
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn effective visible-light driven fumarate production from gaseous CO2 and pyruvate by the cationic zinc porphyrin-based photocatalytic system with dual biocatalysts†
Mika
Takeuchi
a and
Yutaka
Amao
 *ab
*ab
aGraduate School of Science, Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: amao@omu.ac.jp
bResearch Centre of Artificial Photosynthesis (ReCAP), Osaka Metropolitan University, 3-3-138 Sugimoto, Sumiyoshi-ku, Osaka 558-8585, Japan
First published on 27th November 2023
Abstract
Fumaric acid is a useful unsaturated dicarboxylic acid that serves as a precursor for the biodegradable plastics poly(butylene succinate) and poly(propylene fumarate). Currently, fumaric acid is mainly synthesised from petroleum resources such as benzene. It is therefore desirable to develop methods to produce fumaric acid from renewable resources such as those derived from biomass. In this work, an effective visible-light driven fumarate production from gaseous CO2 and pyruvate with the system consisting of triethanolamine, cationic water-soluble zinc porphyrin, zinc tetrakis(4-N,N,N-trimethylaminophenyl)porphyrin, pentamethylcyclopentadienyl coordinated rhodium(III) 2,2′-bipyridyl complex, NAD+, malate dehydrogenase (NAD+-dependent oxaloacetate-decarboxylating) and fumarase was developed.
Fumaric acid, trans-butenedioic acid, is the simplest dicarboxylic acid with a carbon–carbon double bond in the molecule. Fumaric acid is a chemical building block with many applications, including in the polymer materials industry.1–3 Particularly in the field of polymer chemistry, fumaric acid is also useful as a raw material for the biodegradable plastics poly(butylene succinate) (PBS)4 and poly(propylene fumarate) (PPF).5 Currently, fumaric acid is synthesised mainly by petroleum-based chemical production. Conventional fumaric acid is produced by oxidising furfural with chlorate in the presence of a vanadium catalyst.6 Limited fossil resources, rising oil prices and growing interest in the carbon cycle in chemical synthesis have led to increased interest in the development of bio-based fumaric acid from renewable resources and CO2. Fermentation methods produce fumaric acid from glucose via the reductive tricarboxylic acid (TCA) pathway and were used in industry before the development of the petrochemical industry.7 The conventional fumaric acid fermentation has problems of high costs with low product yields and productivity, and produces a wide variety of by-products. In view of these constraints, we devoted to a simple biocatalytic fumaric acid production from CO2 and biomass-derived materials such as pyruvic acid. The biocatalytic fumarate production from bicarbonate or gaseous CO2 and pyruvate viaL-malate as an intermediate with dual biocatalysts (malate dehydrogenase; NAD+-dependent oxaloacetate-decarboxylating; MDH EC 1.1.1.38) and fumarase (FUM; EC 4.2.1.2) in the presence of NADH in an aqueous media has been reported.8,9 After 7 h incubation, the conversion yields of pyruvate to fumarate from bicarbonate or gaseous CO2 as feedstock are estimated to be 13 or 12%, respectively. Since this system uses NADH as a sacrificial reagent, it is necessary to integrate the NAD+ to NADH regeneration system. Thus, the visible-light driven fumarate production from pyruvate and bicarbonate or gaseous CO2 with the combination of NAD+ reduction to NADH system of triethanolamine (TEOA) as an electron donor, water-soluble zinc porphyrin, (ZnP) as a photosensitiser and [Cp*Rh(bpy)(H2O)]2+, and dual biocatalysts (MDH and FUM) as shown in Fig. 1 also has been reported.10,11 In this system, zinc meso-tetra(4-sulfonatophenyl)porphyrin tetrasodium salt (ZnTPPS4−) was used as a photosensitiser.
 | ||
| Fig. 1 Visible-light driven fumarate production from pyruvate and CO2 with the system consisting of TEOA, water-soluble zinc porphyrin (ZnP), [Cp*Rh(bpy)(H2O)]2+, NAD+, MDH and FUM. | ||
By using this system, the conversion yields of pyruvate to fumarate from bicarbonate or gaseous CO2 as feedstock are estimated to be 0.9 or 1.0% after 5 h irradiation, respectively. Using this system, fumarate could be synthesised from pyruvate and gaseous CO2 using visible light energy as the driving force, but the yield of fumarate production remained low compared with that the non-photoredox system consisting of NADH, MDH and FUM. One of the reasons for the low efficiency of fumarate production was predicted to be the direct effect of ZnTPPS4− used as a photosensitiser in the visible light-driven NADH regeneration on the catalytic activity of FUM for the L-malate dehydration. It was reported that the addition of anionic water-soluble zinc porphyrin, ZnTPPS4− directly inhibited the catalytic activity of FUM-catalysed L-malate dehydration to produce fumarate (reduced to about 16% compared to control experiment).12 The sulfonatophenyl-group of ZnTPPS4− binds to the substrate binding site of FUM, thereby inhibiting L-malate binding and decreasing fumarate production.12 It was also reported that the catalytic activity of FUM was not affected by the addition of the cationic water-soluble zinc porphyrin, zinc tetrakis(4-N,N,N-trimethylaminophenyl)porphyrin (ZnTMAP4+; chemical structure as shown in Fig. 2).12
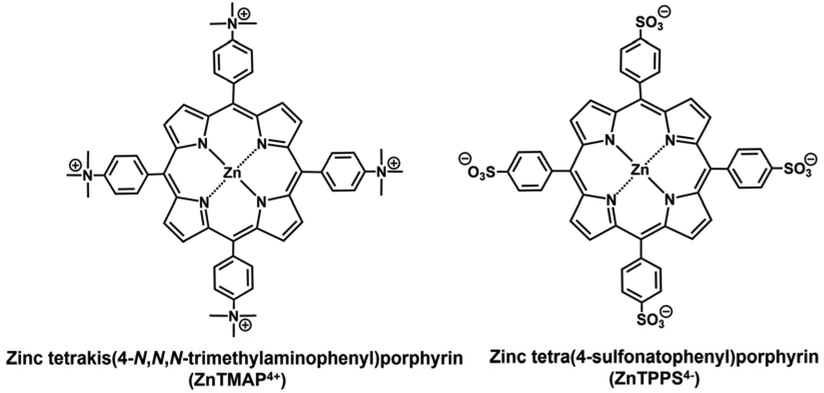 | ||
| Fig. 2 Chemical structures of zinc tetrakis(4-N,N,N-trimethylaminophenyl)porphyrin (ZnTMAP4+) and zinc tetra(4-sulfonatophenyl)porphyrin (ZnTPPS4−). | ||
In this study, the visible-light driven fumarate production from pyruvate and gaseous CO2 with the combination of NAD+ reduction system of TEOA, ZnTMAP4+ and [Cp*Rh(bpy)(H2O)]2+, MDH and FUM was investigated for the purpose of improving fumarate production efficiency.
ZnTMAP4+ was prepared by refluxing metal-free TMAP4+ with about 5 times molar equivalent of zinc acetate in the methanol solution according to the previous report.13,14 Pentamethylcyclopentadienyl(2,2′-bipyridyl)rhodium(III) chloride ([Cp*Rh(bpy)(H2O)]2+) was synthesized with [Cp*RhCl2]2 and 2,2′-bipyridine according to a previous report.15
First, the optimum condition for the visible-light driven NAD+ reduction to NADH was investigated. A solution consisting of TEOA (0.2 M), ZnTMAP4+ (10–100 μM), [Rh(Cp*)(bpy)(H2O)]2+ (10 μM) and NAD+ (0.5 mM) in 5.0 ml of 500 mM 2-[4-(2-hydroxyethyl)-1-piperazinyl]ethanesulfonic acid (HEPES) buffer solution (pH 7.8) was deaerated by freeze–pump–thaw cycles repeated 6 times and the gas phase was introduced argon gas for 10 min. The sample solution was irradiated with a 250 W halogen lamp (Panasonic) as a visible-light source at 30.5 °C. Fig. 3 shows the concentration of ZnTMAP4+ dependence of NADH production rate (v) with the system of TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+ and NAD+ in HEPES buffer solution. The concentration of NADH produced was determined by absorption spectrum change using UV-visible absorption spectroscopy (SHIMADZU, MaltiSpec-1500) with the molar coefficient at 340 nm (ε = 6.22 × 103 cm−1 M−1).16 The NADH production rate was determined from the concentration of NADH produced after 30 min of irradiation. As shown in Fig. 3, the NADH production rate increased generally linearly with increasing ZnTMAP4+ concentrations. The NADH production rate was out of linearity under ZnTMAP4+ concentration of 10 μM. It was reported that NADH production rate is maximized under the condition that the concentration of [Rh(Cp*)(bpy)(H2O)]2+ is equivalent to that of ZnTPPS4−. This suggests that this is a specific phenomenon under conditions of equal concentration of ZnTMAP4+ and [Cp*Rh(bpy)(H2O)]2+.
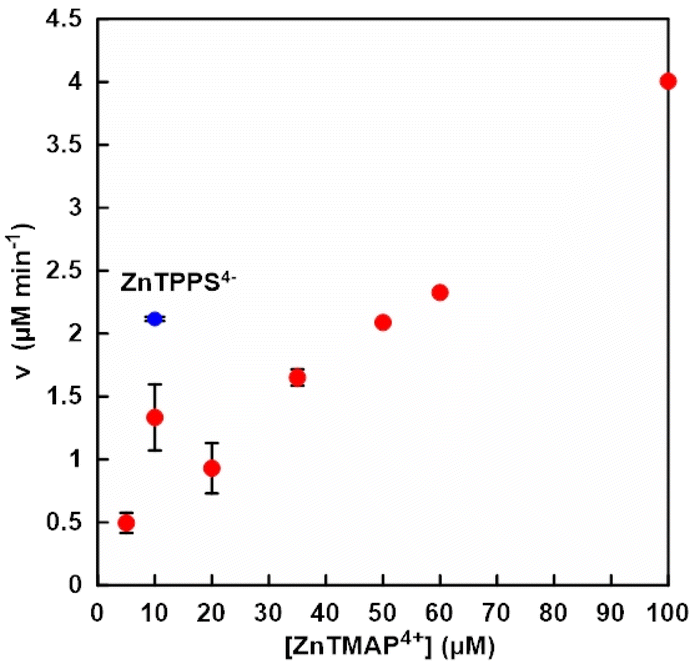 | ||
| Fig. 3 The concentration of ZnTMAP4+ dependence of NADH production rate (v) with the system of TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+ and NAD+ in 5.0 mL of 500 mM HEPES buffer (pH 7.8). Blue circle shows the NADH production rate (v) with the system of TEOA, ZnTPPS4−, [Cp*Rh(bpy)(H2O)]2+ and NAD+ in 5.0 mL of 500 mM HEPES buffer (pH 7.8). Errors were obtained from the average of multiple trials. | ||
The NADH production rate with the system of TEOA (0.2 M), ZnTPPS4− (10 μM), [Cp*Rh(bpy)(H2O)]2+ (10 μM) and NAD+ (0.5 mM) in HEPES buffer solution also was indicated in the Fig. 3 (blue circle). Under water-soluble zinc porphyrin concentration of 10 μM, the NADH production rate in the system with ZnTMAP4+ was about 60% lower than that with ZnTPPS4−. It was reported that the quantum yields of NADH production with ZnTPPS4− and ZnTMAP4+ were estimated to be 1.7% and 1.1% for Soret band with 420 nm, and to be 1.3% and 1.6% for Q-band with 550 nm, respectively.17 The molar absorption coefficients of ZnTPPS4− and ZnTMAP4+ are determined to be 680 and 448 for 420 nm, and 22.1 and 18.2 cm−1 mM−1 for 550 nm, respectively.17 Thus, these results suggested that under conditions of comparable ZnTPPS4− and ZnTMAP4+ concentrations, NADH production rate are consistent with the ordinates of the molar absorption coefficients of the Soret and Q bands of the ZnTPPS4− and ZnTMAP4+. From these results, it was shown that a ZnTMAP4+ concentration of 50 μM is required to equal the previously reported NADH production rate by a system using ZnTPPS4− (10 μM) as a photosensitiser. In subsequent experiments, the ZnTMAP4+ concentration was adjusted at 50 μM.
Next let us focus on the visible-light driven L-malate production with ZnTMAP4+ as a photosensitiser. A sample solution containing sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), TEOA (0.2 M), ZnTMAP4+ (50 μM), [Cp*Rh(bpy)(H2O)]2+ (10 μM), NAD+ (0.5 mM) and MDH from Sulfolobus tokodaii purchased from Thermostable Enzyme Laboratory Co., Ltd (EC 1.1.1.38 MDH-73-01) (0.7 U; ca. 1.6 μM) in 5.0 mL of 500 mM HEPES–NaOH buffer (pH 7.8). The sample solution was deaerated by freeze–pump–thaw cycles repeated 6 times and then introduced in gas phase and balloon with the CO2 gas for 10 min. The sample solution was irradiated with a 250 W halogen lamp as a visible-light source (light intensity: 200 J m−2 s−1) at 30 °C. The reaction used an isobaric system as shown in Fig. S1.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 The total pressure in the reaction system was maintained at 1.01325 × 105 Pa. The system with ZnTPPS4− (10 μM) was used as a control experiment. Fig. S2(a)† shows the chromatogram of sodium L-malate (0–1000 μM) in 50 mM-HEPES buffer (pH 7.0). Inset of Fig. S2† shows the relationship between the L-malate concentration and the detection peak area. The L-malate concentration was calculated from the calibration curve shown in inset of Fig. S2(a)† using the eqn (S1).† After 1 h irradiation, 56.0 μM of L-malate was produced with the system of sodium pyruvate, magnesium chloride, TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+, NAD+ and MDH in gas phase and balloon with the CO2 gas. On the other hand, 43.2 μM of L-malate was produced with the system using ZnTPPS4−. There was no significant difference between both systems with ZnTMAP4+ and ZnTPPS4− in the concentration of L-malate production after 1 h irradiation. Fig. 4 shows the concentration of L-malate production using ZnTMAP4+ or ZnTPPS4− after 5 h irradiation (the ion chromatograph chart during the reaction is shown in Fig. S3 and S4†).
11 The total pressure in the reaction system was maintained at 1.01325 × 105 Pa. The system with ZnTPPS4− (10 μM) was used as a control experiment. Fig. S2(a)† shows the chromatogram of sodium L-malate (0–1000 μM) in 50 mM-HEPES buffer (pH 7.0). Inset of Fig. S2† shows the relationship between the L-malate concentration and the detection peak area. The L-malate concentration was calculated from the calibration curve shown in inset of Fig. S2(a)† using the eqn (S1).† After 1 h irradiation, 56.0 μM of L-malate was produced with the system of sodium pyruvate, magnesium chloride, TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+, NAD+ and MDH in gas phase and balloon with the CO2 gas. On the other hand, 43.2 μM of L-malate was produced with the system using ZnTPPS4−. There was no significant difference between both systems with ZnTMAP4+ and ZnTPPS4− in the concentration of L-malate production after 1 h irradiation. Fig. 4 shows the concentration of L-malate production using ZnTMAP4+ or ZnTPPS4− after 5 h irradiation (the ion chromatograph chart during the reaction is shown in Fig. S3 and S4†).
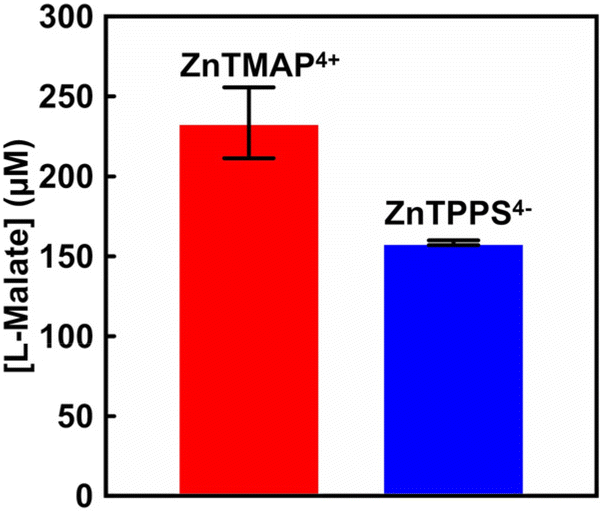 | ||
| Fig. 4 The concentration of L-malate production in the system of sodium pyruvate, magnesium chloride, TEOA, [Cp*Rh(bpy)(H2O)]2+, NAD+, MDH and CO2 gas in the presence of ZnTMAP4+ (red) or ZnTPPS4− (blue) after 5 h irradiation. Errors were obtained from the average of multiple trials. | ||
As shown in Fig. 4, 233.4 (±22.2) μM of L-malate was produced with the system of sodium pyruvate, magnesium chloride, TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+, NAD+ and MDH in gas phase and balloon with the CO2 gas after 5 h irradiation. However, 158.5 (±1.5) μM of L-malate was produced with the system using ZnTPPS4−. By using ZnTMAP4+ as a photosensitiser, approximately 1.5 times more L-malate was produced than in the system with ZnTPPS4−. For the system using ZnTMAP4+, the L-malate production rates after 1 and 5 h irradiation were estimated to be 56.0 and 46.7 μM h−1, respectively. For the system using ZnTPPS4−, the production rates after 1 and 5 h irradiation were estimated to be 43.7 and 31.7 μM h−1, respectively. The rate of L-malate production gradually decreased with irradiation time in the system with ZnTPPS4−. In contrast, no decrease in the rate of L-malate production with irradiation time was observed in the system with ZnTMAP4+. We have reported stable NAD+ reduction NADH in a system of TEOA and [Cp*Rh(bpy)(H2O)]2+ with ZnTMAP4+ as a photosensitiser compared to a system using anionic water-soluble zinc porphyrins such as ZnTPPS4−.17 Thus, it was found that the efficiency of visible-light driven L-malate production was improved by using ZnTMAP4+ as a photosensitiser.
Finally, visible-light driven fumarate production from pyruvate and gaseous CO2 was investigated using the system of with the addition of FUM from porcine heart purchased from Merck Co., Ltd (EC 4.2.1.2; molecular weight: 200 kDa)18 (details of the experiment are described in ESI†). A sample solution containing sodium pyruvate (5.0 mM), magnesium chloride (5.0 mM), TEOA (0.2 M), ZnTMAP4+ (50 μM), [Cp*Rh(bpy)(H2O)]2+ (10 μM), NAD+ (0.5 mM), MDH (0.7 U; ca. 1.6 μM) and FUM (0.5 U; ca. 1.3 nM) in 5.0 mL of 500 mM HEPES–NaOH buffer (pH 7.8). The sample solution was deaerated by freeze–pump–thaw cycles repeated 6 times and then introduced in gas phase and balloon with the CO2 gas for 10 min. The sample solution was irradiated with a 250 W halogen lamp as a visible-light source (light intensity: 200 J m−2 s−1) at 30 °C. The reaction used an isobaric system as shown in Fig. S1.† The total pressure in the reaction system was maintained at 1.01325 × 105 Pa. The system with ZnTPPS4− (10 μM) also was used as a control experiment. The concentration of fumarate produced was measured by an ion chromatography. Fig. S2(b)† shows the chromatogram of sodium fumarate (0–1000 μM) in 50 mM-HEPES buffer (pH 7.0). The inset of Fig. S2(b)† shows the relationship between the fumarate concentration and the detection peak area. The fumarate concentration was calculated from the calibration curve shown in inset of Fig. S3† using the eqn (S2).† After 1 h irradiation, 13.2 μM of fumarate was produced with the system of sodium pyruvate, magnesium chloride, TEOA, ZnTMAP4+, [Cp*Rh(bpy)(H2O)]2+, NAD+, MDH and FUM in gas phase and balloon with the CO2 gas. In contrast, 9.2 μM of fumarate was produced with the system using ZnTPPS4−. By using ZnTMAP4+ as a photosensitiser, approximately 1.4 times more fumarate was produced than in the system with ZnTPPS4−. Fig. 5 shows the concentration of fumarate production using ZnTMAP4+ or ZnTPPS4− after 5 h irradiation (the ion chromatograph chart during the reaction is shown in Fig. S5 and S6†). As shown in Fig. 5, 96.9 (±3.9) μM of fumarate was produced with the system using ZnTMAP4+ after 5 h irradiation. However, 46.9 (±1.8) μM of fumarate was produced with the system using ZnTPPS4−. By using ZnTMAP4+, approximately 2.0 times more fumarate was produced than in the system with ZnTPPS4−. Let's now discuss the argument based on the catalytic efficiency of the FUM in both systems using ZnTMAP4+ and ZnTPPS4−. For the system using ZnTMAP4+, the turnover number (TON) and turnover frequency (TOF) of FUM (1.3 nM) were estimated to be 76769 and 4.3 s−1, respectively. For the system using ZnTPPS4−, in contrast, TON and TOF of FUM were calculated to be 37462 and 2.0 s−1, respectively. Furthermore, 41.0 μM of fumarate was produced in the system with 5-fold equivalent amounts of ZnTPPS4− (50 μM) (the ion chromatograph chart during the reaction is shown in Fig. S7†). Under ZnTPPS4− concentrations of 10 and 50 μM, 191 and 339 μM L-malate were produced as the intermediate after 5 h of irradiation, respectively. No increase in fumarate production was observed under the condition of increased ZnTPPS4− concentrations, even though more of the intermediate L-malate was produced. After 5 h irradiation, the present ratio of L-malate and fumarate under ZnTPPS4− concentrations of 10 and 50 μM was calculated to be 0.25 and 0.12, respectively. These results suggest that ZnTPPS4− also directly inhibits the catalytic activity of FUM in the visible-light driven fumarate production with the system of sodium pyruvate, magnesium chloride, TEOA, ZnTPPS4−, [Cp*Rh(bpy)(H2O)]2+, NAD+, MDH and FUM in the presence of gaseous CO2. Meanwhile, 313 μM of L-malate was produced as the intermediate after 5 h of irradiation using ZnTMAP4+ (50 μM). After 5 h irradiation, the present ratio of L-malate and fumarate was calculated to be 0.30. The results indicate that ZnTMAP4+ acts only as a photosensitiser for visible light-driven NADH and is not involved in the catalytic activity of FUM. By using ZnTMAP4+ as a photosensitiser, therefore, an effective visible-light driven fumarate production from gaseous CO2 and pyruvate with MDH and FUM was accomplished.
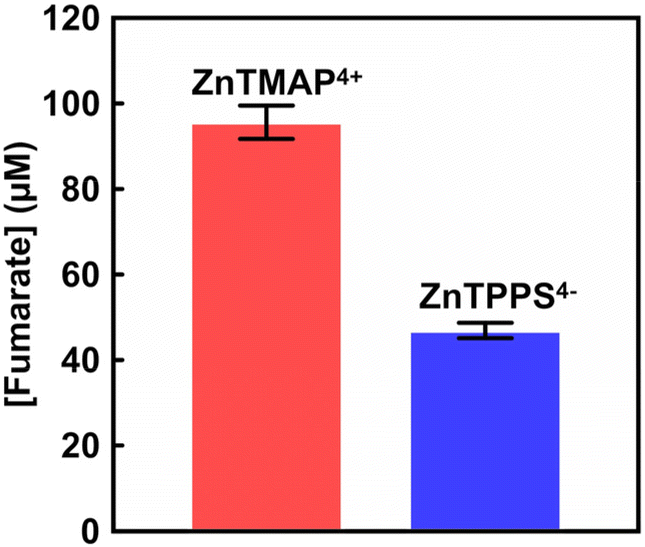 | ||
| Fig. 5 The concentration of fumarate production in the system of sodium pyruvate, magnesium chloride, TEOA, [Cp*Rh(bpy)(H2O)]2+, NAD+, MDH, FUM and CO2 gas in the presence of ZnTMAP4+ (red) or ZnTPPS4− (blue) after 5 h irradiation. Errors were obtained from the average of multiple trials. | ||
In this system, fumarate production has been observed up to 24 h of irradiation; MDH is a thermostable enzyme and durable to prolonged use, so the reaction can proceed again with the addition of pyruvate, whereas FUM shows decreased catalytic activity for longer reaction. This means that the addition of FUM after a certain irradiation time is expected to extend the lifetime of the reaction system.
In conclusion, we have successfully improved the efficiency of visible-light driven fumarate production from pyruvate and gaseous CO2 by using ZnTMAP4+ as a photosensitiser in the system combination of NAD+ reduction with [Cp*Rh(bpy)(H2O)]2+ and dual biocatalysts (MDH and FUM) in the presence of TEOA (approximately 2.0 times more fumarate is produced than in the system using ZnTPPS4−, conventionally photosensitizer). The use of ZnTMAP4+ increased the yield of fumarate production from pyruvate and CO2 without interfering with the catalytic activity of FUM, coupled with the effect of also increasing the yield of visible light driven L-malate production using MDH as a catalyst.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grant-in-Aid for Specially promoted Research (23H05404), Scientific Research (B) (22H01872), (22H01871) and Fund for the Promotion of Joint International Research (Fostering Joint International Research (B)) (19KK0144), and by Institute for Fermentation, Osaka (IFO) (G-2023-3-050).Notes and references
- A. Pellis, A. E. Herrero, L. Gardossi, V. Ferrario and G. M. Guebitz, Polym. Int., 2016, 65, 861 CrossRef CAS.
- N. A. Rorrer, J. R. Dorgan, D. R. Vardon, C. R. Martinez, Y. Yang and G. T. Beckham, ACS Sustainable Chem. Eng., 2016, 4, 6867 CrossRef CAS.
- Y. Jiang, A. J. J. Woortman, G. O. R. Alberda van Ekenstein and K. Loos, Polym. Chem., 2015, 6, 5451 RSC.
- N. Jacquel, F. Freyermouth, F. Fenouillot, A. Rousseau, J. P. Pascault, P. Fuertes and R. Saint-Loup, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5301 CrossRef CAS.
- S. He, M. D. Timmer, M. J. Yaszemski, A. W. Yasko, P. S. Engel and A. G. Mikos, Polymer, 2001, 42, 1251 CrossRef CAS.
- J. Shao, Y. Ni and L. Yan, J. Bioresour. Bioprod., 2021, 6, 39 CrossRef CAS.
- V. Martin-Dominguez, J. Estevez, F. De Borja Ojembarrena, V. E. Santos and M. Ladero, Fermentation, 2018, 4, 33 CrossRef.
- M. Takeuchi and Y. Amao, React. Chem. Eng., 2022, 7, 1931 RSC.
- M. Takeuchi and Y. Amao, RSC Sustainability, 2023, 1, 90 RSC.
- M. Takeuchi and Y. Amao, Sustainable Energy Fuels, 2023, 7, 355 RSC.
- M. Takeuchi and Y. Amao, RSC Sustainability, 2023, 1, 1874 RSC.
- M. Takeuchi and Y. Amao, New J. Chem., 2023, 47, 17679 RSC.
- K. Kalyanasundaram and M. Neumann-Spallart, J. Phys. Chem., 1982, 86, 5163 CrossRef CAS.
- K. Kalyanasundaram, J. Chem. Soc., Faraday Trans. 2, 1983, 79, 1365 RSC.
- U. Kölle, B. S. Kang, P. Infelta, P. Comte and M. Grätzel, Chem. Ber., 1989, 122, 1869 CrossRef.
- R. B. McComb, L. W. Bond, R. W. Burnett, R. C. Keech and G. N. Bowers Jr., Clin. Chem., 1976, 22, 141 CrossRef CAS.
- M. Takeuchi and Y. Amao, Bull. Chem. Soc. Jpn., 2023, 96, 1206 CrossRef CAS.
- S. Beeckmans and E. Van Driessche, J. Biol. Chem., 1998, 273, 31661 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dt03492e |
| This journal is © The Royal Society of Chemistry 2024 |