
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Therapeutic synthetic and natural materials for immunoengineering
Anna
Slezak
a,
Kevin
Chang
 a,
Samir
Hossainy
a,
Aslan
Mansurov
a,
Stuart J.
Rowan
ab,
Jeffrey A.
Hubbell
*a and
Mustafa O.
Guler
*a
a,
Samir
Hossainy
a,
Aslan
Mansurov
a,
Stuart J.
Rowan
ab,
Jeffrey A.
Hubbell
*a and
Mustafa O.
Guler
*a
aThe Pritzker School of Molecular Engineering, The University of Chicago, Chicago, IL 60637, USA. E-mail: jhubbell@uchicago.edu; mguler@uchicago.edu
bDepartment of Chemistry, The University of Chicago, Chicago, IL 60637, USA
First published on 3rd January 2024
Abstract
Immunoengineering is a rapidly evolving field that has been driving innovations in manipulating immune system for new treatment tools and methods. The need for materials for immunoengineering applications has gained significant attention in recent years due to the growing demand for effective therapies that can target and regulate the immune system. Biologics and biomaterials are emerging as promising tools for controlling immune responses, and a wide variety of materials, including proteins, polymers, nanoparticles, and hydrogels, are being developed for this purpose. In this review article, we explore the different types of materials used in immunoengineering applications, their properties and design principles, and highlight the latest therapeutic materials advancements. Recent works in adjuvants, vaccines, immune tolerance, immunotherapy, and tissue models for immunoengineering studies are discussed.
 Anna Slezak | Anna J. Slezak is a graduate student in the Pritzker School of Molecular Engineering at the University of Chicago, working in the lab of Jeffrey Hubbell. Anna received her BS in Chemistry from Duke University in 2020. Her current research focuses on the development of polymeric materials for in situ cancer vaccination. |
 Kevin Chang | Kevin Chang is an MD-PhD student at the University of Chicago doing his PhD in Molecular Engineering with Jeffrey Hubbell. He graduated from Yale University in 2020 with a BS in Molecular Biophysics and Biochemistry. Kevin currently works on developing novel cancer immunotherapies that redirect existing vaccine immunity against tumors. |
 Samir Hossainy | Samir Hossainy received his BS/MS in bioengineering and materials science from the University of California, Berkeley in 2019. He joined the Pritzker School of Molecular Engineering in Autumn 2019 as a PhD student and is currently co-advised by Professors Stuart Rowan and Jeffrey Hubbell. Samir currently designs thermally responsive, polymeric nanoparticles as delivery vehicles for subunit vaccines, inducing immune tolerance and cancer therapies. |
 Aslan Mansurov | Aslan Mansurov graduated from the University of Minnesota in 2017, where he earned his bachelor's degree in chemical engineering with distinction. He then went on to pursue a PhD degree in molecular engineering at the University of Chicago, which he completed in 2021 in the laboratory of Jeffrey Hubbell. His research focuses on improving the therapeutic properties of immunotherapy agents. Aslan is currently a postdoctoral scholar at the University of Chicago. |
 Stuart J. Rowan | Stuart J. Rowan is the Barry L. MacLean Professor of Molecular Engineering and Professor of Chemistry at the University of Chicago. He is the Director of the University of Chicago's Materials Research Science and Engineering Center (MRSEC) and has a staff appointment at Argonne National Labs. He is an ACS Fellow, an ACS POLY Fellow and a Fellow of the Royal Society of Chemistry. His group works on supramolecular polymers, dynamic covalent polymers, self-healing materials, responsive adhesives, sustainable plastics, nanocellulose, polymers for battery applications, biomaterials and developing new synthetic methods for the construction of complex polymeric architectures. |
 Jeffrey A. Hubbell | Jeffrey A Hubbell is Eugene Bell Professor in Tissue Engineering at the Pritzker School of Molecular Engineering of the University of Chicago. Previously, he was on the faculty of the Swiss Federal Institute of Technology Lausanne (EPFL, where he served as Director of the Institute of Bioengineering and Dean of the School of Life Sciences), the Swiss Federal Institute of Technology Zurich and University of Zurich, the California Institute of Technology, and the University of Texas in Austin. He was elected to the US National Academy of Engineering in 2010, the National Academy of Inventors in 2014, the National Academy of Medicine in 2019, and the American Academy of Arts and Sciences in 2021, and he will be inducted into the National Academy of Sciences in its class of 2024. |
 Mustafa O. Guler | Mustafa O. Guler is a Senior Instructional Professor at the Pritzker School of Molecular Engineering at the University of Chicago and a Fellow of the Royal Society of Chemistry (FRSC). He received his PhD from Northwestern University, Department of Chemistry, and worked as a postdoctoral research fellow at the School of Medicine, Institute for Bionanotechnology for Medicine at Northwestern University. Previously, he was a professor of materials science and nanotechnology at Bilkent University. |
1 Introduction
Immunoengineering is an interdisciplinary field that aims to design and develop novel immunomodulatory agents, vaccines, and therapeutics by using the fundamentals of bioengineering, materials science, and immunology. Recently, therapeutic materials for a broad range of diseases, including cancer, infectious diseases, and autoimmune disorders have been developed.1–3 Immunoengineering efforts have been focused on advancing new approaches to modulate the immune system's function, either by enhancing or suppressing it, to achieve targeted therapeutic outcomes.4The development of vaccines relies heavily on the ability to design and engineer antigens that can elicit a robust and specific immune response against a particular pathogenic marker. Immunoengineering has enabled the development of novel vaccine platforms, including protein and RNA vaccines, viral vectors, and nanoparticle-based vaccines, which have shown promising results in preclinical and clinical studies.5 Immunomodulatory materials including adjuvants alter the immune response to a particular antigen and are added to vaccines. They stimulate the immune system to produce a stronger and longer-lasting response to the vaccine. Commonly used adjuvants include aluminum salts, oil-in-water emulsions such as squalines, biologically-derived materials such as saponins, and pattern recognition receptor (PRR) agonists like Toll-like receptor (TLR) agonists.6 The TLR agonists can be used as immunomodulators to stimulate the immune response to a particular antigen.7 Examples of TLR agonists include Toll-like Receptor 7 (TLR7) agonist Imiquimod, TLR9 Agonist CpG, and TLR7 and TLR8 Agonist Resiquimod.8,9
Immunoengineering tools are also used to induce an immune tolerance effect. Immune tolerance induction aims to prevent or treat autoimmune diseases, allergies, and transplant rejection, where the body generates an inappropriate immune response to a non-pathogenic antigen. Strategies to induce immunological tolerance involve the use of engineered cells, nanoparticles, and biomaterials to prevent overactivation of the immune system against self-antigens or non-pathogenic foreign antigens.10
Another important application of biomaterials in immunoengineering is using antibody- and cytokine-containing systems to trigger immune response, particularly against cancer. Cancer immunotherapy involves the modulation of the immune system to target and destroy malignant or transformed cells by monoclonal antibodies and cytokine therapies.11 Monoclonal antibodies are particularly useful in blocking certain immune checkpoints that inhibit the cancer-killing activity of T cells. Immune checkpoints are typically cell–surface molecules that dampen the magnitude of an immune response. Examples of monoclonal antibodies used in immuno-oncology include pembrolizumab, nivolumab, and ipilimumab.12 Cytokines are soluble factors that play a role in regulating the immune response and can be broadly classified into pro- and anti-inflammatory ones. Pro-inflammatory cytokines can be used effectively in immuno-oncology to enhance the activity of T cells or natural killer cells. Examples of cytokines used in immunotherapy include interleukins (e.g., IL-2, IL-12) and interferons (e.g., IFN-α, IFN-γ).13 Protein engineering approaches aimed at improving the therapeutic properties of immune checkpoint inhibitors and cytokines remain an active area of research.14
Nanoparticles can also act as immunomodulators by targeting a particular type of immune cells and delivering antigens, genetic material or other immunomodulators for a variety of disease applications.15,16 Types of nanoparticles that are often employed for developing novel immunotherapies include fully synthetic polymers (i.e., polymersomes), lipid nanoparticles, and inorganic nanoparticles17 More specific examples of each of these are provided in the following chapters of this review. Broadly speaking, nanomaterials refer to structures (either organized or disorganized) with nanometer scale dimensions ideal for cellular recognition, targeting and uptake. Over the past few decades, advancements in nanotechnology have allowed researchers to design synthetic and biologic nanomaterials for applications in immunoengineering, where precise targeting and modulation is necessary at the cellular and sub-cellular level. Owing to their tunability, nanomaterials present a promising approach to tackling challenges in delivery of payloads such as proteins, nucleic acids and peptides. Nanomaterials can be designed using techniques in synthetic biology (in the context of antibody–drug conjugates and other biologics), organic/inorganic chemistry (in the context of nanoparticles) or a combination of both.
Local immunomodulation refers to the targeted modulation of the immune response at specific tissues or organs, aiming to achieve a balanced and controlled immune reaction.18 This approach is valuable for addressing various autoimmune disorders, inflammatory conditions, and localized infections, as it allows for tailored interventions without compromising the overall immune system.19–21 By fine-tuning immune responses at the site of concern, local immunomodulation holds promise for enhancing therapeutic precision and minimizing systemic side effects in the realm of medical interventions.
In this review, we present recent studies in biomaterials for immunoengineering, including the development of novel vaccine platforms, adjuvants, immunotherapies, cellular engineering, and tissue models for immunoengineering studies. We also highlight the challenges and opportunities and discuss the potential impact of immunoengineering on human health.
2 Vaccine technologies
Vaccines are one of the most effective medical interventions to prevent the spread of infectious diseases. They work by priming the immune system to recognize and fight off specific pathogens. Their history dates back to 1796 when Dr. Edward Jenner administered the first vaccine against smallpox.22 Since then, our understanding and subsequent design of vaccines has grown tremendously. At their core, vaccines consist of protein antigens to which the immune response is mounted and adjuvants that activate the immune system. Important components of vaccines and examples are depicted in Fig. 1. In this section, we highlight key components of vaccines and important engineering advances in each component, with suggestions for future research.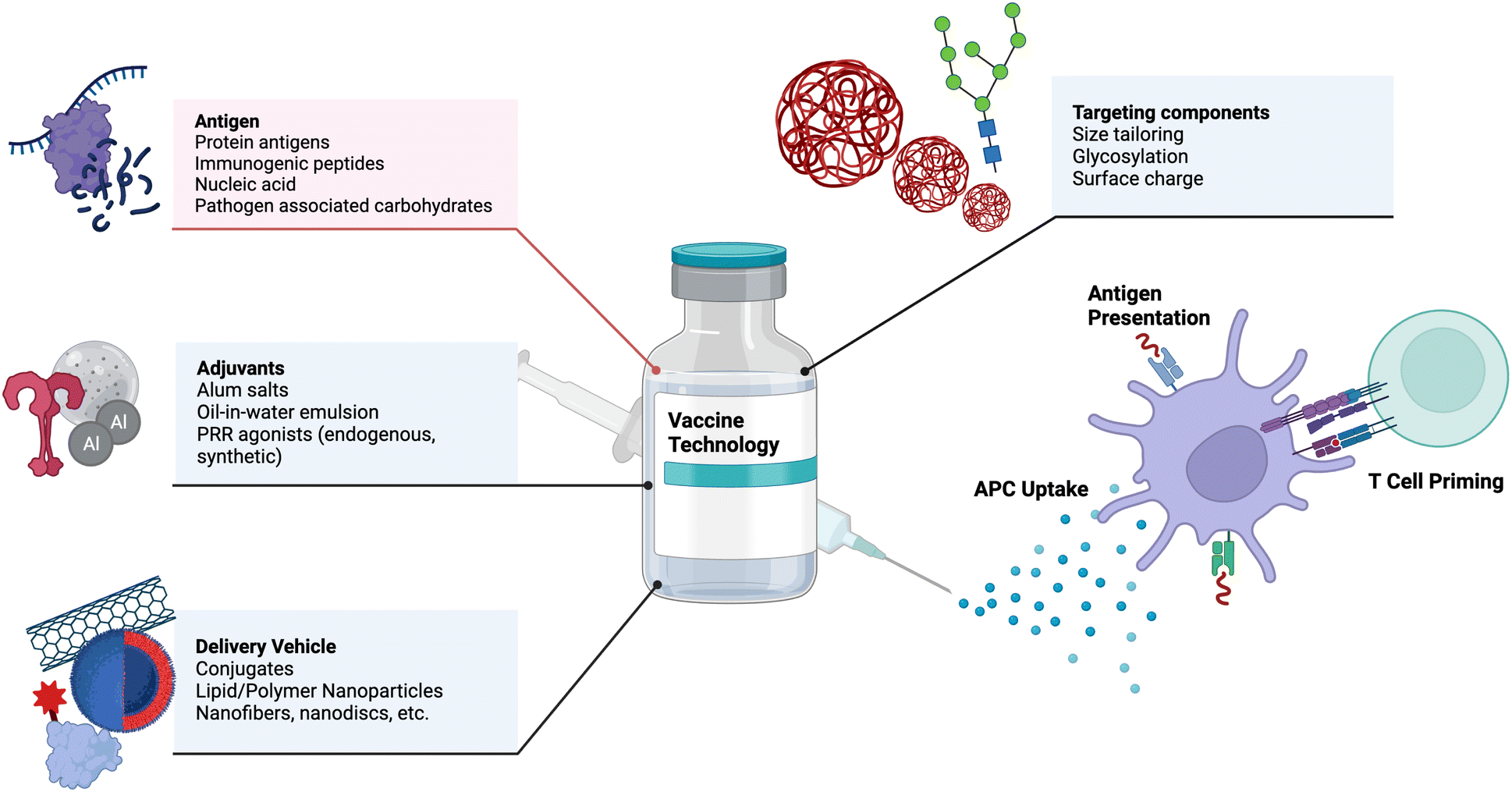 | ||
| Fig. 1 Key components of vaccines are listed as antigens, adjuvants, delivery vehicls, and targeting components, including examples of each. Upon injection, antigen presenting cells (APCs) uptake vaccine components, present the antigen, and prime adaptive immune cells. This process is depicted above. Adapted from “Common Components of Vaccines”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates. | ||
2.1 Adjuvant design and delivery
Adjuvants are added to vaccines to enhance the immune response. They work by stimulating the innate immune system, which in turn enhances the adaptive immune response. Adjuvants can improve the efficacy of vaccines by increasing the magnitude, duration, and specificity of the immune response. They can also reduce the amount of antigen needed in a vaccine, which can lower the cost of production. There are many types of adjuvants including aluminum salts, oil-in-water emulsions, and others. Aluminum salts, also known as alum, are the most commonly used adjuvants in human vaccines and have been used for over 80 years. They work by forming a depot at the site of injection, which slowly releases the antigen and stimulates the immune system. Oil-in-water emulsions, such as MF59 and AS03, are used in some influenza vaccines and have been shown to improve the immune response in elderly populations.23 The design of adjuvants is a complex and evolving field. Adjuvants must be safe and effective in stimulating an appropriate immune response for the specific pathogen or disease being targeted. Recent advances in our understanding of the immune system have led to the identification and development of new adjuvants.24 This includes agonists of innate immune PRRs such as TLRs, NOD-like receptors (NLRs), RIG-I-like receptors (RLRs), C-type lectin receptors (CLRs), and AIM2-like receptors (ALRs). PRRs normally function to recognize specific pathogen- or damage-associated molecular patterns (PAMPs, DAMPs).25 The endogenous ligands to many of these receptors have drug-like properties such as stability and solubility, so it is of great scientific and clinical interest to identify and optimize novel adjuvant formulations.One common way to identify new adjuvants is through the use of high-throughput screening. With a distinct immune activation pathway identified, in vitro assays can be optimized to screen thousands of small molecules for their targeted modulation of pathways of interest. For example, cGAS-STING is a known inflammatory pathway for the development of robust adaptive immunity, but its endogenous ligand, cGAMP (or other cyclic dinucleotides, CDNs), is not suitable for therapeutic applications due to poor drug-like properties. Recent studies have identified non-CDN STING agonists by detecting downstream IFN-γ26 or interferon regulatory factor (IRF) and nuclear factor kappa B (NFκB).27,28 Other groups use structure-based virtual screens to identify potential modulators.29,30 Similar screens have been used to identify agonists of other receptors,31,32 or of synergistic receptor agonizing. Once agonists are identified, their structure can be optimized for potency, specificity, and drug-like properties. The continued discovery, optimization, and evaluation of adjuvants can drive the development of more safe and effective vaccines. Current and future research may focus more on identifying molecules or combinations with unique biological properties, like improved secretion of certain cytokines with beneficial effect without increasing others with negative side effects. Examples of several small molecule PRR agonists are depicted in Fig. 2.
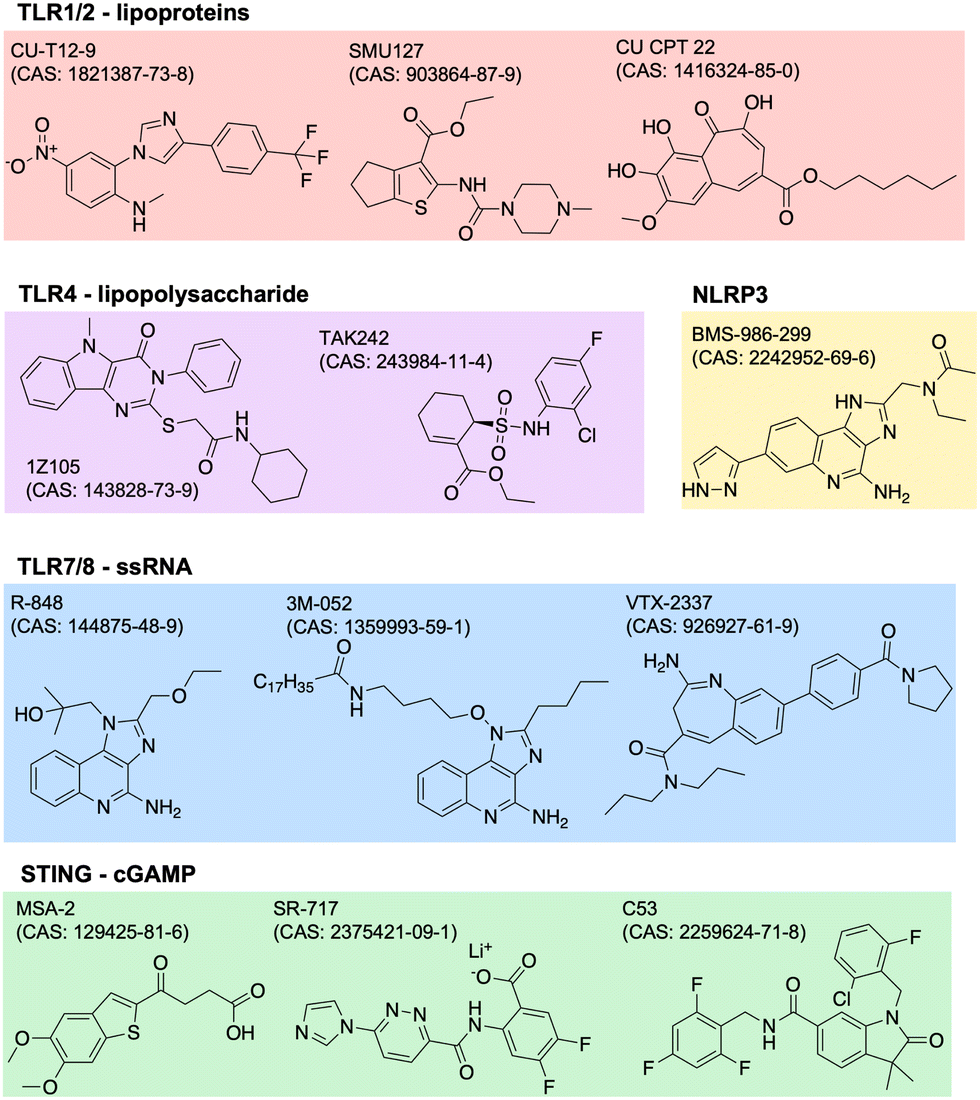 | ||
| Fig. 2 Small molecule agonists of many PRRs have been identified, including TLR1/2, TLR4, TLR7/8, STING, and NLRP3. Key examples of these are shown, grouped by protein target, with associated CAS numbers. PRR: pattern recognition receptor; TLR: toll-like receptors; PRR: pattern recognition receptor; TLR: toll-like receptor; NLRP3: nucleotide oligomerization domain-like receptor family, pyrin domain containing 3; STING: stimulator of interferon genes. | ||
Beyond small molecule adjuvants, many researchers are interested in the immunostimulatory properties of macromolecular materials, including polymers and nanoparticles.33 It is important to note that adjuvants are powerful immunostimulatory molecules and may potentially have consequential off-target effects if not delivered appropriately.34 When appropriately delivered via nanoparticle co-encapsulation with or covalent conjugation to antigen, the effects can be localized and specified.35 The optimization of delivery systems remains an active area of research and is detailed in the sections to follow.
2.2 Antigen presenting cells
APCs, including dendritic cells (DCs), macrophages, and B cells, are specialized immune cells capable of capturing, processing, and presenting antigens to lymphocytes. Their ability to efficiently process antigens, coupled with their unique capacity to activate T cells, makes them central to initiating adaptive immune responses. Upon encountering antigens derived from pathogens or vaccine formulations, APCs internalize the antigens and present them on their cell surface in association with major histocompatibility complex (MHC) molecules. This process, known as antigen presentation, allows T cells to recognize and respond to specific antigens.36The DCs, in particular, are regarded as the most potent, or “professional” APCs due to their exceptional antigen-capturing capabilities and ability to prime naive T cells. They capture antigens in peripheral tissues, migrate to lymphoid organs, and present antigens to T cells within specialized regions known as secondary lymphoid organs. By activating T cells, DCs provide critical signals for the differentiation and expansion of antigen-specific T cells, which are instrumental in mounting robust immune responses.37
The success of vaccines relies on the ability of APCs to efficiently capture, process, and present vaccine antigens to immune cells. By mimicking natural infections, vaccines stimulate APCs, leading to the activation of both the innate and adaptive arms of the immune system. APCs recognize PAMPs through PRRs, as detailed above. This recognition triggers a cascade of immune responses, including the production of pro-inflammatory cytokines and the recruitment of immune effector cells. Moreover, APCs also regulate the balance between tolerance and immunity. They possess mechanisms to induce tolerance towards harmless antigens encountered in non-inflammatory, homeostatic settings, preventing unnecessary immune activation and autoimmunity. By modulating the immune response, APCs contribute to the development of long-lasting immune memory, which is essential for effective vaccination strategies.38 Tolerogenic immune modulation is discussed below in more detail. Therefore, the critical role of APCs in vaccine responses cannot be overstated.39,40 These specialized immune cells are at the forefront of initiating and shaping adaptive immunity by capturing, processing, and presenting antigens to T cells. A comprehensive understanding of APC biology and their interactions with vaccine antigens is pivotal for designing novel vaccines and improving existing ones. Further research into the precise mechanisms governing APC function and the development of targeted strategies to enhance their efficacy will undoubtedly pave the way for more effective and tailored vaccines against infectious diseases and cancer. Recent advances in this space are detailed below.
2.3 Targeted delivery of antigens
Efficient and selective delivery of antigens to APCs is a key strategy in the development of advanced vaccines and immunotherapies. Targeting antigens specifically to APCs holds great promise for enhancing immune responses, improving vaccine efficacy, and enabling the development of personalized medicine approaches. This section highlights the importance of targeted antigen delivery to APCs and explores various strategies employed to achieve this objective.At the most basic level, targeted glycosylation of single protein antigens can change the way the antigen is recognized and processed by the immune system. This approach dates back to at least the late 1990s, where scientists began thinking about the glycosylation of mucin-1 (MUC1), a glycoprotein heavily overexpressed in some cancers.41,42 This approach is still explored today, although with more advanced synthetic techniques. One recent report from Rong et al.43 employed in vitro glycosylation reactions to attach N-acetyl-galactosamine (GalNAc) to SARS-CoV-2 receptor binding domain (RBD) of the spike protein. This specific glycosylation did not lead to an increase in RBD-specific immunoglobulin G (IgG) with respect to native RBD, but, importantly, the serum antibodies exhibited 30–40% higher neutralization capacity. This work highlights the importance in understanding glycosylations to improve vaccinal responses.
More advanced synthetic strategies are also being developed. One recent strategy employed by Wilson et al.44 used a polymeric glyco-adjuvant to elicit antigen-specific humoral and cellular immune responses. The polymer consists of mannose units and a TLR7 agonist. The mannose units are capable of binding mannose receptor and other C-type lectins on the surface and enable receptor-mediated endocytosis by APCs, while the TLR7 agonist powerfully activates the innate immune system upon endosomal receptor binding. The polymer is reversibly conjugated to protein antigens ovalbumin, circumsporozoite protein, or later SARS-CoV-2 spike protein45via a self-immolative linker. Their work demonstrates the importance of antigen and adjuvant targeting in both enhancing the breadth and specificity of the immune response, as well as reducing the side effects of vaccinal immune activation.
The incorporation of glycosylations, synthetic or natural, to enhance antigen targeting offers several advantages. It provides a versatile strategy to precisely direct antigens to APCs, promoting efficient immune recognition and activation. Glycosylation can also confer stability, improve antigen solubility, and prolong antigen circulation time. Moreover, glycosylated antigens can elicit immune responses against specific glycan epitopes, broadening the repertoire of targeted immune responses. However, challenges exist in optimizing glycan structures, ensuring consistency in glycan attachment, and determining the optimal glycosylation sites on antigens.
Further research is needed to elucidate the interactions between glycosylated antigens and APC receptors, allowing for the rational design of glycan modifications to enhance antigen targeting. Additionally, the development of glycoengineering techniques, such as enzymatic glycan remodeling or chemoenzymatic approaches, will enable precise control over glycan structures and facilitate the production of glycosylated antigens on a larger scale. Integration of glycosylations with other antigen delivery systems, such as nanoparticle-based platforms, could further enhance targeted antigen delivery to APCs and improve vaccine efficacy. We believe better understanding of glycobiology and its subsequent manipulation will drive the next generation of vaccine innovation.
In a recent report by Chen et al.,51 the group reported a lymph node targeted lipid nanoparticle to deliver mRNA vaccines. They screened a variety of lipids with differing tail lengths, combinations, and linker chemistries, and identified an optimal lipid for accumulation in the injection draining lymph node. They explored the efficacy of their lipid as compared to those in Comirnaty, the U.S. Food and Drug Administration (FDA) approved COVID-19 mRNA vaccine. This work serves as just one example of the beneficial effects of lymph node targeting and the many ways to achieve accumulation. The topic of lymph node targeting nanoparticles has been reviewed further elsewhere.52,53 Most modern vaccines are developed for intramuscular administration, and we believe design with lymph node targeting could improve immune responses.
2.4 Viral vectors
Viral vectors are cutting-edge tools for vaccine delivery, leveraging the relatively benign nature of harmless viruses to transport antigenic materials into host cells. By modifying the viral vector to carry specific genes encoding vaccine antigens, they can effectively introduce these antigens into the body, triggering a targeted immune response. The viral vector itself does not cause disease but acts as a delivery vehicle, mimicking a natural infection to stimulate the immune system. This approach has shown promise in generating potent and long-lasting immune responses against various infectious diseases and even certain cancers, paving the way for the development of safer and more effective vaccines with broader applicability.54While viral vectors are promising for some applications, they face limitations in immunogenicity.54 There is clear evidence that viral vectors can be dosed repeatedly in immune-privileged tissues like the eye, suggesting that synthetic passivation could expand the use of viral vectors to other tissues.55 Some biomaterials approaches show promise in enhancing the efficacy, safety, and specificity of viral vector-based vaccines.56 For example, conjugation with stealth polymers like polyethylene glycol (PEG)57 or elastin-like polypeptides58 have been shown to reduce adeno-associated virus (AAV)-specific antibody-mediated neutralization, specifically protecting against humoral immunity. Other strategies aimed at reducing cellular immunogenicity using conjugation to immunosuppressive materials like zwitterionic phosphoserine.59 The future of viral vectors holds immense promise for developing new vaccines and personalized medicine. More efficient, targeted, and safer vaccines against a wide range of infectious diseases and cancer can be developed by using them. The ability to precisely control vaccine delivery, enhance immune responses, and tailor treatments to individual patients' needs opens new avenues for tackling challenging health conditions. As we utilize more biomaterials and their interactions with the immune system, we can expect new novel therapeutic strategies beyond traditional vaccination.
2.5 Synthetic vaccines
Synthetic vaccines offer precise manipulation of antigen structures, improved immunogenicity, and tailored immune responses. Here, we highlight the unique capabilities and design parameters of synthetic vaccines to shed light on this exciting frontier and encourage further research and innovation in this field.One way to create a synthetic vaccine involves the selection of an immunodominant T cell epitope to drive cellular immunity against pathogens. An interesting strategy was recently published by Cooney et al.60 using immunogenic peptide epitopes conjugated at high density to a single adjuvant, α-galactosylceramide. Their synthetic route is high-yield and versatile to a variety of peptide antigens. Notably, their work explores the importance of conjugation and antigen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :adjuvant ratio, which they demonstrate impacts the NKT cell response and thus tolerability. The synthetic processes detailed here serve as an example of the creative engineering required to develop next-generation synthetic vaccines.
:adjuvant ratio, which they demonstrate impacts the NKT cell response and thus tolerability. The synthetic processes detailed here serve as an example of the creative engineering required to develop next-generation synthetic vaccines.
Tsoras et al.61 showed peptide subunit vaccines with increased immunogenicity. However, they are limited in their ability to produce long-lived memory immune responses. Their strategy employs peptide nanoclusters (PNCs), which are suspended clusters of crosslinked peptide antigens. They systematically evaluated how the crosslinking and modification affect the induced cellular immune response. This sort of deliberate study provides a mechanistic understanding, paving the way for the development of next-generation vaccines.
Clearly, peptide antigens and their tailored delivery in the form of synthetic vaccines offer promise in the area of infectious disease protection. There are other examples of biomaterials strategies to deliver peptides for cancer vaccination, such as macroporous alginate nanoparticles carrying leukemia antigens,62 coordinative dendrimers carrying antigenic peptides,63 or peptides conjugated to immune-stimulating agonists.64 Further immunological considerations of cancer vaccines are highlighted in the following section.
2.6 Cancer vaccines
Cancer vaccines hold immense promise as a breakthrough in cancer therapy by leveraging the body's immune system to specifically target and eliminate cancer cells. The design of these vaccines includes various components, such as antigen identification, formulation, and adjuvant selection, many of which are familiar from the previous discussion on traditional, antiviral, or antibacterial vaccines. Here, the antigens are tumor-specific or tumor-associated, rather than derived from a particular pathogen. When these antigens can be identified, vaccines can be directed towards them and induce a cancer-antigen-specific immune response.65,66 However, antigens are not always uniquely and consistently expressed by cancer cells, particularly in the case of metastatic cancer.67 Additionally, because cancer grows otherwise uncontrolled, the immune system is chronically exposed to high levels of these antigens, requiring significant adjuvanting to generate an antigen-specific immune response.68 These limitations are important to understand as the scientific community works towards more effective cancer vaccination.One strategy to overcome the issue of antigen selection is by simply applying adjuvant to the tumor site, hoping to stimulate a local innate immune response that can serve to prime the adaptive immune system in an antigen-specific manner against tumor cells and tumor debris.69,70 This strategy can be considered as in situ cancer vaccination. This approach worked well for basal cell carcinoma, a type of skin cancer, that can be successfully treated with topical imiquimod cream, a TLR7/8 agonist.71,72 Many other groups have been interested in similar strategies employing other adjuvants like CpG (TLR9)73,74 or poly I:C (TLR3).75,76 However, next generation adjuvant delivery strategy can help direct the immune response and reduce systemic toxicity associated with adjuvant administration. Recent work in this space expands to other adjuvants as well, e.g., Chen et al. recently reported the identification and characterization of STING-agonist loaded liposomes that enhance antigen cross-presentation and immune activation.77 Other biomaterials innovations help deliver adjuvants intratumorally, including a polypeptide depot that controls release of CpG,78 a polymer that binds tumor cells and debris with a polymeric TLR7 agonist,79 high-density lipoprotein nanodiscs that co-deliver MPLA and CpG,80 and many others that are reviewed extensively elsewhere.81–84
2.7 Tolerogenic vaccination
Tolerogenic vaccination is a type of vaccine designed to induce immune tolerance rather than an effector immune response. Sometimes referred to as inverse vaccination, a term coined by Lawrence Steinman in 2010, these technologies aim to specifically reduce a pathological adaptive autoimmune response and instead induce immunological tolerance to the antigen.85,86 The goal of a tolerogenic vaccine is to re-educate the immune system to recognize self-antigens (components of the body's own tissues) as harmless, thereby reducing or preventing autoimmune reactions. These vaccines often contain specific antigens or proteins associated with the targeted autoimmune disease. By exposing the immune system to these antigens in a controlled manner, tolerogenic vaccines aim to induce immune tolerance and prevent or dampen the immune response against self-antigens. Traditionally, therapies that are used in treating autoimmunity involve broadly immunosuppressive drugs.87 However, such broad suppression of the immune system can lead to opportunistic infections and other side effects. Inverse vaccination, on the other hand, aims to induce tolerance in an antigen-specific manner and bypassing the need for broad immune suppression. In recent years, several interesting materials science approaches have shown promise in the ability to induce antigen-specific immunological tolerance.One such strategy is the use of poly(lactic-co-glycolic acid) (PLGA) particles to carry antigens to APCs. Miller et al. published many works on the material, where they used carboxylated, biodegradable PLGA microparticles to bear encephalitogenic peptides. The specific properties of the particles, including 500 nm size, negative surface charge, and disease-specific antigen cocktail, promote APC uptake by mimicking apoptotic debris in size, deliver tolerogenic signals, and enable the presentation of immunologically relevant antigens. In this work, they demonstrated the particles' ability to delay the onset of experimental autoimmune encephalomyelitis (EAE), a mouse model of multiple sclerosis.88 Since then, the group has gone on to expand the microparticle platform for the treatment of other diseases. This includes the encapsulation of gliadin for the treatment of celiac disease,89 of insulin, GAD65, and chromogranin A proteins for the prevention of autoimmune diabetes,90 and of peanut extract to prevent allergic anaphylaxis.91 This technology has recently been evaluated in human clinical trials and showed favorable safety and suggestive efficacy results in celiac disease patients.92 Many other groups have designed PLGA nanoparticle systems for tolerogenic vaccination based on similar principles and are reviewed in detail elsewhere.93
Other nanoparticle-based strategies have also been employed, the design parameters of which are explained in further detail elsewhere in this review. Recent work by Nguyen et al. combines two nanoparticle technologies to elicit protective tolerance in EAE.94 Primarily, mesoporous silica nanoparticles (MSN) loaded with a high density of antigen (peptide from myelin oligodendrocyte glycoprotein (MOG)) were administered semi-therapeutically (after disease induction but prior to symptom onset), which prevented disease progression. In order to demonstrate efficacy in a fully therapeutic setting, where symptoms have already developed, MSN–MOG particles were functionalized with cerium oxide nanoparticles (CeNP), which function to scavenge reactive oxygen species and promote tolerogenic APC phenotype. Together, this work demonstrates unique materials design to fit the constraints of the complex immunological problems at play in EAE disease progression.
Another approach has recently been developed by our lab involving the use of polymerized sugars to target antigens to the immunosuppressive environment of the liver.44 The materials, dubbed pGal and pGlu, are based on N-acetylgalactosamine and N-acetylglucosamine and can be conjugated to proteins to induce antigen-specific tolerance, here for diabetes. In other works, the lab applied the materials to models of skin transplant95 and further explored subcutaneous administration for enhanced lymph node trafficking, rather than the liver.96 It has remained a question as to whether these approaches could only restrain a developing response, or could contract existing immune aggression, such as in relapsing-remitting multiple sclerosis. Recent work from the lab with pGal-MOG showed that tolerance induction in the face of existing immune aggression was possible.97 Similar materials based on polymerized mannose have more recently been published in models of anti-drug antibody responses98 and food allergy.99 Together, these works provide further evidence for the future of biomaterials in the clinical treatment of autoimmune diseases. With increasing prevalence of autoimmune diseases worldwide, researchers in the coming years will continue to develop a deeper understanding of immunological mechanisms at play and engineer novel therapeutic strategies.
3 Chemical modifications to proteins and nucleic acids
Chemical modification of biologic agents (e.g., recombinant proteins) offers an expanded spectrum of therapeutic applications compared to the use of unmodified biologic agents. Incorporating synthetic elements into biologics is thus an important and quickly evolving field in modern biomedicine. In this section, we will discuss select examples of how synthetic chemistry can be applied to recombinant biologics such as growth factors, cytokines, and antibodies.3.1 Stealth polymers
Clinical translation of recombinant proteins is hindered by their immunogenicity, short in vivo half-life, and stability. In order to enhance the therapeutic properties of proteins, chemical attachment of polyethylene glycol, a process called PEGylation, has been successfully employed over the last 30 years. PEGylation of biologics that do not contain natural half-life extension domains, such as Fc or serum albumin, confers stealth-like properties, limiting their recognition by the immune system, decreasing their clearance rate, and drastically improving pharmacokinetic (PK) and pharmacodynamic (PD) properties. Other major benefits of protein PEGylation are increased protein solubility, higher thermal and chemical stability, and reduced proteolysis.100 Increased protein solubility, enabled by a high degree of hydrophilicity of PEG molecules, is particularly important because it allows for biologic therapeutics to be administered at higher doses in smaller volumes with less risk of protein aggregation.101 A summary of the benefits of PEGylation is depicted in Fig. 3.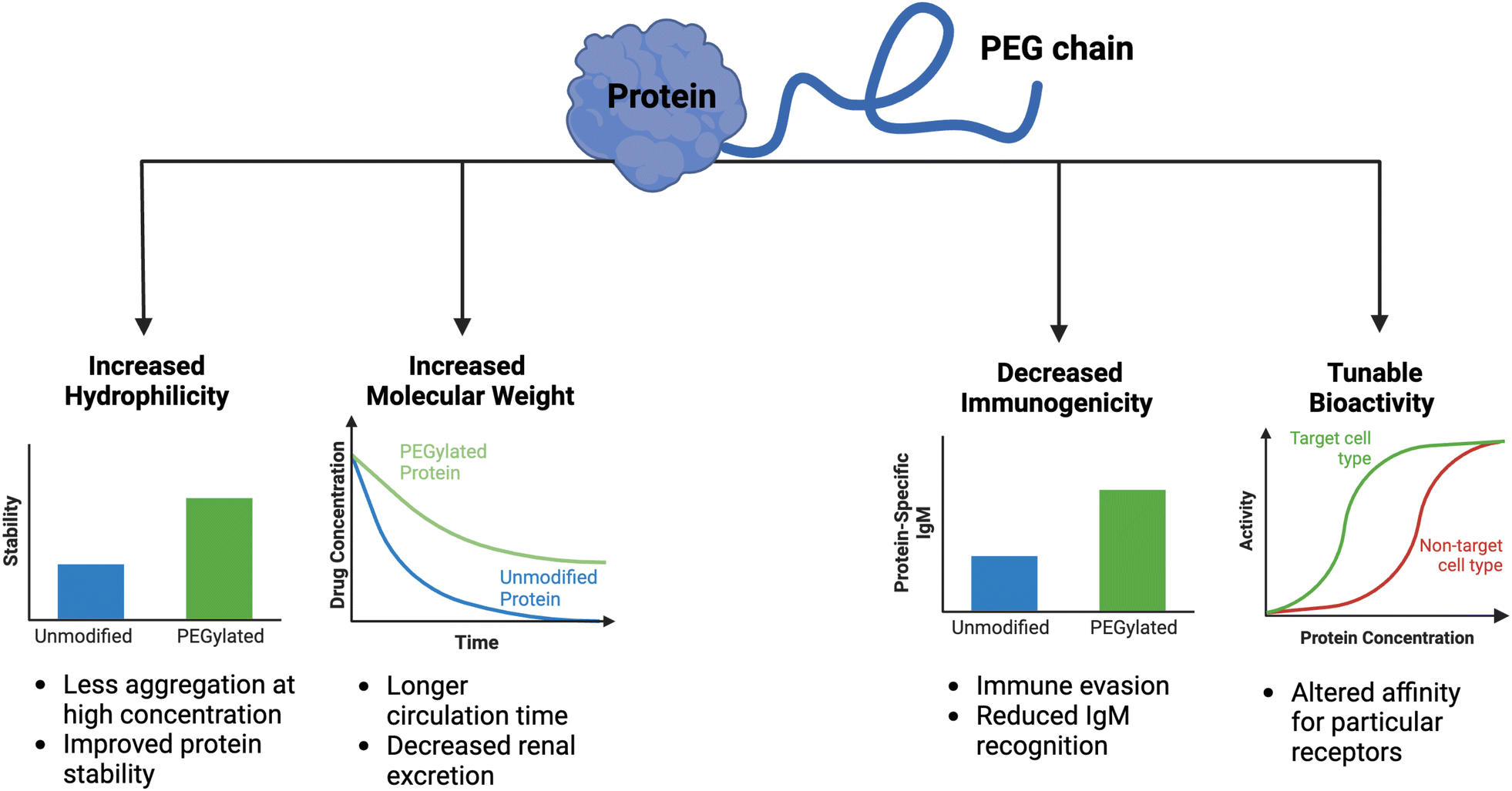 | ||
| Fig. 3 Consequences of PEGylation of biologics. When therapeutic proteins are attached to PEG, several key benefits are conferred, including increased hydrophilicity and molecular weight, decreased immunogenicity, and tunable bioactivity. The practical implications of these alterations are graphically depicted. | ||
There are now approximately 30 FDA-approved PEGylated biologics, spanning a diverse range of indications (Table 1). In the majority of these applications (e.g., granulocyte colony-stimulating factor (G-CSF) and growth hormones), PEG chains primarily serve as a synthetic half-life extension domain to improve the PK profile of the biologic.102 In applications pertaining to enzyme replacement therapies (e.g., alpha-galactosidase A and adenosine deaminase), PEGylation is aimed to reduce the immunogenicity of otherwise ‘non-self’ enzymes.103 The decrease in immunogenicity of the ‘non-self’ proteins is due to the shielding effect enabled by PEG chains, which minimizes immune complex formation. Despite the fact that the modified protein has reduced immunogenicity upon PEGylation, PEG itself may evoke an immune response, especially after repeated dosing with the PEGylated biologic.104 Because PEG is a common food additive and widely used in cosmetics, nearly a quarter of healthy individuals have pre-existing anti-PEG antibodies.105 Such pre-existing anti-PEG immunity is thought to be predominantly mediated by circulating IgM antibodies, but other mechanisms may contribute to the immunogenicity of PEG.106 Although the immunological mechanisms are not fully understood, the impact of the anti-PEG antibodies on the PK properties is profound. The presence of high levels of antibodies against PEG may result in accelerated blood clearance of the intravenously administered PEGylated biologic by the complement system.107 Another serious consequence of anti-PEG antibodies is the risk of anaphylactic shock in patients receiving PEGylated biologics.108
| Entry | Trade name | Active biologic | Manufacturer | Indication | Approval year |
|---|---|---|---|---|---|
| Abbreviations: ADA, adenosine deaminase; G-CSF, granulocyte colony-stimulating factor; IFN, interferon; SCID, severe combined immunodeficiency disease; TNF, tumor necrosis factor; PNH, Paroxysmal nocturnal hemoglobinuria. | |||||
| 1 | Elfabrio | α-Galactosidase-A | Chiesi Group & Protalix Bio | Fabry disease | 2023 |
| 2 | Rolvedon | G-CSF | Spectrum Pharmaceuticals | Febrile neutropenia | 2022 |
| 3 | Stimufend | G-CSF | Fresenius Kabi | Febrile neutropenia | 2022 |
| 4 | Fyletra | G-CSF | Amneal Pharmaceuticals | Febrile neutropenia | 2022 |
| 5 | Besremi | IFN-α-2b | PharmaEssentia | Polycythemia vera | 2021 |
| 6 | Skytrofa | Somatrophin | Ascendis Pharma | Growth hormone deficiency | 2021 |
| 7 | Empaveli | Pegcetacoplan | Apellis Pharmaceuticals | PNH | 2021 |
| 8 | Nyvepria | G-CSF | Pfizer | Febrile neutropenia | 2020 |
| 9 | Ziextenxo | G-CSF | Sandoz | Febrile neutropenia | 2019 |
| 10 | Esperoct | Anti-hemophilic factor | Novo Nordisk | Hemophilia A | 2019 |
| 11 | Udenyca | G-CSF | Coherus BioSciences | Febrile neutropenia | 2018 |
| 12 | Palnyziq | Phenylalanine ammonia lyase | BioMarin | Phenylketonuria | 2018 |
| 13 | Revcovi | Adenosine deaminase | Leadiant Biosciences | ADA-SCID | 2018 |
| 14 | Revcovi | Adenosine deaminase | Leadiant Biosciences | ADA-SCID | 2018 |
| 15 | Fulphila | G-CSF | Mylan GmbH | Febrile neutropenia | 2018 |
| 16 | Asparlas | L-Asparaginase | Servier Pharmaceuticals | Acute lymphoblastic leukemia | 2018 |
| 17 | Jivi | Anti-hemophilic factor | Bayer | Hemophilia A | 2018 |
| 18 | Rebinyn | Coagulation factor IX | Novo Nordisk | Hemophilia B | 2017 |
| 19 | Adynovate | Anti-hemophilic factor | Baxalta | Hemophilia A | 2015 |
| 20 | Plegridy | IFN-α 1a | Biogen | Relapsing multiple sclerosis | 2014 |
| 21 | Omontys | Erythropoietin receptor agonist | Takeda | Anemia – chronic kidney disease | 2012 (withdrawn) |
| 22 | Sylatron | IFN-α 2b | Merck | Melanoma | 2011 |
| 23 | Krystexxa | Uricase | Horizon Pharma | Chronic gout | 2010 |
| 24 | Cimzia | Anti-TNFα | UCB | Rheumatoid arthritis & Crohns | 2008 |
| 25 | Mircera | Erythropoietin | Roche | Anemia – chronic kidney disease | 2007 |
| 26 | Somavert | Growth hormone antagonist | Pfizer | Acromegaly | 2003 |
| 27 | Neulasta | G-CSF | Amgen | Febrile neutropenia | 2002 |
| 28 | Pegasys | IFN-α 2a | Roche | Hepatitis C | 2002 |
| 29 | Oncaspar | Asparaginase | Enzon | Acute lymphoblastic leukemia | 1994 |
| 30 | Adagen | Adenosine deaminase | Enzon | ADA-SCID | 1990 |
To overcome this limitation, alternative strategies to PEGylation are being studied. One such approach is to expand the hydrodynamic volume of a therapeutic with Pro, Ala, and Ser amino acids (typically hundreds) of residues,109 a process termed PASylation.110 A recent study demonstrated that PASylation was equally as effective as PEGylation in improving the half-life of liposomes but displayed reduced immunogenicity.111 Another study that aimed to reduce the immunogenicity of PEG in a peptide conjugate context utilized poly[oligo(ethylene glycol)methyl ether methacrylate] (POEGMA).112 POEGMA conjugated to exendin, a peptide drug used in the clinic to treat type 2 diabetes, displayed prolonged half-live in vivo upon subcutaneous administration and, importantly, did not elicit an immune response against the polymer. In contrast, repeated administration of the PEG-exendin conjugate mounted an antibody response against the PEG moiety, thus significantly reducing its efficacy long-term. Development of alternate synthetic strategies that enhance the PK properties of biologics without causing a neutralizing immune response is warranted.
A majority of the FDA-approved PEGylated biologics were generated using non-site-specific conjugation strategies, resulting in a mixture of polydisperse proteins.113 The reason for such heterogeneity is that a given protein contains multiple potential reactive lysine residues (the amine of which serves a reactive handle for most PEGylated proteins), thereby posing certain challenges in downstream separation processes.114 Therefore, the development of site-specific PEG conjugation strategies is crucial for obtaining well-defined therapeutic products. Heterogeneous products encounter issues with batch-to-batch variability and altered activity profiles and thus are avoided if possible. One method of specifically conjugating PEG is by exploiting a free cysteine that is either genetically inserted at the N-terminus or that naturally exists in a given protein.113 Once a free cysteine is identified, the sulfhydryl (–SH) group can be reacted with a maleimide-containing PEG chain in a specific fashion. This strategy was successfully implemented to produce certolizumab pegol (Cimzia), a long-acting agent that neutralizes tumor necrosis factor-alpha.115
Rather than relying on “click chemistry” to achieve specific PEGylation, enzymatic bioconjugation methods can also be used. One of the enzymes that has been widely used to yield precisely conjugated biomolecules is sortase A from Staphylococcus aureus. Sortase A is a thiol-containing transpeptidase that recognizes an LPXTG (where X can be any amino acid) motif in multiple structurally unrelated substrates.116 This enzyme was used to produce several C-terminal PEGylated growth factors with equipotent activity as their unmodified counterparts. A more recent chemo-enzymatic PEGylation approach was applied to interleukin-4 (IL-4),117 a cytokine that can polarize macrophages towards an anti-inflammatory phenotype but has a short half-life in vivo. In order to specifically PEGylate the C-terminus of IL-4, a known substrate of Factor XIIIa was genetically inserted into the IL-4 sequence. The resulting IL-4 mutant was enzymatically decorated with an azide group, which was reacted in a copper-free manner with a dibenzo cyclooctyne- (DBCO)-functionalized PEG moiety. The resulting PEGylated IL-4 product was homogenous and exhibited a similar in vitro bioactivity profile as unmodified IL-4.
Site-specific PEGylation approaches have recently been applied to interleukin-2 (IL-2), a pleiotropic cytokine that can act both as an immune-activating and immune-suppressing agent, depending on what T cell it activates.118 High-affinity IL-2 binding requires a trimeric receptor complex, consisting of IL-2R-α, IL-2R-β, and the common gamma chain.119 It is hypothesized that increasing the affinity of IL-2 binding towards IL-2R-α and decreasing the binding towards IL-2R-β induces preferential signaling on regulatory T (immune-suppressive) cells over effector cluster of differentiation (CD) 8+ T cells (immune-activating).120 To achieve this, copper-free chemistry was utilized to specifically PEGylate Tyr 31 and Thr 51 in IL-2, residues that interact mostly with the β subunit.121 Such precise conjugation of the PEG chains was enabled by non-natural, azide-bearing amino acids (incorporated via genetic code expansion) and the subsequent orthogonal conjugation of PEG moieties via a click reaction. Preferential binding of the PEGylated IL-2 to its α receptor subunit over β subunit resulted in sustained activation and proliferation of regulatory T cells, which led to improvement in several autoimmune models such as lupus, rheumatoid arthritis, and graft-versus-host disease. A similar site-specific PEGylation approach was applied to IL-2 for the opposite purpose: reduce binding to IL-2R-α and increase the affinity for IL-2R-beta.122 This engineered IL-2, termed THOR-707, predominantly binds to IL-2R-β and the common gamma chain (but not IL-2R-α), which potently activates CD8+ T cells and natural killer (NK) cells for antitumor immunity. In THOR-707, Pro 65 a (residue that interacts with IL-2R-α) is replaced by an azide-modified non-natural amino acid, which reacts with a reactive PEG chain in one step. Importantly, in mice, THOR-707 did not cause proliferation of peripheral regulatory T cells but caused a robust expansion and activation of tumor-infiltrating CD8+ T cells and NK cells. These two examples demonstrate how site-specific PEGylation can be used to achieve a desired pharmacological profile and elicit certain immunological phenotypes.
3.2 Antibody-drug conjugates
Antibody-drug conjugates (ADC) are a promising technology to treat advanced, chemotherapy-refractory solid tumors as well as hematological malignancies by delivering a cytotoxic molecule directly to the cancer cell. Despite being in development for more than 50 years,123 ADCs have only recently begun to demonstrate success in the clinic. Some of the challenges that were faced in the development of initial ADCs were poor antibody properties (e.g., half-life, specificity for the target antigen), suboptimal small molecule conjugation methodologies and cytotoxic agents demonstrating poor activity. The exponential success of ADCs observed in the last decade or so is predominantly thanks to the advances in biochemistry and medicinal chemistry that solved many of these challenges124 encountered in the early days of ADC research. ADCs are composed of three basic elements: a monoclonal antibody that recognizes cancer cell-specific or cancer cell-associated antigen, a cytotoxic payload, and a linker that covalently bridges the small molecule to the antibody. The conjugation chemistry employed is generally grouped into three categories, as depicted in Fig. 4. The number of small molecules attached per antibody is referred to as the drug-to-antibody ratio (DAR) and is a crucial parameter when optimizing a given ADC.125 The basic idea behind the mechanism of action of ADCs is that the administered ADC initially binds to the cancer cell-specific or -overexpressed antigen, which triggers the internalization of the antigen-ADC complex, resulting in the lysosomal degradation and release of the cytotoxic molecules. Because ADCs are typically constructed on an IgG1 backbone,126 they are also potent inducers of effector functions (e.g., antibody-dependent cellular cytotoxicity, complement-dependent cytotoxicity, and antibody-dependent cellular phagocytosis), making them classifiable under the broader immuno-oncology umbrella. This subsection will primarily focus on the advances in the chemistry of ADCs, particularly the cytotoxic payload and the linker. | ||
| Fig. 4 Antibody drug conjugates are generally synthesized through three main chemistries, including lysine (amine) to NHS ester, cysteine (thiol) to maleimide, or non-natural amino acid (azide) to strained alkyne (DBCO or BCN). Each of the three chemistries are shown here with a high-level description of their key favorable or unfavorable properties. NHS: N-hydroxysuccinimide; DBCO: dibenzocyclooctyne; BCN: bicyclononyne; DAR: drug-antibody ratio. | ||
One of the major breakthroughs in ADC research that led to favorable clinical results was the development of highly potent cytotoxic payloads.127 One reason that early ADCs failed to demonstrate any clinical improvement compared to the unconjugated chemotherapeutics was the high half-maximal inhibitory concentration (IC50) value of traditional anticancer drugs.128 The IC50 values of the initial chemotherapeutics (doxorubicin and methotrexate) used in ADCs was in the range of micromolar potency. The next iteration of cytotoxic payloads, such as microtubule inhibitors and DNA-damaging agents, were synthesized to be significantly more potent, with IC50 values in the low nanomolar range.129 Currently, out of 14 FDA-approved ADCs, 6 carry microtubule inhibitors (auristatins) and 3 carry DNA-damaging agents. It is also worthwhile to note that cytotoxic agents with picomolar range potency have also been tested in the clinic but severe adverse events halted their translation.130 This finding illustrates that achieving optimal potency of the payload is important for obtaining favorable results in the clinic.
Instead of synthesizing novel, more potent cytotoxic payloads, researchers have also attempted to combine existing payloads in a single ADC. A dual payload ADC directed against human epidermal growth factor receptor 2 (HER2), a receptor overexpressed in 14–20% of breast cancer patients,131 has recently been engineered to carry both monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF).132 Using microbial transglutaminase (MTGase)-mediated transpeptidation, highly homogenous dual-payload ADCs were engineered with well-defined DARs. This method allowed the authors to avoid very high DAR (>8), which can cause aggregation issues and shorter PK due to increased hydrophobicity of the molecule. Importantly, the dual-drug ADC carrying four copies of MMAE and two copies of MMAF exerted a greater antitumor effect than did the co-administration of two single-drug variants in xenograft mouse models. This strategy highlights the therapeutic potential of the dual-drug ADC format for the treatment of refractory HER2+ breast cancer.
Another promising class of payloads for ADCs are STING agonists. STING agonists are able to induce a broad inflammatory response, particularly type I interferon signature, which leads to activation of T and NK cells.133 Despite very promising preclinical data with STING agonists, systemic administration has shown limited success in the clinic due to intolerable systemic toxicity. Thus, delivering STING agonists using tumor-specific antibodies can greatly improve their tolerability, eliciting a more localized response. To this end, a STING agonist (IMSA172) has been conjugated to an antibody targeting epidermal growth factor receptor (EGFR), which is overexpressed in a wide range of cancer cells.134 The authors demonstrated that STING signaling can be induced in EGFR-expressing cells with a nanomolar-range EC50, but not on EGFR-negative cells. In a syngeneic mouse model of melanoma engineered to express EGFR, the ADC significantly prolonged the survival and was not associated with body weight loss. Importantly, anti-EGFR-IMSA172 also synergized with an immune checkpoint blocking antibody, anti-PD-L1, setting the stage for the clinical development of such STING-carrying ADCs. XMT-2056, a STING agonist conjugated to an anti-HER2 antibody, is already undergoing a Phase I clinical trial in multiple solid tumors.135 A more in-depth review of other ADC payloads is available elsewhere.130
Perhaps one of the major challenges in ADC development and optimization is the selection of the linker.136 The main objective of the linker is to keep the small molecule in the antibody-bound state in circulation (plasma) and release the payload once the ADC is internalized and reaches the endosome/lysosome. Linkers can broadly be classified into cleavable and non-cleavable ones: cleavable linkers release the payload upon specific stimuli such as low intracellular or tumor microenvironment pH (e.g., hydrozone) or proteases (e.g., cathepsin B, which cleaves the valine-citrulline bond).129 Non-cleavable linkers, such as succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) rely on complete degradation of the antibody by cytosolic and lysosomal proteases.137 Furthermore, optimization of not only linker chemistry but also the linker length and linker steric hindrance can influence the in vivo efficacy of ADCs.138 Attaching the linker to a more sterically hindered site of the antibody may limit the extracellular cleavage of the cytotoxic drug and enhance the safety profile of the ADC.139
Another promising strategy to improve the stability of the ADC in circulation is to employ a tandem-cleavage linker design, in which two sequential enzymatic cleavage events trigger the release of the payload.140 By using CD79b-targeted MMAE conjugate as the model system, the authors successfully demonstrated that the dipeptide tandem-cleavage linker significantly outperformed the standard vedotin linker in terms of hematopoietic toxicity in rats. While the ADC is in circulation, the cathepsin cleavage site is sterically protected via a glucuronide moiety, limiting the premature release of MMAE. However, once the ADC is internalized, glucuronidase removes the protective monosaccharide moiety, exposing the cathepsin cleavage site for subsequent MMAE release. These findings indicate the importance of balancing the stability and sensitivity of the linker to achieve optimal antitumor efficacy with minimal adverse effects.
Another avenue of research in the ADC field is finding complementary combination agents for cancer therapy.141 One class of therapeutics that has shown preclinical potential in synergizing with ADCs is checkpoint inhibitor (CPI) antibodies, (anti-programmed cell death protein 1 (PD-1), anti-programmed cell death ligand 1 (PD-L1), and anti-cytotoxic T-lymphocyte associated protein 4 (CTLA-4)).142 ADC treatment can increase the quantity of intratumoral T cells whereas CPIs can reinvigorate the T cells that are becoming exhausted or dysfunctional. ADCs can also be rationally combined with other traditional chemotherapies such as tyrosine kinase inhibitors. Indeed, a combination of Flt3-targeting antibody conjugated to MMAF (20D9-ADC) with midostaurin showed enhanced efficacy in preclinical models of acute myeloid leukemia.143
3.3 Altering antibody biodistribution via peptide conjugation
In ADCs, the antibody serves as a delivery vehicle for the cytotoxic payload. However, chemical modification of therapeutic antibodies has also been pursued to improve the biodistribution of the antibody itself. In their unmodified form, antibodies do not display an inherent preference to accumulate at the site of disease; rather, their biodistribution is predominantly guided by the antigen expression pattern. Expression of the target antigen in non-diseased tissues often gives rise to immune-related adverse events associated with the neutralization or blockade of the target.144 To ameliorate this, antibodies can be engineered to achieve higher drug concentration in the tissue of interest, while sparing the healthy organs. For example, CPIs targeting CTLA-4 and PD-L1 were chemically conjugated to an extracellular matrix-binding peptide derived from placenta growth factor-2.145 In this case, the peptide serves as a retention mechanism for the CPIs that is injected peritumorally. The authors showed that peptide-conjugated CPIs were retained near the tumor and induced minimal off-tumor activity, and significantly decreased the immune-related adverse events. A similar peptide conjugation strategy was applied to a neutralizing antibody against tumor necrosis factor-α (TNF-α), which is used to treat rheumatoid arthritis.146 In this work, a collagen-binding peptide (CBP) derived from decorin was chemically linked to an anti-TNF-α antibody and administered intravenously. Due to the inflammation in the joints, collagen is exposed to the vascular endothelium, enabling CBP-anti-TNF-α to accumulate in the disease site and greatly enhancing the anti-rheumatic efficacy. In the same work, collagen affinity was conferred to an anti-transforming growth factor-β (TGF-β) antibody, which demonstrated profound anti-fibrotic activity in a model of bleomycin-induced pulmonary fibrosis. These results show that the therapeutic efficacy of monoclonal antibodies can be elevated by chemically conjugating them to the extracellular matrix-binding moieties.3.4 Glycosylation
While glycosylation was discussed in detail for vaccines, glycans can also be used to improve the targeting of biologic therapeutics to treat infectious diseases. A report from Ye et al.147 details the development of a small interfering RNA (siRNA) conjugate system. They employ both hexavalent mannose to target macrophages and DCs and N-acetylgalactosamine to target hepatocytes for liver-specific infection to treat Marburg virus (MARV) infection. By optimizing glycan structure and combining targeting approaches, the therapeutic strategy provides promising protection in terms of viremia, body weight loss, and ultimately overall survival. This work highlights the importance of rational design of combinatorial approaches in designing the next generation of therapeutics.4 Nanoparticles
Nanoparticles (NPs) have emerged as a valuable tool in medicine, offering enhanced stability, delivery, and efficacy of therapeutic agents. Due to their unique physical properties, nanometer-scale carriers are capable of encapsulating drugs and biological agents, both within their cores and on their surfaces.148 These carrier platforms are further functionalized with bioactive coatings, enabling improved circulation, disease diagnosis, and targeted delivery applications. This exciting integration of nanomaterials in medicine is commonly referred to as nanomedicine. Notably, the FDA has already approved a wide range of nanomedicines for cancer treatment, showcasing their potential to revolutionize the field and improve patient outcomes.1494.1 Nanoparticle design
NP size greatly influences the properties and behavior of the material in the body. Primarily, it determines the ability of the particles to be uptaken by cells and tissues. Smaller NPs may penetrate tissues more easily, whereas larger ones may be more restricted.151 Similarly, it has been demonstrated that NPs around 25 nm can be transported in interstitial flow to lymph nodes, where they can be delivered to resident cells.48 Once there, APC are the primary target cells. DCs uptake NPs through lectin-mediated endocytosis that are under 100 nm. Alternatively, they can uptake particles larger than 200 nm via phagocytosis.152 Many studies have characterized the influence of size on NP distribution and uptake, which should be an important consideration for the development of new NP-based therapies.153 As a result, researchers have explored novel approaches to predict and modulate nanoparticle sizes using a variety of technologies. One such approach is to use microfluidic devices, which are often used lipid nanoparticle technologies but can be adapted to polymers as well.154 Martin et al. described the key parameters affecting nanoparticle size and stability and demonstrated reproducible particles ranging from 75 to 200 nm simply by altering polymer concentration in the organic phase. As a result, straightforward modifications can be made to adjust nanoparticle size based on intended use e.g. cellular targeting, lymphatic drainage and circulation time.
Surface charge of NPs also has important implications for the uptake, distribution, and stability of the particles. In general, positively charged particles exhibit superior cell uptake due to electrostatic interactions with negatively charged membranes, but phagocytotic cells can also ingest anionic NPs.155 Surface charge, as well as chemistry (discussed later), also has an important role in dictating protein adsorption to the surface of the NPs. The changes in surface charge can locally change solution pH, which can have significant impacts on which proteins adsorb and the conformations they adopt.156 This rapid absorption can alter the stability of NPs in solution and is more significant for positively charged NPs.157 For individual cases, the surface charge can be intentionally manipulated for specific applications. For example, neutral and negatively charged polymer micelles release their payloads in different gut regions after intragastric administration based on interaction with the gut mucosa.158 As is true with NP size, it is important to understand and strategically manipulate the surface charge of therapeutic NPs to optimize drug delivery. In the future, scientists will continue to understand how these properties work together to dictate materials properties in living systems.
The nanoparticles are designed to specifically target and deliver therapeutic agents to desired cells or tissues. One strategy involves the conjugation of ligands on the surface of nanomaterials, enabling them to anchor to the target cells and promote endocytosis. Ligands such as antibodies, peptides, or aptamers can be attached to the nanoparticles, allowing for precise recognition and binding to specific receptors on the cell surface.165 This targeted binding enhances the uptake of nanoparticles by the cells, leading to the efficient delivery of bioactive agents encapsulated within the nanoparticles. Through this approach, the release of therapeutic agents can be enhanced, improving their efficacy and minimizing off-target effects, thereby paving the way for more effective and precise treatments in various biomedical applications.
4.2 Lipid nanoparticles & liposomes
Lipid nanoparticle (LNP) technology has witnessed unprecedented growth in recent years, primarily attributed to the success of mRNA LNP-based SARS-CoV-2 vaccines developed by Moderna and Pfizer-BioNTech. LNP technology has rapidly expanded into numerous application areas and, perhaps most importantly, evolved into the premier method for non-viral gene delivery of nucleic acids, especially RNA. In this section, we review the most important characteristics and applications of LNP technology within the context of immunoengineering, highlighting novel and promising developments in the field. Specifically, we delve into the composition of LNPs, the wide array of RNA payloads LNPs can carry, the significance of different chemical properties of RNA payloads, and the application of LNP technology across different disease areas. These advancements underscore the transformative potential of LNPs as immunomodulatory biomaterials, and our exploration aims to provide an understanding of the current state-of-the-art.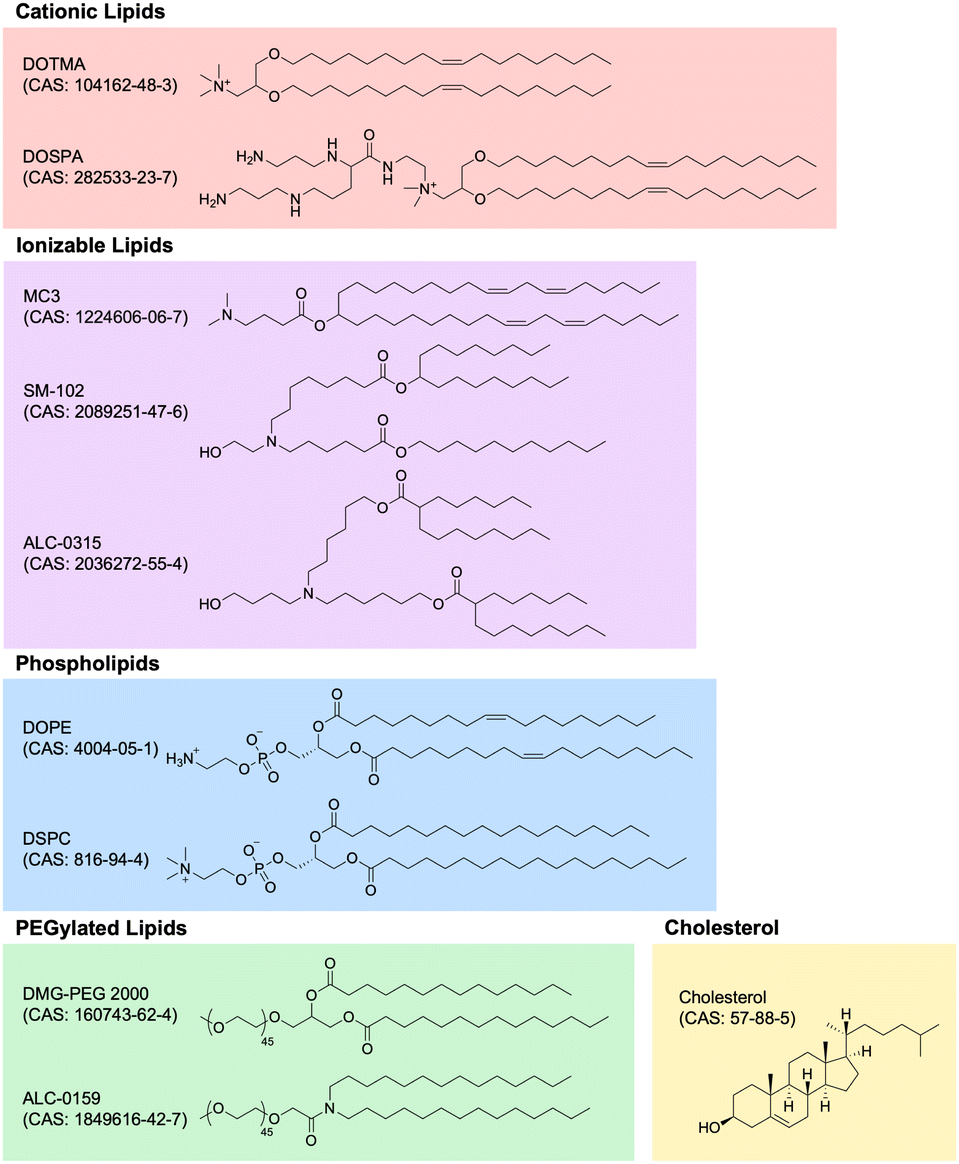 | ||
| Fig. 5 Chemical structures of lipid nanoparticle components. Lipid nanoparticles are generally made of cationic lipids, ionizable lipids, phospholipids, PEGylated lipids, and cholesterol. Key examples in each of these classes are depicted with structures, names, and CAS numbers. | ||
Cationic lipids. Nucleic acids, being negatively charged due to their sugar-phosphate backbone, face significant hurdles in crossing the negatively charged cell membrane, owing to electrostatic repulsion. To overcome this obstacle, cationic lipids with permanent positively charged head groups have been developed, enabling effective encapsulation of nucleic acid payloads within LNPs and facilitating their delivery across the cell membrane.
One of the first cationic lipids to be developed for nucleic acid delivery is 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA). DOTMA is a quaternary ammonium lipid that was first synthesized in 1987 and has since been extensively employed for both in vitro and in vivo nucleic acid delivery across various cell types.169–171 Lipofectin, a widely used commercialized transfection reagent, constitutes a 1:1 (w/w) blend of DOTMA and a fusogenic helper phospholipid called 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE). The incorporation of DOPE enhances stability and delivery efficiency.172
The advancement of research on cationic lipids led to the development of 2,3-dioleyloxy-N-[2-(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propanaminium trifluoroacetate (DOSPA), another noteworthy quaternary ammonium lipid featuring a spermine moiety. DOSPA has been incorporated into Lipofectamine, another widely used commercialized transfection reagent, which comprises a 3:1 (w/w) mixture of DOSPA and DOPE.173 Lipofectamine has proven highly effective in nucleic acid transfection protocols across a diverse range of cell types and is often considered the default standard for comparison against other methods of exogenous nucleic acid delivery into cells.174
Ionizable lipids. Ionizable lipids have emerged as a critical element in the formulation of LNPs due to their potential to address toxicity concerns associated with cationic lipids while retaining their beneficial characteristics. Although cationic lipids are generally considered non-toxic at low concentrations, their toxicity becomes significant at higher levels, limiting their effectiveness and clinical applicability.175 Because the toxicity and immunogenicity of cationic lipids are strongly associated with their positively charged headgroups, lipids with ionizable headgroups have become the predominant component of clinically relevant LNP formulations.176
Ionizable lipids remain neutral at physiological pH but become positively charged when protonated at low pH, which allows them to maintain the advantageous chemical properties of cationic lipids while exhibiting comparatively reduced toxicity. This feature becomes particularly relevant because nucleic acid LNPs are typically formulated in low pH buffers, enabling efficient complexation of the positively charged, protonated ionizable lipids with the negatively charged nucleic acids.177 Moreover, after administration, the ionizable lipids generally remain neutral in the extracellular environment until the LNPs are endocytosed into endosomes, thereby minimizing toxicity.178
As the endosome matures and fuses with the lysosome, the pH of the endolysosomal compartment decreases from 6.8 to 4.5, leading to the protonation of the ionizable lipids within the LNPs.179 The now positively charged ionizable lipid is then thought to play a crucial role in mediating the endolysosomal escape of the nucleic acid payload by fusing with the endolysosomal membrane, which primarily consists of negatively charged lipids. This fusion results in cationic–anionic lipid pairs that drive the lipid membrane into the inverted hexagonal HII phase, causing the formation of non-lamellar structures, disruption of the bilayer integrity, and release of the nucleic acid payload into the cytoplasm.180
The quest for an ideal ionizable lipid for efficient nucleic acid delivery yielded a significant breakthrough with the discovery of 1,2-dilinoleyloxy-N,N-dimethyl-3-aminopropane (DLinDMA).181 DLinDMA was specifically synthesized for the delivery of siRNA and exhibited promising results when incorporated into LNPs and administered intravenously, enabling effective genetic silencing in the hepatocytes of non-human primates.182 Further iterative improvements to the linker and headgroup moieties of DLinDMA led to the development of 2,2-dilinoleyl-4-dimethylaminoethyl-[1,3]-dioxolane (DLin-KC2-DMA), which exhibited significantly enhanced potency and tolerability compared to DLinDMA.183 Subsequent efforts focused on systematic structural modifications to the amine headgroup in order to modulate the pKa of ionizable lipids. These studies culminated in the development of (6Z,9Z,28Z,31Z)-heptatriaconta-6,9,28,31-tetraen-19-yl 4-(dimethylamino) butanoate (DLin-MC3-DMA; MC3), resulting in MC3-based LNPs with more than two orders of magnitude increased potency compared to the original DLinDMA-based LNPs.184
In an extraordinary milestone, these efforts to develop LNPs for nucleic acid delivery led to the United States Food and Drug Administration (FDA) approval of patisiran (brand name Onpattro) in 2018 for the treatment of polyneuropathy in patients with hereditary transthyretin-mediated amyloidosis. Patisiran is the first-ever FDA-approved siRNA-based drug and utilizes MC3-based LNPs containing siRNA that effectively reduces the production of pathogenic transthyretin protein.185–188
Although MC3-based LNPs were initially optimized for siRNA delivery to the liver, their application for the delivery of other nucleic acids, such as mRNA, faced limitations due to concerns regarding MC3's prolonged tissue half-life and associated toxicity issues.168 Thus, researchers sought to innovate the next generation of ionizable lipids with enhanced biodegradability, allowing for rapid metabolism and swift elimination from plasma and tissues while retaining potency. One illustrative example involved introducing ester linkages into MC3's hydrocarbon chain region. The introduction of these ester linkages, prone to enzymatic hydrolysis in vivo but stable under physiological pH, gave rise to di((Z)-non-2-en-1-yl) 9-((4-(dimethylamino)butanoyl)oxy)heptadecanedioate (L319). Remarkably, L319 exhibited substantially improved tolerability and faster clearance compared to MC3, without compromising on potency.189
In a separate study to engineer LNPs specifically tailored for mRNA delivery, researchers synthesized a novel series of amino lipids, chemically inspired by previous studies on MC3, and systematically identified and optimized structural motifs crucial for efficient mRNA delivery.190 This pivotal investigation yielded ionizable biodegradable lipids that, when formulated into mRNA LNPs, exhibited higher delivery efficiency through improved endosomal escape. Moreover, these lipids demonstrated a favorable toxicity profile and, for the first time in the field, enabled safe repeat dosing of mRNA LNPs at therapeutically relevant levels without adverse events in non-human primates. Noteworthy examples from this lipid series include heptadecan-9-yl 8-((2-hydroxyethyl)(8-(nonyloxy)-8-oxooctyl)amino) octanoate (lipid 5) and heptadecan-9-yl 8-((2-hydroxyethyl)(6-oxo-6-(undecyloxy)hexyl)amino) octanoate (lipid 8; lipid H; SM-102).190 The same lipid series was then also further screened and optimized for both delivery efficiency and immunogenicity when formulated as mRNA vaccines and administered intramuscularly.191 Of note, SM-102, a lead ionizable lipid characterized in these studies, ended up becoming a key component of Modernas elasomeran (brand name Spikevax) COVID-19 vaccine, which contains LNP-encapsulated mRNA coding for an engineered version of the spike protein of SARS-CoV-2.192 Likewise, a similar biodegradable ionizable lipid called ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315) is a key component of Pfizer-BioNTechs tozinameran (brand name Comirnaty) COVID-19 vaccine.193–195 At the time of writing, MC3, SM-102, and ALC-0315 are the only three ionizable lipids to have received FDA approval in a formulation for nucleic acid-based therapies.
Other lipid components (cholesterol, phospholipids, PEGylated lipids). In addition to cationic and ionizable lipids, LNP formulations incorporate other lipid components, including cholesterol, phospholipids, and PEGylated lipids, that are also important for their overall characteristics and functionality.
Cholesterol is often the second-most abundant component on a molar ratio basis and is critical for enhancing particle integrity by filling gaps between lipid tails, thus stabilizing the overall structure.196 Furthermore, cholesterol has been shown to play a key role in enhancing the fusogenicity of LNPs, which is important for endosomal escape.197,198 Although most clinically advanced LNPs use cholesterol in their formulations, there has been increasing interest in using other sterol analogs and derivatives instead of cholesterol to rationally design LNPs with improved nucleic acid delivery.199–201
The phospholipid component of most LNPs is another important structural constituent that promotes LNP fusion with cell membranes. As previously mentioned, DOPE is a phosphoethanolamine often used as a helper lipid in LNP formulations due to its tendency to adopt an inverted hexagonal HII phase, which facilitates endosomal escape.202 Another phospholipid commonly used in LNP formulations is 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), which is the phophatidylcholine used as the phospholipid component in both Modernas mRNA-1273 and Pfizer-BioNTechs BNT162b2 COVID-19 vaccines.203
Lastly, the PEGylated lipid component plays an important role in determining both the structural and delivery characteristics of the resulting LNPs. First, the molar proportion of PEGylated lipids in the formulation is a major factor in controlling particle size, with higher levels of PEGylated lipids resulting in smaller LNPs.204 Furthermore, PEGylated lipids also contribute greatly to the stability of LNPs both before administration in vitro, by decreasing particle aggregation in storage, and after administration in vivo, by reducing opsonization by serum proteins and clearance by the kidneys and mononuclear phagocyte system.205–207 Notable examples of PEGylated lipids include 1,2-distearoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DSG-PEG 2000), 1,2-dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 (DMG–PEG 2000), and 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159). DMG-PEG 2000 and ALC-0159 are the PEGylated lipids used in Modernas elasomeran and Pfizer-BioNTechs tozinameran COVID-19 vaccines, respectively.208
Types of RNA payloads. One of the first types of RNA payloads to be explored for use with LNP delivery was siRNA. siRNA comprises of short RNA duplexes, typically 21 nucleotides in length, and can be used to exploit the RNA interference (RNAi) pathway for precisely targeted post-transcriptional gene silencing.209 The RNAi pathway is a natural gene expression regulatory mechanism ubiquitous in eukaryotic cells.210,211 In the first step of RNAi, the enzyme Dicer cleaves precursor double-stranded RNA into shorter siRNAs.212 These siRNAs are subsequently processed and integrated into an RNA-induced silencing complex (RISC) by the RISC-loading complex.213 Of note, when exogenous siRNA is delivered into the cell via LNPs, this first Dicer-mediated step is unnecessary, as the exogenous siRNA can directly associate with RISCs, simplifying the system for therapeutic applications.214 Once loaded onto the RISC, the siRNA duplex unwinds. Only the antisense strand, referred to as the guide strand, is retained as part of the complex while the sense strand, referred to as the passenger strand, is degraded.215 The mature RISC complex then scans for and binds to mRNA molecules harboring complementary sequences to the guide strand. Binding of RISC to perfectly complementary target mRNA sequences triggers cleavage of the mRNA strand by Argonaute, the catalytic endonuclease component of RISC.216 The cleaved target mRNA sequence is subsequently degraded by cellular exonucleases, preventing further translation of the mRNA strand and knocking down expression of the mRNA-encoded gene.217 As previously mentioned, Patisiran, the first ever LNP-delivered siRNA-based drug, leverages the RNAi pathway to combat hereditary transthyretin-mediated amyloidosis by knocking down the production of pathogenic transthyretin protein and, since being FDA-approved in 2018, has kickstarted a flurry of FDA approvals of siRNA-based therapies.218–220
In recent years, mRNA has become one of the most studied payloads for LNPs, especially after the FDA approval of mRNA-based SARS-CoV-2 vaccines. Indeed, there has been much interest in developing mRNA-based therapies ever since the first proof-of-concept study in 1990 showed that mRNA could be used for direct gene transfer in vivo.221 mRNA is a single-stranded RNA molecule that can range anywhere from a few hundred nucleotides to a few thousand nucleotides long. In cells, mRNA is transcribed in the ribosome from a genomic DNA template by RNA polymerases and then exported into the cytoplasm, where the mRNA is then translated by ribosomes.222 Functioning as a transient “messenger molecule,” mRNA provides ribosomes with the correct sequence in which amino acids should be assembled into proteins. There are a few structural components of mRNA that important for regulating translation: the 5′ cap, the 5′ untranslated region (UTR), the coding sequence for the gene of interest, the 3′ UTR, and the 3′ poly(A) tail.223 In cap-dependent translation, recognition of the 5′ cap by eukaryotic translation initiation factor 4E (eIF4E) is a critical first step for assembling the translation preinitiation complex (PIC).224 Proteins in the PIC bound to the 5′ cap are also responsible for binding to the poly(A)-binding proteins (PABP) that coat the 3′ poly(A) tail of the mRNA. This binding interaction circularizes the mRNA, which has been hypothesized to increase translation efficiency via ribosome recycling.225 Once the PIC is fully formed, the small 40S ribosomal subunit then begins scanning through the 5′ UTR in a 5′ to 3′ direction until it encounters the first start codon.226 Then, the large 60S ribosomal subunit is recruited to the small 40S ribosomal subunit, forming the complete 80S ribosome, which will then translate the coding sequence for the gene of interest.227 By delivering exogenous mRNA into a cell, it is thus theoretically possible to instruct a cell to produce any protein using a cells own native translation machinery. Furthermore, delivering exogenous mRNA has numerous advantages over-delivering exogenous DNA.228 First, the mRNA only has to be delivered into the cytoplasm to be translated, whereas DNA requires more challenging delivery into the nucleus in order to be transcribed then translated. Second, mRNA does not integrate into the genome, so there is reduced concern about unintentional mutagenesis resulting from integration, a risk associated with DNA-based gene therapies. Lastly, because mRNA is degraded over time by intracellular nucleases, the mRNA is only transiently active in the cell, which can be beneficial from both a safety and pharmacological perspective.
Although LNP-mediated delivery of mRNA is extremely powerful, there are two additional promising variations on mRNA delivery in development that are worth noting: circular RNA (circRNA) and self-amplifying RNA (saRNA). The circRNA are covalently closed loops of RNA that exhibit much higher stability than linear mRNA due to their circular structure. The two primary pathways for mRNA degradation involve either 3′ to 5′ exonuclease digestion, after the 3′ poly(A) tail has been sufficiently shortened by nonspecific exonuclease digestion, or alternatively, 5′ to 3′ exonuclease digestion, after the 5′ cap has been enzymatically removed by decapping enzymes. Because circRNA lacks both a 5′ and 3′ end, exonucleases are unable to degrade intact circRNAs. Thus, circRNA degradation must be initiated by other cellular mechanisms, such as those involving endonucleases.229 The lack of a 5′ cap, however, also means that translation initiation cannot proceed via the canonical cap-dependent pathway. Instead, circRNAs must rely on cap-independent translation. One of the most commonly employed mechanisms for cap-independent translation is the use of internal ribosome entry sites (IRES), which are RNA elements that fold into secondary and tertiary structures to directly recruit translation initiation factors.230 There are currently two main methods for synthesizing circRNA. One way is to align the two free ends of a linear piece of RNA, either using a DNA splint or engineered RNA secondary structure and then use a ligase to ligate the two ends together. The second way is to encode a ribozyme, such as a self-splicing group I intron, into the linear RNA sequence and have the ribozyme autocatalyze the RNA circularization. Currently, however, both methods have shortcomings such that the large-scale synthesis and purification of circRNA remains one of the main challenges limiting the adoption of the technology.231
The saRNA are mRNA transcripts that utilize the self-replication machinery of RNA viruses, usually RNA alphaviruses, to produce additional copies of the mRNA transcript after being delivered into the cytosol of a cell.232 Typically, the overall structure of saRNA is very similar to that of mRNA. Both contain a 5′ cap, 5′ UTR, 3′ UTR, and a 3′ poly(A) tail. However, within the coding region, rather than only containing the gene of interest, saRNA also encodes for viral machinery used to replicate viral RNA genomes. In wild-type RNA viruses, the genome usually is split into two open reading frames (ORFs). The first ORF contains the genes for the viral replication machinery, while the second ORF contains the genes for the viral structural proteins. In an saRNA molecule, the genes for the viral structural proteins in the second ORF are replaced to instead encode for the desired gene of interest.233 After entering a cell, the first ORF on the saRNA is translated to produce the necessary RNA replication machinery, which includes an RNA-dependent RNA polymerase (RdRp). Next, a complementary negative strand copy of the saRNA is created and used as the template for further transcription by the RdRp to create multiple mRNA copies of the second ORF containing the gene of interest.234 Through this replication process, the intracellular amount of mRNA encoding the gene of interest can be amplified by multiple orders of magnitude. Thus, the dose of saRNA required to achieve a given therapeutic effect is generally lower than that of mRNA. One downside of using saRNA is that the replication process creates double-stranded RNA intermediates that can activate interferon-mediated defense mechanisms, triggering a strong antigen-specific immune response against the gene of interest.235 While this may be advantageous for some uses such as vaccination, it is extremely limiting for other uses such as protein replacement therapy. Another limitation of saRNA is that the inclusion of the viral replication machinery results in RNA molecules that can be prohibitively long. To overcome this downside, some researchers have experimented with using “trans-amplifying” RNA systems that deliver the viral replication machinery ORF and the gene of interest ORF on separate strands of mRNA.236,237
Considerations for synthesizing mRNA molecules. Unlike siRNA, mRNA is too long and complex to be chemically synthesized. Thus, mRNA is typically synthesized using in vitro transcription (IVT), in which the mRNA is transcribed into a linearized DNA template using a cell-free system with an RNAP, usually either the bacteriophage T7 or SP6 RNAP.223 Even though IVT was first developed in the 1980s, the development of mRNA-based therapies struggled for many years largely due to the instability and immunogenicity of IVT mRNA.238 Decades of research later, however, advances in our understanding of RNA chemistry and RNAs interactions with intracellular pathways have led to improved methods for synthesizing more stable and less immunogenic mRNA. Here, we will review the most important considerations to keep in mind when synthesizing mRNA molecules, all of which must account for key structural features depicted in Fig. 6.
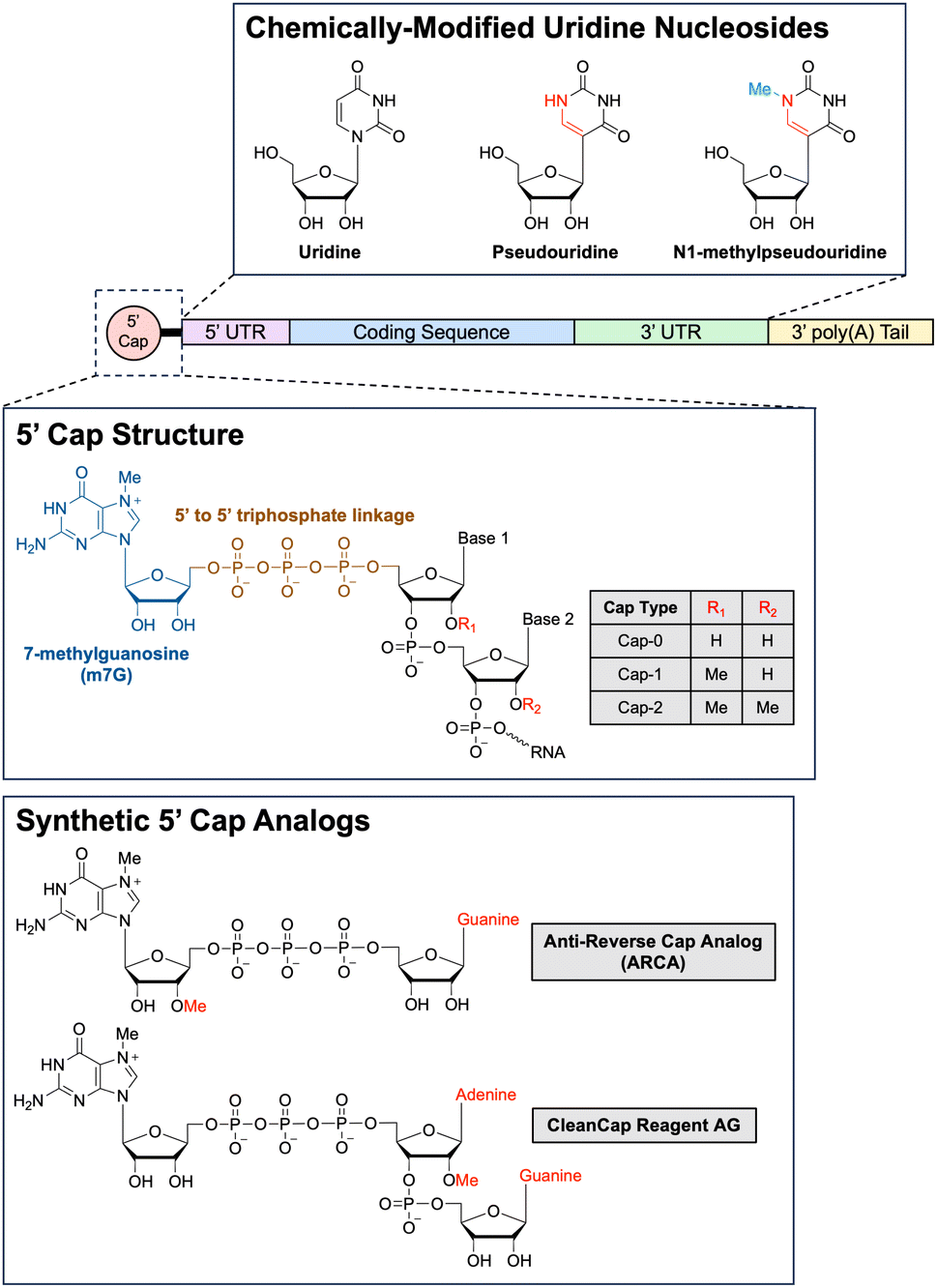 | ||
| Fig. 6 Important structural features of mRNA. mRNA synthesized via in vitro transcription must recapitulate key structural features of naturally-produced mRNA, such as the 5′ cap structure and nucleoside chemical modifications, in order to reduce the immunogenicity of the mRNA and to improve mRNA translation efficacy and stability. These include analogs to uridine in the 5′ UTR, coding sequence, and 3′ UTR; as well as key analogs for the 5′ cap. | ||
As previously mentioned, the 5′ cap is important for binding to eIF4E and recruiting the rest of the PIC. Naturally-produced cellular mRNA is capped co-transcriptionally in a multi-step process in which a guanidine nucleotide is first covalently attached to the 5′ end of the nascent mRNA using a 5′ to 5′ triphosphate linkage and then methylated on the N7 position by methyltransferases.239 IVT mRNA can be capped either post-transcriptionally or co-transcriptionally. One of the earliest methods developed for capping IVT mRNA post-transcriptionally utilizes purified Vaccinia virus capping enzyme to attach a 7-methylguanosine to the 5′ triphosphate end of the mRNA.240–242 Recently, the mRNA capping enzyme from Faustovirus has also been characterized and commercialized for use in post-transcriptional capping, offering advantages over the Vaccinia virus capping enzyme such as improved capping efficiency and stability over a broader range of temperatures.243 Although post-transcriptional capping is highly efficient, the need to run multiple sequential reactions to generate fully functional IVT mRNA can be a disadvantage for some workflows. Thus, co-transcriptional capping in a one-pot reaction has been developed such that a 5′ cap dinucleotide analog is incorporated into the nascent mRNA strand by the RNAP. However, co-transcriptional capping with the standard cap analog m7G(5′)ppp(5′)G can potentially incorporate onto some of the mRNA in a nonfunctional reverse orientation. To avoid this undesirable situation of having a heterogenous pool of IVT mRNA containing a mixture of both functionally and nonfunctionally capped mRNA, the anti-reverse cap analog (ARCA) 3′-O-Me-m7G(5′)ppp(5′)G was developed.244,245 ARCAs are methylated at the 3′ position to prevent incorporation in the reverse orientation and have been shown to result in IVT mRNA preparations with improved translational efficiency.246 The major downside of using ARCAs, however, is that they require the relative concentration of guanosine triphosphate (GTP) to be reduced so that the ARCA can outcompete GTP as the first nucleotide to be incorporated into the mRNA, resulting in an overall capping efficiency of 80% and decreased overall RNA yield. To overcome this drawback, the trinucleotide CleanCap Reagent AG cap analog was developed such that a reduction in GTP concentration is not required. The CleanCap Reagent AG trinucleotide is composed of a m7G linked to an AG dinucleotide via a 5′-5′ triphosphate linkage and allows for IVT with capping efficiencies of >95% and higher RNA yields compared to IVT with ARCAs.247 Of note, Pfizer-BioNTechs tozinameran SARS-CoV-2 vaccine uses the CleanCap Reagent AG.248
Another advantage of using the CleanCap Reagent AG is that it results in the formation of a cap-1 structure due to the methylation already present on the first adenine nucleotide after the 5′ cap. A cap-0 structure corresponds to an mRNA with no methylations on the two nucleotides following the 5′ cap, and a cap-2 structure corresponds to an mRNA with methylations on both of the two nucleotides following the 5′ cap.249 For mRNA that is capped post-transcriptionally, the addition of mRNA cap 2′-O-methyltransferase is necessary to convert a cap-0 structure into a cap-1 structure.250 The presence or absence of methylations on these two first nucleotides adjacent to the 5′ cap has been shown to be an important way in which cells are able to discriminate between self and non-self nucleic acids.251 As such, mRNA molecules with cap-1 structures have been shown to have higher mRNA translation efficiencies by evading cellular innate immune responses against non-self RNA, such as members of the interferon-induced proteins with tetratricopeptide repeats (IFIT) protein family and retinoic acid-inducible gene I (RIG-I).252,253 Recent results suggest that the same may be true for mRNAs with cap-2 structures, but more extensive research and characterization are required to draw definitive conclusions about their impact on mRNA translation efficiency.254
The 3′ poly(A) tail is important for translation to occur efficiently and for conferring stability to mRNA. Poly(A) tails can either be added post-transcriptionally by using an E. coli poly(A) polymerase or encoded in the DNA template.255 Because enzymatic addition of poly(A) tails results in a distribution of varying poly(A) tail lengths and varies batch-to-batch, using a template-encoded poly(A) tail is generally preferred. Based on quantification of mRNA transcripts in HeLa and 3T3 cells, it was found that natural poly(A) tails are, on average, 50–100 nt long and that longer tails typically correlate with having long half-lives but not necessarily higher translation efficiencies.256 Indeed, previous work to optimize poly(A) tails for IVT mRNA reported that a poly(A) tail of at least 30–40 nt is required for translation and that 120 nt was optimal.257 Poly(A) tails longer than 120 nt generally do not appear to yield any significant benefits to translation efficiency but may help nonetheless with increasing the half-life of the mRNA.258 More recently, it has been reported that segmented poly(A) tails are much more stable to maintain in plasmids by reducing unwanted recombination events, without compromising mRNA translation efficiency of half-life.259 As such, a segmented poly(A) tail consisting of a 30 nt poly(A) sequence linked to a 70 nt poly(A) sequence by a 10 nucleotide linker was used in Pfizer-BioNTechs tozinameran COVID-19 vaccine.260
Sequence optimization of the coding region, the 5′ UTR, and the 3′ UTR is a highly nuanced topic and an active area of research. For most applications involving genetic engineering, sequence codon optimization of the coding region is a common practice and often the default strategy.261 While codon optimization has been shown to be helpful in many situations, there are also some important caveats to keep in mind. For example, codon optimization could disrupt naturally evolved mRNA secondary structure, affecting translation rates and preventing proper folding; alter natural recognition sites for post-transcriptional modifications, creating novel unnatural epitranscriptomic patterns; and generate new alternative translation initiation start sites, producing uncharacterized novel peptides.262,263 Nonetheless, there are tangible benefits to codon optimization. In one study, sequence-engineered unmodified mRNA was shown to improve translational efficiency by eliciting any undesired immunostimulatory responses even after repeat dosing, something that was generally thought to require the use of modified nucleosides.264 In another recent study, a novel computer algorithm was designed to generate optimal mRNA sequences encoding vaccine antigens and was shown to vastly improve mRNA half-life and protein expression, allowing for more effective vaccination in mouse models.265 Indeed, codon usage can be thought of as a complex “second genetic code” that will require further investigation to be fully understood and harnessed.266
While the 5′ and 3′ UTRs are non-coding regions of the mRNA, they are nonetheless important for regulating mRNA translation efficiency and stability. mRNA can be regulated by the UTRs in a few different ways. For example, RNA-binding proteins can recognize certain nucleotide primary or secondary structures in the UTRs to regulate subcellular localization and translational control.267 The UTRs also often contain microRNA binding sites that are important for regulating mRNA stability and translation efficiency.268 When producing synthetic IVT mRNA transcripts, there are a few potential UTR engineering approaches if one does not wish to use a genes own native UTR. First, a common strategy that has been demonstrated to work quite well is to swap a genes UTRs with the UTRs of a gene that is known to be highly expressed, such as human or Xenopus beta-globulin.269 Recently, this approach has also been expanded via large-scale cellular library screens of natural UTRs to discover and characterize additional new UTRs with desirable properties.270,271 The second approach is to use algorithms to evaluate and design novel UTR sequences. In one study, by combining experimental data from a library of 5′ UTRs with a deep learning model, researchers were able to precisely engineer novel 5′ UTRs with a range of specified levels of ribosome loading.272 Similar to codon optimization, sequence optimization of UTRs is an active area of ongoing research and further progress in the field is sure to have a large impact on improving mRNA design.
Lastly, perhaps one of the most important advancements in mRNA-based therapy development has been an increased understanding of how to reduce the innate immunogenicity of mRNA.223,260,273 While activation of the innate immune response could be beneficial for some applications, such as vaccination, it can significantly inhibit mRNA translation and be detrimental in other applications. Thus, a detailed understanding of how to modulate the immunogenicity is essential for any mRNA-based therapy. Immune cells have a variety of PRRs specialized for detecting non-self mRNA in the endosomal compartment, which are usually the result of some type of viral infection. Notable examples include members of the TLR family: TLR3 recognizes double-stranded RNA (dsRNA), and TLR7 and TLR8 recognize single-stranded RNA (ssRNA), especially GU-rich sequences.274–277 Non-immune cells also have various PPRs for detecting RNA. Notable examples include members of the RLR family: RIG-I can recognize by short, blunt dsRNA, and melanoma differentiation-associated protein 5′ (MDA5) can recognize long dsRNA. Both RIG-I and MDA5 can also detect the absence of 2′-O-methylation on mRNA.278–281 Activation of these PPRs leads to a strong type I interferon response, which can significantly hamper mRNA translation efficiency and stability due to the upregulation of downstream effectors. Notable effectors include protein kinase R (PKR), which phosphorylates eukaryotic translation initiation factor 2A (eIF2A) to inhibit translation, and 2′-5′-oligoadenylate synthetase (OAS), which degrades intracellular RNA.282,283
One common way to circumvent the activation of these innate immune sensors is through the incorporation of chemically modified nucleosides into IVT mRNA. There are a multitude of natural chemical modifications to mRNA that regulate gene expression.284 It has been found that mRNA containing these naturally modified nucleosides do not activate PPRs as much as an mRNA containing unmodified nucleosides.285–289 Luckily, modified nucleosides are easily incorporated into IVT mRNA using standard IVT protocols.290 Although there have been many chemically modified nucleosides that have been used in IVT mRNA, pseudouridine is notable for being one of the first modified nucleosides shown to reduce the immunogenicity of mRNA. Incorporating pseudouridine into IVT mRNA in place of uridine was found to increase both the translational efficiency and stability of the mRNA.291 Further research has shown that using N1-methylpseudouridine in place of uridine yielded even larger reductions in IVT mRNA immunogenicity compared to pseudouridine.292 A separate study found that uridine depletion in IVT mRNA sequences in combination with using chemically modified nucleosides can yield greater reductions in immunogenicity.293 Of note, the IVT mRNA in both Modernas elasomeran and Pfizer-BioNTechs tozinameran SARS-CoV-2 vaccines were synthesized using N1-methylpseudouridine in place of uridine.192,248
Because dsRNA is often times a contaminant generated during IVT, another way to reduce the immunogenicity of IVT mRNA is to use chromatography methods to purify out any dsRNA.294 Previous work has shown that HPLC purification of chemically modified IVT mRNA can result in up to significantly greater translation efficiency, up to 1000-fold higher, in primary cells.295,296 Recently, a fast, low-cost alternative approach to removing dsRNA contaminants has also been developed using a cellulose-based removal process.297
As previously mentioned, the use of RNA LNP-based therapies to combat infectious diseases has experienced significant growth and is the most clinically advanced applications of mRNA-based treatments. There are two primary approaches for employing RNA-based therapies in the fight against infectious diseases. The first approach involves providing passive immunity to patients by delivering mRNA that encodes for antibodies that target a particular pathogen. This approach is particularly beneficial for immunocompromised individuals who cannot mount their own immune response against pathogens. For example, a previous study has demonstrated that delivering mRNA encoding a broadly neutralizing antibody against the human immunodeficiency virus (HIV) protected mice from subsequent intravenous challenges with HIV.302 Separate work has also explored the delivery of mRNA encoding neutralizing antibodies against various other infectious agents, including respiratory syncytial virus (RSV), Zika virus, and Chikungunya virus.303–305 The second approach aims to stimulate active immunity by delivering mRNA-encoding antigens from the pathogen of interest. When a cell internalizes and starts translating this mRNA into protein, the cell becomes a source of the antigen. APCs, namely DCs,306 can then capture and present the antigen, triggering both cellular and humoral immune responses. This approach can also leverage the flexibility of delivery platforms to introduce multiple mRNA sequences, each encoding a distinct antigen, within a single vaccine formulation, allowing for a more robust immune response against a single pathogen or a broader immune response against multiple pathogens or pathogen variants.307 This strategy could be especially useful for vaccinating against pathogens that are known to mutate rapidly, such as influenza. Lastly, of note, saRNA encoding infectious disease antigens are an exceptionally promising technology to use for vaccination because they have been shown to elicit protection equivalent to conventional mRNA vaccines, but require much lower doses.235,308 Besides SARS-CoV-2, clinical trials utilizing mRNA LNP-based vaccines have begun for a variety of infectious diseases, including cytomegalovirus, Zika virus, Chikungunya virus, rabies virus, influenza, RSV, human metapneumovirus, and parainfluenza.168,273,300
RNA LNP-based therapies have also shown much promise for applications in cancer immunotherapy. There are three main ways in which RNA LNPs are being developed to treat cancer. First, similar to the passive immunity approach for infectious diseases, mRNA-encoding anticancer antibodies can be delivered using LNPs such that anticancer antibodies are produced by the recipients own cellular machinery. This approach to antibody-based cancer treatments could potentially be more cost-effective, flexible, and logistically simple compared to traditional recombinant protein-based approaches, which often struggle with delivery and manufacturing challenges.309,310 Currently, mRNA LNPs encoding for anticancer antibodies are being tested in clinical trials for treating various solid tumors by targeting shared cancer biomarkers such as CLDN6 and CLDN18.2 (NCT04683939, NCT05262530). Second, mRNA can be used in cancer vaccines by encoding either non-mutated or mutated tumor-associated antigens to elicit an antitumor immune response. In one notable example, FixVac is an intravenously administered liposomal RNA cancer vaccine that contains mRNA encoding for four non-mutated antigens prevalent in melanoma. Based on an interim analysis of phase I clinical trial data, FixVac was found to mediate durable antitumor responses in patients with CPI-experienced melanoma, either alone or in combination with CPI therapy, demonstrating that there are benefits to using cancer vaccines against common non-mutant shared tumor antigens.311 Using a completely different cancer vaccine approach that is highly specific to each individual patient, mRNA-4157/V940 is a personalized mRNA-based cancer vaccine that encodes for up to 34 patient-specific tumor neoantigens, each of which are computationally predicted based on sequencing analysis of a patients tumor. Due to promising phase II clinical trial results, in which mRNA-4157/V940 in combination with CPI reduced the risk of recurrence or death by 44% compared to CPI alone in patients with stage III/IV melanoma, mRNA-4157/V940 is moving into phase III clinical trials (NCT05933577). Lastly, LNPs can be used to deliver mRNA encoding cytokines that can reshape the tumor microenvironment from being immunosuppressive to proinflammatory. In one study, the intratumoral administration of an mRNA cocktail encoding for IL-23, IL-36γ, and OX40L resulted in remarkably durable anticancer immunity in mouse models of cancer.312 The therapy has moved into phase I clinical trials as mRNA-2752 for the treatment of various solid tumors (NCT03739931).313 Similarly, intratumoral administration of mRNA encoding IL-12 has also been shown to work well as an anticancer therapy in preclinical models and is now being investigated in clinical trials (NCT03946800, NCT04455620).314
4.3 Polymersomes
Polymersomes were first reported by Discher and Eisenberg in the late 1990's, and have since been researched extensively as vehicles for drug delivery and immune modulation.315,316 Consisting of amphiphilic polymers, these nanoparticles are treated as the macromolecular analogue to LNPs and liposomes. Morphology of polymersomes are contrasted with other lipid and polymer nanoparticles in Fig. 7. Materials commonly used include polystyrene, polybutadiene or hydrophobic poly (meth)acrylates as the membrane forming block and hydrophilic polymers such as polyethylene glycol or acrylic acid as the corona forming segment. The vesicular morphology is attained by keeping the hydrophilic weight fraction between 25 and 45 weight percent of the total polymer, to ensure the proper curvature of the self-assembled unimers.317 Polymers are either processed as a film or dissolved in water-miscible organic solvents such as tetrahydrofuran, dimethylsulfoxide or dimethylformamide, before being transferred into aqueous buffer to induce self-assembly. Polymersomes have been proposed to have enhanced stability on account of chain entanglements and their vesicular structure allows for the entrapment of hydrophilic payloads in the core and hydrophobic molecules in the membrane.318 Furthermore, advances in polymer chemistry have allowed researchers to develop a wide array of bioactive and stimuli-responsive polymersomes, which impart unique properties for many parenteral formulations. Excellent reviews on the development of novel chemistries and design considerations for polymersomes can be found elsewhere.319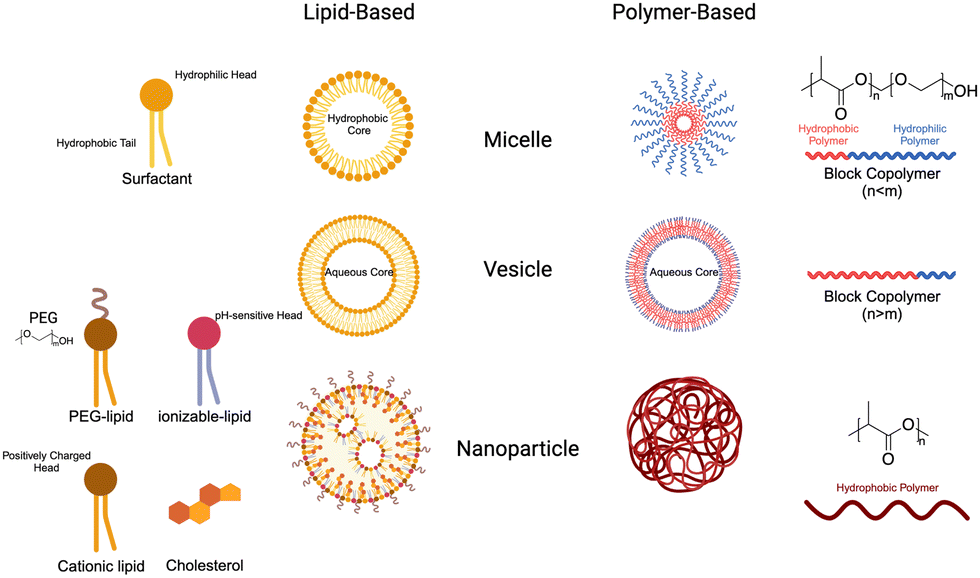 | ||
| Fig. 7 Comparison of morphology and composition of different nanoparticle technologies, which includes both lipid- and polymer-based micelles, vesicles, and nanoparticles, each of which have unique compositions to favor their unique morphology. | ||
Despite their initial promise, there are currently no polymersome formulations used commercially.320 However, ongoing research is working to improve formulations by enhancing payload encapsulation and delivery, while simplifying formulations to allow for large-scale production. Here, we review some of the recent work exploring delivery of protein antigens and nucleic acids in the context of vaccination. As mentioned above, a variety of polymers can be chosen as the hydrophobic and hydrophilic segments, therefore we only focus on recent developments in polymer nanoparticle formulation.
The versatility of polymer nanoparticles is highlighted in work from Zou et al., as they developed “asymmetric” polymersomes consisting of a charged inner surface and inert outer surface.329 This was achieved by controlling the relative weight fractions of each portion in the polymer chains comprising the nanoparticle. Specifically, polyethylenimine was used as the charge-complexing group, but preferentially segregated to the inside of the vesicles to prevent toxicity. As a result of controlled syntheses, the authors were able to formulate a simple, one-component material capable of encapsulating and delivering siRNA. Materials were also designed to be reduction-sensitive as they were cross-linked via disulfide bonds and such synthetic modifications highlight the versatility of polymersomes. Furthermore, cleavable crosslinks are needed to circumvent the high stability of the nanoparticles once they reach their target cells. Importantly, the authors demonstrated tumor regression and enhanced survival in murine cancer models.
Redox-responsive polymersomes have also been developed to exploit the reductive environment of the endosome.332 It is known that the disulfide bond is sensitive to the intracellular influx of reducing agents such as cysteine and glutathione.333 To this extent, Jia et al. developed diblock copolymers consisting of a disulfide bond at the junction between the two polymer segments.334 In this work, the authors designed polymersomes using standard polymerization techniques and demonstrated the enhanced release of payloads compared to non-reduction sensitive analogs. The materials can be made in high yield with facile purification, representing an improvement over previous formulations often difficult to design due to the instability of the disulfide junction. Importantly, enhanced uptake and release of the auto-quenching fluorophore calcein highlights the utility of this system, which may prove beneficial for delivering more complex macromolecules such as protein and nucleic acid.
Polymer nanoparticles can also be designed to be inherently adjuvanting, as demonstrated by materials designed by Luo et al. to stimulate the STING pathway.335 The authors utilized pH-sensitive tertiary amines over a wide range of pKa, proposing that an acidification of the endosome leads to transient protonation of the polymer, followed by endosomal rupture via the proton-sponge effect. This internal stress in turn activates the STING pathway, leading to a type 1 interferon response and robust CD8+ cytotoxic T cell immunity. The upregulation of IDO-1 and CXCL10 was also reported as a validation for STING pathway activation along with phosphorylation of IRF3. To demonstrate its applications, the authors treated B16-OVA, MC38 and TC-1 tumors via subcutaneous injection, and reported flattened tumor growth curves and enhanced survival. Enhanced responses were observed when using CPIs for TC-1 treatment.
Despite requiring fewer components than LNPs, the macromolecular nature of polymers requires carefully controlled parameters for processing the materials in order to form reproducible morphologies. Due to challenges in materials design along with the uncontrolled nature of self-assembly, extensive purification is required to attain materials of desired size and polydispersity. As a result of this post-processing, there is a substantial loss of synthetic material, making the actual efficiency of production lower than what is theoretically possible.
As mentioned in Section 4.1.1, researchers have designed and utilized microfluidic devices to form reproducible, homogeneous nanoparticles.154 In addition to carefully modulating the size of polymersomes, microfluidic mixers allow for minimal purification needed prior to injection. As a result, these techniques are critical in improving the scalability of nanoparticle formulations, while retaining narrow polydispersities needed for clinical approval. Such techniques may prove necessary to implement polymersomes as commercially-relevant formulations.
4.4 Inorganic nanoparticles
Inorganic nanoparticles have emerged as a groundbreaking frontier in medicine, showcasing their remarkable potential to revolutionize diagnostics, therapy, and drug delivery.337 These nanoscale structures, composed of various materials such as metals, metal oxides, and semiconductors, possess unique physicochemical properties that enable them to interact with biological systems in unprecedented ways. Moreover, their ability to be functionalized with ligands and biomolecules allows for precise targeting and interaction with cellular receptors, paving the way for personalized and highly effective therapeutic interventions.338,339 As research in this field continues to advance, the integration of inorganic nanoparticles holds the promise of transforming the landscape of medical treatments, enabling more precise, efficient, and tailored approaches to disease management.Other inorganic nanoparticles such as calcium phosphate, manganese oxide, iron oxide, and carbon nanomaterials have also been used for immunoengineering applications as discussed elsewhere.354
5 Local immunomodulation
Immunomodulatory materials are used to promote the growth and differentiation of new tissue composed of scaffold materials, biochemical cues, and cellular composite systems.355One general example is given in a recent work, where local inflammatory response is suppressed by introducing a combination of immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 and galectin-3 protein binding glycans present at the tissue after local injection.356 The authors studied the fused protein system to block gene expression of inflammatory cytokines IL-6 and IL-12p40 on the disease models caused by chronic inflammation such as osteoarthritis and psoriasis. Several other technologies are explored in the following sections.
5.1 Device coatings
Medical device coatings play a pivotal role in enhancing the performance, biocompatibility, and durability of a wide range of healthcare technologies. From implantable devices to diagnostic tools and drug delivery systems, coatings serve as a crucial interface between the medical device and the biological environment. These coatings are meticulously designed to address multifaceted challenges, including improving biocompatibility, preventing infections, promoting tissue integration, and enabling controlled drug release. As the field of medical devices continues to advance, the development and application of innovative coatings have emerged as a dynamic area of research and development, offering new avenues for improving patient outcomes and revolutionizing the landscape of modern healthcare. Here we discuss recent advances in biomaterial coatings. One primary goal of many device coatings is antimicrobial activity, which can be achieved through a variety of strategies. One recent one, published by Sadrearhami et al., used polydopamine and PEG to encapsulate low amounts of nitric oxide (NO). The coating, which released NO over two days, was able to prevent biofilm attachment in a bacterial culture.357 Another strategy by Piktel et al. involved the fabrication of gold nanoparticles of varying morphology, including rods, peanuts, stars, and spheres. Their materials could be coated onto latex urinary catheters and prevent growth of patient-derived bacteria.358 These works stand out as two recent examples of promising antimicrobial device coatings, but tremendous effort is ongoing in the space and has been reviewed in-depth elsewhere.359,360 While we have come a long way in understand the foreign body response, further research is needed to understand the immunological interface with 21st century bioelectronics.5.2 Hydrogels
Hydrogels can face challenges related to immune rejection when implanted into the native tissue environment.361,362 The immune system's response to foreign materials, including hydrogels, can lead to inflammation, fibrosis, and ultimately failure of the engineered tissue construct. Understanding and mitigating these immune rejection problems is crucial for successful tissue regeneration applications. This inflammatory reaction can lead to the release of pro-inflammatory cytokines, recruitment of immune cells, and degradation of the hydrogel scaffold and destruction of the cells. The immune response can result in the formation of a fibrous capsule around the hydrogel, limiting its integration with the surrounding tissue.363Incorporation of immunomodulatory agents, such as anti-inflammatory drugs or growth factors, into the hydrogel structure can help attenuate the immune response.364 These agents can be released in a controlled manner from the hydrogel, dampening the inflammatory reaction and promoting tissue regeneration. Materials such as these have often been employed to improve outcomes diabetic wound healing, including fibrous mats releasing pioglitazone hydrochloride,365 stimuli-responsive materials releasing metformin,366 or soluble polymeric materials releasing butyrate.367
Co-implantation of immunomodulatory cells with the hydrogel can help regulate the immune response.368 These cells can secrete anti-inflammatory factors and modulate immune cell behavior, promoting a more tolerant environment for the hydrogel. For example, a recent paper by Guo et al.369 demonstrated the utility of injectable, conductive hydrogels that degrade over time to delivery myofibroblasts cells to induce muscle regeneration. Their hydrogel material is based on dextran-graft-aniline tetramer-graft-4-formylbenzoic acid and N-carboxyethyl chitosan, which demonstrate rapid self-healing properties attributable to dynamic Schiff base bonds between the formylbenzoic acid and amine group from N-carboxyethyl chitosan. The engineering of advanced materials such as these hold tremendous potential for the future of cell therapy.
A crosslinkable norbornene-modified collagen type I hydrogel was used to develop a mechanically tunable environment to regulate T cell phenotype.370 Collagen type 1 was used as an extracellular matrix (ECM) constituent and functionalized with covalently crosslinkable units to control matrix stiffness. The AP-1 pathway was used as a marker for determining T cell phenotype. Slowly relaxing the stiffness of the matrix makes T cell to respond antigens faster at an in vitro and in vivo xenograft lymphoma model. Tailorable T cell products are expected to be developed by manipulating the ECM mechanics.
5.3 Microneedles
Microneedles have emerged as a tool at the intersection of immunology and engineering as a new tool for vaccination and immunotherapy delivery. These minuscule, minimally invasive structures offer a new way of applying vaccines and immunomodulatory agents and promising improved efficacy, patient compliance, and enhanced immune responses.371,372 By harnessing precision fabrication techniques, microneedles enable controlled and targeted delivery of antigens, adjuvants, and other bioactive compounds directly into the skin's immune-rich microenvironment. Microneedles have a multitude of design parameters to be altered for various applications across drug delivery. The composition, morphology, length, and stimuli-responsiveness can all be precisely tuned for different applications.373,374 Here, we focus on the recent advances in microneedles for immunomodulatory applications, including vaccination and allergic desensitization.The concept of microneedle vaccination dates back to the early 1970s, but their widespread adoption has been limited for a variety of factors including manufacturing. The latest advances in manufacturing, combined with cutting edge vaccine technologies, are under investigation. For example, recent work by vander Straeten et al.375 reported a 3D printing device capable of fabricating microneedle arrays loaded with LNPs ensapsulating mRNA for SARS-CoV-2 vaccination. The resulting arrays demonstrated both robust efficacy and room temperature stability for up to 6 months. Further, the arrays can be fabricated on-site without the need for trained healthcare personnel. Another work by Shin et al.376 demonstrates the utility of HPLC in the coating process to produce H3N2 influenza vaccinations that perform comparably to traditional intramuscular vaccines. Together, these works present advancement in multiple areas of vaccine technology and supports the future success of microneedle-based vaccination.
Microneedles are also of interest in allergic desensitization. The same skin-resident APCs are capable of presenting antigen in a pro-tolerogenic manner and can support desensitization to allergens. This therapy is often dubbed subcutaneous or epicutaneous immunotherapy (SCIT or EPIT), depending on the penetration depth of the microneedles. A recent work by Landers et al.377 demonstrated the protective effect of peanut antigen-coated steel microneedles in murine models of oral allergic challenge. Interestingly, a similar technology was employed by Shakya et al.378 to deliver the model allergen ovalbumin, which offered protection in a murine model of airway allergy. Finally, more advanced materials science strategies can further improve the therapeutic efficacy and clinical utility of microneedle-based therapies. A recent example by Zhang et al.379 reported implantable microneedles with dissolvable components of silk fibroin hydrogel and polyvinyl alcohol, which can offer prolonged antigen exposure to improve clinical outcomes in food allergy. Many other attempts have been made at microneedle-based immunotherapy and are reviewed in-depth elsewhere.380 Clearly, the skin is a powerful immunologically active tissue and can be used to manipulate the balance of immunity in allergy and infectious disease.
6 Conclusions and future perspectives
In this review, we discussed the recent works in the design and synthesis of new biomaterials interacting with the immune response. The vaccine technologies and therapeutic systems for fighting and treating diseases by exploiting the immune system have recently attracted immense interest among the researchers and we have been witnessing the availability of the clinical methods and materials. New immunomodulatory systems including small molecules, biologics and nanoparticle formulations have been finding more applications in clinics. As we research and learn mechanisms of interactions among the synthetic tools and biological events, we are able to engineer new responsive materials that can precisely affect the immune system and their disease mechanisms. While developing new tools for treating the diseases, it is important to understand the quality by design (QbD) principles and regulations during the research for making these therapeutics more efficient and set the translation goals realistic and timely.381 Reliability of the results and safety of the new methods are vital for producing more precise and efficient therapies.Author contributions
All authors contributed to conceptualization, data curation process, analysis, writing, review and editing of the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BioRender.com was used to create some figures. ChatGPT (OpenAI, 2023) was used to assist with the writing and editing process in some sections.Notes and references
- Z. Cheng, M. Li, R. Dey and Y. Chen, J. Hematol. Oncol., 2021, 14, 1–27 CrossRef.
- P. P. Kalelkar, M. Riddick and A. J. Garca, Nat. Rev. Mater., 2022, 7, 39–54 CrossRef CAS.
- H. Dai, Q. Fan and C. Wang, Exploration, 2022, 20210157 CrossRef PubMed.
- D. Furman, J. Campisi, E. Verdin, P. Carrera-Bastos, S. Targ, C. Franceschi, L. Ferrucci, D. W. Gilroy, A. Fasano and G. W. Miller, et al. , Nat. Med., 2019, 25, 1822–1832 CrossRef CAS.
- P. Yousefpour, K. Ni and D. J. Irvine, Nat. Rev. Bioeng., 2023, 1, 107–124 CrossRef PubMed.
- C. R. Alving, K. K. Peachman, M. Rao and S. G. Reed, Curr. Opin. Immunol., 2012, 24, 310–315 CrossRef CAS.
- T. Kaisho and S. Akira, Biochim. Biophys. Acta, Mol. Cell Res., 2002, 1589, 1–13 CrossRef CAS PubMed.
- J. H. Cho, H.-J. Lee, H.-J. Ko, B.-I. Yoon, J. Choe, K.-C. Kim, T.-W. Hahn, J. A. Han, S. S. Choi and Y. M. Jung, et al. , Oncotarget, 2017, 8, 24932 CrossRef PubMed.
- G. Frega, Q. Wu, J. Le Naour, E. Vacchelli, L. Galluzzi, G. Kroemer and O. Kepp, Oncoimmunology, 2020, 9, 1796002 CrossRef PubMed.
- D. Rotrosen and J. A. Bluestone, et al. , J. Allergy Clin. Immunol., 2002, 110, 17–23 CrossRef CAS.
- A. Mansurov, A. Lauterbach, E. Budina, A. T. Alpar, J. A. Hubbell and J. Ishihara, Am. J. Physiol.: Cell Physiol., 2021, 321, C369–C383 CrossRef CAS.
- H. Sui, N. Ma, Y. Wang, H. Li, X. Liu, Y. Su and J. Yang, J. Immunol. Res., 2018, 2018, 6984948 Search PubMed.
- T. Floros and A. A. Tarhini, Semin. Oncol., 2015, 539–548 CrossRef CAS PubMed.
- D. Liu, J. Hematol. Oncol., 2019, 12, 1–5 CrossRef PubMed.
- G. Gunay, M. Sardan Ekiz, X. Ferhati, B. Richichi, C. Nativi, A. B. Tekinay and M. O. Guler, ACS Appl. Mater. Interfaces, 2017, 9, 16035–16042 CrossRef CAS PubMed.
- M. B. Demircan, S. Tohumeken, N. Gunduz, M. A. Khalily, T. Tekinay, M. O. Guler and A. B. Tekinay, Biotechnol. J., 2020, 15, 2000100 CrossRef CAS PubMed.
- R. Mammadov, G. Cinar, N. Gunduz, M. Goktas, H. Kayhan, S. Tohumeken, A. E. Topal, I. Orujalipoor, T. Delibasi and A. Dana, et al. , Sci. Rep., 2015, 5, 1–15 Search PubMed.
- M. O. Dellacherie, B. R. Seo and D. J. Mooney, Nat. Rev. Mater., 2019, 4, 379–397 CrossRef.
- C. R. Zamecnik, E. S. Levy, M. M. Lowe, B. Zirak, M. D. Rosenblum and T. A. Desai, Biomaterials, 2020, 230, 119626 CrossRef CAS PubMed.
- K. C. Maita, F. R. Avila, R. A. Torres-Guzman, J. P. Garcia, A. S. Eldaly, L. Palmieri, O. S. Emam, O. Ho and A. J. Forte, J. Clin. Transl. Res., 2022, 8, 488 CAS.
- K. Adu-Berchie and D. J. Mooney, Acc. Chem. Res., 2020, 53, 1749–1760 CrossRef CAS PubMed.
- S. Riedel, Baylor University Medical Center Proceedings, 2005, pp. 21–25 Search PubMed.
- S. De Donato, D. Granoff, M. Minutello, G. Lecchi, M. Faccini, M. Agnello, F. Senatore, P. Verweij, B. Fritzell and A. Podda, Vaccine, 1999, 17, 3094–3101 CrossRef CAS PubMed.
- B. Pulendran, P. S. Arunachalam and D. T. OHagan, Nat. Rev. Drug Discovery, 2021, 20, 454–475 CrossRef CAS PubMed.
- D. Li and M. Wu, Signal Transduction Targeted Ther., 2021, 6, 291 CrossRef CAS PubMed.
- B.-S. Pan, S. A. Perera, J. A. Piesvaux, J. P. Presland, G. K. Schroeder, J. N. Cumming, B. W. Trotter, M. D. Altman, A. V. Buevich and B. Cash, et al. , Science, 2020, 369, eaba6098 CrossRef CAS PubMed.
- E. C. Cherney, L. Zhang, J. Lo, T. Huynh, D. Wei, V. Ahuja, C. Quesnelle, G. L. Schieven, A. Futran and G. A. Locke, et al. , J. Med. Chem., 2022, 65, 3518–3538 CrossRef CAS.
- X. Cui, Y. Xie, M. Zhang, J. Gao, X. Zhou, J. Ding, S. Cen and J. Zhou, ChemMedChem, 2022, 17, e202100719 CrossRef CAS PubMed.
- H. Hou, R. Yang, X. Liu, X. Wu, S. Zhang, K. Chen and M. Zheng, Bioorg. Chem., 2020, 100, 103958 CrossRef CAS PubMed.
- S. Zhong, W. Li, Y. Bai, B. Wu, X. Wang, S. Jiang, Y. Zhao, J. Ren, H. Li and R. Jin, PLoS One, 2019, 14, e0216678 CrossRef CAS PubMed.
- A. C. Salyer, G. Caruso, K. K. Khetani, L. M. Fox, S. S. Malladi and S. A. David, PLoS One, 2016, 11, e0149848 CrossRef PubMed.
- Y. Guan, K. Omueti-Ayoade, S. K. Mutha, P. J. Hergenrother and R. I. Tapping, J. Biol. Chem., 2010, 285, 23755–23762 CrossRef CAS PubMed.
- A. M. Weiss, S. Hossainy, S. J. Rowan, J. A. Hubbell and A. P. Esser-Kahn, Macromolecules, 2022, 55, 6913–6937 CrossRef CAS PubMed.
- I. A. Clark and B. Vissel, Semin. Immunopathol., 2017, 505–516 CrossRef CAS PubMed.
- Z.-B. Wang and J. Xu, Vaccines, 2020, 8, 128 CrossRef CAS PubMed.
- K. L. Hilligan and F. Ronchese, Cell. Mol. Immunol., 2020, 17, 587–599 CrossRef CAS PubMed.
- J. Liu, X. Zhang, Y. Cheng and X. Cao, Cell. Mol. Immunol., 2021, 18, 2461–2471 CrossRef CAS PubMed.
- C. R. Hole, C. M. L. Wager, N. Castro-Lopez, A. Campuzano, H. Cai, K. L. Wozniak, Y. Wang and F. L. Wormley Jr, Nat. Commun., 2019, 10, 2955 CrossRef PubMed.
- J. J. Donnelly, M. A. Liu and J. B. Ulmer, Am. J. Respir. Crit. Care Med., 2000, 162, S190–S193 CrossRef CAS PubMed.
- C. Qu, N.-S. Brinck-Jensen, M. Zang and K. Chen, Int. J. Infect. Dis., 2014, 19, 1–5 CrossRef CAS PubMed.
- E. P. Bennett, H. Hassan, U. Mandel, E. Mirgorodskaya, P. Roepstorff, J. Burchell, J. Taylor-Papadimitriou, M. A. Hollingsworth, G. Merkx and A. G. Van Kessel, et al. , J. Biol. Chem., 1998, 273, 30472–30481 CrossRef CAS PubMed.
- S. von Mensdorff-Pouilly, E. Petrakou, P. Kenemans, K. van Uffelen, A. A. Verstraeten, F. G. Snijdewint, G. J. van Kamp, D. J. Schol, C. A. Reis and M. R. Price, et al. , Int. J. Cancer, 2000, 86, 702–712 CrossRef CAS PubMed.
- Y. Rong, X. Wang, W. Mao, M. Chen, S. Wang, P. G. Wang, Y. He and Y. Kong, Chem. Commun., 2023, 59, 1797–1800 RSC.
- D. S. Wilson, S. Hirosue, M. M. Raczy, L. Bonilla-Ramirez, L. Jeanbart, R. Wang, M. Kwissa, J.-F. Franetich, M. A. Broggi and G. Diaceri, et al. , Nat. Mater., 2019, 18, 175–185 CrossRef CAS PubMed.
- L. T. Gray, M. M. Raczy, P. S. Briquez, T. M. Marchell, A. T. Alpar, R. P. Wallace, L. R. Volpatti, M. S. Sasso, S. Cao and M. Nguyen, et al. , Biomaterials, 2021, 278, 121159 CrossRef CAS PubMed.
- A. Aung, A. Cui, L. Maiorino, A. P. Amini, J. R. Gregory, M. Bukenya, Y. Zhang, H. Lee, C. A. Cottrell and D. M. Morgan, et al. , Science, 2023, 379, eabn8934 CrossRef CAS PubMed.
- S. Ols, L. Yang, E. A. Thompson, P. Pushparaj, K. Tran, F. Liang, A. Lin, B. Eriksson, G. B. K. Hedestam and R. T. Wyatt, et al. , Cell Rep., 2020, 30, 3964–3971 CrossRef CAS PubMed.
- S. T. Reddy, A. J. Van Der Vlies, E. Simeoni, V. Angeli, G. J. Randolph, C. P. O'Neil, L. K. Lee, M. A. Swartz and J. A. Hubbell, Nat. Biotechnol., 2007, 25, 1159–1164 CrossRef CAS PubMed.
- S. N. Mueller, S. Tian and J. M. DeSimone, Molecular pharmaceutics, 2015, 12, 1356–1365 CrossRef CAS PubMed.
- J. Lee, S. Kang, H. Park, J. G. Sun, E. C. Kim and G. Shim, Pharmaceutics, 2023, 15, 565 CrossRef CAS PubMed.
- J. Chen, Z. Ye, C. Huang, M. Qiu, D. Song, Y. Li and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2207841119 CrossRef CAS PubMed.
- Y. Chen, S. De Koker and B. G. De Geest, Acc. Chem. Res., 2020, 53, 2055–2067 CrossRef CAS PubMed.
- Y. Ding, Z. Li, A. Jaklenec and Q. Hu, Adv. Drug Delivery Rev., 2021, 179, 113914 CrossRef CAS PubMed.
- T. Travieso, J. Li, S. Mahesh, J. D. F. R. E. Mello and M. Blasi, npj Vaccines, 2022, 7, 75 CrossRef CAS PubMed.
- G. A. Rodrigues, E. Shalaev, T. K. Karami, J. Cunningham, N. K. Slater and H. M. Rivers, Pharm. Res., 2019, 36, 1–20 CrossRef CAS PubMed.
- J. Earley, E. Piletska, G. Ronzitti and S. Piletsky, Trends Biotechnol., 2022, 41, 836–845 CrossRef PubMed.
- G. K. Lee, N. Maheshri, B. Kaspar and D. V. Schaffer, Biotechnol. Bioeng., 2005, 92, 24–34 CrossRef CAS PubMed.
- K. Wang, M. Zheng, C. Askew, X. Zhang, C. Li and Z. Han, Adv. Ther., 2022, 5, 2200128 CrossRef CAS.
- Z. Yuan, B. Li, W. Gu, S. Luozhong, R. Li and S. Jiang, J. Am. Chem. Soc., 2022, 144, 20507–20513 CrossRef CAS PubMed.
- T. R. Cooney, K. Farrand, S. L. Draper, R. J. Anderson, P. M. Rendle, I. F. Hermans, B. J. Compton and G. F. Painter, Bioconjugate Chem., 2023, 34, 799–808 CAS.
- A. N. Tsoras, K. M. Wong, A. K. Paravastu and J. A. Champion, Front. Immunol., 2020, 11, 1547 CrossRef CAS PubMed.
- N. J. Shah, A. J. Najibi, T.-Y. Shih, A. S. Mao, A. Sharda, D. T. Scadden and D. J. Mooney, Nat. Biomed. Eng., 2020, 4, 40–51 CrossRef CAS PubMed.
- Z. Cao, L. Ren, L. Niu, R. Zhao, N. Liu, Q. Zhuang, F. Pan, Z. Liu, Y. Cheng and Y. Yang, et al. , Matter, 2023, 6, 3574–3597 CrossRef CAS.
- G. M. Lynn, C. Sedlik, F. Baharom, Y. Zhu, R. A. Ramirez-Valdez, V. L. Coble, K. Tobin, S. R. Nichols, Y. Itzkowitz and N. Zaidi, et al. , Nat. Biotechnol., 2020, 38, 320–332 CrossRef CAS PubMed.
- R. E. Hollingsworth and K. Jansen, npj Vaccines, 2019, 4, 7 CrossRef PubMed.
- M. Saxena, S. H. van der Burg, C. J. Melief and N. Bhardwaj, Nat. Rev. Cancer, 2021, 21, 360–378 CrossRef CAS PubMed.
- J. Jou, K. J. Harrington, M.-B. Zocca, E. Ehrnrooth and E. E. Cohen, Clin. Cancer Res., 2021, 27, 689–703 CrossRef CAS PubMed.
- S. Cuzzubbo, S. Mangsbo, D. Nagarajan, K. Habra, A. G. Pockley and S. E. McArdle, Front. Immunol., 2021, 11, 615240 CrossRef PubMed.
- K. Vermaelen, Front. Immunol., 2019, 10, 8 CrossRef CAS PubMed.
- A. Kaur, J. Baldwin, D. Brar, D. B. Salunke and N. Petrovsky, Curr. Opin. Chem. Biol., 2022, 70, 102172 CrossRef CAS PubMed.
- D. N. Sauder, J. Am. Acad. Dermatol., 2000, 43, S6–S11 CrossRef CAS PubMed.
- F. Scarf, A. Patrizi, G. Veronesi, M. Lambertini, F. Tartari, M. Mussi, B. Melotti and E. Dika, Dermatol. Ther., 2020, 33, e14165 Search PubMed.
- Y. Shirota, H. Shirota and D. M. Klinman, J. Immunol., 2012, 188, 1592–1599 CrossRef CAS PubMed.
- S. Wang, J. Campos, M. Gallotta, M. Gong, C. Crain, E. Naik, R. L. Coffman and C. Guiducci, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7240–E7249 CAS.
- M. A. Aznar, L. Planelles, M. Perez-Olivares, C. Molina, S. Garasa, I. Etxeberra, G. Perez, I. Rodriguez, E. Bolaños and P. Lopez-Casas, et al. , J. ImmunoTher. Cancer, 2019, 7, 1–16 CrossRef PubMed.
- S. Di, M. Zhou, Z. Pan, R. Sun, M. Chen, H. Jiang, B. Shi, H. Luo and Z. Li, Front. Oncol., 2019, 9, 241 CrossRef PubMed.
- J. Chen, M. Qiu, Z. Ye, T. Nyalile, Y. Li, Z. Glass, X. Zhao, L. Yang, J. Chen and Q. Xu, Sci. Adv., 2021, 7, eabf1244 CrossRef CAS PubMed.
- G. Kelly, J. J. Milligan, E. M. Mastria, S. Kim, S. R. Zelenetz, J. Dobbins, L. Y. Cai, X. Li, S. K. Nair and A. Chilkoti, J. Controlled Release, 2022, 343, 267–276 CrossRef CAS PubMed.
- A. J. Slezak, A. Mansurov, M. M. Raczy, K. Chang, A. T. Alpar, A. L. Lauterbach, R. P. Wallace, R. K. Weathered, J. E. Medellin and C. Battistella, et al. , ACS Cent. Sci., 2022, 8, 1435–1446 CrossRef CAS PubMed.
- R. Kuai, X. Sun, W. Yuan, L. J. Ochyl, Y. Xu, A. H. Najafabadi, L. Scheetz, M.-Z. Yu, I. Balwani and A. Schwendeman, et al. , J. Controlled Release, 2018, 282, 131–139 CrossRef CAS PubMed.
- S. Lim, J. Park, M. K. Shim, W. Um, H. Y. Yoon, J. H. Ryu, D.-K. Lim and K. Kim, Theranostics, 2019, 9, 7906 CrossRef CAS PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 175–196 CrossRef CAS PubMed.
- G. Zhu, F. Zhang, Q. Ni, G. Niu and X. Chen, ACS Nano, 2017, 11, 2387–2392 CrossRef CAS PubMed.
- P. Abdou, Z. Wang, Q. Chen, A. Chan, D. R. Zhou, V. Gunadhi and Z. Gu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1632 Search PubMed.
- E. Dolgin, Nat. Med., 2010, 16, 740–744 CrossRef CAS PubMed.
- L. Steinman, J. Intern. Med., 2010, 267, 441–451 CrossRef CAS PubMed.
- S. Chandrashekara, Indian J. Pharmacol., 2012, 44, 665 CrossRef CAS PubMed.
- D. R. Getts, A. J. Martin, D. P. McCarthy, R. L. Terry, Z. N. Hunter, W. T. Yap, M. T. Getts, M. Pleiss, X. Luo and N. J. King, et al. , Nat. Biotechnol., 2012, 30, 1217–1224 CrossRef CAS PubMed.
- T. L. Freitag, J. R. Podojil, R. M. Pearson, F. J. Fokta, C. Sahl, M. Messing, L. C. Andersson, K. Leskinen, P. Saavalainen and L. I. Hoover, et al. , Gastroenterology, 2020, 158, 1667–1681 CrossRef CAS PubMed.
- J. R. Podojil, S. Genardi, M.-Y. Chiang, S. Kakade, T. Neef, T. Murthy, M. T. Boyne, A. Elhofy and S. D. Miller, J. Immunol., 2022, 209, 465–475 CrossRef CAS PubMed.
- K. R. Hughes, M. N. Saunders, J. J. Landers, K. W. Janczak, H. Turkistani, L. M. Rad, S. D. Miller, J. R. Podojil, L. D. Shea and J. J. O'Konek, Front. Allergy, 2022, 3, 829605 CrossRef PubMed.
- C. P. Kelly, J. A. Murray, D. A. Leffler, D. R. Getts, A. C. Bledsoe, G. Smithson, M. R. First, A. Morris, M. Boyne and A. Elhofy, et al. , Gastroenterology, 2021, 161, 66–80 CrossRef CAS PubMed.
- G. Cappellano, C. Comi, A. Chiocchetti and U. Dianzani, Int. J. Mol. Sci., 2019, 20, 204 CrossRef PubMed.
- T. L. Nguyen, Y. Choi, J. Im, H. Shin, N. M. Phan, M. K. Kim, S. W. Choi and J. Kim, Nat. Commun., 2022, 13, 7449 CrossRef CAS PubMed.
- M. Damo, D. S. Wilson, E. A. Watkins and J. A. Hubbell, Front. Immunol., 2021, 12, 555095 CrossRef CAS PubMed.
- C. D. Maulloo, S. Cao, E. A. Watkins, M. M. Raczy, A. S. Solanki, M. Nguyen, J. W. Reda, H.-N. Shim, D. S. Wilson and M. A. Swartz, et al. , Front. Immunol., 2021, 12, 714842 CrossRef CAS PubMed.
- A. C. Tremain, R. P. Wallace, K. M. Lorentz, T. B. Thornley, J. T. Antane, M. R. Raczy, J. W. Reda, A. T. Alpar, A. J. Slezak and E. A. Watkins, et al. , Nat. Biomed. Eng., 2023, 1–14 Search PubMed.
- R. P. Wallace, K. C. Refvik, J. T. Antane, K. Brünggel, A. C. Tremain, M. R. Raczy, A. T. Alpar, M. Nguyen, A. Solanki and A. J. Slezak, et al. , Cell Rep. Med., 2023, 101345 CrossRef PubMed.
- S. Cao, C. D. Maulloo, M. M. Raczy, M. Sabados, A. J. Slezak, M. Nguyen, A. Solanki, R. P. Wallace, H.-N. Shim and D. S. Wilson, et al. , Cell Rep. Med., 2023, 101346 CrossRef PubMed.
- G. Pasut and F. M. Veronese, J. Controlled Release, 2012, 161, 461–472 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- V. Gupta, S. Bhavanasi, M. Quadir, K. Singh, G. Ghosh, K. Vasamreddy, A. Ghosh, T. J. Siahaan, S. Banerjee and S. K. Banerjee, J. Cell Commun. Signaling, 2019, 13, 319–330 CrossRef CAS PubMed.
- M. Ibrahim, E. Ramadan, N. E. Elsadek, S. E. Emam, T. Shimizu, H. Ando, Y. Ishima, O. H. Elgarhy, H. A. Sarhan and A. K. Hussein, et al. , J. Controlled Release, 2022, 351, 215–230 CrossRef CAS PubMed.
- R. P. Garay, R. El-Gewely, J. K. Armstrong, G. Garratty and P. Richette, Expert Opin. Drug Delivery, 2012, 9, 1319–1323 CrossRef CAS PubMed.
- D. Shi, D. Beasock, A. Fessler, J. Szebeni, J. Y. Ljubimova, K. A. Afonin and M. A. Dobrovolskaia, Adv. Drug Delivery Rev., 2022, 180, 114079 CrossRef CAS PubMed.
- Y. Ju, J. M. Carreño, V. Simon, K. Dawson, F. Krammer and S. J. Kent, Nat. Rev. Immunol., 2023, 23, 135–136 CrossRef CAS PubMed.
- Z.-H. Zhou, C. A. Stone, B. Jakubovic, E. J. Phillips, G. Sussman, J. Park, U. Hoang, S. L. Kirshner, R. Levin and S. Kozlowski, J. Allergy Clin. Immunol.: Pract., 2021, 9, 1731–1733 CAS.
- N. E. Powers, B. Swartzwelter, C. Marchetti, D. M. de Graaf, A. Lerchner, M. Schlapschy, R. Datar, U. Binder, C. K. Edwards and A. Skerra, et al. , J. Biol. Chem., 2020, 295, 868–882 CrossRef PubMed.
- M. Schlapschy, U. Binder, C. Börger, I. Theobald, K. Wachinger, S. Kisling, D. Haller and A. Skerra, Protein Eng., Des. Sel., 2013, 26, 489–501 CrossRef CAS PubMed.
- Q. Zhang, S. Li, W. Wu, X. Xia and J. Zhang, Nanomedicine, 2023, 47, 102622 CrossRef CAS PubMed.
- I. Ozer, A. Slezak, P. Sirohi, X. Li, N. Zakharov, Y. Yao, J. I. Everitt, I. Spasojevic, S. L. Craig and J. H. Collier, et al. , Biomaterials, 2023, 294, 121985 CrossRef CAS PubMed.
- L. H. Belén, C. D. O. Rangel-Yagui, J. F. Beltrán Lissabet, B. Effer, M. Lee-Estevez, A. Pessoa, R. L. Castillo and J. G. Faras, Front. Pharm., 2019, 10, 1450 CrossRef PubMed.
- C. J. Fee and J. M. Van Alstine, Chem. Eng. Sci., 2006, 61, 924–939 CrossRef CAS.
- S. B. Ebrahimi and D. Samanta, Nat. Commun., 2023, 14, 2411 CrossRef CAS PubMed.
- M. W. Popp, S. K. Dougan, T.-Y. Chuang, E. Spooner and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3169–3174 CrossRef CAS PubMed.
- D. Haas, N. Hauptstein, M. Dirauf, M. D. Driessen, M. Ruopp, U. S. Schubert, T. Luhmann and L. Meinel, Bioconjugate Chem., 2021, 33, 97–104 CrossRef PubMed.
- M. E. Raeber, D. Sahin, U. Karakus and O. Boyman, EBioMedicine, 2023, 90, 104539 CrossRef CAS PubMed.
- J. Damoiseaux, Clin. Immunol., 2020, 218, 108515 CrossRef CAS PubMed.
- L. Xu, X. Song, L. Su, Y. Zheng, R. Li and J. Sun, Int. Immunopharmacol., 2019, 72, 322–329 CrossRef CAS PubMed.
- B. Zhang, J. Sun, Y. Wang, D. Ji, Y. Yuan, S. Li, Y. Sun, Y. Hou, P. Li and L. Zhao, et al. , Nat. Biomed. Eng., 2021, 5, 1288–1305 CrossRef CAS PubMed.
- J. L. Ptacin, C. E. Caffaro, L. Ma, K. M. San Jose Gall, H. R. Aerni, N. V. Acuff, R. W. Herman, Y. Pavlova, M. J. Pena and D. B. Chen, et al. , Nat. Commun., 2021, 12, 4785 CrossRef CAS PubMed.
- T. Ghose, M. Path and S. Nigam, Cancer, 1972, 29, 1398–1400 CrossRef CAS PubMed.
- A. M. Wu and P. D. Senter, Nat. Biotechnol., 2005, 23, 1137–1146 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- J. Yu, Y. Song and W. Tian, J. Hematol. Oncol., 2020, 13, 1–10 CrossRef PubMed.
- S. Yaghoubi, M. H. Karimi, M. Lotfinia, T. Gharibi, M. Mahi-Birjand, E. Kavi, F. Hosseini, K. Sineh Sepehr, M. Khatami and N. Bagheri, et al. , J. Cell. Physiol., 2020, 235, 31–64 CrossRef CAS PubMed.
- J. Z. Drago, S. Modi and S. Chandarlapaty, Nat. Rev. Clin. Oncol., 2021, 18, 327–344 CrossRef PubMed.
- A. Beck, L. Goetsch, C. Dumontet and N. Corvaa, Nat. Rev. Drug Discovery, 2017, 16, 315–337 CrossRef CAS PubMed.
- L. Conilh, L. Sadilkova, W. Viricel and C. Dumontet, J. Hematol. Oncol., 2023, 16, 3 CrossRef CAS PubMed.
- S. Loibl and L. Gianni, The Lancet, 2017, 389, 2415–2429 CrossRef CAS PubMed.
- C. M. Yamazaki, A. Yamaguchi, Y. Anami, W. Xiong, Y. Otani, J. Lee, N. T. Ueno, N. Zhang, Z. An and K. Tsuchikama, Nat. Commun., 2021, 12, 3528 CrossRef CAS PubMed.
- J. B. Hines, A. J. Kacew and R. F. Sweis, Curr. Oncol. Rep., 2023, 25, 189–199 CrossRef PubMed.
- Y.-T. Wu, Y. Fang, Q. Wei, H. Shi, H. Tan, Y. Deng, Z. Zeng, J. Qiu, C. Chen and L. Sun, et al. , Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2214278119 CrossRef CAS PubMed.
- Z. Najminejad, F. Dehghani, Y. Mirzaei, A. H. Mer, S. A. Saghi, M. H. Abdolvahab, N. Bagheri, A. Meyfour, A. Jafari and S. Jahandideh, et al. , Mol. Ther., 2023, 31, 1874–1903 CrossRef CAS PubMed.
- Z. Su, D. Xiao, F. Xie, L. Liu, Y. Wang, S. Fan, X. Zhou and S. Li, Acta Pharm. Sin. B, 2021, 11, 3889–3907 CrossRef CAS PubMed.
- K. Tsuchikama and Z. An, Protein Cell, 2018, 9, 33–46 CrossRef CAS PubMed.
- D. Su and D. Zhang, Front. Pharm., 2021, 12, 687926 CrossRef CAS PubMed.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. Galindo Casas, K. Delaria, V. Lui, K. Poulsen and C. Loo, et al. , Bioconjugate Chem., 2015, 26, 650–659 CrossRef CAS PubMed.
- S. Chuprakov, A. O. Ogunkoya, R. M. Barfield, M. Bauzon, C. Hickle, Y. C. Kim, D. Yeo, F. Zhang, D. Rabuka and P. M. Drake, Bioconjugate Chem., 2021, 32, 746–754 CrossRef CAS PubMed.
- J. Fuentes-Antrás, S. Genta, A. Vijenthira and L. L. Siu, Trends Cancer, 2023, 9, 339–354 CrossRef PubMed.
- E. Nicolò, F. Giugliano, L. Ascione, P. Tarantino, C. Corti, S. M. Tolaney, M. Cristofanilli and G. Curigliano, Cancer Treat. Rev., 2022, 106, 102395 CrossRef PubMed.
- M. Roas, B. Vick, M.-A. Kasper, M. Able, H. Polzer, M. Gerlach, E. Kremmer, J. S. Hecker, S. Schmitt and A. Stengl, et al. , Blood, J. Am. Soc. Hematol., 2023, 141, 1023–1035 CAS.
- M. Conroy and J. Naidoo, Nat. Commun., 2022, 13, 392 CrossRef CAS PubMed.
- J. Ishihara, K. Fukunaga, A. Ishihara, H. M. Larsson, L. Potin, P. Hosseinchi, G. Galliverti, M. A. Swartz and J. A. Hubbell, Sci. Transl. Med., 2017, 9, eaan0401 CrossRef PubMed.
- K. Katsumata, J. Ishihara, A. Mansurov, A. Ishihara, M. M. Raczy, E. Yuba and J. A. Hubbell, Sci. Adv., 2019, 5, eaay1971 CrossRef CAS PubMed.
- X. Ye, R. Holland, M. Wood, C. Pasetka, L. Palmer, E. Samaridou, K. McClintock, V. Borisevich, T. W. Geisbert and R. W. Cross, et al. , Mol. Ther., 2023, 31, 269–281 CrossRef CAS PubMed.
- K. A. Ross, T. M. Brenza, A. M. Binnebose, Y. Phanse, A. G. Kanthasamy, H. E. Gendelman, A. K. Salem, L. C. Bartholomay, B. H. Bellaire and B. Narasimhan, J. Controlled Release, 2015, 219, 548–559 CrossRef CAS PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS PubMed.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- S. Barua and S. Mitragotri, Nano Today, 2014, 9, 223–243 CrossRef CAS PubMed.
- G. Lin, J. Wang, Y.-G. Yang, Y. Zhang and T. Sun, Front. Bioeng. Biotechnol., 2023, 11, 1242126 CrossRef PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- A. Martin, P. Lalanne, A. Weber-Vax, A. Mutschler and S. Lecommandoux, Int. J. Pharm., 2023, 642, 123157 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 5577–5591 CrossRef PubMed.
- M. Aramesh, O. Shimoni, K. Ostrikov, S. Prawer and J. Cervenka, Nanoscale, 2015, 7, 5726–5736 RSC.
- M. P. Calatayud, B. Sanz, V. Raffa, C. Riggio, M. R. Ibarra and G. F. Goya, Biomaterials, 2014, 35, 6389–6399 CrossRef CAS PubMed.
- R. Wang, S. Cao, M. E. H. Bashir, L. A. Hesser, Y. Su, S. M. C. Hong, A. Thompson, E. Culleen, M. Sabados and N. P. Dylla, et al. , Nat. Biomed. Eng., 2023, 7, 38–55 CrossRef CAS PubMed.
- X. Jiang, B. Ma, M. Sun, C. Guo, Z. Liu, Y. Du, B. Wang, N. Li, M. Chen and Y. Zhang, et al. , Anal. Chem., 2023, 95(32), 11885–11891 CrossRef CAS PubMed.
- R. Rampado, S. Crotti, P. Caliceti, S. Pucciarelli and M. Agostini, Front. Bioeng. Biotechnol., 2020, 8, 166 CrossRef PubMed.
- Y. Tao, V. Jakobsson, X. Chen and J. Zhang, Acc. Chem. Res., 2023, 3213–3221 Search PubMed.
- J. Mariam, S. Sivakami and P. M. Dongre, Drug Delivery, 2016, 23, 2668–2676 CrossRef CAS PubMed.
- S. Goel, D. G. Duda, L. Xu, L. L. Munn, Y. Boucher, D. Fukumura and R. K. Jain, Physiol. Rev., 2011, 91, 1071–1121 CrossRef CAS PubMed.
- B. Lahooti, R. G. Akwii, F. T. Zahra, M. S. Sajib, M. Lamprou, A. Alobaida, M. S. Lionakis, G. Mattheolabakis and C. M. Mikelis, J. Controlled Release, 2023, 361, 212–235 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Nat. Nanotechnol., 2020, 15, 313–320 CrossRef CAS PubMed.
- X. Wang, S. Liu, Y. Sun, X. Yu, S. M. Lee, Q. Cheng, T. Wei, J. Gong, J. Robinson, D. Zhang, X. Lian, P. Basak and D. J. Siegwart, Nat. Protoc., 2023, 18, 265–291 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northrop, G. M. Ringold and M. Danielsen, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7413–7417 CrossRef CAS PubMed.
- R. W. Malone, P. L. Felgner and I. M. Verma, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6077–6081 CrossRef CAS PubMed.
- T. Ren, Y. K. Song, G. Zhang and D. Liu, Gene Ther., 2000, 7, 764–768 CrossRef CAS PubMed.
- Z. Du, M. M. Munye, A. D. Tagalakis, M. D. I. Manunta and S. L. Hart, Sci. Rep., 2014, 4, 7107 CrossRef PubMed.
- B. Dalby, S. Cates, A. Harris, E. C. Ohki, M. L. Tilkins, P. J. Price and V. C. Ciccarone, Methods, 2004, 33, 95–103 CrossRef CAS PubMed.
- F. Cardarelli, L. Digiacomo, C. Marchini, A. Amici, F. Salomone, G. Fiume, A. Rossetta, E. Gratton, D. Pozzi and G. Caracciolo, Sci. Rep., 2016, 6, 25879 CrossRef CAS PubMed.
- H. Lv, S. Zhang, B. Wang, S. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- S. Cui, Y. Wang, Y. Gong, X. Lin, Y. Zhao, D. Zhi, Q. Zhou and S. Zhang, Toxicol. Res., 2018, 7, 473–479 CrossRef CAS PubMed.
- K. A. Hajj and K. A. Whitehead, Nat. Rev. Mater., 2017, 2, 17056 CrossRef CAS.
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Mol. Ther., 2019, 27, 710–728 CrossRef CAS PubMed.
- J. Huotari and A. Helenius, EMBO J., 2011, 30, 3481–3500 CrossRef CAS PubMed.
- P. R. Cullis and M. J. Hope, Mol. Ther., 2017, 25, 1467–1475 CrossRef CAS PubMed.
- J. Heyes, L. Palmer, K. Bremner and I. MacLachlan, J. Controlled Release, 2005, 107, 276–287 CrossRef CAS PubMed.
- T. S. Zimmermann, A. C. H. Lee, A. Akinc, B. Bramlage, D. Bumcrot, M. N. Fedoruk, J. Harborth, J. A. Heyes, L. B. Jeffs, M. John, A. D. Judge, K. Lam, K. McClintock, L. V. Nechev, L. R. Palmer, T. Racie, I. Röhl, S. Seiffert, S. Shanmugam, V. Sood, J. Soutschek, I. Toudjarska, A. J. Wheat, E. Yaworski, W. Zedalis, V. Koteliansky, M. Manoharan, H.-P. Vornlocher and I. MacLachlan, Nature, 2006, 441, 111–114 CrossRef CAS PubMed.
- S. C. Semple, A. Akinc, J. Chen, A. P. Sandhu, B. L. Mui, C. K. Cho, D. W. Y. Sah, D. Stebbing, E. J. Crosley, E. Yaworski, I. M. Hafez, J. R. Dorkin, J. Qin, K. Lam, K. G. Rajeev, K. F. Wong, L. B. Jeffs, L. Nechev, M. L. Eisenhardt, M. Jayaraman, M. Kazem, M. A. Maier, M. Srinivasulu, M. J. Weinstein, Q. Chen, R. Alvarez, S. A. Barros, S. De, S. K. Klimuk, T. Borland, V. Kosovrasti, W. L. Cantley, Y. K. Tam, M. Manoharan, M. A. Ciufolini, M. A. Tracy, A. de Fougerolles, I. MacLachlan, P. R. Cullis, T. D. Madden and M. J. Hope, Nat. Biotechnol., 2010, 28, 172–176 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- T. Coelho, D. Adams, A. Silva, P. Lozeron, P. N. Hawkins, T. Mant, J. Perez, J. Chiesa, S. Warrington, E. Tranter, M. Munisamy, R. Falzone, J. Harrop, J. Cehelsky, B. R. Bettencourt, M. Geissler, J. S. Butler, A. Sehgal, R. E. Meyers, Q. Chen, T. Borland, R. M. Hutabarat, V. A. Clausen, R. Alvarez, K. Fitzgerald, C. Gamba-Vitalo, S. V. Nochur, A. K. Vaishnaw, D. W. Y. Sah, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2013, 369, 819–829 CrossRef CAS PubMed.
- O. B. Suhr, T. Coelho, J. Buades, J. Pouget, I. Conceicao, J. Berk, H. Schmidt, M. Waddington-Cruz, J. M. Campistol, B. R. Bettencourt, A. Vaishnaw, J. Gollob and D. Adams, Orphanet J. Rare Diseases, 2015, 10, 109 CrossRef PubMed.
- D. Adams, A. Gonzalez-Duarte, W. D. O'Riordan, C.-C. Yang, M. Ueda, A. V. Kristen, I. Tournev, H. H. Schmidt, T. Coelho, J. L. Berk, K.-P. Lin, G. Vita, S. Attarian, V. Planté-Bordeneuve, M. M. Mezei, J. M. Campistol, J. Buades, T. H. Brannagan, B. J. Kim, J. Oh, Y. Parman, Y. Sekijima, P. N. Hawkins, S. D. Solomon, M. Polydefkis, P. J. Dyck, P. J. Gandhi, S. Goyal, J. Chen, A. L. Strahs, S. V. Nochur, M. T. Sweetser, P. P. Garg, A. K. Vaishnaw, J. A. Gollob and O. B. Suhr, N. Engl. J. Med., 2018, 379, 11–21 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros, W. Querbes, Y. K. Tam, S. M. Ansell, V. Kumar, J. Qin, X. Zhang, Q. Wang, S. Panesar, R. Hutabarat, M. Carioto, J. Hettinger, P. Kandasamy, D. Butler, K. G. Rajeev, B. Pang, K. Charisse, K. Fitzgerald, B. L. Mui, X. Du, P. Cullis, T. D. Madden, M. J. Hope, M. Manoharan and A. Akinc, Mol. Ther., 2013, 21, 1570–1578 CrossRef CAS PubMed.
- S. Sabnis, E. S. Kumarasinghe, T. Salerno, C. Mihai, T. Ketova, J. J. Senn, A. Lynn, A. Bulychev, I. McFadyen, J. Chan, O. Almarsson, M. G. Stanton and K. E. Benenato, Mol. Ther., 2018, 26, 1509–1519 CrossRef CAS PubMed.
- K. J. Hassett, K. E. Benenato, E. Jacquinet, A. Lee, A. Woods, O. Yuzhakov, S. Himansu, J. Deterling, B. M. Geilich, T. Ketova, C. Mihai, A. Lynn, I. McFadyen, M. J. Moore, J. J. Senn, M. G. Stanton, O. Almarsson, G. Ciaramella and L. A. Brito, Mol. Ther.--Nucleic Acids, 2019, 15, 1–11 CrossRef CAS PubMed.
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey, R. Novak, D. Diemert, S. A. Spector, N. Rouphael, C. B. Creech, J. McGettigan, S. Khetan, N. Segall, J. Solis, A. Brosz, C. Fierro, H. Schwartz, K. Neuzil, L. Corey, P. Gilbert, H. Janes, D. Follmann, M. Marovich, J. Mascola, L. Polakowski, J. Ledgerwood, B. S. Graham, H. Bennett, R. Pajon, C. Knightly, B. Leav, W. Deng, H. Zhou, S. Han, M. Ivarsson, J. Miller, T. Zaks and COVE Study Group, N. Engl. J. Med., 2021, 384, 403–416 CrossRef CAS PubMed.
- S. M. Ansell and X. Du, 2019.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, L. L. Hammitt, O. Tureci, H. Nell, A. Schaefer, S. Unal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Åžahin, K. U. Jansen, W. C. Gruber and C4591001 Clinical Trial Group, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef CAS PubMed.
- A. B. Vogel, I. Kanevsky, Y. Che, K. A. Swanson, A. Muik, M. Vormehr, L. M. Kranz, K. C. Walzer, S. Hein, A. Güler, J. Loschko, M. S. Maddur, A. Ota-Setlik, K. Tompkins, J. Cole, B. G. Lui, T. Ziegenhals, A. Plaschke, D. Eisel, S. C. Dany, S. Fesser, S. Erbar, F. Bates, D. Schneider, B. Jesionek, B. Sänger, A.-K. Wallisch, Y. Feuchter, H. Junginger, S. A. Krumm, A. P. Heinen, P. Adams-Quack, J. Schlereth, S. Schille, C. Kröner, R. de la Caridad Güimil Garcia, T. Hiller, L. Fischer, R. S. Sellers, S. Choudhary, O. Gonzalez, F. Vascotto, M. R. Gutman, J. A. Fontenot, S. Hall-Ursone, K. Brasky, M. C. Griffor, S. Han, A. A. H. Su, J. A. Lees, N. L. Nedoma, E. H. Mashalidis, P. V. Sahasrabudhe, C. Y. Tan, D. Pavliakova, G. Singh, C. Fontes-Garfias, M. Pride, I. L. Scully, T. Ciolino, J. Obregon, M. Gazi, R. Carrion, K. J. Alfson, W. V. Kalina, D. Kaushal, P.-Y. Shi, T. Klamp, C. Rosenbaum, A. N. Kuhn, O. Türeci, P. R. Dormitzer, K. U. Jansen and U. Sahin, Nature, 2021, 592, 283–289 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- B. G. Tenchov, R. C. MacDonald and D. P. Siegel, Biophys. J., 2006, 91, 2508–2516 CrossRef CAS PubMed.
- D. Pozzi, C. Marchini, F. Cardarelli, H. Amenitsch, C. Garulli, A. Bifone and G. Caracciolo, Biochim. Biophys. Acta, 2012, 1818, 2335–2343 CrossRef CAS PubMed.
- K. Paunovska, C. J. Gil, M. P. Lokugamage, C. D. Sago, M. Sato, G. N. Lando, M. Gamboa Castro, A. V. Bryksin and J. E. Dahlman, ACS Nano, 2018, 12, 8341–8349 CrossRef CAS PubMed.
- Y. Eygeris, S. Patel, A. Jozic and G. Sahay, Nano Lett., 2020, 20, 4543–4549 CrossRef CAS PubMed.
- S. Patel, N. Ashwanikumar, E. Robinson, Y. Xia, C. Mihai, J. P. Griffith, S. Hou, A. A. Esposito, T. Ketova, K. Welsher, J. L. Joyal, O. Almarsson and G. Sahay, Nat. Commun., 2020, 11, 983 CrossRef CAS PubMed.
- K. J. Kauffman, J. R. Dorkin, J. H. Yang, M. W. Heartlein, F. DeRosa, F. F. Mir, O. S. Fenton and D. G. Anderson, Nano Lett., 2015, 15, 7300–7306 CrossRef CAS PubMed.
- I. Koltover, T. Salditt, J. O. Rädler and C. R. Safinya, Science, 1998, 281, 78–81 CrossRef CAS PubMed.
- N. M. Belliveau, J. Huft, P. J. Lin, S. Chen, A. K. Leung, T. J. Leaver, A. W. Wild, J. B. Lee, R. J. Taylor, Y. K. Tam, C. L. Hansen and P. R. Cullis, Mol. Ther.--Nucleic Acids, 2012, 1, e37 CrossRef PubMed.
- A. Akinc, M. Goldberg, J. Qin, J. R. Dorkin, C. Gamba-Vitalo, M. Maier, K. N. Jayaprakash, M. Jayaraman, K. G. Rajeev, M. Manoharan, V. Koteliansky, I. Röhl, E. S. Leshchiner, R. Langer and D. G. Anderson, Mol. Ther., 2009, 17, 872–879 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- R. C. Ryals, S. Patel, C. Acosta, M. McKinney, M. E. Pennesi and G. Sahay, PLoS One, 2020, 15, e0241006 CrossRef CAS PubMed.
- R. Tenchov, J. M. Sasso and Q. A. Zhou, Bioconjugate Chem., 2023, 34, 941–960 CrossRef CAS PubMed.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS PubMed.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS PubMed.
- G. J. Hannon, Nature, 2002, 418, 244–251 CrossRef CAS PubMed.
- R. F. Ketting, S. E. Fischer, E. Bernstein, T. Sijen, G. J. Hannon and R. H. Plasterk, Genes Dev., 2001, 15, 2654–2659 CrossRef CAS PubMed.
- P. D. Zamore, T. Tuschl, P. A. Sharp and D. P. Bartel, Cell, 2000, 101, 25–33 CrossRef CAS PubMed.
- M. M. Zhang, R. Bahal, T. P. Rasmussen, J. E. Manautou and X.-B. Zhong, Biochem. Pharmacol., 2021, 189, 114432 CrossRef CAS PubMed.
- C. Matranga, Y. Tomari, C. Shin, D. P. Bartel and P. D. Zamore, Cell, 2005, 123, 607–620 CrossRef CAS PubMed.
- S. L. Ameres, J. Martinez and R. Schroeder, Cell, 2007, 130, 101–112 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- B. Hu, L. Zhong, Y. Weng, L. Peng, Y. Huang, Y. Zhao and X.-J. Liang, Signal Transduction Targeted Ther., 2020, 5, 101 CrossRef CAS.
- J. J. Rossi and D. J. Rossi, Mol. Ther., 2021, 29, 431–432 CrossRef CAS PubMed.
- E. A. Narasipura, R. VanKeulen-Miller, Y. Ma and O. S. Fenton, Bioconjugate Chem., 2023, 34, 1177–1197 CrossRef CAS PubMed.
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CrossRef CAS PubMed.
- D. D. Licatalosi and R. B. Darnell, Nat. Rev. Genet., 2010, 11, 75–87 CrossRef CAS PubMed.
- N. Pardi, M. J. Hogan, F. W. Porter and D. Weissman, Nat. Rev. Drug Discovery, 2018, 17, 261–279 CrossRef CAS PubMed.
- M. Bhat, N. Robichaud, L. Hulea, N. Sonenberg, J. Pelletier and I. Topisirovic, Nat. Rev. Drug Discovery, 2015, 14, 261–278 CrossRef CAS PubMed.
- R. J. Jackson, C. U. T. Hellen and T. V. Pestova, Nat. Rev. Mol. Cell Biol., 2010, 11, 113–127 CrossRef CAS PubMed.
- A. G. Hinnebusch, Trends Biochem. Sci., 2017, 42, 589–611 CrossRef CAS PubMed.
- A. G. Hinnebusch, Ann. Rev. Biochem., 2014, 83, 779–812 CrossRef CAS PubMed.
- U. Sahin, K. Karikó and O. Tureci, Nat. Rev. Drug Discovery, 2014, 13, 759–780 CrossRef CAS PubMed.
- N. L. Garneau, J. Wilusz and C. J. Wilusz, Nat. Rev. Mol. Cell Biol., 2007, 8, 113–126 CrossRef CAS PubMed.
- K. Leppek, R. Das and M. Barna, Nat. Rev. Mol. Cell Biol., 2018, 19, 158–174 CrossRef CAS PubMed.
- T. Loan Young, K. Chang Wang, A. James Varley and B. Li, Adv. Drug Delivery Rev., 2023, 197, 114826 CrossRef CAS PubMed.
- D. H. Fuller and P. Berglund, N. Engl. J. Med., 2020, 382, 2469–2471 CrossRef PubMed.
- A. K. Blakney, S. Ip and A. J. Geall, Vaccines, 2021, 9, 97 CrossRef CAS PubMed.
- G. Maruggi, J. B. Ulmer, R. Rappuoli and D. Yu, Curr. Top. Microbiol. Immunol., 2022, 440, 31–70 Search PubMed.
- K. Bloom, F. van den Berg and P. Arbuthnot, Gene Ther., 2021, 28, 117–129 CrossRef CAS PubMed.
- A. K. Blakney, P. F. McKay and R. J. Shattock, Front. Mol. Biosci., 2018, 5, 71 CrossRef PubMed.
- T. Beissert, M. Perkovic, A. Vogel, S. Erbar, K. C. Walzer, T. Hempel, S. Brill, E. Haefner, R. Becker, O. Tureci and U. Sahin, Mol. Ther., 2020, 28, 119–128 CrossRef CAS PubMed.
- P. A. Krieg and D. A. Melton, Nucleic Acids Res., 1984, 12, 7057–7070 CrossRef CAS PubMed.
- A. Ramanathan, G. B. Robb and S.-H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 CrossRef PubMed.
- M. J. Ensinger, S. A. Martin, E. Paoletti and B. Moss, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 2525–2529 CrossRef CAS PubMed.
- S. A. Martin, E. Paoletti and B. Moss, J. Biol. Chem., 1975, 250, 9322–9329 CrossRef CAS PubMed.
- B. Moss, A. Gershowitz, C. M. Wei and R. Boone, Virology, 1976, 72, 341–351 CrossRef CAS PubMed.
- S. H. Chan, C. N. Molé, D. Nye, L. Mitchell, N. Dai, J. Buss, D. W. Kneller, J. M. Whipple and G. B. Robb, RNA, 2023, 29, 1803–1817 CrossRef CAS PubMed.
- J. Stepinski, C. Waddell, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2001, 7, 1486–1495 CAS.
- J. Jemielity, T. Fowler, J. Zuberek, J. Stepinski, M. Lewdorowicz, A. Niedzwiecka, R. Stolarski, E. Darzynkiewicz and R. E. Rhoads, RNA, 2003, 9, 1108–1122 CrossRef CAS PubMed.
- M. Mockey, C. Gonçalves, F. P. Dupuy, F. M. Lemoine, C. Pichon and P. Midoux, Biochem. Biophys. Res. Commun., 2006, 340, 1062–1068 CrossRef CAS PubMed.
- J. M. Henderson, A. Ujita, E. Hill, S. Yousif-Rosales, C. Smith, N. Ko, T. McReynolds, C. R. Cabral, J. R. Escamilla-Powers and M. E. Houston, Curr. Protoc., 2021, 1, e39 CrossRef CAS PubMed.
- U. Sahin, A. Muik, E. Derhovanessian, I. Vogler, L. M. Kranz, M. Vormehr, A. Baum, K. Pascal, J. Quandt, D. Maurus, S. Brachtendorf, V. Lörks, J. Sikorski, R. Hilker, D. Becker, A.-K. Eller, J. Grützner, C. Boesler, C. Rosenbaum, M.-C. Kühnle, U. Luxemburger, A. Kemmer-Brück, D. Langer, M. Bexon, S. Bolte, K. Karikó, T. Palanche, B. Fischer, A. Schultz, P.-Y. Shi, C. Fontes-Garfias, J. L. Perez, K. A. Swanson, J. Loschko, I. L. Scully, M. Cutler, W. Kalina, C. A. Kyratsous, D. Cooper, P. R. Dormitzer, K. U. Jansen and O. Tureci, Nature, 2020, 586, 594–599 CrossRef CAS PubMed.
- E. Decroly, F. Ferron, J. Lescar and B. Canard, Nat. Rev. Microbiol., 2011, 10, 51–65 CrossRef PubMed.
- S. W. Lockless, H. T. Cheng, A. E. Hodel, F. A. Quiocho and P. D. Gershon, Biochemistry, 1998, 37, 8564–8574 CrossRef CAS PubMed.
- M. Schlee and G. Hartmann, Nat. Rev. Immunol., 2016, 16, 566–580 CrossRef CAS PubMed.
- S. Daffis, K. J. Szretter, J. Schriewer, J. Li, S. Youn, J. Errett, T.-Y. Lin, S. Schneller, R. Zust, H. Dong, V. Thiel, G. C. Sen, V. Fensterl, W. B. Klimstra, T. C. Pierson, R. M. Buller, M. Gale, P.-Y. Shi and M. S. Diamond, Nature, 2010, 468, 452–456 CrossRef CAS PubMed.
- C. Schuberth-Wagner, J. Ludwig, A. K. Bruder, A.-M. Herzner, T. Zillinger, M. Goldeck, T. Schmidt, J. L. Schmid-Burgk, R. Kerber, S. Wolter, J.-P. Stümpel, A. Roth, E. Bartok, C. Drosten, C. Coch, V. Hornung, W. Barchet, B. M. Kümmerer, G. Hartmann and M. Schlee, Immunity, 2015, 43, 41–51 CrossRef CAS PubMed.
- K. Drazkowska, R. Tomecki, M. Warminski, N. Baran, D. Cysewski, A. Depaix, R. Kasprzyk, J. Kowalska, J. Jemielity and P. J. Sikorski, Nucleic Acids Res., 2022, 50, 9051–9071 CrossRef CAS PubMed.
- G. J. Cao and N. Sarkar, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 10380–10384 CrossRef CAS PubMed.
- H. Chang, J. Lim, M. Ha and V. N. Kim, Mol. Cell, 2014, 53, 1044–1052 CrossRef CAS PubMed.
- S. Holtkamp, S. Kreiter, A. Selmi, P. Simon, M. Koslowski, C. Huber, O. Türeci and U. Sahin, Blood, 2006, 108, 4009–4017 CrossRef CAS PubMed.
- M. S. D. Kormann, G. Hasenpusch, M. K. Aneja, G. Nica, A. W. Flemmer, S. Herber-Jonat, M. Huppmann, L. E. Mays, M. Illenyi, A. Schams, M. Griese, I. Bittmann, R. Handgretinger, D. Hartl, J. Rosenecker and C. Rudolph, Nat. Biotechnol., 2011, 29, 154–157 CrossRef CAS PubMed.
- Z. Trepotec, J. Geiger, C. Plank, M. K. Aneja and C. Rudolph, RNA, 2019, 25, 507–518 CrossRef CAS PubMed.
- E. Fang, X. Liu, M. Li, Z. Zhang, L. Song, B. Zhu, X. Wu, J. Liu, D. Zhao and Y. Li, Signal Transduction Targeted Ther., 2022, 7, 94 CrossRef CAS PubMed.
- C. Gustafsson, S. Govindarajan and J. Minshull, Trends Biotechnol., 2004, 22, 346–353 CrossRef CAS PubMed.
- C. Kimchi-Sarfaty, J. M. Oh, I.-W. Kim, Z. E. Sauna, A. M. Calcagno, S. V. Ambudkar and M. M. Gottesman, Science, 2007, 315, 525–528 CrossRef CAS PubMed.
- V. P. Mauro and S. A. Chappell, Trends Mol. Med., 2014, 20, 604–613 CrossRef CAS PubMed.
- A. Thess, S. Grund, B. L. Mui, M. J. Hope, P. Baumhof, M. Fotin-Mleczek and T. Schlake, Mol. Ther., 2015, 23, 1456–1464 CrossRef CAS PubMed.
- H. Zhang, L. Zhang, A. Lin, C. Xu, Z. Li, K. Liu, B. Liu, X. Ma, F. Zhao, H. Jiang, C. Chen, H. Shen, H. Li, D. H. Mathews, Y. Zhang and L. Huang, Nature, 2023, 621, 396–403 CrossRef CAS PubMed.
- G. Hanson and J. Coller, Nat. Rev. Mol. Cell Biol., 2018, 19, 20–30 CrossRef CAS PubMed.
- F. Mignone, C. Gissi, S. Liuni and G. Pesole, Genome Biol., 2002, 3, REVIEWS0004 CrossRef PubMed.
- J. O'Brien, H. Hayder, Y. Zayed and C. Peng, Front. Endocrinol., 2018, 9, 402 CrossRef PubMed.
- K. Karikó, A. Kuo and E. Barnathan, Gene Ther., 1999, 6, 1092–1100 CrossRef PubMed.
- A. G. Orlandini von Niessen, M. A. Poleganov, C. Rechner, A. Plaschke, L. M. Kranz, S. Fesser, M. Diken, M. Löwer, B. Vallazza, T. Beissert, V. Bukur, A. N. Kuhn, O. Tureci and U. Sahin, Mol. Ther., 2019, 27, 824–836 CrossRef CAS PubMed.
- N. Sultana, Y. Hadas, M. T. K. Sharkar, K. Kaur, A. Magadum, A. A. Kurian, N. Hossain, B. Alburquerque, S. Ahmed, E. Chepurko and L. Zangi, Mol. Ther.--Methods Clin. Dev., 2020, 17, 622–633 CrossRef CAS PubMed.
- P. J. Sample, B. Wang, D. W. Reid, V. Presnyak, I. J. McFadyen, D. R. Morris and G. Seelig, Nat. Biotechnol., 2019, 37, 803–809 CrossRef CAS PubMed.
- E. Rohner, R. Yang, K. S. Foo, A. Goedel and K. R. Chien, Nat. Biotechnol., 2022, 40, 1586–1600 CrossRef CAS PubMed.
- L. Alexopoulou, A. C. Holt, R. Medzhitov and R. A. Flavell, Nature, 2001, 413, 732–738 CrossRef CAS PubMed.
- S. S. Diebold, C. Massacrier, S. Akira, C. Paturel, Y. Morel and C. Reis e Sousa, Eur. J. Immunol., 2006, 36, 3256–3267 CrossRef CAS PubMed.
- H. Tanji, U. Ohto, T. Shibata, M. Taoka, Y. Yamauchi, T. Isobe, K. Miyake and T. Shimizu, Nat. Struct. Mol. Biol., 2015, 22, 109–115 CrossRef CAS PubMed.
- Z. Zhang, U. Ohto, T. Shibata, E. Krayukhina, M. Taoka, Y. Yamauchi, H. Tanji, T. Isobe, S. Uchiyama, K. Miyake and T. Shimizu, Immunity, 2016, 45, 737–748 CrossRef CAS PubMed.
- M. Yoneyama, M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira and T. Fujita, Nat. Immunol., 2004, 5, 730–737 CrossRef CAS PubMed.
- M. Schlee, A. Roth, V. Hornung, C. A. Hagmann, V. Wimmenauer, W. Barchet, C. Coch, M. Janke, A. Mihailovic, G. Wardle, S. Juranek, H. Kato, T. Kawai, H. Poeck, K. A. Fitzgerald, O. Takeuchi, S. Akira, T. Tuschl, E. Latz, J. Ludwig and G. Hartmann, Immunity, 2009, 31, 25–34 CrossRef CAS PubMed.
- Y.-M. Loo and M. Gale, Immunity, 2011, 34, 680–692 CrossRef CAS PubMed.
- R. Züst, L. Cervantes-Barragan, M. Habjan, R. Maier, B. W. Neuman, J. Ziebuhr, K. J. Szretter, S. C. Baker, W. Barchet, M. S. Diamond, S. G. Siddell, B. Ludewig and V. Thiel, Nat. Immunol., 2011, 12, 137–143 CrossRef PubMed.
- S. Balachandran, P. C. Roberts, L. E. Brown, H. Truong, A. K. Pattnaik, D. R. Archer and G. N. Barber, Immunity, 2000, 13, 129–141 CrossRef CAS PubMed.
- R. Hartmann, J. Justesen, S. N. Sarkar, G. C. Sen and V. C. Yee, Mol. Cell, 2003, 12, 1173–1185 CrossRef CAS PubMed.
- W. V. Gilbert and S. Nachtergaele, Ann. Rev. Biochem., 2023, 92, 175–198 CrossRef CAS PubMed.
- K. Karikó, M. Buckstein, H. Ni and D. Weissman, Immunity, 2005, 23, 165–175 CrossRef PubMed.
- K. Karikó and D. Weissman, Curr. Opin. Drug Discovery Dev., 2007, 10, 523–532 Search PubMed.
- S. R. Nallagatla and P. C. Bevilacqua, RNA, 2008, 14, 1201–1213 CrossRef CAS PubMed.
- B. R. Anderson, H. Muramatsu, S. R. Nallagatla, P. C. Bevilacqua, L. H. Sansing, D. Weissman and K. Karikó, Nucleic Acids Res., 2010, 38, 5884–5892 CrossRef CAS PubMed.
- B. R. Anderson, H. Muramatsu, B. K. Jha, R. H. Silverman, D. Weissman and K. Karikó, Nucleic Acids Res., 2011, 39, 9329–9338 CrossRef CAS PubMed.
- N. Pardi, H. Muramatsu, D. Weissman and K. Karikó, Methods Mol. Biol., 2013, 969, 29–42 CrossRef CAS PubMed.
- K. Karikó, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira and D. Weissman, Mol. Ther., 2008, 16, 1833–1840 CrossRef PubMed.
- O. Andries, S. Mc Cafferty, S. C. De Smedt, R. Weiss, N. N. Sanders and T. Kitada, J. Controlled Release, 2015, 217, 337–344 CrossRef CAS PubMed.
- S. Vaidyanathan, K. T. Azizian, A. K. M. A. Haque, J. M. Henderson, A. Hendel, S. Shore, J. S. Antony, R. I. Hogrefe, M. S. D. Kormann, M. H. Porteus and A. P. McCaffrey, Mol. Ther.--Nucleic Acids, 2018, 12, 530–542 CrossRef CAS PubMed.
- D. Weissman, N. Pardi, H. Muramatsu and K. Karikó, Methods Mol. Biol., 2013, 969, 43–54 CrossRef CAS PubMed.
- K. Karikó, H. Muramatsu, J. Ludwig and D. Weissman, Nucleic Acids Res., 2011, 39, e142 CrossRef PubMed.
- J. Nelson, E. W. Sorensen, S. Mintri, A. E. Rabideau, W. Zheng, G. Besin, N. Khatwani, S. V. Su, E. J. Miracco, W. J. Issa, S. Hoge, M. G. Stanton and J. L. Joyal, Sci. Adv., 2020, 6, eaaz6893 CrossRef CAS PubMed.
- M. Baiersdörfer, G. Boros, H. Muramatsu, A. Mahiny, I. Vlatkovic, U. Sahin and K. Karikó, Mol. Ther.--Nucleic Acids, 2019, 15, 26–35 CrossRef PubMed.
- K. S. Corbett, D. K. Edwards, S. R. Leist, O. M. Abiona, S. Boyoglu-Barnum, R. A. Gillespie, S. Himansu, A. Schäfer, C. T. Ziwawo, A. T. DiPiazza, K. H. Dinnon, S. M. Elbashir, C. A. Shaw, A. Woods, E. J. Fritch, D. R. Martinez, K. W. Bock, M. Minai, B. M. Nagata, G. B. Hutchinson, K. Wu, C. Henry, K. Bahl, D. Garcia-Dominguez, L. Ma, I. Renzi, W.-P. Kong, S. D. Schmidt, L. Wang, Y. Zhang, E. Phung, L. A. Chang, R. J. Loomis, N. E. Altaras, E. Narayanan, M. Metkar, V. Presnyak, C. Liu, M. K. Louder, W. Shi, K. Leung, E. S. Yang, A. West, K. L. Gully, L. J. Stevens, N. Wang, D. Wrapp, N. A. Doria-Rose, G. Stewart-Jones, H. Bennett, G. S. Alvarado, M. C. Nason, T. J. Ruckwardt, J. S. McLellan, M. R. Denison, J. D. Chappell, I. N. Moore, K. M. Morabito, J. R. Mascola, R. S. Baric, A. Carfi and B. S. Graham, Nature, 2020, 586, 567–571 CrossRef CAS PubMed.
- Z. Kis, C. Kontoravdi, A. K. Dey, R. Shattock and N. Shah, J. Adv. Manuf. Process., 2020, 2, e10060 CrossRef CAS PubMed.
- N. Chaudhary, D. Weissman and K. A. Whitehead, Nat. Rev. Drug Discovery, 2021, 20, 817–838 CrossRef CAS PubMed.
- S. Qin, X. Tang, Y. Chen, K. Chen, N. Fan, W. Xiao, Q. Zheng, G. Li, Y. Teng, M. Wu and X. Song, Signal Transduction Targeted Ther., 2022, 7, 166 CrossRef CAS PubMed.
- N. Pardi, A. J. Secreto, X. Shan, F. Debonera, J. Glover, Y. Yi, H. Muramatsu, H. Ni, B. L. Mui, Y. K. Tam, F. Shaheen, R. G. Collman, K. Karikó, G. A. Danet-Desnoyers, T. D. Madden, M. J. Hope and D. Weissman, Nat. Commun., 2017, 8, 14630 CrossRef PubMed.
- P. M. Tiwari, D. Vanover, K. E. Lindsay, S. S. Bawage, J. L. Kirschman, S. Bhosle, A. W. Lifland, C. Zurla and P. J. Santangelo, Nat. Commun., 2018, 9, 3999 CrossRef PubMed.
- N. Kose, J. M. Fox, G. Sapparapu, R. Bombardi, R. N. Tennekoon, A. D. de Silva, S. M. Elbashir, M. A. Theisen, E. Humphris-Narayanan, G. Ciaramella, S. Himansu, M. S. Diamond and J. E. Crowe, Sci. Immunol., 2019, 4, eaaw6647 CrossRef CAS PubMed.
- J. H. Erasmus, J. Archer, J. Fuerte-Stone, A. P. Khandhar, E. Voigt, B. Granger, R. G. Bombardi, J. Govero, Q. Tan, L. A. Durnell, R. N. Coler, M. S. Diamond, J. E. Crowe, S. G. Reed, L. B. Thackray, R. H. Carnahan and N. Van Hoeven, Mol. Ther.--Methods Clin. Dev., 2020, 18, 402–414 CrossRef CAS PubMed.
- D. Galati, S. Zanotta, L. Capitelli and M. Bocchino, Allergy, 2022, 77, 100–110 CrossRef CAS PubMed.
- A. W. Freyn, J. Ramos da Silva, V. C. Rosado, C. M. Bliss, M. Pine, B. L. Mui, Y. K. Tam, T. D. Madden, L. C. de Souza Ferreira, D. Weissman, F. Krammer, L. Coughlan, P. Palese, N. Pardi and R. Nachbagauer, Mol. Ther., 2020, 28, 1569–1584 CrossRef CAS PubMed.
- A. B. Vogel, L. Lambert, E. Kinnear, D. Busse, S. Erbar, K. C. Reuter, L. Wicke, M. Perkovic, T. Beissert, H. Haas, S. T. Reece, U. Sahin and J. S. Tregoning, Mol. Ther., 2018, 26, 446–455 CrossRef CAS PubMed.
- C. R. Stadler, H. Bähr-Mahmud, L. Celik, B. Hebich, A. S. Roth, R. P. Roth, K. Karikó, O. Tureci and U. Sahin, Nat. Med., 2017, 23, 815–817 CrossRef CAS PubMed.
- Y. Rybakova, P. S. Kowalski, Y. Huang, J. T. Gonzalez, M. W. Heartlein, F. DeRosa, D. Delcassian and D. G. Anderson, Mol. Ther., 2019, 27, 1415–1423 CrossRef CAS PubMed.
- U. Sahin, P. Oehm, E. Derhovanessian, R. A. Jabulowsky, M. Vormehr, M. Gold, D. Maurus, D. Schwarck-Kokarakis, A. N. Kuhn, T. Omokoko, L. M. Kranz, M. Diken, S. Kreiter, H. Haas, S. Attig, R. Rae, K. Cuk, A. Kemmer-Bruck, A. Breitkreuz, C. Tolliver, J. Caspar, J. Quinkhardt, L. Hebich, M. Stein, A. Hohberger, I. Vogler, I. Liebig, S. Renken, J. Sikorski, M. Leierer, V. Müller, H. Mitzel-Rink, M. Miederer, C. Huber, S. Grabbe, J. Utikal, A. Pinter, R. Kaufmann, J. C. Hassel, C. Loquai and O. Tureci, Nature, 2020, 585, 107–112 CrossRef CAS PubMed.
- S. L. Hewitt, A. Bai, D. Bailey, K. Ichikawa, J. Zielinski, R. Karp, A. Apte, K. Arnold, S. J. Zacharek, M. S. Iliou, K. Bhatt, M. Garnaas, F. Musenge, A. Davis, N. Khatwani, S. V. Su, G. MacLean, S. J. Farlow, K. Burke and J. P. Frederick, Sci. Transl. Med., 2019, 11, eaat9143 CrossRef CAS PubMed.
- M. Patel, A. Jimeno, D. Wang, S. Stemmer, T. Bauer, R. Sweis, R. Geva, S. Kummar, P. Reagan, R. Perets, P. LoRusso, S. Gupta, S. Zacharek, A. Laino, O. Milberg, J. Frederick, S. Chen, S. Pascarella, W. Randolph, P. Aanur, L. Johansen, K. Do, R. Meehan and R. Sullivan, J. ImmunoTher. Cancer, 2021, 9, A569–A569 CrossRef.
- S. L. Hewitt, D. Bailey, J. Zielinski, A. Apte, F. Musenge, R. Karp, S. Burke, F. Garcon, A. Mishra, S. Gurumurthy, A. Watkins, K. Arnold, J. Moynihan, E. Clancy-Thompson, K. Mulgrew, G. Adjei, K. Deschler, D. Potz, G. Moody, D. A. Leinster, S. Novick, M. Sulikowski, C. Bagnall, P. Martin, J.-M. Lapointe, H. Si, C. Morehouse, M. Sedic, R. W. Wilkinson, R. Herbst, J. P. Frederick and N. Luheshi, Clin. Cancer Res., 2020, 26, 6284–6298 CrossRef CAS PubMed.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728–1731 CrossRef CAS PubMed.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- P. V. Pawar, S. V. Gohil, J. P. Jain and N. Kumar, Polym. Chem., 2013, 4, 3160–3176 RSC.
- S. Matoori and J.-C. Leroux, Mater. Horiz., 2020, 7, 1297–1309 RSC.
- A. Stano, E. A. Scott, K. Y. Dane, M. A. Swartz and J. A. Hubbell, Biomaterials, 2013, 34, 4339–4346 CrossRef CAS PubMed.
- A. Sette and S. Crotty, Cell, 2021, 184, 861–880 CrossRef CAS PubMed.
- L. R. Volpatti, R. P. Wallace, S. Cao, M. M. Raczy, R. Wang, L. T. Gray, A. T. Alpar, P. S. Briquez, N. Mitrousis, T. M. Marchell, M. S. Sasso, M. Nguyen, A. Mansurov, E. Budina, A. Solanki, E. A. Watkins, M. R. Schnorenberg, A. C. Tremain, J. W. Reda, V. Nicolaescu, K. Furlong, S. Dvorkin, S. S. Yu, B. Manicassamy, J. L. LaBelle, M. V. Tirrell, G. Randall, M. Kwissa, M. A. Swartz and J. A. Hubbell, ACS Cent. Sci., 2021, 7, 1368–1380 CrossRef CAS PubMed.
- K. Sauer and T. Harris, Front. Immunol., 2020, 11, 581807 CrossRef CAS PubMed.
- S. Iqbal, M. Blenner, A. Alexander-Bryant and J. Larsen, Biomacromolecules, 2020, 21, 1327–1350 CrossRef CAS PubMed.
- X. Jiang, K. Abedi and J. Shi, in Encyclopedia of Nanomaterials, ed. Y. Yin, Y. Lu and Y. Xia, Elsevier, 1st edn, 2023, pp. 555–573 Search PubMed.
- M. Sioud, Cancers, 2019, 11, 176 CrossRef CAS PubMed.
- Y. Chen, Y. Huang, Q. Li, Z. Luo, Z. Zhang, H. Huang, J. Sun, L. Zhang, R. Sun, D. J. Bain, J. F. Conway, B. Lu and S. Li, Nat. Nanotechnol., 2023, 18, 193–204 CrossRef CAS PubMed.
- Y. Zou, M. Zheng, W. Yang, F. Meng, K. Miyata, H. J. Kim, K. Kataoka and Z. Zhong, Adv. Mater., 2017, 29, 1703285 CrossRef PubMed.
- S. Kansz and Y. M. Elçin, Adv. Colloid Interface Sci., 2023, 317, 102930 CrossRef PubMed.
- K. Song, D. C. Nguyen, T. Luu, O. Yazdani, D. Roy, P. S. Stayton and S. H. Pun, J. Controlled Release, 2023, 356, 232–241 CrossRef CAS PubMed.
- J. Yang, H. Chen, I. R. Vlahov, J.-X. Cheng and P. S. Low, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13872–13877 CrossRef CAS PubMed.
- S. Cerritelli, D. Velluto and J. A. Hubbell, Biomacromolecules, 2007, 8, 1966–1972 CrossRef CAS PubMed.
- L. Jia, D. Cui, J. Bignon, A. Di Cicco, J. Wdzieczak-Bakala, J. Liu and M.-H. Li, Biomacromolecules, 2014, 15, 2206–2217 CrossRef CAS PubMed.
- M. Luo, H. Wang, Z. Wang, H. Cai, Z. Lu, Y. Li, M. Du, G. Huang, C. Wang, X. Chen, M. R. Porembka, J. Lea, A. E. Frankel, Y.-X. Fu, Z. J. Chen and J. Gao, Nat. Nanotechnol., 2017, 12, 648–654 CrossRef CAS PubMed.
- M. G. Gouveia, J. P. Wesseler, J. Ramaekers, C. Weder, P. B. V. Scholten and N. Bruns, Chem. Soc. Rev., 2023, 52, 728–778 RSC.
- H. Huang, W. Feng, Y. Chen and J. Shi, Nano Today, 2020, 35, 100972 CrossRef CAS.
- C. T. Turner, S. J. McInnes, N. H. Voelcker and A. J. Cowin, J. Nanomater., 2015, 2015, 2 Search PubMed.
- D. Albulet, D. A. Florea, B. Boarca, L. M. Ditu, M. C. Chifiriuc, A. M. Grumezescu and E. Andronescu, Nanostructures for Cancer Therapy, Elsevier, 2017, pp. 1–21 Search PubMed.
- S. Sarkar, M. Ekbal Kabir, J. Kalita and P. Manna, ChemBioChem, 2023, e202200672 CrossRef CAS PubMed.
- J. Zarubova, X. Zhang, T. Hoffman, M. M. Hasani-Sadrabadi and S. Li, Matter, 2021, 4, 1528–1554 CrossRef CAS PubMed.
- M. O. Dellacherie, A. Li, B. Y. Lu, C. S. Verbeke, L. Gu, A. G. Stafford, E. J. Doherty and D. J. Mooney, Adv. Funct. Mater., 2020, 30, 2002448 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, J. Phys. Chem., 1953, 57, 670–673 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- T. K. Sau and C. J. Murphy, Langmuir, 2004, 20, 6414–6420 CrossRef CAS PubMed.
- D. Seo, J. C. Park and H. Song, J. Am. Chem. Soc., 2006, 128, 14863–14870 CrossRef CAS PubMed.
- M. L. Personick, M. R. Langille, J. Zhang and C. A. Mirkin, Nano Lett., 2011, 11, 3394–3398 CrossRef CAS PubMed.
- S. E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 2, 2182–2190 CrossRef CAS PubMed.
- C. S. Ah, Y. J. Yun, H. J. Park, W.-J. Kim, D. H. Ha and W. S. Yun, Chem. Mater., 2005, 17, 5558–5561 CrossRef CAS.
- S. Ahn, I.-H. Lee, S. Kang, D. Kim, M. Choi, P. E. Saw, E.-C. Shin and S. Jon, Adv. Healthcare Mater., 2014, 3, 1194–1199 CrossRef CAS PubMed.
- X. Chen and C. Gao, Colloids Surf., B, 2017, 160, 372–380 CrossRef CAS PubMed.
- K. R. Trabbic, K. A. Kleski and J. J. Barchi Jr, ACS Bio Med. Chem. Au, 2021, 1, 31–43 CrossRef CAS PubMed.
- D. N. Desai, A. Mahal, R. Varshney, A. J. Obaidullah, B. Gupta, P. Mohanty, P. Pattnaik, N. C. Mohapatra, S. Mishra and V. Kandi, et al. , ACS Omega, 2023, 8, 27953–27968 CrossRef CAS PubMed.
- J. I. Andorko and C. M. Jewell, Bioeng. Transl. Med., 2017, 2, 139–155 CrossRef PubMed.
- E. Bracho-Sanchez, F. G. Rocha, S. K. Bedingfield, B. D. Partain, S. L. Macias, M. A. Brusko, J. M. Colazo, M. M. Fettis, S. A. Farhadi and E. Y. Helm, et al. , Nat. Biomed. Eng., 2023, 1–14 Search PubMed.
- Z. Sadrearhami, F. N. Shafiee, K. K. Ho, N. Kumar, M. Krasowska, A. Blencowe, E. H. Wong and C. Boyer, ACS Appl. Mater. Interfaces, 2019, 11, 7320–7329 CrossRef CAS PubMed.
- E. Piktel, Ł. Suprewicz, J. Depciuch, S. Chmielewska, K. Skŀodowski, T. Daniluk, G. Król, P. Koŀat-Brodecka, P. Bijak and A. Pajor-Swierzy, et al. , Sci. Rep., 2021, 11, 12546 CrossRef CAS PubMed.
- M. Kazemzadeh-Narbat, H. Cheng, R. Chabok, M. M. Alvarez, C. De La Fuente-Nunez, K. S. Phillips and A. Khademhosseini, Crit. Rev. Biotechnol., 2021, 41, 94–120 CrossRef CAS PubMed.
- C. R. Arciola, D. Campoccia and L. Montanaro, Nat. Rev. Microbiol., 2018, 16, 397–409 CrossRef CAS PubMed.
- A. Singh and N. A. Peppas, Adv. Mater., 2014, 26, 6530–6541 CrossRef CAS PubMed.
- K. Sadtler, A. Singh, M. T. Wolf, X. Wang, D. M. Pardoll and J. H. Elisseeff, Nat. Rev. Mater., 2016, 1, 1–17 Search PubMed.
- D. Salthouse, K. Novakovic, C. M. Hilkens and A. M. Ferreira, Acta Biomater., 2023, 155, 1–18 CrossRef CAS PubMed.
- B. Zhang, Y. Su, J. Zhou, Y. Zheng and D. Zhu, Adv. Sci., 2021, 8, 2100446 CrossRef CAS PubMed.
- M. E. Cam, S. Yildiz, H. Alenezi, S. Cesur, G. S. Ozcan, G. Erdemir, U. Edirisinghe, D. Akakin, D. S. Kuruca and L. Kabasakal, et al. , J. Royal Soc. Interface, 2020, 17, 20190712 CrossRef CAS PubMed.
- Y. Liang, M. Li, Y. Yang, L. Qiao, H. Xu and B. Guo, ACS Nano, 2022, 16, 3194–3207 CrossRef CAS PubMed.
- A. L. Lauterbach, A. J. Slezak, R. Wang, S. Cao, M. M. Raczy, E. A. Watkins, C. J. M. Jimenez and J. A. Hubbell, Adv. Healthcare Mater., 2023, 2300515 CrossRef CAS PubMed.
- J. Zhang, X. Huang, H. Wang, X. Liu, T. Zhang, Y. Wang and D. Hu, Stem Cell Res. Ther., 2015, 6, 1–7 CrossRef PubMed.
- B. Guo, J. Qu, X. Zhao and M. Zhang, Acta Biomater., 2019, 84, 180–193 CrossRef CAS PubMed.
- K. Adu-Berchie, Y. Liu and D. K. Y. Zhang, et al. , Nat. Biomed. Eng., 2023, 7, 1374–1391 CrossRef CAS PubMed.
- M. Bilal, S. Mehmood, A. Raza, U. Hayat, T. Rasheed and H. M. Iqbal, Adv. Wound Care, 2021, 10, 204–219 CrossRef PubMed.
- I. Menon, P. Bagwe, K. B. Gomes, L. Bajaj, R. Gala, M. N. Uddin, M. J. Dsouza and S. M. Zughaier, Micromachines, 2021, 12, 435 CrossRef PubMed.
- F. K. Aldawood, A. Andar and S. Desai, Polymers, 2021, 13, 2815 CrossRef CAS PubMed.
- M. R. Prausnitz, Ann. Rev. Chem. Biomol. Eng., 2017, 8, 177–200 CrossRef CAS PubMed.
- A. Vander Straeten, M. Sarmadi, J. L. Daristotle, M. Kanelli, L. H. Tostanoski, J. Collins, A. Pardeshi, J. Han, D. Varshney and B. Eshaghi, et al. , Nat. Biotechnol., 2023, 1–8 Search PubMed.
- Y. Shin, J. Kim, J. H. Seok, H. Park, H.-R. Cha, S. H. Ko, J. M. Lee, M.-S. Park and J.-H. Park, Sci. Rep., 2022, 12, 12189 CrossRef CAS PubMed.
- J. J. Landers, K. W. Janczak, A. K. Shakya, V. Zarnitsyn, S. R. Patel, J. R. Baker Jr, H. S. Gill and J. J. O'Konek, Immunotherapy, 2022, 14, 539–552 CrossRef CAS PubMed.
- A. K. Shakya, C. H. Lee and H. S. Gill, Mol. Pharm., 2020, 17, 3033–3042 CrossRef CAS PubMed.
- E. Zhang, B. Zeng, R. Song, L. Yao and H. Che, Drug Delivery Transl. Res., 2023, 13, 1828–1841 CrossRef CAS PubMed.
- J. L. Paris, L. K. Vora, M. J. Torres, C. Mayorga and R. F. Donnelly, Drug Discovery Today, 2023, 103556 CrossRef CAS PubMed.
- L. X. Yu, G. Amidon, M. A. Khan, S. W. Hoag, J. Polli, G. Raju and J. Woodcock, AAPS J., 2014, 16, 771–783 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |