
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesigned functions of oxide/hydroxide nanosheets via elemental replacement/doping
Kanji
Saito
 ab,
Masashi
Morita
c,
Tomohiko
Okada
d,
Rattanawadee (Ploy)
Wijitwongwan
e and
Makoto
Ogawa
*e
ab,
Masashi
Morita
c,
Tomohiko
Okada
d,
Rattanawadee (Ploy)
Wijitwongwan
e and
Makoto
Ogawa
*e
aDepartment of Materials Science, Graduate School of Engineering Science, Akita University, 1-1 Tegatagakuen-machi, Akita-shi, Akita 010-8502, Japan
bKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0054, Japan
cDepartment of Applied Chemistry, Tokyo University of Agriculture and Technology, Tokyo 184-8588, Japan
dDepartment of Materials Chemistry, and Research Initiative for Supra-Materials, Shinshu University, 4-17-1 Wakasato, Nagano, Nagano-shi 380-8553, Japan
eSchool of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), 555 Moo 1, Payupnai, Wangchan, Rayong 21210, Thailand. E-mail: makoto.ogawa@vistec.ac.th
First published on 7th October 2024
Abstract
Partial replacement of one structural element in a solid with another of a similar size was conducted to impart functionality to the solids and modify their properties. This phenomenon is found in nature in coloured gemstones and clay minerals and is used in materials chemistry and physics, endowing materials with useful properties that can be controlled by incorporated heteroelements and their amounts. Depending on the area of research (or expected functions), the replacement is referred to as “isomorphous substitution”, “doping”, etc. Herein, elemental replacement in two-dimensional (2D) oxides and hydroxides (nanosheets or layered materials) is summarised with emphasis on the uniqueness of their preparation, characterisation and application compared with those of the corresponding bulk materials. Among the 2D materials (graphene, metallenes, transition metal chalcogenides, metal phosphate/phosphonates, MXenes, etc.), 2D oxides and hydroxides are characterised by their presence in nature, facile synthesis and storage under ambient conditions, and possible structural variation from atomic-level nanosheets to thicker nanosheets composed of multilayered structures. The heteroelements to be doped were selected depending on the target application objectively; however, there are structural and synthetic limitations in the doping of heteroelements. In the case of layered double hydroxides (single layer) and layered alkali silicates (from single layer to multiple layers), including layered clay minerals (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 layer), the replacement (commonly called isomorphous substitution) is discussed to understand/design characteristics such as catalytic, adsorptive (including ion exchange), and swelling properties. Due to the variation in their main components, the design of layered transition metal oxide/hydroxide materials via isomorphous substitution is more versatile; in this case, tuning their band structure, doping both holes and electrons, and creating impurity levels are examined by the elemental replacement of the main components. As typical examples, material design for the photocatalytic function of an ion-exchangeable layered titanate (lepidocrocite-type titanate) and a perovskite niobate (KCa2Nb3O10) is discussed, where elemental replacement is effective in designing their multiple functions.
:1 layer), the replacement (commonly called isomorphous substitution) is discussed to understand/design characteristics such as catalytic, adsorptive (including ion exchange), and swelling properties. Due to the variation in their main components, the design of layered transition metal oxide/hydroxide materials via isomorphous substitution is more versatile; in this case, tuning their band structure, doping both holes and electrons, and creating impurity levels are examined by the elemental replacement of the main components. As typical examples, material design for the photocatalytic function of an ion-exchangeable layered titanate (lepidocrocite-type titanate) and a perovskite niobate (KCa2Nb3O10) is discussed, where elemental replacement is effective in designing their multiple functions.
 Kanji Saito | Kanji SAITO received his BS in 2012 and MS in 2014 from Waseda University, supervised by Professor Makoto Ogawa. He received his PhD from Waseda University in 2017 under the direction of Professor Yoshiyuki Sugahara. He was Research Associate at Waseda University from 2015 to 2017. Thereafter, he joined Akita University in 2017 as Specially Appointed Assistant Professor and moved to a permanent position in 2021. He was promoted to Lecturer, his current position, in 2024. His research interest focuses on layered inorganic solids including layered clay minerals and layered transition metal oxides. |
 Masashi Morita | Masashi Morita received his BS (2011) and MS (2013) from Waseda University under the supervision of Professor Makoto Ogawa. From 2013 to 2021, he worked as a researcher at Panasonic Corporation and was promoted to senior researcher (2017–present) and has been working as a project leader (2018–present). In 2021, he received his PhD from Nagoya University under the supervision of Professor Ryotaro Matsuda. After receiving his PhD, he moved to the Division of Applied Chemistry, Institute of Engineering at Tokyo University of Agriculture and Technology as an assistant professor. He was promoted to lecturer in October 2024. His current research interests are the design and synthesis of inorganic–organic hybrid materials focusing on 2D materials and metal complexes for adsorbents and catalysts. |
 Tomohiko Okada | Tomohiko Okada received his PhD from Waseda University in 2004 under the direction of Professor Makoto Ogawa. In 2006, he moved to Shinshu University, Nagano Prefecture Japan as an assistant professor. He is presently an associate professor in the Department of Materials Chemistry, Shinshu University. His research interests are materials chemistry of clay-based adsorbents and catalysts. |
 Rattanawadee (Ploy) Wijitwongwan | Rattanawadee (Ploy) Wijitwongwan received her BEng degree in Petrochemicals and Polymeric Materials Engineering from Silpakorn University with First Class Honors. She completed her PhD in 2023 at Vidyasirimedhi Institute of Science and Technology (VISTEC), Thailand. Currently, she is a postdoctoral researcher under the supervision of Prof. Makoto Ogawa at the School of Energy Science and Engineering (ESE) at VISTEC. Her research interests focus on the synthesis of layered double hydroxides, especially for composition control, for environmental and energy-related applications. |
 Makoto Ogawa | Makoto Ogawa was educated in the Department of Applied Chemistry, Waseda University, supervised by Professor Chuzo Kato. After postdoctoral research at RIKEN, he joined Waseda University and worked on inorganic materials chemistry as a full professor in 2004. In 2015, he moved to Thailand as an opening professor at the School of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), which opened in 2015 in Rayong, Thailand and continues his research activity in materials chemistry. |
1. Introduction
The replacement of one structural element with another one of a similar size is often referred to as “isomorphous substitution”, which is commonly confused with the term “defect”, another form of replacement (concerning vacancies). In nature, there are well-known classes of materials in which isomorphous substitution and/or defects play key roles in their characteristics/functions. The replacement of an element in the structure of semiconductor materials is one of the most common examples, and the term “doping” is used for the intentional introduction of impurities into an intrinsic semiconductor to modulate its electrical, optical and structural properties. Alternatively, since the discovery of cupric oxide-based superconductors in the 1980s by J. G. Bednorz and K. A. Muller,1 changes in the superconducting and magnetic properties of high-temperature oxide-based superconductors as a function of oxygen stoichiometry and cation substitution have been systematically and extensively investigated to find superconducting properties that can be continuously optimised at a specific doping level. Thus, the replacement of a framework element is a versatile way to modify the properties of solids (not only crystalline but also amorphous solids) from insulators to superconductors and impart functions (optical properties as notable examples). In this case, various terms such as doping/dopants, replacement/guests, isomorphous substitution and lattice substitution are used depending on the research field and materials. Doping is commonly used in semiconductor science, where very small quantities of heteroelements can significantly modify the electrical properties of materials. The term “substitution” is commonly used when replacing one element with another in larger quantities ranging from several percentage to several tens of percentage in some cases.Elemental doping/isomorphous substitution is found in nature. For example, the origin of colours in coloured gemstones2 and charges in clay minerals3 is well-known. The replacement in oxide-based crystals such as quartz (crystalline silica), corundum (α-alumina), beryl and chrysoberyl with trace amounts of heteroelements such as iron, titanium, chromium, vanadium and magnesium causes changes in the corresponding colours. The colour of amethyst (Fig. 1), purple-coloured quartz, is explained by the unusual irradiation-induced valence of iron (or other transition metal ions) as impurities in the quartz crystal lattice. Sapphires are described by their colour (blue, green, and yellow, which is dependent on the doped element and its states), where the intense blue of blue sapphire is caused by the replacement of aluminium in corundum with titanium and iron. The synthesis of coloured gemstone crystals (ruby and sapphire as notable examples) in the laboratory has also been examined to obtain materials mimicking the mechanical, chemical, optical, and physical characteristics of natural gemstones for industrial applications, including the jewellery industry. Synthetic gem crystals have been manufactured since the 19th century, and among them, synthetic ruby is one of the first successful examples used in abrasives, communications, electronics and optics. Lasing materials were designed from yttrium aluminium garnet (Y3Al5O12) by the replacement of yttrium with titanium and rare earth ions to obtain varying wavelength emissions (known as YAG laser).4 Intense emission was seen when the Nd content was 3 atomic%. When the Nd content was 6 atomic%, the emission intensity was reduced as a result of the Nd–Nd interactions. Various optical properties were reported for rare earth element-doped oxides.5 In addition to the crystalline host lattice, various transition metal ions and rare earth metal ions have been incorporated in inorganic glasses for optical applications as phosphors and laser since the first demonstration of the laser action of Nd3+ ions (concentration of Nd2O3 in the range of 0.13 to 2.0 wt%) in barium crown glass.6 These examples clearly indicate the importance of the heteroelements and their amounts.
 | ||
| Fig. 1 Colour variation in quartz caused by isomorphous substitution; amethyst is quartz containing a trace amount of iron. Permission for photography was given by Mineral Industry Museum of Akita University. The archive number is 292 and 14277 for quartz and amethyst, respectively. | ||
Thus, the doping (isomorphous substitution) of heteroelements has been applied in many solid-state materials. However, there is no general consensus on the relationship between doping and properties and there are several emerging new classes of materials, and thus doping in numerous materials has become the focus of research. To tune the target properties, parameters such as elemental variation and quantity and distribution of the dopant have been examined. Also, to obtain reliable and reproducible results, the synthetic methods and conditions have been optimised. There are solubility limits depending on the system, the required concentration level is different for each application, and in some applications, an extension of the dopant concentration is expected. Furthermore, the state and location of the doped heteroelements have been investigated using advanced analytical tools with the appropriate atomic resolution.
The materials and material designs described above are based on bulk materials and the solubility limit is applicable to bulk solids. When isomorphous substitution occurs at the surface (not in the bulk), the surface properties are substantially modified, and new surface properties emerge. This occurs in various materials and is efficient, especially for materials with a nanoscopic size and/or nanoporous structures. The isomorphous substitution in layered clay minerals is a representative example, where the isomorphous substitution of the main component with a heteroelement with lower valence represents the primary source of negative charges in silicate-based clay minerals. Alternatively, isomorphous substitution in brucite-type layered hydroxide with an ion of higher valence (namely M2+ is replaced with M3+) leads to a positive charge, providing a class of anion-exchangeable materials, i.e., layered double hydroxides (LDHs, also as anionic clay or hydrotalcite-type compounds).
Another well-known example of isomorphous substitution in nanomaterials is that of the main components such as silicon in zeolites with other tetrahedrally coordinated heteroatoms such as Al3+ and Ga3+ at a few wt%, which has been used to obtain catalysts.7,8 The activities are known to be determined by the coordination states of the heteroelements. Besides trivalent metal cations, Ti4+ has been incorporated into the frameworks of silicates as an isolated tetrahedrally coordinated species. A charge transfer-type excited state is generated under light irradiation. The photocatalytic reduction of carbon dioxide by UV light has been examined using Ti-containing zeolites, mesoporous silicas and metal–organic frameworks.9–11 Initially, this concept was known as “single-site catalyst”, while more recently, it is referred to as “single-atom catalyst”.12
The incorporation of a second metal ion into the nodes of the frameworks of porous coordination polymers (PCPs), which are also called metal–organic frameworks (MOFs), has been reported.13–17 MOFs are a class of porous materials with a very large surface area and high porosity, leading to significant interest for their application in gas storage, separation,18,19 detection,20 catalysis,21 medicine,22etc. MOFs are constructed from inorganic nodes and organic linkers. The partial replacement of the inorganic nodes with other metal ions (the products are called bimetallic or mixed metal MOFs) has been used to control their properties, including adsorptive, catalytic, and optical properties.22–27 These bimetallic (or mixed metal) MOFs have been prepared by metal doping during crystallisation or post-synthetic ion exchange.28
Heteroelement doping/substitution in nanomaterials with lower dimensions (2D, 1D and 0D) has been examined. The replacement of the main components in oxide and hydroxide nanosheets with heteroelements are the topics of this review article because of their presence in nature, facile synthesis and storage, possible structural variation from nanosheet to multilayered structures, and morphological variation from nanodot to large single crystals. Considering their advantageous characteristics, 2D oxides/hydroxides are useful for vast applications ranging from civil engineering to molecular and biomedical applications. Non-oxide 2D materials including graphene and transition metal dichalcogenides have been studied extensively and heteroatom doping has become an effective method to tune their opto-electronic and chemical characteristics for the increasing demands in fields such as optoelectronics and sensing.29–32
After the discovery of a layered zeolite (MCM-22, later designated as MWW) framework in the 1990s,33,34 layered or 2D zeolites, which are stacked nanometer-thick layers or mono-layer assemblies, became an important direction in the development of zeolites for better performances and new applications. One of the advantages of zeolite nanosheets is generating more open pore connections for the diffusion of reactants and products.35,36 Similarly nanosheet (or 2D) MOFs are becoming popular materials.37,38 Nanosheets of bimetallic MOFs have also been published.39
2D materials (layered materials/flat materials) of varying compositions and structures are known, as described above (Fig. 2).40–44 Some layered materials have been dispersed in solvents to realise a single-sheet dispersion (exfoliation) and obtain nanosheets.45,46 Exfoliation has been achieved in some polymers to obtain polymer nanocomposites. Porous nanoarchitectures have been designed by the cross-linking of nanosheets with organic moieties and inorganic (metals, oxides, and chalcogenides) nanoparticles (pillars) by intercalation into the layered materials and by exfoliation and restacking.47 The surface of each nanosheet is exposed by these approaches to utilise it for adsorption/immobilisation and reactions more efficiently. Thus, compared with isomorphous substitution in bulk materials, that in layered materials has a greater impact on the sophisticated materials design.
 | ||
| Fig. 2 Incorporation of heteroelements into 2D materials. | ||
In the present review, we summarise the elemental replacement of the framework elements (isomorphous substitution) in oxide-based and hydroxide-based layered materials (using layered double hydroxides, layered alkali silicates, 2:1 type phyllosilicates, and layered transition metal oxides) and derived nanosheets. Based on the variation in the layer thickness and composition, four groups of layered materials, as summarised in Fig. 3, will be introduced as representative examples of layered materials functionalised by isomorphous substitution. Layered double hydroxides are regarded as the representative example of single layer materials with versatile compositional variation. Layered alkali silicates are chosen because a variation in their layer thickness keeps the main component as silica/silicate. Also, 2:1-type phyllosilicates (smectite group of clay minerals) are important examples of unit layers composed of multiple sheets (composed of two silicate sheets sandwiching one metal hydroxide sheet), where the metal ion in the hydroxide sheet has a compositional variation (Mg, Al, Fe, etc.). Layered transition metal oxides (LTMO) are larger categories including several structural types, offering a wide compositional and structural variation. To simplify the discussion, lepidocrocite-type layered titanates will be discussed as the representative example of single-layer LTMO, and a perovskite niobate (KCa2Nb3O10, a Dion–Jacobsen-type perovskite) as the example of LTMO with possible expandable thickness of the unit layer. The experimental methods for the replacement (or the preparation of the heteroelement-incorporated layered materials and the nanosheets), the characterisation and the possible (and the examined) functions of the products will be introduced to highlight the important roles of the elemental replacement in the design of 2D materials.
 | ||
| Fig. 3 Layered double hydroxides, layered alkali silicates, 2:1 type phyllosilicates, and layered transition metal oxides represent layered materials with varying layer thickness and/or composition. | ||
2. Metal hydroxides and layered double hydroxides
Layered double hydroxides (LDHs) are representative examples of layered materials, where isomorphous substitution plays a key role in their structures, characteristics, and functions. As the most well-known example, the isomorphous substitution of Mg2+ in brucite (magnesium hydroxide) with Al3+ results in layered materials with anion exchange capability. The material containing carbonate as the charge compensating anion in the interlayer space is named hydrotalcite, which was found as a mineral in Sweden in the 19th century and named due its capacity to be crushed into a white powder similar to talc, a 2:1-type layered clay mineral (see Section 4 of this review). The structural concept is applicable to other metal hydroxides and the substitution of the main component with heteroelements produces a series of layered materials named layered double hydroxides (LDHs),48–53 as shown in Fig. 4. LDHs with various compositions such as Mg6Fe2(OH)16CO3·4.5H2O (pyroaurite), Mg6Cr2(OH)16CO3·4H2O (stichtite), Ni6Al2(OH)16CO3OH·4H2O (takovite), and Mg4Fe(OH)10Cl·3H2O (iowaite) have been found as minerals.54–59 Occasionally, “anionic clay”, “hydrotalcite”, and “hydrotalcite-like compounds” are used as alternative terminologies for LDHs in the literature. The synthesis of LDHs (at that time, referred to as hydrotalcite-like compounds) was reported by Feitknecht in the 1940s.60,61
 | ||
| Fig. 4 Structure of layered double hydroxides. The crystal structure was drawn using the VESTA program.44 | ||
Some LDHs are commercially available as an antacid under the product name Talcid® (Bayer Healthcare AG) or a stabiliser in polyvinylchloride (PVC) resins by scavenging chloride.62,63 The properties of LDHs are different depending on their M2+, M3+ and interlayer anions. Therefore, the compositional variation in LDHs leads to their application in many different fields such as adsorbent/anion exchangers,64 catalysts and catalyst supports,65,66 electrodes/capacitors,67 anti-corrosion coatings,68 drug/gene carriers,69,70 and other medical/pharmaceutical applications. In addition, LDHs are used as precursors of oxides upon calcination, and the obtained mixed oxides are used as pigments, functional ceramics, catalysts and ion exchangers (reconstruction method).71–73 The ability to tune their properties through compositional adjustments and possible morphosynthesis has attracted considerable attention in each application.
2.1. Structure of LDHs and their compositional variation
The structure of LDHs is determined by the selection of M2+ and M3+, M3+/(M2+ + M3+) ratio and interlayer anions.The M3+/(M2+ + M3+) ratio in the brucite-like sheet corresponds to the layer charge density, correlating the properties of LDHs. The M3+/(M2+ + M3+) ratio is denoted by x in the chemical formula of LDHs, [M2+1−xM3+x(OH)2]x+[An−]x/n·mH2O and x is commonly found in the range of 0.20–0.33.49–51 When attempting to vary x beyond the common range, impurity phases such as metal hydroxides, metal oxides, and basic salts of the divalent or trivalent metal ion are encountered in most cases, as shown in Fig. 5. The limitation of x is thought to be caused by two factors, i.e., electrostatic repulsion between the neighbouring trivalent cations in the brucite-like sheet and the repulsion between the charge-balancing anions. These repulsive forces prevent higher layer charge densities corresponding to x > 0.33. The difficulty in obtaining a smaller x (x < 0.20) was explained by the large distance between the adjacent interlayer anions, leading to the collapse of the layered structure.
 | ||
| Fig. 5 Common phenomena in the formation of LDHs when different values of x were employed. | ||
In addition to the substitution in the brucite-like sheet, the defects in the sheets are thought to play a role in several functions, especially in photocatalysis,75 where LDHs have been used.76 M3+ is thought to be isolated by M2+ in the brucite-like sheet, and thus the removal of M3+ was examined to generate vacancies in the brucite-like sheet.77,78 Because of the difficulties in identifying and quantifying the vacancies in the brucite-like sheet, the roles of the vacancies in the functions of LDHs have not been well elucidated.79
2.2. Synthetic methods for LDHs
A variety of synthetic methods (and conditions in each method) has been employed to prepare LDHs with the desired composition (metal combinations, interlayer anions, and composition) and particle morphology to satisfy the application requirements.(a) Co-precipitation from an aqueous solution of metal salts by the addition of a basic solution is the most common method for the synthesis of LDHs.48,80,82–84 This method has many advantages, such as ease of scaling-up. The sequence of mixing varies as follows: (a-i) An aqueous solution of NaOH, KOH or NH4OH is slowly added to an aqueous solution (normally acidic) of M2+ and M3+ salts, resulting in a high pH of the mixture, referred to as the titration method.85–87 Given that metal hydroxides precipitate at different pH, the composition of the precipitate is not the same as the M2+ and M3+ ratio in the initial solution when the final pH is not high enough for the complete precipitation of M2+ and M3+.88,89 Co-precipitation at the pH for high supersaturation leads to the formation of less crystalline LDHs and the aggregation of nanometer-size particles. This is a result of the formation of a larger number of nuclei by the rapid nucleation in the initial stage.49,90–93 Thus, a crucial limitation of this sequence (adding base to the acidic solution of metal salts; titration) is the continuous change in the solution pH during the addition. The formation of individual metal hydroxide phases and LDHs also occurs. (a-ii) A basic solution and an acidic solution of metal salts are simultaneously added to a third solution, which contains the anion to be intercalated into the interlayer space of the resulting LDHs (constant pH method). By adjusting the rate of the addition, the pH of the mixture is controlled at a constant value suitable for the formation of the target LDHs. This method provides the condition of lower supersaturation, favouring particle growth over nucleation, resulting in LDHs with relatively higher crystallinity and larger particle sizes. A limitation of the constant pH method is the large volume of the solvent required to control the pH. With the large volume of solvent used in the process, the separation of the precipitate is difficult.49 Alternatively, this method has a frequent acquisition of the desired composition as an advantage. Generally, LDHs formed by the co-precipitation are finite particles with a broad particle size distribution, and aggregation of the finite particles is observed. Several conditions and methods such as different types of mixing conditions and post-synthetic ageing have been introduced to achieve homogeneity and higher crystallinity and to reduce particle aggregation.
(b) Co-precipitation from a homogeneous solution of metal salts during the hydrolysis of urea, hexamethylenetetramine, ammonium carbonate, etc. Co-precipitation of LDHs is possible by the decomposition of urea at elevated temperatures. This process involves the hydrolysis of urea, releasing carbonate and ammonium, which results in an increase in pH. The gradual release of ammonium leads to the growth of LDH crystals with high crystallinity. The LDHs prepared by the urea method have a narrower particle size distribution compared to the LDHs prepared by the titration method using a basic solution and constant pH method.94–101 Because of the generation of the carbonate anion from urea, the LDHs prepared by this method are commonly carbonate type. The use of hexamethylenetetramine (HMT) as the precipitating reagent of LDH enables the possibility to obtain LDHs with an interlayer anion other than carbonate given that the formaldehyde released from the hydrolysis of HMT is less plausible to be intercalated.101–103
The formation of organic anion-intercalated LDHs by mechanochemical reactions has also been reported.111,112 MgAl-LDH intercalated with p-toluene sulfonate (p-TS-LDH), malonate (M-LDH), and oxalate anions (O-LDH) was synthesised by milling Mg(OH)2 and Al(OH)3 in a planetary ball mill for 1 h and subsequent milling with organic anions for another 1 h. The d values were 1.77, 0.88, and 0.86 nm for p-TS-LDH, M-LDH, and O-LDH, respectively. The aggregated particles with a size of over 700 nm were observed from the TEM image, together with small disk-shaped particles with a lateral size of ca. 150 nm. p-TS-LDH was used as the starting material for the anion exchange reaction with dodecyl sulphate anion. The X-ray diffraction (XRD) patterns showed an increase in the basal spacing from 1.77 nm to 3.09 nm, and the FT-IR results also confirmed the successful ion exchange.112
The mechanochemical synthesis of LDHs has the following advantages: (i) solid–liquid separation is not necessary, (ii) carbonate contamination is less plausible, and (iii) the starting materials are not expensive compared with that used for the conventional synthesis starting from an aqueous solution of metal salts. However, the variation in the metal combination, composition, and morphology of LDHs achieved by the mechanochemical method is still limited.
2.3. Characterisation of LDHs
The composition of the products is determined by X-ray fluorescence (XRF) for solid samples and inductively coupled plasma emission spectrometry (ICP) after dissolving the products in an acidic solution. An energy dispersive X-ray fluorescence spectrometer (EDS) equipped with SEM or TEM is used to determine the distribution of metal cations and anions in the samples. Also, X-ray photoelectron spectroscopy (XPS) is employed to analyse the elemental composition and oxidation states of the components.
2.4. Synthetic efforts to vary composition of LDHs
For the functional design of LDHs, the main component (M2+) and the second component (M3+) are selected to satisfy the required function. In addition to the selection of the M2+/M3+ combination, the M3+ content (or (M3+/(M2+ + M3+) or x) is also an important parameter to determine the properties of the resulting LDHs. However, there are several requirements in the selection of M2+ and M3+ from the preparation viewpoints including the solubility and stability of the available starting materials and solubility and stability of their hydroxides. Taking advantage of the facile synthesis of LDHs from solution and in the solid-state, as mentioned above, various synthetic methods and conditions have been examined to prepare LDHs with desired compositions.48–53,74,80,81,106 Hereafter, the reported examples of LDHs with varying (M3+/(M2+ + M3+)) are summarised. To investigate the effects of x on the properties of LDHs, the preparation of LDHs with varying x is examined. LDHs with x of 0.20–0.33 have commonly been reported as pure LDH phase.49,51 Even when x in the products was less than 0.20, the products were found to be a mixture of LDHs and impurity phases (M2+(OH)2) in many cases including MgAl-,118,119 MgGa-,120,121 NiAl-,122,123 CoFe-,124,125 CaAl-,126 and CaFe-LDHs.127 The limitation of x was claimed to be caused by the large distance between the adjacent interlayer anion in the interlayer space and the difference in the ionic radii of the divalent and trivalent metal cations in the brucite-like sheet.48–52 However, the reasons for this limitation are not clearly understood. Therefore, the preparation of LDHs with varying x is still worth investigating.In the case of co-precipitation using the titration method, the effect of the final pH of the suspension on x in the product was examined.128 To control x in MgAl-LDHs intercalated with carbonate, the acidic solution containing Mg(NO3)2 and Al(NO3)3 with varying M3+/(M2+ + M3+) ratios (x′) from 0.05 to 0.50 were used as starting solutions. An aqueous solution of NaOH containing NaHCO3 was slowly added to the starting solution until the pH reached 9.5–11. Then, the resulting suspension was aged and dried for the crystallisation of LDHs. It was shown that x in the products was larger than x′ (in the starting solution) when the final pH of the suspension was 9.5, indicating that a portion of magnesium in the starting solution did not precipitate at this pH. It was reported that the mechanism for the formation of MgAl-LDH by the titration method involves aluminium hydroxide precipitation in the pH range of 3.4–7.1, followed by magnesium precipitation (and to be incorporated in the pre-formed aluminium hydroxide) at pH above 8.5.89,91 To enhance the precipitation of magnesium to achieve x in the products closer to x′, titration was carried out to reach a pH in the range of 10–11. Although the pH was found to affect x in the products, the products were mixtures of LDHs and impurity phases when x in the product was 0.05 and 0.07.128 It should be noted here that x in the product determined by ICP is not x of the LDH phase when the product is a mixture and it is difficult to determine the composition of the LDH phase in the mixture.
Miyata reported the preparation of MgAl-LDHs using a starting solution of metal chlorides at x′ of 0.10–0.60 by co-precipitation at a constant pH.118 The reaction was conducted at 40 °C with a constant pH of 10. The x in products was determined by chelatometric titration after dissolution with dilute HCl in the range of 0.25–0.33. Phase separation to LDHs and impurity phases such as MgAl-LDH and boehmite was often observed when x = 0.38–0.60 in the product, MgAl-LDH and hydromagnesite when x = 0.16–0.20, and MgAl-LDH, hydromagnesite and magnesium hydroxide when x = 0.10.118 The constant pH method has been widely used to prepare LDHs such as MgFe-, NiAl-, NiFe-, ZnAl-, CuAl-, CoFe-, and CaAl-LDHs with varying x. However, single-phase LDHs were available for x in 0.20–0.33, and phase separation into LDH and impurity phase was observed when x < 0.20 or x > 0.30 in many cases.
Taking advantage of the size matching between Mg2+ (0.072 nm) and Ga3+ (0.062 nm),129 the possibility of the extension of x was investigated in MgGa-LDHs (Table 1).120,121 MgGa-LDHs with x of 0.12–0.33 in the product were obtained by co-precipitation at a constant pH in the range of 11–12, using acidic solutions of Mg(NO3)2 and Ga(NO3)3 and basic solutions of NaOH and Na2CO3. The product with x of 0.07 was found to consist of a mixture of LDH and magnesium hydroxide. It was observed that MgGa-LDH was prepared without phase separation at a smaller x compared to MgAl-LDH, as reported by Miyata.118 More recently, the preparation of MgGa-LDHs with iodide (MgGa-I-LDHs) with x of 0.06–0.24 was examined by co-precipitation from aqueous solutions of sodium iodide, magnesium nitrate and gallium nitrate, using the constant pH method, where the bulky I− ion was thought to play a role in suppressing their collapse when x was 0.06.130 The relationship between the lattice parameter a and x in the reported MgGa-LDHs is summarised in Fig. 6. There was a linear relationship between a and x, confirming the successful quantitative incorporation of Ga.130–133
| M3+ | M2+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.069 | 0.072 | 0.073 | 0.074 | 0.075 | 0.078 | 0.083 | 0.1 | ||
| Ni | Mg | Cu | Zn | Co | Fe | Mn | Ca | ||
| 0.054 | Al | 22 | 26 | 27 | 28 | 28 | 31 | 36 | 47 |
| 0.055 | Co | 21 | 24 | 25 | 26 | 27 | 30 | 34 | 46 |
| 0.062 | Cr | 11 | 15 | 16 | 17 | 17 | 21 | 26 | 39 |
| 0.062 | Ga | 10 | 14 | 15 | 16 | 17 | 21 | 25 | 38 |
| 0.065 | Fe | 6 | 10 | 12 | 13 | 13 | 17 | 22 | 36 |
| 0.08 | In | –16 | –11 | –10 | –8 | –7 | –3 | 4 | 20 |
 | ||
| Fig. 6 Relationships between the lattice parameter a and composition x in the reported MgGa-LDHs. Diamonds are for iodide-type MgGa-LDHs130 and circles are for the carbonate-type MgGa-LDHs.131–134 | ||
Dioctyl sulfosuccinate was used for the preparation of NiFe-LDHs with x of 0.05–0.25.134 NiFe-LDHs with a wide range of x were expected because of the similar ionic size of Ni2+ (0.069 nm) and Fe3+ (0.064 nm), while phase separation has been reported. By using dioctyl sulfosuccinate as the interlayer anion, NiFe-LDHs with x < 0.20 were obtained by co-precipitation at a constant pH. The interlayer dioctyl sulfosuccinate was replaced with the carbonate anion by a common ion exchange method using an aqueous solution of sodium carbonate to obtain NiFe-LDH carbonates. The basal spacings of the NiFe-LDH carbonates varied depending on x, as shown in Fig. 7.134–143 NiFe-LDHs with x of 0.05–0.25 were obtained by co-precipitation from an aqueous solution of nickel nitrate and iron nitrate containing glycerol.144
 | ||
| Fig. 7 (A) XRD patterns of NiFe–CO3-LDH/x, where x is 0.04, 0.11, 0.16, and 0.25. (B) Relationship between the basal spacing and x. Diamonds are values for NiFe–CO3-LDH/x,134 closed circles are values reported for NiFe–CO3-LDHs,135–140 and open circles are values reported for MgAl–CO3-LDHs,118,141 MgGa–CO3-LDHs,133 MgFe–CO3-LDHs,142 and NiAl-CO3-LDHs.143 | ||
Co-precipitation during the hydrolysis of urea and HMT has also been employed to prepare LDHs with varying x. Most of the reported studies focused on the common range of x (0.20–0.33). Carbonate-type NiAl-LDHs with x of 0.22–0.33 and NiFe-LDHs with x of 0.21–0.34 were prepared by the hydrolysis of urea or HMT. The synthesis for NiAl-LDHs was conducted at 180 °C for 72 h in air, while for NiFe-LDH, it was conducted at 100 °C for 48 h.142,145 NiFe-LDH and CoAl-LDH with varying x were prepared using an aqueous solution of metal salts with x′ < 0.20 by the hydrolysis of urea, while the composition of the products was not reported.142,146
The preparation from a slurry of metal oxides/hydroxides has been examined. The preparation of ZnAl-LDHs intercalated with the benzene sulfonate anion (BS) was investigated under hydrothermal conditions using ZnO and Al(OH)3 as the starting materials. The formation of single-phase LDH was observed at x′ of 0.4, while the starting materials were detected by XRD as impurity phases when x′ was 0.20, 0.25, and 0.33. The remaining amount of the starting materials decreased when the amount of BS increased, resulting in single-phase ZnAl-LDH with x = 0.33. Alternatively, the ZnO remained when starting slurries with the Zn:Al:BS ratios of 4:1:1 and 4:1:2 (x′ = 0.20) were employed.147 In the case of MgAl-LDH with deoxycholate, the basal spacing varied depending on the x in the products.105
The preparation of LDHs with varying x by mechanochemical reaction has been reported.106 Mg(OH)2 and Al(OH)3 were mixed with x′ = 0.14–0.25 in a planetary ball mill for 15 min, resulting in the formation of MgAl-LDH with x of 0.25.126 A part of Mg(OH)2 was unreacted when x′ was 0.14 and 0.20. Cl−-, NO3−-, and SO42−-intercalated MgAl-LDHs were prepared by using planetary ball mills to mix Mg(OH)2 and aluminium salts (AlCl3, Al(NO3)3 and Al2(SO4)3) for 3–15 min, followed by washing the product with deionised water.148 However, although the formation of an LDH with the targeted interlayer anion was confirmed, the Al3+/(Mg2+ + Al3+) ratio of the product was found to be smaller than the initial ratio of precursors. Accordingly, the importance of the precursor composition in the formation of LDHs by mechanochemical milling was studied. Mg(OH)2 and Al(OH)3 (at Al3+/(Mg2+ + Al3+) ratios of the precursor = 0.14–0.25) were mixed in a planetary ball mill for 15 min at room temperature. The results yield MgAl-LDH when the initial Al3+/(Mg2+ + Al) ratio was 0.25, whereas Mg(OH)2 was unreacted when the initial Al3+/(Mg2+ + Al3+) ratio was 0.14 and 0.20.
Redox reaction has been employed to vary x in CoFe-, CoNi-, and CoCo-LDHs by using metal hydroxides as the starting materials.124,149–151 Hydroxides of divalent transition metal cations (TM2+(OH)2) were prepared by precipitation using the hydrolysis of HMT under nitrogen gas bubbling. Some portions of divalent transition metal cations in the brucite were oxidised to trivalent form by oxidation using iodine (I2) or bromine (Br2). Meanwhile, the oxidising agent was reduced to iodide (I−) or bromide (Br−), and then intercalated into the interlayer space after the reaction. LDHs consisting of Co2+ and Fe2+ with varying Fe2+/(Co2+ + Fe2+) ratios of 0.17–0.33 were oxidised using iodine for 168 h under nitrogen gas protection, resulting in the formation of Co2+Fe3+-LDHs intercalated with iodide for the sample with an Fe3+/(Co2+ + Fe3+) ratio of 0.33. However, the phase separation to Co2+Fe3+-LDH and Co2+ hydroxide was observed when the Fe3+/(Co2+ + Fe3+) ratios of the starting mixture were in the range of 0.17–0.20.124,151 Green rust is another example of an LDH obtained by the redox process with the general formula of [Fe2+1–xFe3+x(OH)2]x+·(x/n)[An−]x−·m[H2O] (An−: interlayer anions, H2O: interlayer water molecules).152 Because of the poor stability, the application of green rust was limited.153 However, green rust with remarkable stability was obtained from Fe3+ chloride and glycerol by the partial reduction and precipitation during the hydrothermal reaction.154 The remarkable stability was explained by the less defective particle surface and dense interlayer structure, which suppress the diffusion of oxygen to oxidize Fe2+ in the hydroxide sheet.
The preparation of LDHs with x < 0.20 or x > 0.30 has been attempted thus far, as summarised in Table 2.
| M2+ | M3+ | Interlayer anion | x′ | x | By-product | Preparation | Objective | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Method | Temp. (°C) | Aging time | Atmosphere | ||||||||
| Note: x′ is the M3+/(M2+ + M3+) ratio of the initial solution of metal salts, and x is the M3+/(M2+ + M3+) ratio of the product. “n.r.” indicates “not reported”, and “n.d.” indicates “not detected”. | |||||||||||
| Mg | Al | Cl− and CO32− | 0.29 | 0.29 | n.d. | 2.2.1 (a-ii) (pH = 10) | 60 | 24 h | Air | Nitrate ion exchanger | 155 |
| CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 25 °C) | 75 | Air | Precursor of metal oxide for perchlorate adsorption | 156 | |||
| Cl− and CO32− | 0.20–0.33 | 0.20–0.33 | n.d. | 2.2.1 (a-ii) (pH = 10) | 70 | 24 h | N2 for Cl-type | Adsorption of norfloxacin | 157 | ||
| CO32− | 0.02–0.50 | 0.33–0.50 | n.d. | 2.2.1 (a-i) (pH = 9.5, 10, 10.5, 11) | Air | Fundamental study | 88 | ||||
| CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-i) (pH = 10.5) | 30 | 1 h | Air | Precursor of metal oxide for antimonate removal | 158 | ||
| CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 40 °C, pH = 10–11) | 65 | 18 h | Air | Adsorption of nitrate and nitrite ions | 159 | ||
| NO3− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 65 °C for 30 min) | 120 | 24 h | Air | Adsorption of chloride ions | 160 | ||
| NO3− | 0.20, 0.33 | 0.18, 0.33 | n.d. | 2.2.1 (a-ii) (at 30 °C, pH = 10.5) | N2 | Adsorption of arsenate and iron ions | 161 | ||||
| Terephthalate and benzoate | 0.18–0.43 | 0.18–0.50 | n.d. | 2.2.1 (a-ii) (pH = 10) | 55 | 18 h | Air | Fundamental study | 162 | ||
| Mg | Al | Cl− | 0.10–0.61 | 0.10–0.60 | Boehmite (x = 0.38–0.60) | 2.2.1 (a-ii) (at 40 °C, pH = 10) | Fundamental study | 118 | |||
| n.d. (x = 0.25–0.33) | |||||||||||
| Hydromagnesite (x = 0.16–0.20) | |||||||||||
| Hydromagnesite + Mg(OH)2 (x = 0.10) | |||||||||||
| CO32− | 0.17–0.33 | 0.17–0.33 | n.d. | 2.2.1 (a-ii) | Air | Catalyst for aromatic nitrile hydrolysis | 131 | ||||
| OH− and Cl− | 0.14–0.33 | n.r. | Mg(OH)2 (x′ = 0.14, 0.20, 0.25 and 0.33) | 2.2.2 | 110 | 5–10 days | Air | Fundamental study | 163 | ||
| CO32− | 0.14 and 0.20 | n.r. | Mg(OH)2 + MgO (x′ = 0.14 and 0.20) | 2.2.2 | 110 | 5–10 days | Air | ||||
| CO32− | n.d. (x = 0.25) | 2.2.3 | Milling for 15 min | Fundamental study | 119 | ||||||
| 0.14–0.25 | 0.14–0.25 | Mg(OH)2 (x = 0.14–0.20) | |||||||||
| Mg | Ga | CO32− | 0.07–0.33 | 0.07–0.33 | n.d. (x = 0.12–0.33) | 2.2.1 (a-ii) (at 40 °C, pH = 11–12) | N2 | Fundamental study | 120 and 121 | ||
| Mg(OH)2 (x = 0.07) | |||||||||||
| NO3− and CO32− | 0.18 | 0.18 | n.d. | 2.2.1 (a-ii) (pH = 9.5) | 25 | 18 h | Air | Fundamental study | 164 | ||
| CO32− | 0.14–0.25 | 0.12–0.25 | n.d. | 2.2.1 (a-ii) (pH = 9–10) | N2 | Catalyst for aromatic nitrile hydrolysis | 131 | ||||
| Mg | Fe | Cl− and CO32− | 0.16 | 0.16 | n.d. | 2.2.1 (a-ii) (pH = 10) | 25 | 24 h | Air | Nitrate ion exchanger | 155 |
| CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 25 °C) | 75 | Air | Precursor of metal oxide for perchlorate adsorption | 156 | |||
| CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (pH = 10) | 80 | 24 h in oven | Air | Adsorption of copper, cobalt, and cadmium ions | 165 | ||
| NO3− | 0.17–0.33 | n.r. | n.d. | 2.2.1 (a-i) (at 25 °C for 30 min) | 110 | 10 h | Air | Adsorption of fluoride and arsenate ions | 143 | ||
| Cl− | 0.20–0.33 | n.r. | n.d. (x′ = 0.33; reaction at 25–100 °C) | 2.2.1 (a-ii) (pH = 10) | 25–150 | 48 h | N2 | Fundamental study | 166 | ||
| n.d. (x′ = 0.25; reaction at 25–125 °C) | |||||||||||
| n.d. (x′ = 0.20; reaction at 25–150 °C) | |||||||||||
| Ni | Al | CO32− | 0.17–0.33 | 0.17–0.33 | n.d. | 2.2.1 (a-i) (pH = 10.5) | 40 | 1 h | Air | Catalyst for oxidation of ethylbenzene | 65 |
| CO32− | 0.20–0.33 | 0.22–0.33 | n.d. (x = 0.33) | 2.2.1 (b) | 180 | 72 h | Air | Pseudocapacitor | 145 | ||
| NO3− | 0.10–0.33 | 0.10–0.33 | Ni(OH)2 (x = 0.10, 0.17) | 2.2.1 (a-ii) (pH = 10) | 75–80 | 16 h | N2 | Characterisation of electrochemical behavior | 122 | ||
| CO32− | 0.09–0.33 | 0.09–0.33 | n.d. (x = 0.17–0.33) | 2.2.1 (a-i) (at 25 °C, pH = 11–12) | Air | Precursor of metal oxide using as catalyst for reforming of methane | 123 | ||||
| Ni(OH)2 (x = 0.09, 0.11) | |||||||||||
| Cl− | 0.09–0.33 | 0.09–0.33 | n.d. | 2.2.1 (a-i) | 100 | 20 h | Air | Fundamental study | 135 | ||
| Ni | Fe | Cl− and CO32− | 0.21 | 0.21 | n.d. | 2.2.1 (a-ii) (pH = 10) | 120 | 24 h | Air | Nitrate ion exchanger | 155 |
| Cl− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (b) | 6 h under reflux condition | Air | Electrocatalyst for water splitting | 66 | |||
| CO32− | 0.09–0.17 | n.r. | n.d. | 2.2.1 (b) | 150 | 48 h | Air | Electrocatalyst for oxygen evolution | 115 | ||
| CO32− | 0.20–0.33 | 0.21–0.34 | n.d. | 2.2.1 (b) (at 25 °C for 24 h) | 100 | 48 h | Air | Electrochemical glucose sensing | 137 | ||
| Zn | Al | CO32− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 25 °C) | 75 | Air | Precursor of metal oxide for perchlorate adsorption | 156 | |
| NO3− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 65 °C for 30 min) | 120 | 24 h | Air | Adsorption of chloride ion | 131 | ||
| Benzene sulfonate | 0.20–0.40 | n.r. | n.d. (x′ = 0.40) | 2.2.2 | 150 | 24 h | Air | Fundamental study | 147 | ||
| ZnO (x′ = 0.20, 0.25 and 0.33) | |||||||||||
| Cu | Al | CO32− | 0.20–0.67 | n.r. | Malachite | 2.2.1 (a-ii) (at 40 °C) | 40 | 15 min | Air | Fundamental study | 167 |
| Co | Al | CO32− | 0.14–0.80 | n.r. | n.d. (x′ = 0.33) | 2.2.1 (b) (in methanol) | 150 | 12 h | Air | Precursor of metal oxide using as catalyst for 4-nitrophenol reduction | 168 |
| Co | Fe | I− | 0.17–0.33 | 0.33 | n.d. (x′ = 0.33) | 2.2.2. | 25 | 168 h | N2 | Fundamental study | 124 and 125 |
| Co(OH)2 (x′ = 0.20–0.25) | |||||||||||
| Cl− and CO32− | 0.26 | 0.26 | n.d. | 2.2.1 (a-ii) (pH = 10) | 25 | 24 h | Air | Nitrate ion exchanger | 155 | ||
| Ca | Al | Cl− | 0.14–0.33 | 0.20–0.33 | n.d. (x′ = 0.20–0.33) | 2.2.1 (a-i) | 25 | 1 h | Air | Removal of copper, nickel, zinc, chromium, and phosphate ions | 126 |
| Ca(OH)2 + Ca(CO)3 (x′ = 0.14, 0.17) | |||||||||||
| NO3− | 0.20–0.33 | n.r. | n.d. | 2.2.1 (a-ii) (at 65 °C for 30 min) | 120 | 24 h | Air | Adsorption of chloride ion | 160 | ||
| Ca | Fe | Cl− | 0.14–0.33 | 0.33 | n.d. (x′ = 0.33) | 2.2.1 (a-i) (pH = 13) | 25 | N2 | Fundamental study | 127 | |
| Ca(OH)2 (x′ = 0.14–0.25) | |||||||||||
2.5. Characteristics and application of LDHs
The surface charge of drug delivery materials is a factor affecting their cellular uptake given that they should interact with negatively charged cellular membranes.186 Controlling the x of LDH resulted in varying charges of the LDH surface to modify their cellular interactions.187–189
2.6. Summary and perspectives
As mentioned for the incorporation of a useful isotope, Co-57, into the LDH framework for diagnostic applications,17 multiple (more than three) components of layered hydroxides have been prepared for application as catalysts including (photo)electrocatalysts and adsorbents. A third component has been incorporated by the post-synthetic treatment of pre-synthesised LDHs and co-precipitation. Single atomic Ru, Rh and Au were reported to be anchored on sheets of ZnCr-, NiFe-, CoFe- and FeCoNi-LDHs,193–197 while some ions were reported to be located in the brucite-like sheets such as Fe2+ in NiFe-LDH198,199 and Fe3+ in MgAl-LDH.200 The incorporation of Bi in ZnAl-LDH and V in NiFe-LDH was reported, where the important roles played vacancies were pointed out.201,202The incorporation of La was reported to affect the adsorption of metal oxo-anions such as arsenate and tungstate onto CuMgFe- and MgFe-LDHs.203,204 The incorporation of La3+ in brucite-like sheets by isomorphous substitution was not favoured. The size of the octahedra in the LDH sheets is determined by the size of the divalent cations, such as Mg2+ (ionic radius of 0.072 nm). The Fe3+ ions have an even smaller ionic radius (0.065 nm) and their preferentially adopted coordination number is 6. Thus, because of the larger ionic radius of La3+ (0.136 nm) with higher coordination numbers ranging from 7 to 10, the isomorphous substitution of Fe3+ with La3+ in MgFe-LDH is not favoured. The formation of carbonate and oxy-hydroxide phases of La3+ on the surface of the LDH sheets was seen in the attempt to incorporate La3+ in MgFe-LDH. Recently, it was reported that the incorporation of La3+ in the brucite-like sheet was possible by optimising the synthetic parameters using an ammonia alkaline solution for the preparation.205 Thermally activated purple-to-blue luminescence was reported for CoMgAl-LDHs, which were prepared by co-precipitation.206 Thus, the incorporation of a third (or the fourth) component into LDHs and preparation of layered triple (quadruple) hydroxides are a direction for materials design. In this direction, synthetic efforts are still necessary to achieve the quantitative introduction of multiple components into the final products. Appropriate characterisation to discuss the location of the incorporated elements is another challenge, which needs to be addressed.
To satisfy the requirements for the above-mentioned versatile applications (anion exchangers, catalysts, polymer additives, and drug/gene carriers), as well as to find novel applications, the hybridisation of LDHs with various functional particles has been reported to design materials with modified, improved, and multiple functions.207 In the extended application by hybridisation, the precise design of the composition of LDHs has important roles, and consequently their detailed characterisation will be more challenging.
3. Layered alkali silicates
A variety of crystalline and amorphous silicas exist, which are composed of an [SiO4]4− tetrahedron with varying connections (Fig. 8), where the elemental substitution of Si with heteroelements is possible. As a known example in nature, the substitution of Si in quartz with iron is the origin of the purple-colour of amethyst (Fig. 1). Silicate glasses doped with rare earth (for example Sm2+ and Eu3+) and transition metal ions (for example Mn3+ and Cu2+) exhibit useful optical (including luminescent) properties, showing possible application in phosphors (including lasers), optical recording, sensors, etc.208,209 | ||
| Fig. 8 Classification of crystalline and amorphous silica/silicates. | ||
The connection of [SiO4]4− tetrahedron leads to silica/silicate of varying three-dimensional structures. Silica/silicate-based nanoporous materials (silica gels, mesoporous silicas and zeolites) with varying pore sizes have been investigated for adsorption/separation and catalysis. To impart catalytic functions and modify the surface properties for adsorption, the introduction of heteroelements such as Al3+, B3+, Ga3+, Fe3+, Ti4+, V4+, Sn4+, and Zr4+ into the frameworks of silicas has been conducted.210 For example, Ti-containing mesoporous silicas and zeolites have been prepared by the isomorphous substitution of Si4+ with Ti4+ during the preparation of silicas/silicates and used as (photo)catalysts. The incorporated Ti exists as isolated tetrahedrally coordinated species in the silica/silicate frameworks at a low Ti content (the upper limit of its framework incorporation was Ti/Si > 30/1).11 Metal oxide may segregate out of their frameworks when the loading of the heteroelement increases.
Phyllosilicates are abundant in nature, which are commonly found as aluminium silicates and magnesium silicates, where gibbsite and brucite-like hydroxide sheets play a key role in directing layered structures (these materials will be introduced in Section 4 of this review). Alternatively, some layered silicic acids and their alkaline salts are composed of [SiO4]4− tetrahedrons.43,211,212 These layered silicic acids/silicates with varying structures (varying connection of [SiO4]4− tetrahedron resulting in varying layer thicknesses) are available and the notable examples are introduced in Table 3.213–217 Some of them are found in nature, for example, kenyaite (Na2Si20O41·nH2O) and magadiite (Na2Si14O29·nH2O) were found at Lake Magadi, Kenya in 1967.218 The layered alkali silicates introduced in Table 3 have been synthesised by hydrothermal reactions in the laboratory for different applications.219–222
| Layered silicatea |
|
Thickness of layer/nm | Ideal cation exchange capacity (meq g−1) | Ref. |
|---|---|---|---|---|
| a Water molecules are omitted for clarity. | ||||
| Kenyaite Na16[Si160O320(OH)16]·64H2O |

|
1.6 | 1.4 | 213 |
| Magadiite Na2[Si14O28(OH)2]·8H2O |

|
1.1 | 2.0 | 214 |
| Octosilicate Na8[Si32O64(OH)8]·32H2O |
|
0.74 | 2.8 | 215 |
| Kanemite NaH[Si2O5]·3H2O |

|
0.49 | 4.7 | 216 |
| HUS-1 Si10O24H6·2[(CH3)4N] |

|
0.90 | 2.4 | 217 |

The isomorphous substitution of Si4+ with heteroelements may lead to novel functional layered silicates. Here, the synthetic methods for the incorporation of heteroelements, examples of the heteroelements and their amount, characterisation, and the expected (achieved) functions reported thus far are introduced. The isomorphous substitution of Si4+ in layered alkali silicates with heteroelements (Al3+, Ti4+, etc.) has been reported,212 as summarised in Table 4.223–248 One of the motivations for substitution is the application of heteroelement-containing layered alkali silicates as the precursors of zeolites and mesoporous silicas as catalysts and adsorbents with designed material performances connected with the incorporated heteroelements.232–238 Due to the similarity in the local structure of the [SiO4]4− network, some layered alkali silicates have been converted to zeolites topochemically.249–251 It is worth noting that some zeolites (framework types: NSI, CDO, RWR, RRO, etc.) are available only by topotactic conversion of layered silicates such as Nu-6(1), PLS-1, octosilicate, and RUB-39.252–255
| Layered silicate | Composition | HE | Effective ionic radiia/nm | HE source | HE/Si atomic ratio (initial) | HE/Si atomic ratio (products) | Preparationb | Characterisation | Objective | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a The ionic radii of tetrahedrally coordinated heteroelements by the isomorphous substitution of Si4+ (0.026 nm) in layered alkali silicates, according to Shannon et al.129
b The type of synthetic method and reaction time are indicated, where hydrothermal reaction and solid-state reaction are abbreviated as HTR and SSR, respectively. Value in () indicates reaction temperature.
|
||||||||||||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al2O3 | — | 1/17.1–1/53.4 | HTR (130 °C) and 120 h | 27Al MAS NMR | Characterisation of acid sites | 223 | ||
| Magadiite | Na2Si14O29 | Ga3+ | 0.047 | Ga2(SO4)3·13H2O | — | 1/21.3–1/23.6 | HTR (130 °C) and 120 h | FT-IR spectra of adsorbed pyridine | Characterisation of acid sites | 223 | ||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15–1/60 | 1/15–1/69 | HTR (150 °C) and 12 or 24 h | 27Al and 29Si MAS NMR, and FT-IR spectra of adsorbed CO | Characterisation of acid sites | 224 | ||
| Magadiite | Na2Si14O29 | Sn4+ | 0.055 | SnCl4·5H2O | 1/67–1/143 | 1/51–1/108 | HTR (150 °C) and 48 h | 29Si MAS NMR and H2-TPR | Characterisation of acid sites | 225 | ||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | AlOOH | 1/15–1/40 | 1/13.9–1/42.5 | HTR (150 °C) and 72 h | 27Al and 29Si MAS NMR | Characterisation of acid sites | 226 | ||
| Al2(SO4)3 | 1/15–1/40 | 1/22.4–1/64.2 | ||||||||||
| Al[OCH(CH3)2]3 | 1/15–1/40 | 1/11.8–1/32.8 | ||||||||||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/64 | — | HTR (150 °C) and 72 h | 29Si MAS NMR and NH3-TPD | Characterisation of acid sites | 227 | ||
| Co3+ | — | Co(NO3)2·6H2O | Catalytic butyraldehyde conversion | |||||||||
| Er3+ | — | Er(NO3)3·5H2O | ||||||||||
| Kenyaite | Na2Si20O41 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/64 | — | HTR (180 °C) and 96 h | 29Si MAS NMR and NH3-TPD | Characterisation of acid sites | 227 | ||
| Co3+ | — | Co(NO3)2·6H2O | Catalytic butyraldehyde conversion | |||||||||
| Er3+ | — | Er(NO3)3·5H2O | ||||||||||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15 | 1/9.25 | HTR (150 °C) and 66 h | 27Al MAS NMR and NH3-TPD | Characterisation of acid sites | 228 | ||
| Catalytic ethanol conversion | ||||||||||||
| Magadiite | Na2Si14O29 | V5+ | 0.0355 | VOSO4·4.2H2O | 1/33 | 1/80.91 | HTR (150 °C) and 66 h | UV-Vis absorption spectroscopy | Characterisation of acid sites | 228 | ||
| Catalytic ethanol conversion | ||||||||||||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15 | 1/28.3–1/29.4 | HTR (150 °C) and 66 h | 27Al and 29Si MAS NMR and NH3-TPD | Characterisation of acid sites | 229 | ||
| Catalytic ethanol conversion | ||||||||||||
| Magadiite | Na2Si14O29 | Al3+ and V5+ | 0.039 and 0.0355 | Al[OCH(CH3)2]3 | 1/15 | 1/42.0–1/54.7 | HTR (150 °C) and 66 h | 27Al and 29Si MAS NMR and NH3-TPD | Characterisation of acid sites | 229 | ||
| VOSO4·4.2H2O | 1/33 | 1/71.1–1/86.4 | Catalytic ethanol conversion | |||||||||
| Magadiite | Na2Si14O29 | Sn4+ | 0.055 | Na2SnO3, SnCl4·5H2O | 1/67–1/333 | — | HTR (150 °C) and 72 h | FT-IR and Raman and UV-Vis absorption spectroscopy | Characterisation of acid sites | 230 | ||
| Octosilicate | Na2Si8O17 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15–1/60 | 1/22–1/47 | HTR (100 °C) and 336 h | 27Al and 29Si MAS NMR | Characterisation of acid sites | 231 | ||
| Catalytic ethanol conversion | ||||||||||||
| Kanemite | NaHSi2O5 | Al3+ | 0.039 | Al(NO3)3·9H2O, NaAlO2 | 1/2.5–1/100 | 1/7.2–1/188 | SSR (700 °C) and 6 h | 27Al and 29Si MAS NMR | Precursor for mesoporous silica | 232 | ||
| Kanemite | NaHSi2O5 | Al3+ | 0.039 | NaAlO2 | 1/20–1/100 | 1/20–1/100 | SSR (700 °C) | 27Al MAS NMR | Precursor for mesoporous silica | 233 | ||
| Kanemite | NaHSi2O5 | Ga3+ | 0.047 | GaOOH | 1/100–1/200 | — | SSR (700 °C) | 71Ga MAS NMR | Precursor for mesoporous silica | 233 | ||
| Kanemite | NaHSi2O5 | Al3+ | 0.039 | NaAlO2 | 1/20–1/100 | — | SSR (800 °C) | 27Al MAS NMR | Precursor for mesoporous silica | 234 | ||
| Kanemite | NaHSi2O5 | Ti4+ | 0.042 | Ti[O(CH2)3CH3]4 | 1/100–1/500 | 1/96–1/435 | SSR (675 °C) and 3 h | UV-Vis absorption spectroscopy | Precursor for mesoporous silica | 235 | ||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al2O3 | 1/18.5 | — | HTR (130 °C) and 120 h | 27Al MAS NMR | Precursor for zeolite | 236 | ||
| Magadiite | Na2Si14O29 | Co2+ | 0.058 | Co(CH3COO)2·4H2O | 1/50 | — | HTR (150 °C) and 72 h | — | Precursor for zeolite | 237 | ||
| Magnetism | ||||||||||||
| Magadiite | Na2Si14O29 | Co2+ | 0.058 | Co(CH3COO)2·4H2O | 1/50 | — | HTR (150 °C) and 72 h | UV-Vis absorption and XPS spectroscopy | Precursor for zeolite | 238 | ||
| Photocatalysts for water splitting | ||||||||||||
| Octosilicate | Na2Si8O17 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15–1/60 | 1/22–1/42 | HTR (100 °C) and 336 h | 27Al and 29Si MAS NMR | Precursor for pillared materials | 239 | ||
| Kanemite | NaHSi2O5 | Ga3+ | 0.047 | Ga2(SO4)3·13H2O | 1/18.5–1/37 | 1/19.4–1/31.5 | SSR (700 °C) and 6 or 156 h | 71Ga and 29Si MAS NMR | Fundamental study | 240 | ||
| Kenyaite | Na2Si20O41 | B3+ | 0.011 | B2O3 | 1/0.83 | — | HTR (150 °C) and 168 h | 11B MAS NMR | Fundamental study | 241 | ||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al2O3 | 1/22.5–1/180 | — | HTR (100–200 °C) and 30–259 h | 27Al MAS NMR | Fundamental study | 242 | ||
| Kenyaite | Na2Si20O41 | B3+ | 0.011 | B2O3 | 1/0.83–1/1.5 | — | HTR (175 °C) and 48–168 h | 11B MAS NMR | Fundamental study | 242 | ||
| Magadiite | Na2Si14O29 | B3+ | 0.011 | B2O3 | 1/2.5 | — | HTR (125/150 °C) and 552/72 h | 11B, 23Na and 29Si MAS NMR | Fundamental study | 243 | ||
| Kenyaite | Na2Si20O41 | |||||||||||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/15 | 1/14.5 | HTR (150 °C) and 12 h | 27Al and 29Si MAS NMR | Fundamental study | 244 | ||
| Magadiite | Na2Si14O29 | Ti4+ | 0.042 | TiCl4 | 1/50–1/200 | — | HTR (150 °C) and 48 h | UV-Vis absorption and XAFS spectroscopy | Fundamental study | 245 | ||
| Octosilicate | Na2Si8O17 | Ti4+ | 0.042 | TiCl4 | 1/50–1/200 | 1/54–1/204 | HTR (100 °C) and 240 h | UV-Vis absorption and XAFS spectroscopy | Fundamental study | 245 | ||
| Octosilicate | Na2Si8O17 | Sn4+ | 0.055 | SnCl4·5H2O | 1/800–1/2000 | 1/893–1/3703 | HTR (100 °C) and 504–672 h | FT-IR spectroscopy and H2-TPR | Fundamental study | 246 | ||
| Magadiite | Na2Si14O29 | Al3+ | 0.039 | Al[OCH(CH3)2]3 | 1/120–1/480 | 1/112–1/420 | HTR (150 °C) and 72 h | 27Al MAS NMR | Adsorbent for Pb2+ and methylene blue | 247 | ||
| Octosilicate | Na2Si8O17 | Al3+ | 0.039 | Al2O3 | 1/40 | 1/22 | HTR (105 °C) and 216 h | 27Al and 29Si MAS NMR | Ion exchange with Cu2+ | 248 | ||
In addition to substitution in the silicate layer, defects as microchannels in the silicate layer are considered to be the key for some functions. Magadiite showed the selective adsorption of benzoic acid from acetonitrile solution, where its microchannel was thought to play a role in its selectvity.256 Layered silicic acid/alkali silicates are characterised by regularly arranged reactive silanol groups on the layer surface, which can be used for cation exchange and grafting to obtain intercalation compounds of controlled nanostructures.257–261 The chemical/thermal stabilities and reactivity of layered silicates/silicic acids vary depending on the structures (derived from the composition), and thus the development of novel layered alkali silicates has been reported.262 For example, layered silicates named Hiroshima University Silicate (HUS) series were synthesised by hydrothermal reactions as adsorbents and catalyst precursors.217,263,264
3.1. The amount of the heteroelement to be doped in the silicate layer and the structural transformation
The acidity generated by the isomorphous substitution of Si4+ with heteroelements of lower valency and similar sizes, such as Al3+, Ga3+, and B3+ has been investigated.223,224,226–229,231–234,236,239–244,247,248 Layered silicic acid/alkali silicates have weak Brønsted acid sites owing to their silanol groups on the layer surface.249 Al is the element conveniently used to substitute Si in the silicate framework, as seen in many crystalline silicates (zeolites) and amorphous silicas (including mesoporous silicas). To impart acidity, Al-containing magadiites have been prepared by the hydrothermal reaction starting from Si and Al sources (sodium metasilicate and aluminium tri(isopropoxide); initial Al/Si = 1/15–1/60), respectively.224 The FT-IR spectra of Al-containing magadiite after the adsorption of CO at 100 K indicated the presence of Brønsted acidity, which was comparable with that of acidic zeolites (H-mordenite zeolite). As the Al content increased (Al/Si = 1/15–1/30), the population of acidic sites also increased. However, some of the Al was in octahedral coordination (probably extra framework aluminium oxide or (oxy)hydroxide), suggesting that the upper limit of the isomorphous substitution of Si with Al was lower than Al/Si = 1/30.Ti-containing kanemite was synthesised from silicon tetraethoxide and titanium(IV) tetrabutoxide via the sol–gel process and subsequent crystallisation by calcination. It was possible to transform it into Ti-containing mesoporous silicas (designated as KSW-2).235 The UV-Vis absorption spectra of the Ti-containing kanemite showed absorption bands at 210–220 nm when the Si/Ti ratio was larger than 300, indicating that the tetrahedrally coordinated titanium oxide species existed in the framework of kanemite. As the Ti content increased (Ti/Si = 1/100–1/200), the absorption band became broader because of the co-existence of octahedrally coordinated titanium oxide species. The upper limit of the incorporation of tetrahedrally coordinated Ti in the framework was as low as Ti/Si = 1/250. Recently, Ti-containing octosilicates and magadiites were synthesised via hydrothermal reactions using TiCl4 as the Ti source in the starting mixture (silica gel and NaOH) for the preparation of magadiite and octosilicate.245 The UV-Vis absorption and X-ray absorption near edge structure (XANES) spectra suggested that tetrahedrally coordinated titanium oxide species existed in the silicate frameworks. The XANES spectra of the Ti-containing silicates are shown in Fig. 9. In the Ti K-edge XANES spectra of the Ti-containing magadiites (Ti/Si = 1/100 and 1/50), a weak pre-edge peak was observed (Fig. 9A), indicating that the titanium oxide species existed mainly in tetrahedral coordination in the framework of magadiite. Alternatively, that of the Ti-containing octosilicate (Ti/Si = 1/200) showed a weak but single pre-edge peak (Fig. 9B), indicating that some of the titanium oxide species were in tetrahedral coordination in the framework of octosilicate. As the content of Ti increased (Ti/Si = 1/100 and 1/50), the XANES spectra of the Ti-containing octosilicates (Fig. 9B) were observed to be similar to that of rutile and anatase, indicating the presence of an isolated tetrahedrally coordinated titanium oxide species in the framework of octosilicate together with extra framework octahedrally coordinated titanium oxide species. These results suggested that the upper limit of the framework incorporation of Ti was Ti/Si = 1/50 and 1/100 for magadiite and octosilicate, respectively. In addition, as the Si/Ti ratio increased (Ti/Si = 1/10 and 1/20 for magadiite and octosilicate; Fig. 10), the XRD patterns of the products showed no crystalline phase and morphologies different from the Ti-containing silicates and aggregated TiO2 particles were seen. The capacity of the incorporation of Ti into the silicate framework was higher for the silicates with thicker silicate layers (magadiite > octosilicate > kanemite, Table 3).235,245 The amount of tetrahedrally coordinated titanium oxide species can be increased for kenyaite. To further understand the relationship between the incorporated amount of heteroelements in the silicate frameworks and emerging functions such as (photo)catalysis and adsorption, synthetic methods and the conditions need to be developed to prepare materials with a higher heteroelement content.
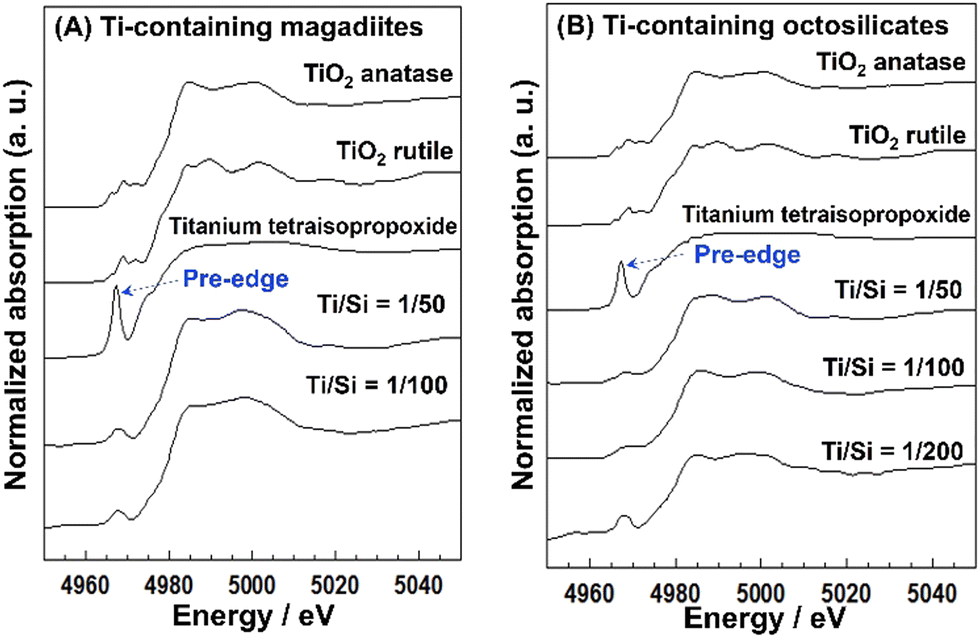 | ||
| Fig. 9 (A) XANES spectra of the Ti-containing magadiites and reference. (B) XANES spectra of the Ti-containing octosilicates and ref. 245. | ||
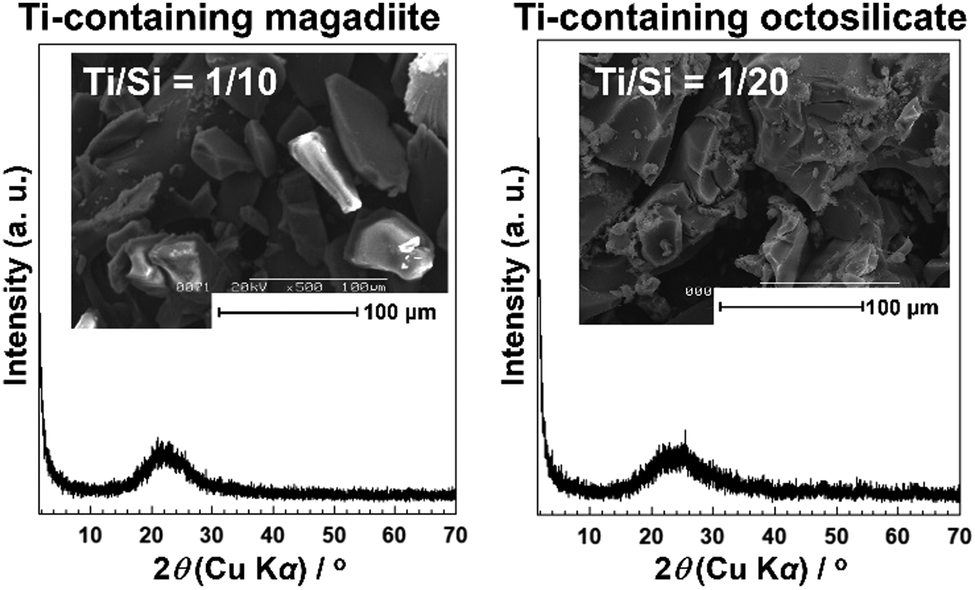 | ||
| Fig. 10 XRD pattern and SEM image of the Ti-containing magadiites and octosilicates (Ti/Si = 1/10 and 1/20).245 | ||
Layered silicic acid/alkali silicates have been used as precursors of zeolites and mesoporous silicas.249,250 In particular, some zeolites are only obtained by topotactic conversion through the interlayer condensation of layered silicic acid/alkali silicates (Fig. 11).251 The conversion of the Ti-containing layered silicic acid/alkali silicates into zeolites is worth investigating.
 | ||
| Fig. 11 Schematic of the topotactic conversion of magadiite into a silica zeolite (RWZ-1) during refluxing in N-methylformamide (NMF) and subsequent calcination. Reprinted from ref. 251 with permission from John Wiley & Sons. | ||
3.2. Variation in the heteroelements to be incorporated in the layered alkali silicates
The incorporation of other heteroelements besides Al3+ such as Co2+/Co3+, Er3+, Ti4+, Sn4+ and V5+ into the silicate framework has also been investigated given that these heteroelements are expected to play roles as isolated active sites for magnetic and catalytic properties.225,227–230,235,237,238,245,246 The ionic radius of tetrahedrally coordinated heteroelements by the isomorphous substitution of Si4+ (0.026 nm) in layered alkali silicates and the experimental conditions of the examples are summarised in Table 4.129 The effective ionic radius is one of the important factors determining the isomorphous substitution. The isomorphous substitution with the elements such as Ni2+ (0.055 nm), Fe2+/Fe3+ (0.063/0.049 nm), Cu2+ (0.057 nm) and Eu3+ (0.0947 nm) seems to be possible because of the size matching.3.3. Synthesis of heteroelement-containing layered alkali silicates
Most of the heteroelement-containing layered alkali silicates have been prepared via the hydrothermal reaction of the starting material of layered alkali silicates in the presence of molecular species (alkoxides and metal salts) as the source of heteroelements. The amount of heteroelement incorporated in the framework of layered alkali silicates is as low as a few wt% (oxide base). The type of heteroelement source plays an important role in the incorporation. Al-containing magadiite was prepared with different types of Al sources (aluminium oxyhydroxide, aluminium triisopropoxide and aluminium sulfate), and its acidic sites evaluated in the catalytic cracking of cumene.226 The XRD patterns of Al-containing magadiite with different types of Al sources are shown in Fig. 12. Although aluminium oxyhydroxide led to the formation of crystallised Al-containing magadiite (Al/Si = 1/15–1/40), the formation of mordenite impurities was observed using aluminium triisopropoxide at Al/Si = 1/40. With a higher Al/Si = 1/15, the XRD patterns of the products using aluminium triisopropoxide and aluminium sulfate showed no crystalline phase, suggesting that the anions affected the crystallisation. Thus, the amount of heteroelement in the heteroelement-containing layered alkali silicates is limited (detail in Section 4).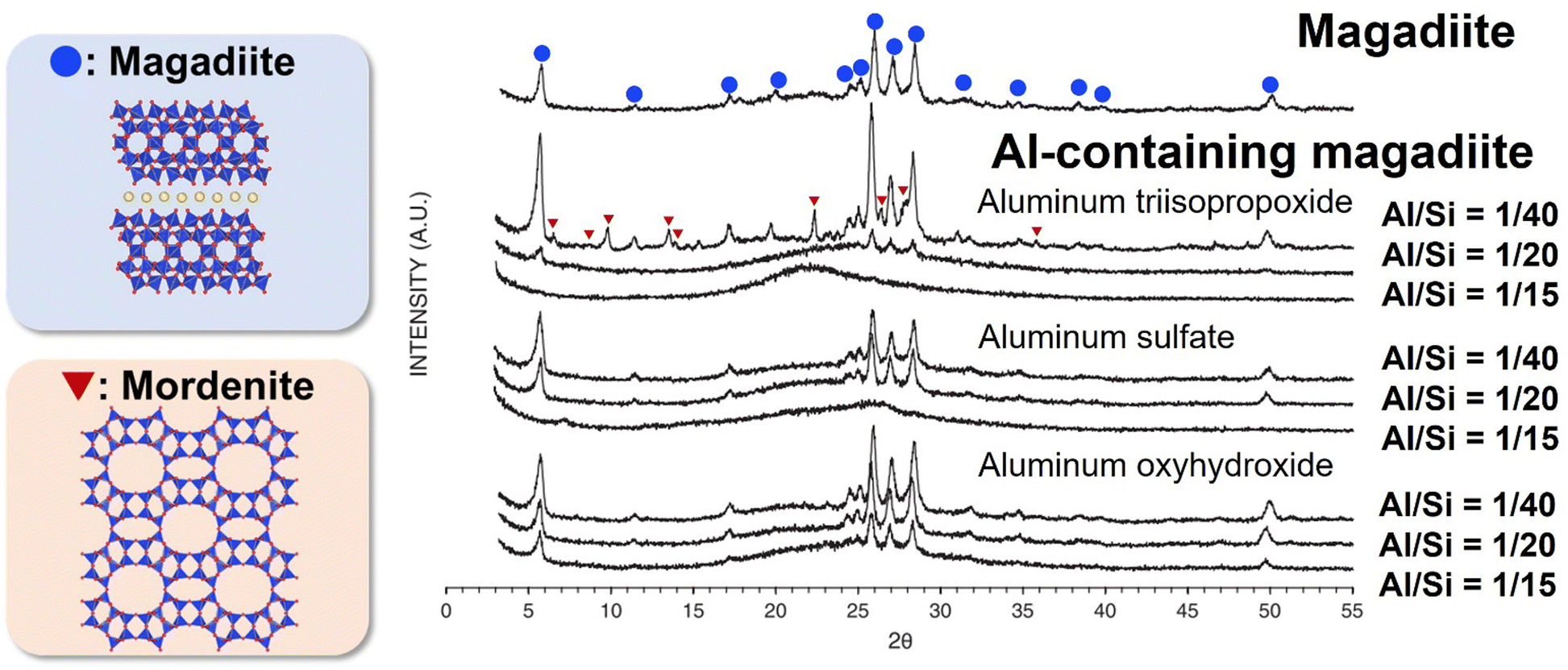 | ||
| Fig. 12 XRD patterns of magadiite and Al-containing magadiites using aluminum triisopropoxide, aluminum sulfate and aluminum oxyhydroxide (Al/Si = 1/40, 1/20 and 1/15) as the aluminium source.226 | ||
3.4. Characterisation of the states of the incorporated heteroelements
The states of the heteroelements in silica/silicate frameworks can be evaluated various spectroscopic techniques, including NMR, UV-Vis absorption, photoluminescence and X-ray absorption (XANES and EXAFS) spectroscopy.To gain further insight into the chemical state of the heteroelements in the silica/silicate framework, solid-state magic angle spinning (MAS) NMR spectroscopy is a key technique. As a typical example, the 27Al and 29Si MAS NMR spectra of Al-containing magadiite using aluminium oxyhydroxide are shown in Fig. 13.226 The coordination state of the Al sites in either tetrahedral or octahedral coordination is characterised by 27Al MAS NMR measurement, showing signals at ca. 55 ppm or 0 ppm, respectively. These samples (Al/Si = 1/15–1/40) showed only a signal at around 55 ppm (Fig. 13A), corresponding to the tetrahedral coordination, indicating the isomorphous substitution of Si with Al. In the 29Si MAS NMR spectrum of magadiite (pure silicate; Fig. 13B), four peaks were observed at ca. −100 ppm, which were attributed to Q3 (HOSi(OSi)3), and ca. −110, −112 and −114 ppm, corresponding to Q4 (Si(OSi)4). The 29Si MAS NMR spectrum of Al-containing magadiite (Al/Si = 1/40) was similar to that of magadiite, whereas the spectrum became broader with an increase in the Al content (Al/Si = 1/20), with the appearance of a broad peak at −107 ppm which was attributed to amorphous silica. The incorporation of Al in the silicate framework affects the chemical environment of Si, i.e., not only first coordination but also second or third coordination environment, resulting in low-field shifts (about 5 ppm: Q4(1Al) i.e., Si(OSi)3(OAl)) and the broadening of the signal in the 29Si MAS NMR spectrum.265,266 As the content of Al increased (Al/Si = 1/15), two broad peaks were observed at −112 and −102 ppm, corresponding to Q4(0Al) and Q4(1Al)/Q3(0Al), respectively, which suggested that most of this product existed as an amorphous phase.
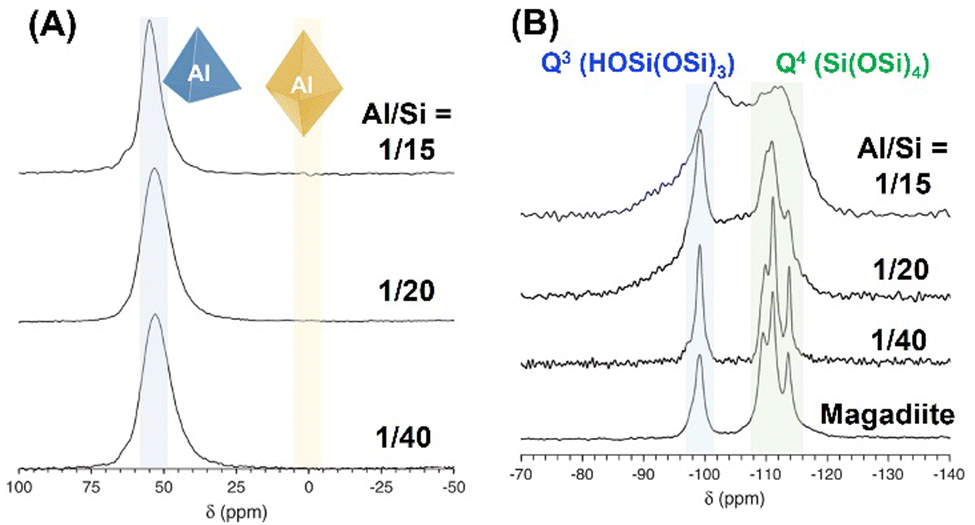 | ||
| Fig. 13 (A) 27Al and (B) 29Si MAS NMR spectra of Al-containing magadiite using aluminum oxyhydroxide.226 | ||
The incorporation of Ti into various silicas/silicates has been examined for the design of (photo)catalysts. Titanium silicalite-1 (TS-1), which is a representative titanosilicate zeolite with isomorphous substitution of Si4+ with Ti4+ in the MFI zeolite framework, exhibited selective oxidation of various hydrocarbons with H2O2.267,268 Regarding the states of the incorporated titanium oxide species in zeolites and mesoporous silica/silicate frameworks, UV-Vis absorption, photoluminescence and XAFS spectroscopy (Section 3.1) are powerful measurement tools to determine if the titanium oxide species exist as isolated tetrahedrally coordinated species in the silica/silicate framework.235,245,269–273 Tetrahedrally coordinated titanium oxide species in silicas/silicates showed absorption bands with the maximum at 220–280 nm due to the charge transfer excited state ([Ti4+–O2−] → [Ti3+–O−]*). In addition, these samples exhibited photoluminescence at around 450–550 nm upon excitation at 220–280 nm, which is ascribed to the charge transfer process.
3.5. Summary and perspectives
The incorporated heteroelements in layered silicates/silicic acids are exposed to the surface of silicate nanosheets, affecting their surface characteristics and associated functions, while their function has not been fully explored to date. Layered alkali silicates accommodate various guest species in their interlayer space for functional materials. Organic modification with cationic surfactants and silane coupling reagents has been done to design materials for applications in environment, energy and life science-related fields through molecular recognition.43,249,260–264,274–284 Recently, the site-specific organic modification of octosilicate led to the fabrication of a floating adsorbent for the collection of metal ions from water.283 Selective adsorption on layered alkali silicates (with defects in the silicate layer) and their silylated products has been reported.256,285–287 These material designs will be examined using heteroelement-doped layered silicates.Regarding synthetic aspects, the incorporation of heteroelements into the silicate frameworks is done during the preparation of layered silicates via the hydrothermal reaction, where phase separation of the heteroelements and the formation of different phases (such as zeolites) may occur depending on the composition. However, alternative synthesis methods are worth examining to extend the compositional variation. Heteroelement-containing silicate zeolites, such as zeolites with Ti4+, Al3+ and Fe3+, have been synthesised via mechanochemically assisted hydrothermal reactions using metal oxy(hydroxide) (e.g., TiO2 (anatase), γ-AlO(OH), α-Fe2O3, and α-FeOOH) and silica (fumed silica).288–291 The synthesis of magadiite, octosilicate, and kanemite via the solid-state reaction at 50–900 °C has been reported.292 In addition to the application of these reported procedures, the development of novel synthetic methods (including post-synthetic immobilisation293,294) is worth examining to extend the compositional variation.
4. 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 type phyllosilicates
:1 type phyllosilicates
Layered clay minerals, namely 2:1-type phyllosilicates (Fig. 14), are materials where isomorphous substitution plays a key role in their characters/functions. The useful adsorptive, ion exchange, swelling and catalytic functions of the clay minerals are known.295,296 Silicate minerals of various structures and compositions are widely distributed in nature and clay minerals are known to be formed by the hydrothermal alteration or weathering of these silicate minerals. Metal (Mg2+ and Al3+) hydroxide sheets are thought to play a role in directing the layered structure, given that layered silicic acids/silicates (given in Section 3) are less common from the aspects of mineral quantity in Earth's crust.
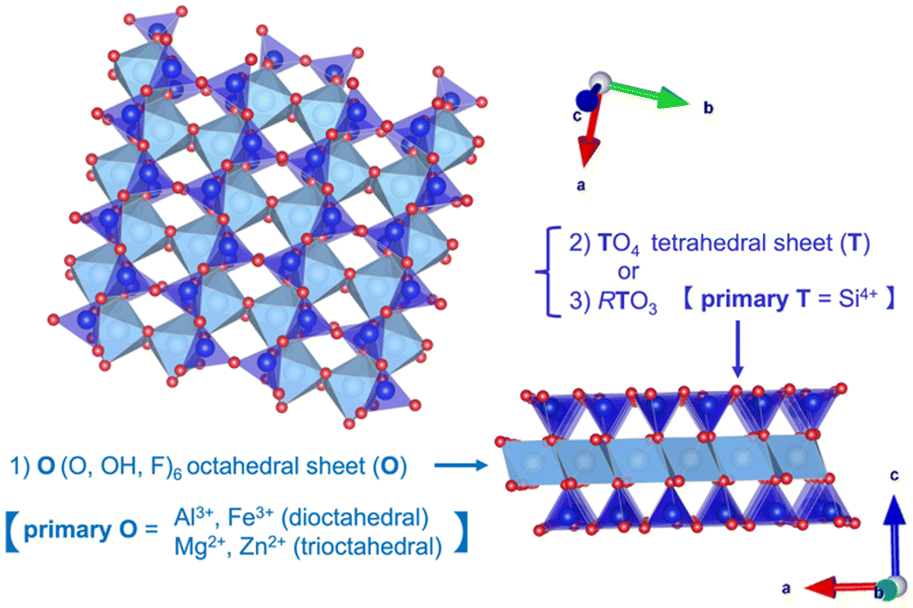 | ||
| Fig. 14 Structure of a 2:1-type phyllosilicate, two TO4 (the primary element of T = Si4+) tetrahedral sheets (T) on each side of an O (O, OH, F) octahedral sheet (O) occupied by octahedrally coordinated cations (e.g., Mg2+, Zn2+/Al3+, Fe3+, etc.), forming a T–O–T layer (prepared using VESTA software).44 | ||
4.1. Structural versatility: combination of an LDH structure with SiO4 tetrahedra and interlayer cations
The compositional variation of 2:1-type phyllosilicates by isomorphous substitution has led to versatile applications (Fig. 14). The layered clay minerals, which belong to the hydroxylated phyllosilicates, include T–O (1:1) and T–O–T (2:1) phyllosilicates.3Table 5 shows the classification of phyllosilicate clay minerals. They can be classified based on their sheet stacking (1:1 or 2:1), layer charge (x + y: or charge per unit cell of Si8O20(OH)4), and type of interlayer into eight major groups. Further subdivision into sub-groups and species is made based on the type of octahedral sheet (dioctahedral and trioctahedral, which will be explained later), chemical composition and the geometry of the superposition of individual layers and interlayers. The names used for groups, sub-groups, and mineral species are those approved by the AIPEA Nomenclature Committee or the IMA Commission on New Minerals and Mineral Names.
| Layer type | Group (x + y = charge per unit cella) | Sub-group | Speciesb |
|---|---|---|---|
|
a x + y refers to an Si8O20(OH)4 unit cell for smectite, vermiculite, mica, and brittle mica.
b Only few examples are given.
|
|||
| 1:1 |
Serpentine–kaolin | Serpentine | Chrysotile, antigorite, lizardite, amesite |
| (x + y ∼ 0) | Kaoline | Kaolinite, dickite, nacrite | |
| 2:1 |
Talc–pyrophyllite | Talc | Talc, willemseite |
| (x + y ∼ 0) | Pyrophyllite | Pyrophyllite | |
| Smectite | Saponite | Saponite, hectorite, sauconite | |
| (x + y ∼ 0.4–1.2) | Montmorillonite | Montmorillonite, beidellite, nontronite | |
| Vermiculite | Trioctahedral vermiculite | Trioctahedral vermiculite | |
| (x + y ∼ 1.2–1.8) | Dioctahedral vermiculite | Dioctahedral vermiculite | |
| Mica | Trioctahedral mica | Phlogopite, biotite, lepidolite | |
| (x + y ∼ 2.0) | Dioctahedral mica | Muscovite, paragonite | |
| Brittle mica | Trioctahedral brittle mica | Clintonite, anandite | |
| (x + y ∼ 4.0) | Dioctahedral brittle mica | Margarite | |
| Chlorite | Trioctahedral chlorite | Clinochlore, chamosite, nimite | |
| (x + y variable) | Dioctahedral chlorite | Donbassite | |
| Di, trioctahedral chlorite | Cookeite, sudoite | ||
| 2:1 inverted ribbons |
Sepiolite–palygorskite (x + y variable) | Sepiolite | Sepiolite, loughlinite |
| Palygorskite | Palygorskite | ||
Isomorphous substitution of the main components in the silicate layer as Si4+ and/or M2+/3+ with cations of similar size and lower valency gives a net negative charge. The quantity of the isomorphous substitution is expressed as x and y in the unit cell of (Si, Al)8O20(OH)4 (e.g., [Si8−xMtetrax]IV [Al4−yMoctay]VIO20(OH)4, where Mtetra and Mocta are heteroelements in the SiO4 tetrahedral sheet (T) and octahedral sheet (O), respectively. The compositional variation of 2:1-type phyllosilicates arises from three structural aspects, as follows: (1) Two types of octahedral sheets (O; dioctahedral and trioctahedral types composed of M3+ and M2+ hydroxides, respectively) are represented, as shown in Fig. 14, with a wide variety of octahedrally coordinated cations to replace M3+ and M2+, which influence their functions similar to LDHs (the structures and functions were described in Section 2). (2) Substitution of Si4+ with M3+ in the tetrahedral sheet (T) affords acidic character, as observed in zeolites, mesoporous silicas and other silicas in addition to the generation of negative charge. (3) Covalent attachment of organic groups (R) with the silicon atom in the SiO4 tetrahedra in T provides organically modified silicates with a type of self-assembled monolayer, which can form multilayers composed of alternatively stacked silicate layers and organic layers. When the silicate possesses a permanent net negative charge as a result of the isomorphous substitution, cations are interposed in the interlamellar region between the silicate layers. The amount and the location of the isomorphous substitution influence the surface characteristics, which are correlated with the colloidal, catalytic and adsorptive properties of the phyllosilicates. The charge per unit cell (x + y) is an important value in determining the characteristics of phyllosilicates. Among the phyllosilicates, smectites have been used most extensively in industrial applications as well as scientific research. Smectite is the name of a group of swellable 2:1-type phyllosilicates (0.4 ≤ x + y ≤ 1.2).3,297 Depending on the composition and the location of the isomorphous substitution, smectite is classified as summarised in Table 6. The interlayer cations (e.g., Na+ and Ca2+) are usually hydrated and are exchangeable.
| Sub-group | Species | T | O | Chemical formulaa | ||
|---|---|---|---|---|---|---|
| Main | Mtetra | Main | Mocta | |||
|
a An+: exchangeable cation.
|
||||||
| Dioctahedral smectites | Montmorillonite | Si4+ | — | Al3+ | Mg2+ | An+y/n[Si8]IV[Al4−yMgy]VIO20(OH)4 |
| Beidellite | Si4+ | Al3+ | Al3+ | — | An+x/n[Si8−xAlx]IV[Al4]VIO20(OH)4 | |
| Nontronite | Si4+ | Al3+ | Fe3+ | — | An+x/n[Si8−xAlx]IV[Fe4]VIO20(OH)4 | |
| Trioctahedral smectites | (Fluoro)hectorite | Si4+ | — | Mg2+ | Li+ | An+y/n[Si8]IV[Mg6−yLiy]VIO20(OH, F)4 |
| Saponite | Si4+ | Al3+ | Mg2+ | — | An+x/n[Si8−xAlx]IV[Mg6]VIO20(OH)4 | |
| Sauconite | Si4+ | Al3+ | Zn2+ | — | An+x/n[Si8−xAlx]IV[Zn6]VIO20(OH)4 | |
Swelling occurs when the hydration energy overcomes the attractive forces between the silicate layers and is sufficient to cause osmotic swelling in excess water.298 The spontaneous swelling of smectites in water leads to the formation of a stable thixotropic gel or suspension.45,46,298,299 Smectites occasionally form liquid crystalline phase phyllosilicates under specific conditions with careful preparation.300–302 Structural colour emerged when nanosheets were stacked with submicrometer periodicity in an aqueous suspension (Fig. 15).302 Gravity and an electric field led to different alignments of the nanosheets in poly(N-isopropylacrylamide) composite gel films. The combined two-layer gels exhibited three-dimensional deformations (e.g., jumping motion), which were triggered by temperature changes with controlled curvature (Fig. 16).303
 | ||
| Fig. 15 (A) Photographs of nanosheet colloids (average nanosheet diameter of 1.8 μm) with various nanosheet concentrations c. (B) Photographs and UV-Vis spectra of the nanosheet colloid with the average nanosheet diameter of (a) 0.14, (b) 0.16 (c) 0.98, and (d) 1.8 μm. The spectra were recorded at an observing angle of 10°.302 | ||
 | ||
| Fig. 16 Jumping motion of poly(N-isopropylacrylamide) composite gel films in water in response to thermal stimulus. Photographs and corresponding illustrations show the time course of the gel films after immersion in hot water. The heights from the ground surface to the red point on the gel are also shown. The red arrows indicate the direction of movement of each part. Reprinted with permission from ref. 303. Copyright 2022, the American Chemical Society. | ||
When the solvents in the suspension are evaporated on a flat plate, plate particles pile up with their ab plane parallel to the substrate to form an oriented film.304,305 A similar procedure in the presence of a water-soluble polymer afforded multi-functionalised inorganic–organic hybrid films with transparent gas barrier,306 thermal stability, water resistance, water-induced self-healing behaviour, and adhesion.307 The Langmuir–Blodgett308 and layer-by-layer deposition techniques (alternating adsorption of a cation and an anionic silicate layer) from exfoliated platelets were used to obtain inorganic–organic multilayer thin films.309–311
In addition to the swelling property and the associated film-forming ability from a colloidal suspension, isomorphous substitution is the origin of cation exchange ability and acidic character. The cation exchange capability was used to modify the properties of smectites for application as fillers in clay-polymer hybrids,312–317 rheology-controlling reagents,318,319 reaction media to control photochemical reactions,320–323 optics,324 selective adsorption,325–327 chiral discrimination,328,329 and catalysis.330
4.2. Bentonite and purified bentonite
Bentonite (coarse samples, a natural resource containing smectites as the main component) contains accessory minerals such as crystalline SiO2 (e.g., quartz, cristobalite), amorphous SiO2, feldspar, and goethite FeO(OH), as presented in Table 7.331,332 Depending on the application, purified bentonites, which are commercially available, are used.| T | O | |||||
|---|---|---|---|---|---|---|
| Si | Altetra | Alocta | Fe | Mg | ||
| a SAz-1: (Cheto, AZ), a magnesium rich montmorillonite. b ST (Stebno): an iron-rich beidellite from the Czech Republic, which contains about 21% of total iron bound in goethite. c SWa-1: a ferruginous smectite from The Clay Mineral Repository of the Clay Minerals Society. d JP: hydrothermal, aluminium-rich montmorillonite from the Kremnica mountains in central Slovakia. e JCSS3101: montmorillonite, a reference clay sample of the Clay Science Society of Japan. f JCSS3501: a synthetic saponite, a reference clay sample of the Clay Science Society of Japan. g NIMS: analysed by the National Institute for Materials Science, Japan. h NMNS: analysed by the National Museum of Nature and Science, Japan. | ||||||
| SAz-1a331 |
8.00 | 0.00 | 2.67 | 0.15 | 1.20 | |
| STb331 |
7.22 | 0.78 | 1.96 | 1.60 | 0.58 | |
| SWa-1c331 |
7.33 | 0.67 | 0.91 | 2.86 | 0.28 | |
| JPd331 |
7.71 | 0.29 | 3.00 | 0.38 | 0.63 | |
| JCSS3101 | NIMSg | 7.66 | 3.37 (Altetra+octa) | 0.21 | 0.64 | |
|
332
|
NMNSh | 7.56 | 3.47 (Altetra+octa) | 0.25 | 0.69 | |
| JCSS3501 | NIMS | 7.19 | 0.81 (Altetra+octa) | 0.00 | 5.99 | |
|
332
|
NMNS | 7.02 | 0.71 (Altetra+octa) | 0.00 | 6.26 | |
Purification is possible by the following dispersion-sedimentation method. Coarse samples were suspended in deionised water, treated several times with 1.0 M aqueous CaCl2, washed until free of chloride, and centrifuged. After the nominally <2 μm sample was collected, drying at 60 °C and subsequent grounding to <0.2 mm were performed.331 As a dissolution method, the alkaline fusion technique was employed, melting the sample with BO3 and Li2CO3, followed by dissolution in acidic HCl solution.332 The purification was confirmed by XRF and ICP.
Bentonite, purified bentonites (non-swelling impurities were removed from bentonite by dispersion-sedimentation method), and synthetic smectites are commercially available and some of them are authorised by academic societies (The Clay Minerals Society333 and The Clay Science Society of Japan334). However, their quantitative characterisation is difficult because of the fine grain-size of clay minerals and the association with accessory minerals. The compositional variation is seen even for samples from the same deposits.335 The Clay Science Society of Japan authorizes reference clay samples with their chemical formula/compositions.332 Some additional data are available, as shown in Table 7. JCSS3101 includes only a small amount of quartz as a result of careful purification. The associated minerals make it difficult to determine the accurate compositions of the smectite phase by bulk analysis. The location of isomorphous substitution is another difficulty given that the quantity is normally low and clay minerals are commonly poorly crystalline.
4.3. Site of isomorphous substitution in 2:1-type phyllosilicates, heteroelements and their amount
Available range of Al/Si ratio in T–O–T (2:1)-type phyllosilicate is discussed employing the “pyrophyllite” structure as the original structure, as represented by the following formula (eqn (1)).| [Si8]IV[Al4]VIO20(OH)4 | (1) |
| [Si6Al2]IV[Al4]VIO20(OH)4 | (2) |
| [Si8]IV[Al3.33Mg0.67]VIO20(OH)4 | (3) |
| [Si7.33Al0.67]IV[Mg6]VIO20(OH)4 | (4) |
Compared with other 2:1-type phyllosilicates, smectites have high interlayer accessibility for various inorganic and organic species. The cation exchange capacity (CEC) of smectites, which is directly correlated with the amount of isomorphous substitution, is usually expressed in the unit of milliequivalent (meq) of exchangeable cations per 100 g of smectite (clay). The CECs of smectites are empirical values ranging from 60 to 120 meq/100 g of clay. Several techniques have been used for determining the CEC using ammonium acetate,338 triethanolamine buffered barium chloride,339–341 calcium chloride,342 methylene blue,343,344 silver thiourea,345 and copper(II) ethylenediamine.346 It should be noted that some organic and organometallic cations may be adsorbed over the CEC by van der Waals interactions (intersalation).347,348 The CECs of smectites are governed by permanent charge and independent of the ionic strength electrolyte concentration and pH.
Smectites and vermiculites are distinguished at the layer charge of x + y = 1.2 per unit cell.3 The identification is commonly made by swelling in water and in glycerol; when saturated with Mg2+ ions, the basal spacing of vermiculite expands to approximately 1.5 nm in water and glycerol, whereas smectites expand to 1.7–1.8 nm in glycerol. The synthesis of smectites with an extremely large layer charge (e.g., x + y = 2.66) resulted in the formation of a non-expandable phase even in the presence of Na+ ions.349 The question arises if any division can be made between smectites (x + y = 0.4–1.2) with high layer charge and vermiculite (x + y = 1.2–1.8) with a low layer charge? Méring and Perdo350 suggested that the order or disorder of the isomorphous substitutions may be another feature of these minerals.
Here, the amount of isomorphous substitution (x + y value or CEC) will be discussed for both sites (T and O) in 2:1-type phyllosilicate [T8]IV[O4]VIO20(OH)4 and [T8]IV[O6]VIO20(OH)4, where the isomorphous substitution occurs as [Si8−xMtetrax]IV[Al4−yMoctay]VIO20(OH)4 and [Si8−xMtetrax]IV[Mg6−yMoctay]VIO20(OH)4, respectively (Table 8).
:1-type phyllosilicates and synthesis conditions
| Formula of anionic form | Heteroelement | Concentration x′, y′ (nominal) x, y (found) | Preparation | Specific characterisation | Objective | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| Mtetra | Mocta | Methoda | Temp. (°C) | |||||
| a RE: reflux, HTR: hydrothermal reactions, SSR: solid-state reactions. b The chemical formula of Na0.05Mg6.52Li1.12Si7.93O21.91F2.09. | ||||||||
| [Si8−xMtetrax]IV[Al4−yMoctay]VIO20(OH)4 | Al3+ | (y = 0) | 0.34 ≤ x ≤ 1.34 | HTR | 300 | Thermodynamics of the formation | 349 | |
| (y = 0) | x = 0.66 | HTR | 260–430 | Thermodynamics of the formation | 349 | |||
| (y′ = 0) | 0.80 ≤ x′ ≤ 1.56 | HTR | 220 | Thermodynamics of the formation | 351 | |||
| Fe3+ | x = 1.15, y = 1.53 | RE | 95 | Thermodynamics of the formation | 438 | |||
| [Si8−xMtetrax]IV[Mg6−yMoctay]VIO20(OH)4 | Al3+ | Al3+ |
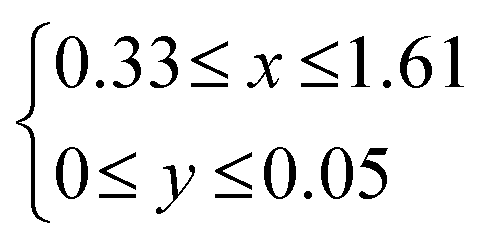
|
HTR | 300 | UV-Vis, 27Al-NMR | Dye organisation | 353 |
| Fe3+ |

|
HTR | 100–250 | IR, XRD(060) | Fundamental study | 354 | ||
| (y = 0) | 0.34 ≤ x ≤ 2.68 | HTR | 260–450 | 27Al, 29Si-NMR, Al-EXAFS | Thermodynamics of the formation | 349 | ||
| (y = 0) | x = 0.66 | HTR | 550 | Thermodynamics of the formation | 355 | |||
| (y = 0) | x ≤ 1.88 | HTR | 400 | Thermodynamics of the formation | 356 | |||
| (y′ = 0) | 0.2 ≤ x′ ≤1.2 | RE | 90 | Solid-acid catalysis | 357 and 358 | |||
| [Si8−xMtetrax]IV[Co6]VIO20(OH)4 | Al3+ | — | 0.2 < x′ < 0.9 | RE | 90 | 27Al, 29Si-NMR, | Solid-acid catalysis | 359 |
| [Si8−xMtetrax]IV[Zn6]VIO20(OH)4 | — | 0.2 ≤ x′ ≤ 2.4 | RE | 90 | Al-EXAFS | Solid-acid catalysis | 359 | |
| [Si8−xMtetrax]IV[(Ni, Co)6]VIO20(OH)4 | — | x′ = 2 | HTR | 100–250 | IR, XRD(060) | Fundamental study | 354 | |
| [Si8]IV[Al4−yMoctay]VIO20(OH,F)4 | — | Mg2+ | 0.40 ≤ y′ ≤ 0.80 | HTR | 220 | 19F-NMR | Fundamental study | 413 |
| Zn2+ | y′ = 0.4, 0.8 | HTR | 220 | 19F-NMR, Zn-EXAFS | Fundamental study | 360 | ||
| [Si8]IV[Fe3+4−yMoctay]VIO20(OH)4 | — | Mg2+ | 0.1 ≤ y ≤ 2 | HTR | 200 | XRD(060) | Fundamental study | 410 |
| [Si8]IV[Mg6−yMoctay]VIO20(OH)4 | — | Fe3+ | 0.6 ≤ y ≤ 2 | HTR | 200 | XRD(060) | Fundamental study | 410 |
| [Si8]IV[Mg6−yMoctay]VIO20(OH, F)4 | — | Li+ | y′ = 0.57 | RE | — | SAXS | Fundamental study | 426 |
| Li+ | 0.07 ≤ y′ ≤ 1.4 | RE | — | XRD(001) | Varying layer charge for adsorbent design | 430,431 | ||
| Li+ | SSR | 1650 | XRD (single-crystal refinement) | Homogeneous layer charge for homogeneous pore and nanocomposite | 460 | |||
| [Si8]IV[Ni6−y]VIO20(OH, F)4 | — | Ni2+ | y′ = 0.6 | HTR | 200 | Precursor of catalyst | 361 | |
| [Si8]IV[Mn6−yMoctay]VIO20(OH, F)4 | — | Li+ | y′ = 0.8 | HTR | 150 | Fundamental study | 411 | |
| [Si8]IV[Zn6−yMoctay]VIO20(OH, F)4 | Li+ | y′ = 0.8 | HTR | 150 | Fundamental study | 383 | ||
4.4. Roles of isomorphous substitution in the functions of smectites
The phase diagram of smectites was summarised by Kloprogge et al.370 The dioctahedral smectite was obtained as single phase at 0.34 ≤ x ≤ 1.34 when the synthesis conditions were 300 °C and 100 MPa (Table 8).349 Single-phase smectite with the x of 0.66 was exemplified at the temperature range 260–430 °C (Table 8).349 Instead of Al3+ in O, three Mg2+ ions are incorporated in O in half of the unit cell with the Si4+–Al3+ substitution in T (“trioctahedral smectites”), where the formula is expressed as [Si8−xAlx]IV[Mg6]VIO20(OH)4. When x was 0.66, a single phase was achieved in the temperature range of 260–450 °C. In the case of 0.34 ≤ x ≤ 1.34, 550 °C was required to obtain a single phase (Table 8).355 A high concentration of Al3+ in the initial mixture and high temperature were preferred for the Si4+–Al3+ substitution in T (100 MPa).369
The Si4+–Al3+ substitution in T was discussed for trioctahedral smectites using a reference clay (JCSS3501, synthetic saponite) for application in solid-acid catalysis371 and dye immobilisation.372–374 The synthesis and characteristics of saponite (one of trioctahedral smectites) were summarised in a recent review.375 The maximum x value was 1.88 (Table 8), when the preparation was conducted at 450 °C and at x′ = 2.0 (x′ corresponds to x in the initial mixture for the preparation).356 When the preparation was conducted at 300 °C and 8.5 MPa for 6 h, x in [Si8−xAlx]IV[Mg6−yAly]VIO20(OH)4 was in the range of 0.33 to 1.61 (Table 9). A small amount of Al3+ was substituted in O.353 It was confirmed experimentally that the CEC varied depending on x. Cationic porphyrins were immobilised without aggregation as a result of matching the distances of the positive charge of the porphyrins with that of the negative charge on the silica layer, giving highly emissive materials (dye aggregation usually deactivates the excited state via energy transfer between the dyes: Fig. 17).
| {[Si8−xAlx]IV[Mg6−yAly]VIO20(OH)4}−(x−y) | CEC/meq (100 g)−1 | Inter-charge distance/nm | ||
|---|---|---|---|---|
| x | y | x–y | ||
| 1.61 | 0.05 | 1.56 | 195 | 0.83 |
| 1.32 | 0 | 1.32 | 168 | 0.92 |
| 1.11 | 0 | 1.11 | 142 | 1.00 |
| 1.03 | 0 | 1.03 | 132 | 1.04 |
| 0.79 | 0.01 | 0.78 | 100 | 1.19 |
| 0.56 | 0.03 | 0.53 | 69 | 1.45 |
| 0.45 | 0 | 0.45 | 59 | 1.57 |
| 0.33 | 0.03 | 0.30 | 39 | 1.92 |
 | ||
| Fig. 17 (A) Structures of cationic porphyrins H2TMPyP4+ and H2TMAP4+, where the distances between quaternary ammonium (arrow) are 1.05 and 1.31 nm, respectively and (B) relationship between the inter-charge distance on the trioctahedral smectites shown in Table 8 and the position of the absorption maximum for ● H2TMPyP4+ and ▲ H2TMAP4+ on the smectites. Reprinted with permission from ref. 353. Copyright 2011, the American Chemical Society. | ||
Solid-acid catalysts were obtained by Si4+–Al3+ substitution in T.331,371,376 Acid-treatment activated smectites to be Brønsted acids because the protons in the Si4+–O–Al3+-OH bonds participated as Brønsted acidic sites, as seen in aluminosilicate zeolites. Pyridine has been employed as a probe for evaluating the acidic sites (IR and temperature-programmed desorption (TPD) are used to distinguish physisorption and chemisorption at Brønsted and Lewis acid sites, and by hydrogen bonding). The desorption behaviour of pyridine depended on the particle size (60% of montmorillonite JCSS3101 particles are >0.2 μm, whereas 90% of synthetic smectite JCSS3501 was <0.2 μm).371 The acid-treatment of smectites with high magnesium contents (trioctahedral smectites, O = Mg2+) more readily leached Mg2+ than that containing a higher proportion of Al3+ as Mocta, giving more acidic sites. This difference was explained by the difference in the bond energy of Mg–O (362 kJ mol−1) from that of Al–O (511 kJ mol−1). Some of the cations leached from O to migrate into the interlayer space as exchangeable cations, which also participated as catalytically active species.
 | ||
| Fig. 18 Octahedral layer of Na0.8[Si8]IV[Al3.2Mg0.8]VIO20(OH)4 viewed normal to the surface. The average distribution of Mg defects (highlighted) is shown. Four types of “defect” distribution: (1) a total of 35% of the Al atoms is surrounded by three Al atoms and no Mg; (2) 55% of the Al atoms is surrounded by two Al atoms and one Mg atom; (3) 10% of the Al atoms is surrounded by one Al atom and two Mg atoms; and (4) none of the Al atoms are surrounded by three Mg neighbours. Reprinted with permission from ref. 377. Copyright 2005, the American Chemical Society. | ||
Both Lewis (octahedral) and Brønsted (tetrahedral or interlayer cations) acidic sites have been investigated for application as solid-acid catalysts.330,379,380 Bifunctional catalysts were designed when basic nanoparticles (e.g., Pt) were deposited.381 The Lewis acidic sites emerged from Zn2+ in O were effective in a catalytic reaction (Friedel–Crafts alkylation of benzene) with the aid of the Brønsted acidic sites, which were derived from the Si4+–Al3+ substitution in T. The molar Al/Si ratio was varied in [Si8−xAlx]IV[O6]VIO20(OH)4 as 0.2 ≤ x′ ≤ 1.2 (O = Mg2+), 0.2 ≤ x′ ≤ 2.4 (O = Zn2+), and 0.2 ≤ x′ ≤ 0.9 (O = Co2+) (Table 8)357,358 to examine the catalytic activity in n-alkane cracking above 200 °C in an inert atmosphere.359 The interlayer cation was exchanged with Al3+ to enhance the acidic properties. However, the reduction of Ni2+ in O by hydrogen resulted in the formation of metallic Ni particles, causing deactivation by coke formation. The substitution of Ni2+ with Co2+ in O suppressed the coke formation.382
Transition metal ions such as Ni2+, Co2+, Mn2+, Fe3+, and Zn2+ have been incorporated in O (Table 8).383,384 Hydrothermal reactions led the incorporation of Zn2+ in O as compared to Cu2+, Co2+ or Ni2+.358 Ti4+ was incorporated as octahedrally coordinated species, when Zn2+ was incorporated in O of a (fluoro)smectite.385
The incorporation of iron with varying oxidation states has been studied. Iron in O has been recognised as an electron donor/acceptor386–392 and energy acceptor to deactivate the excited state of the adsorbed luminophores.393–396 The luminescence quenching is not favoured for designing photoluminescent materials, and thus synthetic 2:1-type phyllosilicates have been used. Alternatively, the quenching of the excited state by iron was used to stabilise dyes upon photoirradiation.396 When rhodamine 6G was adsorbed on 2:1-type phyllosilicates with varying iron contents, the dye stabilities were proportional to the quenching efficiency, which was correlated with the iron content (Fig. 19). This is an important phenomenon given that dye stability is required for the development of dye-clay pigments.397 Along this line, a bentonite with a low iron content was reported recently.398 The mechanical properties of a biopolymer hydrogel were improved using the bentonite, where synthetic hectorites did not have positive effects. Due to the low iron content, the photoluminescence from the adsorbed cyanine dyes on the clay in the hydrogel was seen and a unique photoluminescence mechanochromism was reported.373,399
 | ||
| Fig. 19 (A) Photograph of an organic luminophore (Rhodamine 6G) and (B) relationship of the photodecolorisation rate constant with the normalised photoluminescence intensity (black) and the photoluminescence quantum efficiency (red) of SWF-R6G, SA-R6G, LPN-R6G, and KF-R6G suspensions.396 | ||
A bentonite with high iron content was also found and the state of the iron was extensively characterised to be octahedrally coordinated Fe2+ probably in T. The bentonite was shown to be a photocatalyst for H2 evolution from aqueous methanol (Fig. 20).386 The layer charge varied in response to the content of Fe3+ and Fe2+, affecting the hydration of the interlayer cations and swelling pressure.400–402 Iron-containing smectites have been synthesised,403–408 where a reducing agent (hydrazine or sodium dithionite) was required to obtain Fe2+-rich (ferruginous) smectites. Ferruginous smectites (nontronite) included iron as Fe2+2.8(Si, Al)8O20 for the SWa-1 sample (Grant County, Washington, USA, Clay Mineral Society),367 and Fe2+3.6(Si, Al)8O20 for Garfield nontronite (Spokane, Washington, USA, API-no. 33-A).409 The syntheses were performed at 90 °C for 2–3 weeks, and the amounts of Fe were Fe2+2.8(Si, Al)8O20408 and Fe2+1.5(Si, Al)8O20.362 According to the IR absorption band due to the Fe3+–Al–OH bonds, Al3+ and Fe3+ were miscible (not Fe2+ because the ionic radius of Fe2+ was too large to be miscible with Al3+) in O.362 Mg2+ and Fe3+ were miscible in O (Table 8).354,410 The incorporation of Mn in O of smectite has scarcely been reported, which is probably because of the difficulty in controlling the oxidation state of Mn.411
 | ||
| Fig. 20 (A) Polyhedral model of the smectite structure from the [100] direction and [001] direction, (B) Fe 2p XPS spectrum of Fe-clay, and (C) photocatalytic H2 evolution of Fe-clay (red circle), Bengel next LU (black square), and LAPONITE® (blue triangle) from aqueous MeOH.386 | ||
Torii et al.361 prepared [Si8]IV[Ni6−y]VIO20(OH, F)4 in the presence of HF at an atomic F/Si ratio in the range of 0 to 1/2 in the initial compositions. When the HF amount was larger, a smaller amount of vacancy was shown by the methylene blue adsorption. Replacing OH with F resulted in lower viscosity in the aqueous slurry. Al-rich fluorophyllosilicates ([Si8]IV[Al4−yZny]VIO20(OH, F)4, y′ = 0.4 and 0.8) were synthesised at 220 °C in the presence of HF.360,413 Despite the large difference in the ionic radius between Al3+ and Zn2+, they were adjacent to each other (miscible) and bridged by oxygen in O.
Dioctahedral-type 2:1 phyllosilicates (montmorillonite, [Si8]IV[Al4−yMgy]VIO20(OH)4 in the most common example) have vacancies in O. Interlayer Li+ is migrated to O to decrease the layer charge. This phenomenon is named the Hoffman–Klemen effect.414 This is a method for the post-adjustment of the y value in this smectite system. The reduced charged silicates have been used415 as a clay-based gas barrier film,416 adsorbents for a cationic dye,417 a polymer,418 and organic contaminants.419–422
LAPONITE® is a synthetic hectorite, in which Li+ is fixed in O: [Si8]IV[Mg6−yLiy]VIO20(OH)4, and its reported formula is Na0.7[Si8]IV[Mg5.5Li0.3]VIO20(OH)4.363,423 Its synthesis, characteristics, and applications were previously summarised in a review article.424 A hectorite analogue was prepared using LiF, Mg(OH)2, and silica sol under reflux condition425 at the molar ratio of Li:Mg:Si = 1.4:5.3:8426 in the starting mixture (y′ = 0.7 in [Si8]IV[Mg6−yLiy]VIO20(OH, F)4, where the amount of Li was twice in the formula). Fluoride in LiF was proposed to be an accelerator for crystallisation.425 The structural OH group was partially substituted with F. Tetraethylammonium was used to assist crystallisation and incorporated as an exchangeable cation.427 A small amount of actinoids (Am3+364 and Cm3+365) and lanthanoids (Eu3+)428 in the range of 10−3–10−5/Mg2+ was doped in O of the tetraethylammonium form. Another example was proposed for preparing analogues at 180 °C by Torii and co-workers, where the atomic ratios of Li:M:Si were 0.6:5.4:8 (M = Ni2+)361 and 0.8:5.2:8 (M = Mg2+)429 in the starting mixtures with the formula of [Si8]IV[M6−yLiy]VIO20(OH)4.
The limitation of the Li+-fixation in O was examined when hectorite was prepared using LiF, Mg(OH)2, and SiO2 by varied Li:Mg:Si ratio in the starting mixture (Table 8).430,431 The variation of y has been frequently discussed by CEC. The CEC varied empirically in the range of 40 to 90 meq/100 g, when y′ ranges from 0.07 to 1.4 in [Si8]IV[Mg6−yLiy]VIO20(OH)4. The high solubility of Li+ in water and uncontrollable formation of vacancies in the MgO6 octahedral sheet make it difficult to precisely control the amount of Li+-fixation in O. However, the Li+/Mg2+ ratio in the starting mixture was dominant in determining the CEC, which was directly influenced by the Li+-fixation in O. An excess amount of silica sol did not influence the layer charge density of the hectorite analogue.432 Microwave and ultrasonication were used to promote the migration of Li+ toward the vacancy in O to decrease the CEC.433,434 It was difficult to obtain pure hectorite giving the remaining amorphous silica in the product.
The mechanism of Li+-fixation in O is still controversial and two schemes were proposed, as follows: (1) migration of Li+ from the interlamellar to O upon heat treatment435 and (2) substitution or Li+-fixation into O during the hydrothermal reactions (Fig. 21).427 Regarding the formation of 2:1-type phyllosilicate, the mechanism in the initial stage was kinetically discussed using an in situ SAXS technique during the reactions for 48 h at 100 °C.427 The SAXS data showed a progressive increase in the power law value with time from 2.39 to 3.36 recorded at a low Q (nm−1) region, indicating condensation into more dense structures. The 29Si MAS NMR spectra displayed a Q3 signal, referring to the number of branching units typical of sheet silicate structures, which appeared after 1 h and intensified after 4 h, the time at which a (001) reflection (1.50 nm) at a high Q (nm−1) region was observed in the SAXS pattern.
 | ||
| Fig. 21 Route for the formation of hectorite: (A) substitution or fixation of Li into brucite Mg(OH)2 sheet, (B) adsorption of silicate anions on the octahedral sheet and (C) progress of the silicate anions to form the 2:1 type. phyllosilicate.427 | ||
4.5. Synthetic methods for 2:1-type phyllosilicates
2:1-type phyllosilicates have been synthesised370 and the methods can be classified into three groups, as follows: (i) precipitation from an aqueous solution at room temperature and 0.1 MPa for a long time (Section 4.5.1), (ii) hydrothermal reactions of a stoichiometric mixture of amorphous and/or crystalline oxide solids (Section 4.5.2), and (iii) melting of oxides in metal fluorides as a flux via solid-state reactions (Section 4.5.3).370 Part (i) evokes chemical weathering including lateritic weathering condition as precipitation of amorphous Al(OH)3 with coprecipitating goethite and silica.436 Homogeneously mixed glasses are preferred in (ii), which are evoked from quenching magma/lava by water.368,369,437 (iii) High-purity reagents are necessary for stoichiometry in a fluorine medium at a high temperature.352
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1-type phyllosilicates under ambient conditions.
Smectite was precipitated from low-concentration solutions of SiO2 (30 ppm) and Al2O3 (3 ppm) at 20 °C for 3 months at a high Mg2+ concentration and higher pH. Its formation mechanism was explained as the initial formation of Mg(OH)2, followed by the polycondensation of silica on the brucite layers.438 Under alkaline conditions (pH 9–10), it was possible to precipitate10 ppm Mg, whereas at pH 8, 1250 ppm Mg (seawater concentration) was necessary.438 Subsequently, the polycondensation with silica sol was followed by forming a T–O–T layer. Even at a dilute Zn2+ concentration (1–2.5 ppm), a smectite containing Zn2+ in O was formed at 20 °C by aging for 1 year.
:1-type phyllosilicates under ambient conditions.
Smectite was precipitated from low-concentration solutions of SiO2 (30 ppm) and Al2O3 (3 ppm) at 20 °C for 3 months at a high Mg2+ concentration and higher pH. Its formation mechanism was explained as the initial formation of Mg(OH)2, followed by the polycondensation of silica on the brucite layers.438 Under alkaline conditions (pH 9–10), it was possible to precipitate10 ppm Mg, whereas at pH 8, 1250 ppm Mg (seawater concentration) was necessary.438 Subsequently, the polycondensation with silica sol was followed by forming a T–O–T layer. Even at a dilute Zn2+ concentration (1–2.5 ppm), a smectite containing Zn2+ in O was formed at 20 °C by aging for 1 year.
The effective ionic radius of the components are important factors in designing 2:1-type phyllosilicates with a controlled composition (Table 10). The presence of Fe2+ enabled the formation of O.436 Harder also suggested that the small size of Al3+ made it difficult to be installed in O, whereas appropriate sizes ranging 0.078 to 0.082 nm436 or 0.069 to 0.078 nm129 were necessary to form O. This is a reason for the formation of an Fe2+-rich (ferruginous) phyllosilicate under reducing conditions. It became possible to crystallise tetrahedrally coordinated Al3+ in 2:1-type phyllosilicates with the help of divalent cations, which enable the direct formation of phyllosilicates. The mechanism for the primary formation of O seems to be different from that for the formation of Mg–Al/LDHs (uptake of Mg2+ by the Al hydroxide is the possible reaction for the formation of the LDH).
:1-type phyllosilicates at surface temperatures from chemically pure solutions
| Effective radius of ion in octahedral coordinationab/nm | Synthesis of 2:1-type phyllosilicatesa |
||
|---|---|---|---|
| a H. Harder, Clay Miner., 1977, 12, 281–288.436 b R. D. Shannon, Acta Cryst., A, 1976, 32, 751–767.129 | |||
| Mg2+ | 0.078 | 0.0720 | Possible |
| Ni2+ | 0.078 | 0.0690 | Possible |
| Co2+ | 0.082 | 0.0745 (HS: high spin) | Possible |
| Zn2+ | 0.083 | 0.0740 | Possible |
| Fe2+ | 0.082 | 0.0780 (HS) | Possible |
| Fe3+ | 0.067 | 0.0645 (HS) | Not possible under oxidative condition |
| Cu2+ | 0.070 | 0.073 | Difficult in chemically pure solutions |
| V3+ | 0.065 | 0.0640 | Difficult in chemically pure solutions |
| Cr3+ | 0.064 | 0.0615 | Difficult in chemically pure solutions |
| Mn2+ | 0.091 | 0.0830 (HS) | Probably too large for clay minerals |
| Mn3+ | 0.070 | 0.0645 (HS) | Difficult |
| Al3+ | 0.057 | 0.0535 | Not possible in chemically pure solutions at low temperatures |
Organosilanes were reacted at room temperature with metal chlorides to form 2:1-type phyllosilicate derivatives,439,440 which were later named “aminoclay”441,442 and “organotalc”,443 after the pioneering study by Fukushima and Tani on the preparation of 2:1-type 3-(methacryloxy)propyl phyllosilicate with O = Mg2+, Ni2+ for a polymer additive.444,445 The amphiphilic nature of RSiO3 (R = C8H17) also assisted the formation of a T–O–T layer at room temperature; this type of octylsilyl derivative of 2:1 phyllosilicate is based on the structural formula of [Si8−xAlx]IV[Al4]VIO20(OH)4446,447 and [Si8−xAlx]IV[Mg6]VIO20(OH)4.447–449 Reflux or hydrothermal conditions were employed to prepare various functionalised phyllosilicate derivatives (R = CH3,450 C6H6,450 3-(methacryloxy)propyl),451 and 3-acryloxypropyl451) based on the structure of [Si8]IV[Mg6−yLiy]VIO20(OH)4. It is worth examining the incorporation of heteroelements in the T of these organic derivatives of 2:1 phyllosilicates.
:1-type phyllosilicates.
The apparatus and the conditions for the preparation of smectites are listed in Table 11 with the upper limits of pressure, temperature, and mass of product for each apparatus.452 A hectorite analogue, [Si8]IV[Mg6−yLiy]VIO20(OH, F)4, was obtained, as described in Section 4.4.3, and the dosage of organic molecules or cations assisted its crystallisation.453 The nominal compositions for Mg and Si in the initial mixture were maintained after the crystallisation, whereas a larger amount was required for the Li dosage because of its high solubility in water. (The reaction mechanism was already introduced in the Section 4.4.3.) The reactions were performed using Morey-type autoclave, where the pressure is often autogenous water pressure. Researchers prepared materials of desired structures and compositions based on the phase diagram under controlled pressure, if available (Section 4.4.1). Smectite single crystals of several tens of micrometers in lateral size were successfully synthesised at pressure higher than 3 GPa (up-to 5.5 GPa) and temperature of 1000 °C using a belt-type high-pressure apparatus.454 The smectite crystals were considered to be quenched crystals metastably formed from the hydrous silicate melts formed at high pressures and temperatures.
| Reactions | Reactor | Maximum pressure | Maximum temperature (°C) | Scale of production (mL) |
|---|---|---|---|---|
| Precipitation | Beaker | Atmospheric | 100 | No upper limit |
| Hydrothermal reactions | Morey type | 50 MPa | 350 | <1000 |
| Autoclave | 200 MPa | 600 | <1000 | |
| Test tube | 1 GPa | 1100 | <0.5 | |
| Internal heat type | 1 GPa | 1400 | <30 | |
| Pin cylinder type | 6 GPa | 1800 | <0.1 | |
| Belt type | 8 GPa | 2500 | <1000 | |
| Bridgman anvil | 30 GPa | 3000 | <0.01 | |
| Diamond anvil | 200 GPa | 3000 | 10−5 | |
| Solid-state reactions | Electronic furnace | Vacuum ∼ atmospheric | 1700 | <50 |
:1-type phyllosilicate from melt.
Solid-state and melt syntheses were applied to prepare 2:1-type phyllosilicates, where the OH groups in smectites were substituted with fluorine. The temperature and pressure in these syntheses were below 1700 °C and normal pressure, respectively (Table 11). Careful melt synthesis was investigated by Breu to prepare fluorosmectites with nominal compositions of Na1.0[Si8]IV[Mg5.0Li1.0]VIO20F4352 and K0.6[Si8]IV[Mg5.4Li0.3]VIO20F4 with a homogeneous charge distribution.455 High-purity reagents of LiF, MgF2 (>99.9%), MgO (99.95%), NaF (99.99%), and washed and calcined SiO2 (99.9%) were used.352 To avoid gravity segregation in the melt, the crucible was aligned horizontally with rotation by 60 rpm during heating to 1550 °C for 1 h, and finally to 1650 °C for another 15 min. This method is advantageous for the large-scale production (0.85 kg352) of the product with a composition similar to the initial composition of K0.96[Si8]IV[Mg5.09Li0.86]VIO20F4.455 High structural order was found for the synthetic fluorohectorite in contrast to smectites, and a huge aspect ratio of 20k was realised.456 Accordingly, structural coloured nanosheet suspensions366 and gas barrier films prepared from suspension were developed.457–459 A stretchable nanocomposite barrier film for improving water vapour permeability was obtained by sandwiching poly(ethylene glycol) (PEG)-fluorohectorite crystalline (Bragg stack) barrier films between plasticised poly(vinyl alcohol) (PVOH) (Fig. 22).
 | ||
| Fig. 22 (A) Sketch of the tortuous path (white) of a gas permeate dodging impermeable clay nanosheets incorporated in a polymer matrix (blue). (B) Tortuous path of a clay nanocomposite barrier film upon uniaxial stretching and relaxation. (C) Sketch of the sequential coating process to produce a self-standing barrier laminate. (D) Schematic structure and SEM micrographs of a cross section of the self-standing barrier laminate. Elemental mapping of Si (cyan) via EDX spectroscopy indicates no interdiffusion of fluorohectorite nanosheets into the poly(vinyl alcohol) (PVOH) layers.457 | ||
A synthetic strategy was developed to obtain pillared clays with a homogeneous micropore distribution.460–463 The homogeneous charge distribution was confirmed by the two-dimensional 3a × b super-cell (Fig. 23A and B) in Me2DABCO2+(N,N-dimethyl-1,1-diazabicyclo[2.2.2]octane dication)-460 and 2H-DABCO (diprotonated 1,4-diazabicyclo[2.2.2]octane)-exchanged fluorohectorites.461
 | ||
| Fig. 23 Schematic of pillaring process using Me2DABCO2+ (N,N-dimethyl-1,1-diazabicyclo[2.2.2]octane dication) and tris(2,2′-bipyridine)rhodium(III) ([Rh(bpy)3]3+). The layer charge was reduced by the migration of Mg2+, which was exchanged with K+ in the pristine fluorohectorite.455 (A) Powder XRD pattern of 2H-DABCO-ecchanged fluorosmectite. Asterisks (*) mark reflections due to the two-dimensional 3a × b super-cell. (B) Scheme of the a × b super-cell of pillars in 2H-DABCO-fluorosmectite. The unit cell of the parent fluorohectorite is shown as dotted lines.460 (C) Results of the X-ray single-crystal refinement (tetrahedral sheet of the layered silicate and Me2DABCO2+ in the interlayer space).463 | ||
Charge homogeneity was also shown in a high-charged phyllosilicate, an iron-containing Cs-exchanged mica (Cs1.96[Si8]IV[Fe2+3.86Li2.02]VIO20F4).462Fig. 23C illustrates the orientation of the pillar (Me2DABCO2+) with its C3 axis tilted by approximately 24° against the ab plane. Although two of the C2H4 handles protruded into the silicate layers, the third one was in the hexagonal plane of the interlayer space.463 The layer charge was reduced as CEC from 84 to 59 meq/100 g (Fig. 23) by the migration of interlayer Mg2+, which was exchanged with interlayer K+ in the pristine fluorohectorite.455 The sizes and volume of the micropores created by Me2DABCO2+ and tris(2,2′-bipyridine)rhodium(III) ([Rh(bpy)3]3+) increased by the reduction in the layer charge.
4.6. Characterisation of 2:1-type phyllosilicate
 | ||
| Fig. 24 (A) Room-temperature XRD patterns of K-saturated and ethylene glycol-solvated LiSAz-1 samples pre-heated in the range of 60–135 °C for 24 h. Layer charges (e−/(Si, Al)4O10(OH)2) calculated according to ref. 465 are shown in parentheses. The inset shows the patterns of the corresponding samples heated at higher temperatures and up to 300 °C. Note the appearance of a new peak ∼9.5 Å, which indicates collapsed layers. (B) Dependence of CEC on the heating temperature. The inset compares the layer charge and CEC data of the same samples. Reprinted with permission from ref. 466. Copyright 2013, Springer. | ||
Quantitative ion exchange of the interlayer cation with a series of alkylammonium ions using varying alkyl chain lengths has been proposed as a method for the determination of the layer charge density.467 The d001 spacing should increase with the layer charge of 2:1-type phyllosilicates (Fig. 25), corresponding to the inclination of the alkyl chains. Owing to their adsorption selectivity in 2:1-type phyllosilicates, alkylammonium ions are a good indicator of estimating the layer charge, even when accessory minerals are present. After the ion exchange, interactions of another guest (solvent) with the intercalated alkylammonium became a dominant driving force for the guest inclusion to expand the interlayer space468,469 in several applications as rheology-controlling agents318,469 and plastic fillers.470 Purification and classification were necessary to optimise the performances of the materials.471–473
 | ||
| Fig. 25 Schematic of the variation in the arrangements of the intercalated alkylammonium ions in 2:1-type phyllosilicates depending on layer charge density. | ||
| X = (2ν01 − ν02)/2 | (5) |
The migration of Li+ into montmorillonite was investigated by 7Li NMR.474 Heat-treatment up to 250 °C split the signals at −0.16 and −0.60 ppm. The former signal, which was observed before heating, was attributed to the tetrahedrally to pentahedrally coordinated Li+ located in the interlayer space in a symmetrical environment due to the presence of water. The spectral shift to −0.60 ppm upon heating at 250 °C indicated a more strongly bonded cation in the octahedral coordination. Combined with the 19F-MAS NMR result, the Li+ migration occurred in two steps, as follows: at 110 °C, 86% of Li+ moved to the hexagonal cavity of the basal surface in T. The remaining interlayer Li+ migrated progressively to this cavity upon heating at 170 °C to 250 °C.
The chemical shift of the resonances in the 19F NMR spectra is determined by the average electronegativity of the groups attached to F. Therefore, it was employed to determine the elements in O.439 The larger electronegativity of Zn2+ than Mg2+ resulted in a less (shielded) chemical shift in a fluorohectorite. The incorporation of Li+ into vacancies of O was reflected in the resonances; it was ascribed as F bound to three elements in O, either containing two Zn2+ and one Li+ or one Zn2+ and two Li+.
:1-type phyllosilicate.
The coordination numbers of the heteroelements are a clue to monitor their location. Although 27Al-MAS NMR spectroscopy is a method for identifying coordination, the quantitative analysis of the 27Al spectra is difficult475 because of the strong 27Al resonances in Al in T. The resonances due to Al3+ in O are considerably weaker than that expected based on chemical analysis. An amorphous aluminium (hydro)oxide would also interrupt the accuracy. 29Si MAS NMR spectra provided supporting evidence for the Al occupancy.476 The 29Si spectra of the synthetic [Si8−xAlx]IV [Mg,Zn6]VIO20(OH)4 displayed three Q-type signals at approximately −94, −90 and −85 ppm, corresponding to Q3Si(0Al), Q3Si(1Al) and Q2S, respectively.384
Given that the d spacing of the (060) reflection in XRD reflects the ionic radius of the main components in O,477 it has been used to identify dioctahedral (Al3+–Al3+-vacancy) or trioctahedral (Mg2+–Mg2+–Mg2+) type. Depending on the amount of heteroelements in O, the d060 spacing changes.354 Although the similar ionic radius of Ni2+ and Mg2+ made it difficult to identify (d060 = 0.1520 and 0.1523 nm), the difference in the ionic radius between Ni2+ and Co2+ caused a change in the d060 spacing, respectively. This obeyed Vegard's law, which is interpreted as direct proof of a solid-solution. In the case of Fe3+ and Mg2+, because of the small difference in their ionic radius, a linear change was observed in the relationship between the composition and the d060 spacing. On the contrary, in the case of the Mg2+–Al3+ combination, the relation was not linear, indicating the immiscibility of Al3+ and Mg2+ in O. The IR absorption band of OH stretching in O is useful as an alternative to d060 when the ionic radius of two cations is similar.354
19F NMR was used to discuss the states of the heteroelements. Zn2+ and Al3+ were miscible in O of a fluorophyllosilicate ([Si8]IV[Al4−yZny]VIO20(OH, F)4, y′ = 0.4 and 0.8), as indicated by the splitting of the signal in its 19F NMR spectrum, where one was attributed Zn2+–Al3+ (−142 ppm) and the other Al3+–Al3+ (−133 ppm).360 The former signal was intense when the y value increased from 0.4 to 0.8.
Extended X-ray absorption fine structure (EXAFS) spectroscopy has been used to monitor the local structures of O. A fluorophyllosilicate ([Si8]IV[Al4−yZny]VIO20(OH, F)4, y′ = 0.4 and 0.8 in nominal) was examined to determine its interatomic distances and the apparent (average) coordination number of Al3+.360 In addition to the interatomic Zn2+–Zn2+ distance of 0.311 nm, a short distance of 0.298 nm was obtained, indicating neighbouring Zn2+ with Al3+ between the oxygen in O. The coordination number was four, indicating the presence of vacancy in O, as well as Si4+–Al3+ substitution in T.
Mössbauer spectra have been used to examine the valence, coordination number, and structural symmetry of Fe in smectites.478 Given that the quadrupole shift (QS) and the isomer shift (IS) of high-spin Fe3+ are smaller than that of high-spin Fe2+ and their values are defined, the Fe2+/Fe3+ ratio can be determined. The QS of Fe-containing clays is reversibly changed by the oxidation/reduction of Fe.367,479,480
Dithionite (S2O42−) and hydrazine (N2H4) were used to reduce Fe in smectites.481 The reduction of the structural Fe3+ was proposed482 based on the literature400 using sodium dithionite in a citrate-bicarbonate buffer solution as the reducing agent. The layer charge increased upon the reduction of Fe3+. Stucki and Roth483 proposed that a couple of hydroxyl groups bound to Fe2+ and Fe3+ in O was removed by releasing H2O, followed by combining H+ in the aqueous solution with the remaining oxygen atoms in O to form a hydroxyl group. The resulting OH group was predicted to bind to Fe2+ and Fe3+ as pentahedral coordination (later this prediction was modified to Fe maintains octahedral coordination after complete reduction through Fe-EXAFS studies409). Gan et al. used a two-step reduction for the reduction of Fe3+,484 generation of SO2−· radical from sodium dithionate and the reduction of Fe3+ by combining with H+ in the solution, which compensated the charge generated by the removal of the hydroxyl group in O.
Fe2+ in a reduced Fe-rich smectite was examined to apply the products of varying layer charges obtained by Li+-fixation.367 Mössbauer spectroscopy proved that more than 20% of the total Fe was stabilised as Fe2+ by heat treatment in N2 at 260 °C for 24 h.
4.7. Summary and perspectives
In O of montmorillonite, [Si8]IV[Al4−yMgy]VIO20(OH)4, the two cations with oxygen between are not necessarily next to each other (described in Section 4.4.2). After the formation of the brucite Mg(OH)2 layer, the condensation of silicate anions on the layer gave 2:1-type phyllosilicate (T–O–T layer: Fig. 21). This reaction was rapid (preferential adsorption of silicate anions to carbonate ones) compared to substituting Al3+ for Mg2+. Dissolved CO2436,438 and carbonate anions485,486 did not affect the formation of trioctahedral smectite.486 The large difference in ionic radii between Mg2+ and Al3+ is the reason preventing Al3+ substitution in O.438 Therefore, the expression of the isomorphous substitution, [Al4−yMgy]VI, in the formula of montmorillonite should be apparent or nominal, and the negative charge should originate from the vacancy in O.
A high concentration of Al3+ in the starting mixtures or a high temperature was necessary to obtain a dioctahedral smectite, beidellite ([Si8−xAlx]IV[Al4]VIO20(OH)4). Al3+ and Zn2+ co-existed in O of fluorophyllosilicate ([Si8]IV[Al4−yZny]VIO20(OH, F)4, y′ = 0.4 and 0.8 in nominal) (Section 4.4.3).360
According to Loewenstein's rule, the amount of Si4+–Al3+ substitution in T is limited within Al/Si = 1/3, as shown in Table 8 (A recent 17O-MAS NMR study should be noted as being in disagreement with Lowenstein's rule with Si4+–Al3+ substitution in T of synthetic micas487). The charge per unit cell (x + y) of smectites is in the range of 0.4 ≤ x + y ≤ 1.2, which is approved by the AIPEA Nomenclature Committee or the IMA Commission on New Minerals and Mineral Names. This range resulted from the limitation of Si4+–Al3+ substitution in T, in addition to the amount of vacancy in O. The amount of vacancy in O should be influenced by the solution pH during the formation, given that vacancies may arise if the sheet expansion rate of T is higher than that of O (Section 4.4.3). Further kinetic study on the balance of the sheet expansion will optimised the amount of vacancies. The migration of Li+ from the interlayer space toward the vacancy (Hoffman–Klemen effects) reduces the negative charge by heating at around 250 °C, losing hydrated water around Li+ (Section 4.6). The mechanism of the incorporation of Li+ in O of hectorite during the synthesis (during precipitation or drying) is still unclear (Section 4.4.3).
The synthesis of 2:1-type phyllosilicates with various heteroelements at 220 °C or lower temperatures was overviewed (Section 4.4.2 and Table 8). The cations with similar ionic radii, 0.07 nm, in O (e.g., O = Ni2+–Co2+354 and Mg2+–Fe3+410) were miscible in O based on Vegard's law (Section 4.6). As schematically shown in Fig. 26, the Ni2+ and Mg2+ cations at a 1:1 atomic ratio became miscible in a 2:1-type phyllosilicate, when the synthesis temperature was increased from 100 °C to 300 °C.488 However, hydrothermal synthesis at 300 °C may be difficult to prevent the thermal decomposition of the target mineral and cogeneration of accessory minerals. Although water is a commonly used flux for crystallising 2:1-type phyllosilicates, the location and the amount of heteroelements in the products were not critically controlled. The fluorohectorite prepared by solid-state reaction at 1650 °C (Section 4.5.3) had a highly homogeneous charge distribution.352,366,455–458
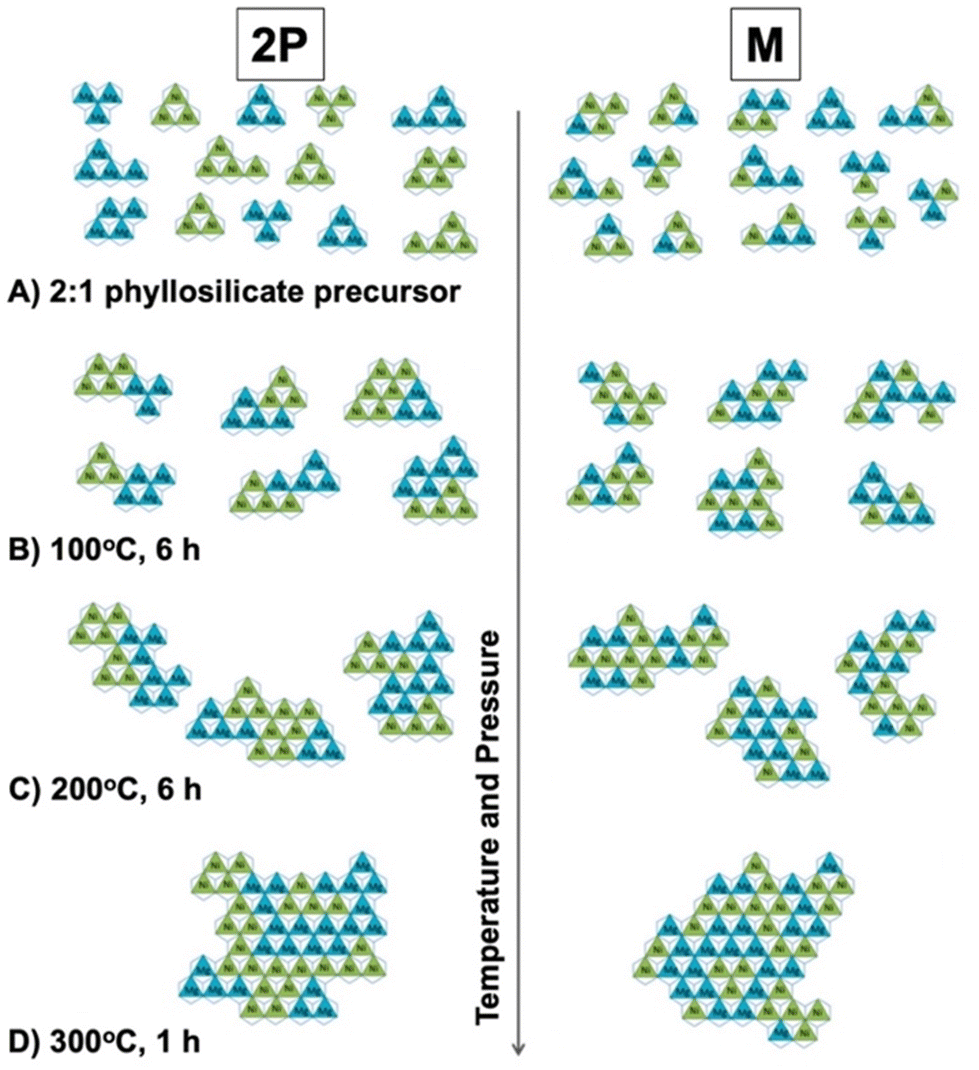 | ||
| Fig. 26 Proposed model of the locational changes in Mg2+ (blue) and Ni2+ (green) at 1:1 atomic ratio under hydrothermal conditions in the octahedral sheet of a phyllosilicate. Conditions are (A) after precursor precipitation, after synthesis at (B) 100 °C, (C) 200 °C, and (D) 300 °C for 6 h. The model was also compared based on the difference in the precursor preparation (2P and M), where 2P and M designate subsequent and simultaneous dosages of Mg2+ and Ni2+, respectively. Reprinted with permission from ref. 488. Copyright 2015, the American Chemical Society. | ||
In addition to precise control of the charge distribution, incorporating functional units in the silicate layers is a challenging task for designing multiply-functionalised materials, as shown by the redox ability in Fe-containing bentonite386,401 (Sections 4.4.2 and 4.6.4). The aforementioned hybridisation of LDHs with functional particles (Section 2.6) was also reported in a 2:1-type phyllosilicate structure for modifying and improving its performances.489–491 Huge crystals (high aspect ratio)456 exhibit superior functionalities including optical457 and gas barrier properties (Section 4.5.3).458 The structural and morphological variation in 2:1-type phyllosilicates will satisfy the requirements in photochemical reactions, optics, selective adsorption, and catalysis and lead to unexplored applications.
5. Layered transition metal oxides
Besides the layered oxides and hydroxides of main elements, layered transition metal oxides with interlayer reactivity are known (hereafter abbreviated as LTMO) (Fig. 27).492–498 LTMOs are distinguished from the layered oxides/hydroxides of main elements by the variation in their electric, electronic, optical, and magnetic characteristics. Similar to bulk transition metal oxides, the characteristics of LTMOs are designed by isomorphous substitution.
Simple metal oxides with layered structures such as RuO2, V2O5 and MnO2 are known.499–502 These LTMOs accommodate cations through redox reactions. For example, a layered MnO2 (birnessite) accommodates alkali or alkaline earth metal ions in its interlayer space for compensating the negative charge of the host layer, which is caused by the difference in the oxidation state of manganese ions. The layer charge is variable depending on the redox reaction employed, and the average oxidation state of the manganese in birnessite was reported to be between 3.4 and 3.99.502 Although V2O5 and MnO2 have been synthesised through solution routes,501,502 some metal salts of LTMOs (layered alkali titanates and layered alkali niobates are known examples) with a defined stoichiometry (e.g., A2TinO1+2n) are obtained by solid-state reactions. The incorporation of heteroelements in LTMOs and their characteristics such as chemical, electric, electronic, magnetic, and optical properties have been examined thus far.
Ion exchange reaction is used to tune the characteristics of LTMOs. For example, the protonated forms of LTMOs, which are obtained by the acid treatment of LTMOs, exhibit characteristics different from that of LTMOs. Protonated forms of LTMO are exfoliated into nanosheets through an ion exchange reaction with the appropriate alkylammonium salts.503–506 The obtained nanosheet is regarded as an anisotropic single crystal of transition metal oxide with an atomic-order thickness and bulk lateral size. As a unique magneto-optical property, an LTMO nanosheet with a high-aspect ratio was shown to orient along the magnetic field and its orientation was switchable by photoirradiation, which reduced Ti4+ in the LTMO nanosheet and Ti3+ was oxidised in the dark spontaneously, suggesting that the LTMO-based nanosheet is likely to be applicable as an optical switch operated by light and magnet (Fig. 28).507 The nanosheets were deposited on a substrate by drop casting,508–510 Langmuir–Blodgett method,308 layer-by-layer assembly510,511 and spin-coating512,513 for device design.514 The nanosheets of LTMO were also used as building blocks of hetero-assemblies with other nanoparticles to design porous hetero-structures for application as electrodes, catalysts, photocatalysts, and adsorbents.47,515–517 The layered structure with expandable interlayer space was useful for molecular recognition photocatalytic reactions.518–520 Thus, the preparation and the modification of various LTMOs have been investigated for a wide variety of applications.
 | ||
| Fig. 28 Schematics of the magnetic orientation; Ti4+-based nanosheet (TiIVNS; left) and its reduced form containing Ti3+ species (TiIV/IIINS; right) orient their 2D planes orthogonal and parallel to the applied magnetic field, respectively. Reprinted from ref. 507. Copyright (2018), the American Chemical Society. | ||
Fig. 29 shows the characteristics of LTMOs designed by isomorphous substitution. Isomorphous substitution has also been used to impart functional units in the LTMO lattice. For example, light-emitting materials by the emission from the rare earth elements in LTMOs have been reported.521–524 Surface characteristics including soft-hard acid (surface ion mobility), swelling (dispersion) and acidity, which are correlated with application in solid-electrolytes, fillers (UV-absorber using the band gap absorption) and solid-acid catalysts, have been designed by isomorphous substitution. Different from the common targets reported for main element oxides/hydroxides (Sections 2–4), the band structure of LTMOs has been tuned by isomorphous substitution (doping), resulting in the designed electrical conductivity, magnetic and optical properties for a wide range of applications starting from superconductor to dielectric525–527 applications. Among the many functions of LTMOs designed by isomorphous substitution, catalytic functions are a representative example. The substitution has been used to control the adsorptive properties and band structure as well as incorporate single-atom catalysts. In this review article, lepidocrocite-type layered titanates and a layered perovskite (KCa2Nb3O10) (Fig. 30) will be introduced as representative examples of LTMOs to explain the important effects of the incorporation of heteroelements in the structure of LTMO on their characteristics.
 | ||
| Fig. 29 Roles of heteroelements and the functions/applications designed by isomorphous substitution in LTMOs. | ||
 | ||
| Fig. 30 Crystal structures of (A) lepidocrocite-type layered titanate K0.8Ti1.73Li0.27O4 and (B) Dion–Jacobson-type layered perovskite KCa2Nb3O10. The structural parameters were referred from ref. 528 and 529, respectively. The crystal structure was drawn using the VESTA program.44 | ||
5.1. Crystal structures of lepidocrocite-type layered titanates and a layered perovskite (KCa2Nb3O10)
Lepidocrocite-type layered titanates are characterised by s single-layer titanium oxide sheet, where the Ti in the octahedrally coordinated TiO6 site is replaced with heteroelements. Layered perovskites have multilayered structures with varying compositions, where several sites can be replaced with heteroelements. Perovskites with varying layer thicknesses have been designed, showing their extended possibilities. To make the discussion simple, examples of KCa2Nb3O10 will be mainly introduced. (A′, interlayer cation; MHE, vacancy (h) or heteroelements such as Li, Zn, Ni, Co and Fe; x = a/(4 − n), where a and n represent the number of interlayer cations per unit formula and the valence of the MHE cation, respectively) (Fig. 30).528–534 The 2D structure of the titanium oxide layer is formed by the assembly of 1D chains of edge-shared TiO6 octahedron through edge-sharing along the a direction.528 The negative charge in the titanium oxide layer arises from isomorphous substitution as Ti4+ → vacancy or Mn+HE (n < 4). The origin of the permanent negative charge is similar to that for smectite. Ti is substituted with MHE with the x of up to ∼0.8 (equivalent to 2/5 of all the Ti sites), making its compositional variation wide. The functions of lepidocrocite-type layered titanate, which are designable by heteroelement and MHE/MOE (the original element, Ti) ratio, are summarised in Table 12. One application of lepidocrocite-type layered titanate taking advantage of its compositional variation is to construct a multilayer film of diluted magnetic semiconductors, which is useful in the field of spintronics.535–539 The a value, which is equivalent to the number of interlayer cations per Ti2−xMHExO4 unit, was correlated with the interlayer cation species, and the typical value was reported to be 0.70, 0.75, and 0.80 for Cs, Rb, and K, respectively.528 The low charge density (2.63–4.00(−) nm−2) compared with other LTMOs, which is likely favourable in accommodating guest species into the interlayer space based on relaxed the electrostatic interactions between the titanium oxide layer and interlayer cation, is another characteristic of lepidocrocite-type layered titanate. Consequently, lepidocrocite-type layered titanate has been used as a host material for designing nanostructured composites or exfoliated into titanate nanosheets through ion-exchange reaction with the tetrabutylammonium ion.503,504,540
(A′, interlayer cation; MHE, vacancy (h) or heteroelements such as Li, Zn, Ni, Co and Fe; x = a/(4 − n), where a and n represent the number of interlayer cations per unit formula and the valence of the MHE cation, respectively) (Fig. 30).528–534 The 2D structure of the titanium oxide layer is formed by the assembly of 1D chains of edge-shared TiO6 octahedron through edge-sharing along the a direction.528 The negative charge in the titanium oxide layer arises from isomorphous substitution as Ti4+ → vacancy or Mn+HE (n < 4). The origin of the permanent negative charge is similar to that for smectite. Ti is substituted with MHE with the x of up to ∼0.8 (equivalent to 2/5 of all the Ti sites), making its compositional variation wide. The functions of lepidocrocite-type layered titanate, which are designable by heteroelement and MHE/MOE (the original element, Ti) ratio, are summarised in Table 12. One application of lepidocrocite-type layered titanate taking advantage of its compositional variation is to construct a multilayer film of diluted magnetic semiconductors, which is useful in the field of spintronics.535–539 The a value, which is equivalent to the number of interlayer cations per Ti2−xMHExO4 unit, was correlated with the interlayer cation species, and the typical value was reported to be 0.70, 0.75, and 0.80 for Cs, Rb, and K, respectively.528 The low charge density (2.63–4.00(−) nm−2) compared with other LTMOs, which is likely favourable in accommodating guest species into the interlayer space based on relaxed the electrostatic interactions between the titanium oxide layer and interlayer cation, is another characteristic of lepidocrocite-type layered titanate. Consequently, lepidocrocite-type layered titanate has been used as a host material for designing nanostructured composites or exfoliated into titanate nanosheets through ion-exchange reaction with the tetrabutylammonium ion.503,504,540
| Function | MHE | MHE/MOE (Ti) atomic ratio |
|---|---|---|
| Photocatalysis (Vis.) | Fe, Ni | 1/5–2/5 |
| Magnetism | Fe, Co, Mn | 1/20–2/5 |
| Solid acidity | Zn, vacancy | 8.3/100–17.5/100 |
Table 13 summarises the functions of the Dion–Jacobson phase with m = 3 depending on the heteroelement and the atomic ratio, MHE/MOE. The MHE/MOE ratio in both the A and B sites has been varied to tune the characteristics of layered perovskites.554–561 The number of interlayer cations per unit formula (A′) is variable, in accordance with the change in the charge density in the perovskite layer upon the substitution of MOE by MHE with different valence.555
| Function | Isomorphous substitution | |||||
|---|---|---|---|---|---|---|
| A site | B site | |||||
| MOE | MHE | MHE/MOE atomic ratios | MOE | MHE | MHE/MOE atomic ratio | |
| Electric conducting | Ca | La, Sr, Nd, Sm, Gd, Se | 1/50–1/5 | Nb | — | — |
| Dielectric | Ca, Sr | Sr, Bi | 1/10–1/2 | Nb | Ta | 6.7/100–1/3 |
| Photocatalysis | Ca | Sr | 1/8–3/4 | Nb | Mo, Ta | –1 |
| Solid acidity | Ca | La | 1/4–1 | Nb | Ti | 8.3/100–2/3 |
| Electrocatalysis | Ca | — | — | Nb | Vacancy | 3.3/100–13.3/100 |
5.2. The roles of heteroelements in the characteristics of LTMOs
The roles of isomorphous substitution are divided into two, as follows: (i) the control of band edges and (ii) the creation of localised energy states in the band gap. The position of the band edges (the valence band maximum and the conduction band minimum) determines the photo-absorption and the redox potentials of the semiconductors, which are important in energy conversion, energy storage, and energy transfer applications. Alternatively, excitation from the localised energy states to the conduction band minimum often imparts visible-light absorption to semiconductors whose band gap energy is equivalent to UV light. As another case, visible-light absorption associated with the colour centre created by oxygen vacancy formed for compensating positive charge deficiency was reported.564 Compared with the control of the band edge positions, the amount of the heteroelement required for the localised energy state creation is generally small.559,565
The Ti in lepidocrocite-type layered titanate has been substituted by heteroelements with the MHE/MOE value of ∼2/5, and the compositional variation has been correlated with the band structure modification.566 As shown in Fig. 31, the absorption edge for a colloidal suspension of K0.8Ti1.2Fe0.8O4 nanosheet was located at the wavelength of ca. 400 nm, which is remarkably longer than that for the titanate nanosheet obtained from a lepidocrocite-type layered titanate containing vacancy, Cs0.7Ti1.825h0.175O4.567 This indicated a decrease in the band gap energy by the Fe-incorporation. Also, the tail of the absorption edge was observed in the wavelength range of 400 nm ≤ in both cases of K0.8Ti1.6Ni0.4O4 and K0.8Ti1.2Fe0.8O4, resulting in colouration of the suspensions. The light absorption in the visible-light region is presumably correlated with the energy transition between the localised energy states formed by the transition metal ions and the conduction band minimum. The Fe- and Ni-substituted titanate nanosheets were used as photocatalysts to decompose the poly(diallyldimethylammonium) (PDDA) located between the nanosheets in titanate nanosheet/PDDA alternative multilayers by visible-light, indicating the visible-light responsive photocatalytic activity taking advantage of the isomorphous substitution in the lepidocrocite-type layered titanate.566
 | ||
| Fig. 31 Example of the functionalisation of lepidocrocite-type layered titanate by isomorphous substitution; development of visible-light absorption and red-shift in the light absorption edge. The absorption spectra correspond to an aqueous colloidal suspension (0.16 g dm−3) of nanosheets derived from the protonated forms of (a) K0.8Ti1.2Fe0.8O4 and (b) K0.8Ti1.6Ni0.4O4.566 | ||
The layer charge density, which is correlated with the isomorphous substitution in the metal oxide layer, is a parameter that affects photocatalytic reactions through the diffusion of reactants/solvents/products into the interlayer. The layer charge density of LTMO is determined by its structures, and its modification has been examined by the introduction of heteroelements in the metal oxide layer. Post-synthetic treatment is a way to control the layer charge density.568,569 For example, the composition of a lepidocrocite-type layered titanate, K0.8Ti1.73Li0.27O4, was controlled by dilute HCl treatment and subsequent annealing process. The interlayer K+ of the product with a decreased layer charge density was replaced with Na+ for the selective decomposition of benzene in a ternary aqueous solution of benzene, phenol, and 4-buthyl phenol under UV-light irradiation (Fig. 32). This molecular recognition photocatalytic reaction was not observed when K0.8Ti1.73Li0.27O4 or bulk TiO2 was applied.519 The lepidocrocite-type layered titanate with reduced layer charge density was also used as a photocatalyst for the selective conversion of benzene to phenol under visible-light irradiation after the deposition of gold nanoparticles in the interlayer space.520 These results reinforce the roles of isomorphous substitution in the photocatalytic activity of LTMOs through their controlled layer charge density and the band structure.
 | ||
| Fig. 32 Schematic of molecular recognition photocatalytic reaction induced by Na+-type lepidocrocite-type layered titanate with reduced layer charge density.519 | ||
The band structure of layered perovskites has been controlled by isomorphous substitution with the elemental variation in the A and the B sites of the perovskite layer.565,570 To correlate the element in the A or the B site with the band edge position in KCa2Nb3O10, restacked-nanosheets of KCa2−xSrxNb3O10 (0 ≤ x ≤ 1.5) and KCa2Nb3−yTayO10 (0 ≤ y ≤ 1.5) were obtained through exfoliation and re-assembly, and their optical properties and photocatalytic activity were investigated.570 The more the A site element was substituted, the light absorption wavelength became longer. Alternatively, the opposite trend was observed for the case of the substitution of the B site element. These facts indicated that the band gap energies decreased and increased with an increase in Sr and Ta contents. The amounts of Sr (x) and Ta (y) were optimised at 0 and 1, respectively, for the photocatalytic H2 evolution reaction, where photoexcited electrons reduced water, while photogenerated holes were consumed to oxidise an aqueous organic compound. Given that the valence band of d0-transition metal cation-based oxides consists of O 2p orbitals571 and the valence band maximum position is constant regardless of the composition,572 the difference in the band gap energy indicated different conduction band minimum positions. Mallouk and co-workers applied the Mott–Schottky method to multilayers of Ca2−xSrxNb3O10 (x = 0 or 2) or CaNb3−yTayO10 (0.75 ≤ y ≤ 1.5) nanosheets assembled on a substrate by the layer-by-layer method to directly measure their flat-band potential, and successfully correlated the conduction band minimum position with the band gap energies (Fig. 33).573 The synthesis of perovskite-based mixed anion compounds has been reported, and control of their valence band maximum position is also possible.574–580 These results indicate the benefits of the compositional variation of Dion–Jacobson-type layered compounds for designing nanosheet-based opto/electronic materials with optimised band edge positions depending on the target applications.
 | ||
| Fig. 33 Controlling the flat band position of perovskite nanosheet by isomorphous substitution of A or B site. The flat band potential of the nanosheets with varied compositions on a tin oxide-coated glass was measured by Mott–Schottky method under valid pH.573 The measured values are in good agreement with the Fermi level values estimated based on Matsumoto's empirical correlation between the conduction band-edge positions and band gap energies for metal oxide semiconductors (solid lines).572 | ||
Rh was incorporated into the Ti site in a lepidocrocite-type layered titanate containing vacancy581 and the Nb site in KCa2Nb3O10.582 The nominal MHE/MOE ratio of ∼1/20 and 1/100 for the Ti site in a lepidocrocite-type layered titanate and the Nb site in KCa2Nb3O10, respectively, was demonstrated for high photocatalytic activity in H2 evolution from an aqueous solution of methanol compared with that loaded with Rh. Considering the amount of heteroelement, there is a possibility that the doped Rh contributed to the development of visible-light absorption properties by creating localised energy states in the band gap, while the role of Rh as a co-catalyst to suppress the charge recombination in the semiconductor nanosheets is also plausible.
The heteroelements in the titanium oxide layer of lepidocrocite-type layered titanate often leach upon acid-treatments,528,583,584 and a defect site is generated to correlate the structural transformation of titanium oxide layer into rutile during the drying process in the air at room temperature with the aid of remaining Cl− from the treatment with dilute hydrochloric acid.585 A rutile nanoparticle-decorated protonated layered titanate exhibited high photocatalytic activity for the H2 evolution reaction, benefiting from the created heterojunction, which enabled inter-particle electron transfer for retarding charge-recombination. The sensitising effect by rutile with a narrower band gap was also pointed out. This result demonstrates the beneficial aspect of defects originating from heteroelements in the design of lepidocrocite-type layered titanate-based photocatalysts.
Given that proton exchange of LTMOs with varying layer charge densities leads layered solid-acids with varying H+ (Brønsted site) densities, the correlation of the layer charge density of LTMOs with their performances as solid-acid catalyst is of interest. HCa2Nb3O10 with controlled amounts of acidic sites was obtained by the acid-treatment of K1−xCa2−xLaxNb3O10 (0 < x ≤ 1.0), whose interlayer cation amount was controlled by the substitution of Ca2+ with La3+.555 Because lower charge density leads to weaker electrostatic interactions between the layer and the interlayer cation, the correlation between the catalytic activity and reactant diffusion into the interlayer is also of interest. The layer charge density was controlled by the isomorphous substitution of a Ruddlesden–Popper layered perovskite, A2La2Ti3O10 (A = K or Rb).556 The replacement of Ti4+ with Nb5+ resulted in A2−xLa2Ti3−xNbxO10 (A = K, Rb) (0 ≤ x ≤ 1.0). In the range of 0 ≤ x ≤ 0.75, all the products were the Ruddlesden–Popper phase, while the Dion–Jacobson phase was obtained in the case of x = 1.0. The product with the Dion–Jacobson structure spontaneously hydrated under ambient conditions, which was related to the acidity of the products. Alternatively, H2−xLa2Ti3−xNbxO10 (0 ≤ x ≤ 0.75) did not show hydration properties for all the x values, suggesting the importance of the crystal structure in addition to the composition556,557 in the acidity of layered perovskites.
The solid-acidity of LTMO containing alkali metal cations in its interlayer space has been investigated. The lepidocrocite-type layered titanates CsaTi2−xhxO4 (a = 0.67 or 0.70; h represents vacancy) and CsaTi2−xZnxO4 (a = 0.70) were demonstrated to exhibit Lewis acidity.586,587 The layer structure of the lepidocrocite-type layered titanate whose interlayer alkali cation was replaced by a proton generally collapsed at <400 °C.528 Alternatively, these layered solid-acids with interlayer Cs maintained their structure at above 400 °C, which was beneficial for the ethanol conversion reaction at 380 °C. The acidity of CsaTi2−xhxO4 (a = 0.67) treated at 400 °C, which was evaluated by NH3-TPD, was higher than that of HZSM-5 treated in the same manner. The structure of the titanate layer and the incorporated Zn or vacancy in it, in addition to the interlayer cation, were related to the acidity.
5.3. Synthetic method for heteroelement-incorporated LTMOs
To incorporate heteroelements into LTMOs, solid-state synthesis has been used, where the source of the heteroelements is mixed in the starting mixture. Given that a homogeneous distribution of the heteroelement is required, the starting mixtures were carefully prepared. The starting materials were mixed for a long period (20 h) using a ball mill for synthesising the layered perovskite applicable to the dielectric application.525,526 Although the mixing time and the instrument used for the mixing are important to achieve homogeneity of the components, the mixing time has been scarcely described.525,526,588The starting mixture is often pelletised for an intimate connection between the components to facilitate the solid-state reaction. For applications that require a higher surface area such as photocatalysts, calcination in the powder form at a lower temperature is desirable for suppressing sintering,589,590 where the possibility of an inhomogeneous distribution of components in the final products is plausible. Alternatively, the unconventionally low synthetic temperature is worth applying given that a new composition is likely to be found according to an undeveloped phase diagram. In the case of low-temperature synthesis, careful mixing of the starting materials will be beneficial for the better distribution of heteroelements in the products. The synthesis of lepidocrocite-type layered titanates K0.8Ti1.73Li0.27O4589 and KCa2Nb3O10590 at a temperature lower than that reported previously528,541,591 is applicable for isomorphous substitution, where the phase separation of the added heteroelements into by-products may be suppressed.
The atmosphere in the solid-state reaction is selected depending on the objective of isomorphous substitution. Although K1−xCa2−xLaxNb3O10 (0 < x ≤ 1.0) was obtained through solid-state reaction in air,555 an inert atmosphere was applied for the synthesis of KCa2−xLaxNb3O10 whose interlayer K+ number per Ca2Nb3O10 unit was fixed to 1.554 This is because the excess positive charge arising from the isomorphous substitution was expected to be compensated by the reduction of Nb5+ for the desired electric properties.554
Increasing attention has been directed to the design of layered perovskite-type compounds with more complicated composition/structures (Fig. 34). Regarding nanosheets derived from KCam−1NbmO3m+1 (m = 3–6), it was found that the ferroelectric properties are correlated with the thickness of the perovskite layer.547,592 Layered lead halide perovskites have attracted attention in versatile fields, including dye-sensitised solar cells, and Dion–Jacobson lead perovskite iodide with varying perovskite layer thicknesses.593 It was demonstrated that the interlayer aromatic diammonium cations affected the structure and electric properties. Also, layered perovskite oxyhalides with a controlled layer thickness were reported, and iodide and three-layers were found to be the best among the tested samples for visible-light-induced photocatalytic water splitting.594 Bulk perovskite oxides with more complicated compositions such as LaMn0.2Fe0.2Co0.2Ni0.2Cu0.2O3 were reported.595 These results indicate that LTMOs with more complicated compositions and structures by means of carefully optimised synthetic conditions are likely to be a future intriguing topic.
 | ||
| Fig. 34 Structural and compositional variation in perovskite-type layered compounds; oxides,547,551 hallides593 and oxyhalides.594 | ||
5.4. Characterisation
The formation of LTMO with the desired composition is judged by confirming that all the reflections in its XRD pattern are assigned to the target product. The successful isomorphous substitution is confirmed by the changes in the lattice constant. An example of the correlation of the d value and x is shown in Fig. 35.554 The products with heteroelements are sometimes accompanied with by-products and used in the target application although the by-products may affect the performance of the main product. Alternatively, the required amounts of the heteroelement were relatively low (MHE/MOE ≤ ∼1/20) in the development of visible-light absorption and luminescence properties,521,522,565 making it difficult to confirm the formation of by-products by XRD. Even this small amount of impurity, which cannot be identified by XRD, will affect the materials properties. Successful doping can be confirmed by observation using instruments such as high-resolution (HR) TEM, high-angle annular dark-field scanning transmission electron microscope (HAADF STEM) and EDS.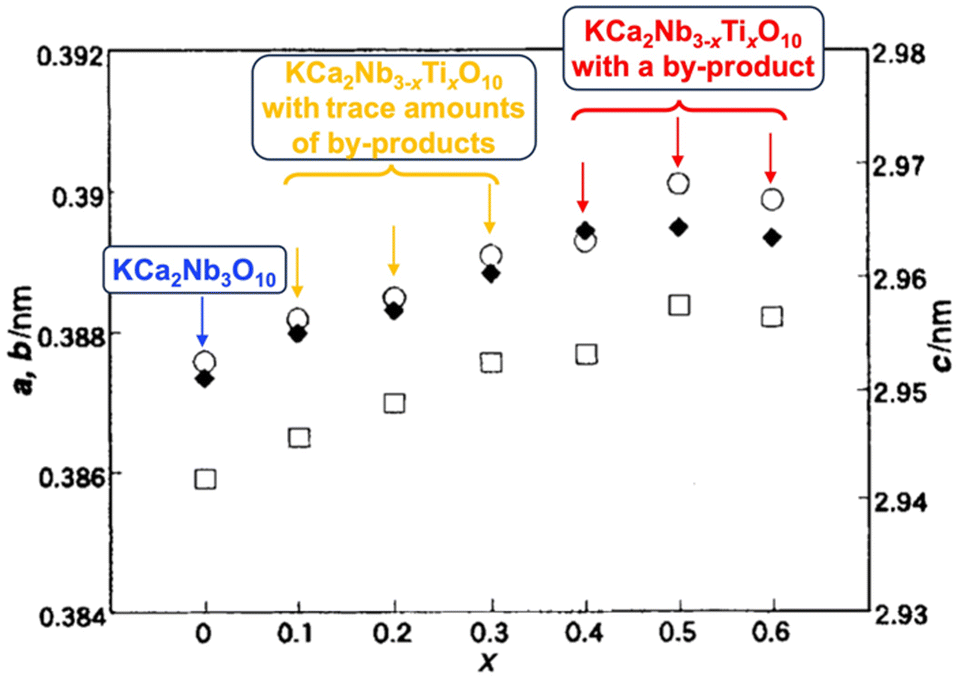 | ||
| Fig. 35 Variation in the lattice parameters of KCa2Nb3−xTixO10 by isomorphous substitution, where an increase in the lattice parameters of KCa2Nb3−xTixO10 are correlated with x, according to Vegard's law.554 | ||
Besides characterisation in the solid-state, the characterisation of the dispersion of LTMOs in solvents and in polymers is worth examining. A periodic arrangement in the distance between the nanosheets in solvent or polymer matrices has been developed. Interests have been focused on the assembly of nanospheres (e.g., nanoparticles and surfactant micelles) for developing structural colours inspired by nature, and LTMO-based nanosheets were demonstrated to orientate for developing structural colour.596,597 An aqueous suspension of titanate nanosheet, which was obtained by the exfoliation of a lepidocrocite-type layered titanate, exhibited structural colour, depending on the concentration of the titanate nanosheet.596 Also, the structural colour was controlled by external stimuli such as temperature, pH and magnetic field. The inter-nanosheet distances estimated by the UV-Vis spectra of the suspensions was in good agreement with that determined by SAXS. An aqueous colloidal suspension of perovskite nanosheets also demonstrated concentration-dependent structural colour, which was successfully explained by the variation in the inter-nanosheet distances estimated using UV-Vis spectroscopy and SAXS (Fig. 36).597 The polymer hydrogel containing the perovskite nanosheets showed reversible mechanochromic responses, which are applicable to mechano-sensors and displays. The development of evaluation methods for understanding the characteristics of LTMO-based nanosheets will facilitate the design of novel LTMO-based materials.
 | ||
| Fig. 36 Structural color of the liquid crystalline perovskite nanosheet suspension. (A)–(D) UV-Vis reflectance spectra and photographs of the colloids with the nanosheet concentration of (A) 0.1, (B) 1, (C) 2 and (D) 3 wt%. (E) Small-angle X-ray scattering and 2D patterns of the colloids with 1, 2 and 3 wt% of nanosheets. Reprinted from ref. 597. Copyright (2021), John Wiley & Sons. | ||
5.5. Summary and perspectives
In addition to the common topics on the electric, electronic, magnetic, and optical properties of LTMOs by the incorporation of heteroelements, efforts to design surface properties, which are connected to hydration/swelling, adsorption, and catalysis, have been reported. The incorporation of heteroelements has been examined using a common ceramic process (solid-state reaction), where efforts have been made to homogenise the starting mixture by careful mechanical mixing and applying a sol–gel reaction. Post-synthetic treatments have also been examined for directing the lepidocrocite-type layered alkali titanate with varying layer charge densities, which is designed by the amount of heteroelement in the titanate sheet.568,569 Conventional synthetic methods are worth revisiting to optimise the detailed conditions for each target function, which may extend the amount of heteroelements incorporated in LTMOs. For example, the synthesis of LTMOs by solid-state reactions at temperatures lower than the commonly reported ones589,590 is a possible candidate.Materials design that benefits from defects/vacancies, which are formed by the leaching of the heteroelement from the lepidocrocite-type titanate framework,598 is also an intriguing topic. An anionic titanate nanosheet with Ti vacancy (sub-nanometric pore) let cations pass through selectively, benefitting from the electrostatic repulsion with anionic species, enabling its application as a coating material in a membrane separator for alkali metal batteries.599 A metal-deficient titanate nanosheet was also demonstrated to be useful for electricity generation from water evaporation, where the density of vacancies was crucial for controlling the water-solid interactions that govern the material performance.600 The important role of the Ti vacancy on the magnetic properties of lepidocrocite-type titanate was also pointed out.601 The structural transformation of defective titanium oxide thin-layer into titanium oxide species including rutile,584,585,602 brookite,603,604 and Magneli phase605 has been reported. Thus, the design of a titanate layer-TiO2 heterojunction that is useful in electric/electronic applications is of interest.
6. Conclusion and future perspective
The partial replacement of one structural element in 2D oxides and hydroxides (nanosheets or layered materials) with heteroelements was summarised with emphasis on the correlation of the basic chemistry of their preparation and characterisation with their application. Depending on the area of research, the phrase referring to the replacement varies (“isomorphous substitution”, “doping”, etc). The application of the resulting materials includes adsorbents, catalysts, drug carriers, pigments, electrodes, and other advanced optical/magnetic/dielectric/semiconducting/superconducting materials. This concept (replacement of one structural element) has been extended to the partial replacement of two structural elements (two sites) in 2:1-type phyllosilicates and mixed oxides (including perovskite transition metal oxides), and the replacement of one structural element (one site) with multiple heteroelements, as seen in layered double hydroxides. Thus, the incorporation of multiple heteroelements is a way to further and more precisely modify the properties of these materials. The incorporation of multiple metal cations with unique oxidation states is thought to be chromophores of some gemstones (blue sapphire as a known example), which are fascinating examples in nature. Thus, it is necessary to prepare and characterise the materials with this compositional complexity more carefully and extensively. The important roles of vacancies have been pointed out, which will be examined further for their effects on the properties, interactions with the incorporated heteroelements, and as possible hosts to accommodate heteroelements.
The heteroelements to be incorporated and their amount have been selected objectively for each target function. Here, it should be noted that in some studies, the composition of the products was not determined experimentally, and the discussion on the properties was based on the starting composition. Thus, for the reliable discussion on the composition–property relationship, the appropriate characterisation to confirm the structural incorporation of the heteroelement is necessary. The recent development of the preparation589,590 and modification of layered materials547,606–608 will extend the elemental and compositional variation of heteroelements to tune the properties further. To extend the compositional variation (the heteroelement and the amount), there are two important factors, as follows: (1) crystallographic aspects and (2) synthetic methods. (1) The size of the ions for replacement (replacing elements and elements being replaced) is a factor based on structural aspects. (2) The availability and chemical reactivity of the starting materials containing the target heteroelements, and the synthetic conditions by which the main structures form without transformation or formation to/of other phases. The new and modified synthetic methods may lead to extended compositional variation (extended dopant concentration).
The hybridisation (or designing heterojunction) of 2D oxides/hydroxides207,609–612 is a current topic in designing photo/electrocatalysts, adsorbents and other advanced functional materials. Therefore, the variation in 2D materials designed by the incorporation of heteroelements discussed herein will be useful for the extension of the combinations to design new/improved functions through the synergistic effects achieved by hybridisation.
Author contributions
The present work was written through the contributions of all authors. All authors have given approval to the final version of the review.Data availability
All the data given in the present review article is based on the already published data as indicated in the text.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Distinguished Professor Grant (Grant number N41A640072) from the National Research Council of Thailand (NRCT), JSPS KAKENHI (Grant-in-Aid for Scientific Research), Grant numbers of 20K05661 and 24K08057 (T. O.), 21K14660 and 24K17711 (K. S.) and 24K17752 (M. M.), the 37th research grant from Proterial Materials Science Foundation (M. M.), and a joint research program of the Institute of Materials and Systems for Sustainability, Nagoya University. All the authors acknowledge Mr. Kosei Ogawa (University of British Columbia) for the proof reading. R. P. W. acknowledges Vidyasirimedhi Institute of Science and Technology (VISTEC) for the Postdoctoral Fellowship.Notes and references
- J. G. Bednorz and K. A. Muller, ZPhys-e.B: Condens. Matter, 1986, 64, 189–193 Search PubMed.
- R. Hansen, Gemstones: A Concise Reference Guide, Princeton University Press, 2020 Search PubMed.
- G. W. Brindley and G. Brown, Crystal Structure of Clay Minerals and their X-ray Identifications, Miner. Soc., London, 1980 Search PubMed.
- J. E. Geusic, H. M. Marcos and L. G. Van Uitert, Appl. Phys. Lett., 1964, 4, 182–184 CrossRef.
- A. A. Haider, H. Zhao, Y. Zi, Z. Xu, X. Bai, Y. Cun, Y. Liu, H. Babeker, A. Saeed, Z. Song, J. Qiu, A. Huang, J. Liao and Z. Yang, Adv. Opt. Mater., 2024, 12, 2302265 CrossRef.
- E. Snitzer, Phys. Rev. Lett., 1961, 7, 444–446 CrossRef.
- R. M. Barrer, Hydrothermal Chemistry of Zeolites, Academic Press, London, 1982 Search PubMed.
- R. Szostak, Molecular Sieves: Principles of Synthesis and Identification, Blackie Academic and Profession, London, 1998 Search PubMed.
- M. Matsuoka and M. Anpo, J. Photochem. Photobiol., C, 2003, 3, 225–252 CrossRef.
- M. Anpo, J. CO2 Util., 2013, 1, 8–17 CrossRef.
- H. Yamashita, K. Mori, Y. Kuwahara, T. Kamegawa, M. Wen, P. Verma and M. Che, Chem. Soc. Rev., 2018, 47, 8072–8096 RSC.
- H. Fei, J. Dong, D. Chen, T. Hu, X. Duan, I. Shakir, Y. Huang and X. Duan, Chem. Soc. Rev., 2019, 48, 5207–5241 RSC.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS.
- H. Furukawa, K. E. Cordova, M. Keeffe and O. M. Yaghi, Science, 2013, 341, 974–986 CrossRef CAS.
- Q. Wang and D. Astruc, Chem. Rev., 2019, 120, 1438–1511 CrossRef PubMed.
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC.
- M. Wen, K. Mori, Y. Kuwahara, T. An and H. Yamashita, Chem. – Asian J., 2018, 13, 1767–1779 CrossRef CAS.
- M. Woellner, S. Hausdorf, N. Klein, P. Mueller, M. W. Smith and S. Kaskel, Adv. Mater., 2018, 30, 1704679 CrossRef.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef.
- K. Lu, T. Aung, N. Guo, R. Weichselbaum and W. Lin, Adv. Mater., 2018, 30, 1707634 CrossRef.
- L. Chen and Q. Xu, Matter, 2019, 1, 57–89 CrossRef.
- T. Simon-Yarza, A. Mielcarek, P. Couvreur and C. Serre, Adv. Mater., 2018, 30, 1707365 CrossRef PubMed.
- X. Yang and Q. Xu, Cryst. Growth Des., 2017, 17, 1450–1455 CrossRef.
- A. D. Burrows, CrystEngComm, 2011, 13, 3623–3642 RSC.
- M. Y. Masoomi, A. Morsali, A. Dhakshinamoorthy and H. Garcia, Angew. Chem., Int. Ed., 2019, 58, 15188–15205 CrossRef.
- L. Feng, K.-Y. Wang, G. S. Day and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4823–4853 RSC.
- Z. Liang, R. Zhao, T. Qiu, R. Zou and Q. Xu, EnergyChem, 2019, 1, 100001 CrossRef.
- H. Lin, Y. Xu, B. Wang, D.-S. Li, T. Zhou and J. Zhang, Small Struct., 2022, 3, 2100176 CrossRef.
- H. Zhu, X. Gan, A. McCreary, R. Lv, Z. Lin and M. Terrones, Nano Today, 2020, 30, 100829 CrossRef.
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, K. Maegawa, W. K. Tan, G. Kawamura, K. K. Kar and A. Matsuda, Mater. Today, 2020, 39, 47–65 CrossRef.
- Y. Zhou, X. Xu, B. Shan, Y. Wen, T. Jiang, J. Lu, S. Zhang, D. P. Wilkinson, J. Zhang and Y. Huang, Energy Storage Mater., 2015, 1, 103–111 CrossRef.
- S. Ippolito, F. Urban, W. Zheng, O. Mazzarisi, C. Valentini, A. G. Kelly, S. M. Gali, M. Bonn, D. Beljonne, F. Corberi, J. N. Coleman, H. I. Wang and P. Samorì, Adv. Mater., 2023, 35, 2211157 CrossRef.
- M. E. Leonowicz, J. A. Lawton, S. L. Lawton and M. K. Rubin, Science, 1994, 264, 1910–1913 CrossRef.
- M. Shamzhy, B. Gil, M. Opanasenko, W. J. Roth and J. Čejka, ACS Catal., 2021, 11, 2366–2396 CrossRef.
- W. J. Roth and J. Čejka, Catal. Sci. Technol., 2011, 1, 43–53 RSC.
- M. V. Opanasenko, W. J. Roth and J. Čejka, Catal. Sci. Technol., 2016, 6, 2467–2484 RSC.
- M. Hmadeh, Z. Lu, Z. Liu, F. Gándara, H. Furukawa, S. Wan, V. Augustyn, R. Chang, L. Liao, F. Zhou, E. Perre, V. Ozolins, K. Suenaga, X. Duan, B. Dunn, Y. Yamamoto, O. Terasaki and O. M. Yaghi, Chem. Mater., 2012, 24, 3511–3513 CrossRef.
- M. Wang, R. Dong and X. Feng, Chem. Soc. Rev., 2021, 50, 2764–2793 RSC.
- J.-M. Li, Q. C. Lin, N. Li, Z. H. Li, G. Tan, S. J. Liu, L. H. Chung, W. M. Liao, L. Yu and J. He, Adv. Funct. Mater., 2023, 33, 2210717 CrossRef.
- G. Alberti and T. Bein, Comprehensive Supramolecular Chemistry, Pergamon, Oxford, 1996 Search PubMed.
- M. S. Whittingham and A. J. Jacobson, Intercalation Chemistry, Academic Press, New York, 1982 Search PubMed.
- W. Müller-Warmuth and R. Schöllhorn, Progress in Intercalation Research, Kluwer Academic, Dordrecht, 1994 Search PubMed.
- S. M. Auerbach, K. A. Carrado and P. K. Datta, Handbook of Layered Materials, Marcel Dekker, New York, 2004 Search PubMed.
- B. K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- V. Dudko, O. Khoruzhenko, S. Weiß, M. Daab, P. Loch, W. Schwieger and J. Breu, Adv. Mater. Technol., 2023, 8, 2200553 CrossRef.
- J. L. Gunjakar, I. Y. Kim, J. M. Lee, Y. K. Jo and S. J. Hwang, J. Phys. Chem. C, 2014, 118, 3847–3863 CrossRef.
- W. T. Reichle, Solid State Ionics, 1986, 22, 135–141 CrossRef.
- D. G. Evans and R. C. T. Slade, in Layered Double Hydroxides, ed. X. Duan and D. G. Evans, Springer-Verlag, Berlin Heidelberg, 2005, vol. 119, pp. 1–87 Search PubMed.
- A. I. Khan and D. O'Hare, J. Mater. Chem., 2002, 12, 3191–3198 RSC.
- V. Rives, Layered Double Hydroxides: Present and Future, Nova Science Publishers, New York, 2001 Search PubMed.
- H. Yi, S. Liu, C. Lai, G. Zeng, M. Li, X. Liu, B. Li, X. Huo, L. Qin, L. Li and M. Zhang, Adv. Energy Mater., 2021, 11, 2002863 CrossRef.
- C. Taviot-Guého, V. Prévot, C. Forano, G. Renaudin, C. Mousty and F. Leroux, Adv. Funct. Mater., 2018, 28, 1703868 CrossRef.
- R. Allmann, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1968, 24, 972–977 CrossRef CAS.
- D. Bish and G. Brindley, Am. Mineral., 1977, 62, 458–464 CAS.
- E. S. Zhitova, N. V. Chukanov, I. V. Pekov, A. A. Zolotarev, V. V. Shilovskikh and V. N. Bocharov, Appl. Clay Sci., 2023, 243, 107070 CrossRef CAS.
- C. Frondel, Am. Mineral., 1941, 26, 295–315 CAS.
- H. F. W. Taylor, Mineral. Mag., 1973, 39, 377–389 CrossRef CAS.
- H. F. W. Taylor, Mineral. Mag., 1969, 37, 338–342 CrossRef CAS.
- W. Feitknecht and M. Gerber, Helv. Chim. Acta, 1942, 25, 131–137 CrossRef CAS.
- W. Feitknecht, Helv. Chim. Acta, 1942, 25, 555–569 CrossRef.
- A. C. Playle, S. R. Gunning and A. F. Llewellyn, Pharm. Acta Helv., 1974, 49, 298–302 Search PubMed.
- L. van der Ven, M. van Gemert, L. Batenburg, J. Keern, L. Gielgens, T. Koster and H. Fischer, Appl. Clay Sci., 2000, 17, 25–34 CrossRef.
- T. Hibino, Eur. J. Inorg. Chem., 2018, 722–730 CrossRef.
- S. K. Jana, P. Wu and T. Tatsumi, J. Catal., 2006, 240, 268–274 CrossRef.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef PubMed.
- K. Ma, J. P. Cheng, M. Li, F. Liu and X. Zhang, Electrochim. Acta, 2016, 198, 231–240 CrossRef CAS.
- A. C. Bouali, M. Serdechnova, C. Blawert, J. Tedim, M. G. S. Ferreira and M. L. Zheludkevich, Appl. Mater. Today, 2020, 21, 100857 CrossRef.
- V. A. Shirin, R. Sankar, A. P. Johnson, H. V. Gangadharappa and K. Pramod, J. Controlled Release, 2021, 330, 398–426 CrossRef.
- T. Hu, Z. Gu, G. R. Williams, M. Strimaite, J. Zha, Z. Zhou, X. Zhang, C. Tan and R. Liang, Chem. Soc. Rev., 2022, 51, 6126–6176 RSC.
- T. Hibino and A. Tsunashima, Chem. Mater., 1998, 10, 4055–4061 CrossRef CAS.
- N. Dewangan, W. M. Hui, S. Jayaprakash, A. R. Bawah, A. J. Poerjoto, T. Jie, A. Jangam, K. Hidajat and S. Kawi, Catal. Today, 2020, 356, 490–513 CrossRef CAS.
- J. Hong, C. Chen, A. Siriviriyanun, D. G. Crivoi, P. Holdway, J. C. Buffet and D. O'Hare, RSC Adv., 2021, 11, 27267–27275 RSC.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- L. Yang, P. Yu, J. Liu, X. Li, H. Li, C. Liu, Y. Pan, B. Ye, B. Pan, C. Xiao and Y. Xie, J. Phys. Chem. C, 2023, 127, 7172–7183 CrossRef CAS.
- L. Tan, Z. Wang, Y. Zhao and Y. F. Song, Chem. – Asian J., 2020, 15, 3380–3389 CrossRef.
- J. An, T. Shen, W. Chang, Y. Zhao, B. Qi and Y. F. Song, Inorg. Chem. Front., 2021, 8, 996–1004 RSC.
- Q. Xie, Z. Cai, P. Li, D. Zhou, Y. Bi, X. Xiong, E. Hu, Y. Li, Y. Kuang and X. Sun, Nano Res., 2018, 11, 4524–4534 CrossRef.
- Y. Zhao, G. Chen, T. Bian, C. Zhou, G. I. Waterhouse, L. Z. Wu, C. H. Tung, L. J. Smith, D. O’Hare and T. Zhang, Adv. Mater., 2015, 27, 7824–7831 CrossRef PubMed.
- P. S. Braterman, Z. P. Xu and F. Yarberry, in Handbook of Layered Materials, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker, New York, 2004, pp. 373–474 Search PubMed.
- R. P. Wijitwongwan, S. G. Intasa-ard and M. Ogawa, ChemEngineering, 2019, 3, 68 CrossRef.
- G. S. Intasa-ard, S. Bureekaew and M. Ogawa, J. Ceram. Soc. Jpn., 2019, 127, 11–17 CrossRef.
- G. S. Intasa-ard and M. Ogawa, J. Solid State Chem., 2023, 317, 123664 CrossRef.
- G. S. Intasa-ard and M. Ogawa, Appl. Clay Sci., 2022, 228, 106615 CrossRef.
- S. K. Yun and T. J. Pinnavaia, Chem. Mater., 1995, 7, 348–354 CrossRef CAS.
- N. Kobylinska, L. Puzyrnaya and G. Pshinko, RSC Adv., 2022, 12, 32156–32172 RSC.
- Q. Xiao, Y. Yuan, J. Zhu, Z. Shi, Z. Li and J. Zhu, J. Alloys Compd., 2022, 916, 165391 CrossRef CAS.
- J. Y. Lee, G. H. Gwak, H. M. Kim, T. I. Kim, G. J. Lee and J. M. Oh, Appl. Clay Sci., 2016, 134, 44–49 CrossRef CAS.
- A. Seron and F. Delorme, J. Phys. Chem. Solids, 2008, 69, 1088–1090 CrossRef CAS.
- S. Miyata, Clays Clay Miner., 1983, 31, 305–311 CrossRef CAS.
- J. W. Boclair and P. S. Braterman, Chem. Mater., 1999, 11, 298–302 CrossRef CAS.
- J. T. Kloprogge, L. Hickey and R. L. Frost, J. Solid State Chem., 2004, 177, 4047–4057 CrossRef CAS.
- Z. Gu, A. C. Thomas, Z. P. Xu, J. H. Campbell and G. Q. Lu, Chem. Mater., 2008, 20, 3715–3722 CrossRef CAS.
- U. Costantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1439–1446 CrossRef CAS.
- M. Ogawa and H. Kaiho, Langmuir, 2002, 18, 4240–4242 CrossRef CAS.
- M. Adachi-Pagano, C. Forano and J. P. Besse, J. Mater. Chem., 2003, 13, 1988–1993 RSC.
- M. Kayano and M. Ogawa, Bull. Chem. Soc. Jpn., 2006, 79, 1988–1990 CrossRef CAS.
- J. D. Yong, R. Valdez, M. Á. Armenta, N. Arjona, G. Pina-Luis and A. Olivas, RSC Adv., 2022, 12, 16955–16965 RSC.
- L. B. Staal, S. S. C. Pushparaj, C. Forano, V. Prevot, D. B. Ravnsbæk, M. Bjerring and U. G. Nielsen, J. Mater. Chem. A, 2017, 5, 21795–21806 RSC.
- Z. Liu, R. Ma, M. Osada, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2006, 128, 4872–4880 CrossRef CAS.
- J. Qu, F. Li, M. Wang, S. Subakti, M. Deconinck, G. Chen, Y. Li, L. Liu, X. Wang, M. Yu and D. Wolf, Adv. Mater. Interfaces, 2022, 9, 2200973 CrossRef CAS.
- F. Geng, Y. Matsushita, R. Ma, H. Xin, M. Tanaka, F. Izumi, N. Iyi and T. Sasaki, J. Am. Chem. Soc., 2008, 130, 16344–16350 CrossRef CAS.
- J. Zhao, X. R. Wang, F. W. Chen, C. He, X. J. Wang, Y. P. Li, R. H. Liu, X. M. Chen, Y. J. Hao, M. Yang and F. T. Li, Inorg. Chem. Front., 2020, 7, 737–745 RSC.
- S. P. Newman, W. Jones, P. O’Connor and D. N. Dtamires, J. Mater. Chem., 2002, 12, 153–155 RSC.
- M. Ogawa and S. Asai, Chem. Mater., 2000, 12, 3253–3255 CrossRef CAS.
- S. G. Intasa-ard, K. J. Imwiset, S. Bureekaew and M. Ogawa, Dalton Trans., 2018, 47, 2896–2916 RSC.
- K. R. Poeppelmeier and S. J. Hwu, Inorg. Chem., 1987, 26, 3297–3302 CrossRef CAS.
- A. N. Ay, B. Zümreoglu-Karan and L. Mafra, Anorg. Allg. Chem., 2009, 635, 1470–1475 CrossRef CAS.
- Z. Ferencz, M. Szabados, M. Ádok-Sipiczki, Á. Kukovecz, Z. Kónya, P. Sipos and I. Pálinkó, J. Mater. Sci., 2014, 49, 8478–8486 CrossRef CAS.
- E. Conterosito, W. V. Beek, L. Palin, G. Croce, L. Perioli, D. Viterbo, G. Gatti and M. Milanesio, Cryst. Growth Des., 2013, 13, 1162–1169 CrossRef CAS.
- K. Kuramoto, S. G. Intasa-ard, S. Bureekaew and M. Ogawa, J. Solid State Chem., 2017, 253, 147–153 CrossRef CAS.
- K. Kuramoto and M. Ogawa, Bull. Chem. Soc. Jpn., 2011, 84, 675–677 CrossRef CAS.
- E. Conterosito, V. Gianotti, L. Palin, E. Boccaleri, D. Viterbo and M. Milanesio, Inorg. Chim. Acta, 2018, 470, 36–50 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS.
- X. Long, S. Xiao, Z. Wang, X. Zheng and S. Yang, Chem. Commun., 2015, 51, 1120–1123 RSC.
- L. Jin, X. Zhou, F. Wang, X. Ning, Y. Wen, B. Song, C. Yang, D. Wu, X. Ke and L. Peng, Nat. Commun., 2022, 13, 6093 CrossRef CAS PubMed.
- U. G. Nielsen, Annual Reports on NMR Spectroscopy, 2021, vol. 104, pp. 75–140 Search PubMed.
- S. Miyata, Clays Clay Miner., 1980, 28, 50–56 CrossRef CAS.
- V. P. Khusnutdinov and V. P. Isupov, Inorg. Mater., 2008, 44, 263–267 CrossRef CAS.
- E. López-Salinas, M. Garcia-Sanchez, J. A. Montoya, D. R. Acosta, J. A. Abasolo and I. Schifter, Langmuir, 1997, 13, 4748–4753 CrossRef.
- E. Lopez-Salinas, M. García-Sánchez, M. L. Ramón-García and I. Schifter, J. Porous Mater., 1996, 3, 169–174 CrossRef CAS.
- G. A. Caravaggio, C. Detellier and Z. Wronski, J. Mater. Chem., 2001, 11, 912–921 RSC.
- F. Touahra, M. Sehailia, W. Ketir, K. Bachari, R. Chebout, M. Trari, O. Cherifi and D. Halliche, Appl. Petrochem. Res., 2016, 6, 1–13 CrossRef CAS.
- R. Ma, J. Liang, X. Liu and T. Sasaki, J. Am. Chem. Soc., 2012, 134, 19915–19921 CrossRef CAS PubMed.
- R. Ma, J. Liang, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2011, 133, 613–620 CrossRef CAS PubMed.
- J. L. Milagres, C. R. Bellato, R. S. Vieira, S. O. Ferreira and C. Reis, J. Environ. Chem. Eng., 2017, 5, 5469–5480 CrossRef CAS.
- M. Sipiczki, E. Kuzmann, Z. Homonnay, J. Megyeri, I. Pálinkó and P. Sipos, J. Mol. Struct., 2013, 1044, 116–120 CrossRef CAS.
- J. Y. Lee, G. H. Gwak, H. M. Kim, T. I. Kim, G. J. Lee and J. M. Oh, Appl. Clay Sci., 2016, 134, 44–49 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- R. Wijitwongwan, S. Intasa-ard and M. Ogawa, Nanomater., 2021, 11, 1206 CrossRef CAS.
- R. Prihod’ko, M. Sychev, I. Kolomitsyn, P. J. Stobbelaar, E. J. Hensen and R. A. van Santen, Microporous Mesoporous Mater., 2002, 56, 241–255 CrossRef.
- M. Bellotto, B. Rebours, O. Clause, J. Lynch, D. Bazin and E. Elkaïm, J. Phys. Chem., 1996, 100, 8527–8534 CrossRef CAS.
- E. López-Salinas, M. Garcia-Sanchez, J. A. Montoya, D. R. Acosta, J. A. Abasolo and I. Schifter, Langmuir, 1997, 13, 4748–4753 CrossRef.
- R. P. Wijitwongwan, T. K. Saothayanun and M. Ogawa, Dalton Trans., 2023, 52, 4692–4699 RSC.
- C. Carteret, B. Grégoire and C. Ruby, Solid State Sci., 2011, 13, 146–150 CrossRef CAS.
- X. Long, S. Xiao, Z. Wang, X. Zheng and S. Yang, Chem. Commun., 2015, 51, 1120–1123 RSC.
- S. Moolayadukkam, S. Thomas, R. C. Sahoo, C. H. Lee, S. U. Lee and H. R. Matte, ACS Appl. Mater. Interfaces, 2020, 12, 6193–6204 CrossRef CAS PubMed.
- X. Zhao, X. Zhao, I. Ullah, L. Gao, J. Zhang and J. Lu, Catal. Lett., 2021, 151, 1683–1692 CrossRef CAS.
- J. Sun, Y. Li, C. Chen, T. Qi, D. Xia, W. Mao, T. Yang, L. Chen, W. Shen and S. Tang, Talanta, 2018, 187, 265–271 CrossRef CAS PubMed.
- M. Wilhelm, A. Bastos, C. Neves, R. Martins and J. Tedim, Mater. Des., 2021, 212, 110188 CrossRef CAS.
- G. Abellan, E. Coronado, C. Marti-Gastaldo, J. Waerenborgh and A. Ribera, Inorg. Chem., 2013, 52, 10147–10157 CrossRef CAS.
- A. Mantilla, G. Jácome-Acatitla, G. Morales-Mendoza, F. Tzompantzi and R. Gómez, Ind. Eng. Chem. Res., 2011, 50, 2762–2767 CrossRef CAS.
- D. Kang, X. Yu, S. Tong, M. Ge, J. Zuo, C. Cao and W. Song, Chem. Eng. J., 2013, 228, 731–740 CrossRef CAS.
- R. P. Wijitwongwan and M. Ogawa, Langmuir, 2024, 40, 1408–1417 CrossRef CAS PubMed.
- G. Li, X. Zhang, D. Qiu, Z. Liu, C. Yang, C. B. Cockreham, B. Wang, L. Fu, J. Zhang, B. Sudduth and X. Guo, Adv. Electron. Mater., 2019, 5, 1900215 CrossRef.
- X. Wang, R. Cao, S. Zhang, P. Hou, R. Han, M. Shao and X. Xu, J. Mater. Chem. A, 2017, 5, 23999–24010 RSC.
- M. Ogawa and Y. Sugiyama, J. Ceram. Soc. Jpn., 2009, 117, 179–184 CrossRef CAS.
- V. Isupov, L. Chupakhina and R. Mitrofanova, J. Mater. Synth. Process., 2000, 8, 251–253 CrossRef CAS.
- R. Ma, Z. Liu, K. Takada, N. Iyi, Y. Bando and T. Sasaki, J. Am. Chem. Soc., 2007, 129, 5257–5263 CrossRef CAS PubMed.
- R. Ma, K. Takada, K. Fukuda, N. Iyi, Y. Bando and T. Sasaki, Angew. Chem., Int. Ed., 2008, 47, 86–89 CrossRef CAS.
- J. Liang, R. Ma, N. Iyi, Y. Ebina, K. Takada and T. Sasaki, Chem. Mater., 2010, 22, 371–378 CrossRef CAS.
- H. C. Noltimier and P. A. Colinvaux, Nature, 1976, 259, 197–200 CrossRef CAS.
- R. M. Taylor, Clay Miner., 1984, 19, 591–603 CrossRef CAS.
- R. Tahawy, E. Doustkhah, E. A. Abdel-Aal, M. Esmat, F. E. Farghaly, H. El-Hosainy, N. Tsunoji, F. I. El-Hosiny, Y. Yamauchi, M. H. N. Assadi and Y. Ide, Appl. Catal., B, 2021, 286, 119854 CrossRef CAS.
- S. Tezuka, R. Chitrakar, A. Sonoda, K. Ooi and T. Tomida, Green Chem., 2004, 6, 104–109 RSC.
- Y. Lin, Q. Fang and B. Chen, J. Environ. Sci., 2014, 26, 493–501 CrossRef CAS PubMed.
- M. Sui, Y. Zhou, L. Sheng and B. Duan, Chem. Eng. J., 2012, 210, 451–460 CrossRef CAS.
- T. Kameda, M. Honda and T. Yoshioka, Sep. Purif. Technol., 2011, 80, 235–239 CrossRef CAS.
- D. Wan, H. Liu, R. Liu, J. Qu, S. Li and J. Zhang, Chem. Eng. J., 2012, 195, 241–247 CrossRef.
- M. Chen, F. Wu, L. Yu, Y. Cai, H. Chen and M. Zhang, CrystEngComm, 2019, 21, 6790–6800 RSC.
- X. Yang, M. T. Rahman, T. Kameda, Y. Masaki, Y. Saito, S. Kumagai and T. Yoshioka, Mine Water Environ., 2020, 39, 881–887 CrossRef CAS.
- F. Kooli, I. C. Chisem, M. Vucelic and W. Jones, Chem. Mater., 1996, 8, 1969–1977 CrossRef CAS.
- P. Z. Xu and Q. G. Lu, Chem. Mater., 2005, 17, 1055–1062 CrossRef.
- D. Tichit, A. Rolland, F. Prinetto, G. Fetter, M. De Jesus Martinez-Ortiz, M. A. Valenzuela and P. Bosch, J. Mater. Chem., 2002, 12, 3832–3838 RSC.
- L. N. Puzyrnaya, G. N. Pshinko, V. Y. Zub and O. V. Zuy, Bull. Mater. Sci., 2020, 43, 3 CrossRef CAS.
- K. Morimoto, K. Tamura, H. Yamada, T. Sato and M. Suzuki, Appl. Clay Sci., 2016, 121, 71–76 CrossRef.
- Y. Lwin, M. A. Yarmo, Z. Yaakob, A. B. Mohamad and W. R. W. Daud, Mater. Res. Bull., 2001, 36, 193–198 CrossRef CAS.
- X. Wang, R. Cao, S. Zhang, P. Hou, R. Han, M. Shao and X. Xu, J. Mater. Chem. A, 2017, 5, 23999–24010 RSC.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- M. Chen, R. Zhu, X. Lu, J. Zhu and H. He, Inorg. Chem., 2018, 57, 7299–7313 CrossRef CAS PubMed.
- S. Mandal, S. Natarajan, S. Raja, N. Vijayalakshmi, C. Muralidharan and A. B. Mandal, Key Eng. Mater., 2013, 571, 57–69 Search PubMed.
- D. Wan, H. Liu, R. Liu, J. Qu, S. Li and J. Zhang, Chem. Eng. J., 2012, 195, 241–247 CrossRef.
- D. Wan, Y. Liu, S. Xiao, J. Chen and J. Zhang, Colloids Surf., A, 2015, 469, 307–314 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- V. K. Diez, C. R. Apesteguia and J. I. Di Cosimo, J. Catal., 2003, 215, 220–223 CrossRef CAS.
- V. L. Constantino and T. J. Pinnavaia, Inorg. Chem., 1995, 34, 883–892 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., 2005, 287, 183–190 CrossRef CAS.
- H. S. Chavan, C. H. Lee, A. I. Inamdar, J. Han, S. Park, S. Cho, N. K. Shreshta, S. U. Lee, B. Hou, H. Im and H. Kim, ACS Catal., 2022, 12, 3821–3831 CrossRef CAS.
- W. Jiang, A. Y. Faid, B. F. Gomes, I. Galkina, L. Xia, C. M. S. Lobo, M. Desmau, P. Borowski, H. Hartmann, A. Maljusch and A. Besmehn, Adv. Funct. Mater., 2022, 32, 2203520 CrossRef CAS.
- S. Lee, L. Bai and X. Hu, Angew. Chem., 2020, 132, 8149–8154 CrossRef.
- H. Zhang, D. Pan, K. Zou, J. He and X. Duan, J. Mater. Chem., 2009, 19, 3069–3077 RSC.
- M. Del Arco, S. Gutierrez, C. Martí, V. Rives and J. Rocha, J. Solid State Chem., 2000, 151, 272–280 CrossRef CAS.
- L. X. Zhang, J. Hu, Y. B. Jia, R. T. Liu, T. Cai and Z. P. Xu, Nanoscale, 2021, 13, 7533–7549 RSC.
- G. Su, H. Zhou, Q. Mu, Y. Zhang, L. Li, P. Jiao, G. Jiang and B. Yan, J. Phys. Chem. C, 2012, 116, 4993–4998 CrossRef CAS.
- J. M. Oh, S. J. Choi, S. T. Kim and J. H. Choy, Bioconjugate Chem., 2006, 17, 1411–1417 CrossRef CAS PubMed.
- J. M. Oh, S. J. Choi, G. E. Lee, J. E. Kim and J. H. Choy, Chem. – Asian J., 2009, 4, 67–73 CrossRef CAS PubMed.
- T. Yamaguchi, H. M. Kim, B. C. Jung, Y. S. Kim and J. M. Oh, Appl. Clay Sci., 2022, 225, 106549 CrossRef CAS.
- A. W. Musumeci, T. L. Schiller, Z. P. Xu, R. F. Minchin, D. J. Martin and S. V. Smith, J. Phys. Chem. C, 2010, 114, 734–740 CrossRef CAS.
- H. J. Kim, J. Y. Lee, T. H. Kim, G. H. Gwak, J. H. Park and J. M. Oh, Appl. Clay Sci., 2020, 186, 105454 CrossRef CAS.
- M. S. Usman, M. Z. Hussein, A. U. Kura, S. Fakurazi, M. J. Masarudin and F. F. A. Saad, Mater. Chem. Phys., 2020, 240, 122232 CrossRef CAS.
- T. H. Kim, J. Y. Lee, M. K. Kim, J. H. Park and J. M. Oh, RSC Adv., 2016, 6, 48415–48419 RSC.
- D. G. Jeung, T. H. Kim and J. M. Oh, Nanomater., 2021, 11, 44 CrossRef CAS PubMed.
- S. Mansingh, D. P. Sahoo, L. Paramanik, M. Sahoo and K. Parida, Inorg. Chem. Front., 2022, 9, 559–576 RSC.
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS.
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2021, 11, 2002816 CrossRef CAS.
- P. Li, M. Wang, X. Duan, L. Zheng, X. Cheng, Y. Zhan, Y. Kuang, Y. Li, Q. Ma, Z. Feng, W. Liu and X. Sun, Nat. Commun., 2019, 10, 1711 CrossRef PubMed.
- B. Zhang, C. Zhu, Z. Wu, E. Stavitski, Y. H. Lui, T.-H. Kim, H. Liu, L. Huang, X. Luan, L. Zhou, K. Jiang, W. Huang, S. Hu, H. Wang and J. S. Francisco, Nano Lett., 2020, 20, 136–144 CrossRef CAS.
- X. Meng, J. Han, L. Lu, G. Qiu, Z. L. Wang and C. Sun, Small, 2019, 15, 1902551 CrossRef CAS PubMed.
- Z. Cai, D. Zhou, M. Wang, S.-M. Bak, Y. Wu, Z. Wu, Y. Tian, X. Xiong, Y. Li, W. Liu, S. Siahrostami, Y. Kuang, X.-Q. Yang, H. Duan, Z. Feng, H. Wang and X. Sun, Angew. Chem., Int. Ed., 2018, 130, 9536–9540 CrossRef.
- Q. Li, G. Liu, Z. Wu, X. Sun, W. Chen and Y. F. Song, Chem. Eng. J., 2024, 2024, 154029 CrossRef.
- L. Wang, Z. Xiang, H. Zhang, Y. Deng, J. Wang, H. Xiao, W. Wang and X. Song, New J. Chem., 2024, 48, 5681–5695 RSC.
- P. Li, X. Duan, Y. Kuang, Y. Li, G. Zhang, W. Liu and X. Sun, Adv. Energy Mater., 2018, 8, 1703341 CrossRef.
- Y. Guo, Z. Zhu, Y. Qiu and J. Zhao, J. Hazard. Mater., 2012, 15, 239–240 Search PubMed.
- H. Jun, Z. Zhiliang, L. Hongtao and Q. Yanling, RSC Adv., 2014, 4, 5156–5164 RSC.
- E. M. Seftel, J. Spooren, M. Kun, P. Cool and B. Michielsen, J. Environ. Chem. Eng., 2024, 12, 112196 CrossRef CAS.
- B. R. Gevers, E. Roduner, A. Leuteritz and F. J. W. J. Labuschagné, Nanoscale, 2024, 16, 6449–6454 RSC.
- R. P. Wijitwongwan, S. G. Intasa-ard and M. Ogawa, Dalton Trans., 2024, 53, 6144–6156 RSC.
- M. Nogami, Y. Abe, K. Hirao and D. H. Cho, Appl. Phys. Lett., 1995, 66, 2952–2954 CrossRef.
- M. Nogami, X. H. Le and X. Q. Vu, J. Non-Cryst. Solids, 2019, 503–504, 260–267 CrossRef CAS.
- A. Tuel, Microporous Mesoporous Mater., 1999, 27, 151–169 CrossRef CAS.
- G. Lagaly, Adv. Colloid Interface Sci., 1979, 11, 105–148 CrossRef CAS.
- T. Selvam, A. Inayat and W. Schwieger, Dalton Trans., 2014, 43, 10365–10387 RSC.
- B. Marler, I. Grosskreuz and H. Gies, J. Solid State Chem., 2021, 300, 122215 CrossRef CAS.
- Y. Krysiak, M. Maslyk, B. N. Silva, S. Plana-Ruiz, H. M. Moura, E. O. Munsignatti, V. S. Vaiss, U. Kolb, W. Tremel, L. Palatinus, A. A. Leitão, B. Marler and H. O. Pastore, Chem. Mater., 2021, 33, 3207–3219 CrossRef CAS.
- S. Vortmann, J. Rius, S. Siegmann and H. Gies, J. Phys. Chem. B, 1997, 101, 1292–1297 CrossRef CAS.
- S. Vortmann, J. Rius, B. Marler and H. Gies, Eur. J. Mineral., 1999, 11, 125–134 CrossRef CAS.
- T. Ikeda, Y. Oumi, K. Honda, T. Sano, K. Momma and F. Izumi, Inorg. Chem., 2011, 50, 2294–2301 CrossRef CAS PubMed.
- H. P. Eugster, Science, 1967, 157, 1177–1180 CrossRef CAS.
- K. Kosuge, A. Yamazaki, A. Tsunashima and R. Otsuka, J. Ceram. Soc. Jpn., 1992, 100, 326–331 CrossRef CAS.
- K. Beneke and G. Lagaly, Am. Mineral., 1983, 68, 818–826 CAS.
- K. Endo, Y. Sugahara and K. Kuroda, Bull. Chem. Soc. Jpn., 1994, 67, 3352–3355 CrossRef CAS.
- T. G. Dos Santos, G. C. De Assis, A. O. S. Da Silva and S. M. P. Meneghetti, ACS Appl. Mater. Interfaces, 2023, 15, 43234–43250 CrossRef CAS.
- G. Pal-Borberly and A. Auroux, Stud. Surf. Sci. Catal., 1995, 94, 55–62 CrossRef.
- G. B. Superti, E. C. Oliveira, H. O. Pastore, A. Bordo, C. Bisio and L. Marchese, Chem. Mater., 2007, 19, 4300–4315 CrossRef CAS.
- W. Supronowicz, F. Roessner, W. Schwieger, M. Meilikhov and D. Eskenm, Clays Clay Miner., 2012, 60, 254–264 CrossRef CAS.
- Y. Bi, J. Blanchard, J. F. Lambert, Y. Millot, S. Casale, S. Zeng, H. Nie and D. Li, Appl. Clay Sci., 2012, 57, 71–78 CrossRef CAS.
- M. E. R. Oliveira, E. C. da Silva Filho, J. M. Filho, S. S. Ferreira, A. C. Oliveira and A. F. Campos, Chem. Eng. J., 2015, 263, 257–267 CrossRef CAS.
- G. L. Paz, E. C. O. Munsignatti and H. O. Pastore, J. Mol. Catal. A: Chem., 2016, 422, 43–50 CrossRef CAS.
- R. K. S. Almeida, G. L. Paz, G. B. Báfero and H. O. Pastore, Microporous Mesoporous Mater., 2019, 284, 1–9 CrossRef CAS.
- T. G. Santos, A. O. S. Silva and S. M. P. Meneghetti, Appl. Clay Sci., 2019, 183, 105293 CrossRef CAS.
- G. B. Báfero, B. N. N. Silva, A. A. Leitão and H. O. Pastore, Mol. Catal., 2023, 535, 112870 CrossRef.
- S. Inagaki, Y. Yamada and Y. Fukushima, Stud. Surf. Sci. Catal., 1997, 105, 109–116 CrossRef.
- Q. Kan, V. Fornés, F. Rey and A. Corma, J. Mater. Chem., 2000, 10, 993–1000 RSC.
- T. Shigeno, K. Inoue, T. Kimura, N. Katada, M. Niwa and K. Kuroda, J. Mater. Chem., 2003, 13, 883–887 RSC.
- T. Kimura, M. Suzuki, T. Ikeda, K. Kato, M. Maeda and S. Tomura, Microporous Mesoporous Mater., 2006, 95, 146–153 CrossRef CAS.
- G. Pal-Borberly, H. Beyer, Y. Kiyozumi and F. Mizukami, Microporous Mesoporous Mater., 1998, 22, 57–68 CrossRef.
- E. M. Barea, V. Fornés, A. Corma, P. Bourges, E. Guillon and V. F. Puntes, Chem. Commun., 2004, 1974–1975 RSC.
- S. Neatu, M. Puche, V. Fornes and H. Garcia, Chem. Commun., 2014, 50, 14643–14646 RSC.
- F. S. O. Ramos and H. O. Pastore, Dalton Trans., 2017, 46, 11728–11737 RSC.
- M. D. Alba, P. Chain and E. Pavon, Microporous Mesoporous Mater., 2006, 94, 66–73 CrossRef CAS.
- W. Schwieger and E. Brunner, Colloid Polym. Sci., 1992, 270, 935–938 CrossRef CAS.
- W. Schwieger, K. Pohl, U. Brenn, C. A. Fyfe, H. Grondey, G. Fu and G. T. Kokotailo, Stud. Surf. Sci. Catal., 1995, 94, 47–54 CrossRef CAS.
- C. A. Fyfe, J. Skibsted and W. Schwieger, Inorg. Chem., 2001, 40, 5906–5912 CrossRef CAS.
- H. M. Moura, F. A. Bonk, R. C. G. Vinhas, R. Landers and H. O. Pastore, CrystEngComm, 2011, 13, 5428–5438 RSC.
- M. Morita, Y. Horiuchi, M. Matsuoka and M. Ogawa, Cryst. Growth Des., 2022, 22, 1638–1644 CrossRef CAS.
- W. Supronowicz and F. Roessner, Clays Clay Miner., 2011, 59, 95–105 CrossRef CAS.
- Q. Sun, X. Guo, B. Guo, Q. Tang, W. Yu, Q. Wan and Y. An, Appl. Clay Sci., 2023, 231, 106745 CrossRef CAS.
- C. T. G. V. M. T. Pires, J. R. Costa and C. Airoldi, Microporous Mesoporous Mater., 2012, 163, 1–10 CrossRef CAS.
- N. Takahashi and K. Kuroda, J. Mater. Chem., 2011, 21, 14336–14353 RSC.
- W. J. Roth, P. Nachtigall, R. E. Morris and J. Čejka, Chem. Rev., 2014, 114, 4807–4837 CrossRef CAS.
- M. Koike, I. Grosskreuz, Y. Asakura, R. Miyawaki, H. Gies, H. Wada, A. Shimojima, B. Marler and K. Kuroda, Chem. – Eur. J., 2023, e202301942 CrossRef CAS PubMed.
- S. Zanardi, A. Alberti, G. Cruciani, A. Corma, V. Fornés and M. Brunelli, Angew. Chem., Int. Ed., 2004, 43, 4933–4937 CrossRef CAS PubMed.
- T. Ikeda, Y. Akiyama, Y. Oumi, A. Kawai and F. Mizukami, Angew. Chem., Int. Ed., 2004, 43, 4892–4896 CrossRef CAS.
- B. Marler, N. Ströter and H. Gies, Microporous Mesoporous Mater., 2005, 83, 201–211 CrossRef CAS.
- Y. X. Wang, H. Gies, B. Marler and U. Müller, Chem. Mater., 2005, 17, 43–49 CrossRef CAS.
- Y. Ide, S. Tominaka, H. Kono, R. Ram, A. Machida and N. Tsunoji, Chem. Sci., 2018, 9, 8637–8643 RSC.
- G. Lagaly, K. Beneke and A. Weiss, Am. Mineral., 1975, 60, 642–649 CAS.
- G. Lagaly, K. Beneke and A. Weiss, Am. Mineral., 1975, 60, 650–658 CAS.
- G. Lagaly, Solid State Ionics, 1986, 22, 43–51 CrossRef CAS.
- M. Ogawa, K. Saito and M. Sohmiya, Dalton Trans., 2014, 43, 10340–10354 RSC.
- C. Tirayaphanitchkul, K. Imwiset and M. Ogawa, Bull. Chem. Soc. Jpn., 2021, 94, 678–693 CrossRef CAS.
- E. Doustkhah and Y. Ide, New J. Chem., 2020, 44, 9957–9968 RSC.
- N. Tsunoji, T. Ikeda, Y. Ide, M. Sadakane and T. Sano, J. Mater. Chem., 2012, 22, 13682–13690 RSC.
- N. Tsunoji, M. Fukuda, K. Yoshida, Y. Sasaki, T. Ikeda, Y. Ide, M. Sadakane and T. Sano, J. Mater. Chem. A, 2013, 1, 9680–9688 RSC.
- G. J. Ray, B. L. Meyers and C. L. Marshall, Zeolites, 1987, 7, 307–310 CrossRef CAS.
- C. A. Fyfe, G. C. Gobbi, W. J. Murphy, R. S. Ozubko and D. A. Slack, Chem. Lett., 1983, 1547–1550 CrossRef CAS.
- B. Notari, Adv. Catal., 1996, 41, 253–334 CAS.
- C. Perego, A. Carati, P. Ingallina, M. A. Mantegazza and G. Bellussi, Appl. Catal., A, 2001, 221, 63–72 CrossRef CAS.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321–323 CrossRef CAS PubMed.
- A. Corma, M. T. Navarro and J. P. Pariente, J. Chem. Soc., Chem. Commun., 1994, 147–148 RSC.
- K. Koyano and T. Tatsumi, Chem. Commun., 1996, 145–146 RSC.
- W. Zhang, M. Fröba, J. Wang, P. T. Tanev, J. Wong and T. J. Pinnavaia, J. Am. Chem. Soc., 1996, 118, 9164–9171 CrossRef CAS.
- M. Ogawa, K. Ikeue and M. Anpo, Chem. Mater., 2001, 13, 2900–2904 CrossRef CAS.
- K. J. Imwiset and M. Ogawa, Inorg. Chem., 2021, 60, 9563–9570 CrossRef CAS.
- T. Yamaguchi, K. J. Imwiset, M. G. Choi, J. M. Oh, S. Y. Lee and M. Ogawa, Appl. Clay Sci., 2023, 236, 106882 CrossRef CAS.
- E. Ruiz-Hitzky and J. M. Rojo, Nature, 1980, 287, 28–30 CrossRef CAS.
- E. Ruiz-Hitzky, J. M. Rojo and G. Lagaly, Colloid Polym. Sci., 1985, 263, 1025–1030 CrossRef CAS.
- M. Ogawa, S. Okutomo and K. Kuroda, J. Am. Chem. Soc., 1998, 120, 7361–7362 CrossRef CAS.
- Y. Ide and M. Ogawa, Bull. Chem. Soc. Jpn., 2007, 80, 1624–1629 CrossRef CAS.
- E. Ruiz-Hitzky, P. Aranda, M. Darder and M. Ogawa, Chem. Soc. Rev., 2011, 40, 801–828 RSC.
- M. Yatomi, M. Koike, N. Rey, Y. Murakami, S. Saito, H. Wada, A. Shimojima, D. Portehault, S. Carenco, C. Sanchez, C. Carcel, M. Wong Chi Man and K. Kuroda, Eur. J. Inorg. Chem., 2021, 1836–1845 CrossRef CAS.
- M. Nomi, M. Morita, A. Kondo and K. Maeda, Inorg. Chem., 2022, 61, 5255–5261 CrossRef CAS.
- D. Sruamsiri, A. Shimojima and M. Ogawa, ACS Appl. Mater. Interfaces, 2023, 15, 41130–41140 CrossRef CAS PubMed.
- E. Doustkhah, N. Tsunoji, M. H. N. Assadi and Y. Ide, Adv. Mater. Interfaces, 2023, 10, 2202368 CrossRef CAS.
- Y. Ide, N. Ochi and M. Ogawa, Angew. Chem., Int. Ed., 2011, 50, 654–656 CrossRef CAS.
- N. Homhuan, S. Bureekaew and M. Ogawa, Langmuir, 2017, 33, 9558–9564 CrossRef CAS.
- D. Sruamsiri, T. Sirinakorn and M. Ogawa, Clays Clay Miner., 2021, 69, 416–424 CrossRef CAS.
- K. Yamamoto, S. E. Borjas Garcia, F. Saito and A. Muramatsu, Chem. Lett., 2006, 35, 570–571 CrossRef CAS.
- K. Yamamoto, S. E. Borjas García and A. Muramatsu, Microporous Mesoporous Mater., 2007, 101, 90–96 CrossRef CAS.
- S. E. Borjas Garcia, K. Yamamoto and A. Muramatsu, J. Mater. Sci., 2008, 43, 2367–2371 CrossRef.
- M. Yabushita, H. Kobayashi, R. Osuga, M. Nakaya, M. Matsubara, S. Maki, K. Kanie and A. Muramatsu, Ind. Eng. Chem. Res., 2021, 60, 2079–2088 CrossRef CAS.
- I. A. Crone, K. R. Franklin and P. Graham, J. Mater. Chem., 1995, 5, 2007–2011 RSC.
- M. Yatomi, T. Hikino, S. Yamazoe, K. Kuroda and A. Shimojima, Dalton Trans., 2023, 52, 18158–18167 RSC.
- H. Inada, M. Morita and K. Maeda, Dalton Trans., 2024, 53, 13756–13763 RSC.
- R. E. Grim, Applied Clay Mineralogy, McGraw-Hill Inc, US, 1962 Search PubMed.
- F. Bergaya, B. K. G. Theng and G. Lagaly, Handbook of Clay Science, Elsevier Science, Amsterdam, 2006 Search PubMed.
- B. K. G. Theng, The Chemistry of Clay-Organic Reactions, 2nd edn, CRC Press, Boca Raton, 2024 Search PubMed.
- H. van Olphen, An Introduction to Clay Colloid Chemistry, Wiley-Interscience, New York, 1977 Search PubMed.
- H. Iwase, M. Kubota, T. Itoh, T. Ogura, T. Ebina, H. Ohtani, K. Kurosawa and Y. Fukushima, Langmuir, 2021, 37, 6435–6441 CrossRef CAS PubMed.
- N. Miyamoto and T. Nakato, Isr. J. Chem., 2012, 52, 881–894 CrossRef CAS.
- N. Yamaguchi, S. Anraku, E. Paineau, C. R. Safinya, P. Davidson, L. J. Michot and N. Miyamoto, Sci. Rep., 2018, 8, 4367 CrossRef.
- N. Miyamoto and S. Yamamoto, ACS Omega, 2022, 7, 6070–6074 CrossRef CAS.
- T. Inadomi, K. Urayama and N. Miyamoto, ACS Appl. Polym. Sci., 2022, 4, 4664–4672 CAS.
- M. Ogawa, M. Takahashi, C. Kato and K. Kuroda, J. Mater. Chem., 1994, 4, 519–523 RSC.
- R. A. Schoonheydt, Appl. Clay Sci., 2014, 96, 9–21 CrossRef CAS.
- P. Das, J.-M. Malho, K. Rahimi, F. H. Schacher, B. Wang, D. E. Demco and A. Walther, Nat. Commun., 2015, 6, 5967 CrossRef.
- A. P. Teepakakorn and M. Ogawa, Langmuir, 2021, 37, 12887–12896 CrossRef CAS.
- K. Inukai, Y. Hotta, M. Taniguchi, S. Tomura and A. Yamagishi, J. Chem. Soc., Chem. Commun., 1994, 959 RSC.
- E. R. Kleinfeld and G. S. Ferguson, Science, 1994, 265, 370–373 CrossRef CAS.
- J. R. Kovacs, C. Liu and P. T. Hammond, ACS Appl. Mater. Interfaces, 2015, 7, 13375–13383 CrossRef CAS PubMed.
- B. V. Lotsch and G. A. Ozin, Adv. Mater., 2008, 20, 4079–4084 CrossRef CAS.
- Y. Fukushima and S. Inagaki, J. Inclusion Phenom., 1987, 5, 473–482 CrossRef CAS.
- K. Haraguchi and H. J. Li, Macromolecules, 2006, 39, 1898–1905 CrossRef CAS.
- K. Haraguchi, T. Takada and R. Haraguchi, ACS Appl. Nano Mater., 2018, 1, 418–425 CrossRef CAS.
- T. Ebina, Chem. Rec., 2018, 18, 1020–1032 CrossRef CAS.
- C. Zhao, P. Zhang, J. Zhou, S. Qi, Y. Yamauchi, R. Shi, R. Fang, Y. Ishida, S. Wang, A. P. Tomsia, M. Liu and L. Jiang, Nature, 2020, 58, 210–215 CrossRef.
- T. Aizawa, M. Kubota and T. Ebina, Appl. Clay Sci., 2022, 226, 106571 CrossRef CAS.
- T. R. Jones, Clay Miner., 1983, 18, 399–410 CrossRef CAS.
- M. Minase, T. Hayakawa, M. Oya, K. Fujita and M. Ogawa, Bull. Chem. Soc. Jpn., 2019, 92, 1329–1334 CrossRef CAS.
- T. Okada, M. Sohmiya and M. Ogawa, Struct. Bonding, 2015, 166, 177–211 CrossRef CAS PubMed.
- T. Yamaguchi, J.-M. Oh and M. Ogawa, Struct. Bonding, 2020, 183, 251–320 CrossRef CAS.
- T. Yamaguchi and M. Ogawa, Sci. Technol. Adv. Mater., 2022, 23, 796–844 CrossRef CAS.
- S. Takagi, T. Shimada, Y. Ishida, T. Fujimura, D. Masui, H. Tachibana, M. Eguchi and H. Inoue, Langmuir, 2013, 29, 2108–2119 CrossRef CAS PubMed.
- Y. Suzuki, Y. Tenma, Y. Nishioka and J. Kawamata, Chem. – Asian J., 2012, 7, 1170–1179 CrossRef CAS.
- W. Soontornchaiyakul, K. Takada, T. Kaneko and M. Ogawa, Inorg. Chem., 2024, 63, 2787–2792 CrossRef CAS.
- T. Okada, Y. Ide and M. Ogawa, Chem. – Asian J., 2012, 7, 1980–1992 CrossRef CAS PubMed.
- A. Phuekphong, K. Imwiset and M. Ogawa, J. Hazard. Mater., 2020, 399, 122888 CrossRef.
- A. Yamagishi, K. Tamura, M. Kamon, J. Yoshida and H. Sato, Appl. Clay Sci., 2024, 251, 107290 CrossRef CAS.
- A. Yamagishi and H. Sato, Clays Clay Miner., 2012, 60, 411–419 CrossRef CAS.
- T. J. Pinnavaia, Science, 1983, 220, 365–371 CrossRef CAS.
- C. Breen, F. D. Zahoor, J. Madejová and P. Komadel, J. Phys. Chem. B, 1997, 101, 5324–5331 CrossRef CAS.
- R. Miyawaki, T. Sano, F. Ohashi, M. Suzuki, T. Kogure, T. Okumura, J. Kameda, T. Umezome, T. Sato, D. Chino, K. Hiroyama, H. Yamada, K. Tamura, K. Morimoto, S. Uehara and T. Hatta, Nendo Kagaku, 2010, 48, 158–198 CAS.
- The Clay Minerals Society, https://www.clays.org/source-and-special-clays/, (accessed March 2024).
- The Clay Science Society of Japan, https://www.cssj2.org/english/reference_clay_e/, (accessed March 2024).
- E. A. Warren and B. Ransom, Clay Miner., 1992, 27, 193–209 CrossRef CAS.
- W. Loewenstein, Am. Mineral., 1954, 39, 92–96 CAS.
- C. S. Ross and S. B. Hendricks, in Minerals of the montmorillonite group, their origin and relation to soils and clays, ed. C. S. Ross and S. B. Hendricks, U.S. Geol. Survey Prof. Paper, 1945, ch. 205-B, pp. 23–79 Search PubMed.
- D. R. Lewis, in Analytical data on reference clay materials, ed. P. F. Kerr, Columbia University, New York, Preliminary Rept. no. 7, Reference Clay Minerals, A.P.I. Project 49, 1950, pp. 91–124 Search PubMed.
- A. Mehlich, Soil Sci., 1948, 66, 429–446 CrossRef CAS.
- C. L. Bascomb, J. Sci. Food Agric., 1964, 15, 821–823 CrossRef CAS.
- R. Dohrmann, Appl. Clay Sci., 2006, 34, 31–37 CrossRef CAS.
- M. Ogawa, Y. Nagafusa, K. Kuroda and C. Kato, Appl. Clay Sci., 1992, 7, 291–302 CrossRef CAS.
- P. T. Hang and G. W. Brindley, Clays Clay Miner., 1970, 10, 203–212 CrossRef.
- ASTM International, ASTM C837-09(2019), https://www.astm.org/c0837-09r19.html, (accessed March 2024).
- R. Dohrmann, Appl. Clay Sci., 2006, 34, 38–46 CrossRef CAS.
- F. Bergaya and M. Vayer, Appl. Clay Sci., 1997, 12, 275–280 CrossRef CAS.
- R. E. Grim, W. H. Allaway and F. L. Cuthbert, J. Am. Ceram. Soc., 1947, 30, 137–142 CrossRef CAS.
- M. F. Traynor, M. M. Mortland and T. J. Pinnavaia, Clays Clay Miner., 1978, 26, 318–326 CrossRef.
- M. Koizumi and R. Roy, Am. Mineral., 1959, 44, 788–805 CAS.
- J. Méring and G. Perdo, Bull. Groupe Fr. Argiles, 1969, 21, 1–30 CrossRef.
- J.-F. Joly, L. Huve, P. Martin, R. Le Dred, D. Saehr and J. Baron, Eur. Pat., 92/400522.6, 1992 Search PubMed.
- J. Breu, W. Seidl, A. J. Stoll, K. G. Lange and T. U. Probst, Chem. Mater., 2001, 13, 4213–4220 CrossRef CAS.
- T. Egawa, H. Watanabe, T. Fujimura, Y. Ishida, M. Yamato, D. Masui, T. Shimada, H. Tachibana, H. Yoshida, H. Inoue and S. Takagi, Langmuir, 2011, 27, 10722–10729 CrossRef CAS PubMed.
- A. Decarreau, O. Grauby and S. Petit, Appl. Clay Sci., 1992, 7, 147–167 CrossRef CAS.
- J. T. Iiyama and R. Roy, Clay Miner. Bull., 1963, 5, 161–171 CrossRef CAS.
- H. Suquet, J. T. Iiyama, H. Kodama and H. Pezerat, Clays Clay Miner., 1977, 25, 231–242 CrossRef CAS.
- R. J. M. J. Vogels, T. J. Kloprogge, J. W. Geus and A. W. F. Beers, Am. Mineral., 2005, 90, 945–953 CrossRef CAS.
- R. J. M. J. Vogels, T. J. Kloprogge and J. W. Geus, Am. Mineral., 2005, 90, 931–944 CrossRef CAS.
- R. J. M. J. Vogels, T. J. Kloprogge and J. W. Geus, J. Catal., 2005, 231, 443–452 CrossRef CAS.
- M. Reinholdt, J. Miehe-Brendle, L. Delmotte, M. H. Tuilier, R. le Dred, R. Cortes and A. M. Flank, Eur. J. Inorg. Chem., 2001, 2831–2841 CrossRef CAS.
- K. Torii and T. Iwasaki, Chem. Lett., 1988, 2045–2048 CrossRef CAS.
- V. C. Farmer, Clays Clay Miner., 1997, 45, 591–597 CrossRef CAS.
- BYK Additives & Instruments, https://www.byk.com/en/products/additives-by-name/laponite-rd, (accessed March 2024).
- N. Finck, K. Dardenne and H. Geckeis, Chem. Geol., 2015, 409, 12–19 CrossRef CAS.
- H. Brandt, D. Bosbach, P. J. Panak and T. Fanghänel, Geochim. Cosmochim. Acta, 2007, 71, 145–154 CrossRef CAS.
- P. H. Michels-Brito, V. Dudko, D. Wagner, P. Markus, G. Papastavrou, L. Michels, J. Breu and J. O. Fossum, Sci. Adv., 2022, 8, eabl8147 CrossRef.
- P. Komadel, J. Madejova and J. W. StuckIi, Clays Clay Miner., 1999, 47, 458–465 CrossRef CAS.
- H. Yamada, K. Yoshioka and H. Nakazawa, Mineral. J., 1991, 15, 300–308 CrossRef CAS.
- H. Yamada, H. Nakazawa, K. Yoshioka and T. Fujita, Clay Miner., 1991, 26, 359–369 CrossRef CAS.
- J. T. Kloprogge, S. Komarneni and J. E. Amonetie, Clays Clay Miner., 1999, 47, 529–554 CrossRef CAS.
- K. Urabe, H. Sakurai and Y. Izumi, J. Chem. Soc., Chem. Commun., 1988, 1520–1521 RSC.
- M. Ogawa, R. Kawai and K. Kuroda, J. Phys. Chem. B, 1996, 100, 16218–16221 CrossRef CAS.
- P. Saengdet and M. Ogawa, Chem. Commun., 2022, 58, 3278–3281 RSC.
- Y. Ishida, T. Shimada, H. Tachibana, H. Inoue and S. Takagi, J. Phys. Chem. A, 2012, 116, 12065–12072 CrossRef CAS PubMed.
- C. H. Zhou, Q. Zhou, Q. Q. Wu, S. Petit, X. C. Jiang, S. T. Xia, C. S. Li and W. H. Yu, Appl. Clay Sci., 2018, 168, 136–154 CrossRef.
- R. S. Varma, Tetrahedron, 2002, 58, 1235–1255 CrossRef CAS.
- H. Heinz, H. Koerner, K. L. Anderson, R. A. Vaia and B. L. Farmer, Chem. Mater., 2005, 17, 5658–5669 CrossRef CAS.
- W. Shen, L. Li, H. Zhou, Q. Zhou, M. Chen and J. Zhu, Appl. Clay Sci., 2016, 159, 10–15 CrossRef.
- K. Ohtsuka, Chem. Mater., 1997, 9, 2039–2050 CrossRef CAS.
- P. Laszlo, Science, 1987, 235, 1473–1477 CrossRef CAS PubMed.
- M. Arai, S. L. Guo, M. Shirai, Y. Nishiyama and K. Torii, J. Catal., 1996, 161, 704–712 CrossRef CAS.
- B. Zhoufeng and K. Sibudjing, J. CO2 Util., 2017, 18, 345–352 CrossRef.
- S. Higashi, K. Miki and S. Komarneni, Clays Clay Miner., 2002, 50, 299–305 CrossRef CAS.
- C. Q. Zhang, H. P. He, Q. Tao, S. C. Ji, S. Y. Li, L. Y. Ma, X. L. Su and J. X. Zhu, Appl. Clay Sci., 2017, 135, 282–288 CrossRef CAS.
- V. Luca, D. J. MacLachlan, R. F. Howe and R. Bramley, J. Mater. Chem., 1995, 5, 557–564 RSC.
- A. Phuekphong, T. Hayakawa and M. Ogawa, Chem. Commun., 2022, 58, 12661–12664 RSC.
- M. Ogawa, T. Matsutomo and T. Okada, Bull. Chem. Soc. Jpn., 2009, 82, 408–412 CrossRef CAS.
- T. B. Hofstetter, R. P. Schwarzenbach and S. B. Haderlein, Environ. Sci. Technol., 2003, 37, 519–528 CrossRef CAS PubMed.
- J. R. White and A. J. Bard, J. Electroanal. Chem., 1986, 197, 233–244 CrossRef CAS.
- B. K. G. Theng, Clays Clay Miner., 1971, 19, 383–390 CrossRef CAS.
- D. T. B. Tennakoon, J. M. Thomas, M. J. Tricker and J. O. Williams, J. Chem. Soc., Dalton Trans., 1974, 20, 2207–2211 RSC.
- Y. Qian, A. C. Scheinost, S. Grangeon, A. Hoving, S. V. Churakov and M. M. Fernandes, Geochim. Cosmochim. Acta, 2024, 377, 19–33 CrossRef CAS.
- F. Bergaya and H. V. Damme, J. Chem. Soc., Faraday Trans., 1983, 79, 505–518 RSC.
- A. Habti, D. Keravis, P. Levitz and H. van Damme, J. Chem. Soc., Faraday Trans. 2, 1984, 80, 67–83 RSC.
- R. A. Schoonheydt, P. De Pauw, D. Vliers and F. C. De Schrijver, J. Phys. Chem., 1984, 88, 5113–5118 CrossRef CAS.
- P. A. Teepakakorn, S. Bureekaew and M. Ogawa, Langmuir, 2018, 34, 14069–14075 CrossRef.
- A. P. Teepakakorn, T. Yamaguchi and M. Ogawa, Chem. Lett., 2019, 48, 398–409 CrossRef CAS.
- A. Nag, T. Hayakawa, M. Minase and M. Ogawa, Langmuir, 2022, 38, 2979–2985 CrossRef CAS PubMed.
- P. M. Saengdet and M. Ogawa, Langmuir, 2024, 40, 1016–1023 CrossRef CAS.
- J. W. Stucki, D. C. Golden and C. B. Roth, Clays Clay Miner., 1984, 32, 350–356 CrossRef CAS.
- J. W. Stucki, P. F. Low, C. B. Roth and D. C. Golden, Clays Clay Miner., 1984, 32, 357–362 CrossRef CAS.
- J. W. Stucki, J. Wu, H. Gan, P. Komadel and A. Banin, Clays Clay Miner., 2000, 48, 290–298 CrossRef CAS.
- S. Petit, F. Baron and A. Decarreau, Clay Miner., 2017, 52, 469–483 CrossRef CAS.
- S. Caillere, S. Henin and J. Esquevin, J. Bull. Soc. Francaise Miner. Cristallogr., 1955, 78, 227–241 CAS.
- H. Harder, Clays Clay Miner., 1978, 26, 65–72 CrossRef CAS.
- A. Decarreau, D. Bonnin, D. Badaut-Trauth, R. Couty and P. Kaiser, Clays Clay Miner., 1987, 22, 207–223 CrossRef CAS.
- K. Tomita, H. Yamane and M. Kawano, Clays Clay Miner., 1993, 41, 655–661 CrossRef CAS.
- V. C. Farmer, W. J. McHardy, F. Elsass and M. Robert, Clays Clay Miner., 1994, 42, 180–186 CrossRef CAS.
- A. Manceau, V. A. Drits, B. Lanson, D. Chateigner, J. Wu, D. Huo, W. P. Gates and J. W. Stucki, Am. Mineral., 2000, 85, 153–172 CrossRef CAS.
- O. Grauby, S. Petit, A. Decarreau and A. Baronnet, Eur. J. Mineral., 1994, 6, 99–112 CrossRef CAS.
- S. Higashi, K. Miki and S. Komarneni, Appl. Clay Sci., 2007, 38, 104–112 CrossRef CAS.
- N. J. Tosca and A. L. Masterson, Clay Miner., 2014, 49, 165–194 CrossRef.
- M. Reinholdt, J. Miehe-Brendle, L. Delmotte, M. H. Tuilier and R. le Dred, Clay Miner., 2001, 40, 177–190 CrossRef.
- U. Hofmann and R. Klemen, Z. Anorg. Chem., 1950, 262, 95–99 CrossRef CAS.
- P. Komadel, J. Madejova and K. Bujdak, Clays Clay Miner., 2005, 53, 313–334 CrossRef CAS.
- K. Takahashi, R. Ishii, T. Nakamura, A. Suzuki, T. Ebina, M. Yoshida, M. Kubota, T. T. Nge and T. Yamada, Adv. Mater., 2017, 29, 1606512 CrossRef.
- J. Bujdak and N. Iyi, Clays Clay Miner., 2002, 50, 446–454 CrossRef CAS.
- J. Bujdak, J. Phys. Chem. C, 2015, 119, 12016–12022 CrossRef CAS.
- W. F. Jaynes and S. A. Boyd, Clays Clay Miner., 1991, 39, 428–436 CrossRef CAS.
- R. Zhu, L. Zhu and L. Xu, Colloids Surf., A, 2007, 294, 221–227 CrossRef CAS.
- R. Zhu, J. Zhao, F. Ge, L. Zhu, J. Zhu, Q. Tao and H. He, Appl. Clay Sci., 2014, 88–89, 73–77 CrossRef CAS.
- A. L. Barrientos-Velázquez, A. M. Cardona, L. Liu, T. Phillips and Y. Deng, Appl. Clay Sci., 2016, 132–133, 281–289 CrossRef.
- S. Jatav and Y. M. Joshi, Appl. Clay Sci., 2014, 97–98, 72–77 CrossRef CAS.
- J. Zhang, C.-H. Zhou, S. Petit and H. Zhang, Appl. Clay Sci., 2019, 177, 114–138 CrossRef CAS.
- W. T. Granquist and S. S. Pollack, Clays Clay Miner., 1960, 8, 150–169 CAS.
- K. A. Carrado, P. Thiyagarajan and K. Song, Clay Miner., 1997, 32, 29–40 CrossRef CAS.
- K. A. Carrado, L. Xu, D. M. Gregory, K. Song, S. Seifert and R. E. Botto, Chem. Mater., 2000, 12, 3052–3059 CrossRef CAS.
- N. Finck, T. Stumpf, C. Walther and D. Bosbach, J. Contam. Hydrol., 2008, 102, 253–262 CrossRef CAS.
- T. Iwasaki, M. Reinikainen, Y. Onodera, H. Hayashi, T. Ebina, T. Nagase, K. Torii, K. Kataja and A. Chatterjee, Appl. Surf. Sci., 1998, 130–132, 845–850 CrossRef CAS.
- M. Ogawa, T. Matsutomo and T. Okada, J. Ceram. Soc. Jpn., 2008, 116, 1309–1313 CrossRef CAS.
- T. Okada, T. Matsutomo and M. Ogawa, J. Phys. Chem. C, 2010, 114, 539–545 CrossRef CAS.
- T. Okada, A. Suzuki, S. Yoshido and H. M. Minamisawa, Microporous Mesoporous Mater., 2015, 215, 168–174 CrossRef CAS.
- T. Sánchez, P. Salagre and Y. Cesteros, Microporous Mesoporous Mater., 2013, 171, 24–34 CrossRef.
- G. Wang, J. Wei, G. Liang, Y. Chen, S. Ma, J. Zhu and H. Liu, Colloids Surf., A, 2024, 687, 133475 CrossRef CAS.
- S. Petit, D. Righi and A. Decarreau, Clays Clay Miner., 2008, 56, 645–654 CrossRef CAS.
- H. Harder, Clay Miner., 1977, 12, 281–288 CrossRef CAS.
- K. Tomita, H. Yamane and M. Kawano, Clays Clay Miner., 1993, 41, 655–661 CrossRef CAS.
- H. Harder, Chem. Geol., 1976, 18, 169–180 CrossRef CAS.
- M. Jaber and J. Miehé-Brendlé, Microporous Mesoporous Mater., 2008, 107, 121–127 CrossRef CAS.
- J. Brendlé, Dalton Trans., 2018, 47, 2925–2932 RSC.
- A. J. Patil, E. Muthusamy and S. Mann, Angew. Chem., Int. Ed., 2004, 43, 4928–4933 CrossRef CAS PubMed.
- K. K. R. Datta, A. Achari and M. Eswaramoorthy, J. Mater. Chem. A, 2013, 1, 6707–6718 RSC.
- M. Claverie, A. Dumas, C. Carême, M. Poirier, C. Le Roux, P. Micoud, F. Martin and C. Aymonier, Chem. – Eur. J., 2018, 24, 519–542 CrossRef CAS PubMed.
- Y. Fukushima and M. Tani, J. Chem. Soc., Chem. Commun., 1995, 241–242 RSC.
- Y. Fukushima and M. Tani, Bull. Chem. Soc. Jpn., 1996, 69, 3667–3671 CrossRef CAS.
- M. Jaber and J. Miehé-Brendlé, J. Mater. Sci., 2004, 39, 1489–1490 CrossRef CAS.
- T. Okada, N. Taguchi and S. Shimomura, Colloids Surf., A, 2023, 676, 132135 CrossRef CAS.
- M. Jaber, J. Miehé-Brendlé, L. Delmotte and R. Le Dred, Microporous Mesoporous Mater., 2003, 65, 155–163 CrossRef CAS.
- M. Jaber, J. Miehé-Brendlé, M. Roux, J. Dentzer, R. Le Dred and J. Guth, New J. Chem., 2002, 26, 1597–1600 RSC.
- M. Yamauchi and T. Okada, Clays Clay Miner., 2018, 66, 104–113 CrossRef CAS.
- K. A. Carrado, L. Xu, R. Csencsits and J. V. Muntean, Chem. Mater., 2001, 13, 3766–3773 CrossRef CAS.
- H. Yamada, Nendo Kagaku, 2001, 41, 87–96 CAS.
- K. A. Carrado, Appl. Clay Sci., 2000, 17, 1–23 CrossRef CAS.
- H. Yamada, H. Nakazawa and H. Hashizume, Clays Clay Miner., 1994, 42, 674–678 CrossRef CAS.
- M. M. Herling, H. Kalo, S. Seibt, R. Schobert and J. Breu, Langmuir, 2012, 28, 14713–14719 CrossRef CAS PubMed.
- M. Stöter, D. A. Kunz, M. Schmidt, D. Hirsemann, H. Kalo, B. Putz, J. Senker and J. Breu, Langmuir, 2013, 29, 1280–1285 CrossRef.
- M. Röhrl, R. L. Timmins, S. Rosenfeldt, D. D. Schuchardt, F. Uhlig, S. Nürmberger and J. Breu, ACS Appl. Mater. Interfaces, 2023, 15, 22524–22531 CrossRef.
- D. D. Schuchardt, S. Rosenfeldt, H. Kalo and J. Breu, Nanoscale, 2023, 15, 7044–7050 RSC.
- C. Habel, E. S. Tsurko, R. L. Timmins, J. Hutschreuther, R. Kunz, D. D. Schuchardt, S. Rosenfeldt, V. Altstädt and J. Breu, ACS Nano, 2020, 14, 7018–7024 CrossRef CAS PubMed.
- M. Stöcker, W. Seidl, L. Seyfarth, J. Senker and J. Breu, Chem. Commun., 2008, 629–631 RSC.
- M. Stöcker, L. Seyfarth, D. Hirsemann, J. Senker and J. Breu, Appl. Clay Sci., 2010, 48, 146–153 CrossRef.
- R. Mariychuk, A. Baumgartner, F. E. Wagner, A. Lerf, A. Dubbe, R. Moos and J. Breu, Chem. Mater., 2007, 19, 5377–5387 CrossRef CAS.
- A. Baumgartner, K. Sattler, J. Thun and J. Breu, Angew. Chem., Int. Ed., 2008, 47, 1640–1644 CrossRef.
- R. Greene-Kelly, J. Soil Sci., 1953, 4, 233–237 CAS.
- E. N. Skoubris, G. D. Chryssikos, G. E. Christidis and V. Gionis, Clays Clay Miner., 2013, 61, 83–97 CrossRef CAS.
- G. E. Christidis and D. D. Eberl, Clays Clay Miner., 2013, 61, 644–655 Search PubMed.
- A. R. Mermut, Layer Charge Characteristics of 2:1 Silicate Clay Minerals, CMS workshop lectures, Clay Miner. Society, Aurora, USA, 1994, vol. 6 Search PubMed.
- J. W. Jordan, J. Phys. Colloid Chem., 1950, 54, 294–306 CrossRef PubMed.
- G. Lagaly, Clay Miner., 1981, 16, 1–16 CrossRef CAS.
- K. Imwiset, A. Teepakakorn, P. Saengdet, C. Tirayaphanitchkul and M. Ogawa, in Concepts and Design of Materials Nanoarchitectonics, ed. O. Azzaroni and K. Ariga, The Royal Society of Chemistry, Croydon, 2022, ch. 12, pp. 247–278 Search PubMed.
- BYK Additives & Instruments, Cloisite, https://www.byk.com/en/product/additive-guide/cloisite, (accessed March 2024).
- HOJUN Co., Ltd, Organophilic bentonites, https://www.hojun.co.jp/eng/e29_f_chemicals_no.html, (accessed March 2024).
- T. Hayakawa, M. Minase, K. Fujita and M. Ogawa, Clays Clay Miner., 2016, 64, 275–282 CrossRef CAS.
- P. R. Bodart, L. Delmotte, S. Rigolet, J. Brendlé and R. D. Gougeon, Appl. Clay Sci., 2018, 157, 204–211 CrossRef CAS.
- M. Lipsicas, R. H. Raythatha, T. J. Pinnavaia, I. D. Johnson, R. F. Giese, P. M. Costanzo and J. L. Robert, Nature, 1984, 309, 604–607 CrossRef CAS.
- D. Plee, F. Borg, L. Gatineau and J. J. Fripiat, J. Am. Chem. Soc., 1985, 107, 2362–2369 CrossRef CAS.
- G. W. Brindley, in X-Ray Identification and Crystal Structures of Clay Minerals, ed. G. W. Brindley, Taylor and Francis, London, 1951, pp. 159–164 Search PubMed.
- L. Heller and I. Rozenson, Phys. Chem. Miner., 1981, 7, 223–238 CrossRef.
- I. Rozenson and L. Herrer-Kallai, Clays Clay Miner., 1976, 24, 283–288 CrossRef CAS.
- J. D. Russel, B. A. Goodman and A. R. Fraser, Clays Clay Miner., 1979, 27, 63–71 CrossRef.
- J. W. Stucki, G. W. Bailey and H. Gan, Appl. Clay Sci., 1996, 10, 417–430 CrossRef CAS.
- T. B. Hofstetter, A. Neumann and R. P. Schwarzenbach, Environ. Sci. Technol., 2006, 40, 235–242 CrossRef CAS.
- J. W. Stucki and C. B. Roth, Clays Clay Miner., 1977, 41, 808–814 CAS.
- H. Gan, J. W. Stucki and G. W. Bailey, Clays Clay Miner., 1992, 40, 659–665 CrossRef CAS.
- C. P. Ponce and J. T. Kloprogge, Life, 2020, 10, 168 CrossRef CAS PubMed.
- T. Okada, M. Sueyoshi and H. M. Minamisawa, Langmuir, 2015, 31, 13842–13849 CrossRef CAS PubMed.
- E. Pavón, F. J. Osuna, M. D. Alba and L. Delevoye, Solid State Nucl. Magn. Reson., 2019, 100, 45–51 CrossRef PubMed.
- A. Dumas, M. Mizrahi, F. Martin and F. G. Requejo, Cryst. Growth Des., 2015, 15, 5451–5463 CrossRef CAS.
- T. Okada, A. Kumasaki, K. Shimizu, A. Yamagishi and H. Sato, J. Chromatogr. Sci., 2016, 54, 1238–1243 CAS.
- T. Okada, S. Hosoyamada, C. Takada and C. Ohta, ChemPhotoChem, 2021, 5, 32–35 CrossRef CAS.
- Y. Nakauchi, H. Minamisawa and T. Okada, Dalton Trans., 2024, 53, 2558–2564 RSC.
- V. Shklover, T. Haibach, F. Ried, R. Nesper and P. Novák, J. Solid State Chem., 1996, 123, 317–323 CrossRef CAS.
- J. E. Post and D. R. Veblen, Am. Mineral., 1990, 75, 477–489 CAS.
- M. Catti, I. Pinus and A. Scherillo, J. Solid State Chem., 2013, 205, 64–70 CrossRef CAS.
- J. Kwiatkowska, I. E. Grey, I. C. Madsen and L. A. Bursill, Acta Crystallogr., 1987, B43, 258–265 CAS.
- S. Anderson and A. D. Wadsley, Acta Crystallogr., 1961, 14, 1245–1249 CrossRef.
- A. D. Wadsley, Acta Crystallogr., 1964, 17, 623–628 CrossRef CAS.
- M. Gasperin and M. T. le Bihan, J. Solid State Chem., 1982, 43, 346–353 CrossRef CAS.
- D. B. Rogers, R. D. Shannon, A. W. Sleight and J. L. Gillson, Inorg. Chem., 1969, 8, 841–849 CrossRef CAS.
- W. D. Ryden, A. W. Lawson and C. C. Sartain, Phys. Rev. B: Condens. Matter Mater. Phys., 1970, 1, 1494–1500 CrossRef.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- S. L. Brock, N. Duan, Z. R. Tian, O. Giraldo, H. Zhou and S. L. Suib, Chem. Mater., 1998, 10, 2619–2628 CrossRef CAS.
- T. Taniguchi, L. Nurdiwijayanto, R. Ma and T. Sasaki, Appl. Phys. Rev., 2022, 9, 021313 CAS.
- F. Geng, R. Ma, A. Nakamura, K. Akatsuka, Y. Ebina, Y. Yamauchi, N. Miyamoto, Y. Tateyama and T. Sasaki, Nat. Commun., 2013, 4, 1632 CrossRef PubMed.
- R. Uppuluri, A. S. Gupta, A. S. Rosas and T. E. Mallouk, Chem. Soc. Rev., 2018, 47, 2401–2430 RSC.
- P. Wattanapaphawong, P. Reubroycharoen, C. Samart, L. Kou, J. Song, J. Su and K. Kajiyoshi, Appl. Ceram. Technol., 2024, 21, 750–758 CrossRef CAS.
- X. Wang, X. Li, S. Aya, F. Araoka, Y. Ishida, A. Kikkawa, M. Kriener, Y. Taguchi, Y. Ebina, T. Sasaki, S. Koshiya, K. Kimoto and T. Aida, J. Am. Chem. Soc., 2018, 140, 16396–16401 CrossRef CAS.
- Y. Shi, M. Osada, Y. Ebina and T. Sasaki, ACS Nano, 2020, 14, 15216–15226 CrossRef CAS PubMed.
- K. Awaya, A. Takashida, T. Taniguchi, M. Koinuma, T. Ishihara and S. Ida, Chem. Commun., 2019, 55, 4586–4588 RSC.
- Y. Shi, E. Yamamoto, M. Kobayashi and M. Osada, ACS Appl. Mater. Interfaces, 2023, 15, 22737–22743 CrossRef CAS.
- T. Sasaki, Y. Ebina, T. Tanaka, M. Harada, M. Watanabe and G. Decher, Chem. Mater., 2001, 13, 4661–4667 CrossRef CAS.
- K. Matsuba, C. Wang, K. Saruwatari, Y. Uesusuki, K. Akatsuka, M. Osada, Y. Ebina, R. Ma and T. Sasaki, Sci. Adv., 2017, 3, e1700414 CrossRef PubMed.
- K. Awaya and S. Ida, Chem. Commun., 2020, 56, 9811–9814 RSC.
- N. Sakai and T. Sasaki, Acc. Mater. Res., 2024, 5, 752–760 CrossRef CAS.
- S. J. Hwang, T. Zhang and I. Chung, Coord. Chem. Rev., 2021, 430, 213762 CrossRef CAS.
- N. H. Kwon, K.-G. Lee, H. K. Kim and S. J. Hwang, Mater. Chem. Front., 2021, 5, 3549–3575 RSC.
- K. Ament, D. R. Wagner, T. Götsch, T. Kikuchi, J. Kröhnert, A. Trunschke, T. Lunkenbein, T. Sasaki and J. Breu, ACS Catal., 2021, 11, 2754–2762 CrossRef CAS PubMed.
- B. Ohtani, S. Ikeda, H. Nakayama and S. Nishimoto, Phys. Chem. Chem. Phys., 2000, 2, 5308–5313 RSC.
- Y. Ide, Y. Nakasato and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 3601–3604 CrossRef CAS PubMed.
- Y. Ide, M. Matsuoka and M. Ogawa, J. Am. Chem. Soc., 2010, 132, 16762–16764 CrossRef CAS PubMed.
- A. Kudo and T. Sakata, J. Phys. Chem., 1995, 99, 15963–15967 CrossRef CAS.
- A. Kudo, Chem. Mater., 1997, 9, 664–669 CrossRef CAS.
- J. Du, K. Li, R. Van Deun, D. Poelman and H. Lin, Adv. Opt. Mater., 2022, 10, 2101714 CrossRef CAS.
- Q. Du, J. Ueda and S. Tanabe, J. Mater. Chem. C, 2023, 11, 16225–16233 RSC.
- H. Yim, S. Y. Yoo, Y. H. Kim, K. H. Chae, Y.-H. Kim, S. K. Kim, S.-H. Baek, C.-H. Lee and J.-W. Choi, Chem. Mater., 2021, 33, 8685–8692 CrossRef CAS.
- H. Yim, S. Y. Yoo, H. Choi, H. J. Chang, S.-J. Hwang, S. Nahm, M. Osada and J.-W. Choi, J. Alloys Compd., 2022, 925, 166606 CrossRef CAS.
- N. N. Rabin, S. Ida, M. R. Karim, Md. S. Islam, R. Ohatni, M. Nakamura, M. Koinuma, L. Lindoy and S. Hayami, ACS Omega, 2018, 3, 2074–2083 CrossRef CAS.
- T. Sasaki, F. Kooli, M. Iida, Y. Michiue, S. Takenouchi, Y. Yajima, F. Izumi, B. C. Chakoumakos and M. Watanabe, Chem. Mater., 1998, 10, 4123–4128 CrossRef CAS.
- A. J. Jacobson, J. W. Johnson and J. T. Lewandowski, Mater. Res. Bull., 1987, 22, 45–51 CrossRef CAS.
- A. F. Reid, W. G. Mumme and A. D. Wadsley, Acta Crystallogr., 1968, B24, 1228–1232 CrossRef.
- D. Groult, C. Mercey and B. Raveau, J. Solid State Chem., 1980, 32, 289–296 CrossRef CAS.
- M. Hervieu and B. Raveau, Rev. Chim. Miner., 1981, 18, 642–649 CAS.
- I. E. Grey, C. Li, I. C. Madsen and J. A. Watts, J. Solid State Chem., 1987, 66, 7–19 CrossRef CAS.
- T. Sasaki, Y. Komatsu and Y. Fujiki, J. Chem. Soc., Chem. Commun., 1991, 817–818 RSC.
- M. Osada, Y. Ebina, K. Takada and T. Sasaki, Adv. Mater., 2006, 18, 295–299 CrossRef CAS.
- M. Osada, Y. Ebina, K. Fukuda, K. Ono, K. Takada, K. Yamaura, E. Takayama-Muromachi and T. Sasaki, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 153301 CrossRef.
- X. Dong, M. Osada, H. Ueda, Y. Ebina, Y. Kotani, K. Ono, S. Ueda, K. Kobayashi, K. Takada and T. Sasaki, Chem. Mater., 2009, 21, 4366–4373 CrossRef CAS.
- M. Osada, S. Yoguchi, M. Itose, B.-W. Li, Y. Ebina, K. Fukuda, Y. Kotani, K. Ono, S. Ueda and T. Sasaki, Nanoscale, 2014, 6, 14227–14236 RSC.
- F. Kishimoto, Y. Takamura, S. Nakagawa and Y. Wada, Adv. Mater. Interfaces, 2016, 3, 1600509 CrossRef.
- Y. Ide, M. Sadakane, T. Sano and M. Ogawa, J. Nanosci. Nanotechnol., 2014, 14, 2135–2147 CrossRef CAS.
- A. J. Jacobson, J. W. Johnson and J. T. Lewandowski, Inorg. Chem., 1985, 24, 3727–3729 CrossRef CAS.
- T. Tokumitsu, K. Toda, T. Aoyagi, D. Sakuraba, K. Uematsu and M. Sato, J. Ceram. Soc. Jpn., 2006, 114, 795–797 CrossRef CAS.
- H. Suzuki, O. Tomita, M. Higashi and R. Abe, Catal. Sci. Technol., 2015, 5, 2640–2648 RSC.
- T. Oshima, T. Yokoi, M. Eguchi and K. Maeda, Dalton Trans., 2017, 46, 10594–10601 RSC.
- S. Atri, M. Pokhriyal and S. Uma, Inorg. Chem., 2020, 59, 8044–8053 CrossRef CAS PubMed.
- S. Pal and S. Uma, Ceram. Int., 2024, 50, 31540–31547 CrossRef CAS.
- H. J. Kim, S. Morita, K.-N. Byun, Y. Shi, T. Taniguchi, E. Yamamoto, M. Kobayashi, Y. Ebina, T. Sasaki and M. Osada, Nano Lett., 2023, 23, 3788–3795 CrossRef CAS PubMed.
- B.-W. Li, M. Osada, Y. Ebina, T. C. Ozawa, R. Ma and T. Sasaki, Appl. Phys. Lett., 2010, 96, 182903 CrossRef.
- T. B. N. Le, C.-W. Chang and Y.-H. Su, Surf. Interfaces, 2023, 36, 102623 CrossRef CAS.
- R. Liu, B. Feng, Y. Ma, H. Liu, J. Cui, D. Yang and H. Yuan, Mater. Lett., 2020, 281, 128635 CrossRef CAS.
- Y. Zhou, T. Wen, X. Zhang, B. Chang, W. Kong, Y. Guo, B. Yang and Y. Wang, Chem. – Asian J., 2017, 12, 2727–2733 CrossRef CAS.
- M. R. Aziza, C.-W. Chang, A. Mohapatra, C.-W. Chu, C.-C. Kaun and Y.-H. Su, ACS Appl. Nano Mater., 2020, 3, 6367–6375 CrossRef CAS.
- W. Zhang and M. Yashima, Chem. Commun., 2023, 59, 134–152 RSC.
- D. Hamada, M. Machida, Y. Sugahara and K. Kuroda, J. Mater. Chem., 1996, 6, 69–72 RSC.
- S. Uma and J. Gopalakrishnan, J. Solid State Chem., 1993, 102, 332–339 CrossRef CAS.
- S. Uma, A. R. Raju and J. Gopalakrishnan, J. Mater. Chem., 1993, 3, 709–713 RSC.
- J. Gopalakrishnan, S. Uma and V. Bhat, Chem. Mater., 1993, 5, 132–136 CrossRef CAS.
- D. Hamada, M. Machida, Y. Sugahara and K. Kuroda, J. Ceram. Soc. Jpn., 1997, 105, 284–287 CrossRef CAS.
- Y. Huang, J. Li, Y. Wei, Y. Li, J. Lin and J. Wu, J. Hazard. Mater., 2009, 166, 103–108 CrossRef CAS PubMed.
- M. Osada and T. Sasaki, Int. J. Ceram. Technol., 2012, 9, 29–36 CrossRef CAS.
- T.-H. Wang, C.-L. Chiang, P.-C. Li, Y.-K. Hsieh and C.-F. Wang, Chem. Eng. J., 2014, 244, 243–251 CrossRef CAS.
- Y. Matsumoto, S. Ida and T. Inoue, J. Phys. Chem. C, 2008, 112, 11614–11616 CrossRef CAS.
- M. Khodabakhsh, B. Yilmaz, S. Firoozi, D. F. Haghshenas and U. Unal, ACS Omega, 2023, 8, 10607–10617 CrossRef CAS PubMed.
- S. Dong, J. Feng, M. Fan, Y. Pi, L. Hu, X. Han, M. Liu, J. Sun and J. Sun, RSC Adv., 2015, 5, 14610–14630 RSC.
- Y. Huang, Y. Li, Y. Wei, M. Huang and J. Wu, Sol. Energy Mater. Sol. Cells, 2011, 95, 1019–1027 CrossRef CAS.
- M. Harada, T. Sasaki, Y. Ebina and M. Watanabe, J. Photochem. Photobiol., A, 2002, 148, 273–276 CrossRef CAS.
- T. Sasaki and M. Watanabe, J. Phys. Chem. B, 1997, 101, 10159–10161 CrossRef CAS.
- R. Hiroi, S. S. Ray, M. Okamoto and T. Shiroi, Macromol. Rapid Commun., 2004, 25, 1359–1364 CrossRef CAS.
- Y. Fuse, Y. Ide and M. Ogawa, Bull. Chem. Soc. Jpn., 2008, 81, 767–772 CrossRef CAS.
- K. Maeda, M. Eguchi and T. Oshima, Angew. Chem., Int. Ed., 2014, 53, 13164–13168 CrossRef CAS.
- D. E. Scaife, Sol. Energy, 1980, 25, 41–54 CrossRef CAS.
- Y. Matsumoto, J. Solid State Chem., 1996, 126, 227–234 CrossRef CAS.
- P. Xu, T. J. Milstein and T. E. Mallouk, ACS Appl. Mater. Interfaces, 2016, 8, 11539–11547 CrossRef CAS PubMed.
- M. Lv, X. Sun, S. Wei, C. Shen, Y. Mi and X. Xu, ACS Nano, 2017, 11, 11441–11448 CrossRef CAS.
- Y. Tang, K. Kato, T. Oshima, H. Mogi, A. Miyoshi, K. Fujii, K. Yanagisawa, K. Kimoto, A. Yamakata, M. Yashima and K. Maeda, Inorg. Chem., 2020, 59, 11122–11128 CrossRef CAS.
- Y. Zhou, T. Wen, Y. Guo, B. Yang and Y. Wang, RSC Adv., 2016, 69, 64930–64936 RSC.
- K. Maeda, Y. Tokunaga, K. Hibino, K. Fujii, H. Nakaki, T. Uchiyama, M. Eguchi, D. Lu, S. Ida, Y. Uchimoto and M. Yashima, ACS Appl. Energy Mater., 2018, 1, 1734–1741 CrossRef CAS.
- H. Wakayama, K. Hibino, K. Fujii, T. Oshima, K. Yanagisawa, Y. Kobayashi, K. Kimoto, M. Yashima and K. Maeda, Inorg. Chem., 2019, 58, 6161–6166 CrossRef CAS PubMed.
- T. Oshima, T. Ichibha, K. Oqmhula, K. Hibino, H. Mogi, S. Yamashita, K. Fujii, Y. Miseki, K. Hongo, D. Lu, R. Maezono, K. Sayama, M. Yashima, K. Kimoto, H. Kato, M. Kakihana, H. Kageyama and K. Maeda, Angew. Chem., Int. Ed., 2020, 59, 9736–9743 CrossRef CAS PubMed.
- Y. Shiroma, H. Mogi, T. Mashiko, S. Yasuda, S. Nishioka, T. Yokoi, S. Ida, K. Kimoto and K. Maeda, J. Mater. Chem. A, 2023, 11, 9485–9492 RSC.
- S. Ida, N. Kim, E. Ertekin, S. Takenaka and T. Ishihara, J. Am. Chem. Soc., 2015, 137, 239–244 CrossRef CAS.
- Y. Okamoto, S. Ida, J. Hyodo, H. Hagiwara and T. Ishihara, J. Am. Chem. Soc., 2011, 133, 18034–18037 CrossRef CAS PubMed.
- T. Gao, H. Fjellvåg and P. Norby, J. Mater. Chem., 2009, 19, 787–794 RSC.
- K. Saito, S. Orikasa, Y. Asakura, Y. Ide, Y. Sugahara, M. Ogasawara, S. Yin and S. Kato, Int. J. Photoenergy, 2021, 2021, 8847956 CrossRef.
- K. Saito, S. Tominaka, S. Yoshihara, K. Ohara, Y. Sugahara and Y. Ide, ACS Appl. Mater. Interfaces, 2017, 9, 24538–24544 CrossRef CAS.
- T. Maluangnont, B. Wuttitham, P. Hongklai, P. Khunmee, S. Tippayasukho, N. Chanlek and T. Sooknoi, Inorg. Chem., 2019, 58, 6885–6892 CrossRef CAS.
- T. Maluangnont, N. Chanlek, O. Khamman, W. Vittayakorn and T. Sooknoi, Inorg. Chem., 2021, 60, 16326–16336 CrossRef CAS.
- G. Zhang, J. Gong, H. Gan and F. Lü, J. Alloys Compd., 2011, 509, 9791–9797 CrossRef CAS.
- M. Ogawa, M. Morita, S. Igarashi and S. Sato, J. Solid State Chem., 2013, 206, 9–13 CrossRef CAS.
- S. Igarashi, S. Sato, T. Takashima and M. Ogawa, Ind. Eng. Chem. Res., 2013, 52, 3329–3333 CrossRef CAS.
- M. Ohashi, Mol. Cryst. Liq. Cryst., 1998, 311, 51–56 CrossRef.
- B. W. Li, M. Osada, Y. H. Kim, Y. Ebina, K. Akatsuka and T. Sasaki, J. Am. Chem. Soc., 2017, 139, 10868–10874 CrossRef CAS.
- X. Li, W. Ke, B. Traoré, P. Guo, I. Hadar, M. Kepenekian, J. Even, C. Katan, C. C. Stoumpos, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2019, 141, 12880–12890 CrossRef CAS PubMed.
- M. Ogawa, H. Suzuki, K. Ogawa, O. Tomita and R. Abe, Solid State Sci., 2023, 141, 107221 CrossRef CAS.
- J. Liu, C. Deng, X. Liu, S. Shao, P. Zheng, L. Chen, P. Wu, H. Li, H. Ji and W. Zhu, Inorg. Chem., 2023, 62, 11044–11055 CrossRef CAS PubMed.
- K. Sano, Y. S. Kim, Y. Ishida, Y. Ebina, T. Sasaki, T. Hikima and T. Aida, Nat. Commun., 2016, 7, 12559 CrossRef CAS.
- W. Yang, S. Yamamoto, K. Sueyoshi, T. Inadomi, R. Kato and N. Miyamoto, Angew. Chem., Int. Ed., 2021, 60, 8466–8471 CrossRef.
- M. Ohwada, K. Kimoto, T. Mizoguchi, Y. Ebina and T. Sasaki, Sci. Rep., 2013, 3, 2801 CrossRef PubMed.
- P. Xiong, F. Zhang, X. Zhang, Y. Liu, Y. Wu, S. Wang, J. Safaei, B. Sun, R. Ma, Z. Liu, Y. Bando, T. Sasaki, X. Wang, J. Zhu and G. Wang, Nat. Commun., 2021, 12, 4184 CrossRef CAS.
- C. Liu, C. Ye, Y. Wu, Y. Liu, Z. Liu, Z. Chen, R. Ma, N. Sakai, L. Xue, J. Sun, W. Zhang, W. Zhang, X. Wang, T. Sasaki, P. Xiong and J. Zhu, Nano Energy, 2023, 110, 108348 CrossRef CAS.
- H. Dai, X. Cai, X. Li, C. Wang and Y. Hou, Solid State Sci., 2022, 129, 106907 CrossRef CAS.
- E. Doustkhah, M. H. N. Assadi, K. Komaguchi, N. Tsunoji, M. Esmat, N. Fukata, O. Tomita, R. Abe, B. Ohtani and Y. Ide, Appl. Catal., B, 2021, 297, 120380 CrossRef CAS.
- T. C. Ozawa and T. Sasaki, Inorg. Chem., 2010, 49, 3044–3050 CrossRef CAS PubMed.
- T. C. Ozawa and T. Sasaki, Dalton Trans., 2014, 43, 14902–14908 RSC.
- M. Ohwada, K. Kimoto, K. Suenaga, Y. Sato, Y. Ebina and T. Sasaki, J. Phys. Chem. Lett., 2011, 2, 1820–1823 CrossRef CAS.
- K. Saito, K. Inaguma, M. Ogawa, P. T. Ha, H. Akiyama, S. Yamaguchi, H. Minokoshi, M. Ogasawara and S. Kato, ACS Appl. Nano Mater., 2022, 5, 9053–9062 CrossRef CAS.
- T. Maluangnont, P. Pulphol, K. Klangvijit, K. Bowornthommatadsana, N. Chanlek, M. Ogawa and W. Wongwiriyapan, Dalton Trans., 2023, 52, 11815–11825 RSC.
- K. Nakase, S. Ichihara, J. Matsumoto, S. Koh, M. Mizuno and T. Okada, Langmuir, 2022, 38, 6076–6085 CrossRef CAS.
- J. Di, J. Xiong, H. Li and Z. Liu, Adv. Mater., 2018, 30, 1704548 CrossRef.
- T. Saothayanun and M. Ogawa, Chem. Commun., 2021, 57, 10003–10006 RSC.
- T. K. Saothayanun, R. P. Wijitwongwan and M. Ogawa, Inorg. Chem., 2022, 61, 20268–20274 CrossRef CAS.
- Q. Zhong, Y. Li and G. Zhang, Chem. Eng. J., 2021, 409, 128099 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |