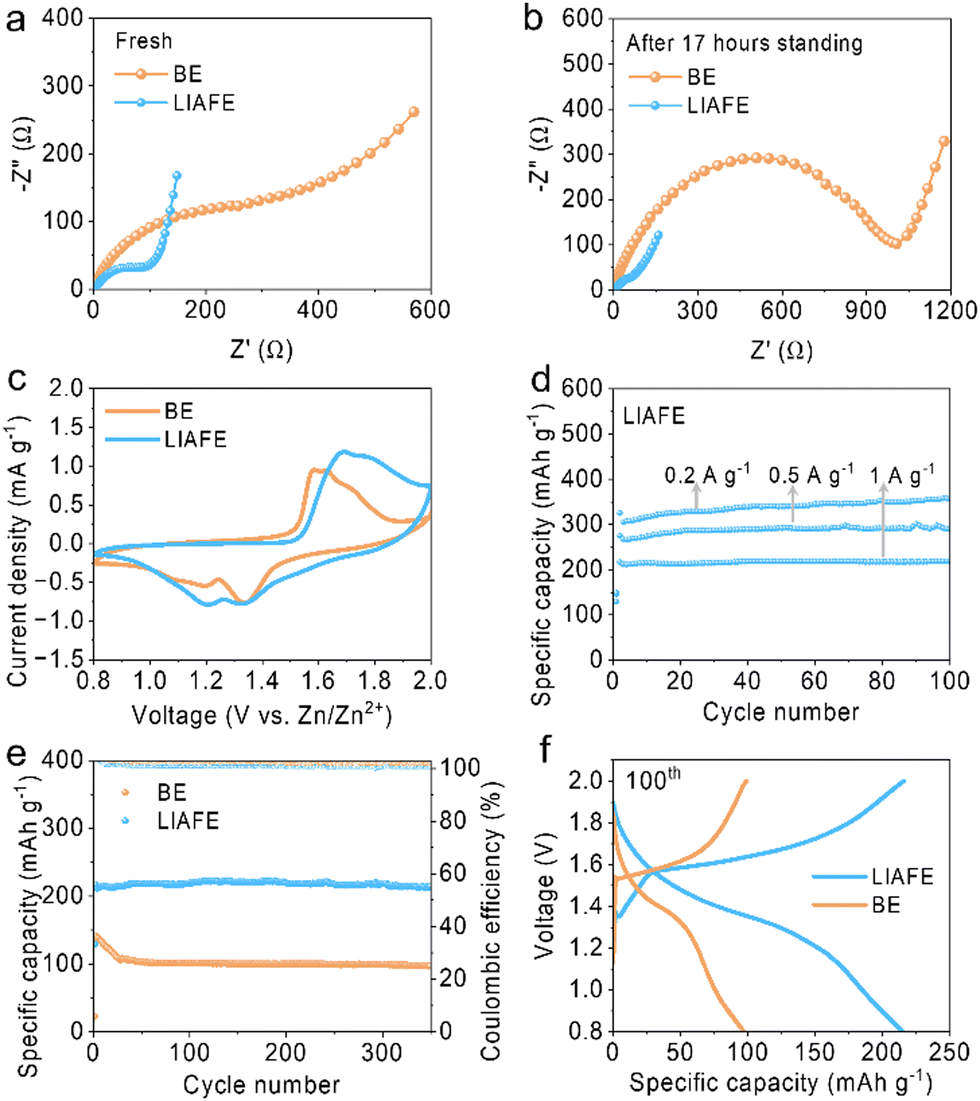 | ||
| Fig. 4 Electrochemical performance of Zn/MnO2 full cells. EIS spectra of the full cells with BE and LIAFE after standing (a) 0 hour; (b) 17 hours. (c) CV profiles of full cells. (d) Rate performance of cells using LIAFE. (e) Long cycles of full cells at a current density of 1 A g−1. (f) The corresponding charge–discharge curves. | ||
To evaluate the practical application of LIAFE, Zn/MnO2 full cells were tested at various current densities within a broadened voltage range of 0.8–2.0 V (Fig. 4d). The cell using LIAFE exhibits a high specific capacity of 325 mA h g−1 and maintains non-attenuation to 100 cycles at a current density of 0.1 A g−1. At a higher current density of 0.5 A g−1, cells with LIAFE demonstrate a specific capacity of 289 mA h g−1. Furthermore, the Zn/MnO2 full cell delivers an exceptional specific capacity of 219.4 mA h g−1 at a current density of 1 A g−1 with a discharge time of 13 minutes (Fig. S11, ESI†). Additionally, the long-term cycle stability of full cells is tested at 1 A g−1. The specific capacity of the cell with BE decayed from 141 mA h g−1 to 106 mA h g−1 after only 30 cycles, and the specific capacity is only 97 mA h g−1 after 350 cycles. In sharp contrast, the cell using LIAFE exhibits a high initial specific capacity of 216.8 mA h g−1, and maintains 214.7 mA h g−1 after 350 cycles with an excellent capacity retention of 99% (Fig. 4e). This suggests the highly competitive performance of LIAFE compared to previously reported works (Table S1, ESI†). The high cycling stability and good capacity retention is attributed to the ability of LIAFE to maintain the interface, preventing the occurrence of side reactions and the growth of dendrites. The corresponding charge–discharge curves for the 100th cycle demonstrate the significantly improved specific capacity of the cell with LIAFE (Fig. 4f).
In summary, a LIAFE electrolyte, prepared by infiltrating liquid electrolytes into pressed Al2O3 tablets, significantly enhances the electrochemical performance of aqueous zinc-ion batteries. LIAFE inhibits the activity of water molecules, thus inhibiting HER side reactions. More importantly, LIAFE induces a dense water-deficient ZHS layer on the surface of zinc anodes, which not only prevents zinc dendrite formation but also eliminates direct contact between the electrolyte and electrode, stabilizing the interface. This work presents a new approach for the development of advanced electrolytes to extend the lifespan of zinc anodes.
The authors acknowledge support from National Natural Science Foundation of China (No. 52172264), the science and technology innovation Program of Hunan Province (No. 2023RC1016), the Royal Society (URF\R\221018), and the facilities and expertise of the University of Warwick's X-ray Diffraction (Dr Jie Liu) and Electron Microscopy (Andrew Unsworth) Research Technology Platforms, and Anton Paar for the loan of the XRDynamic 500 instrument used for the XRD measurements.
Data availability
Data for this article, including electrochemical data, diffractograms, and micrographs, are available at the Warwick Research Archive Portal at https://wrap.warwick.ac.uk/id/eprint/188201/.Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- Y. Zhu, G. Liang, X. Cui, X. Liu, H. Zhong, C. Zhi and Y. Yang, Energy Environ. Sci., 2024, 17, 369–385 RSC.
- C. Nie, G. Wang, D. Wang, M. Wang, X. Gao, Z. Bai, N. Wang, J. Yang, Z. Xing and S. Dou, Adv. Energy Mater., 2023, 13, 2300606 CrossRef CAS.
- Y. Zhu, H. Li, Y. Rao, S. Guo and H. Zhou, Adv. Energy Mater., 2024, 14, 2303928 CrossRef CAS.
- C. Li, S. Jin, L. A. Archer and L. F. Nazar, Joule, 2022, 6, 1733–1738 CrossRef.
- G. Zampardi and F. La Mantia, Nat. Commun., 2022, 13, 687 CrossRef CAS PubMed.
- S. Wu, Z. Hu, P. He, L. Ren, J. Huang and J. Luo, eScience, 2023, 3, 100120 CrossRef.
- Y.-H. Lee, Y. Jeoun, J. H. Kim, J. Shim, K.-S. Ahn, S.-H. Yu and Y.-E. Sung, Adv. Funct. Mater., 2024, 34, 2310884 CrossRef CAS.
- L. Yuan, J. Hao, B. Johannessen, C. Ye, F. Yang, C. Wu, S.-X. Dou, H.-K. Liu and S.-Z. Qiao, eScience, 2023, 3, 100096 CrossRef.
- D. Luo, J. Wu, W. Deng, G. Zou, H. Hou and X. Ji, Chem. Commun., 2024, 60, 7370–7373 RSC.
- Z. Hu, F. Zhang, Y. Zhao, H. Wang, Y. Huang, F. Wu, R. Chen and L. Li, Adv. Mater., 2022, 34, 2203104 CrossRef CAS PubMed.
- W. Ge, H. Peng, J. Dong, G. Wang, L. Cui, W. Sun, X. Ma and J. Yang, Chem. Commun., 2024, 60, 750–753 RSC.
- F. Ming, Y. Zhu, G. Huang, A.-H. Emwas, H. Liang, Y. Cui and H. N. Alshareef, J. Am. Chem. Soc., 2022, 144, 7160–7170 CrossRef CAS PubMed.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 RSC.
- Z. Lv, Y. Kang, G. Chen, J. Yang, M. Chen, P. Lin, Q. Wu, M. Zhang, J. Zhao and Y. Yang, Adv. Funct. Mater., 2024, 34, 2310476 CrossRef CAS.
- H. Zhang, X. Gan, Z. Song and J. Zhou, Angew. Chem., Int. Ed., 2023, 62, e202217833 CrossRef CAS PubMed.
- Y. Pan, Z. Liu, S. Liu, L. Qin, Y. Yang, M. Zhou, Y. Sun, X. Cao, S. Liang and G. Fang, Adv. Energy Mater., 2023, 13, 2203766 CrossRef CAS.
- H. Xia, G. Xu, X. Cao, C. Miao, H. Zhang, P. Chen, Y. Zhou, W. Zhang and Z. Sun, Adv. Mater., 2023, 35, 2301996 CrossRef CAS PubMed.
- R. Deng, J. Chen, F. Chu, M. Qian, Z. He, A. W. Robertson, J. Maier and F. Wu, Adv. Mater., 2024, 36, 2311153 CrossRef CAS PubMed.
- J. Chen, R. Deng, J. Zhou, Z. Jiang, M. Qian and F. Wu, Batteries Supercaps, 2024, e202400404 CrossRef.
- R. Deng, Z. He, F. Chu, J. Lei, Y. Cheng, Y. Zhou and F. Wu, Nat. Commun., 2023, 14, 4981 CrossRef CAS PubMed.
- T. Zhang, J. Li, X. Li, R. Wang, C. Wang, Z. Zhang and L. Yin, Adv. Mater., 2022, 34, 2205575 CrossRef CAS PubMed.
- C. Guo, K. Du, R. Tao, Y. Guo, S. Yao, J. Wang, D. Wang, J. Liang and S.-Y. Lu, Adv. Funct. Mater., 2023, 33, 2301111 CrossRef CAS.
- L. Hong, X. Wu, C. Ma, W. Huang, Y. Zhou, K.-X. Wang and J.-S. Chen, J. Mater. Chem. A, 2021, 9, 16814–16823 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04928d |
| This journal is © The Royal Society of Chemistry 2024 |