
Metal nitrides for seawater electrolysis
Huashuai
Hu
a,
Xiaoli
Wang
a,
J. Paul
Attfield
 b and
Minghui
Yang
*a
b and
Minghui
Yang
*a
aSchool of Environmental Science and Technology, Dalian University of Technology, Dalian 116024, China. E-mail: myang@dlut.edu.cn
bCentre for Science at Extreme Conditions and School of Chemistry, University of Edinburgh, King's Buildings, Mayfield Road, Edinburgh, UK
First published on 29th November 2023
Abstract
Electrocatalytic high-throughput seawater electrolysis for hydrogen production is a promising green energy technology that offers possibilities for environmental and energy sustainability. However, large-scale application is limited by the complex composition of seawater, high concentration of Cl− leading to competing reaction, and severe corrosion of electrode materials. In recent years, extensive research has been conducted to address these challenges. Metal nitrides (MNs) with excellent chemical stability and catalytic properties have emerged as ideal electrocatalyst candidates. This review presents the electrode reactions and basic parameters of the seawater splitting process, and summarizes the types and selection principles of conductive substrates with critical analysis of the design principles for seawater electrocatalysts. The focus is on discussing the properties, synthesis, and design strategies of MN-based electrocatalysts. Finally, we provide an outlook for the future development of MNs in the high-throughput seawater electrolysis field and highlight key issues that require further research and optimization.
 Huashuai Hu | Huashuai Hu is currently working on his PhD degree at the School of Environmental Science and Technology, Dalian University of Technology (DUT) under the supervision of Professor Minghui Yang. His research focuses on the utilization of metal nitrides for electrocatalytic reactions, such as hydrogen production through seawater electrolysis and oxygen electrode reactions. |
 Xiaoli Wang | Xiaoli Wang is pursuing her MS degree at Dalian University of Technology (DUT) under the supervision of Professor Minghui Yang. Her current research interests focus on the synthesis of metal nitrides-based nanostructured composite catalysts and their applications in the field of electrocatalysis. |
 J. Paul Attfield | John Paul Attfield, FRS, holds a Chair in Materials Science at Extreme Conditions at the School of Chemistry, University of Edinburgh and he is the Director of the Centre for Science at Extreme Conditions. He received BA and DPhil degrees from Oxford University, and he was a Co-Director of the Interdisciplinary Research Centre in Superconductivity at the University of Cambridge during 1991–2003. He received the Royal Society of Chemistry's Meldola and Corday-Morgan medals and Peter Day award, and he was elected a Fellow of the Royal Society of Edinburgh in 2006 and of the Royal Society in 2014. His early research contributions included pioneering resonant X-ray scattering experiments of cation and valence ordering, and studies of disorder effects in functional oxides. His current research is centered on electronic and magnetic materials. |
 Minghui Yang | Minghui Yang, FRSC, is a Professor at Dalian University of Technology (DUT). He obtained his MChem degree from the University of Liverpool and completed his PhD in Materials Chemistry under the esteemed guidance of Professor John Paul Attfield at the University of Edinburgh. This was followed by a postdoctoral appointment in the Department of Chemistry and Chemical Biology at Cornell University, where he worked on nanostructured metal nitrides with Professor Francis John DiSalvo. He currently serves as the group leader for the Solid State Functional Materials Lab at DUT. His research group focuses on the exploration and development of solid state functional materials, specifically for catalysis and sensing applications. |
1. Introduction
With the rapid development of the world economy, the problems of energy shortage and storage are becoming more prominent.1–4 With the advantages of high energy density, and clean and efficient combustion, hydrogen (H2) is considered to be one of the most promising fuels to replace fossil fuels in the future.5–8 At present, most industrial H2 is based on steam methane reforming, and the H2 obtained in this way is not only of low purity, but also has residual methane, carbon monoxide and other harmful gases, making the utilization of H2 extremely limited.8–10 Among alternative hydrogen production technologies, water electrolysis has attracted widespread attention and has broad potential applications due to high purity hydrogen production and versatile operability.11–13 The main energy consumption of this process is electrical energy which accounts for about 80% of the total cost. Water electrolysis consists of two half-reactions: water reduction to produce hydrogen at the cathode (hydrogen evolution reaction, HER) and water oxidation to produce oxygen at the anode (oxygen evolution reaction, OER).12,14,15 To achieve the overall water splitting (OWS) reaction, a minimum theoretical thermodynamic voltage of 1.23 V is required under standard conditions (25 °C, 1.0 atm pressure).12 However, the sluggish multi-electron transport process in the system and consequent slow kinetics result in additional potential depletion.16–18 Therefore, it is important to enhance the reaction kinetics of electrocatalysts to achieve high efficiency and low energy loss. Noble metals (Pt) and noble metal oxides (RuO2 and IrO2) are considered as the most effective HER and OER electrocatalysts, respectively.19–21 However, their scarcity, high price and poor stability impede widespread application in industrial water electrolysis.22–24 In addition, these noble metal electrocatalysts have high catalytic activity at small current densities, but often show rapid catalytic performance decay at the high current densities required for industrial H2 production.25,26 Therefore, it is important to develop efficient and stable electrocatalysts for water electrolysis to reduce energy consumption and accelerate the development of industrial water electrolysis.Seawater constitutes 96.5% of the total water on Earth and so is an abundant resource.27–29 Against the backdrop of an increasingly urgent demand for clean energy in human society, researchers have shifted their focus towards obtaining green hydrogen from seawater.30–32 This not only alleviates the pressure on freshwater usage but also expands the utilization of marine resources. However, compared to electrolyzing pure water, seawater electrolysis faces additional challenges and bottlenecks. Natural seawater contains abundant salts and impurities, with a relatively high concentration of Cl− which leads to the generation of chlorine gas at the anode during the electrolysis process, adversely affecting the stability of both the catalyst and the electrolytic cell.33–36 Additionally, cations such as Ca2+ and Mg2+ as well as microbial impurities in seawater tend to accumulate on the electrode surface during electrolysis, forming fouling layers that reduce electrolysis efficiency.35–37 The fouling layers also increase the maintenance and cleaning costs of the electrolytic equipment. In addition, achieving industrial-scale seawater hydrogen production requires electrocatalysts with exceptional durability to withstand the high current densities demanded by industrial electrolysis processes. In industrial alkaline water electrolysis (AWE), catalysts are required to reach current densities of at least 500 and up to 1000 mA cm−2.38,39 For proton exchange membrane water electrolysis (PEMWE), the operating current density tends to be higher, 1000–2000 mA cm−2.40 At such high current density, a large number of bubbles will be instantaneously generated and accumulated on the electrode surface, which will hinder the adsorption of reactants and the release of H2 and O2 bubbles to a certain extent.19 A significant portion of the bubbles will rupture and collapse the initial catalyst nanostructure, resulting in a decrease in active area and hence deactivation of the catalyst.37 Conventional nano-powder catalysts have exhibited satisfactory performance at low current densities, however they require the support of organic polymer binders such as Nafion and polytetrafluoroethylene (PTFE).26,41–43 This can significantly reduce the electron and mass transfer efficiencies and thus have a negative effect on the performance of electrocatalysts and at high current densities,44 as continuous and rapid bubbles can cause the catalyst layer to separate from the electrode surface.44 Therefore, the capacity to sustain high current densities while maintaining long-term efficiency and stability becomes an essential prerequisite for electrocatalysts.
To address the mentioned challenges, a series of high-performance seawater electrocatalysts has been developed using inexpensive and widely available non-precious metals, such as metal hydroxides,11,45–48 phosphides,26,49–52 nitrides,53–58 and more,59,60 with a particular emphasis on tackling the issue of Cl−.61–64 For instance, an effective approach involves coating carbon materials with high corrosion resistance around the catalytic sites.65 Additionally, the construction of abundant heterogeneous interfaces significantly increases the number of active sites, effectively retarding the encroachment of Cl− on the catalysts.66 In order to achieve active adsorption of intermediates and reactants, an advanced three-dimensional (3D) nanostructure design strategy has been proposed.67 This not only increases the electrochemically active surface area, but also enhances the structural robustness of the catalyst,8,68 for example, by using inter-crosslinked nanorod-nanowire superstructures,69 and sandwich structures with double-sided nanosheets encapsulation.70 In addition, electrolyte substitution is also a highly effective strategy, primarily focused on the anode.68,71–73 Utilizing small molecule organic electrolytes with thermodynamic advantages as a replacement for seawater not only reduces energy costs but also avoids the harmful corrosion effects of Cl− on catalytic materials.74–76 It is worth noting that the introduction of organic electrolytes may have an impact on the membrane, thus necessitating the development of durable and compatible membrane materials, which may further advance seawater electrolysis industry.
Metal nitrides (MNs) are usually crystalline solids compounds with strong metal-nitrogen bonds,77,78 which endow them with high structural stability and chemical inertness. MNs, such as CrN,79 TiN,80 ZrN,81 and NbN,82 demonstrate high performance in challenging conditions (high temperatures, corrosive and frictional environments, etc.).82,83 Many nitrides, e.g. Ni, Co, and Fe-based materials, are used in batteries and energy conversion devices due to their excellent catalytic properties.58,65,84 Compared to other catalysts, MNs are less prone to precipitation, corrosion, or structural changes during electrochemical processes, allowing them to participate in reactions with prolonged stability and maintain high catalytic activity. For example, Co4N forms a thin oxide shell (∼2 nm) on the surface during the oxygen reduction reaction (ORR), preventing oxidation of the conducting nitride core inside.84 A similar phenomenon has been observed in the study of ternary nitrides for OER, where Ni2Mo3N rapidly forms a surface-oxide-rich activation layer (SOAL).85 Secondary Mo atoms on the surface act as electron pumps, stabilizing oxygen-containing species and promoting continuous reactions, while the internal MNs core provides efficient pathways for electron transfer. The inherent high stability of MNs makes them suitable for seawater electrolysis, enabling better resistance to corrosion and the influence of impurities in seawater.
In recent years, significant research progress and extensive attention have been devoted to the development of high-performance self-supported MN seawater electrocatalysts under high current density conditions.67,86–89 Consequently, a comprehensive review of the key findings from this research is timely to provide future guidance. In this review, we summarize recent research progress in MNs as catalysts for high throughput seawater electrolysis (Scheme 1). We first summarize the chemical reactions involved in electrochemical seawater splitting, including the anodic OER and chlorine evolution reaction (CER), as well as the cathodic HER and the challenges posed by metal corrosion due to precipitation and Cl−. The chlor-alkali process is also included. Furthermore, we provide a detailed overview of activity descriptors and catalytic mechanisms in seawater splitting, categorizing them into activity parameters, kinetic parameters, and stability. We discuss the selection of self-supported substrates for high current density catalysts, analyzing their advantages and limitations. Next, we introduce the properties and surface reconstruction of MNs and their popular synthesis methods, including the solvothermal method, magnetron sputtering, template synthesis, and electrodeposition techniques. Subsequently, we emphasize current strategies of seawater electrolysis and for optimizing high-throughput seawater hydrogen production from nanostructured MNs. Finally, we provide an outlook on the challenges and development directions of MN seawater electrocatalysts for hydrogen production, with the ultimate goal of promoting the widespread adoption of industrial seawater hydrogen production.
 | ||
| Scheme 1 Schematic diagram of the topics covered in this review. | ||
2. Fundamentals of seawater electrolysis
Seawater electrolysis is considerably more complex than the electrolysis of pure water. In this section, we will provide an overview from the perspectives of the reactions, evaluation parameters, and substrate material selection:2.1. Overview and chemical reactions involved in seawater electrolysis
Electrochemical water splitting breaks down water into hydrogen and oxygen by applying an electrical voltage (Fig. 1a). The electrolytic water device comprises two primary electrodes, the cathode and the anode, immersed in an electrolyte solution. Upon connecting an external power source to the electrolytic cell, H2O molecules undergo dissociation into hydrogen (H+) and hydroxide (OH−) ions. The H+ migrate towards the cathode, while the OH− migrate towards the anode. At the cathode, electrons combine with H+ to yield H2, while simultaneously, O2 is generated at the anode. The mechanism of the water decomposition reaction varies depending on the acid–base properties of the electrolyte solution. Nevertheless, the general reaction equation can be expressed as follows:| 2H2O (l) → 2H2 (g) + O2 (g). |
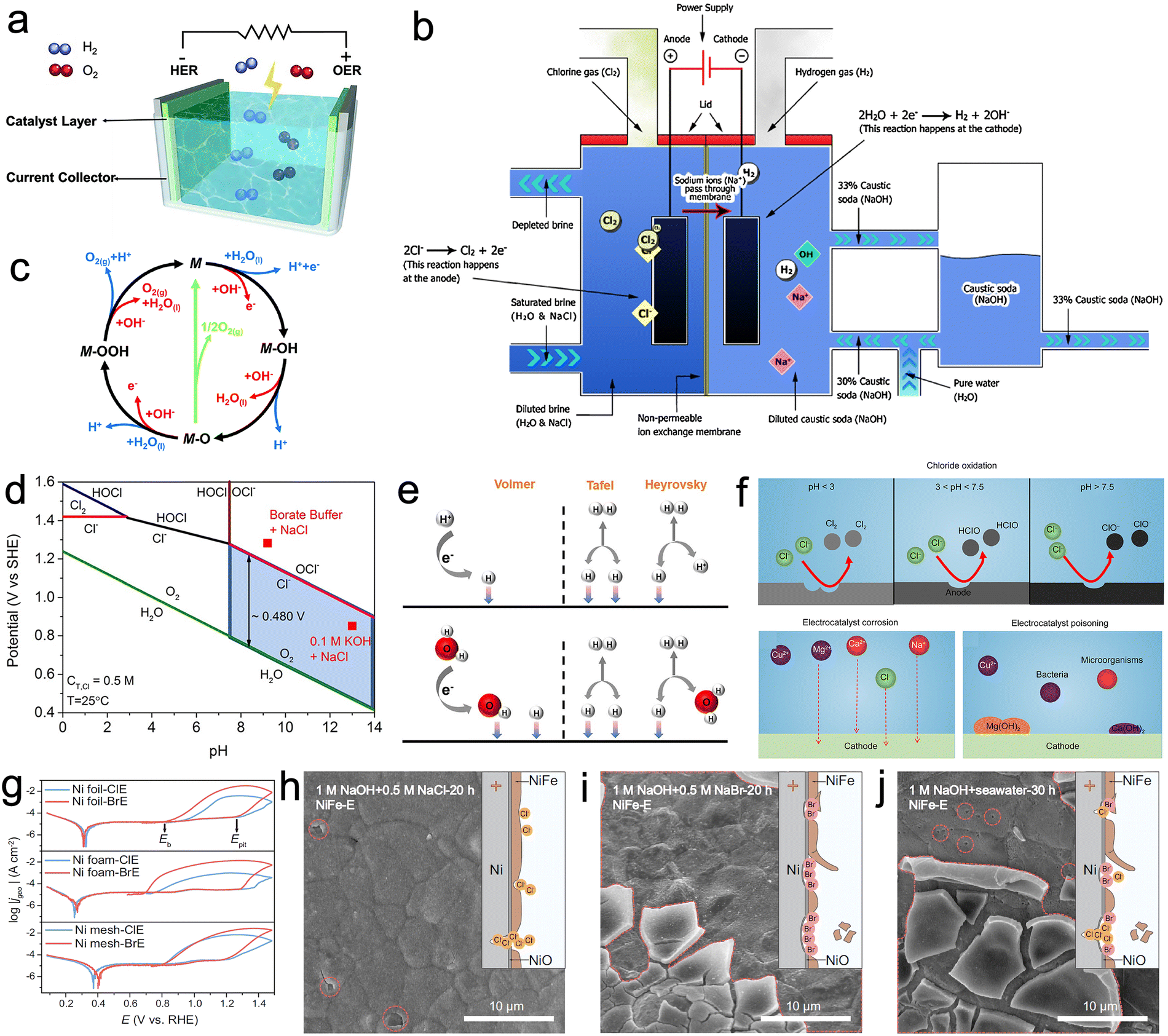 | ||
| Fig. 1 (a) Schematic diagram of electrolysis of water in which hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) occur at the cathode and anode, respectively. Reproduced with permission.8 Copyright 2021, Wiley-VCH. (b) Process flow of the membrane cell process. Reproduced with permission.90 Copyright 2013, Springer Nature Publishing Group. (c) The OER mechanism for acid (blue line) and alkaline (red line) conditions. Reproduced with permission.91 Copyright 2017, The Royal Society of Chemistry. (d) Pourbaix diagram for the artificial seawater model. Reproduced with permission.92 Copyright 2016, Wiley-VCH. (e) The mechanism of HER in acidic (top) and alkaline or neutral solutions (bottom). Reproduced with permission.93 Copyright 2022, Elsevier. (f) Challenges for designing seawater-based electrocatalysts. Chloride oxidation, electrocatalyst corrosion and electrocatalyst poisoning. Reproduced with permission.94 Copyright 2023, Nonferrous Metals Society of China. (g) Cyclic polarization curves (CPCs) of Ni substrates tested in Cl−-containing electrolyte (ClE) and Br−-containing electrolyte (BrE) at a scan rate of 10 mV s−1. Scanning electron microscopy (SEM) images of NiFe-E observed after 20 h operation in (h) 1 M NaOH + 0.5 M NaCl, (i) 1 M NaOH + 0.5 M NaBr, and 30 h operation in (j) 1 M NaOH + seawater. Reproduced with permission.95 Copyright 2023, Springer Nature Publishing Group. | ||
The theoretical voltage required for water decomposition under standard conditions is 1.23 V.8,96
In contrast to freshwater splitting, the intricate composition of seawater poses a significant challenge to its decomposition. Seawater contains approximately 3.5% Cl− and various metal ions,92 so that the oxidation of Cl− competes strongly with the OER. Cl− can produce hypochlorite, which corrodes the active sites of the catalyst. Another crucial aspect to consider is the accumulation of Cl− as H2O molecules continue to decompose during seawater electrolysis. This progressive accumulation can lead to saturation levels of Cl− so the chlor-alkali reaction becomes competitive with the water electrolysis reaction. The chemical reactions involved in these processes are as follows:
| 2NaCl (aq) + 2H2O (l) → 2NaOH (aq) + Cl2 (g) + H2 (g) |
Various techniques are employed in the chlor-alkali process, including diaphragm cells, mercury cells and membrane cells. The first two methods employ toxic materials such as asbestos and mercury, posing severe environmental and health risks. In contrast, membrane cell technology, using ion-exchange membranes, has emerged as a more environmentally friendly and efficient alternative (Fig. 1b).90 Ion-exchange membranes play a pivotal role in membrane cell technology. They permit the transport of cations and anions between compartments while preventing the passage of electrons. In the cathode compartment, water undergoes electrolysis to yield H2 and NaOH. Simultaneously, in the anode compartment, Cl− undergo electrolysis to generate Cl2. Ion-exchange membranes are used to prevent product mixing.
Although the membrane cell process is beneficial for the chlor-alkali industry, hurdles still exist. The reliance on precious metal based-catalytic materials significantly increases production costs. Additionally, the preparation and maintenance of these membranes incur relatively high expenses, which may exert economic pressure when applied on an industrial scale. Ion-exchange membranes often lack long-term durability, especially under conditions of high salt concentration and high temperatures.90 While the membrane cell technology improves energy efficiency compared to alternative methods, it still demands a substantial amount of electrical energy. Consequently, similar to the water electrolysis, there is an urgent need for the development of cost-effective and highly efficient catalysts. Furthermore, the quest for high-stability membrane materials is pressing. These problems are likely to be addressed and alleviated with continuing technological advances.
Acidic:
| 2H2O → 4H+ + O2 + 4e− |
| 4OH− → 2H2O + O2 + 4e− |
The Pourbaix diagram (Fig. 1d) summarizes the electrochemical oxidation process of chlorides in seawater in a simple and clear manner. Considering that the total salinity of natural seawater is 3.5%, an aqueous solution of 0.5 M NaCl is usually used as a substitute for natural seawater. With changes in pH and applied potential, reactions such as chlorine precipitation reactions, hypochlorous acid formation and hypochlorite formation occur. As CER is a 2-electron process with only a single intermediate Cl* involved, under acidic conditions CER is more thermodynamically favorable than OER as illustrated by the relevant reactions:74,92
pH < 3:
| 2Cl− → Cl2 + 2e− E0 = 1.36 V |
| Cl− + H2O → HClO + H+ + 2e− E0 = (1.494–0.0295 pH) V |
| Cl− + 2OH− → ClO− + H2O + 2e− E0 = (1.795–0.0591 pH) V |
Under neutral or alkaline conditions, the potential difference between hypochlorite formation and OER can reach a maximum of 480 mV (Fig. 1d). From a thermodynamic perspective, the competition between OER and hypochlorite formation reactions is most favorable to the latter at this point. This is one of the primary reasons why industrial electrolysis of seawater is typically conducted under alkaline conditions. However, even under alkaline conditions the CER can dominate over the kinetically slow OER. It is important to acknowledge the significant variations in local pH of the electrode material during electrolysis, as these fluctuations have a non-negligible impact.74 This observation was substantiated by Miyazaki et al., where they devised a method to assess the in situ local pH of an electrode using a ring-disk electrode modified with iridium oxide.101
The accumulation of H+ in the vicinity of the anode can promote the preferential formation of hypochlorite or hypochlorite over the OER, resulting in substantial degradation in the performance of the electrode material. This phenomenon highlights the significance of managing and controlling local pH levels to mitigate adverse effects and maintain optimal electrode functionality. In summary, mitigation of the detrimental effects of Cl− is crucial for enhancing the efficiency and stability of seawater electrolysis catalysts.
Acidic solution reactions are:
| Volmer reaction: H+ + e− + M → M − H* |
| Heyrovsky reaction: M − H* + H+ + e− → M + H2 |
| Tafel reaction: 2M − H* → 2M + H2 |
In the Volmer step, water molecules initially adsorb to the active site, leading to the dissociation of water and the formation of OH− and M − H*. The Tafel step involves the combination of M − H* in the adsorbed state, formed in the previous step, to form an H–H bond. Conversely, the Heyrovsky step distinguishes itself from the Tafel step by the combination of adsorbed M − H* and H+ from the electrolyte, resulting in the generation of H2 in the presence of electrons.
In neutral or alkaline media, the same Volmer–Tafel mechanism observed in acidic media applies to the HER mechanism of catalysts.102 Since protons are relatively scarce in the electrolyte under neutral or alkaline conditions, the Volmer–Heyrovsky mechanism involves the participation of H2O in the H2 generation reaction instead of H+.
Neutral or alkaline solution reactions are:
| Volmer reaction: M + H2O + e− → M − H* + OH− |
| Heyrovsky reaction: M − H* + H2O + e− → M + OH− + H2 |
| Tafel reaction: 2M − H* → 2M + H2 |
Based on the reaction mechanism, a catalyst designed for HER should exhibit a lower overpotential in acidic media compared to alkaline media. However, transition metal-based catalysts often face limitations in terms of their service life due to their susceptibility to corrosion by acidic electrolytes. This limitation contributes to the reliance on precious metals in cathode catalysts. A high-performance HER catalyst should possess favorable characteristics such as a low barrier for water dissociation, optimized water adsorption energy, hydrogen binding energy, and OH− adsorption energy.
The high concentration of Cl− in seawater presents an obstacle to the development of seawater electrolysis, as it not only generates hypochlorite species but also directly corrodes the active metal sites of the catalyst. Cl− in seawater initially adsorb onto the metal sites of the catalyst, forming intermediate species that further combine with Cl− to form MClx− complexes. The presence of OH− in the electrolyte facilitates the conversion of these complexes into metal hydroxides or precipitates through the following reactions, resulting in the loss of catalyst performance;94
| M + Cl− → MClads + e− |
| MClads + Cl− → MClx− |
| MClx− + OH− → M(OH)x + Cl− |
Numerous studies have indicated that catalysts exhibit shorter lifespans in real seawater compared to simulated seawater electrolytes. An often overlooked factor is the presence of Br− ions, which can accumulate to concentrations as high as 0.5 M after long periods of uninterrupted operation. Lu et al. conducted a systematic investigation focusing on the most widely used Ni-based electrodes to study the corrosion behavior of Cl− and Br−.95 Results from cyclic polarization curves indicate that the Ni-based substrate exhibits faster corrosion kinetics and low pitting potential in Br−-containing electrolytes (Fig. 1g). In contrast to Cl−, which tend to corrode localized Ni-based substrates, Br− have a propensity for causing widespread corrosion, resulting in shallow yet broad pits and extensive detachment of loaded catalysts (Fig. 1h–j). This study sheds light on the frequently overlooked corrosion behavior of Br− and offers vital insights for the subsequent development of highly stable seawater electrolysis catalysts.
These factors collectively pose challenges to the efficiency and stability of the seawater electrolysis process, necessitating improvements in material selection, optimization of electrolysis conditions, and the implementation of other measures.
2.2. Performance parameters
When evaluating the catalytic properties of electrocatalysts, the following common performance parameters are employed: onset potential (Eonset) and overpotential (η), mass or specific activity (MA or SA), electrochemical active surface area (ECSA), turnover frequency (TOF), Faraday efficiency (FE), Tafel slope, exchange current density (j0), electrochemical impedance spectrum (EIS) and long-term stability.(a) Onset potential and overpotential. The Eonset is the potential where Faraday reaction current initiates in linear sweep voltammetry (LSV) curves of HER, OER, and OWS (Fig. 2).91 However, accurate Eonset determination is challenging due to material properties and instrument sensitivity. To address this, η (derived from eqn (2.1) and (2.2)) at 10 mA cm−2 is introduced to assess electrocatalyst activity. It is defined as the potential deviation between the potential corresponding to 10 mA cm−2 at the electrode and the theoretical equilibrium potential.
 | (2.1) |
| η = E − Eeq | (2.2) |
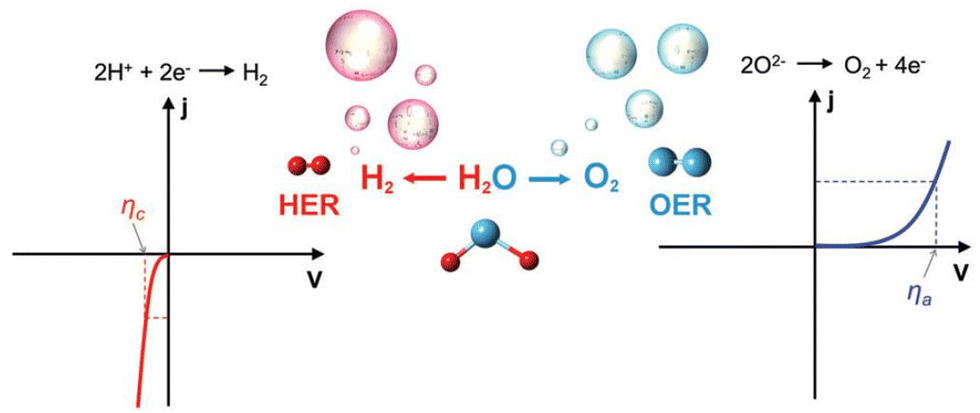 | ||
| Fig. 2 The typical linear sweep voltammetry (LSV) curves of hydrogen evolution reaction (HER) (left) and oxygen evolution reaction (OER) (right). The ηc and ηa represent the overpotential (η) of cathode and anode at the given current density (j). Reproduced with permission.91 Copyright 2017, The Royal Society of Chemistry. | ||
(b) Mass and specific activity. MA represents catalytic activity per electrocatalyst mass unit. High loading typically boosts activity, i.e., lower η for same current density. However, η does not always decrease with increasing loading. Excessive loading hinders charge and mass transfer, resulting in a decreasing trend in activity. For self-supported electrocatalysts at high current density, MA is less crucial due to the interference of conductive substrate in active catalyst mass determination, especially for low-cost non-precious metal electrocatalysts.
SA assesses intrinsic activity, normalized by electrode current to ECSA or Brunauer–Emmett–Teller (BET) surface area.103 SA eliminates active site number and mass loading effects, yielding intrinsic activity per catalytic site. Hence, researchers increasingly use SA to assess electrocatalyst intrinsic activity rather than MA.104
(c) Electrochemical active surface area. Accurate estimate of the electrochemical active surface area of catalysts is of considerable importance for an exact assessment of the number of active sites.104,105 The double capacitance electro-layer values are obtained in the non-Faraday potential range where charge transfer does not occur but chemisorption and desorption can proceed normally.104 The Cdl value can be used to estimate the number of activity sites as it correlates positively with the ECSA. The larger ECSA value represents more exposed electrocatalytic active sites and higher intrinsic activity.26
(d) Turnover frequency. For the same metal particles on carriers with different surface areas, specific activity varies but is not reflected in the SA. To more precisely describe the intrinsic activity of catalysts, Boudart introduced the concept of TOF in 1966.106,107 The concept of TOF originally stemmed from the field of enzyme catalysis but has found application in various catalytic processes. It is important to note that the focus of SA emphasizes the “carrier” of the catalyst, while TOF focuses more on the “active site”, which provides a better reflection of the intrinsic activity of the catalyst. However, it is worth emphasizing that accurately identifying true active sites, especially in heterogeneous catalysts with complex structures and rough surfaces, remains a significant challenge with current characterization techniques. The TOF value for an electrocatalyst can be calculated using the following eqn (2.3):
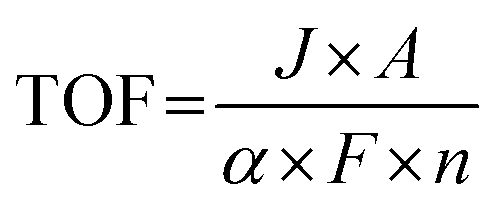 | (2.3) |
(e) Faraday efficiency and hypochlorite detection. FE measures charge transfer efficiency in electrochemical reactions, essential for energy conversion efficiency assessment. There are two common methods of calculating FE: the water–gas displacement method and the gas chromatography (GC) method.110 A sufficient volume of gas is required for both of these methods to reduce measurement errors.108
Due to the oxidation of Cl− in seawater at the anode, hypochlorite and hypochlorous acid are generated, causing severe corrosion to electrode materials. Therefore, these unwanted byproducts must be maintained at low levels, and their detection is essential and preferably in a continuous manner. Currently, the most commonly used methods for this purpose are a combination of test strips, ion chromatography, the orthotolidine method, and UV-Visible spectroscopy.32,94
(a) Tafel slope and exchange current density. In electrochemical reaction systems, a small η can cause a large increase in current density and is an important indicator for assessing the kinetics of electrocatalysts (Fig. 3a).91,111–114 The most common way of obtaining the Tafel slope value is by fitting the linear part of the Tafel curve (η vs. log|j|) derived from the LSV curve (j vs. E).115 As shown in Fig. 3b, compared to Tafel slope (a), Tafel slope (b) means that less overpotential is required to increase the same magnitude of current density, which implies that the electrocatalyst has faster reaction kinetics. In addition, the Tafel slope plays a key role in revealing reaction mechanisms and identifying rate-determining steps. There has been a significant amount of work combining it with in situ characterization techniques to probe the reaction mechanism.116–118
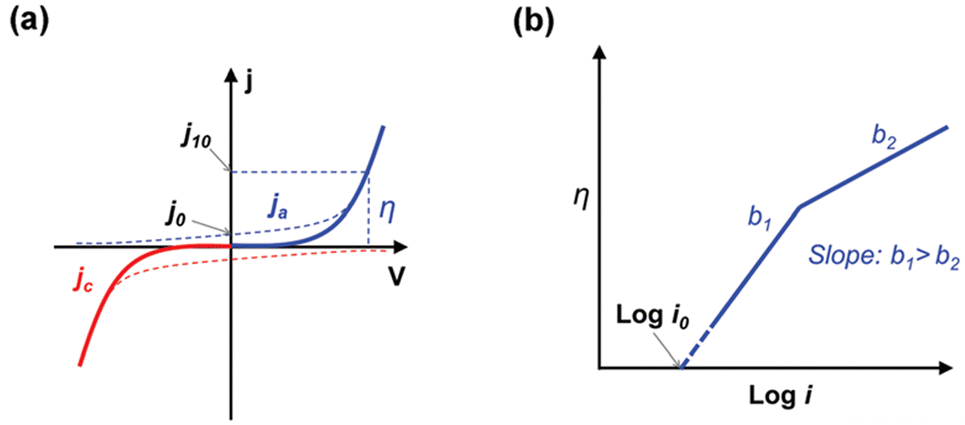 | ||
| Fig. 3 Typical cathode (a, red line), anode (a, blue line) and (b) Tafel curves. Reproduced with permission.91 Copyright 2017, The Royal Society of Chemistry. | ||
j 0 is another critical indicator for evaluating the inherent kinetic properties of an electrocatalyst and can be obtained by extrapolating the Tafel curve in the x-axis direction (Fig. 3b). It is important to note that the j0 only has practical significance in the same electrode reaction. An electrode reaction with a high j0 means that the required driving force (external current density) is lower, in other words that the electrochemical reaction proceeds more easily.
(b) Electrochemical impedance spectrum. EIS is an electrochemical measurement method that uses a small amplitude sinusoidal potential (or current) as the perturbation signal. During the HER, OER and OWS, a constant potential is selected where a significant electrochemical reaction occurs and the test parameters include amplitude and frequency. The selection of the frequency range greatly affects the information obtained from the EIS spectra. For instance, the resistance values obtained in the high-frequency region (where no Faraday reaction takes place) can reflect the solution resistance of the catalytic material with the electrolyte, noted as Rs. The low-frequency region contains much richer kinetic information, which visualizes the speed of charge transfer from electrocatalyst surface to reactant, i.e. the charge transfer resistance (Rct).103 A smaller Rct indicates a fast kinetic process, suggesting excellent catalytic activity of the electrocatalyst.
2.3. Substrate materials for high current seawater electrolysis
3D conductive substrates are a new class of materials are widely used in the fields of aerospace, petrochemical, electrode plates, heat exchangers and electrocatalysis due to their high porosity, superb electron conductivity, controllable flexibility and large surface area.8 In the process of in situ growth, nanomaterials on a 3D substrate tend to form arrays of nanowires, nanorods, nanosheets, and other structures. Consequently, when utilized as electrocatalytic carriers, such 3D materials not only maximize the density of active sites but also possess advantages in streamlining the assembly process and lowering production costs.25,119 In this section, we will provide a detailed introduction to commonly used substrate materials of different types.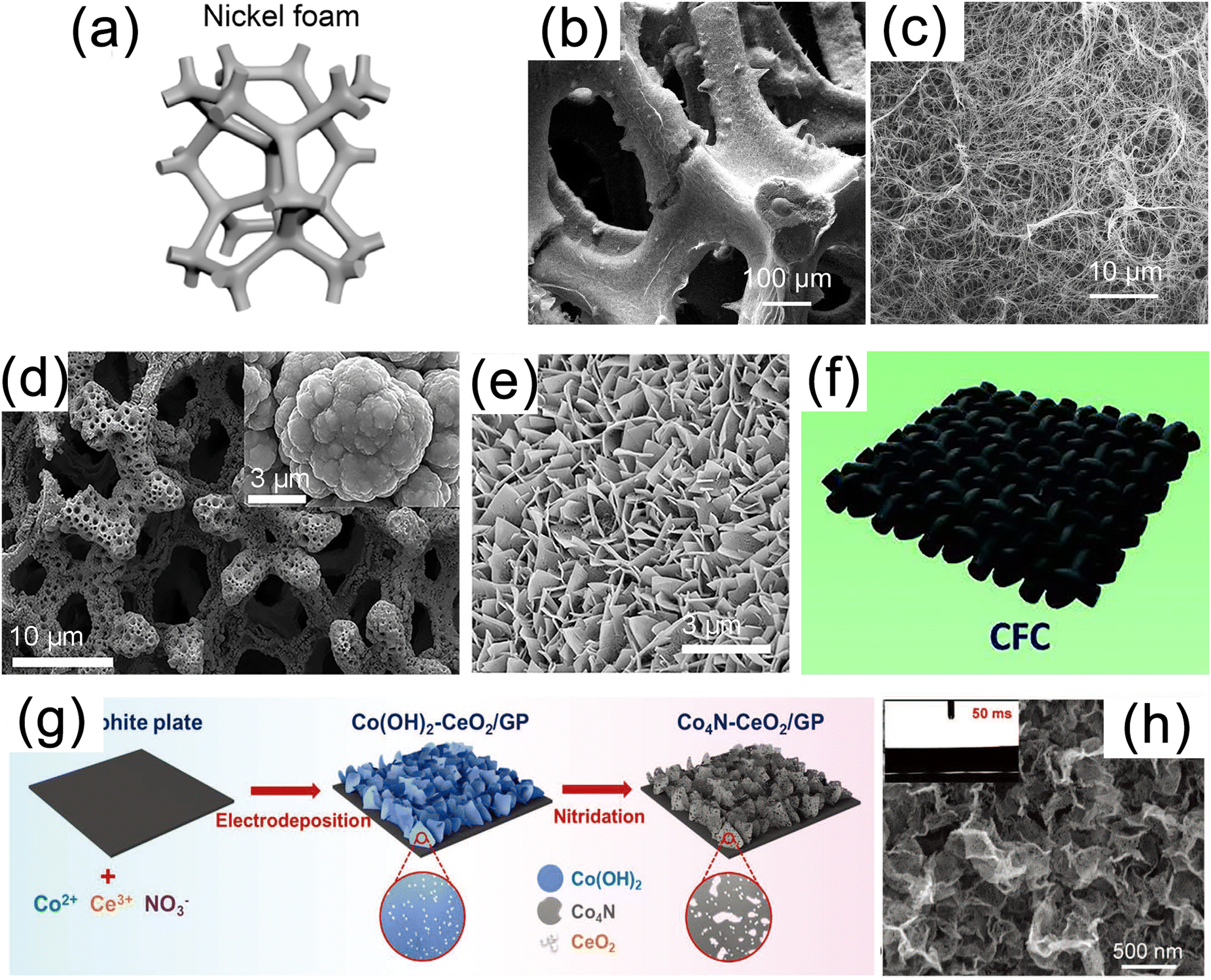 | ||
| Fig. 4 (a) Schematic diagram of nickel foam (NF). (b) Low and (c) high magnification scanning electron microscopy (SEM) images of MoS2/Ni3S2 NW-NF. Reproduced with permission.43 Copyright 2020, Elsevier. (d) SEM image of Ni3N/Ni/NF. Reproduced with permission.120 Copyright 2018, Springer Nature Publishing Group. (e) SEM image of mesoporous networks of NiCo@C nanosheets on electrode surface. Reproduced with permission.121 Copyright 2021, Springer Nature Publishing Group. (f) Schematic diagram of carbon fiber paper (CFP) substrate. Reproduced with permission.127 Copyright 2019, Wiley-VCH. (g) Schematic illustration for synthesis and (h) SEM image of the Co4N–CeO2/GP electrode. Reproduced with permission.58 Copyright 2020 Wiley-VCH. | ||
 | ||
| Fig. 5 (a) Schematic and (b) and (c) scanning electron microscopy (SEM) images of the synthesis of nickel nanowires deposited on nickel mesh (NM). Reproduced with permission.130 Copyright 2021, Elsevier. (d) Schematic, optical photograph and SEM of in situ growth of CrO2–0.16IrO2 catalyst on porous Ti plate. Reproduced with permission.131 Copyright 2023, The Royal Society of Chemistry. (e) and (f) Schematic and SEM image of prepared metal-doped MoSe2 deposited on Si substrate. Reproduced with permission.60 Copyright 2019, American Chemical Society. (g) Photograph of indium doped tin oxide glass (ITO) substrate loaded with DR-MoS2 catalyst and (h)–(j) SEM images. Reproduced with permission.132 Copyright 2017,Tsinghua University Press. (k) Schematic of Ni–Mo–B HF (hollow foam) electrode. SEM images of (l) bare melamine sponge, (m) activated melamine sponge, (n) Ni–Mo–B foam electrode, and (o) Ni–Mo–B HF electrode. Reproduced with permission.133 Copyright 2021, Wiley-VCH. | ||
3. Engineering strategy for seawater splitting
In order to avoid or minimize the detrimental effects of high concentrations of Cl− and insoluble precipitates on the performance of seawater electrolysis catalysts, several strategies have been designed. Firstly, the development of high-performance OER catalysts based on the Pourbaix diagram aims to achieve industrial-level OER current density at 1.71 V by maximizing the thermodynamic potential difference between OER and CER in alkaline environments. Selective OER active sites are achieved by optimizing the Gibbs free energy of OER intermediates, allowing for rational modulation or design of catalysts with OER specificity. Furthermore, the establishment of Cl− protection layers prevents the adsorption of high concentrations of Cl− on the active sites of the catalyst. An electrolyte substitution strategy, involving the replacement of electrolyte solutions on the anode side, utilizes small molecule oxidation reactions with thermodynamic advantages to avoid CER, thereby achieving dual effects of energy saving and hydrogen production. Lastly, the self-driven phase transition migration strategy ingeniously combines physical and mechanical processes such as molecular diffusion and interfacial equilibrium with electrochemical reactions to enable direct electrolysis of seawater without desalination for hydrogen production.3.1. Developing high performance OER catalysts
The development of high-performance OER catalysts is currently the most common strategy for achieving efficient hydrogen production.91,136–139 This strategy has been successfully applied in the electrolysis of freshwater, where a wide range of high-performance OER catalysts, including metal hydroxides, nitrides, phosphides, sulfides, and other materials, have been developed. These catalysts enable efficient and stable hydrogen evolution at high current densities. According to Pourbaix diagrams, for these materials to operate in seawater with long-term stability, achieving industrial-level current density below 1.71 V is required to avoid CER. For instance, Huang et al. reported a novel heterostructure catalyst (Ni3N@2M-MoS2) composed of cubic Ni3N and monoclinic MoS2 (Fig. 6a), exhibiting high current density and efficient hydrogen production performance from alkaline freshwater.140 The 2M-MoS2 component acts as an electron reservoir, modulating the electronic states of Ni3N and enhancing catalytic activity through charge transfer and the formation of heterointerfaces (Fig. 6b). The catalyst overcomes the spatial hindrance of hydroxyl/water adsorption/dissociation and optimizes the free energies of various intermediates (H*, OH*, O*, OOH*), demonstrating outstanding catalytic performance at high current densities. Remarkably, OER and HER current densities of 1000 mA cm−2 are achieved at remarkably low overpotentials of 327 and 155 mV, respectively (Fig. 6c and d). Assembled membrane electrolyzers operate at a current density of 1000 mA cm−2 for at least 300 hours with a cell voltage as low as 1.65 V (Fig. 6e). Furthermore, carbon-coated nickel–iron MN alloy nanospheres (FeNi3–N) prepared through phase engineering exhibit remarkable OER activity and stability (Fig. 6f).141 Electrochemical measurements conducted at 1.6 V potential maintain a current density of approximately 775 mA cm−2 with a retention rate of 90.3%, whereas RuO2/C undergoes rapid decay (36%) (Fig. 6g). These findings indicate that operating at ultra-low voltages can prevent the formation of chlorate and perchlorate, providing potential for efficient seawater electrolysis. Nevertheless, it should be noted that excess impurity ions in seawater can form insoluble precipitates that adsorb onto the catalyst surface, blocking active sites and resulting in performance degradation. Thus, high-performance alkaline freshwater OER catalysts are not fully applicable for seawater electrolysis.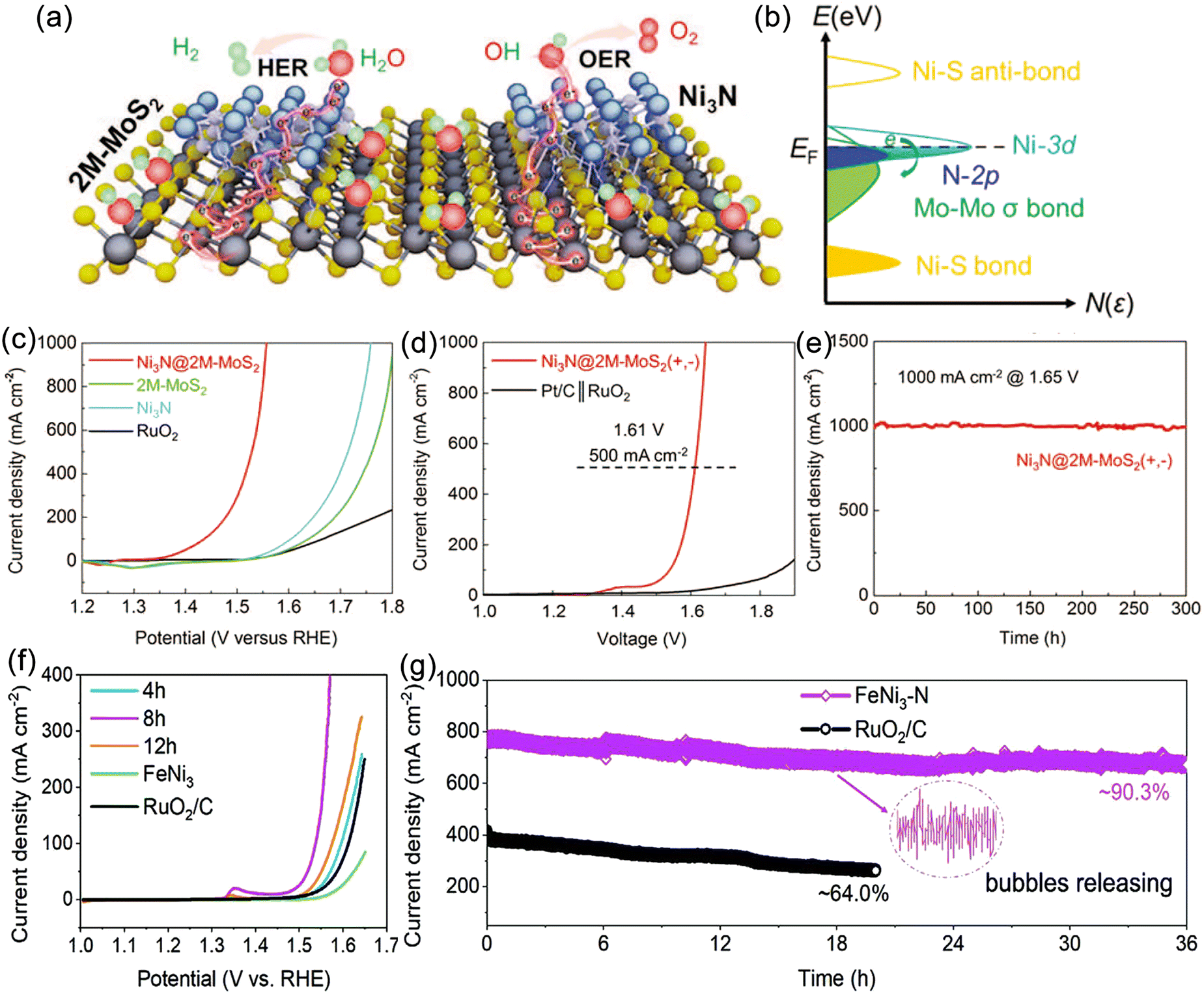 | ||
| Fig. 6 (a) Schematic illustration of water splitting process of the Ni3N@2M-MoS2. (b) Proposed charge regulation effect on manipulating active electronic states in Ni3N@2M-MoS2. (c) Linear sweep voltammetry (LSV) curves of Ni3N@2M-MoS2, 2M-MoS2, Ni3N, and RuO2 in N2-saturated 1 M KOH. (d) Overall water splitting performance of Ni3N@2M-MoS2‖Ni3N@2M-MoS2 and Pt/C‖RuO2 in N2-saturated 1 M KOH solution with a two-electrode configuration. (e) Chronoamperometry (CA) curves of Ni3N@2M-MoS2‖Ni3N@2M-MoS2 at cell voltage of 1.65 V (vs. RHE). Reproduced with permission.140 Copyright 2022, Wiley-VCH. (f) LSV curves (iR-corrected) of FeNi3–N and commercial RuO2/C in 1 M KOH. (g) CA test under a constant potential of 1.60 and 1.65 V (vs. RHE) for FeNi3–N and RuO2/C. Reproduced with permission.141 Copyright 2022, Wiley-VCH. | ||
3.2. Engineering selective OER sites
Optimizing the free energy of reaction intermediates and active sites is an effective strategy to develop catalysts with either a single OER active site or highly selective OER active sites, which can suppress CER. However, the significant intrinsic kinetic advantage of CER has led to slow progress in related research areas. Rossmeisl et al. demonstrated a linear relationship between Cl− and oxygen adsorption energies on MO2 (M = Ir, Ru, Pt, Ti) surfaces.142 The surface phase diagram and Sabatier volcano curve revealed that the overpotential required for CER is generally lower than that for OER. Additionally, the volcano plot of oxygen adsorption energies indicated similar Gibbs free energy losses for OER and CER, implying that improving the selectivity of active sites for OER is impractical when CER dominates. Thus, designing catalysts with a single OER selectivity can break this linear relationship and provide a reliable solution. Patolsky et al. developed a direct seawater electrolysis catalyst with intrinsic chloride-repelling properties, obtained by single-step solid-state thermal treatment of Ni foil in the presence of a sulfur source, resulting in a surface rich in nickel sulfides (Fig. 7a).143 Under neutral conditions, this catalyst exhibited significantly enhanced catalytic activity and stability compared to commercial IrO2 (Fig. 7b). At various current densities, its OER selectivity approached perfection, with no detectable chloride generation (Fig. 7c). During the OER process, rapid reconstruction occurred on the NiS2pSsurfacex, leading to the formation of vinylidene-like sulfur-rich polysulfide species that encapsulate the metal nickel sites (pSn2−/S2− = 2.1), further enhancing chloride resistance. The small S–S bond distance and free space diameter in the NiS2pSx catalyst hindered the adsorption of larger Cl− onto the Ni active sites while allowing the entry of smaller water molecules, resulting in a significant improvement in OER selectivity (Fig. 7d). This work offers a new perspective on the atomic structural design for developing highly OER-selective electrode surfaces. Recently, Sargent et al. improved the OER selectivity of Co3O4 by introducing a series of strong proton-adsorbing cations, including Pd, aiming to facilitate proton adsorption.144In situ synchrotron X-ray absorption spectroscopy (XAS) results showed stable Pd sites during the OER process, indicating that Pd improved the kinetic process of Co sites and reduced the hydrolysis activation barrier, which was further confirmed by deuterium-labelled hydrolysis experiments (Fig. 7e). Benefiting from the strong proton-adsorbing capability of Pd, this catalyst demonstrated stable operation in neutral seawater at a current density of 200 mA cm−2 for 450 hours, with an OER selectivity close to 95% (Fig. 7f).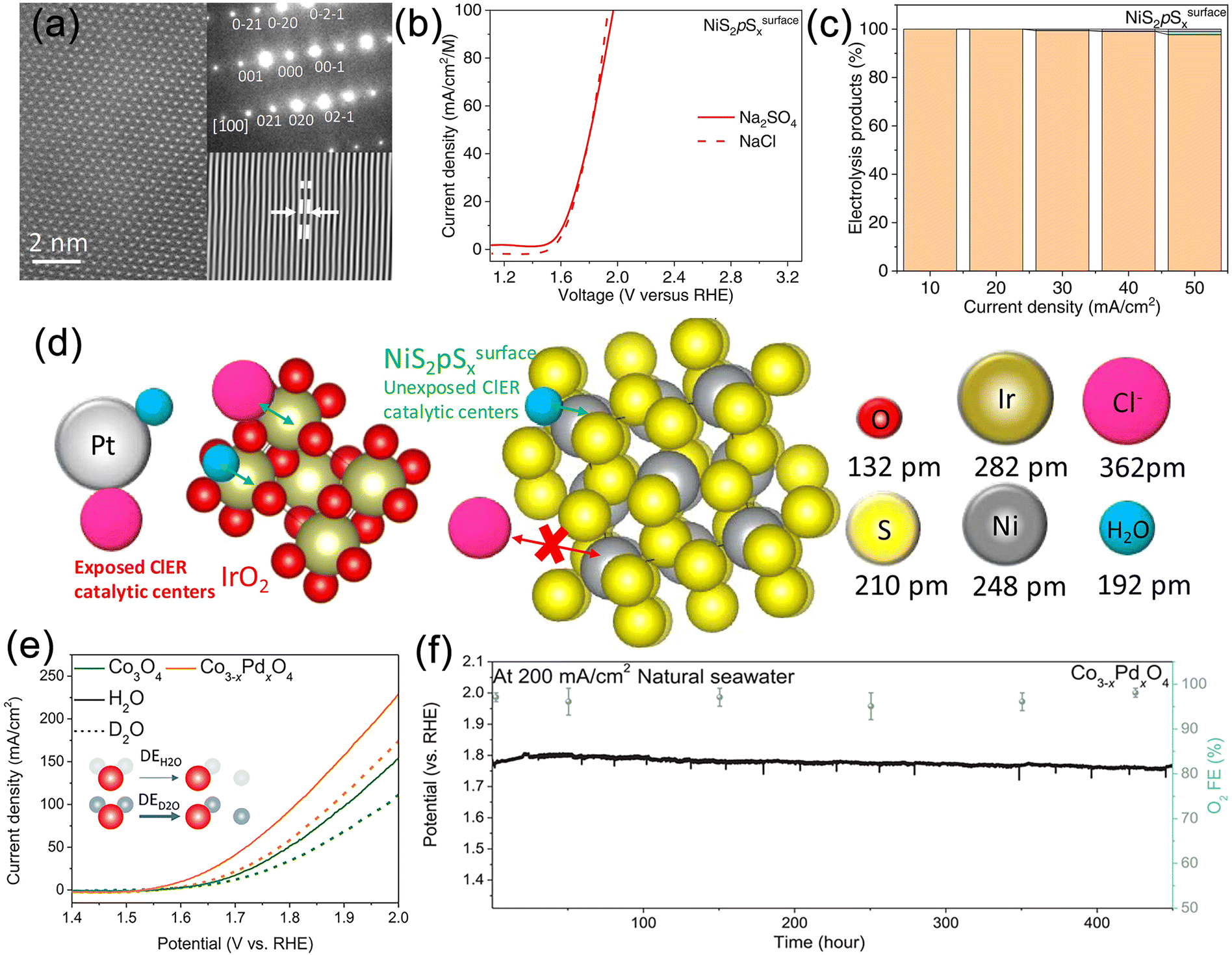 | ||
| Fig. 7 (a) High-resolution transmission electron microscopy (HRTEM) image of NiS2pSsurfacex. (b) Ionic strength normalized comparative oxygen evolution reaction (OER) polarization curves using Na2SO4 and NaCl as electrolytes for NiS2pSsurfacex. (c) Percentage of oxygen, chlorine and hypochlorous acid produced using NiS2pSsurfacex. (d) Schematics showing the possible interaction of Cl− and H2O species with the crystal structure of IrO2 and NiS2pSsurfacex. Reproduced with permission.143 Copyright 2023, Elsevier. (e) Linear sweep voltammetry (LSV) polarization performance obtained in H2O and D2O electrolytes. (f) Stability tests of Co3−xPdxO4 catalysts on MnO2 protected Ni foam substrates at current densities of 200 mA cm−2. Reproduced with permission.144 Copyright 2023, Wiley-VCH. | ||
3.3. Establishing the Cl− protective layers
Building a protective layer on the catalyst surface, without affecting its activity, to prevent Cl− corrosion of active sites or substrate materials has proven to be highly effective. Numerous studies have demonstrated the feasibility of this strategy and showcased good activity and long-term stability on laboratory-scale electrolysis platforms.30,145,146 Various strategies have been developed, including corrosion-resistant coatings, localized environment regulation, anionic charge repulsion, and charge repulsion additives.In 2021, Chen et al. reported a study on enhancing the Cl− corrosion resistance of metal electrodes by adding sulfate salts to the electrolyte.147 The results showed that corrosion resistance increased gradually with the addition of sulfate salts (Fig. 8a). During the reaction, SO42− were adsorbed onto the catalyst surface, forming an electrostatic repulsion layer to hinder the entry of Cl−. Molecular dynamics simulations indicated that the Cl− concentration within 1 nm of the catalyst surface decreased to 0.99 mol m−2 with the introduction of sulfate salts, compared to 3.46 mol m−2 without sulfate salts, further confirming the repulsive effect of sulfate ions. In situ electrochemical infrared and Raman spectroscopy results demonstrated that the introduced sulfate anions did not hinder the OER reaction and provided sufficient active sites at high potentials. Furthermore, this strategy can be extended to other material systems and can significantly enhance their durability in seawater (Fig. 8b and c). A study on IrO2 reveals that coating its surface with a layer of MnOx effectively prevents the diffusion of Cl− into the intermediate layer of the Ir substrate while maintaining water permeability.148 Similarly, a protective effect is also achieved by coating common OER catalysts (CoOx and NiFeOx) with a Cl− selective CeOx layer.149
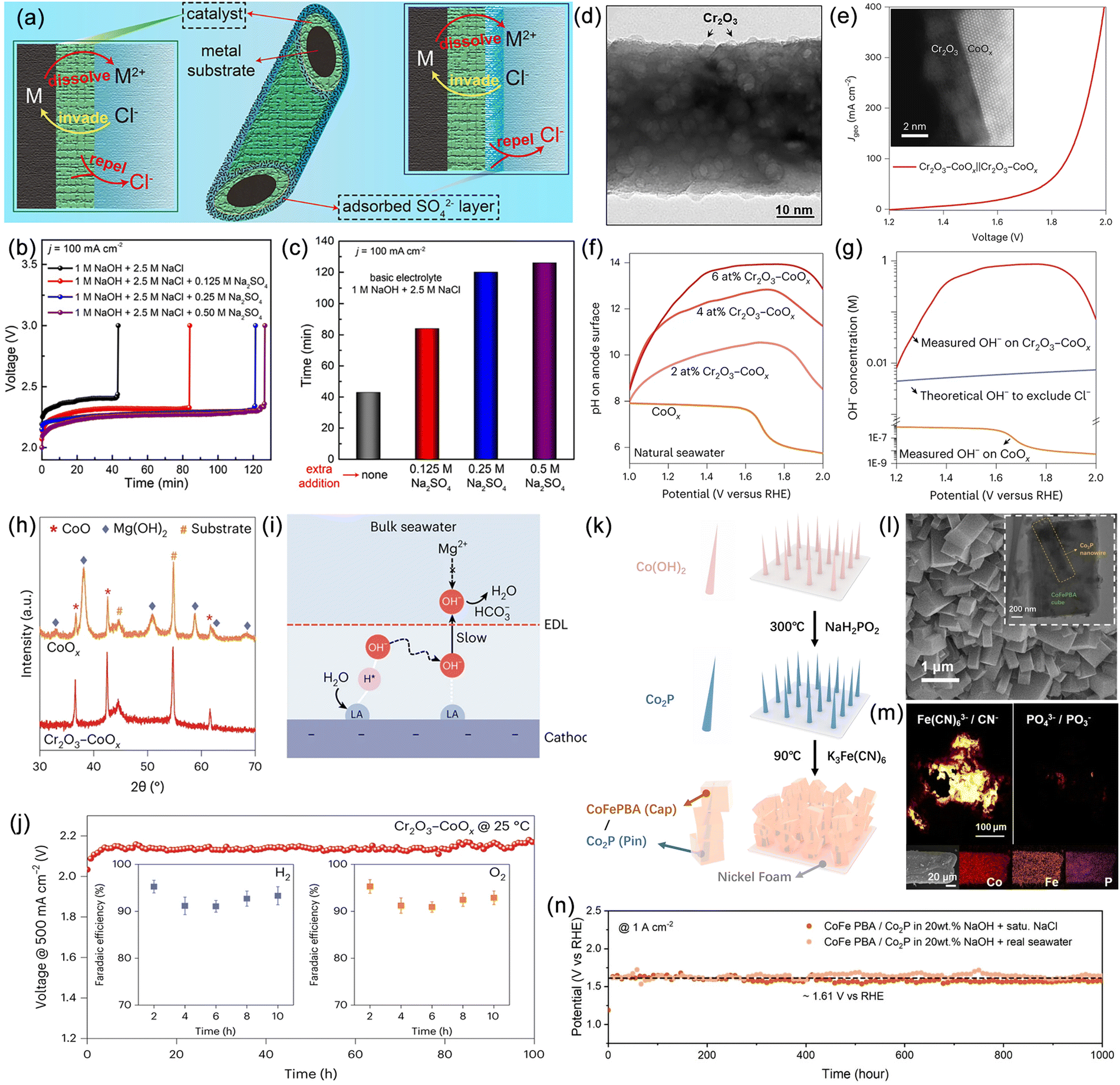 | ||
| Fig. 8 (a) Schematic of the optimization of the catalyst and electrolyte to protect the metal substrate from Cl− corrosion. (b) Stability tests of pure Ni foam in different ratios of Na2SO4 electrolyte. (c) The durability time of Ni foam in different electrolytes. Reproduced with permission.147 Copyright 2021, Wiley-VCH. (d) Transmission electron microscopy (TEM) images and (e) overall seawater splitting performance of Cr2O3–CoOx. (f) Measured pH values on CoOx and Cr2O3–CoOx anode surfaces at different potentials. (g) Measured OH− concentration and theoretical concentration of excess OH− required to resist Cl−. (h) X-ray diffractometry (XRD) spectra of CoOx, Cr2O3–CoOx and (i) Schematic diagram of local alkaline microenvironment generation. (j) Chronopotentiometry (CP) curve of the Cr2O3–CoOx seawater electrolyzer, with the inset showing the faraday efficiency (FE) of H2 and O2. Reproduced with permission.31 Copyright 2023, Springer Nature Publishing Group. (k) Schematic route for the synthesis of CoFePBA/Co2P electrode. (l) Scanning electron microscopy (SEM) and TEM images of CoFePBA/Co2P. (m) Time of Flight Secondary Ion Mass Spectrometry (TOF-SIMS) mapping for Fe(CN)63−/CN− and PO43−/PO3−, and energy dispersive spectroscopic (EDS) elemental mapping. (n) Chronopotentiometry (CP) response at 1 A cm−2 in 20 wt % NaOH + saturated NaCl and 20 wt % NaOH + real seawater. Reproduced with permission.150 Copyright 2023, Wiley-VCH. | ||
Recent work by Ling and Qiao et al. represents a significant advancement in the Cl− protection layer strategy.31 They achieved localized modulation of the catalyst double layer by introducing a Lewis acid layer on the surface of CoOx (Fig. 8d and e), enabling dynamic splitting of water molecules and capturing hydroxide anions. In a neutral seawater electrolyte, the presence of the Lewis acid layer led to the enrichment of OH− on the catalyst surface, with a concentration much higher than that of Cl−, forming a charge repulsion layer (Fig. 8f and g). The strong binding between the Lewis acid layer and OH− suppressed the generation of OH− within the double layer, preventing the formation of insoluble precipitates, while a small amount of desorbed OH− was neutralized by buffering ions (Fig. 8h and i). Assembled flow-type seawater electrolysis devices could achieve an industrial-level current density of 500 mA cm−2 at 25 °C (Fig. 8j). This strategy of manipulating the local microenvironment of the catalyst provides a new direction for preventing the corrosion of catalysts by Cl−. During seawater electrolysis, the concentration of Cl− gradually increases, leading to a significant degradation in catalytic performance. Targeting this problem, Sun et al. synthesized an anode material, cobalt ferrocyanide/cobalt phosphide (CoFePBA/Co2P) with a Cap/Pin structure (Fig. 8k), which demonstrates stable operation at a current density of 2 A cm−2.150 The PO43− and Fe(CN)63− densely covering the catalyst surface drastically reduced the adsorption of Cl- through electrostatic repulsion (Fig. 8l and m), thus achieving an ultra-long stability of 1000 hours (Fig. 8n). The aforementioned strategy of incorporating an anti-chloride layer thus holds significant importance in seawater electrolysis for hydrogen production. This strategy is characterized by its well-defined mechanism, substantial impact, and straightforward application.151,152
3.4. Alternating electrolyte strategy
Hydrazine hydrate, urea, sulfur, furfural, and alcohols are small organic molecules that possess thermodynamic potentials greater than that of OER. Therefore, replacing the anodic OER reaction in seawater electrolysis can not only reduce energy consumption but also avoid the negative effects caused by Cl−. Moreover, coupling these alternative reactions with pollutant degradation and the conversion of low-value chemicals can provide additional environmental and economic benefits. However, this approach imposes higher requirements on the membrane electrode, especially in terms of ion permeability, chemical inertness, and lifespan. Qiu et al. proposed a novel seawater-hydrazine hydrate coupled hydrogen production system using NiCo/Mxene as the catalytic electrode.121 In comparison with conventional OER, the voltage drop for HzOR at a current density of 500 mA cm−2 was reduced by 1.543 V (Fig. 9a). The coupled dual-electrode hydrogen production system efficiently generated hydrogen at an applied voltage range of 0.7–1.0 V, achieving a hydrogen production rate of 9.2 mol h−1 g−1 of catalyst. Furthermore, the system exhibited excellent energy efficiency with an electrical consumption of only 52% compared to commercial alkaline water electrolysis (Fig. 9b and c). After a continuous hydrogen production test at an industrial-level current density for 140 hours (Fig. 9d), no chloride generation was detected. Additionally, the system realized self-driven mixed seawater hydrogen production by utilizing green electricity (Fig. 9b). Subsequently, by replacing OER with the sulfide oxidation reaction (SOR), which exhibited a thermodynamic electrode potential of only −0.48 V (Fig. 9e), large-current continuous hydrogen production was achieved at a significantly lower voltage than 1.23 V, simultaneously facilitating sulfide degradation.153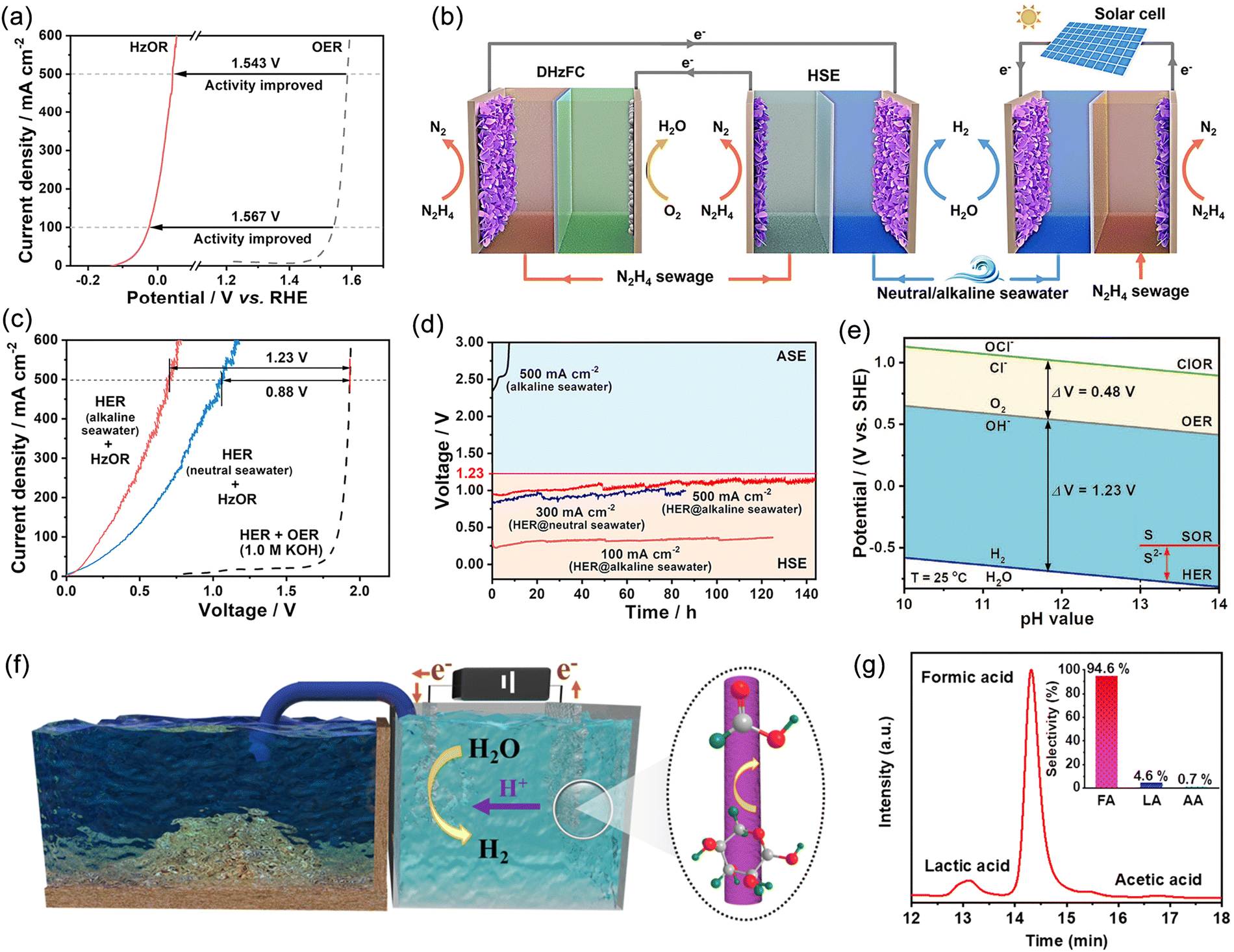 | ||
| Fig. 9 (a) Comparison of anodic potentials of hydrazine oxidation reaction (HzOR) and oxygen evolution reaction (OER) in 1 M KOH. (b) Schematic diagram of a self-powered hydrogen production system integrating a hybrid seawater electrolyzer (HSE) with a low voltage direct hydrazine fuel cell (DHzFC) or solar cell. (c) Comparison of linear sweep voltammetry (LSV) curves between HSE and alkaline seawater electrolyzer (ASE) using neutral or alkaline seawater as electrolyte. (d) Endurance test of HSE under different current and electrolyte conditions. Reproduced with permission.121 Copyright 2021, Springer Nature Publishing Group. (e) Pourbaix plots of sulfion oxidation reaction (SOR), hydrogen evolution reaction (HER), OER and chlorine evolution reaction (CER) under alkaline conditions. Reproduced with permission.153 Copyright 2022, Wiley-VCH. (f) A schematic illustration of the hybrid seawater electrolysis system. (g) High performance liquid chromatography (HPLC) traces of products; the inset exhibits the selectivities of formic acid (FA), lactic acid (LA), and acetic acid (AA). Reproduced with permission.154 Copyright 2023, The Royal Society of Chemistry. | ||
Wang et al. reported a bipolar hydrogen production system via low-potential anodic oxidation of biomass-derived aldehydes coupled with cathodic HER.155 This system can produce hydrogen simultaneously at the cathode and anode at an ultra-low cell voltage of ∼0.1 V. Notably, the aldehydes used (5-hydroxymethylfurfural and furfural) are abundant and readily obtainable biomass-derived compounds that can be extracted from various sustainable biomass feedstocks such as starch, cellulose, and hemicellulose. The oxidation products (corresponding carboxylic acids) serve as high-value chemicals and important precursors for a range of products, including pharmaceutical molecules, fragrances, and sustainable polymers. The energy input per cubic meter of hydrogen in this system is only about 0.35 kW h, which is approximately 1/14th of the conventional water electrolysis value (around 5 kW h). This work provides a promising pathway for the safe, efficient, and scalable production of high-purity hydrogen through electrolyte substitution strategies. Based on this design principle, a hybrid electrolyzer coupling the xylose oxidation reaction (XOR) has also been developed, achieving a remarkable 94.6% selectivity for formic acid with exceptional durability (Fig. 9f and g).154 These studies cleverly circumvent the detrimental effects of Cl− in seawater on the electrodes and offer new insights for the sustainable utilization of marine hydrogen resources.
3.5. Self-driven phase transition migration
In 2022, Xie et al. proposed a novel approach that combines physical mechanics with electrochemistry to address the challenges of direct seawater electrolysis for hydrogen production.32 This innovative concept establishes a new principle and technology for on-site hydrogen generation from seawater without desalination (Fig. 10a). The method employs concentrated KOH (30 wt%) as the self-wetting electrolyte and utilizes a hydrophobic porous PTFE membrane as the gas pathway interface. Water vapor generated by seawater evaporation diffuses through the gas-permeable membrane to the Saturated Depolarizer Electrode (SDE) side under pressure gradient and is absorbed and liquefied. The porous structure of the PTFE membrane plays a crucial role in the seawater electrolysis process by providing a micrometer-scale gas diffusion path for directional water vapor transport while completely preventing liquid penetration. The fluorinated structure of PTFE with low surface energy forms superhydrophobic isolation domains that effectively inhibit the migration of ions such as Cl−, Mg2+, and Ca2+. The electrolysis process maintains a pressure differential between the two sides, where water consumption equals the migration volume, establishing a new thermodynamic equilibrium across the PTFE membrane, thereby providing the required freshwater for electrolysis (Fig. 10b and c). In a high-throughput seawater electrolyzer assembled in Shenzhen Bay, a hydrogen production current density of 400 mA cm−2 was achieved at 2.3 V. Dynamic GC analysis detected no Cl− and nearly 100% faradaic efficiency for OER. Long-term stability tests revealed negligible impurity ions detected in the SDE. COMSOL simulation results demonstrated the system adaptability to seasonal variations and various ocean depths, remaining self-driven even at 0 °C and a depth of 2000 meters. A demonstration device comprising 11 units with a geometric area of 3696 cm2 underwent stable operation for 3200 hours in Shenzhen Bay without electrode corrosion or membrane wetting (Fig. 10d). This technology has been successfully implemented, and collaborations with institutions such as Dongfang Electric Corporation have been established to develop the first offshore wind-powered on-site seawater electrolysis platform. Despite the groundbreaking progress achieved by this technology, there is still a long way to go for its large-scale industrial implementation. There are many challenges that need to be addressed, such as surface modification, improvement of tensile strength, impact resistance, fatigue resistance, enhanced corrosion resistance, and inhibition of marine biofouling. | ||
| Fig. 10 (a) Schematic diagram of a typical seawater electrolysis system (SES). (b) Migration mechanisms for the corresponding water purification and migration processes. (c) Optical image of a scaled-up SES for direct seawater electrolysis. (d) Durability test for the scaled-up seawater electrolysis system at 250 mA cm−2. Reproduced with permission.32 Copyright 2022, Springer Nature Publishing Group. | ||
4. MNs electrocatalysts
4.1. Structural and compositional properties of MNs
MNs are usually crystalline solids.78 In general, the most common structures for simple MNs include face centred cubic (fcc), hexagonal close packed (hcp) and simple hexagonal (hex) structures.156 The crystal structure, which refers to the periodic arrangement of atoms in a solid material, plays a crucial role in determining its properties.157–159 However, the effect of crystal structure on the intrinsic catalytic activity of MNs has not been extensively explored. This is primarily due to the fact that, once the constituent elements and compositions of MNs are fixed, most compounds have only one thermodynamically favorable structure that remains stable over a wide range of temperatures and/or pressure conditions.160 In this section, we will review the physicochemical properties and catalytic activity of MNs, focusing on their crystal structure.WN possesses advantageous properties such as high conductivity, thermal stability, corrosion resistance, and exceptional hardness.161 It has a typical fcc structure,162 characterized by high packing density, which provides a greater number of active sites and enhances the probability of reactant adsorption. It has been demonstrated that a biphasic nitride catalyst, Co4N–WNx, exhibits high electrical conductivity and abundant active sites, surpassing the catalytic performance of Pt.163 Chou et al. conducted a comprehensive investigation of the phase transition process of MoNx by subjecting bulk 2H-MoS2 to nitridation and exfoliation under high-temperature ammonia gas conditions.164 The study revealed a strong correlation between the resulting products and the reaction temperature. When the temperature exceeds 820 °C, Mo5N6 began to decompose into δ-MoN. As the temperature increases, MoN underdoes the following phase transition process: δ-MoN → γ-Mo2N → β-Mo2N → Mo. In compared with γ-Mo2N, δ-MoN has higher catalytic activity.165–167 The fcc crystalline system of the δ-MoN structure typically has higher electron density and electron-rich effects. These electron-rich effects can enhance the interaction between the catalyst and the reactants, leading to a reduction in the activation energy of the reaction and thus improving catalytic activity. In recent years, Group VIII elements Fe, Co, and Ni have exhibited remarkable performance in electrochemical water splitting and have been extensively studied.168 The nitridation of iron sources results in various iron nitride phases, such as Fe2N, Fe3N, Fe4N, and Fe16N2. Among these, the hcp ε-Fe3N has been extensively studied and demonstrates outstanding catalytic performance and magnetism.169,170 Nitriding cobalt-containing precursors can produce various phases of cobalt nitrides, such as tetragonal Co2N, δ-Co3N, and cubic γ-Co4N. The electrical conductivity and OER catalytic activity of these phases follow the order Co2N < Co3N < Co4N, with Co4N exhibiting superior OER activity due to its high conductivity.171
Compared to binary MNs, ternary MNs possess more favorable electronic modulation mechanisms and are able to achieve high current densities of 1000 mA cm−2, which is comparable to performance achieved through heterostructure tuning.172 For example, Ni3FeN, has the anti-perovskite structure (a type of fcc) with iron atoms occupying the corners of the crystal cell and nitrogen atoms filling the octahedral voids, resulting in a compact arrangement and high coordination of metal and nitrogen atoms. This structure provides the catalyst with abundant active sites and a dense surface structure.173–175 Similar structures include Ni3CuN and Ni3lnN.176 There are also bimetallic alloy structures where different metal atoms alloy with the host metal to form alloyed nitrides. Due to the differences in lattice strain, atomic radius, and electronegativity of different metal atoms, it is possible to adjust the electronic structure and metal conductivity of the material while affecting the adsorption behavior of intermediate products.177–179 Designing new synthesis routes for these phases and studying their properties and structural polymorphism is an active area of current research.
Capturing the charge transfer and fleeting intermediates during electrocatalytic reactions poses significant challenges. This leads to uncertainty regarding active sites and ambiguity in the corresponding catalytic mechanisms. Therefore, the combined approach of experimental characterization and density functional theory (DFT) calculations to identify the active sites involved in various electrocatalytic reactions can be considered a relatively direct strategy. Descriptors such as binding energy, Gibbs free energy (ΔG), d-band center, charge difference, and density of states (DOS) are combined to provide a comprehensive description that effectively illustrates the relationship between the structure of the MNs and their performance.180,181
4.2. Quantification and role of N
As early as 1967, Meyer et al. reported the quantitative determination of N in refractory MNs using a combination of tube furnace oxidation and GC.182 N type and content can be determined semi-quantitatively by N 1s spectroscopy. It is essential to note that X-ray photoelectron spectroscopy (XPS) is a near surface analysis technique and the accuracy of the results is highly dependent on instrument calibration and sample preparation.183,184 The quantitative determination of N content can also be obtained through Rietveld analysis, which involves fitting crystal structure parameters to acquire precise crystallographic information.185However, since the scattering coefficients of N and O atoms are so close that X-ray techniques cannot accurately distinguish between them, neutron diffraction techniques are required.186 Although this method has been demonstrated, few studies have provided specific numerical values for the N content of MNs, possibly due to stringent sample crystallinity and size requirements. Recently, Li and colleagues employed a two-step method involving acid digestion and HPLC/UV-vis (Ultraviolet-visible) analysis to quantify the N content in commercial metal-based nitride reduction catalysts (Bi2O3 and Fe).187 This method also provides a valuable reference for the accurate quantification of N content in MNs.
The impact of the degree of N substitution on the oxidation state and electronic structure of V was explored by An et al.188 XPS results revealed that VN-900 (NH3 treatments at 900 °C) exhibited a lower V oxidation state, and electron transfer occurred when N replaced O. This electron transfer process resulted in a reduction of V oxidation state. Furthermore, through extended X-ray absorption fine structures (EXAFS) analysis, a significant difference in the coordination environments was observed between VN-550 (NH3 treatments at 550 °C) and VN-900. The coordination environment in VN-550 is distorted due to N substitution, exhibiting lower symmetry, whereas the coordination environment in VN-900 displays higher symmetry. In the Co–N system, as the N content decreases, the intrinsic conductivity of Co2N, Co3N, and Co4N increases. Lower N content may lead to fewer electron scattering or confinement, making it easier for electrons to transfer from Co to N, thus enhancing conductivity.189
4.3. Surface reconstruction of MNs during reactions
Numerous studies have shown that the surfaces of MNs, and related metal phosphides (MPs), metal sulphides (MSs), metal borides (MBs) and metal carbides (MCs) undergoes significant change during reactions, especially at high potentials.190 The reconstructed surface typically consists of oxides or hydroxides, which are considered to be the true sites of catalytic activity. In the case of electrocatalysis, a typical heterogeneous process, the adsorption and desorption behavior of intermediates critically depend on the compositional structure of the catalyst surface. Consequently, a comprehensive analysis of reconstructed surfaces and an exploration of the active origin at the molecular and atomic levels are of importance. For MNs, the onset of oxide peaks is typically observed in the range of 1.0–1.5 V vs. RHE, corresponding to the initiation potential for water oxidation, which is considered indicative of surface reconstruction.191,192 Low-valent metal species are oxidized to higher valent states and exhibit genuine catalytic activities.Yang et al. successfully synthesized a novel ternary MN, Ni2Mo3N, with excellent OER activity through the soft-urea method.85 In the synthesis process, urea, as the nitrogen source, formed covalent bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O → M) with the metals in the gel matrix (Fig. 11a–e). Ni2Mo3N exhibited lower overpotential and Tafel slope compared to commercial RuO2 and other binary MNs (Mo2N and Ni3N), indicating superior kinetic behavior. Moreover, this catalyst displayed promising practical application potential as it was coupled with FePc (Fe pythalocyanine) to construct a rechargeable zinc–air battery (ZAB), showing outstanding long-term cyclic stability (Fig. 11f). Characterization of the post-test samples revealed the formation of an amorphous oxide layer of approximately 2–5 nm thickness on the catalyst surface (Fig. 11g and h). This layer was oxygen-rich compared to the interior region and had lower Mo content. Due to the high potential of OER, an amorphous SOAL (surface-oxide-rich activation layer) formed in situ on the Ni2Mo3N surface, preventing further oxidation of the internal nitride while providing potential active sites. Ar plasma etching XPS further confirmed this, showing high-valent Ni species on the surface after OER. Ni reappeared after 30 s of etching, suggesting the thinness of the generated SOAL. In contrast, Mo–N bonds remained, indicating that only Ni underwent noticeable oxidation. DFT results indicated that the surface Mo atoms acted as electron pumps for the oxide layer, transferring charges to the surface and ensuring the continuity of OER. The multifunctional electrocatalyst Co3Mo3N, demonstrating high OER, HER, and ORR activity, exhibited similar characteristics (Fig. 11i).193
O → M) with the metals in the gel matrix (Fig. 11a–e). Ni2Mo3N exhibited lower overpotential and Tafel slope compared to commercial RuO2 and other binary MNs (Mo2N and Ni3N), indicating superior kinetic behavior. Moreover, this catalyst displayed promising practical application potential as it was coupled with FePc (Fe pythalocyanine) to construct a rechargeable zinc–air battery (ZAB), showing outstanding long-term cyclic stability (Fig. 11f). Characterization of the post-test samples revealed the formation of an amorphous oxide layer of approximately 2–5 nm thickness on the catalyst surface (Fig. 11g and h). This layer was oxygen-rich compared to the interior region and had lower Mo content. Due to the high potential of OER, an amorphous SOAL (surface-oxide-rich activation layer) formed in situ on the Ni2Mo3N surface, preventing further oxidation of the internal nitride while providing potential active sites. Ar plasma etching XPS further confirmed this, showing high-valent Ni species on the surface after OER. Ni reappeared after 30 s of etching, suggesting the thinness of the generated SOAL. In contrast, Mo–N bonds remained, indicating that only Ni underwent noticeable oxidation. DFT results indicated that the surface Mo atoms acted as electron pumps for the oxide layer, transferring charges to the surface and ensuring the continuity of OER. The multifunctional electrocatalyst Co3Mo3N, demonstrating high OER, HER, and ORR activity, exhibited similar characteristics (Fig. 11i).193
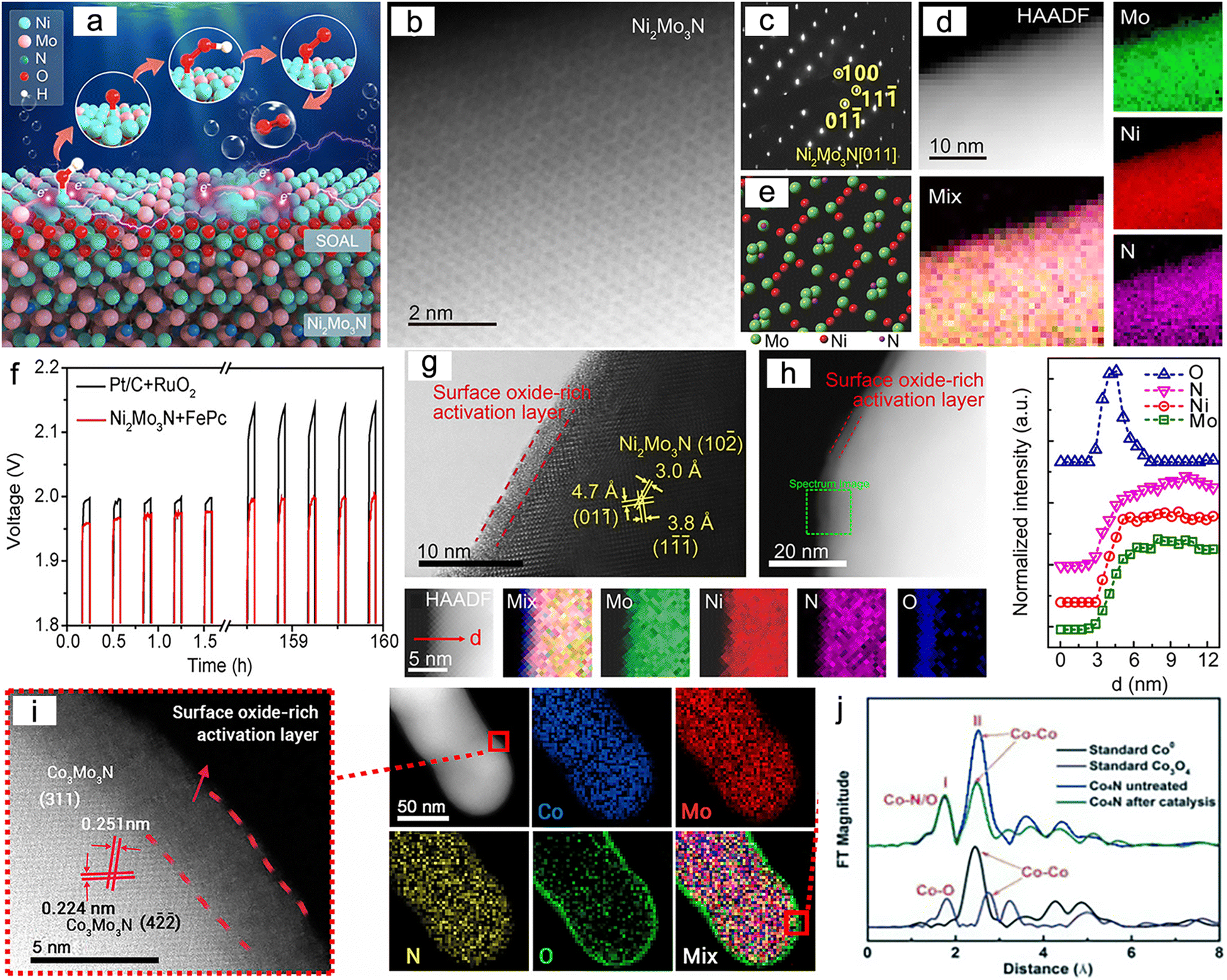 | ||
| Fig. 11 (a) Schematic of the surface-oxide-rich activation layer (SOAL). (b) High-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image, (c) Selected-area electron diffraction (SAED) pattern, (d) atomic structure and (e) corresponding electron energy-loss spectroscopy (EELS) elemental mapping of Ni2Mo3N. (f) Charge/discharge cycling of Zn–air battery based on Ni2Mo3N + FePc (Fe pythalocyanine) and Pt/C + RuO2 at 2 mA cm−2. (g) High-resolution transmission electron microscopy (HRTEM) image of Ni2Mo3N after oxygen evolution reaction (OER). (h) HAADF-STEM images with EELS mapping and line-scan profiles of the SOAL layer. Reproduced with permission.85 Copyright 2020, Wiley-VCH. (i) HRTEM image and EELS element mapping of Co3Mo3N after the OER. Reproduced with permission. Copyright 2021, Elsevier. (j) Fourier transform-extended X-ray absorption fine structure (FT-EXAFS) spectra of Co, Co3O4, and Co4N before and after 1000 potential cycles. Reproduced with permission.194 Copyright 2015, Wiley-VCH. | ||
Wu et al. achieved in situ growth of Co4N nanowire arrays on a CC substrate for OER.194 The authors proposed a two-step reconstruction process for Co4N during the reaction. Initially, the surface Co species undergo partial oxidation to trivalent species (CoOOH). Subsequently, as the reaction progresses, CoOOH is further oxidized to form CoO2, a more favorable active species for the OER process. In situ XAFS spectra indicated the stable maintenance of the internal Co4N structure. However, a significant decrease in the intensity of the FT (fourier transform) of the Co–Co shell layer might result from the oxidation of the first shell layer Co atoms to form cobalt oxide/oxyhydroxide (CoO/CoOOH) (Fig. 11j). Multi-cycle CV experiments demonstrated that the thickness of the outer CoOx layer does not continuously increase, confirming the protective role of the external oxide layer on the nitride core. The surface of this trimetallic MN OER catalyst undergoes a transformation towards metal hydroxides, forming nanospheres composed of FeOOH and CoOOH.195 After the reaction, the XPS spectrum of the catalyst did not exhibit a peak associated with V, which could be attributed to the conversion of V–N bonds into alkali-soluble oxides (VO2 and V2O5), resulting in its disappearance. In the absence of V, there was no significant surface reconstruction observed for CoFeN. Thus, the authors propose that the leaching of V is pivotal in promoting the surface reconstruction of CoVFeN. Wang et al. proposed the in situ growth of hydroxide layers on the surface of Ni3N@Ni3VN through pre-electrochemical reconstruction.196 The resulting catalysts (NiVN@OOH), benefiting from atomic doping and interface effects, exhibited excellent OER performance. The in situ formation of amorphous oxide layers on the surface of MNs not only provides an abundance of active sites but also serves as a protective shield to prevent further oxidation of the core, thereby facilitating rapid electron transfer pathways.84 Electrochemical pre-activation strategies enable the swift development of highly active hydroxide oxide layers on the surface, resulting in exceptional catalytic performance.87,195,196 Hence, MNs serve as pre-catalysts to some extent, but the high electron transfer ability of their core is also essential to their activity.
Surface reconstruction of MNs mostly occurs during high-potential reactions. Recently, Wang et al. observed bulk-phase reconstruction of perovskite materials on the cathode side for HER, demonstrating good activity and stability.197 However, there has been a lack of research focusing on the surface reconstruction of MNs under low-potential conditions. Utilizing in situ characterization techniques to deeply understand material transformations and identify active sites holds significant implications for the rational design of high-performance MNs electrocatalysts.
4.4. Factors influencing the synthesis of MNs
Numerous metal precursors, including oxides, sulfides, layer-double hydroxide (LDH), metal–organic framework (MOF), can react with NH3 to produce MNs. The chemical stability of metal salts is of paramount consideration. For instance, V2O5 is generally unsuitable for VN synthesis due to its higher surface oxygen content during the nitriding process, leading to the dissolution of VN in the electrolyte.198 In contrast, VCl4 and NH4VO3 serve as more favorable precursors. VCl4 can decompose at lower temperatures, while NH4VO3 is rich in ammonium, facilitating the smooth conversion of precursors into VN. The oxidation state of the metal in the precursor also plays a key role. Hydrated nitrates with oxidizing counterions are preferred because of their high oxidizing power (negative charge), high solubility in water or polar organic solvents, low decomposition temperatures, and consequently higher product purity.199 In addition, the use of amorphous precursors can facilitate the reaction process and reduce the likelihood of chemical separation.200 The pyrolysis temperature and calcination time significantly influence the grain size and crystallinity of Co–N. Excessively high temperatures can lead to particle aggregation, which is detrimental to maintaining a porous structure.201 The work of Chou et al. confirmed that the physical phase also changes with the increase of temperature.164 In conclusion, the above factors affect the morphology and physical phase of the synthesized MNs by influencing the crystal growth, the formation of crystal nuclei and the rate of crystal growth.202 Therefore, optimization of these factors allows tailoring of MNs.4.5. Preparation strategies of MNs
In the past decade, significant progress has been made in the synthesis of nanostructured MNs, particularly with the development of new synthesis routes. The general synthesis pathway of MNs involves the thermal treatment of various metal precursors in the presence of different nitrogen sources, such as NH3, N2, or urea. Currently, the most commonly used method is a two-step synthesis, where the precursor materials are first pre-treated using specific processes (such as hydro/solvothermal methods, template methods, sol–gel methods, magnetron sputtering, and electrodeposition) to obtain the corresponding precursors while simultaneously controlling the surface of the products. Subsequently, thermal treatment is carried out in an atmosphere of N2 or NH3. In the second step, the transition metal oxide (TMO) or hydroxide (TMHO) precursors are reduced by NH3x species and converted into the corresponding MNs.203 This section provides a brief discussion on the synthesis strategies employed for various MNs nanostructures.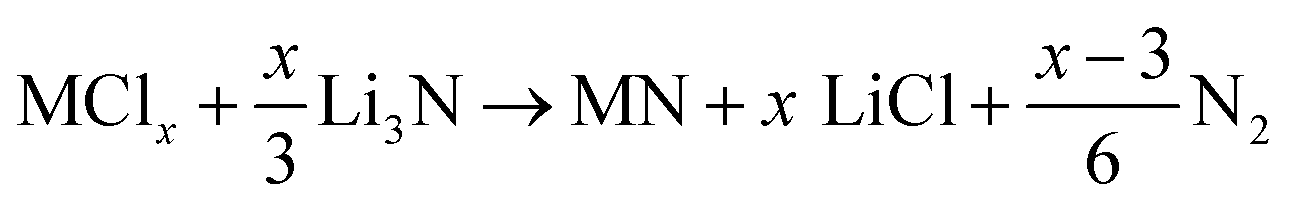 | (4.1) |
VN is obtained through hydrolysis of VCl3 in ammonia solution followed by annealing at 200 °C for 20 hours. The synthesized VN nanoparticles have a quasi-hexagonal shape with an average particle size of approximately 60 nm. Using NH4VO3 as the V source, a 500–600 nm VOOH hollow nanosphere precursor can be obtained through a hydrothermal process at 160 °C for 8 hours, and its macroscopic morphology remains unchanged after annealing (Fig. 12a–c).208 The control of product morphology through hydrothermal parameter variations is a common strategy.209 Different nanostructures of MNs, ranging from zero-dimensional (0D) to 3D structures, have been explored, such as nanowires,210 nanorods,211 nanosheets,212 and nanocages.213
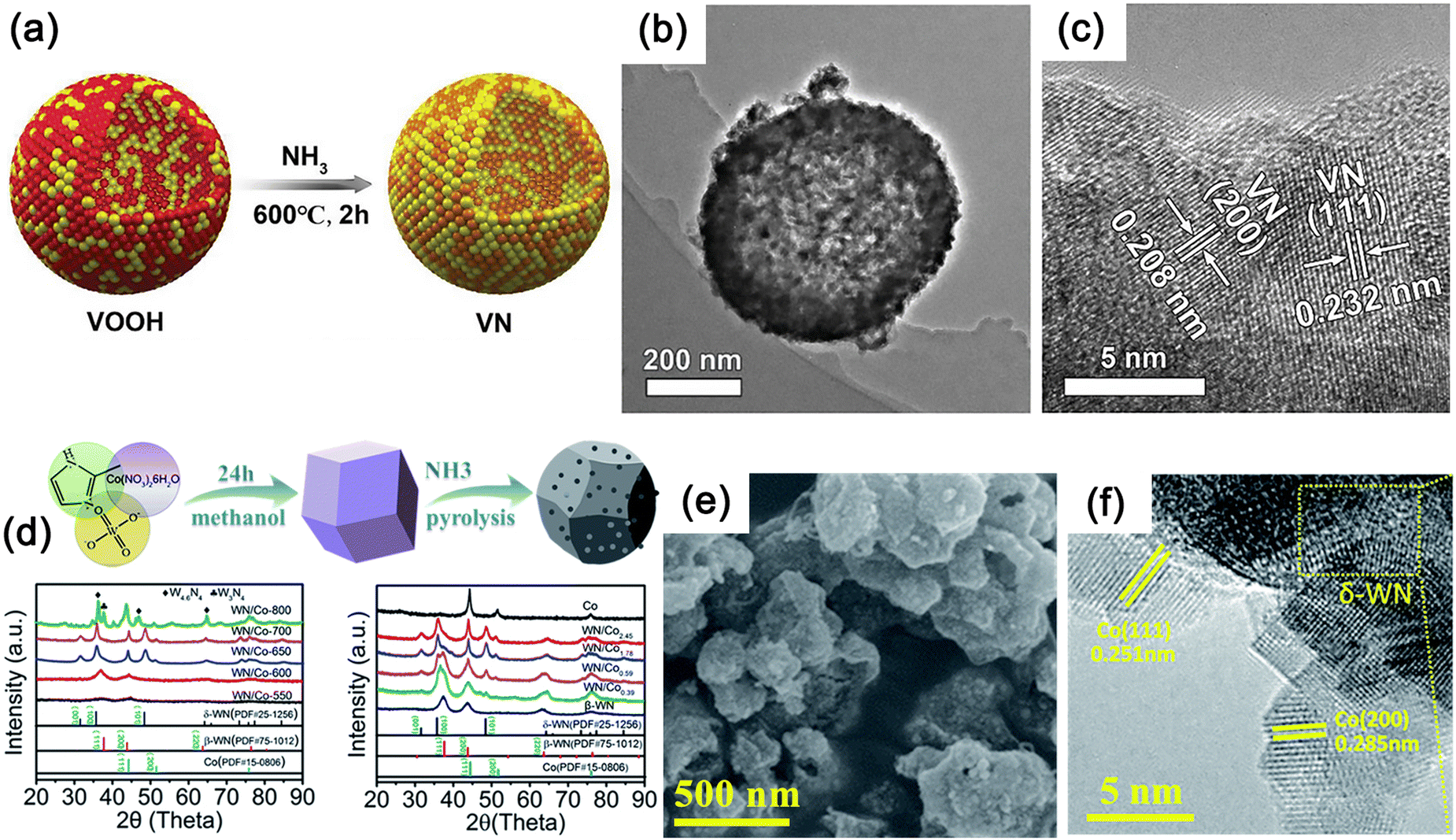 | ||
| Fig. 12 (a) Schematic of the preparation method used for VN and Pd/VN. (b) Transmission electron microscopy (TEM) image showing the 3D VN hollow nanosphere morphology. (c) High-resolution transmission electron microscopy (HRTEM) image of VN. Reproduced with permission.208 Copyright 2019, Wiley-VCH. (d)–(f) The synthesis process and X-ray diffractometry (XRD) patterns of WN/Co hybrids. (e) Scanning electron microscopy (SEM) and (f) HRTEM images of δ-WN/Co2.45 hybrids. Reproduced with permission.178 Copyright 2018, The Royal Society of Chemistry. | ||
WN with a d-band electron configuration similar to Pt shows promising potential as a HER catalyst.178 Micron-sized Co4W6O21(OH)2·4H2O polyhedra anchored to NF were synthesized by a simple hydrothermal method using ammonium paratungstate as W source and cobalt nitrate as Co source. The precursors were then annealed in NH3 atmosphere to transform them into micro/nanostructured Co/WN polyhedra. The choice of W source is crucial for synthesizing highly crystalline precursor of Co4W6O21(OH)2·4H2O.214 WN synthesized by direct ammonolysis at high temperatures is the most common β-WN, while the δ-WN at high temperatures is unstable but may exhibit higher activity. The introduction of metallic Co can stabilize the δ-WN phase by manipulating the phase transition of β-WN. By introducing a Co-based metal organic framework (MOF, ZIF-67) into the tungsten precursor through solvothermal methods, truncated octahedral morphology can be formed due to the MOF container effect, and subsequent NH3 calcination can transform the mixture into δ-WN/Co composite materials (Fig. 12d–f).178
Using direct current reactive sputtering, nanocrystalline nickel nitride (NiN) films with a thickness of 130 nm were successfully synthesized on polished glassy carbon electrodes coated with a titanium adhesion layer (Fig. 13a–c).218 Scanning electron microscopy (SEM) images revealed a closely packed nanorod pyramidal morphology of the thin film. A series of molybdenum nitride thin films, including Mo3N2, Ag–Mo3N2, V–Mo3N2, and Cu–Mo3N2, were fabricated using magnetron sputtering.219 Silicon and stainless steel (SS-304) plates served as substrates for thin film preparation, while Ar and N2 gases were employed as sputtering and reactive gases, respectively (Fig. 13d and e).219 Magnetron sputtering not only enabled the uniform deposition of catalyst thin films over large surface areas but also maintained highly consistent crystal structures and phase purity. In contrast, existing synthetic methods often result in heterogeneous structures with multiple dispersed sizes and compositions.220,221 By adjusting the deposition pressure during magnetron sputtering, it is also possible to control the crystallinity and morphology of CrN, which has a thickness of about 1.1 μm and is strongly correlated to the deposition time.222,223
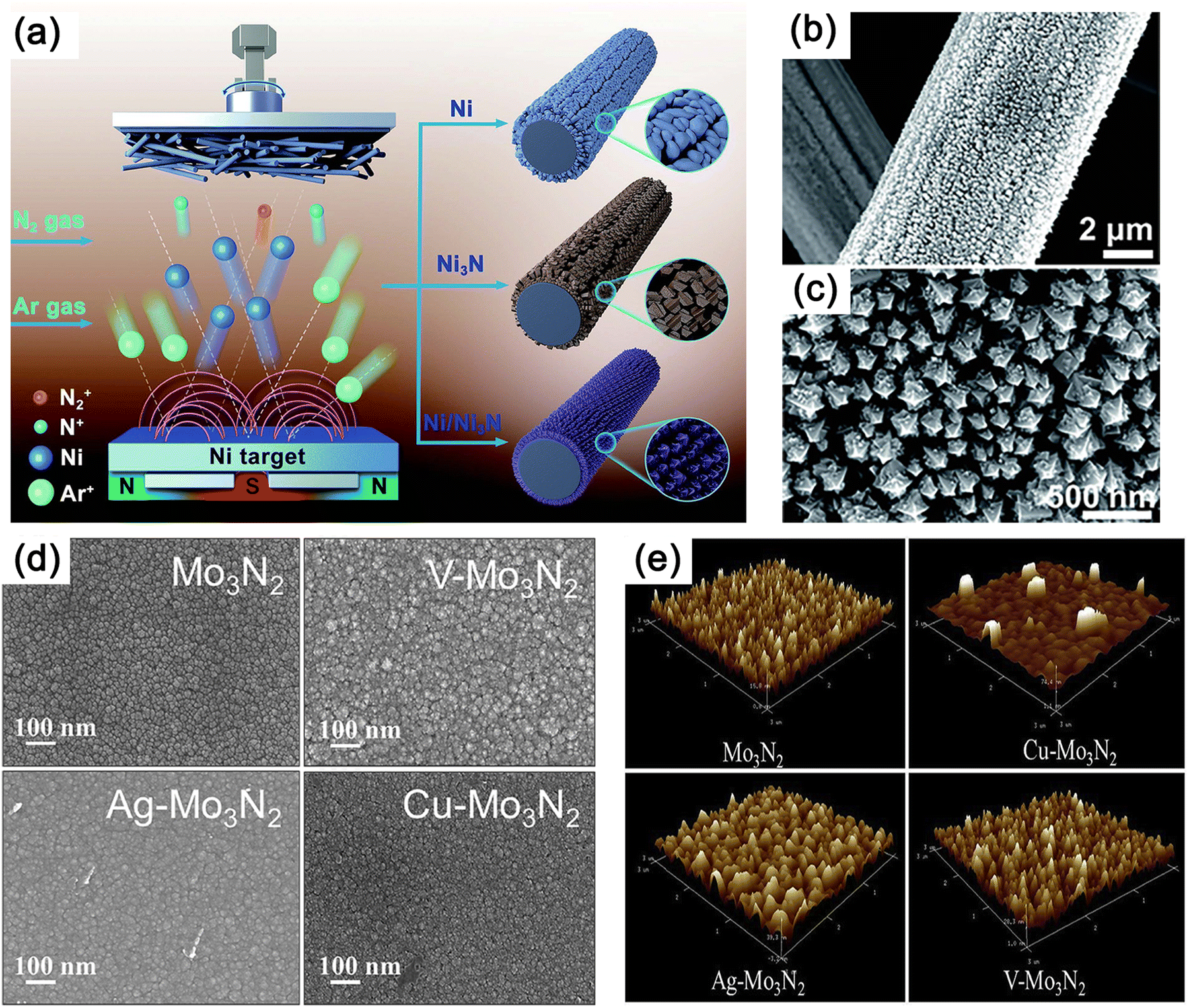 | ||
| Fig. 13 (a) Illustration of synthesis of Ni, Ni3N, and Ni/Ni3N thin films on carbon paper (CP) through magnetron sputtering. (b) Scanning electron microscopy (SEM) and (c) transmission electron microscopy (TEM) images of Ni/Ni3N thin films grown on CP. Reproduced with permission.218 Copyright 2023, Tsinghua University Press. (d) SEM and (e) 3D atomic force microscope (AFM) images of Mo3N2, V–Mo3N2, Ag–Mo3N2 and Cu–Mo3N2. Reproduced with permission.219 Copyright 2018, Elsevier. | ||
Using ion magnetron sputtering, efficient electrocatalysts can be produced by depositing minute quantities of catalytically active materials through a controllable high-purity process. Additionally, from the perspective of green chemistry, the application of ion magnetron sputtering avoids the use of toxic chemical reagents. However, depositing catalytically active particles with specific sizes, structures, and surface properties onto dispersed or porous carriers presents a series of complex challenges. In each case, finding the optimal combination of different process parameters such as power, working gas pressure, distance between the target and substrate, and bias potential on the substrate is necessary. These parameters influence the kinetic energy of deposited particles, i.e., the energy of gas ions acting on the surface.224,225
 | ||
| Fig. 14 (a) Schematic illustration of the synthesis strategy for ordered mesoporous bimetal Ni3FeN. Transmission electron microscopy (TEM) images of (b) KIT-6 silica template and (c) Ni3FeN. Reproduced with permission.174 Copyright 2020, Springer Nature Publishing Group. (d) Preparation of 2D nitrogen rich mesoporous carbon nitride (MCN) and nanoporous GaN (NP-GaN). Reproduced with permission.231 Copyright 2016, Wiley-VCH. | ||
Soft templates lack fixed rigid structures and are formed in situ through intermolecular or intramolecular interactions during the reaction process. Small molecule surfactants or block copolymers (BCPs) are used as structure-directing agents (SDAs) and templates. The composite materials are transformed into mesoporous materials while removing the BCPs through calcination or solvent extraction.232 Currently, there are relatively few reports on the synthesis of MNs using soft templates. A strategy for synthesizing CrN and VN233 catalysts involves using the surfactant Pluronic triblock copolymer as a soft template and phenolic resin as a carbon precursor. During the self-assembly process, these materials form an organized organic polymer intermediate phase, which is subsequently transformed into ordered mesoporous carbon through calcination under an inert atmosphere.234 In the initial step of preparation, the addition of transition metal salts leads to the formation of nanostructured composites of MNs and ordered carbon. Further thermal treatment in an NH3 atmosphere converts them into MN/C materials.
He et al. utilized chronoamperometric deposition to electrodeposit nanoflower NiCo LDHs on hydrophilic carbon fiber paper (CFP), applying an applied potential of −1.0 V (vs. Ag/AgCl) and a total charge of −3.2 C cm−2. After NH3 annealing, they obtained NiCo2N nanosheets with a retained porous and layered structure and a thickness of approximately 20 nm (Fig. 15a and b).177 The conventional synthesis of MNs from TMOs precursors at elevated temperatures (600–800 °C) typically involves the use of hazardous and toxic NH3 as a nitrogen source.177,178,208,214 However, Zhang et al. have introduced a novel approach that circumvents these challenges. They employed a lower reaction temperature (450 °C) and shorter reaction duration (15 minutes) by utilizing N2 plasma treatment on electro-deposited NiMo alloy films (Fig. 15c). This innovative technique resulted in the successful synthesis of a 3D porous NiMoN bimetallic nitride on a CC substrate (Fig. 15d–f). After subjecting the material to 7200 pulse cycles, the CC surface became entirely covered with dense NiMo alloy particles, forming a remarkable 3D porous branched nanostructured bimetallic nitride film (Fig. 15g and h).179
 | ||
| Fig. 15 (a) Schematic representations of synthesis route. (b) Scanning electron microscopy (SEM), and transmission electron microscopy (TEM) images of NiCo2N. Reproduced with permission.177 Copyright 2021, Elsevier. (c) Schematics of the fabrication process of 3D hierarchical porous NiMoN. SEM images of (d) Crabon cloth (CC), (e) NiMoN, and (f) NiMo alloy. (g) TEM image, and (h) X-ray diffractometry (XRD) patterns of Ni3N and NiMoN. Reproduced with permission.179 Copyright 2016, Wiley-VCH. | ||
Niederberger et al. have effectively transformed various types of metal oxides into nanoscale MNs using urea or cyanamide as nitrogen sources.236 The morphology and structure of precursor nanomaterials play a critical role in determining the characteristics of the final MNs, potentially undergoing dissolution–recrystallization processes. Subsequently, Giordano introduced a simple route known as “urea-glass” to bypass the preparation of metal oxides.237,238 This method involves the homogeneous mixing of metal chlorides with urea and anhydrous alcohols, forming a polymer-like, glassy phase. During this process, urea serves a dual role as a stabilizer for the dispersion of nanoparticles and nitrogen source. The resulting W2N and Mo2N exhibit high crystallinity and purity. Moreover, by varying the ratio of metal precursors to urea, directional control of particle size and specific surface area can be achieved. Yang et al. employed this synthesis method to fabricate ZrN nanoparticles with ORR performance surpassing benchmark Pt/C catalysts.81 The ZrN nanoparticles have an average diameter of approximately 45 nm, are protected by a thin oxide layer on the surface, and possess rapid electron transfer capability. LSV curves reveal that the onset and half-wave potentials (E1/2) of ZrN are 0.89 V and 0.80 V, respectively, which are comparable to those of Pt/C and superior to other reported MN-based catalysts. Importantly, ZrN exhibits only a minimal performance degradation of 3 mV after 1000 continuous cycles, far lower than the 39 mV degradation observed with Pt/C, demonstrating outstanding potential for practical applications.
The solid-state method offers several advantages for the synthesis of MNs materials, including its simplicity, cost-effectiveness, and scalability. However, this method poses challenges, such as the requirement for high temperature and pressure, demanding careful control of reaction conditions to facilitate the desired formation of MNs while mitigating undesired side reactions.
5. Design strategy for MNs in seawater electrolysis
As mentioned above, the hybrid bonding characteristics of MNs result in high melting points, exceptional hardness, and superior corrosion resistance, enabling them to withstand the effects of Cl− and the degradation caused by metal corrosion in seawater. Consequently, MNs emerge as highly promising candidates for electrocatalysts in seawater splitting (Table 1). In this section, we will discuss comprehensive strategies aimed at optimizing the seawater electrolysis performance of MNs.| Catalysts | Substrate | Electrolyte | Reaction | Tafel slope (mV dec−1) | j (mA cm−2)@η (mV) | Stability | Ref. |
|---|---|---|---|---|---|---|---|
| a Note: catalyst data for simulated seawater. b KOH. | |||||||
| NiVN/NF | NF | 0.5 M H2SO4 + 0.6 M NaCl | HER | 56.7 | 10@32 | 50 mA cm−2@10 h | 240 |
| 1 M KOH + 0.6 M NaCl | 24.5 | 10@37 | |||||
| NiCrN/NF | 0.5 M H2SO4 + 0.6 M NaCl | HER | 90.2 | 10@70 | 50 mA cm−2@10 h | ||
| 1 M KOH + 0.6 M NaCl | 88.5 | 10@115 | |||||
| NiCoHPi@Ni3N/NF | NF | 1 M KOH | OER | 79.8 | 100@365 | 200 mA cm−2@120 h | 241 |
| 500@425 | |||||||
| HER | 77.5 | 100@182 | 200 mA cm−2@120 h | ||||
| 500@281 | |||||||
| 1 M KOH + seawater | OER | — | 100@396 | 200 mA cm−2@120 ha | |||
| 500@474 | |||||||
| HER | — | 100@182 | — | ||||
| 500@281 | |||||||
| OWS | — | 500@2.1 V | 200 mA cm−2@40 ha | ||||
| MoN–Co2N/NF | NF | 1 M KOH + seawater | OER | 35.6b | 100@357 | — | 242 |
| 500@432 | |||||||
| HER | 82.1b | 100@304 | |||||
| 500@451 | |||||||
| OWS | — | 100@1.7 V | 100 mA cm−2@64 h | ||||
| NiCoN|NixP|NiCoN//S-(Ni,Fe)OOH | NF | Natural seawater | HER | 132.9 | 10@165 | 10 mA cm−2@24 h | 70 |
| OWS | — | 10@1.81 V | 10 mA cm−2@24 h | ||||
| Mo5N6 | — | Natural seawater | HER | — | 10@257 | 20 mA cm−2@100 h | |
| Fe2P/Ni1.5Co1.5N/N2P | NF | 1 M KOH + seawater | HER | 29.1b | 100@88 | 100 mA cm−2@40 h | 66 |
| 500@141 | |||||||
| OER | 43.4b | 100@255 | 100 mA cm−2@40 h | ||||
| 500@307 | |||||||
| OWS | — | 100@1.624 V | 50 mA cm−2@40 h | ||||
| 500@1.742 V | 200 mA cm−2@40 h | ||||||
| NC–Ni3N21 | NF | 2 M KOH + seawater | HER | 98.7 | 100@280 | 400 mA cm−2@200 h | 243 |
| OER | 57.9 | 100@270 | 400 mA cm−2@500 h | ||||
| OWS | — | 400@2.36 V | 400 mA cm−2@50 h | ||||
| Ni-SN@C | — | 1 M KOH + seawater | HER | 39b | 10@28b | 12 mA cm−2@40 h | 244 |
| Ni–MoN | CF | 1 M KOH + seawater | OWS | — | 500@1.783 V | 100 mA cm−2@100 h | 67 |
| 500@1.885 V | 500 mA cm−2@100 h | ||||||
| h-MoN@BNCNT | — | Natural seawater | HER | 128 | 10@135b | 20 mA cm−2@24 h | 245 |
| Ni3N/W5N4 | NF | 1 M KOH + seawater | HER | 43 | 10@36 | 100 mA cm−2@150 h | 246 |
| 200 mA cm−2@150 h | |||||||
| DI + plastics | Plastics upgrading | — | 10@1.32 V | 200 mA cm−2@45 h | |||
| 1 M KOH + seawater + plastics | OWS | — | 50@1.47 V | — | |||
| NiTe–NiCoN | NF | 1 M KOH + seawater | HER | 48b | 400@270 | 10, 20, 50, 100 mA cm−2@40 h | 247 |
| NiTe–NiFeN | OER | 39.9b | 400@350 | 10, 20, 50, 100 mA cm−2@40 h | |||
| NiTe–NiCoN//NiTe–NiFeN | WOS | — | 100@1.69 V | 100 mA cm−2@100 h | |||
| Ni3N/Co4N | NF | 1 M KOH + 0.5 M NaCl | HER | 95 | 10@85 | 10 mA cm−2@15 h | 86 |
| 100@282 | |||||||
| Fe0.01–Ni&Ni0.2Mo0.8N | NF | 1 M KOH + seawater | HER | — | 10@33 | 50, 100, 200, 400 mA cm−2@200 h | 87 |
| 500@144 | |||||||
| Fe0.01&Mo–NiO | OER | 24.5 | 10@211 | 10000 CV | |||
| 500@297 | |||||||
| Fe0.01–Ni&Ni0.2Mo0.8N//Fe0.01&Mo–NiO | OWS | — | 688@1.7 V | 50, 100, 200, 400 mA cm−2@200 h | |||
| NC@CrN/Ni | NF | 1 M KOH + seawater | HER | 40b | 10@24b | 10, 100, 1000 mA cm−2@150 h | 88 |
| 1000@284b | |||||||
| Ni3FeN@C/NF | NF | 1 M KOH + seawater | OWS | — | 100@314 | 100 mA cm−2@100 h | 65 |
| 500@394 | 500 mA cm−2@100 h | ||||||
| NiMo0.75N/PNCT | CC | 1 M KOH + 0.5 M NaCl | HER | 172 | 10@129 | 10, 40, 100 mA cm−2@30 h | 248 |
| NiNS//NiNS | NF | Natural seawater | OWS | — | 48.3@1.8 V | 15 mA cm−2@12 h | 249 |
| HW-NiMoN-2h | NF | 1 M KOH + seawater | HER | 32.3 | 100@40 | 500 mA cm−2@100 h | 69 |
| 500@91 | 1000 mA cm−2@70 h | ||||||
| 1000@130 | |||||||
| NiMoN@NiFeN | NF | 1 M KOH + seawater | HER | 45.6 | 100@82 | 100, 500 mA cm−2@48 h | 250 |
| 500@160 | |||||||
| 1000@218 | |||||||
| NiFeN | OER | 58.6 | 100@307 | 100, 500 mA cm−2@48 h | |||
| 500@369 | |||||||
| 1000@398 | |||||||
| NiMoN@NiFeN//NiFeN | OWS | — | 500@1.608 V | 100 mA cm−2@100 h | |||
| 1000@1.709 V | 500 mA cm−2@100 h | ||||||
| Fe2P/Ni3N | NF | 1 M KOH + seawater | HER | — | 100@109 | 100 mA cm−2@40 h | 251 |
| OER | 45.5 | 100@252 | 100 mA cm−2@40 h | ||||
| 500@302 | 500 mA cm−2@40 h | ||||||
| 1000@340 | |||||||
| OWS | — | 100@1.645 V | 100 mA cm−2@70 h | ||||
| 500@1.768 V | 500 mA cm−2@40 h | ||||||
| Pt–Ni@NiMoN | NF | 1 M KOH | HER | 22.9 | 10@7 | 500, 1000 mA cm−2@110 h | 89 |
| 100@34 | |||||||
| 500@90 | |||||||
| 1 M KOH + seawater | HER | 30.6 | 10@11 | 200, 500 mA cm−2@130 h | |||
| 100@47 | |||||||
| 500@109 | |||||||
5.1. Heterostructuring
Heterostructure engineering is an important strategy for modulating the properties and functionalities of nanomaterials.252–254 By optimizing the morphology, composition, crystal structure, and interface characteristics of heterostructures, key properties of materials, such as electronic transport, chemical activity, and stability can be improved. Therefore, heterostructure engineering provides a powerful approach for achieving high-performance catalytic materials and devices, with broad prospects in catalytic energy applications.255,256 MN-based heterostructures have been widely explored and the resulting catalysts have shown excellent performance. For example, Qiao et al. fabricated heterogeneous electrodes with Ni3N and Ni3S2 interfaces in the presence of thiourea (Fig. 16a),249 where the enriched heterogeneous interface served as active sites for enhanced water dissociation and adsorption, demonstrating superior seawater electrolysis performance over Ir–C and Pt–C (Fig. 16b and c). The unique intrinsic activity of Ni3N enables its composites with other materials, such as (Fe1−xCox)2P/Ni3N (Fig. 16d–f),251 Co4N/Ni3N,86 NiMoN/Ni3N,257 and NiFeP/Ni3N258 to achieve desirable performances. The NiCoHPi@Ni3N/NF catalyst demonstrates excellent seawater splitting performance. This is attributed to the hydrogen phosphate ions anchored on the surface, which not only repel Cl− but also enhance the OH− adsorption capacity.241 It should be noted that lattice distortion often occurs due to lattice mismatch in heterostructures. This phenomenon was observed in a novel copper-nickel nitride (Cu1Ni2–N) catalyst designed by Mai et al.,127 which exhibited abundant Cu4N/Ni3N interfaces (Fig. 16g and h). The large lattice spacing difference resulted in distorted crystal structures, promoting intermediate adsorption and desorption (Fig. 16i).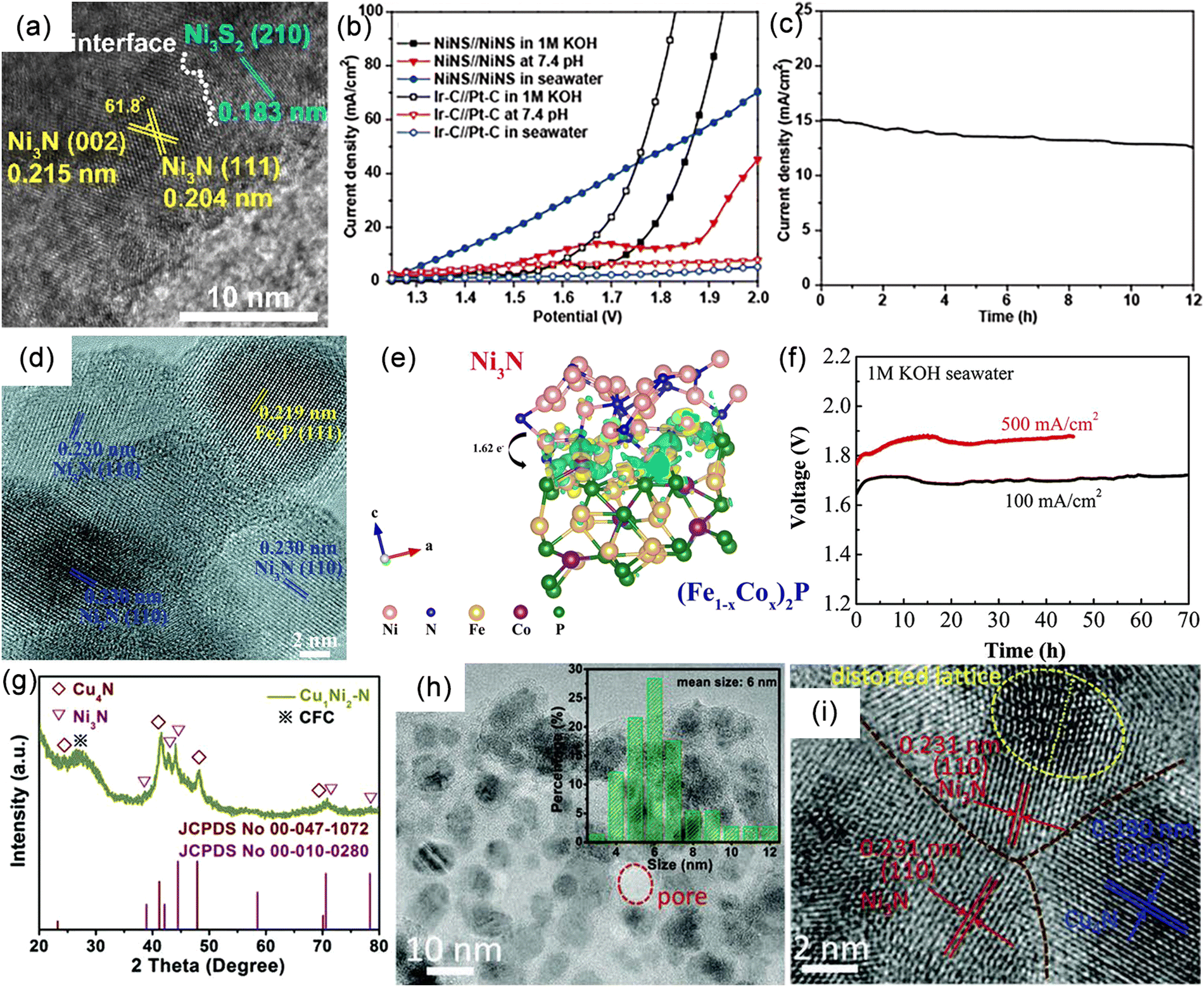 | ||
| Fig. 16 (a) High-resolution transmission electron microscopy (HRTEM) images of NiNS. (b) Linear sweep voltammetry (LSV) curves of NiNS//NiNS and Ir–C//Pt–C overall water splitting systems in 1 M KOH, buffer solution (pH = 7.4) and seawater. (c) Chronoamperometry (CA) curve of NiNS//NiNS for overall water splitting. Reproduced with permission.249 Copyright 2019, The Royal Society of Chemistry. (d) HRTEM images of Fe2P/Ni3N catalyst. (e) Charge difference density (CDD) of the interface of (Fe1−xCox)2P/Ni3N. (f) Chronopotentiometry (CP) curves of Fe2P/Ni3N at constant current densities in 1 M KOH seawater electrolytes. Reproduced with permission.251 Copyright 2023, Wiley-VCH. (g) XRD, (h) TEM, and (i) HRTEM images of Cu1Ni2–N/CFC. Reproduced with permission.127 Copyright 2019, Wiley-VCH. | ||
Furthermore, the utilization of common metal–organic framework (MOF) materials as precursors for the synthesis of heterogeneous MNs has also garnered significant attention.259–261 Li et al. employed ZIF-67 as a sacrificial template to fabricate nitrogen-doped carbon nanosheets loaded with cobalt molybdenum nitride for seawater electrolysis.242 This strategy capitalizes on the corrosion resistance and high conductivity advantages of carbon materials, demonstrating excellent corrosion resistance under real seawater conditions.
Li et al. reported a well-defined organic-polyoxometalate co-crystals (O-POCs) precursor and successfully synthesized structurally and porosity controllable molybdenum-based carbide/nitride catalysts.262 In their work, the silica template served as nucleation site for O-POCs structure, leading to the formation of flower-like pP-Mo/silica with nanosheets. Additionally, the silica template played a role in preventing molybdenum aggregation during high-temperature carburization. Meanwhile, increasing the acid amount in the reaction conditions altered the quantity of protonated nitrogen species and the content of Mo–oxygen clusters in the structure. Single-crystal structures revealed the consumption of organic components within the eutectic with pH decrease, possibly due to gradual replacement of organic counterions by hydrated hydrogen ions, thereby affecting the composition and structure of the eutectic. HRTEM showed a good match between the atomic arrangement in the crystal structure and the simulated crystal structures of Mo2C(101) and Mo2N(111) surfaces. The resulting electrocatalyst exhibited high catalytic activity and good long-term stability. This work opens up a new pathway for the controlled synthesis of molybdenum-based catalysts with multilevel hierarchical pore structures.
5.2. 3D Nanostructure design
3D nanostructures are composed of various forms of nanomaterials, including nanoparticles, nanowires, nanotubes, nanopores, and nanosheets.56,263–265 These nanostructures can be assembled and arranged in different ways to form complex morphologies and structures. By controlling the size, shape and composition of the nanostructures, it is possible to tailor the surface area, proton transport channels and electron transfer properties to optimize electrocatalytic performance.266 3D nanostructures provide abundant active surface area and optimized proton transport channels, enhancing catalytic activity and electron transfer efficiency. Additionally, 3D nanostructures can inhibit oxidation and corrosion processes and improve the stability and durability of catalysts, which is particularly important in the field of hydrogen production from seawater electrolysis.122Ren's group has conducted extensive research and made a series of progress in the field of electrocatalytic seawater hydrogen production. They reported a sandwich-structured catalyst composed of nickel phosphide (NixP) microsheets anchoring NiCoN nanoparticles (Fig. 17a–c), exhibiting excellent HER activity.70 This stacked structure has a high surface area and good electron conductivity. XPS results showed strong charge transfer between the components (Fig. 17d), optimizing H* adsorption energy and enhancing catalytic activity. NixP and NiCoN act as the ‘skeleton’ and ‘armor’ in the catalyst, enhancing seawater corrosion resistance. TOF results demonstrated that at an overpotential of 300 mV, the TOF of NiCoN|NixP|NiCoN is 0.771 s−1, 3.1 times that of NixP (0.252 s−1), and 1.4 times that of NiCoN (0.557 s−1) (Fig. 17e and f). Recently, they designed a novel nanoparticle-nanorod (0D/1D) multidimensional hybrid structure consisting of Ni particles loaded with amorphous MoN. TEM confirmed uniform dispersion and close contact of Ni nanoparticles (Fig. 17g and h).67 Both Ni and MoN derived from NiMoO4 precursor form a closely connected heterogeneous structure, favorable for interface electron transfer. The resulting catalyst exhibited a low Tafel slope value of 35.5 mV dec−1, indicating rapid HER kinetics. The adsorption energy of H2O at the Mo sites was found to be −1.082 eV, crucial for subsequent dissociation. After 100 hours of seawater electrolysis at a current density of 500 mA cm−2, the electrolyte showed no color change, indicating no formation of hypochlorite. A commercial thermoelectric (TE) module was used to test the electrolytic cell, and a large amount of hydrogen gas bubbles were generated on the catalyst surface at a temperature difference of 61 °C, effectively converting waste heat into valuable hydrogen energy, demonstrating promising application prospects (Fig. 17i). HW-NiMoN-2h, a stacked interconnected structure with nanorods and nanowires, exhibits excellent HER activity (Fig. 17j).69 The nanowires effectively connect independent nanorods (Fig. 17k and l), enhancing the structural stability and demonstrating outstanding durability at an ultra high current density of 1 A cm−2. Zeta potential testing shows that HW-NiMoN-2h possesses a negative Zeta potential of −16.0 mV, which enables efficient repulsion of Cl− and reduces the corrosion on active sites. Ji et al. also reported an unique structure where Ni nanoparticles are anchored on MoN nanosheets, demonstrating good alkaline overall water splitting performance.267
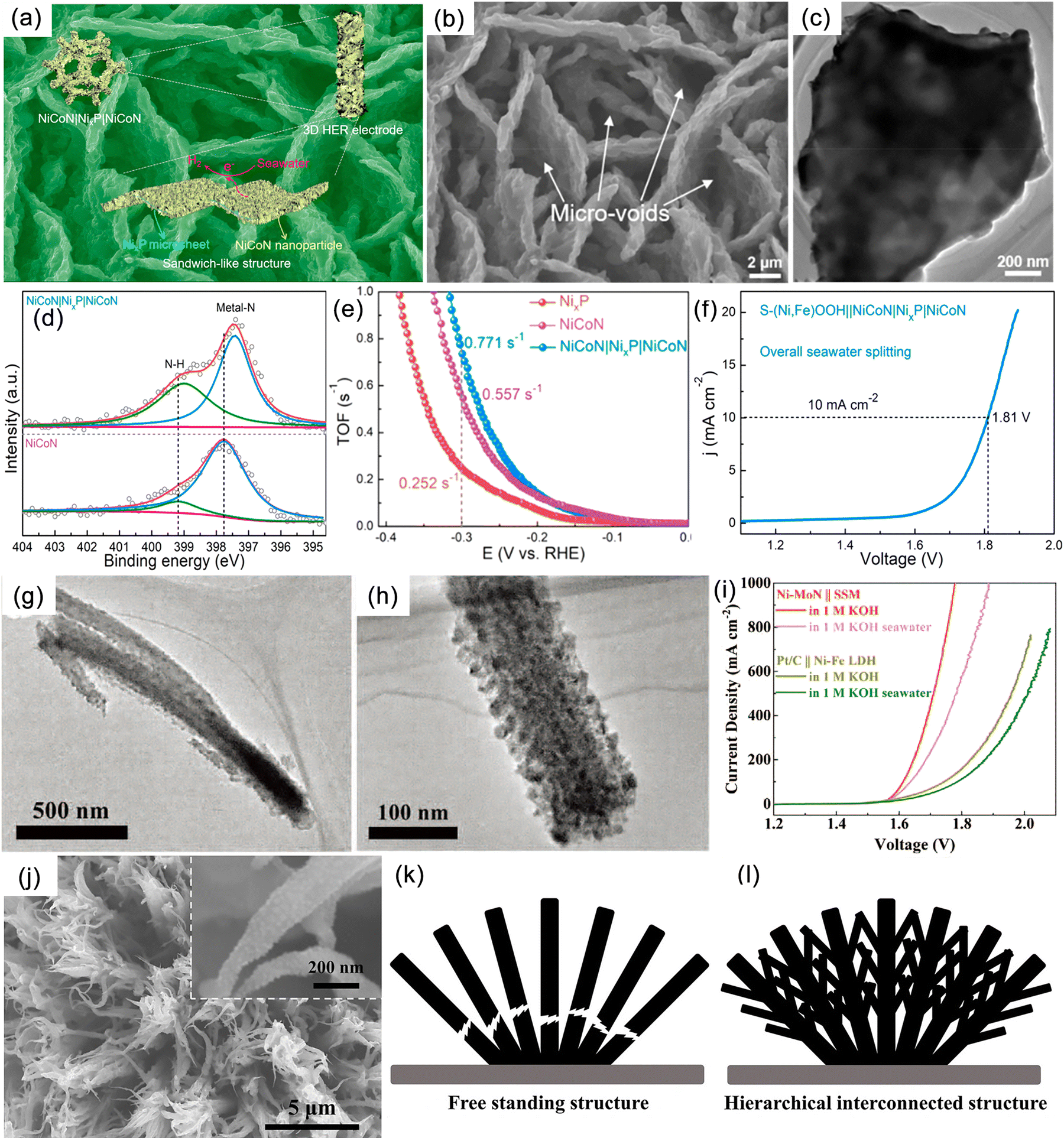 | ||
| Fig. 17 (a) and (b) Scanning electron microscopy (SEM) and (c) transmission electron microscopy (TEM) images of NiCoN|NixP|NiCoN. (d) High-resolution N 1s X-ray photoelectron spectroscopy (XPS) spectra of the NiCoN|NixP|NiCoN and NiCoN. (e) Turnover frequency (TOF) plots. (f) Polarization curve of the S–(Ni,Fe)OOH‖NiCoN|NixP|NiCoN electrolyzer. Reproduced with permission.70 Copyright 2020, American Chemical Society. (g) and (h) TEM images of Ni–MoN. (i) Overall water/seawater electrolysis performance of Ni–MoN‖SSM and Pt/C‖Ni–Fe LDH pairs in 1 M KOH and 1 M KOH seawater electrolytes. Reproduced with permission.67 Copyright 2022, Wiley-VCH. (j) SEM image of HW-NiMoN-2h. Schemes of (k) free-standing and (l) hierarchical interconnected nanocatalyst structures. Reproduced with permission.69 Copyright 2023, Springer Nature Publishing Group. | ||
5.3. Carbon-coating strategy
The carbon-coating strategy is a widely used nanotechnology aimed at enhancing the properties and functionalities of nanomaterials by adding a layer of carbon material to their surfaces.268–271 This approach can be applied to various nanomaterials, including nanoparticles, nanowires, and nanotubes to improve their stability, conductivity, catalytic activity, and chemical reactivity. Organic compounds, carbon black, and graphene are commonly used carbon sources for this purpose. The carbonization methods employed include chemical vapor deposition, pyrolysis, and carbothermal reduction. By carefully controlling the carbonization conditions, the thickness and structural arrangement of the carbon-coating layer can be optimized.272 This strategy enhances the stability of nanomaterials in seawater electrolysis catalysts, effectively preventing oxidation or Cl− corrosion during the reaction.Cheng et al. successfully synthesized a high-performance seawater electrolysis catalyst consisting of multicomponent oxide-derived hexagonal molybdenum nitride (MXene) nanoparticles embedded in N and B doped carbon nanotubes (CNTs) (Fig. 18a).245 This structure effectively mitigates the aggregation of MoN NPs and the corrosion from Cl− (Fig. 18b). The effect between doped CNTs and confined h-MoN NPs creates active HER sites, leading to a low overpotential of 78 mV and a Tafel slope of 46 mV dec−1 (Fig. 18c). The catalyst also exhibits strong adaptability to complex pH environments.
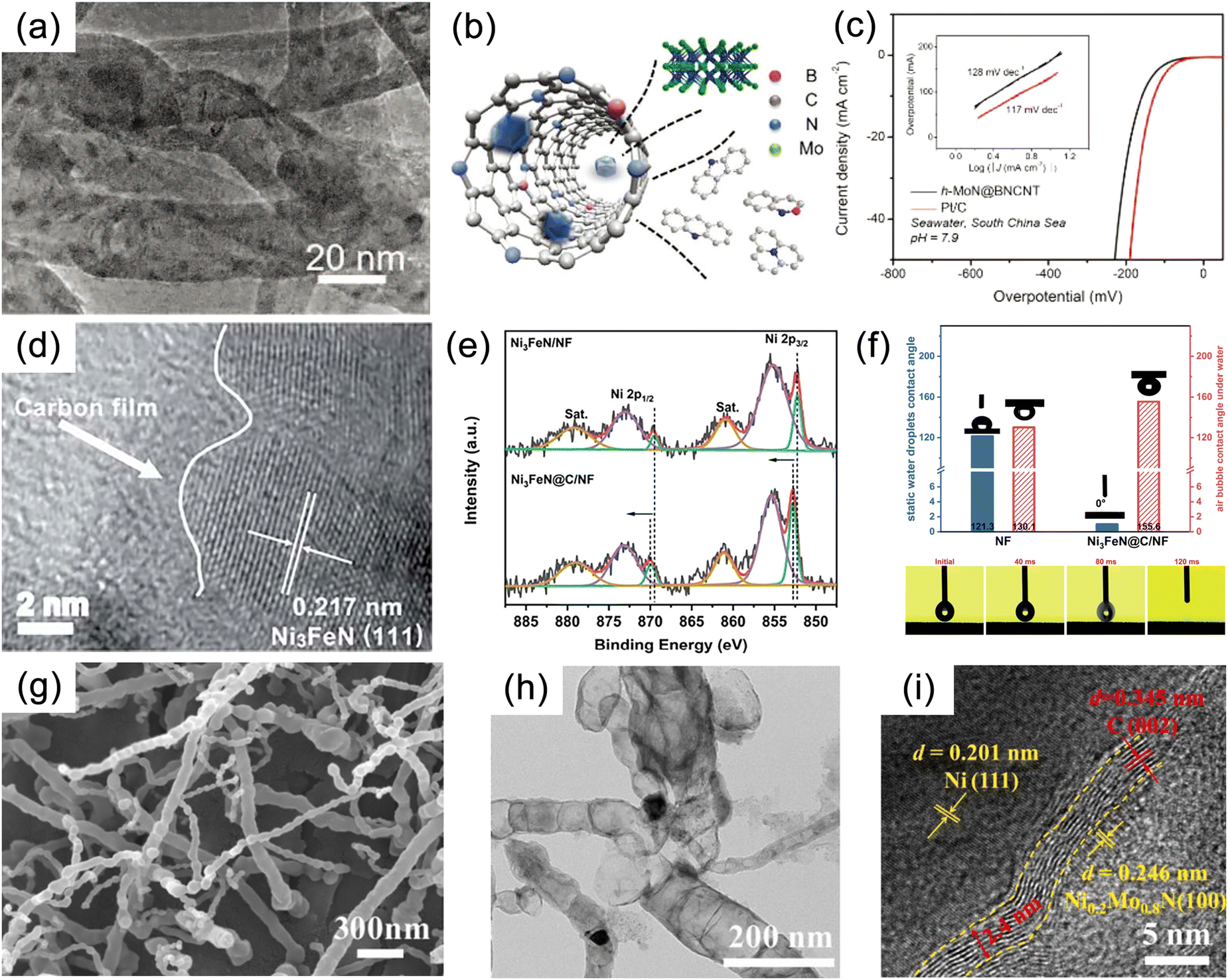 | ||
| Fig. 18 (a) Transmission electron microscopy (TEM) image of h-MoN@BNCNT (B and N doped carbon nanotube). (b) Schematic representation of N atoms and B atoms (graphitic, pyridinic, pyrrolic N and C–B, N–B) in BNCNT and top view of h-MoN. (c) Linear sweep voltammetry (LSV) curves of h-MoN@BNCNT and 20% Pt/C in seawater. Inset: The corresponding Tafel plots. Reproduced with permission.245 Copyright 2019, Wiley-VCH. (d) High-resolution TEM (HRTEM) images of Ni3FeN@C microsheets. (e) Ni 2p X-ray photoelectron spectroscopy (XPS) spectra of Ni3FeN@C/NF and Ni3FeN/NF. (f) Static and dynamic contact angle testing of Ni3FeN@C/NF and NF. Reproduced with permission.65 Copyright 2021, The Royal Society of Chemistry. (g) Scanning electron microscopy (SEM), (h) TEM and (i) HRTEM images NiMo0.75N/PNCT. Reproduced with permission.248 Copyright 2023, Elsevier. | ||
In another study, glucose was utilized as a carbon source to facilitate polymerization and nitridation, leading to the preparation of NixFeyN@C/NF catalysts under high-temperature conditions (Fig. 18d).65 The incorporation of an external carbon layer optimizes the localized electronic structure of metal sites (Fig. 18e). Moreover, the catalyst features a porous structure and highly hydrophilic surface (Fig. 18f), which enhance the gas–liquid interaction at active sites. As a result, the dual-electrode seawater electrolysis cell achieved an impressive high current density of 500 mA cm−2, requiring only a low applied voltage of 1.91 V, while demonstrating remarkable stability over continuous 500 hours of operation. Jiang et al. reported a NiMoxN catalyst coated with porous N-doped carbon nanotubes (PNCT) (Fig. 18g).248 The selection of different nitriding reagents enabled the control of exposed crystal facets of NiMoxN, and using 3-amino-1,2,4-triazole (AT) as nitrogen source resulted in the prevalence of NiMoN(100) facets. The HRTEM image clearly shows a bamboo-like carbon layer encapsulating NiMoN with a thickness of about 2.4 nm (Fig. 18h and i). DFT calculations demonstrated a synergistic effect between the highly active NiMoN(100) facets and PNCT, resulting in a small ΔGH* of 0.05 eV. Interestingly, Umaprasana and colleagues introduced a green synthesis method,243 applicable to various single-metal and bimetallic systems, which involved the use of polyacryloyl hydrazide as reducing and capping agent for coating Ni3N/Fe3N nanosheets with carbon nanotubes. Collectively, the above works illustrate the potential of carbon-coated MNs as a promising approach for developing high performance and durable seawater electrocatalysts.
5.4. Interface engineering
In contrast to the heterostructure strategy, interfacial engineering places greater emphasis on optimizing the interfacial properties among various phases within nanomaterials. Due to the small size and high specific surface area of nanomaterials, interfaces between phases with distinct lattice structures or chemical compositions play a pivotal role in determining material properties. By precisely adjusting the structure and composition of these interfaces, it is possible to effectively tailor critical material properties, including electron transport, chemical reactivity, and mechanical strength.Wang's team employed an interface engineering strategy to fabricate alkaline seawater electrocatalysts with abundant interfaces and optimized structures.247 For the HER and OER electrodes, NiTe–NiCoN and NiTe–NiFeN were utilized, respectively (Fig. 19a and b). The authors observed that temperature regulation influenced the chemical transformation of tellurides, elucidating the crucial role of NH3 gas in the interfacial formation process. The prepared catalysts exhibited rich and closely contacted interfaces, including Ni3Te2(201) with NiCo2N(200) (Fig. 19c) and Ni3FeN(111) (Fig. 19d). X-ray absorption near-edge structure (XANES) analysis of Ni revealed that the introduction of bimetallic nitrides corrected the d-band center, lowering the bonding strength of catalytic intermediates (Fig. 19e). This is further supported by the work function (WF) test, demonstrating abundant electron transfer pathways provided by the interfacial structure. Additionally, the influence of electric fields on ionic distribution in the Helmholtz layer was investigated, revealing that a high-curvature interfacial structure generates stronger field strengths. The presence of “hot spots” near the electrodes leads to rapid charge accumulation, subsequently affecting anion concentration and hindering Cl− erosion (Fig. 19f). This work, combined with COMSOL simulations, offers a new perspective on the mechanism of MN-based seawater electrocatalysts.
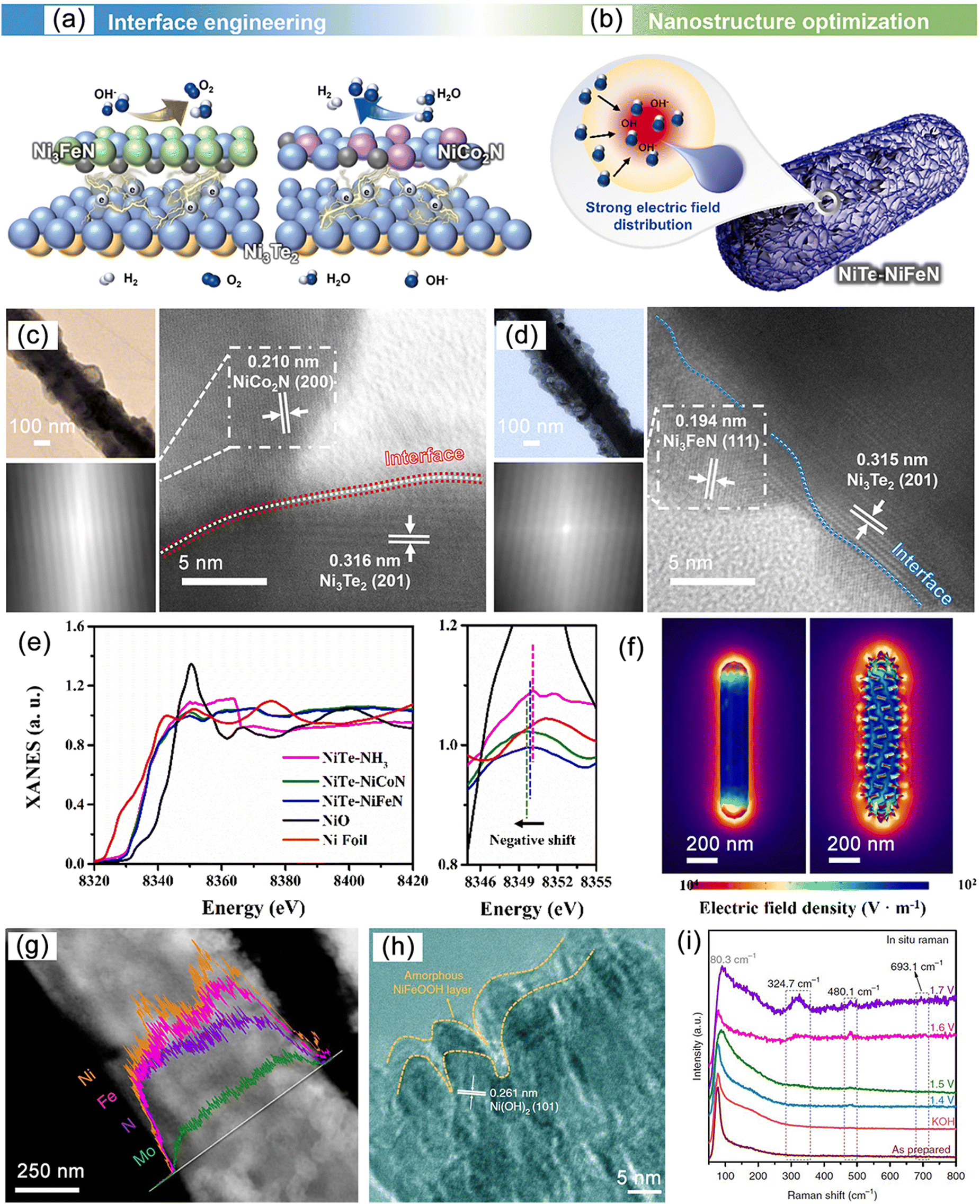 | ||
| Fig. 19 (a) and (b) Electrocatalytic activity improvement mechanism of the heterostructured nickel-based tellurium and nitrides. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of (c) NiTe–NiCoN and (d) NiTe–NiFeN. (insets: partial Fourier transforms). (e) Ni K-edge X-ray absorption near-edge spectroscopy (XANES) spectra for Ni foil, NiO, NiTe–NH3, NiTe–NiCoN, and NiTe–NiFeN. (f) Comparison of electric field distributions with different nanostructures. Reproduced with permission.247 Copyright 2022, Elsevier. (g) Energy dispersive spectroscopic (EDS) line scan of the NiMoN@NiFeN catalyst. (h) HRTEM image of the NiMoN@NiFeN catalyst after oxygen evolution reaction (OER) tests. (i) In situ Raman spectra of the NiMoN@NiFeN catalyst at various potentials for the OER process. Reproduced with permission.250 Copyright 2021, Springer Nature Publishing Group. | ||
A 3D core–shell MNs catalyst, with NiFeN nanoparticles uniformly anchored on NiMoN nanorods supported by Ni foam, was also employed for seawater electrolysis.250 The abundant interface between the outer NiFeN nanoparticles and inner NiMoN nanorods acted as highly efficient reaction sites. EDS images revealed significantly higher Ni and Fe content at the shell, confirming the successful assembly of the nanostructure (Fig. 19g). The catalyst exhibited remarkable activity and stability, with low applied voltages of only 1.608 and 1.709 V required to drive 500 and 1000 mA cm−2 current densities, respectively, and it operated stably at room temperature for 500 hours. HRTEM after durability testing confirmed the formation of NiFeOOH (Fig. 19h), as also verified by in situ Raman spectroscopy (Fig. 19i). This in situ generated high valent amorphous layer not only served as catalytically active sites but also protected the inner catalyst from corrosion by Cl− in seawater. Zhou et al. reported a strategy that involves engineering a multifunctional catalytic interface, developing a porous metal nitride/phosphide array anchored on the conductive Ni2P surface.66 This interface plays a positive role in improving both the HER and OER. It enhances the water adsorption/dissociation and hydrogen adsorption behavior of active Fe sites, increases the adsorption of oxygen-containing species, and forms a passivation layer to resist corrosion from chlorine-containing substances.
5.5. Other strategies
In addition to the aforementioned more general strategies, methods such as heteroatom doping and incomplete nitriding have been employed to enhance the electrolytic seawater performance of MNs. Nitrogen enrichment or incomplete nitridation of nanomaterials can be achieved by manipulating the stoichiometric ratio of the nitrogen source, thus adjusting the proportion of nitrogen in the metal matrix.273,274 Unlike nitrogen-enriched processes with slow thermodynamics and harsh preparation conditions, incomplete nitridation promotes the formation of metal/MNs interfaces by limiting the incorporation of metal–nitrogen bonding in the substrate. Qiao et al. successfully synthesized a catalyst (Ni-SN@C) with Ni surface nitride encapsulated in a carbon shell using Ni and ethylenediaminetetraacetic acid (EDTA) coordination complex under NH3 atmosphere at 500 °C (Fig. 20a and b).244 The outermost carbon layer prevents further nitridation of internal Ni particles. The unsaturated Ni-SN@C exhibits Pt-like HER behavior by promoting the generation of hydronium ions under alkaline conditions (Fig. 20c). In a dual-electrode flow electrolyzer for HzOR, a current density of 1 A cm−2 is achieved with a cell voltage of only 0.7 V (Fig. 20d).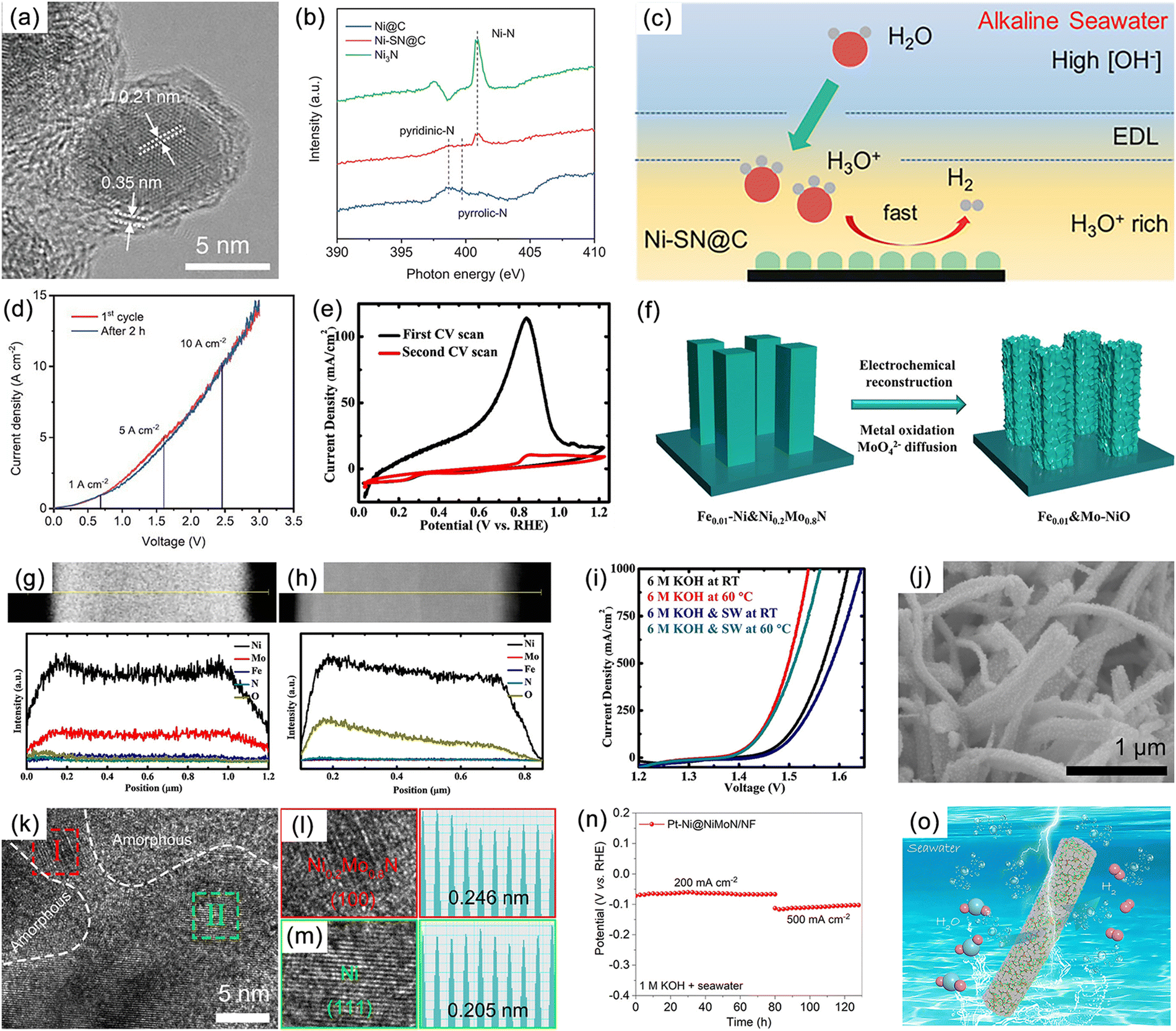 | ||
| Fig. 20 (a) High-resolution transmission electron microscopy (HRTEM) images. (b) N K-edge of Ni @C, Ni-SN@C and Ni3N catalysts. (c) Illustration of the hydronium ion generation mechanism on Ni-SN@C. EDL: electric double layer. (d) Linear sweep voltammetry (LSV) curves using the flow-electrolyzer with Ni-SN@C as the anode and cathode. Reproduced with permission.244 Copyright 2021, Wiley-VCH. (e) Cyclic voltammogram (CV) scans for Fe0.01–Ni&Ni0.2Mo0.8N showing its electrochemical reconstruction. (f) Illustration of the electrochemical reconstruction of Fe0.01–Ni&Ni0.2Mo0.8N into Fe0.01&Mo–NiO. Energy dispersive spectroscopic (EDS) line scans of (g) Fe0.01–Ni&Ni0.2Mo0.8N and (h) Fe0.01&Mo–NiO. (i) Alkaline water and seawater electrolysis performance of Fe0.01–Ni&Ni0.2Mo0.8N‖Fe0.01&Mo–NiO under industrial conditions. Reproduced with permission.87 Copyright 2022, The Royal Society of Chemistry. (j) Scanning electron microscopy (SEM), (k)–(m) HRTEM images and (n) chronopotentiometry (CP) curves of Pt–Ni@NiMoN/NF. (o) Diagram of Pt–Ni@NiMoN electrolysis for hydrogen production from seawater. Reproduced with permission.89 Copyright 2023, The Royal Society of Chemistry. | ||
The incorporation of heteroatoms can significantly alter the electronic and lattice structures, as well as the chemical reactivity of the substrate material, allowing for precise manipulation of material properties. By incorporating trace amounts of Fe into the Ni&Ni0.2Mo0.8N substrate, a highly active seawater electrolysis catalyst was constructed.87 Under anodic potential, Fe–Ni&Ni0.2Mo0.8N underwent irreversible reconstruction into a Fe and Mo co-doped NiO (Fe&Mo–NiO) after only 1 CV scan (Fig. 20e and f). During this process, most of the Mo rapidly leached into the alkaline electrolyte, while Ni and Fe were oxidized to +2 and +3 states, respectively. EDS analysis provided strong evidence of catalyst reconstruction before and after the process (Fig. 20g and h). The reconstructed catalyst demonstrated outstanding seawater splitting performance under industrial conditions, offering promising prospects for the industrial application of seawater electrolysis (Fig. 20i). Just recently, Yang et al. introduced trace amounts of Pt (0.07 wt%) to construct a Pt–Ni@NiMoN/NF catalyst with a multidimensional nanostructure (Fig. 20j). The catalyst consists of 1D NiMoN nanorods/3D Ni nanoparticles, exposing a rough surface with numerous active sites (Fig. 20k–m). It can operate without decay for at least 130 hours in alkaline seawater at 200 and 500 mA cm−2, achieving the current best catalytic activity (Fig. 20n and o).89
6. Conclusions and perspectives
Electrolysis of seawater to produce high-purity hydrogen is beneficial for preserving freshwater resources and energy sustainability. However, the inherent complexity of seawater compositions presents challenges for their electrolysis for hydrogen production. High concentrations of salts and impurities, especially Cl−, lead to corrosion of the catalyst and the electrolysis cell due to the generation of Cl2 gas from the anode during the electrolysis process. MNs have gained widespread application in field of seawater electrolysis due to their exceptional chemical stability, thermal stability, and high intrinsic catalytic activity, attributed to their unique bonding characteristics. In this review, we have summarized the reactions involved in the seawater splitting process, the evaluation parameters of the electrocatalyst and the selection criteria and types of conducting substrate materials. The properties of MNs and standard preparation methods are described in detail. Furthermore, we provide an overview of recent research advancements and cutting-edge processes in seawater electrolysis for hydrogen production. We present comprehensive design strategies for advanced MNs electrocatalysts, aiming to elucidate the intricate relationship between their nanostructure and performance. Despite the progress made in this field, the current technologies and electrocatalysts still fall short of meeting the practical requirements for large-scale industrial hydrogen production through seawater electrolysis. Therefore, we propose that future research in this area should concentrate on the following critical aspects:(1) Innovative preparation and batch synthesis of MNs
MNs have drawn extensive attention in the field of seawater electrolysis for hydrogen production due to their high stability and catalytic performance. However, from a broad perspective, traditional synthesis routes may not suffice to achieve large-scale production of MNs with consistent quality and performance. Therefore, to realize the wide-ranging applications of MNs, future studies should focus on exploring novel technologies and methodologies for achieving efficient and low-cost batch production of MNs. These approaches may involve customized synthesis conditions, template-assisted strategies, and the utilization of eco-friendly precursors. High-entropy metal alloy nitrides exhibit higher potential for efficient and multifunctional electrochemical energy conversion compared to low-entropy MNs.275–277 Nevertheless, the facile synthesis of high-entropy MNs remains a highly challenging subject. Furthermore, the integration of advanced detection techniques, such as in situ spectroscopy and microscopy, to gain insights into the formation mechanisms and nanostructures of MNs during the synthesis process will facilitate the design and optimization of synthesis routes. Appropriate methods should be chosen to achieve simple and high-quality large-scale production.(2) Development of high performance MNs seawater electrocatalysts
The complex composition of seawater leads to different research priorities for seawater electrolysis catalysts compared to pure water electrolysis. Conventional strategies, such as increasing active sites or modulating local electronic structures, often fail to address the challenges posed by seawater. For large-scale industrial seawater hydrogen production, corrosion resistance and long-term stability at high current densities of the catalyst are crucial aspects, which are frequently overlooked. The current stability of MNs is insufficient, and their inherent chemical inertness may negatively impact catalytic activity. Furthermore, the choice of electrode substrate also influences the overall performance of the assembled electrolyzer, necessitating the consideration of multiple factors such as conductivity, corrosion resistance, porosity, and mechanical strength. Protecting the electrode surface without compromising active sites or performance remains a challenging task. Innovative approaches have been explored, including in situ or ex situ deposition of super-selective layers on electrocatalysts or using corrosion inhibitors, to achieve sustainable performance without compromising catalytic activity. Therefore, MNs catalyst modulation is essential to enhance corrosion resistance, utilizing strategies such as 3D structure design, heterointerfaces, Cl− blocking layers, and heteroatom doping.(3) Strengthening research on seawater electrolysis reaction mechanisms
Despite significant progress in understanding the mechanisms of electrocatalytic reactions, a full understanding and theoretical prediction of real seawater electrolysis reactions remain lacking and such reactions are not replicated by using simulated seawater. The rapid development of in situ characterization techniques, such as in situ electron microscopy, in situ synchrotron radiation spectroscopy, and in situ Raman spectroscopy provides opportunities for real-time monitoring of actual catalytic processes of catalysts in seawater. These in situ techniques allow for a deeper understanding of the adsorption of intermediate species at active sites and the reaction mechanisms, which is particularly crucial for OER, HER, CER, and other catalytic reactions. Furthermore, DFT calculations provide insights into the adsorption characteristics of catalysts in seawater, explore the formation and transformation of reaction intermediates, and quantitatively calculate the activation energy of reactions. DFT calculations offer crucial guidance for optimizing the reaction pathways of OER, HER, and other catalytic reactions. Combining in situ techniques and DFT calculations enables a comprehensive understanding of catalyst behavior and performance during seawater electrolysis for hydrogen production, thereby further advancing the development of high-performance electrolysis catalysts for seawater.(4) Development of seawater electrolysis reactors
The design of specific seawater electrolysis cells is currently not receiving sufficient attention, and to achieve large-scale seawater hydrogen production, we must fully consider the rational design of reactors. Specific seawater reactors require extremely high corrosion resistance, which can be achieved by applying corrosion-resistant coatings. Electrocatalytic water splitting is a typical solid–liquid–gas three-phase interfacial reaction, so gas–liquid transport is also one of the factors that need to be carefully considered. The simulation and modeling of engineering factors at the interface, such as fluid mechanics and membrane electrode thickness, play a crucial role in system integration and maximizing efficiency. Furthermore, considering the substitution of OER with small molecule organic oxidation reactions that offer thermodynamic advantages is feasible. However, this approach requires the adoption of asymmetric electrolyte feeding, which places higher demands on the design and performance of membranes. It is also important to improve the Cl− resistance of existing membranes or to develop new membrane materials.(5) Economic and environmental considerations
Compared to traditional fossil fuel-based methods of hydrogen production, current seawater electrolysis for hydrogen generation requires higher energy inputs. To ensure its economic viability, it is crucial to implement measures aimed at reducing energy costs. One important strategy is to utilize abundant offshore renewable energy sources, such as solar and wind power, to lower energy consumption and enhance the economic viability.278 Besides economic aspects, a comprehensive evaluation of the sustainability is crucial, such as raw material sourcing, energy consumption, and waste management. Catalyst design and reactor development must prioritize environmentally friendly materials and technologies to minimize negative impacts on seawater and marine ecology.(6) Coupling hydrogen production from seawater with marine resource utilization
With the development of offshore wind power, the trend of moving towards the deep sea has become a future direction in the energy market. As a valuable resource base, the ocean holds great potential for tidal energy, algae and sea salt. However, achieving the coupling and utilization of seawater electrolysis and marine resources poses challenges. In future research, it will be necessary to investigate and monitor the symbiotic development of seawater electrolysis and marine resources, aiming to offer new possibilities for marine chemical plants and the advancement of clean energy without damaging the natural environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 61971405) and the Fundamental Research Funds for the Central Universities (grant no. DUT22RC(3)050). J. P. A. acknowledges EPSRC (UK) for support.Notes and references
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- M. P. Browne, Z. Sofer and M. Pumera, Energy Environ. Sci., 2019, 12, 41–58 RSC.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Gratzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829 CrossRef CAS.
- S. Cobo, J. Heidkamp, P. A. Jacques, J. Fize, V. Fourmond, L. Guetaz, B. Jousselme, V. Ivanova, H. Dau, S. Palacin, M. Fontecave and V. Artero, Nat. Mater., 2012, 11, 802–807 CrossRef CAS PubMed.
- H. Sun, Z. Yan, F. Liu, W. Xu, F. Cheng and J. Chen, Adv. Mater., 2020, 32, e1806326 CrossRef PubMed.
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS.
- Y. Yao, S. Hu, W. Chen, Z. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- Z. Lin, Z. Wang, J. Gong, T. Jin, S. Shen, Q. Zhang, J. Wang and W. Zhong, Adv. Funct. Mater., 2023, 2307510 CrossRef CAS.
- S. Hao, L. Chen, C. Yu, B. Yang, Z. Li, Y. Hou, L. Lei and X. Zhang, ACS Energy Lett., 2019, 4, 952–959 CrossRef CAS.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao Horn, Adv. Mater., 2019, 31, e1806296 CrossRef PubMed.
- D. S. Raja, X. F. Chuah and S. Y. Lu, Adv. Energy Mater., 2018, 8, 1801065 CrossRef.
- H. Liao, X. Guo, Y. Hou, H. Liang, Z. Zhou and H. Yang, Small, 2020, 16, e1905223 CrossRef PubMed.
- Y. Zhang, Y. Wang, H. Jiang and M. Huang, Small, 2020, 16, e2002550 CrossRef PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- F. Yan, C. L. Zhu, C. Y. Li, S. Zhang, X. T. Zhang and Y. J. Chen, Electrochim. Acta, 2017, 245, 770–779 CrossRef CAS.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Q. Wen, K. Yang, D. Huang, G. Cheng, X. Ai, Y. Liu, J. Fang, H. Li, L. Yu and T. Zhai, Adv. Energy Mater., 2021, 11, 2102353 CrossRef CAS.
- H. Shi, T. Dai, W. Wan, Z. Wen, X. Lang and Q. Jiang, Adv. Funct. Mater., 2021, 31, 2102285 CrossRef CAS.
- C. W. Liang, P. C. Zou, A. Nairan, Y. Q. Zhang, J. X. Liu, K. W. Liu, S. Y. Hu, F. Y. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- R. Zeng, Y. Yang, X. Feng, H. Li, L. M. Gibbs, F. J. DiSalvo and H. D. Abruña, Sci. Adv., 2022, 8, eabj1584 CrossRef CAS PubMed.
- Y. Zhang, B. Huang, G. Luo, T. Sun, Y. Feng, Y. Wang, Y. Ma, Q. Shao, Y. Li, Z. Zhou and X. Huang, Sci. Adv., 2020, 6, eaba9731 CrossRef CAS PubMed.
- J. Wang, J. Zhang, Y. Hu, H. Jiang and C. Li, Sci. Bull., 2022, 67, 1890–1897 CrossRef CAS PubMed.
- X. Zhang, W. Yu, J. Zhao, B. Dong, C. Liu and Y. Chai, Appl. Mater. Today, 2021, 22, 100913 CrossRef.
- H. S. Hu, Y. Li, Y. R. Shao, K. X. Li, G. Deng, C. B. Wang and Y. Y. Feng, J. Power Sources, 2021, 484, 229269 CrossRef CAS.
- P. Ren, T. Zhang, N. Jain, H. Y. V. Ching, A. Jaworski, G. Barcaro, S. Monti, J. Silvestre Albero, V. Celorrio, L. Chouhan, A. Rokicińska, E. Debroye, P. Kuśtrowski, S. Van Doorslaer, S. Van Aert, S. Bals and S. Das, J. Am. Chem. Soc., 2023, 143, 16584–16596 CrossRef PubMed.
- W. J. Dong, Y. Xiao, K. R. Yang, Z. Ye, P. Zhou, I. A. Navid, V. S. Batista and Z. Mi, Nat. Commun., 2023, 14, 179 CrossRef CAS PubMed.
- H. Shi, T. Wang, J. Liu, W. Chen, S. Li, J. Liang, S. Liu, X. Liu, Z. Cai, C. Wang, D. Su, Y. Huang, L. Elbaz and Q. Li, Nat. Commun., 2023, 14, 3934 CrossRef CAS PubMed.
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H. M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed.
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed.
- P. Li, S. Wang, I. A. Samo, X. Zhang, Z. Wang, C. Wang, Y. Li, Y. Du, Y. Zhong, C. Cheng, W. Xu, X. Liu, Y. Kuang, Z. Lu and X. Sun, Research, 2020, 2020, 2872141 CAS.
- D. Liu, H. Ai, M. Chen, P. Zhou, B. Li, D. Liu, X. Du, K. H. Lo, K. W. Ng, S. P. Wang, S. Chen, G. Xing, J. Hu and H. Pan, Small, 2021, 17, e2007557 CrossRef PubMed.
- D. Wu, D. Chen, J. Zhu and S. Mu, Small, 2021, 17, e2102777 CrossRef PubMed.
- X. Xiao, L. Yang, W. Sun, Y. Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, e2105830 CrossRef PubMed.
- Q. Hu, Z. Wang, X. Huang, Y. Qin, H. Yang, X. Ren, Q. Zhang, J. Liu, M. Shao and C. He, Appl. Catal., B, 2021, 286, 119920 CrossRef CAS.
- M. S. P. Rodney, D. L. Smith, R. D. Fagan, Z. Zhang, P. A. Sedach, S. T. Man Kit Jack Siu and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef PubMed.
- Y. T. Luo, L. Tang, U. Khan, Q. M. Yu, H. M. Cheng, X. L. Zou and B. L. Liu, Nat. Commun., 2019, 10, 269 CrossRef PubMed.
- D. Zhang, Z. Wang, X. Wu, Y. Shi, N. Nie, H. Zhao, H. Miao, X. Chen, S. Li, J. Lai and L. Wang, Small, 2022, 18, e2104559 CrossRef PubMed.
- X. Xiao, X. K. Wang, X. X. Jiang, S. W. Song, D. K. Huang, L. Yu, Y. Zhang, S. Chen, M. K. Wang, Y. Shen and Z. F. Ren, Small Methods, 2020, 4, 1900796 CrossRef CAS.
- S. Xue, Z. B. Liu, C. Q. Ma, H. M. Cheng and W. C. Ren, Sci. Bull., 2020, 65, 123–130 CrossRef CAS PubMed.
- X. Zhang, F. Li, R. Fan, J. Zhao, B. Dong, F. Wang, H. Liu, J. Yu, C. Liu and Y. Chai, J. Mater. Chem. A, 2021, 9, 15836–15845 RSC.
- X. D. Cheng, J. X. Yuan, J. H. Cao, C. J. Lei, B. Yang, Z. J. Li, X. W. Zhang, C. Yuan, L. C. Lei and Y. Hou, J. Colloid Interface Sci., 2020, 579, 340–346 CrossRef CAS PubMed.
- N. G. Deshpande, D. S. Kim, C. H. Ahn, S. H. Jung, Y. B. Kim, H. S. Lee and H. K. Cho, ACS Appl. Mater. Interfaces, 2021, 13, 18772–18783 CrossRef CAS PubMed.
- G. Deng, J. Zhao, H. S. Hu, X. Y. Wang, M. Zhu and Y. Y. Feng, Int. J. Hydrogen Energy, 2021, 46, 37333–37339 CrossRef CAS.
- L. Chen, Y. Wang, X. Zhao, Y. Wang, Q. Li, Q. Wang, Y. Tang and Y. Lei, J. Mater. Sci. Technol., 2021, 110, 128–135 CrossRef.
- G. Li, J. Yu, J. Jia, L. Yang, L. Zhao, W. Zhou and H. Liu, Adv. Funct. Mater., 2018, 28, 1801332 CrossRef.
- M. T. Jin, X. Zhang, R. Shi, Q. Lian, S. Z. Niu, O. W. Peng, Q. Wang and C. Cheng, Appl. Catal., B, 2021, 296, 120350 CrossRef CAS.
- X. Wu, R. Wang, W. Li, B. Feng and W. Hu, ACS Appl. Nano Mater., 2021, 4, 3369–3376 CrossRef CAS.
- A. Kumar, V. Q. Bui, J. Lee, A. R. Jadhav, Y. Hwang, M. G. Kim, Y. Kawazoe and H. Lee, ACS Energy Lett., 2021, 6, 354–363 CrossRef CAS.
- H. Shen, S. Liang, S. Adimi, X. Guo, Y. Zhu, H. Guo, T. Thomas, J. P. Attfield and M. Yang, J. Mater. Chem. A, 2021, 9, 19669–19674 RSC.
- X. Gao, X. Liu, W. Zang, H. Dong, Y. Pang, Z. Kou, P. Wang, Z. Pan, S. Wei, S. Mu and J. Wang, Nano Energy, 2020, 78, 105355 CrossRef CAS.
- Z. Chen, Y. Ha, Y. Liu, H. Wang, H. Yang, H. Xu, Y. Li and R. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 7134–7144 CrossRef CAS PubMed.
- Z. Dan, W. Liang, X. Gong, X. Lin, W. Zhang, Z. Le, F. Xie, J. Chen, M. Yang, N. Wang, Y. Jin and H. Meng, ACS Mater. Lett., 2022, 4, 1374–1380 CrossRef CAS.
- H. Jin, X. Liu, A. Vasileff, Y. Jiao, Y. Zhao, Y. Zheng and S. Z. Qiao, ACS Nano, 2018, 12, 12761–12769 CrossRef CAS PubMed.
- H. Sun, C. Tian, G. Fan, J. Qi, Z. Liu, Z. Yan, F. Cheng, J. Chen, C. Li and M. Du, Adv. Funct. Mater., 2020, 30, 1910596 CrossRef CAS.
- Y. Yang, H. Fei, G. Ruan, C. Xiang and J. M. Tour, ACS Nano, 2014, 8, 9518–9523 CrossRef CAS PubMed.
- D. Vikraman, S. Hussain, K. Akbar, K. Karuppasamy, S. H. Chun, J. Jung and H. S. Kim, ACS Sustainable Chem. Eng., 2018, 7, 458–469 CrossRef.
- B. Cui, Y. Shi, G. Li, Y. Chen, W. Chen, Y. Deng and W. Hu, Acta Phys. Chim. Sin., 2021, 6, 2106010 Search PubMed.
- X. Wang, X. Zhou, C. Li, H. Yao, C. Zhang, J. Zhou, R. Xu, L. Chu, H. Wang, M. Gu, H. Jiang and M. Huang, Adv. Mater., 2022, 34, e2204021 CrossRef PubMed.
- W. Zang, T. Sun, T. Yang, S. Xi, M. Waqar, Z. Kou, Z. Lyu, Y. P. Feng, J. Wang and S. J. Pennycook, Adv. Mater., 2021, 33, e2003846 CrossRef PubMed.
- S. H. Hsu, J. Miao, L. Zhang, J. Gao, H. Wang, H. Tao, S. F. Hung, A. Vasileff, S. Z. Qiao and B. Liu, Adv. Mater., 2018, 30, e1707261 CrossRef PubMed.
- B. Wang, M. Lu, D. Chen, Q. Zhang, W. Wang, Y. Kang, Z. Fang, G. Pang and S. Feng, J. Mater. Chem. A, 2021, 9, 13562–13569 RSC.
- F. Zhang, Y. Liu, F. Yu, H. Pang, X. Zhou, D. Li, W. Ma, Q. Zhou, Y. Mo and H. Zhou, ACS Nano, 2023, 17, 1681–1692 CrossRef CAS PubMed.
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou, X. Xing, T. Tong, Y. Yao, J. Bao, L. Yu, S. Chen and Z. Ren, Adv. Mater., 2022, 34, e2201774 CrossRef PubMed.
- X. F. Lu, S. L. Zhang, W. L. Sim, S. Gao and X. W. D. Lou, Angew. Chem., Int. Ed., 2021, 60, 22885–22891 CrossRef CAS PubMed.
- M. Ning, Y. Wang, L. Wu, L. Yang, Z. Chen, S. Song, Y. Yao, J. Bao, S. Chen and Z. Ren, Nano-Micro Lett., 2023, 15, 157 CrossRef CAS PubMed.
- L. Yu, L. Wu, S. Song, B. McElhenny, F. Zhang, S. Chen and Z. Ren, ACS Energy Lett., 2020, 5, 2681–2689 CrossRef CAS.
- Y. Wang, Y. Du, Z. Fu, M. Wang, Y. Fu, B. Li and L. Wang, Int. J. Hydrogen Energy, 2023, 48, 23412–23424 CrossRef CAS.
- X. Zhai, Q. Yu, J. Chi, X. Wang, B. Li, B. Yang, Z. Li, J. Lai and L. Wang, Nano Energy, 2023, 105, 108008 CrossRef CAS.
- X. Xu, X. Wei, L. Xu, M. Huang and A. Toghan, J. Energy Chem., 2023, 85, 116–125 CrossRef CAS.
- S. Khatun, S. Pal, N. Sinha, C. Das, T. Ahmed and P. Roy, Chem. Commun., 2023, 59, 4578–4599 RSC.
- S. Pal, K. Shimizu, S. Khatun, S. Singha, S. Watanabe and P. Roy, J. Mater. Chem. A, 2023, 11, 12151–12163 RSC.
- N. Sinha and P. Roy, Inorg. Chem., 2023, 62, 3349–3357 CrossRef CAS PubMed.
- J. Shi, B. Jiang, C. Li, F. Yan, D. Wang, C. Yang and J. Wan, Mater. Chem. Phys., 2020, 245, 122533 CrossRef CAS.
- H. Wang, J. Li, K. Li, Y. Lin, J. Chen, L. Gao, V. Nicolosi, X. Xiao and J. M. Lee, Chem. Soc. Rev., 2021, 50, 1354–1390 RSC.
- Z. Yang, C. Chen, Y. Zhao, Q. Wang, J. Zhao, G. I. N. Waterhouse, Y. Qin, L. Shang and T. Zhang, Adv. Mater., 2023, 35, 2208799 CrossRef CAS PubMed.
- S. Yang, J. Kim, Y. J. Tak, A. Soon and H. Lee, Angew. Chem., Int. Ed., 2016, 55, 2058–2062 CrossRef CAS PubMed.
- Y. Yuan, J. Wang, S. Adimi, H. Shen, T. Thomas, R. Ma, J. P. Attfield and M. Yang, Nat. Mater., 2020, 19, 282–286 CrossRef CAS PubMed.
- X. Xiao, H. Wang, W. Bao, P. Urbankowski, L. Yang, Y. Yang, K. Maleski, L. Cui, S. J. L. Billinge, G. Wang and Y. Gogotsi, Adv. Mater., 2019, 31, e1902393 CrossRef PubMed.
- Z. Y. Al Balushi, K. Wang, R. K. Ghosh, R. A. Vilá, S. M. Eichfeld, J. D. Caldwell, X. Qin, Y. Lin, P. A. DeSario, G. Stone, S. Subramanian, D. F. Paul, R. M. Wallace, S. Datta, J. M. Redwing and J. A. Robinson, Nat. Mater., 2016, 15, 1166–1171 CrossRef CAS PubMed.
- Y. Yang, R. Zeng, Y. Xiong, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2019, 141, 19241–19245 CrossRef CAS PubMed.
- Y. Yuan, S. Adimi, X. Guo, T. Thomas, Y. Zhu, H. Guo, G. S. Priyanga, P. Yoo, J. Wang, J. Chen, P. Liao, J. P. Attfield and M. Yang, Angew. Chem., Int. Ed., 2020, 59, 18036–18041 CrossRef CAS PubMed.
- L. Zhang, H. Wei, H. Jiu, C. Wang, Y. Qin, S. Che, Z. Guo and Y. Han, Dalton Trans., 2022, 51, 16733–16739 RSC.
- M. Ning, F. Zhang, L. Wu, X. Xing, D. Wang, S. Song, Q. Zhou, L. Yu, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2022, 15, 3945–3957 RSC.
- Z. Sun, B. Chu, S. Wang, L. Dong, Q. Pang, M. Fan, X. Zhang, H. He, B. Li and Z. Chen, J. Colloid Interface Sci., 2023, 646, 361–369 CrossRef CAS PubMed.
- H. Hu, Z. Zhang, Y. Zhang, T. Thomas, H. Du, K. Huang, J. P. Attfield and M. Yang, Energy Environ. Sci., 2023, 16, 4584–4592 RSC.
- S. Lakshmanan and T. Murugesan, Clean Technol. Environ. Policy, 2013, 16, 225–234 CrossRef.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- X. Zhang, J. Xie, Y. Ma, B. Dong, C. Liu and Y. Chai, Chem. Eng. J., 2022, 430, 132312 CrossRef CAS.
- J. Sun, Z. Zhao, J. Li, Z. Li and X. Meng, Rare Met., 2022, 42, 751–768 CrossRef.
- S. Zhang, Y. Wang, S. Li, Z. Wang, H. Chen, L. Yi, X. Chen, Q. Yang, W. Xu, A. Wang and Z. Lu, Nat. Commun., 2023, 14, 4822 CrossRef CAS PubMed.
- B. Xu, J. Liang, X. Sun and X. Xiong, Green Chem., 2023, 25, 3767–3790 RSC.
- J. Crook and A. Mousavi, Environ. Forensics, 2016, 17, 211–217 CrossRef CAS.
- M. Xiao, Q. Wu, R. Ku, L. Zhou, C. Long, J. Liang, A. Mavric, L. Li, J. Zhu, M. Valant, J. Li, Z. Zeng and C. Cui, Nat. Commun., 2023, 14, 5356 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- G. Liu, J. Electroanal. Chem., 2022, 923, 116805 CrossRef CAS.
- Y. Yokoyama, K. Miyazaki, Y. Miyahara, T. Fukutsuka and T. Abe, ChemElectroChem, 2019, 6, 4750–4756 CrossRef CAS.
- L. Li, P. Wang, Q. Shao and X. Huang, Chem. Soc. Rev., 2020, 49, 3072–3106 RSC.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, e2007100 CrossRef PubMed.
- D. Voiry, M. Chhowalla, Y. Gogotsi, N. A. Kotov, Y. Li, R. M. Penner, R. E. Schaak and P. S. Weiss, ACS Nano, 2018, 12, 9635–9638 CrossRef CAS PubMed.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- M. Boudart, A. Aldag, J. E. Benson, N. A. Dougharty and C. Girvin Harkins, J. Catal., 1966, 6, 92–99 CrossRef CAS.
- J. Yang, X. Du and B. Qiao, Chem. Catal., 2023, 3, 100424 CrossRef CAS.
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13, 12788–12817 RSC.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- S. Parvin, A. Kumar, A. Ghosh and S. Bhattacharyya, Chem. Sci., 2020, 11, 3893–3902 RSC.
- Y. Y. Liu, Y. N. Dou, S. Li, T. Xia, Y. Xie, Y. Wang, W. Zhang, J. P. Wang, L. H. Huo and H. Zhao, Small Methods, 2021, 5, 2000701 CrossRef CAS PubMed.
- T. T. Liu, W. B. Gao, Q. Q. Wang, M. L. Dou, Z. P. Zhang and F. Wang, Angew. Chem., Int. Ed., 2020, 59, 20423–20427 CrossRef CAS PubMed.
- C. Y. Jian, Q. Cai and W. Liu, Chem. Commun., 2021, 57, 11819–11822 RSC.
- Y. Shi and B. Zhang, Chem. Soc. Rev., 2016, 45, 1529–1541 RSC.
- J. T. Mefford, A. R. Akbashev, M. Kang, C. L. Bentley, W. E. Gent, H. D. Deng, D. H. Alsem, Y. S. Yu, N. J. Salmon, D. A. Shapiro, P. R. Unwin and W. C. Chueh, Nature, 2021, 593, 67–73 CrossRef CAS PubMed.
- Y. Zou and S. Wang, Adv. Sci., 2021, 8, 2003579 CrossRef CAS PubMed.
- J. Zhang, W. Xu, Y. Liu, S. F. Hung, W. Liu, Z. Lam, H. B. Tao, H. Yang, W. Cai, H. Xiao, H. Chen and B. Liu, Nano Lett., 2021, 21, 7753–7760 CrossRef CAS PubMed.
- L. M. Cao, Y. W. Hu, S. F. Tang, A. Iljin, J. W. Wang, Z. M. Zhang and T. B. Lu, Adv. Sci., 2018, 5, 1800949 CrossRef PubMed.
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef PubMed.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC.
- L. Liu, Y. Wang, Y. Zhao, Y. Wang, Z. Zhang, T. Wu, W. Qin, S. Liu, B. Jia, H. Wu, D. Zhang, X. Qu, M. Chhowalla and M. Qin, Adv. Funct. Mater., 2022, 32, 2112207 CrossRef CAS.
- C. Sun, H. Wang, S. Ji, X. Wang, V. Linkov, X. Tian, L. Yao, J. Zhao and R. Wang, Sustainable Energy Fuels, 2021, 5, 2747–2752 RSC.
- X. Guo, M. Li, L. He, S. Geng, F. Tian, Y. Song, W. Yang and Y. Yu, Nanoscale, 2021, 13, 14179–14185 RSC.
- L. Zhang, Q. Liang, P. Yang, Y. Huang, W. Chen, X. Deng, H. Yang, J. Yan and Y. Liu, Int. J. Hydrogen Energy, 2019, 44, 24209–24217 CrossRef CAS.
- Z. Wang, L. Xu, F. Huang, L. Qu, J. Li, K. A. Owusu, Z. Liu, Z. Lin, B. Xiang, X. Liu, K. Zhao, X. Liao, W. Yang, Y. Cheng and L. Mai, Adv. Energy Mater., 2019, 9, 1900390 CrossRef.
- J. Deng, P. Ren, D. Deng and X. Bao, Angew. Chem., Int. Ed., 2015, 54, 2100–2104 CrossRef CAS PubMed.
- Q. Liu, J. Tian, W. Cui, P. Jiang, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 6710–6714 CrossRef CAS PubMed.
- D. Gao, J. Ren, H. Wang, X. Wang, Y. Liu and R. Wang, J. Power Sources, 2021, 516, 230635 CrossRef CAS.
- S. Ge, R. Xie, B. Huang, Z. Zhang, H. Liu, X. Kang, S. Hu, S. Li, Y. Luo, Q. Yu, J. Wang, G. Chai, L. Guan, H. Cheng and B. Liu, Energy Environ. Sci., 2023, 16, 3734–3742 RSC.
- J. Xie, H. Qu, J. Xin, X. Zhang, G. Cui, X. Zhang, J. Bao, B. Tang and Y. Xie, Nano Res., 2017, 10, 1178–1188 CrossRef CAS.
- H. Liu, X. Li, L. Chen, X. Zhu, P. Dong, M. O. L. Chee, M. Ye, Y. Guo and J. Shen, Adv. Funct. Mater., 2021, 32, 2107308 CrossRef.
- X. Xu, G. Fu, Y. Wang, Q. Cao, Y. Xun, C. Li, C. Guan and W. Huang, Nano Lett., 2023, 23, 629–636 CrossRef CAS PubMed.
- J. Zhang, L. Chen, B. Lu and Y. Guo, ChemSusChem, 2022, 15, e202200937 CrossRef CAS PubMed.
- J. Du, F. Li and L. Sun, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- S. Dresp, T. Ngo Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
- K. Wang, Y. Wang, B. Yang, Z. Li, X. Qin, Q. Zhang, L. Lei, M. Qiu, G. Wu and Y. Hou, Energy Environ. Sci., 2022, 15, 2356–2365 RSC.
- R. Zhang, L. Pan, B. Guo, Z. F. Huang, Z. Chen, L. Wang, X. Zhang, Z. Guo, W. Xu, K. P. Loh and J. J. Zou, J. Am. Chem. Soc., 2023, 145, 2271–2281 CrossRef CAS PubMed.
- T. Wu, E. Song, S. Zhang, M. Luo, C. Zhao, W. Zhao, J. Liu and F. Huang, Adv. Mater., 2022, 34, e2108505 CrossRef PubMed.
- Q. Chen, N. Gong, T. Zhu, C. Yang, W. Peng, Y. Li, F. Zhang and X. Fan, Small, 2022, 18, e2105696 CrossRef PubMed.
- H. A. Hansen, I. C. Man, F. Studt, F. Abild-Pedersen, T. Bligaard and J. Rossmeisl, Phys. Chem. Chem. Phys., 2010, 12, 283–290 RSC.
- G. Bahuguna, B. Filanovsky and F. Patolsky, Nano Energy, 2023, 111, 108439 CrossRef CAS.
- N. Wang, P. Ou, S. F. Hung, J. E. Huang, A. Ozden, J. Abed, I. Grigioni, C. Chen, R. K. Miao, Y. Yan, J. Zhang, Z. Wang, R. Dorakhan, A. Badreldin, A. Abdel Wahab, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, Adv. Mater., 2023, 35, e2210057 CrossRef PubMed.
- F. Yang, Y. Luo, Q. Yu, Z. Zhang, S. Zhang, Z. Liu, W. Ren, H. M. Cheng, J. Li and B. Liu, Adv. Funct. Mater., 2021, 31, 2010367 CrossRef CAS.
- J. Chi, L. Guo, J. Mao, T. Cui, J. Zhu, Y. Xia, J. Lai and L. Wang, Adv. Funct. Mater., 2023, 2300625 CrossRef CAS.
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang, Z. Tian, Z. Lu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 22740–22744 CrossRef CAS PubMed.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. M. Koper, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS PubMed.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- W. Liu, J. Yu, M. G. Sendeku, T. Li, W. Gao, G. Yang, Y. Kuang and X. Sun, Angew. Chem., Int. Ed., 2023, 62, e202309882 CrossRef CAS PubMed.
- C. Huang, Q. Zhou, D. Duan, L. Yu, W. Zhang, Z. Wang, J. Liu, B. Peng, P. An, J. Zhang, L. Li, J. Yu and Y. Yu, Energy Environ. Sci., 2022, 15, 4647–4658 RSC.
- J. Chang, G. Wang, Z. Yang, B. Li, Q. Wang, R. Kuliiev, N. Orlovskaya, M. Gu, Y. Du, G. Wang and Y. Yang, Adv. Mater., 2021, 33, e2101425 CrossRef PubMed.
- L. Zhang, Z. Wang and J. Qiu, Adv. Mater., 2022, 34, e2109321 CrossRef PubMed.
- Y. Yang, R. Zou, J. Gan, Y. Wei, Z. Chen, X. Li, S. Admassie, Y. Liu and X. Peng, Green Chem., 2023, 25, 4104–4112 RSC.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2021, 5, 66–73 CrossRef.
- J. Zheng, W. Zhang, J. Zhang, M. Lv, S. Li, H. Song, Z. Cui, L. Du and S. Liao, J. Mater. Chem. A, 2020, 8, 20803–20818 RSC.
- Q. Yun, Q. Lu, C. Li, B. Chen, Q. Zhang, Q. He, Z. Hu, Z. Zhang, Y. Ge, N. Yang, J. Ge, Y.-B. He, L. Gu and H. Zhang, ACS Nano, 2019, 13, 14329–14336 CrossRef CAS PubMed.
- Z. Li, M. Saruyama, T. Asaka, Y. Tatetsu and T. Teranishi, Science, 2021, 373, 332–337 CrossRef CAS PubMed.
- M. Zhou, J. Liu, C. Ling, Y. Ge, B. Chen, C. Tan, Z. Fan, J. Huang, J. Chen, Z. Liu, Z. Huang, J. Ge, H. Cheng, Y. Chen, L. Dai, P. Yin, X. Zhang, Q. Yun, J. Wang and H. Zhang, Adv. Mater., 2022, 34, 2106115 CrossRef CAS PubMed.
- Q. Zhang, K. Kusada, D. Wu, T. Yamamoto, T. Toriyama, S. Matsumura, S. Kawaguchi, Y. Kubota and H. Kitagawa, J. Am. Chem. Soc., 2022, 144, 4224–4232 CrossRef CAS PubMed.
- D. Meng, S. Zhang, T. Thomas, C. Huang, J. Zhao, R. Zhao, Y. Shi, F. Qu and M. Yang, Sens. Actuators, B, 2020, 307, 127686 CrossRef.
- F. Gao, Q. Wan, J. Yuan, R. Lei, S. Lin and P. Liu, Appl. Catal., B, 2021, 299, 120692 CrossRef CAS.
- S. Liu, W. Kuang, X. Meng, W. Qi, S. Adimi, H. Guo, X. Guo, E. Pervaiz, Y. Zhu, D. Xue and M. Yang, Chem. Eng. J., 2021, 416, 129116 CrossRef CAS.
- G. D. Sun, G. H. Zhang and K. C. Chou, J. Am. Ceram. Soc., 2018, 101, 2796–2808 CrossRef CAS.
- B. Cao, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, Inorg. Chem., 2015, 54, 2128–2136 CrossRef CAS PubMed.
- M. E. Kreider, M. B. Stevens, Y. Liu, A. M. Patel, M. J. Statt, B. M. Gibbons, A. Gallo, M. Ben-Naim, A. Mehta, R. C. Davis, A. V. Ievlev, J. K. Nørskov, R. Sinclair, L. A. King and T. F. Jaramillo, Chem. Mater., 2020, 32, 2946–2960 CrossRef CAS.
- J. Qi, L. Jiang, Q. Jiang, S. Wang and G. Sun, J. Phys. Chem. C, 2010, 114, 18159–18166 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- R. Qiang, H. Wang, K. Xu, Q. Yuan, Y. Yu, L. Li, J. Wang, L. Zheng, P. C. Sherrell, J. Chen and X. Bi, Adv. Mater. Interfaces, 2021, 8, 2100070 CrossRef CAS.
- A. Leineweber, H. Jacobs and S. Hull, Inorg. Chem., 2001, 40, 5818–5822 CrossRef CAS PubMed.
- P. Chen, K. Xu, Y. Tong, X. Li, S. Tao, Z. Fang, W. Chu, X. Wu and C. Wu, Inorg. Chem. Front., 2016, 3, 236–242 RSC.
- H. Zhang, J. Wang, Y. Meng, F. Lu, M. Ji, C. Zhu, J. Xu and J. Sun, Energy Mater. Adv., 2022, 2022, 0006 CrossRef.
- G. Fu, Z. Cui, Y. Chen, L. Xu, Y. Tang and J. B. Goodenough, Nano Energy, 2017, 39, 77–85 CrossRef CAS.
- A. Saad, H. Shen, Z. Cheng, R. Arbi, B. Guo, L. S. Hui, K. Liang, S. Liu, J. P. Attfield, A. Turak, J. Wang and M. Yang, Nano-Micro Lett., 2020, 12, 79 CrossRef CAS PubMed.
- W. Qi, C. Wang, J. Yu, S. Adimi, T. Thomas, H. Guo, S. Liu and M. Yang, ACS Appl. Energy Mater., 2022, 5, 6155–6162 CrossRef CAS.
- J. Zhang, L. Zhang, L. Du, H. L. Xin, J. B. Goodenough and Z. Cui, Angew. Chem., Int. Ed., 2020, 59, 17488–17493 CrossRef CAS PubMed.
- S. He, Y. Chen, M. Wang, K. Liu, P. Novello, X. Li, S. Zhu and J. Liu, Nano Energy, 2021, 80, 105528 CrossRef CAS.
- H. Jin, H. Zhang, J. Chen, S. Mao, Z. Jiang and Y. Wang, J. Mater. Chem. A, 2018, 6, 10967–10975 RSC.
- Y. Zhang, B. Ouyang, J. Xu, S. Chen, R. S. Rawat and H. J. Fan, Adv. Energy Mater., 2016, 6, 1600221 CrossRef.
- X. Liao, R. Lu, L. Xia, Q. Liu, H. Wang, K. Zhao, Z. Wang and Y. Zhao, Energy Environ. Mater., 2021, 5, 157–185 CrossRef.
- Y. Feng, N. Ran, X. Wang, Q. Liu, J. Wang, L. Liu, K. Suenaga, W. Zhong, R. Ma and J. Liu, Adv. Energy Mater., 2023, 2302452 CrossRef CAS.
- R. A. Meyer, E. P. Parry and J. H. Davis, Anal. Chem., 1967, 39, 1321–1323 CrossRef CAS.
- M. Shekhirev, C. E. Shuck, A. Sarycheva and Y. Gogotsi, Prog. Mater. Sci., 2021, 120, 100757 CrossRef CAS.
- X. Zhou, W. Xu, Z. Gui, C. Gu, J. Chen, J. Xie, X. Yao, J. Dai, J. Zhu, L. Wu, E. J. Guo, X. Yu, L. Fang, Y. Zhao, L. Huang and S. Wang, Adv. Sci., 2023, 10, 2205479 CrossRef CAS PubMed.
- F. Tessier, Materials, 2018, 11, 1331 CrossRef PubMed.
- J. M. Joubert, Intermetallics, 2022, 146, 107580 CrossRef CAS.
- Y. Chen, H. Liu, N. Ha, S. Licht, S. Gu and W. Li, Nat. Catal., 2020, 3, 1055–1061 CrossRef CAS.
- S. Tan, Y. Dai, Y. Jiang, Q. Wei, G. Zhang, F. Xiong, X. Zhu, Z. Y. Hu, L. Zhou, Y. Jin, K. Kanamura, Q. An and L. Mai, Adv. Funct. Mater., 2021, 31, 2008034 CrossRef CAS.
- J. Masa and W. Schuhmann, ChemCatChem, 2019, 11, 5842–5854 CrossRef CAS.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428–8469 RSC.
- C. Xia, Q. Jiang, C. Zhao, M. N. Hedhili and H. N. Alshareef, Adv. Mater., 2016, 28, 77–85 CrossRef CAS PubMed.
- X. Cui, P. Xiao, J. Wang, M. Zhou, W. Guo, Y. Yang, Y. He, Z. Wang, Y. Yang, Y. Zhang and Z. Lin, Angew. Chem., Int. Ed., 2017, 56, 4488–4493 CrossRef CAS PubMed.
- Y. Yuan, S. Adimi, T. Thomas, J. Wang, H. Guo, J. Chen, J. P. Attfield, F. J. DiSalvo and M. Yang, Innovation, 2021, 2, 100096 CAS.
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- D. Liu, H. Ai, J. Li, M. Fang, M. Chen, D. Liu, X. Du, P. Zhou, F. Li, K. H. Lo, Y. Tang, S. Chen, L. Wang, G. Xing and H. Pan, Adv. Energy Mater., 2020, 10, 2002464 CrossRef CAS.
- X. Gao, X. Li, Y. Yu, Z. Kou, P. Wang, X. Liu, J. Zhang, J. He, S. Mu and J. Wang, Nano Energy, 2021, 85, 105961 CrossRef CAS.
- G. Lin, Z. Zhang, Q. Ju, T. Wu, C. U. Segre, W. Chen, H. Peng, H. Zhang, Q. Liu, Z. Liu, Y. Zhang, S. Kong, Y. Mao, W. Zhao, K. Suenaga, F. Huang and J. Wang, Nat. Commun., 2023, 14, 280 CrossRef CAS PubMed.
- Y. Zhou, W. Guo and T. Li, Ceram. Int., 2019, 45, 21062–21076 CrossRef CAS.
- E. Carlos, R. Martins, E. Fortunato and R. Branquinho, Chem. – Eur. J., 2020, 26, 9099–9125 CrossRef CAS PubMed.
- P. Wang, J. Bai, B. Zhao, H. Ma, W. Li, X. Zhu and Y. Sun, ACS Appl. Mater. Interfaces, 2022, 14, 38875–38886 CrossRef CAS PubMed.
- W. Yuan, S. Wang, Y. Ma, Y. Qiu, Y. An and L. Cheng, ACS Energy Lett., 2020, 5, 692–700 CrossRef CAS.
- S. A. Rasaki, B. Zhang, K. Anbalgam, T. Thomas and M. Yang, Prog. Solid State Chem., 2018, 50, 1–15 CrossRef CAS.
- H. S. Gujral, G. Singh, A. V. Baskar, X. Guan, X. Geng, A. V. Kotkondawar, S. Rayalu, P. Kumar, A. Karakoti and A. Vinu, Sci. Technol. Adv. Mater., 2022, 23, 76–119 CrossRef CAS PubMed.
- K. Sue, M. Suzuki, K. Arai, T. Ohashi, H. Ura, K. Matsui, Y. Hakuta, H. Hayashi, M. Watanabe and T. Hiaki, Green Chem., 2006, 8, 634–638 RSC.
- P. Chirico, A. L. Hector and B. Mazumder, Dalton Trans., 2010, 39, 6092–6097 RSC.
- S. I. U. Shah, A. L. Hector and J. R. Owen, J. Power Sources, 2014, 266, 456–463 CrossRef CAS.
- B. Mazumder, P. Chirico and A. L. Hector, Inorg. Chem., 2008, 47, 9684–9690 CrossRef CAS PubMed.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- S.-x Yan, S.-h Luo, M.-z Sun, Q. Wang, Y.-h Zhang and X. Liu, Int. J. Energy Res., 2021, 45, 20186–20198 CrossRef CAS.
- M.-S. Balogun, W. Qiu, J. Jian, Y. Huang, Y. Luo, H. Yang, C. Liang, X. Lu and Y. Tong, ACS Appl. Mater. Interfaces, 2015, 7, 23205–23215 CrossRef CAS PubMed.
- J. Jia, M. Zhai, J. Lv, B. Zhao, H. Du and J. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 30400–30408 CrossRef CAS PubMed.
- D. M. Kim, S. J. Kim, Y. W. Lee, D. H. Kwak, H. C. Park, M. C. Kim, B. M. Hwang, S. Lee, J. H. Choi, S. Hong and K. W. Park, Electrochim. Acta, 2015, 163, 132–139 CrossRef CAS.
- J. Sheng, L. Wang, L. Deng, M. Zhang, H. He, K. Zeng, F. Tang and Y. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 7191–7200 CrossRef CAS PubMed.
- A. Wu, Y. Gu, B. Yang, H. Wu, H. Yan, Y. Jiao, D. Wang, C. Tian and H. Fu, J. Mater. Chem. A, 2020, 8, 22938–22946 RSC.
- S. B. Ghantasala and S. Sharma, Phys. Status Solidi A, 2023, 220, 2200229 CrossRef CAS.
- S. Prasad, G. Durai, D. Devaraj, M. S. AlSalhi, J. Theerthagiri, P. Arunachalam, M. Gurulakshmi, M. Raghavender and P. Kuppusami, RSC Adv., 2018, 8, 8828–8835 RSC.
- D. Zhang, H. Li, A. Riaz, A. Sharma, W. Liang, Y. Wang, H. Chen, K. Vora, D. Yan, Z. Su, A. Tricoli, C. Zhao, F. J. Beck, K. Reuter, K. Catchpole and S. Karuturi, Energy Environ. Sci., 2022, 15, 185–195 RSC.
- Z. Qi, Y. Zeng, Z. Hou, W. Zhu, B. Wei, Y. Yang, B. Lin and H. Liang, Nano Res., 2023, 16, 4803–4811 CrossRef CAS.
- A. P. Murthy, D. Govindarajan, J. Theerthagiri, J. Madhavan and K. Parasuraman, Electrochim. Acta, 2018, 283, 1525–1533 CrossRef CAS.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- Z. Feng, P. Ke and A. Wang, J. Mater. Sci. Technol., 2015, 31, 1193–1197 CrossRef CAS.
- B. Wei, H. Liang, D. Zhang, Z. Wu, Z. Qi and Z. Wang, J. Mater. Chem. A, 2017, 5, 2844–2851 RSC.
- H. Liang, J. Xu, D. Zhou, X. Sun, S. Chu and Y. Bai, Ceram. Int., 2016, 42, 2642–2647 CrossRef CAS.
- O. K. Alexeeva and V. N. Fateev, Int. J. Hydrogen Energy, 2016, 41, 3373–3386 CrossRef CAS.
- A. L. M. Sandhya, P. Pleskunov, M. Bogar, X. Xie, P. A. Wieser, M. Orság, T. N. Dinhová, M. Dopita, R. Taccani, H. Amenitsch, A. Choukourov, I. Matolínová and I. Khalakhan, Surf. Interfaces, 2023, 40, 103079 CrossRef CAS.
- Z. Cheng, A. Saad, H. Guo, C. Wang, S. Liu, T. Thomas and M. Yang, J. Alloys Compd., 2020, 838, 155375 CrossRef CAS.
- A. Saad, Z. Cheng, X. Zhang, S. Liu, H. Shen, T. Thomas, J. Wang and M. Yang, Adv. Mater. Interfaces, 2019, 6, 1900960 CrossRef CAS.
- K. Suzuki, T. Kaneko, H. Yoshida, H. Morita and H. Fujimori, J. Alloys Compd., 1995, 224, 232–236 CrossRef CAS.
- X. Zhang, Y. Zhao, X. Jia, Y. Zhao, L. Shang, Q. Wang, G. I. N. Waterhouse, L. Wu, C. H. Tung and T. Zhang, Adv. Energy Mater., 2018, 8, 1702780 CrossRef.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S. Yu and H. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- S. N. Talapaneni, D. H. Park, J. H. Choy, K. Ramadass, A. Elzatahry, A. S. Al Balawi, A. M. Al Enizi, T. Mori and A. Vinu, ChemistrySelect, 2016, 1, 6062–6068 CrossRef CAS.
- M. F. Sarac, W. Wu and J. B. Tracy, Chem. Mater., 2014, 26, 3057–3064 CrossRef CAS.
- H. Qi, X. Roy, K. E. Shopsowitz, J. K.-H. Hui and M. J. MacLachlan, Angew. Chem., Int. Ed., 2010, 49, 9740–9743 CrossRef CAS PubMed.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, H. Yang, Z. Li, C. Yu, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2005, 44, 7053–7059 CrossRef CAS PubMed.
- A. K. Tareen, G. S. Priyanga, S. Behara, T. Thomas and M. Yang, Prog. Solid State Chem., 2019, 53, 1–26 CrossRef CAS.
- J. Buha, I. Djerdj, M. Antonietti and M. Niederberger, Chem. Mater., 2007, 19, 3499–3505 CrossRef CAS.
- C. Giordano, C. Erpen, W. Yao and M. Antonietti, Nano Lett., 2008, 8, 4659–4663 CrossRef CAS PubMed.
- C. Giordano, C. Erpen, W. Yao, B. Milke and M. Antonietti, Chem. Mater., 2009, 21, 5136–5144 CrossRef CAS.
- M. Yang and F. J. DiSalvo, Chem. Mater., 2012, 24, 4406–4409 CrossRef CAS.
- A. Badreldin, A. Nabeeh, Z. K. Ghouri, J. Abed, N. Wang, Y. Wubulikasimu, K. Youssef, D. Kumar, M. K. Stodolny, K. Elsaid, E. H. Sargent and A. Abdel-Wahab, ACS Appl. Mater. Interfaces, 2021, 13, 53702–53716 CrossRef CAS PubMed.
- H. Sun, J. Sun, Y. Song, Y. Zhang, Y. Qiu, M. Sun, X. Tian, C. Li, Z. Lv and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 22061–22070 CrossRef CAS PubMed.
- X. Wang, X. Han, R. Du, C. Xing, X. Qi, Z. Liang, P. Guardia, J. Arbiol, A. Cabot and J. Li, ACS Appl. Mater. Interfaces, 2022, 14, 41924–41933 CrossRef CAS PubMed.
- A. Tewary, S. Mandal, Z. Alam, A. S. K. Sinha and U. Ojha, ACS Sustainable Chem. Eng., 2023, 11, 6556–6566 CrossRef CAS.
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S. Z. Qiao, Adv. Mater., 2021, 33, e2007508 CrossRef PubMed.
- J. Miao, Z. Lang, X. Zhang, W. Kong, O. Peng, Y. Yang, S. Wang, J. Cheng, T. He, A. Amini, Q. Wu, Z. Zheng, Z. Tang and C. Cheng, Adv. Funct. Mater., 2018, 29, 1805893 CrossRef.
- F. Ma, S. Wang, X. Gong, X. Liu, Z. Wang, P. Wang, Y. Liu, H. Cheng, Y. Dai, Z. Zheng and B. Huang, Appl. Catal., B, 2022, 307, 121198 CrossRef CAS.
- R. Li, Y. Li, P. Yang, P. Ren, D. Wang, X. Lu, R. Xu, Y. Li, J. Xue, J. Zhang, M. An, J. Ma, B. Wang, H. Liu and S. Dou, Appl. Catal., B, 2022, 318, 121834 CrossRef CAS.
- B. Jiang, J. Li, Y. Cui, S. Shi, N. Jiang and J. Guan, J. Alloys Compd., 2023, 958, 170371 CrossRef CAS.
- Y. Zhao, B. Jin, A. Vasileff, Y. Jiao and S.-Z. Qiao, J. Mater. Chem. A, 2019, 7, 8117–8121 RSC.
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef PubMed.
- W. Ma, D. Li, L. Liao, H. Zhou, F. Zhang, X. Zhou, Y. Mo and F. Yu, Small, 2023, 19, e2207082 CrossRef PubMed.
- R. Li, X. Zhou, H. Shen, M. Yang and C. Li, ACS Nano, 2019, 13, 10049–10061 CrossRef CAS PubMed.
- Y. Pei, B. Rezaei, X. Zhang, Z. Li, H. Shen, M. Yang and J. Wang, Mater. Chem. Front., 2020, 4, 2665–2672 RSC.
- S. Liang, M. Jing, T. Thomas, J. Liu, H. Guo, J. P. Attfield, A. Saad, H. Shen and M. Yang, Sustainable Energy Fuels, 2020, 4, 6245–6250 RSC.
- T. Wu, S. Xu, Z. Zhang, M. Luo, R. Wang, Y. Tang, J. Wang and F. Huang, Adv. Sci., 2022, 9, e2202750 CrossRef PubMed.
- R. Li, H. Peng, Q. Wu, X. Zhou, J. He, H. Shen, M. Yang and C. Li, Angew. Chem., Int. Ed., 2020, 59, 12129–12138 CrossRef CAS PubMed.
- M. Batool, A. Hameed and M. A. Nadeem, Coord. Chem. Rev., 2023, 480, 215029 CrossRef CAS.
- J. Li, M. Song, Y. Hu, Y. Zhu, J. Zhang and D. Wang, Small Methods, 2023, 7, 2201616 CrossRef CAS PubMed.
- S. Lai, L. Xu, H. Liu, S. Chen, R. Cai, L. Zhang, W. Theis, J. Sun, D. Yang and X. Zhao, J. Mater. Chem. A, 2019, 7, 21884–21891 RSC.
- Y. Xue, N. N. T. Pham, G. Nam, J. Choi, Y. Y. Ahn, H. Lee, J. Jung, S. G. Lee and J. Lee, Chem. Eng. J., 2021, 408, 127305 CrossRef CAS.
- L. Xu, C. Wang, D. Deng, Y. Tian, X. He, G. Lu, J. Qian, S. Yuan and H. Li, ACS Sustainable Chem. Eng., 2020, 8, 343–350 CrossRef CAS.
- S. Li, Z. Zhao, T. Ma, P. Pachfule and A. Thomas, Angew. Chem., Int. Ed., 2022, 61, e202112298 CrossRef CAS PubMed.
- Y. Chen, J. Yu, J. Jia, F. Liu, Y. Zhang, G. Xiong, R. Zhang, R. Yang, D. Sun, H. Liu and W. Zhou, Appl. Catal., B, 2020, 272, 118956 CrossRef CAS.
- W. Tian, J. Ren and Z. Yuan, Appl. Catal., B, 2022, 317, 121764 CrossRef CAS.
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS.
- S. Liang, H. Hu, J. Liu, H. Shen, Q. Li, N. Qiu, H. Guo, X. Guo, S. Du, Y. Zhu, J. Liu, J. P. Attfield and M. Yang, Appl. Catal., B, 2023, 337, 123008 CrossRef CAS.
- Z. Wang, J. Lu, S. Ji, H. Wang, X. Wang, B. G. Pollet and R. Wang, J. Alloys Compd., 2021, 867, 158983 CrossRef CAS.
- F. Yang, Y. Chen, G. Cheng, S. Chen and W. Luo, ACS Catal., 2017, 7, 3824–3831 CrossRef CAS.
- X. Yan, S. Che, F. Yang, Z. Xu, H. Liu, C. Li, L. Yan, N. Ta, S. Sun, Q. Wei, L. Fang and Y. Li, ACS Appl. Mater. Interfaces, 2022, 14, 20358–20367 CrossRef CAS PubMed.
- C. Dai, R. Li, H. Guo, S. Liang, H. Shen, T. Thomas and M. Yang, Dalton Trans., 2021, 50, 5416–5419 RSC.
- R. Wang, X. Y. Dong, J. Du, J. Y. Zhao and S. Q. Zang, Adv. Mater., 2018, 30, 1703711 CrossRef PubMed.
- Y. Xu, M. Liu, S. Wang, K. Ren, M. Wang, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2021, 298, 120493 CrossRef CAS.
- A. Salamat, A. L. Hector, P. Kroll and P. F. McMillan, Coord. Chem. Rev., 2013, 257, 2063–2072 CrossRef CAS.
- B. You, X. Liu, G. Hu, S. Gul, J. Yano, D. Jiang and Y. Sun, J. Am. Chem. Soc., 2017, 139, 12283–12290 CrossRef CAS PubMed.
- Y. Xu, G. Li and Y. Xia, Appl. Surf. Sci., 2020, 523, 146529 CrossRef CAS.
- J. Hou, W. Song, L. Lan and J. Qiao, J. Mater. Sci. Technol., 2020, 48, 140–145 CrossRef CAS.
- L. Zhu, C. Li, R. Zheng, W. Cheng, Y. He, C. Gong, M. Liu, Y. Huang, X. Zhu and Y. Sun, Int. J. Hydrogen Energy, 2023 DOI:10.1016/j.ijhydene.2023.07.011.
- W. Zhuang, G. Pan, W. Gu, S. Zhou, Q. Hu, Z. Gu, Z. Wu, S. Lu and H. Qiu, Energy Environ. Sci., 2023, 16, 2014–2029 RSC.
| This journal is © The Royal Society of Chemistry 2024 |