
The indanone N–H type excited-state intramolecular proton transfer (ESIPT); the observation of a mechanically induced ESIPT reaction†
Jhu-Jyun
You‡
a,
Yan-Ding
Lin‡
b,
Chao-Hsien
Hsu
b,
Jiun-Wei
Hu
a,
Ying-Yi
Tsai
b,
Hao-Ting
Qu
b,
Kew-Yu
Chen
 *a and
Pi-Tai
Chou
*b
*a and
Pi-Tai
Chou
*b
aDepartment of Chemical Engineering, Feng Chia University, 40724 Taichung, Taiwan. E-mail: kyuchen@fcu.edu.tw
bDepartment of Chemistry, National Taiwan University, 10617 Taipei, Taiwan. E-mail: chop@ntu.edu.tw
First published on 4th October 2024
Abstract
We report the design and synthesis of indanone derivatives 1–4 with RR′N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C intramolecular hydrogen bonds, in which ESIPT takes place and its dynamics and thermodynamics correlate with H-bond strength, facilitated by electron-withdrawing R′ groups. Compound 4 (R′ = COCF3) shows mechanically induced ESIPT for the first time, where –CF3⋯HN– interaction plays a key role in the non-centrosymmetric crystal packing.
C intramolecular hydrogen bonds, in which ESIPT takes place and its dynamics and thermodynamics correlate with H-bond strength, facilitated by electron-withdrawing R′ groups. Compound 4 (R′ = COCF3) shows mechanically induced ESIPT for the first time, where –CF3⋯HN– interaction plays a key role in the non-centrosymmetric crystal packing.
Proton transfer is one of the chemistry core reactions, among which excited-state intramolecular proton transfer (ESIPT) has garnered considerable attention in both fundamental investigation and applications.1–3 ESIPT relies on the excited-state electronic coupling between the reactants (normal state) and products (proton-transfer tautomer state) along an intramolecular hydrogen bond (H-bond). Therefore, fundamentally, the H-bond strength must play a crucial role in ESIPT. In the early days, most of the studied ESIPT compounds were categorized as the case of a strong H-bond, and the electron coupling matrix between normal and proton-transfer tautomer states was large. Therefore, for most of the systems, ESIPT is virtually barrierless and highly exergonic, for which proton transfer is induced by those vibration modes that are associated with changes of angle and/or distance of the H-bond. Since early ESIPT systems commonly consist of the –OH group as the proton donor, and carbonyl oxygen or nitrogen as the proton acceptor, directly modifying the –OH site to adjust H-bond strength is not feasible. Recently, the development of R′RN–H (proton donor) type ESIPT offers greater flexibility and versatility in molecular structure, allowing for direct tuning of H-bond strength. The acidity of secondary amines R′RNH, where R is commonly denoted as the carbon element of the corresponding moiety, while R′ can be functionalized with either an electron-donating or electron-withdrawing group, provides a case in point to fine-tune the H-bond strength. These R′RNH-associated H-bond systems have demonstrated a wide range of ESIPT dynamics and thermodynamics, spanning from endergonic to equilibrium and exergonic types of reaction pattern, and hence the establishment of correlation among H-bond strength, ESIPT dynamics, and thermodynamics.4 Accordingly, the emission can be broadly tuned by altering the electronic properties of R′. An example is given by a single-site amino derivatization in 10-aminobenzo[h]quinoline, where the tautomer emission can span from 590 nm to 770 nm. Recently, the R′RN–H ESIPT system has been successfully applied in bio-imaging and sensing.5
Herein, we report the strategic design and synthesis of a new series of single-benzene type RR′N–H⋯OC intramolecular H-bond indanone derivatives 1–4 (Scheme 1) where R′ is functionalized by electron-withdrawing groups. We applied indanone as a new core structure, where the formation of a six-membered ring H-bond is not in a hexagon whose interior angles are all 120 degrees, to probe its influence on the RR′N–H type ESIPT. Comprehensive spectroscopic and dynamic studies support the correlation between H-bond strength and ESIPT properties. In this study, in serendipity, we observed mechanically induced ESIPT in compound 4. Previous studies have indicated that the single benzene ESIPT systems would exhibit mechanochromism in the crystalline states, where mechanical deformation of the crystals can alter the spectral ratio between normal and tautomeric forms by reducing intermolecular interactions through specific molecular design.6 However, in our work, similar single benzene ESIPT frameworks do not display this phenomenon in 1–3, whereas 4 exhibits a bright, prominent tautomer emission maximized at 560 nm. This phenomenon is new and is attributed to the non-centrosymmetric packing mode of compound 4, a feature absent in its analogs 1 (R′ = H), 2 (R′ = COCH3), and 3 (R′ = tosyl), highlighting the critical role of its unique packing mode (vide infra).
 | ||
| Scheme 1 The synthetic routes for compounds 1–4. | ||
The synthetic routes of 2–4 are depicted in Scheme 1. Details of the synthesis and characterization are elaborated in the ESI.† In brief, the synthesis of compounds 2–4 was carried out by mixing the commercially available starting material, 7-amino-2,3-dihydro-1H-inden-1-one (1), with different reagents. Compound 2 was obtained using acetic anhydride in CH2Cl2, compound 4 using trifluoroacetic anhydride in CH2Cl2, and compound 3 using 4-methylbenzenesulfonyl chloride in pyridine. The mixtures were stirred for 3 hours, followed by solvent vaporization and subsequent purification via silica gel column chromatography. Detailed synthesis steps can be found in the ESI.† Also, the corresponding 1H NMR and 13C NMR spectra are presented in Fig. S1–S8 (ESI†). For all of 1–4, by slow evaporation from the saturated CH2Cl2 solution, colorless crystals suitable for crystallographic studies could be obtained, and the resulting X-ray data are presented and discussed below.
The molecular structure and the crystal packing alignment of 1 and 4 are illustrated in Fig. 1, while those for 2 and 3 are displayed in Fig. S9 of the ESI.† Since one might challenge the hydrogen atom being artificially placed in the simulation of X-ray structure, we used the N⋯OC distance as a ruler to assess the H-bond distance and found that its distance is in the order of 1 (2.9524(17) Å) > 2 (2.830(2) Å) > 3 (2.796(2) Å) > 4 (2.779(6) Å) (Fig. S10, ESI†). Additionally, the asymmetric N–H stretching signals for compounds 1–4 are as follows: 1 (3426 cm−1) > 2 (3291 cm−1) > 3 (3175 cm−1) > 4 (3165 cm−1) (see Fig. S11–S14, ESI†). The results indirectly reveal the increasing H-bond strength to be in the order of 1 < 2 < 3 < 4, consistent with the electron-withdrawing strength being in the order of 1 (R′ = H) < 2 (R′ = COCH3) < 3 (R′ = tosyl) < 4 (R′ = COCF3). Moreover, it is worth noting that, as shown in the unit cell (see Fig. 1 and Fig. S9, Table S1–S4, ESI†), 4 presents a non-centrosymmetric space group of P21, while those of 1, 2 and 3 are centrosymmetric with space groups of I41/a, P21/n, and P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , respectively (vide infra).7 Careful examination indicates that compound 4 displays an intermolecular CF3⋯H–NRR′ distance of 2.92 Å (Fig. 1), possibly due to the weak static interaction, showing its distinction from 1–3 in terms of the crystal structure (vide infra).
, respectively (vide infra).7 Careful examination indicates that compound 4 displays an intermolecular CF3⋯H–NRR′ distance of 2.92 Å (Fig. 1), possibly due to the weak static interaction, showing its distinction from 1–3 in terms of the crystal structure (vide infra).
 | ||
| Fig. 1 Crystal packing structures of 1 (left) and 4 (right) where 1 is in a centrosymmetric arrangement (the inversion point is denoted by an orange dot), and 4 is in a non-centrosymmetric arrangement. | ||
The steady-state absorption and emission spectra of 1–4 in cyclohexane at room temperature are shown in Fig. 2, and relevant data are listed in Table 1. All studied compounds reveal similar electronic absorption features, where each shows an intense absorption band with a maximum in the range from 316 to 337 nm. Because the highest occupied molecular orbital (HOMO) of 1–4 is calculated to be all in the –H–NRR′ proton donor part (see Table S5 and Fig. S15, ESI†), replacing one of the N–H protons in 1 by an electron-withdrawing group, forming 2–4, would decrease the HOMO energy. Thus, the lowest-lying absorption energy gap (HOMO–LUMO) is predicted to be in the order of 1 < 2 < 3 < 4, consistent with the blue shift of the absorption spectrum from 1 to 4 shown in Fig. 2.
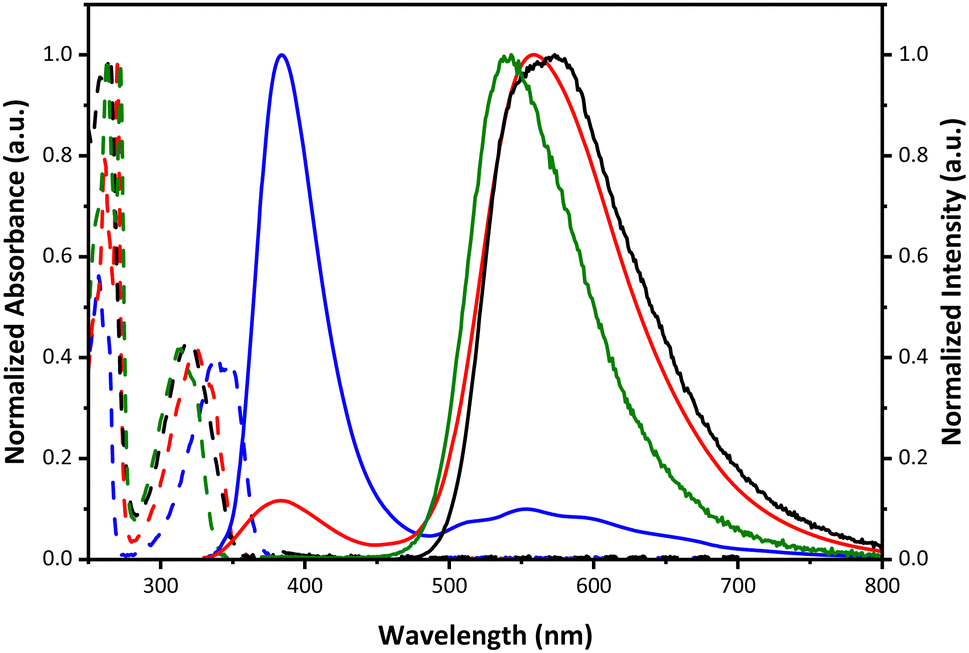 | ||
| Fig. 2 The absorption (dashed line) and emission (solid line) spectra of 1 (blue), 2 (red), 3 (black), and 4 (green) in cyclohexane at 295 K. The excitation wavelengths are at the lowest-lying absorption band. | ||
| Compound | λ abs/nm | λ em/nma | Q.Y.b | τ exp/psc (pre-exp. factor) | K eq |
|---|---|---|---|---|---|
| a N* denotes the normal form, T* denotes the tautomer form. b Q.Y. represents the quantum yield. c Rise time obtained by fluorescence up-conversion. d Population decay time constant was applied for the fitting by sub-nanosecond TCSPC. | |||||
| 1 | 257, 337 | N*: 380a | N*: 0.017a | 400 nm: 3.14 (0.30), 203d (0.70) | 0.43 |
| T*: 552a | T*: 0.002a | 600 nm: 3.72 (−0.34), 203d (0.66) | |||
| 2 | 270, 323 | N*: 371a | N*: 0.001a | 400 nm: 1.84 (0.63), 300d (0.37), 2357d | 1.70 |
| T*: 559a | T*: 0.004a | 600 nm: 2.31 (−0.31), 300d (0.69), 2357d | |||
| 3 | 265, 321 | 573 | 0.028 | 400 nm: 0.51 (0.98) | — |
| 600 nm: 0.60 (−0.52), 2081d (0.38) | |||||
| 4 | 261, 316 | 543 | 0.086 | 400 nm: 0.31 (0.99) | — |
| 600 nm: 0.44 (−0.50), 632d (0.50) | |||||
The increase of electron-withdrawing R′ also increases the N–H acidity and, hence, the H-bond strength, which essentially affects the driving force and thermodynamics of ESIPT.4,8–10 Upon photoexcitation, dual emission bands of 1, specified as F1 (380 nm) and F2 (552 nm) bands, are clearly observed, in which the F1 band intensity is dominant. The excitation spectra monitored at the two emission bands are nearly identical and superimpose with the absorption spectra, corroborating the same ground-state origin of the two emission bands. The F1 and F2 emission bands are thus reasonably attributed to the normal and proton-transfer tautomer emissions, respectively.4 Dual emission is also observed for 2 maximized at 373 nm (F1) and 550 nm (F2). In comparison to 1, the F2 band becomes dominant (Fig. 2). 2 (R′ = COCH3) has a stronger electron withdrawing group and hence a stronger H-bond than 1 (R′ = H). Therefore, despite both undergoing an equilibrium type of ESIPT (vide infra), the excited-state equilibrium is in favor of tautomer in 2, rationalizing its major F2 band. Further enhancing the electron-withdrawing ability of RR′NH, 3 (R′ = tosyl) and 4 (R′ = COCF3) exhibit exclusively the F2 tautomer emission maximized at 573 and 543 nm, respectively, indicating a fast and highly exergonic type ESIPT for 3 and 4. This viewpoint is supported, in a qualitative manner, by the calculation of the energy difference in the lowest lying excited state (singlet manifold) between normal (S1) and tautomer  states (see Table S5, ESI†). The steady-state results thus pave a facile way to fine-tune the ratiometric emission between the normal and tautomer forms in 1–4.
states (see Table S5, ESI†). The steady-state results thus pave a facile way to fine-tune the ratiometric emission between the normal and tautomer forms in 1–4.
To gain further insight into the ESIPT dynamics, time-resolved measurements were carried out. Fig. 3a and b show the early relaxation dynamics of emissions for representatives 1 and 4 using the femtosecond fluorescence up-conversion technique, while the inset shows the population decays acquired by the time-correlated single photon counting (TCSPC) measurement (see ESI† for experimental details). All pertinent time-resolved data for 1–4 are listed in Table 1. For 1, shown in Fig. 3a, the early relaxation dynamics monitored at e.g., 400 nm (the F1 band) exhibit a fast decay component of approximately 3.1 ps, accompanied by a rather slow population decay component, remaining nearly constant throughout the acquisition window of 20 ps; the latter is further resolved to be 203 ps by TCSPC (see inset). On the other hand, the tautomer emission (F2) monitored at 600 nm shows an approximately 3.7 ps rise component and a 203 ps population decay time (see inset of Fig. 3a). The rise of the F2 band is equal to the decay time constant of the F1 band, while both the F1 and F2 population decay time constants are identical. The results thus show an equilibrium type of ESIPT pattern for 1, and the equilibrium constant (Keq) can be derived from the corresponding kinetic expression shown in the ESI,†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 giving a value of 0.43. One may be sceptical as to why this ratio is different from the observed steady-state emission ratio shown in Fig. 2. This is mainly due to the unknown radiative decay rate kr for both normal and tautomer emissions. According to the much smaller steady-state F2/F1 ratio shown in Fig. 2 (cf. Keq = 0.43), kr for F1 must be much larger than that of F2. Fig. 3b depicts the time-dependent results of 4. Upon monitoring at 400 nm where the steady-state emission of 4 is negligible (Fig. 2), a fast decay component is resolved with a time constant of ∼200 fs close to the limit of system response time, which correlates well with the system-limited rise time monitored at 600 nm tautomer emission (Fig. 3b). It is worth noting that when monitored at 400 nm, we detected a gradual growth of the component with a decay around 2.5–10 ps. For 3 and 4 with strong electron withdrawing groups, they are labile to the photolysis during the up-conversion experiment, resulting in an artifact signal. This conclusion is supported by steady-state spectra, where exposure of the sample to a 300 nm Xe lamp (<1 mW) for < 10 minutes clearly shows the formation of impurities that exhibit emission around 400 nm (see Fig. S16, ESI† for 3 and 4). Nevertheless, 3 (Fig. S17, ESI†) and 4 undergo a population decay of 2.0 ns and 632 ps, respectively, at 600 nm. The results thus support the ultrafast, highly exergonic ESIPT in 3 and 4, manifesting their stronger H-bonds among 1–4, resulting in solely 600 nm tautomer emission.
4 giving a value of 0.43. One may be sceptical as to why this ratio is different from the observed steady-state emission ratio shown in Fig. 2. This is mainly due to the unknown radiative decay rate kr for both normal and tautomer emissions. According to the much smaller steady-state F2/F1 ratio shown in Fig. 2 (cf. Keq = 0.43), kr for F1 must be much larger than that of F2. Fig. 3b depicts the time-dependent results of 4. Upon monitoring at 400 nm where the steady-state emission of 4 is negligible (Fig. 2), a fast decay component is resolved with a time constant of ∼200 fs close to the limit of system response time, which correlates well with the system-limited rise time monitored at 600 nm tautomer emission (Fig. 3b). It is worth noting that when monitored at 400 nm, we detected a gradual growth of the component with a decay around 2.5–10 ps. For 3 and 4 with strong electron withdrawing groups, they are labile to the photolysis during the up-conversion experiment, resulting in an artifact signal. This conclusion is supported by steady-state spectra, where exposure of the sample to a 300 nm Xe lamp (<1 mW) for < 10 minutes clearly shows the formation of impurities that exhibit emission around 400 nm (see Fig. S16, ESI† for 3 and 4). Nevertheless, 3 (Fig. S17, ESI†) and 4 undergo a population decay of 2.0 ns and 632 ps, respectively, at 600 nm. The results thus support the ultrafast, highly exergonic ESIPT in 3 and 4, manifesting their stronger H-bonds among 1–4, resulting in solely 600 nm tautomer emission.
 | ||
| Fig. 3 Fluorescence up-conversion lifetimes for (a) 1 and (b) 4 in cyclohexane. The data points (blue and red) were obtained by monitoring with the emission wavelengths at 400, 600 nm, and IRF (black line). Lines depict the best exponential fits. Inset: Pico-nanosecond time-resolved decays recorded for 1 monitored at 400 nm (blue line), 600 nm (red line), and IRF (black line) (λex = 300 nm). | ||
During the spectroscopic measurements, in serendipity, we discovered that 4 revealed prominent triboluminescence (TL) upon slightly scratching or grinding the crystal (Fig. 4). The emission resembles steady-state emission of 4 in solution as well as in a powder (polycrystalline). The lack of any normal emission indicates that TL originates from proton-transfer emission. TL, in theory, is initiated by a piezoelectric-like effect. A piezoelectric crystal is known to accumulate charges on the crack surfaces.11 When these crystals are mechanically stressed, charge separation (or electric polarization) occurs due to the piezoelectric effect, creating an electric field strong enough to cause electrons to accelerate and consequently bombard the material itself or nearby molecules. This electron bombardment excites the molecules, causing them to emit light as they return to their ground state, with specific wavelengths depending on the nature of the electronic transitions involved.12 Previous research has demonstrated that permanent dipolar structures and non-centrosymmetric crystal arrangements are equally crucial for displaying TL.13–15 As shown in Fig. 1 and Fig. S9 (ESI†), the crystal structures of 1–3 are in the centrosymmetric arrangement (the inversion point is denoted by an orange dot), while 4 is apparently non-centrosymmetric (Fig. 1). Note that non-centrosymmetry is a necessary condition for TL.11 This result explains why TL is not observed in 1–3. According to the X-ray analysis, the CF3 functional group in 4 plays a role in its distinct intermolecular arrangement compared with that of 1–3, where the fluorine atom in CF3 of the RR′N–H group forms an H-bond-like static interaction with the RR′N–H proton of its adjacent moiety (vide supra), resulting in a parallel head-to-head (or parallel tail-to-tail) packing. In other words, physically, they cannot be in a centrosymmetric arrangement. Conversely, the rest of compounds 1–3 undergo parallel head-to-tail arrangement and hence centrosymmetric packing. This, together with the intrinsic polar feature for 4, meets the criteria necessary for the occurrence of TL. To our knowledge, this is, for the first time, the observation of mechanic force induced ESIPT, resulting in proton-transfer TL. The slightly different emission spectral features between TL and steady-state emission in the crystal (Fig. 4) is because the origin of TL is from the surface where the electric polarized field is created, while the steady-state emission is from both the surface and interior of the crystal.
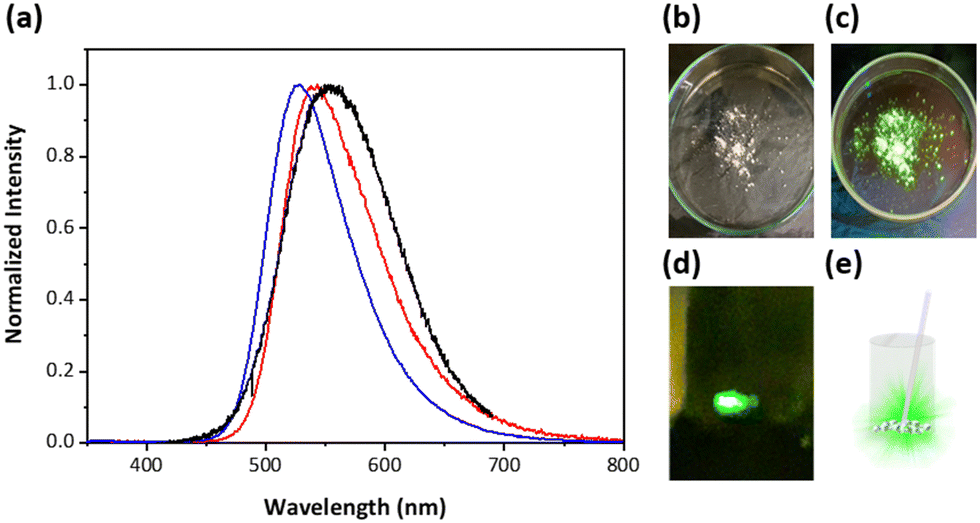 | ||
| Fig. 4 (a) Emission spectra of compound 4, showcasing photoluminescence in cyclohexane (1 × 10−5 M, red line) and in powder form (blue line), as well as TL in crystal form (black line). (b) Compound 4 under ambient light and (c) under UV irradiation at 365 nm. (d) TL image of the compound 4 crystal ground in a sample vial using a spatula. (e) Schematic diagram of TL corresponding to (d). | ||
In summary, through the strategic design and synthesis of a series of 7-amino-2,3-dihydro-1H-inden-1-one (RR′N–H⋯OC)-type intramolecular H-bond derivatives, we have elucidated the spectroscopy and dynamics of N–H type ESIPT. We reaffirm the previously reported correlation among H-bond strength,4 ESIPT dynamics, and thermodynamics, stating that the stronger the H-bond strength is, the more exergonic as well as faster ESIPT is. Accordingly, ESIPT is under thermal equilibrium between normal and tautomer in 1 and 2, while it is highly exergonic in 3 and 4. Among 1–4, compound 4 exhibits remarkable TL properties, which is attributed to its unique crystal structure with a polar non-centrosymmetric arrangement. This, together with the piezoelectric effect, provides the first demonstration of mechanically induced ESIPT.
Author contributions
K.-Y. C. and P.-T. C. conceived the project and supervised the work. J.-J. Y. and J.-W. H. were responsible for the synthesis. Y.-D. L., Y.-Y. T., and H.-T. Q. conducted the photophysical measurements. Y.-D. L. and C.-H. H. wrote the original draft and editing. All authors have given approval to the final version of the manuscript.Data availability
The data that support the findings of this study are available in the ESI† of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P.-T. C. and K.-Y. C. acknowledge the National Science and Technology Council (grant no. NSTC-113-2639-M-002-001-ASP and grant no. NSTC 113-2113-M-035-002) for their generous financial support. The authors acknowledge Prof. Liang-Yan Hsu for providing the supercomputing system at the Institute of Atomic and Molecular Sciences, Academia Sinica.Notes and references
- K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS PubMed.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- Z. Zhang, Y.-A. Chen, W.-Y. Hung, W.-F. Tang, Y.-H. Hsu, C.-L. Chen, F.-Y. Meng and P.-T. Chou, Chem. Mater., 2016, 28, 8815–8824 CrossRef CAS.
- H.-W. Tseng, J.-Q. Liu, Y.-A. Chen, C.-M. Chao, K.-M. Liu, C.-L. Chen, T.-C. Lin, C.-H. Hung, Y.-L. Chou, T.-C. Lin, T.-L. Wang and P.-T. Chou, J. Phys. Chem. Lett., 2015, 6, 1477–1486 CrossRef CAS PubMed.
- Y. Kim, H. Kim, J. B. Son, M. Filatov, C. H. Choi, N. K. Lee and D. Lee, Angew. Chem., Int. Ed., 2023, 62, e202302107 CrossRef CAS PubMed.
- Q. Huang, Q. Guo, J. Lan, R. Su, Y. Ran, Y. Yang, Z. Bin and J. You, Mater. Horiz., 2021, 8, 1499–1508 RSC.
- G. E. Hardy, W. C. Kaska, B. P. Chandra and J. I. Zink, J. Am. Chem. Soc., 1981, 103, 1074–1079 CrossRef CAS.
- M.-W. Chung, T.-Y. Lin, C.-C. Hsieh, K.-C. Tang, H. Fu, P.-T. Chou, S.-H. Yang and Y. Chi, J. Phys. Chem. A, 2010, 114, 7886–7891 CrossRef CAS PubMed.
- H.-W. Tseng, T.-C. Lin, C.-L. Chen, T.-C. Lin, Y.-A. Chen, J.-Q. Liu, C.-H. Hung, C.-M. Chao, K.-M. Liu and P.-T. Chou, Chem. Commun., 2015, 51, 16099–16102 RSC.
- Y.-A. Chen, F.-Y. Meng, Y.-H. Hsu, C.-H. Hung, C.-L. Chen, K.-Y. Chung, W.-F. Tang, W.-Y. Hung and P.-T. Chou, Chem. – Eur. J., 2016, 22, 14688–14695 CrossRef CAS PubMed.
- B. Chandra, V. Chandra and P. Jha, J. Lumin., 2013, 135, 139–153 CrossRef CAS.
- R. S. Fontenot, K. N. Bhat, C. A. Owens, W. A. Hollerman and M. D. Aggarwal, J. Lumin., 2015, 158, 428–434 CrossRef CAS.
- L. M. Sweeting, A. L. Rheingold, J. M. Gingerich, A. W. Rutter, R. A. Spence, C. D. Cox and T. J. Kim, Chem. Mater., 1997, 9, 1103–1115 CrossRef CAS.
- H. Nakayama, J.-i Nishida, N. Takada, H. Sato and Y. Yamashita, Chem. Mater., 2012, 24, 671–676 CrossRef CAS.
- S. Xu, T. Liu, Y. Mu, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, Y. Zhang, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 874–878 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2371431–2371433 and 2375635. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cp02862g |
| ‡ Equal contribution. |
| This journal is © the Owner Societies 2024 |