
Cation induced changes to the structure of cryptophane cages†
Oscar H.
Lloyd Williams
 a,
Claudia S.
Cox
a,
Meng Yuan
Zhang
a,
Martina
Lessio
a,
Olivia
Rusli
a,
William A.
Donald
a,
Lachlan
Jekimovs
b,
David L.
Marshall
c,
Michael C.
Pfrunder
bd,
Berwyck L. J.
Poad
bc,
Thierry
Brotin
e and
Nicole J.
Rijs
*a
a,
Claudia S.
Cox
a,
Meng Yuan
Zhang
a,
Martina
Lessio
a,
Olivia
Rusli
a,
William A.
Donald
a,
Lachlan
Jekimovs
b,
David L.
Marshall
c,
Michael C.
Pfrunder
bd,
Berwyck L. J.
Poad
bc,
Thierry
Brotin
e and
Nicole J.
Rijs
*a
aSchool of Chemistry, UNSW Sydney, Sydney, NSW 2052, Australia. E-mail: n.rijs@unsw.edu.au
bSchool of Chemistry and Physics, Queensland University of Technology, Brisbane 4001, Queensland, Australia
cCentral Analytical Research Facility, Queensland University of Technology, Brisbane, 4001 Australia
dCentre for Materials Science, Queensland University of Technology, Brisbane, 4001 Australia
eENS Lyon, CNRS, Laboratoire de Chimie, UMR 5182, 46 Allée d'Italie, 69364 Lyon, France
First published on 26th September 2024
Abstract
Here the monocation complexes of seven anti-cryptophanes are examined with high-resolution ion-mobility mass spectrometry. The relative size of the [cation + cryptophane]+ complexes were compared based on their measured mobilities and derived collisional cross sections. A paradoxical trend of structural contraction was observed for complexes of increasing cation size. Density functional theory confirmed encapsulation occurs for cation = Na+, K+, Rb+, Cs+ and NH4+. However, cation = Li+ preferred oxygen coordination at a linker over encapsulation within the cavity, leading to a slightly larger gas phase structure overall. Protonated cryptophanes yielded much larger collision cross sections via imploded cryptophane structures. Thus, competing physical effects led to the observed non-periodic size trend of the complexes. Trends in complexation from isothermal titration calorimetry and other condensed phase techniques were borne out by the gas phase studies. Further, predicted cavity sizes compared with the gas phase experimental findings reveal more about the encapsulation mechanisms themselves.
Introduction
Host–guest recognition unlocks the ability to chemoselectively react, detect or sequester a target.1 For example, a host designed to preferentially bind a toxic guest can reduce the toxin's free concentration in solution, and, consequently, its harmful effects.2,3 Molecular recognition is a central tenet of biology, where enzymes selectively react specific molecules through enthalpic (e.g. binding) and entropic (e.g. shape and solvation) means.4 Synthetic efforts have targeted achieving similar levels of recognition. To this end, macrocyclic hosts, such as calixarenes,5 cucurbiturils,6 pillararenes,7 and cryptophanes,8,9 have been developed.10 Different interactions contribute to the stability of the host–guest complex. Like enzymes, an incorrectly recognized molecule can disrupt or damage a synthetic host's ability to interact with the target guest. Understanding the interactions and structural changes that govern encapsulation are vital to designing effective hosts.Cryptophanes, first synthesized by Gabard and Collet in the early 1980s,11 can have an anti or a syn configuration,12–14 depending on the arrangement of the three linkers connecting the cyclotribenzylene (CTB) units (Chart 1). They can encapsulate guests within their hydrophobic cavity. The solubility and binding of cryptophanes is tunable by substitution. For example, cryptophanes with phenolic groups are water-soluble and show affinity for rubidium, cesium and thallium cations in aqueous solution.15,16 Functionalization of the aromatic rings of CTB is also possible.17,18
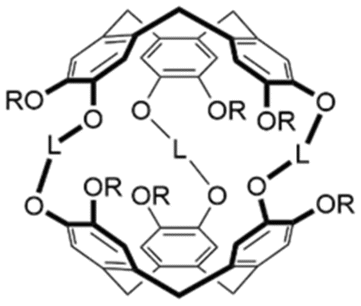 | ||
| Chart 1 Anti-cryptophane; L = alkyl or functionalized linker, and R = H, Me, CH2CO2H, etc. | ||
Cryptophane host–guest behaviors are inextricably linked to their structure and competitive energetics. Isothermal titration calorimetry (ITC) helps determine the latter,19,20 encompassing solvent, counter ion, and competition effects. For example, the different thermodynamics of inclusion versus exclusion alkylammonium complexes.21 However it does not provide detailed structural information. The gas phase structural characterization of host–guest complexes by mass spectrometry is a complementary approach, as these structures may not be accessible to bulk characterization techniques, such as crystallography, chiroptical spectroscopy and nuclear magnetic resonance (NMR) spectroscopy.9,16,22,23–26 Recent electrospray ionization mass spectrometry (ESI-MS) studies have explored Rb and Cs encapsulation by related cyclotricatechylene assemblies.27
Interpretation of intermolecular interactions,28–30 relative binding, and even solvent effects, can be gained in controlled gas phase studies.31,32 For example, Armentrout used guided ion beam tandem mass spectrometry to interrogate the bond energies of cation-crown ether complexes, confirming the inclusion of the cation within the crown ether.33 While this approach can be challenged by the coexistence of isomers,34 isomers can be separated in the gas phase using ion mobility mass spectrometry (IM-MS).35–38 IM-MS can also provide a collision cross-section (Ω) which can categorize the potential “guest@host” species as either inclusion or exclusion complexes. Collision cross-sections can also be used to monitor changes to the size and shape of hosts due to encapsulation or other reactions,28,39–43 giving mechanistic insight. For example, Schröder, Wollschläger and Schalley synthesized an azobenzene “lasso” that self-threads when reduced. The threaded conformer has a larger collision cross-section owing to a more unfolded structure overall compared to the non-threaded species; a structural change which can be detected by IM-MS.44
Herein we probe the encapsulation of cations by anti-cryptophanes’: (i) categorizing inclusion versus exclusion complexes; (ii) monitoring structural changes occurring due to encapsulation or complexation; and (iii) gaining insight into reaction mechanisms. The molecular recognition of seven anti-cryptophanes (1–7, Chart 2) toward protons (H+), alkali metal cations (Li+, Na+, K+, Rb+ and Cs+) and the ammonium cation (NH4+) are examined using IM-MS.
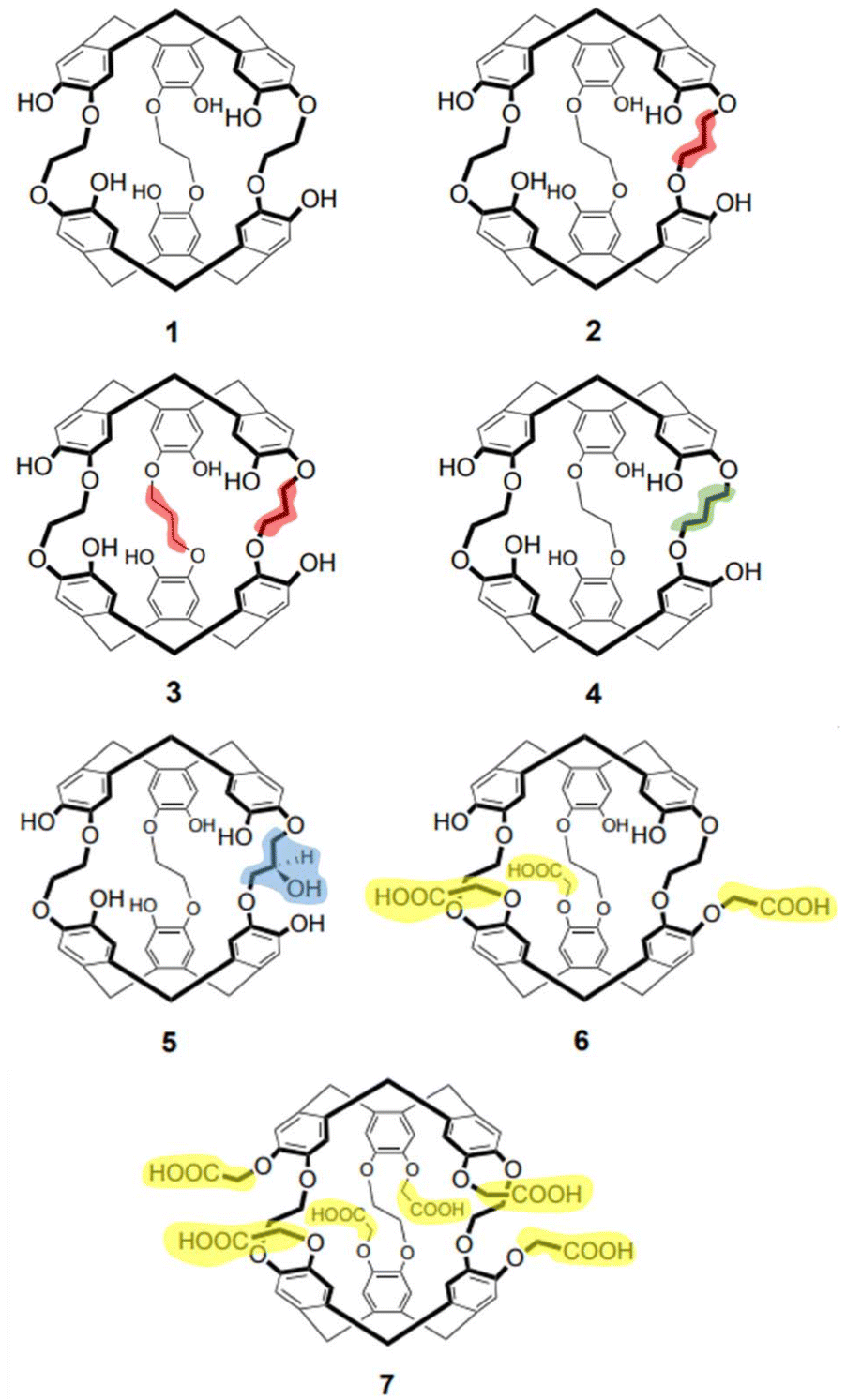 | ||
| Chart 2 Anti-cryptophanes 1–7, highlighting linker length and functionalization. | ||
Structurally, the length of the linker varies in cryptophanes 1–4 (Chart 2, differences highlighted, 1, cryptophane-222(OH)6; 2, cryptophane-223(OH)6; 3, cryptophane-233(OH)6; and 4, cryptophane-224(OH)6). As 4 possesses two ethylenedioxy and one butanedioxy linker (Chart 2, green); it is more flexible than compounds 1 to 3, and can adopt a larger cavity in solution than compounds 1 to 3, simply due to the greater length of the butanedioxy linker.45 Cryptophanes 5–7 (Chart 2) are functionalized with either an additional hydroxyl moiety (cryptophane-233(OH)7, Chart 2, 5, blue) or carboxylic moieties (cryptophane-222(CH2COOH)3(OH)3 and cryptophane-222(CH2COOH)6, Chart 2, 6 and 7 respectively, yellow). For reference, the functionalized cryptophanes have non-functionalized equivalents (cf.: linkers of 2versus5; and 1versus6 or 7).
Results and discussion
The experimental workflow and Methods are summarized in ESI, (Fig. S1 and Pages S3–S8†).8,16,24–26,45–57Isothermal titration calorimetry of cryptophane 4 complexes
As cryptophane 4 is newly synthesized, ITC in LiOH/H2O and NaOH/H2O was performed (ESI, Pages S12 and S13†). In solution, these two cationic species (Li+ and Na+) do not interact with the cavity of these cryptophanes. These conditions mirror those used previously for cryptophanes 1–3 and 6–7,16,25 enabling direct comparison. Binding affinity trends were consistent between 4 and these, where Tl+ > Cs+ ≫ Rb+. In all cases a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with Tl+, Cs+ and Rb+ has been clearly established with compounds 1–6 (compound 7 does not bind cationic species in solution). Previously, slightly higher binding constants were noted in NaOH/D2O, and, consistently, 4 also gives rise to higher binding constants in NaOH/D2O in the presence of Rb+, Cs+ and Tl+.
:1 complex with Tl+, Cs+ and Rb+ has been clearly established with compounds 1–6 (compound 7 does not bind cationic species in solution). Previously, slightly higher binding constants were noted in NaOH/D2O, and, consistently, 4 also gives rise to higher binding constants in NaOH/D2O in the presence of Rb+, Cs+ and Tl+.
Quantitatively, it is difficult to accurately estimate cavity size in solution, but differences in cavity size are anticipated. These anticipated differences could reasonably be 20–25% in solution based on the differences in X-ray structures previously obtained for 1 (i.e. three ethylenedioxy bridges),58versus cryptophane-111 (three methylenedioxy bridges).59 However, despite the anticipated differences in solution phase cavity volume of up to 20% between them, the association constants of 1 and 4 differ by only about one order of magnitude. The similarity between the association constants may be surprising, however, the –OCH2CH2CH2CH2O– linker in 4 significantly increases the degrees of freedom of the compound, as it allows 4 to adopt different overall conformations depending on the gauche versus trans conformation of the linkers. Thus, while difficult to measure, the solution phase cavity size of compound 4 has an allowable change based on linker length and can potentially approach the size of compound 1 upon complexation. This trend in flexibility and allowable cavity size potentially explains the similar association constant. If this trend occurs analogous trends could be expected to be observed in the gas phase.
Ion mobility-mass spectrometry of cryptophane complexes
Cryptophane 1–7 with each cation = H+, Li+, Na+, K+, Rb+, Cs+ and NH4+ formed complexes (eqn (1)) which were measured by two types of IM-MS, travelling wave (TWIM) and cyclic (cIM). The latter allows for extended path lengths for increased resolution. Each [cation + cryptophane]+ ion formed had a single arrival time distribution with TWIM (ESI, Fig. S16†), consistent with a single uniform complex, confirmed using cIM with the benefit of extended path lengths (limiting the possibility of unresolved isoforms contributing to the single observed peak, ESI, Pages S16 and S17†).| cryptophane + cation+ → [cation + cryptophane]+ | (1) |
The measured arrival times of each [cation + cryptophane]+ were used to derive experimental collision cross sections (Ωexp, Table 1) (see ESI, Methods and Page S19†).60
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ω exp | m/z | Ω exp | m/z | Ω exp | m/z | Ω exp | m/z | Ω exp | m/z | Ω exp | m/z | Ω exp | m/z | |
| H+ | 258.5 | 811.3 | 263.4 | 825.3 | 266.3 | 839.3 | 266.6 | 839.3 | 263.4 | 841.3 | 291.8 | 985.3 | 307.7 | 1159.3 |
| Li+ | 246.3 | 817.3 | 253.5 | 831.3 | 258.2 | 845.3 | 256.7 | 845.3 | 251.6 | 847.3 | 268.2 | 991.3 | 293.0 | 1165.3 |
| Na+ | 241.5 | 833.3 | 248.0 | 863.2 | 255.2 | 861.3 | 250.8 | 861.3 | 251.9 | 862.3 | 264.3 | 1007.3 | 292.9 | 1181.3 |
| K+ | 241.5 | 849.2 | 247.4 | 863.2 | 253.0 | 877.3 | 250.7 | 877.5 | 251.4 | 876.3 | 260.6 | 1023.2 | 294.0 | 1197.3 |
| Rb+ | 241.5 | 895.2 | 247.6 | 909.2 | 252.8 | 923.2 | 249.8 | 923.8 | 249.4 | 925.2 | 261.4 | 1069.2 | 292.0 | 1243.2 |
| Cs+ | 241.4 | 943.2 | 248.1 | 957.2 | 253.2 | 971.2 | 251.5 | 971.2 | 249.3 | 972.2 | 262.8 | 1117.2 | 303.3 | 1291.2 |
| NH4+ | 243.5 | 828.3 | 249.6 | 842.3 | 254.2 | 856.3 | 253.2 | 856.3 | 250.5 | 858.8 | 262.6 | 1002.6 | 304.5 | 1176.3 |
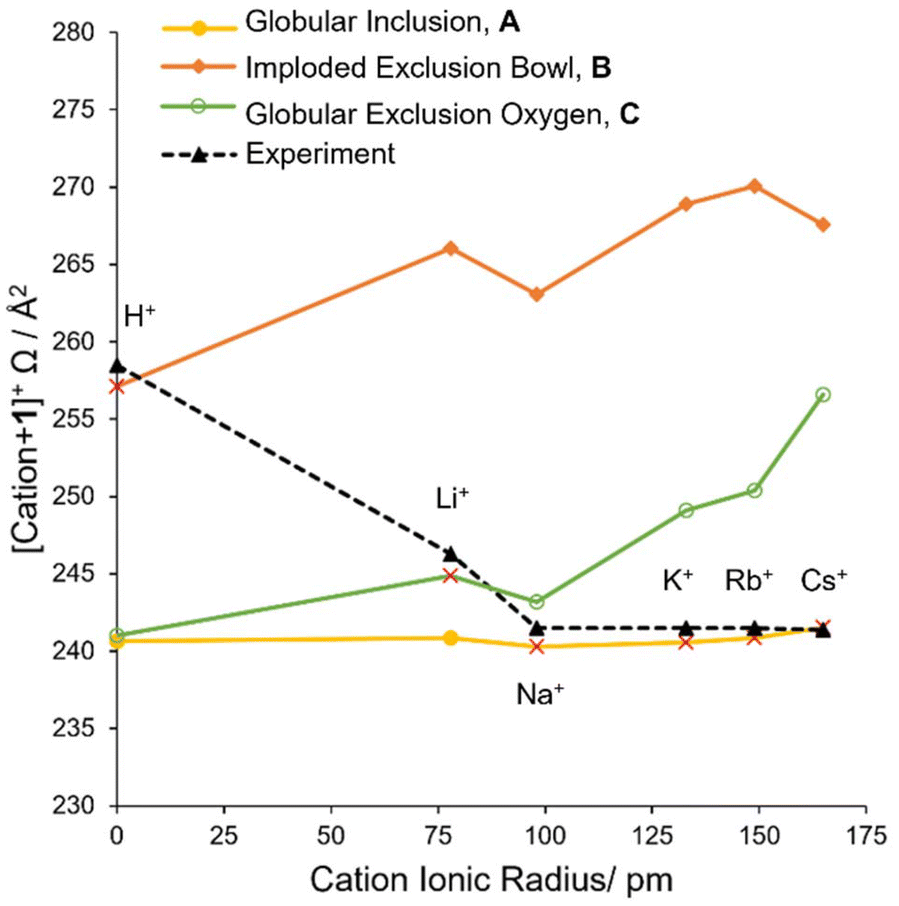
The Ωexp depended significantly on both the type of cryptophane and the cation. Based on the experimental collisional cross sections (Ωexp, Table 1), structural trends were apparent (Fig. 1). All trends were consistent between cIM (ESI Pages S16 and S17†) and TWIM (Table 1) and they held at extended cIM path lengths when ions were stored for up to 130 ms, thus, are not influenced by instrumental conditions. It should be noted that nonspecific dimers, complexes of other stoichiometries, salt clusters or solvent adducts were not observed in any of the mass spectra. This is as expected for a 1:1 association (cage:cation), and is consistent with the ITC stoichiometries.
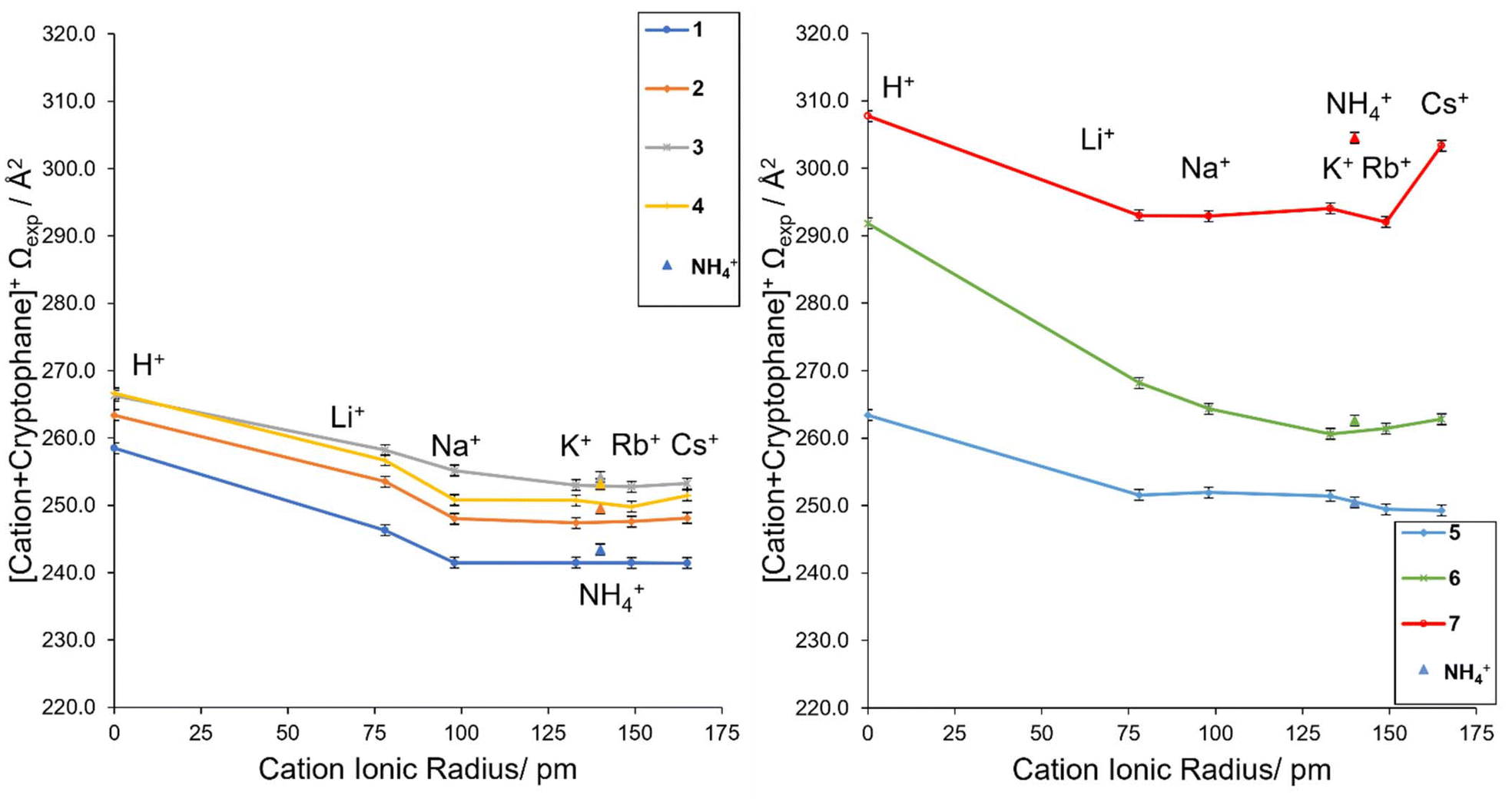 | ||
| Fig. 1 The Ωexpversus cation ionic radius for complexes with (a) cryptophanes 1–4 and (b) functionalized cryptophanes 5–7. | ||
In solution, it is very difficult to experimentally deduce whether the cation remains hydrated once inside the cavity.61 So far, despite efforts, it has not been possible to produce X-ray quality single crystals. However, in the gas phase, after the electrospray ionization process, only species where the cations are not hydrated are present, as solvent adducts are not observed. Under special source,62 or mobility conditions,40,63,64 such species, if present, might be observable. However, to date, only molecular dynamics have been used successfully to explore the potential for hydration with xenon.65,66
The Ωexp differences in 1–4 are large enough for trends to be observed (Fig. 1). For example, [K + 1]+ and [K + 2]+ differ by a single methylene group (cf.1versus2, Chart 2, mass difference 14 Da), but their Ωexp are 241.5 Å2 and 247.4 Å2, respectively, revealing [K + 2]+ is larger than [K + 1]+. Thus, relatively small modifications to the cryptophane, such as an additional CH2, result in an IM-MS measurable change in size and shape. An exception are [H + 3]+ and [H + 4]+, despite their different structures (Chart 2), both have an identical Ωexp of 266 Å2. Generally, 3 > 4 > 2 > 1 is the relative gas phase size order of complexes according to IM-MS (Fig. 1a). This ordering indirectly shows the high flexibility of host 4.
Functionalization as in cryptophanes 5–7 results in an overall increase in the collision cross section (Fig. 1b). For example, a small general increase in Ωexp is observed for 5 compared to 2 for all [cation + cryptophane]+ species, except Li+ (cf.Fig. 1a, orange series and Fig. 1b, blue series). A small increase in Ωexp is expected for 5, with both the small increase in linker size and charge due to the extra OH. The additional OH should not significantly interfere with the encapsulation process and in some cases, may self-encapsulate, further avoiding an increase in cross section.67 The cryptophanes functionalized with carboxylic moieties, 6 and 7, displayed a far more significant increase in collision cross section compared to 1 (cf.Fig. 1b, green and red series and Fig. 1a, blue series). This is expected, as the additional carboxylic groups will inhabit far more space and have more degrees of freedom than the hydrogens they replace, resulting in extended structures. Thus, 7 ≫ 6 ≫ 5 ≥ 3 > 4 > 2 > 1 is the relative size order of the cryptophane complexes, with key outliers as mentioned above for cation = H+ and Li+.
In a relative sense, cryptophanes complexed with cations Na+, K+, Rb+ and Cs+ are approximately 93–95% of the size of the protonated cryptophanes, which is unexpected. Li+ complexes exist at an intermediary size, without the full relative contraction of the lower Group 1 cations, but smaller than the protonated cryptophanes. One exception to this is the large “jump” exhibited by the [Cs + 7]+ species (Fig. 1b), a feature that implies the cesium ion is not fully encapsulated within 7. This is consistent with previously noted internal hydrogen bonding within the cryptophane, resulting in a more extended structure and increased steric hinderance as measured by 1H-NMR and ECD.15,26 The “jump” trend is also observed for NH4+. This suggests NH4+ is also not fully encapsulated in 7, which was not previously known. The ammonium cation's mass of 18 Da lies between Li+ (7 Da) and Na+ (23 Da), whilst its ionic radius of 140 pm lies between K+ (133 pm) and Rb+ (149 pm).69 Experimentally, NH4+ complexes have Ωexp equal to or larger than Cs+ complexes (Fig. 1), including the previously noted “jump” for 7. This suggests that while the anti-cryptophanes are encapsulating NH4+, non-spherical species may not induce the same rearrangement of the cryptophane structure as metal atoms. That is, their coordination may be less generalized and more directional, resulting in a larger structure.70 The flat trend that dominates 1–5 also varies in 6, suggesting that like 7 the encapsulation is being affected by the functionalization, including an uptick of cation = Cs+ that may be a less dramatic “jump” due to incomplete encapsulation of the cation.
Overall, H+ and Li+ are outliers of the cation trends observed, being much larger than expected.
To account for the observed decrease (size of complex of H+ ≫ Li+ ≫ Na+) and the flat trend (size of complex of Na+ ≈ K+ ≈ Rb+ ≈ Cs+ ≈ NH4+) within a given cryptophane series, which does not simply follow the periodic trend in the ionic radii of the cations, several locations of the cation relative to the cryptophane are possible:
• the cation forms an inclusion complex within the globular cryptophane, i.e. is encapsulated (Fig. 2, A);
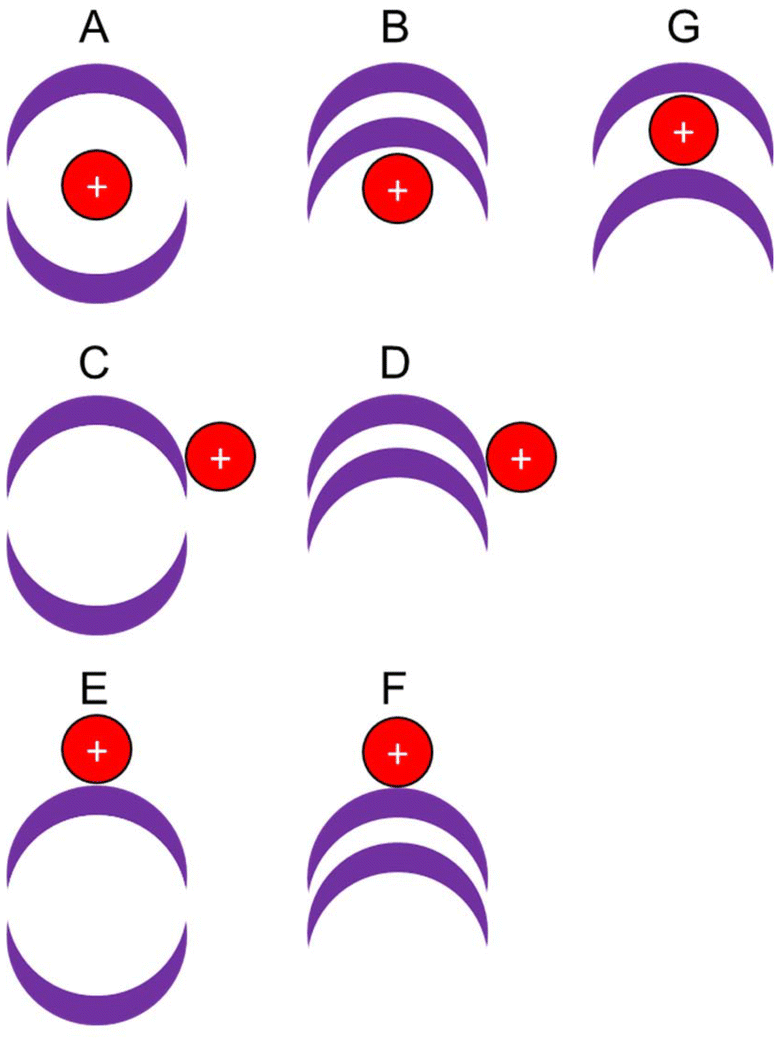 | ||
| Fig. 2 Potential cation isomers in globular and imploded cryptophane forms. | ||
• the cation is adducted to the exterior of the globular cryptophane, i.e. an exclusion complex, (Fig. 2, C or E);
• a non-globular cryptophane with an adducted cation, i.e. an imploded exclusion complex, (Fig. 2, B, D or F);
• a non-globular cryptophane with an interstitial cation between the two CTB bowls i.e. an imploded inclusion complex (Fig. 2, G).
For complexes with external cations, size trends should correlate with the cation's radius and buffer gas interactions due to the external charge distribution. Encapsulated complexes size trends’ will not be as strongly influenced by the cation's radius or charge, as it is shielded within. However, the cryptophane may exhibit structural changes dependent on the guest@host interaction. Due to their rearrangement, imploded non-globular structures will differ in collisional cross sections from globular ones.
As mentioned, experimentally the largest cations (Rb+ and Cs+) counterintuitively yield smaller complexes than the smallest cations (H+ and Li+). Considering the potential trends, a single trend of those mentioned above does not apply. DFT was used to interrogate the potential isomeric complexes.
DFT predicted structures of cryptophane complexes
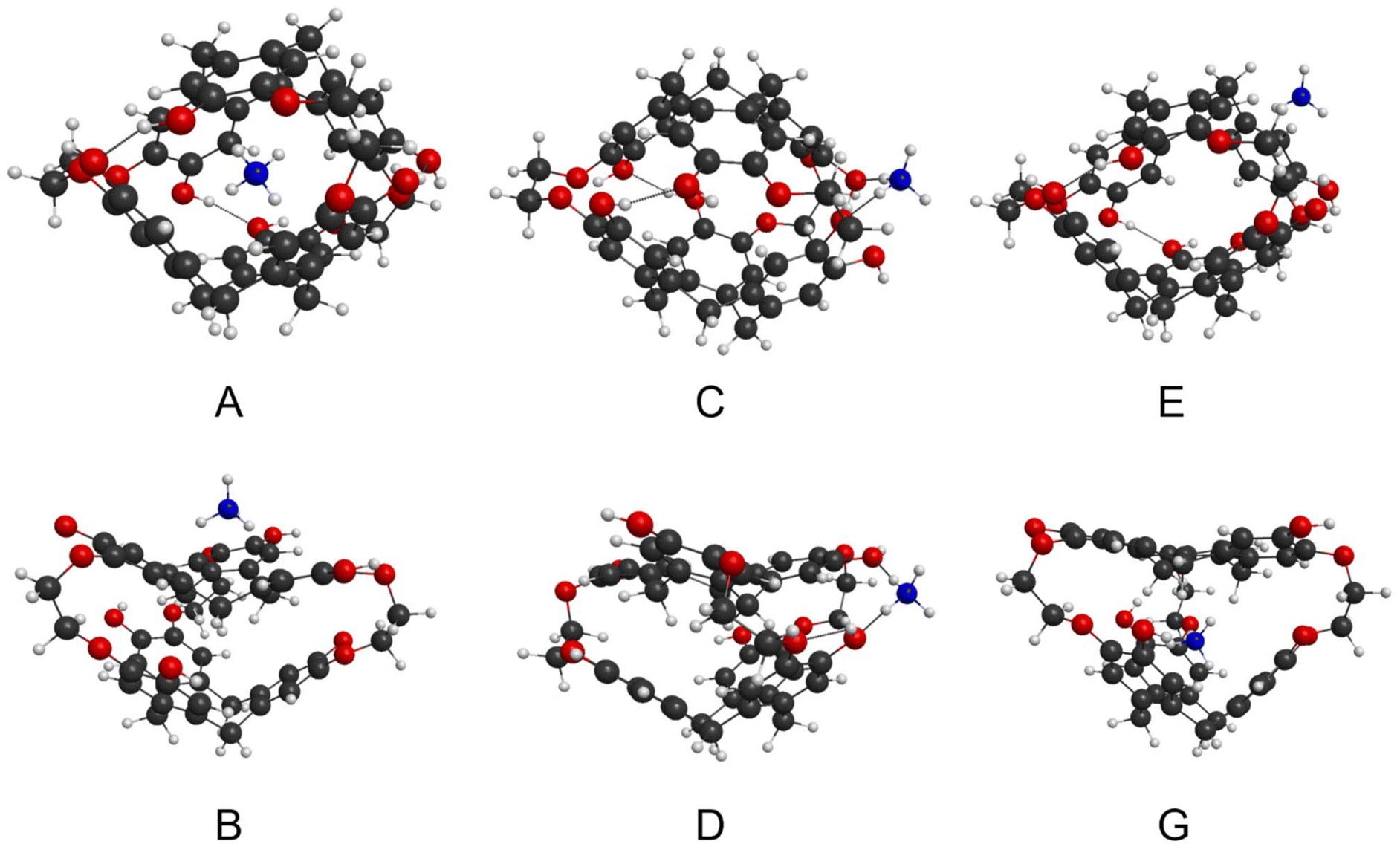 | ||
| Fig. 3 The six optimized structures for [NH4 + 1]+: globular complexes A, C, and E, where the NH4+ is either an inclusion complex (encapsulated) or an exclusion complex in two different locations; imploded complexes B, D and G with NH4+ in different locations. | ||
The globular inclusion complex, A, are the lowest energy structures for cation = Na+, K+, Rb+, Cs+ and NH4+ (Table 2). The energy of A is significantly lower than all the other structures (>70 kJ mol−1 lower), which strongly supports that the single isoform structures experimentally observed for these cations are globular inclusion complexes. The Ωcalc of these globular inclusion complexes very closely matches Ωexp (Fig. 4). This is further support for encapsulation occurring for these cations, without other competitive complexes forming.
 | ||
| Fig. 4 Predicted cross sections, Ωcalc changes with the cation for [cation + 1]+ ions. | ||
| Cation | A | B | C | D | E | G | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Globular inclusion | Imploded exclusion bowl | Globular exclusion oxygen | Imploded exclusion oxygen | Globular exclusion aromatic | Imploded inclusion | |||||||
| ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | |
| a Proton ΔE values are significantly larger in magnitude as the complexes formed are covalent cryptophane carbocations and are not structural analogues to other cationic association complexes and cation–π complexes. b Optimization led to a distorted structure that cannot be described as an inclusion complex. | ||||||||||||
| H+ | −969a | 240.7 | −721a | 257.1 | −962a | 241.0 | −659a | 264.2 | −889a | 244.3 | −658a | 265.9 |
| Li+ | −344 | 240.8 | −70 | 266.0 | −314 | 244.9 | −46 | 260.5 | −207 | 248.9 | −47 | 262.8 |
| Na+ | −321 | 240.3 | 20 | 263.1 | −248 | 243.2 | 21 | 261.8 | −133 | 249.6 | 26 | 262.8 |
| K+ | −277 | 240.6 | 28 | 268.9 | −168 | 249.1 | 74 | 262.7 | −93 | 252.6 | 101 | 264 |
| Rb+ | −254 | 240.8 | 42 | 270.1 | −149 | 250.4 | 94 | 262.6 | −81 | 254.3 | 144 | 262.1 |
| Cs+ | −226 | 241.6 | 98 | 267.6 | −68 | 256.6 | 108 | 266.1 | −76 | 253.7 | n/ab | n/ab |
| NH4+ | −283 | 241.3 | 18 | 293 | −129 | 249.5 | 70 | 298 | −99 | 256.8 | 133 | 300 |
On the other hand, the Li+ cation is predicted to be energetically competitive (Table 2) between globular inclusion, A (−344 kJ mol−1) and globular exclusion oxygen, C (−314 kJ mol−1). The collisional cross section comparison (Fig. 4) reveals the Li+ structure matches a globular exclusion oxygen position. Thus, with the single isoform present, [Li + 1]+ is found to form isomer C. Thus, the Li cation prefers interaction with oxygen over π–cation interactions, the favorability of which may be underestimated by the DFT modelling.
Energetically, cation = H+ is an outlier, with significantly lower energy complexes (ca. 600–700 kJ mol−1 lower) compared with the other cations, (Table 2). These predicted bond energies are commensurate with the proton affinities of similar molecules.71 The formation of C–H covalent bonds is thermodynamically favorable compared to association or cation–π complexes. As such, these are not true structural analogues to the other alkali metals’ complexes, instead with formation of a cryptophane carbocation, with a different electrostatic potential distribution (ESI Page S25 and Fig. S19†). Also, while all structures A–E and G are predicted as thermodynamically accessible, presumably important kinetic barriers govern the selectivity between the structures. Based on predicted versus measured collisional cross sections and presence of a single isoform, the cryptophane's globular structure is not indicated by the experiment (Fig. 4). An imploded exclusion bowl complex, B, is instead assigned. The ordering of energetics is at odds with this assignment, which we suggest is due to kinetic control of the formation reaction. It also should not be assumed an imploded form exists in solution. As evidenced by other experimental analysis, smaller cryptophanes exist exclusively in the globular form in the solution phase.72
Energies (ΔE, Table 3) for alkali metals Na–Cs and NH4, show that a [cation@cryptophane]+ inclusion complex (orange bars, Fig. 5) is favorable. Consistent with 1, for 2–4, an oxygen exclusion complex is energetically competitive with encapsulation for Li+ (Table 3, Fig. 5, green and orange bars).
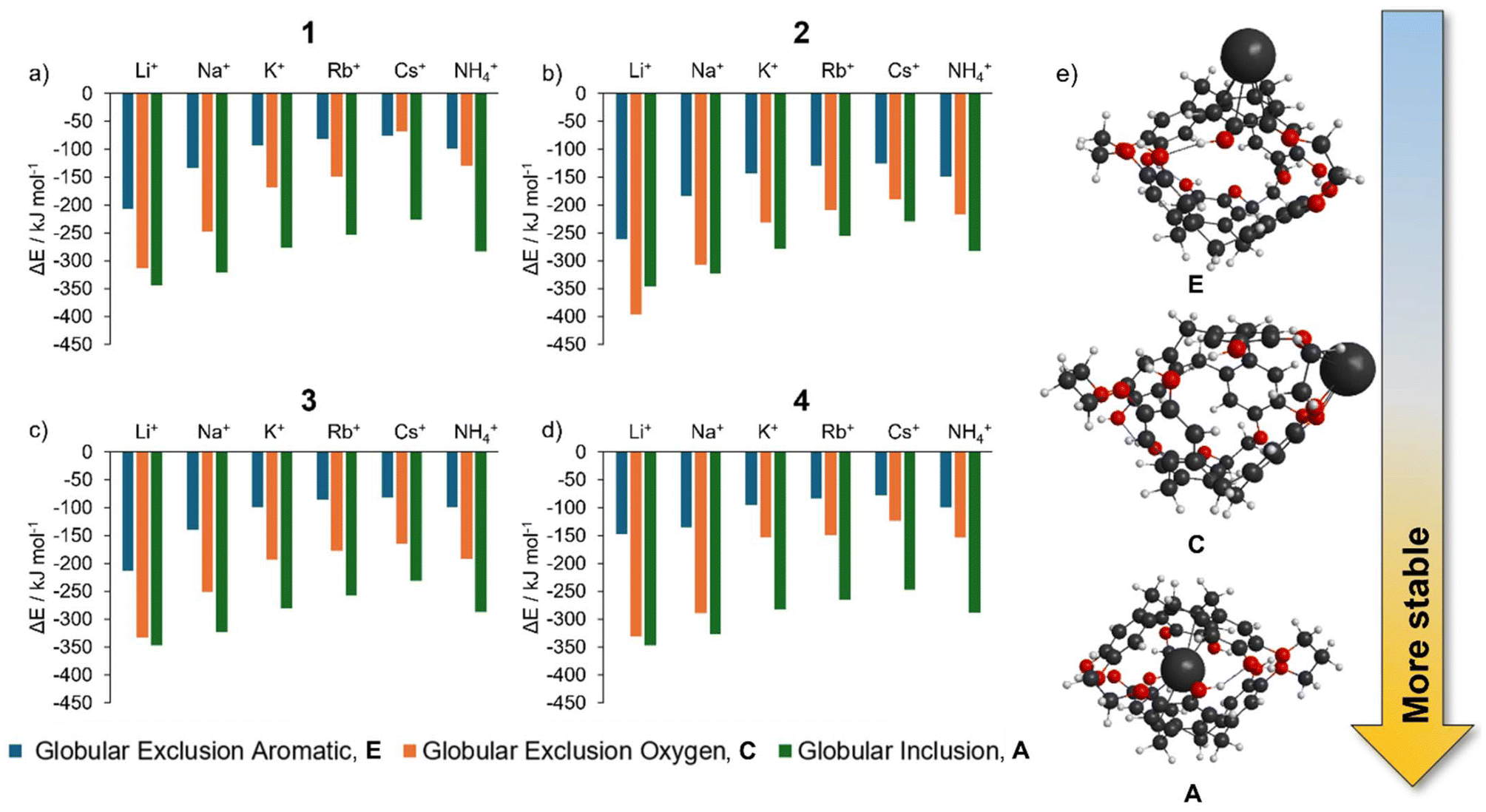 | ||
| Fig. 5 (a–d) The cation position (A, C or E) versus stability of the globular complexes cryptophanes 1–4, respectively, (e), large alkali metals (K–Cs) and NH4 are more stable encapsulated (A) compared to association complexes with exterior positions (exclusion oxygen, C or exclusion aromatic, E). | ||
| Cryptophane | Cation | A | C | E | |||
|---|---|---|---|---|---|---|---|
| Globular inclusion | Globular exclusion oxygen | Globular exclusion aromatic | |||||
| ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ΔE/kJ mol−1 | Ω calc/Å2 | ||
| 2 | Li+ | −346 | 243.5 | −396 | 243.6 | −262 | 248.8 |
| Na+ | −323 | 243.4 | −307 | 244.1 | −184 | 249.9 | |
| K+ | −279 | 243.5 | −231 | 246.1 | −143 | 253.4 | |
| Rb+ | −256 | 243.8 | −209 | 247.3 | −129 | 254.4 | |
| Cs+ | −229 | 244.4 | −190 | 249.3 | −125 | 254.4 | |
| NH4+ | −282 | 244 | −216 | 249.9 | −149 | 256.5 | |
| 3 | Li+ | −347 | 246.7 | −332 | 250.9 | −214 | 253.6 |
| Na+ | −323 | 246.4 | −251 | 251.9 | −140 | 254.6 | |
| K+ | −280 | 246.8 | −193 | 254.1 | −99 | 258.3 | |
| Rb+ | −258 | 246.9 | −177 | 254.6 | −86 | 258.6 | |
| Cs+ | −231 | 247.7 | −164 | 255.2 | −82 | 260.6 | |
| NH4+ | −287 | 247.1 | −192 | 255.4 | −99 | 261.3 | |
| 4 | Li+ | −347 | 246.0 | −330 | 245.4 | −148 | 259.0 |
| Na+ | −327 | 246.1 | −289 | 246.1 | −135 | 256.0 | |
| K+ | −282 | 245.6 | −153 | 252 | −95 | 259.6 | |
| Rb+ | −266 | 246.9 | −149 | 252.9 | −83 | 259.1 | |
| Cs+ | −247 | 247.9 | −123 | 254 | −78 | 258.7 | |
| NH4+ | −288 | 246.2 | −154 | 254.2 | −100 | 259.2 | |
Between the cryptophanes, the size trends observed from experiment (Ωexp, Table 1 and Fig. 1) are reproduced in Ωcalc with 3 ≳ 4 > 2 > 1 (Tables 2 and 3).
Based on Ωexp (i.e. the much larger structures from the experiments) globular structures are not predicted for protonated 2–4, but rather imploded species. For completeness bond energies were calculated (ESI Table S13 and Page S26†) and as previous, these were commensurate with C–H bond formation observed for analogous structures.71
Compellingly, the globular inclusion Ωcalc values for 2–4 have a flat trend from Na+ to Cs+ (Fig. 6a and Table 3) as previous for 1 (Fig. 6a and Table 2). For 1–4 the globular inclusion complexes’ predicted reaction energies, combined with the flat trend in measured collisional cross section between Na–Cs, strongly supports assignment of encapsulated alkali metal cations Na–Cs and NH4 (Fig. 5). This is notable for Na+, as encapsulation has not been observed with any of 1–4 in solution.
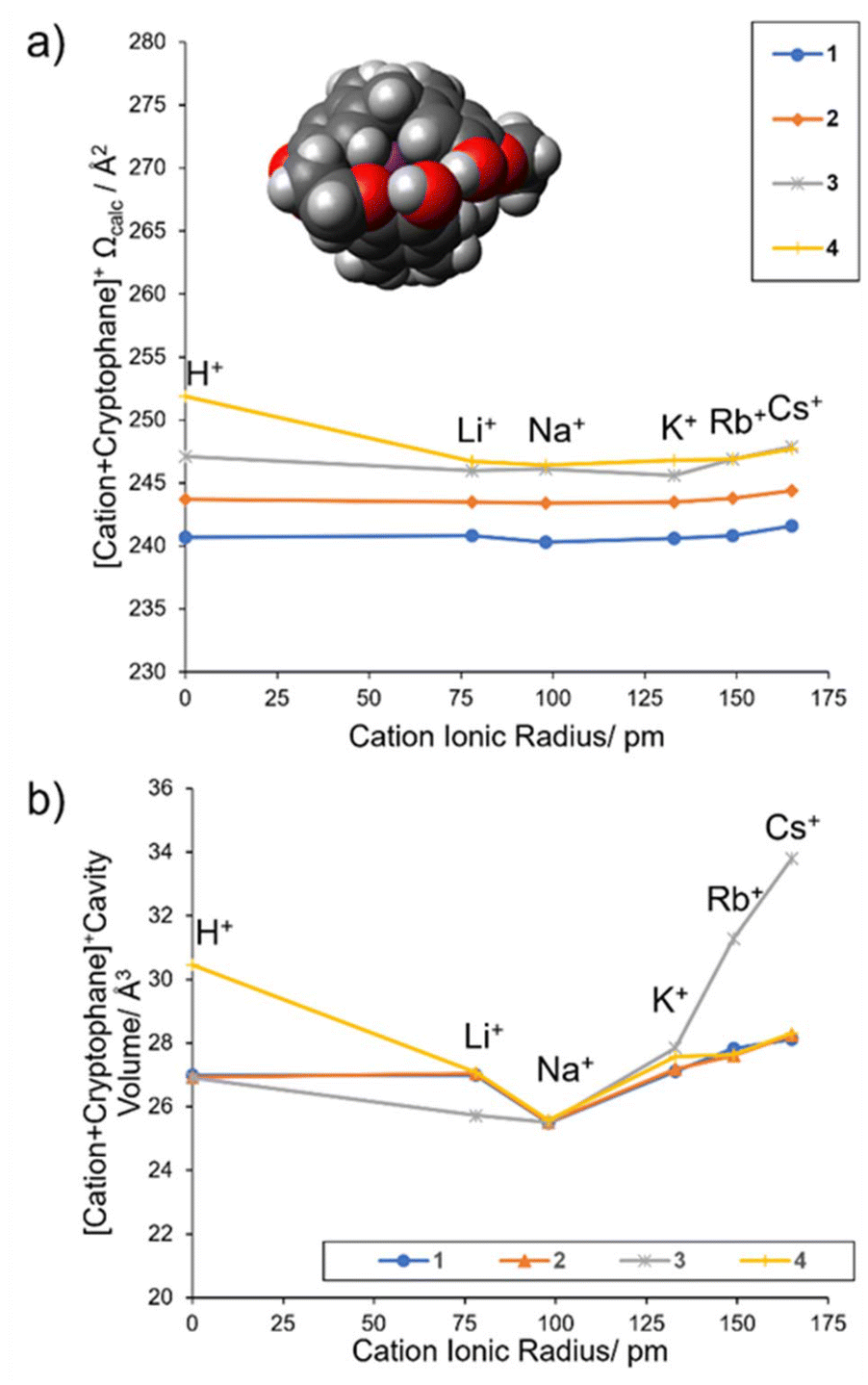 | ||
| Fig. 6 (a) Predicted collision cross sections for globular complexes, A, of 1–4, (b) calculated volume of the cryptophane cavity. | ||
Globular inclusion size trends should be the most immune to variation in cationic radii. Other types of structures if present would show a trend of increasing Ω proportional to the cation's ionic radius (ESI Page S26†). For Na+ to Cs+ complexed with 2–4, the cations fit “snugly” within the central cavity, encouraging a favorable cation–π interaction. This induces contraction and conformational rearrangement of the cryptophane linkers to maximize interaction and minimize the distance between the cation and the electron-rich structure. However, the relatively “rigid” bowls of the cryptophane define a lower bound to this contraction, which creates the flat collisional cross section trend observed. To understand how this trend related to the internal cavity volume, a geometric approach was used (Methods, ESI Pages S5 and S27–S28†). The volume of the internal cavities of both a globular inclusion A, and globular aromatic exclusion complex E were assessed for cryptophanes 1–4 with all alkali metals (Table 4). The globular aromatic exclusion complex E cavity volume does not contract and does not change between metals (consistently being ∼27 Å3, Table 4), as expected for a lack of encapsulation, being an external association complex only (ESI Page S28†). Comparing the volume of this cavity and that of the encapsulated complex allows us to judge the % difference cavity volume upon encapsulation (Table 4).
| Aromatic exclusion (E) | Inclusion (A) | ||||
|---|---|---|---|---|---|
| Cryptophane | Cation | Cavity vol./Å3 | Cavity vol./Å3 | Packing coeff. (%) | % diff. cavity vol. |
| 1 | H+ | 27.9 | 27.0 | 0.0 | −3.2 |
| Li+ | 27.1 | 27.0 | 11.3 | −0.4 | |
| Na+ | 27.3 | 25.5 | 25.7 | −6.6 | |
| K+ | 27.2 | 27.1 | 54.3 | −0.4 | |
| Rb+ | 27.2 | 27.8 | 68.8 | 2.2 | |
| Cs+ | 27.2 | 28.1 | 69.4 | 3.3 | |
| 2 | H+ | 27.8 | 26.9 | 0.0 | −3.2 |
| Li+ | 27.1 | 27.0 | 11.3 | −0.4 | |
| Na+ | 27.2 | 25.5 | 25.6 | −6.3 | |
| K+ | 27.2 | 27.2 | 54.1 | 0.0 | |
| Rb+ | 27.3 | 27.6 | 69.4 | 1.1 | |
| Cs+ | 27.2 | 28.3 | 69.0 | 4.0 | |
| 3 | H+ | 27.7 | 26.9 | 0.0 | −2.9 |
| Li+ | 27.2 | 25.7 | 11.9 | −5.5 | |
| Na+ | 27.1 | 25.5 | 25.6 | −5.9 | |
| K+ | 27.1 | 27.8 | 52.8 | 2.6 | |
| Rb+ | 27.1 | 31.3 | 61.3 | 15.5 | |
| Cs+ | 27.0 | 33.8 | 57.7 | 25.2 | |
| 4 | H+ | 32.2 | 30.5 | 0.0 | −5.3 |
| Li+ | 27.3 | 27.1 | 11.3 | −0.7 | |
| Na+ | 27.4 | 25.6 | 25.6 | −6.6 | |
| K+ | 27.3 | 27.6 | 53.3 | 1.1 | |
| Rb+ | 27.2 | 27.6 | 69.3 | 1.5 | |
| Cs+ | 27.3 | 28.3 | 68.9 | 3.7 | |
It was found for globular inclusion complexes A that the gas phase cavity volume varied with cation. A minimum volume occurred for all cryptophanes’ complexes 1–4 when cation = Na+ (mean cavity volume = 27.6 Å3, σ = 1.9). Put another way, the cavity contracts around the cation, with Na+ causing the largest contraction. Cation = Na consistently caused the cavity volumes ∼6% smaller upon encapsulation (% diff. cavity vol. ∼ −6%, Table 4). On the other hand, cation = Rb and Cs caused the cavities to get larger (∼2% and ∼4% respectively) for 1, 2 and 4, while cryptophane 3 was an outlier, with the largest cavity differences, with Rb being ∼15% and Cs ∼25% larger, respectively (Table 4). Cation = Li barely altered the cavity (% diff. cavity vol. < −1%) except in the case of 3, where it decreased the cavity ∼6%, similar to the change for Na. The % diff. cavity vol. for cation = K is variable but minimal, being 0.4% smaller for 1 and not changing for 2 (% diff. cavity vol = 0), and then increasing in size by only 1% in 4 and only 2.6% in 3. This is consistent with K being a “good fit”.
Calculating the volume of cations as spheres reveals the proportion of the cavity filled, with larger cations filling proportionally more cavity (Table 4). The calculated proportions align with the “55% rule” by Mecozzi and Rebek,73 which suggests optimal binding occurs when the packing coefficient is around 0.55. Among the four cryptophanes, K+ most closely adheres to this rule with a packing coefficient of 0.536, indicating it's the most suitable guest. Conversely, the extreme values for Cs+ and H+ suggest they are weaker guests, deviating significantly from the rule. This correlation supports the geometric approach used to determine cavity size.
For the metals, the internal cavity volume (Fig. 6b) do not significantly correlate to the Ωcalc trend (Fig. 6a), suggesting that e.g. whilst the cavity volume is contracting, the linkers are displaced, offsetting any large change in collision cross section area. Likewise, a large increase in cavity volume as in 3 with Rb and Cs is presumably offset in the collisional cross section, though 3 does have the largest measured collisional cross sections of 1–4, there is no significant uptick to correlate Ωexp with the trend in Fig. 6b (gray line). However, the % diff. cavity volumes do indicate that 4 is behaving like 1 and 2.
For H+, the external carbocations, E, have larger cavities than internal carbocations, A (Table 4). But these cavity sizes cannot account for the larger than anticipated measured collisional cross sections (Fig. 1). The Ωcalc for 1 (Table 2) clearly reveal only imploded structures are predicted.
Conclusions
Cryptophanes’ host–guest complexes can be observed by IM-MS. This is especially relevant to complexes such as these which have proven difficult to crystallize. The larger cations, Na+ to Cs+ and NH4+, were confirmed as globular inclusion complexes with cryptophanes 1–7 (excluding 7 with Cs+ and NH4+) in the gas phase. Electronic structure calculations reveal these cations energetically favor encapsulation within globular cryptophanes 1–4. The experimental cross section for [Cs + 7]+ is noticeably larger, suggesting full encapsulation does not occur, consistent with Bouchet and coworkers’ earlier findings.26 A similar size increase suggests the same occurs for NH4+. Several other predictions from the condensed phase were borne out for specific cryptophanes in the gas phase, such as the smaller structure due to the flexibility of 4. On the other hand, we observed encapsulation of Na+, which so far has not been observed in the condensed phase.Deviating from other alkali metals, Li+ catonization resulted in an experimentally larger overall structure. While DFT energetics were competitive, collisional cross sections confirmed the Li+ cations preferred to associate with oxygen at a linker, and that the cryptophanes remained in the globular form.
Perhaps most surprisingly, the protonated cryptophanes had the largest experimental structures overall. These larger structures more closely matched cross sections of a non-globular “imploded” form. Just as native conditions allow proteins to be studied in a non-collapsed state,74 this suggests that analogous supramolecular-native conditions may be worthwhile for studying some macromolecular cages. In the future, such conditions may even allow for exploration of hitherto analytically inaccessible hydrated states.
Predicted gas phase cavity volumes indicate structural changes within the cryptophane cage upon encapsulation, revealing mechanistic traits. Additional properties and structural insights now present themselves to be derived experimentally from the IM, such as the reactivity of the guests,75 encapsulation over time,76,77 photophysics of isolated macromolecular complexes,78 and the automated categorization of molecular recognition processes,79 to name a few.
Author contributions
The manuscript was written through contributions of all authors: conceptualization: OHLW and NJR; formal analysis: OHLW, CSC, TB; funding acquisition: NJR; investigation: OHLW, CSC, MYZ, OR, LJ, DLM, MCP, TB, NJR; methodology: OHLW, CSC, TB, BLJP, NJR; project administration: NJR; supervision: ML, WAD, BLJP, NJR; writing – original draft: OHLW, TB, NJR. All authors have edited and given approval to the final version of the manuscript.Data availability
Python scripts are available at https://zenodo.org/doi/10.5281/zenodo.11174559. Detailed synthetic and analytical methods, and supplementary figures as mentioned in the text and additional literature citations58,73,79–82 are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge: the Australian Research Council via grants DE170100677 and LE220100031; subsidized access to the Bioanalytical Mass Spectrometry Facility within the Mark Wainwright Analytical Centre of the University of New South Wales; the Queensland University of Technology, Central Analytical Research Facility (CARF, QUT); both the UNSW Resource Allocation Scheme and Katana managed by Research Technology Services at UNSW Sydney, with the assistance of resources and services from the National Computational Infrastructure (NCI, gy60, bb55), which is supported by the Australian Government. Preliminary work was carried out with the support of the Karlsruhe Nano Micro Facility (KNMF, https://www.knmf.kit.edu), a Helmholtz Research Infrastructure at Karlsruhe Institute of Technology. Finally, we thank Jessica Holmes, Felix Rizzuto, Tyren Dodgen, Niklas Geue, Perdita Barran, and Terry Frankcombe for helpful discussions.References
- K. Ariga, H. Ito, J. P. Hill and H. Tsukube, Chem. Soc. Rev., 2012, 41, 5800–5835 RSC.
- C. Jonkergouw, N. K. Beyeh, E. Osmekhina, K. Leskinen, S. M. Taimoory, D. Fedorov, E. Anaya-Plaza, M. A. Kostiainen, J. F. Trant, R. H. A. Ras, P. Saavalainen and M. B. Linder, Nat. Commun., 2023, 14, 2141 CrossRef CAS PubMed.
- T. Brotin, P. Berthault, K. Chighine and E. Jeanneau, ACS Omega, 2022, 7, 48361–48371 CrossRef CAS PubMed.
- A. Ciferri, Soft Matter, 2021, 17, 8585–8589 RSC.
- D. J. Cram, Nature, 1992, 356, 29–36 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC.
- T. Ogoshi, S. Kanai, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Am. Chem. Soc., 2008, 130, 5022–5023 CrossRef CAS PubMed.
- A. Collet, Tetrahedron, 1987, 43, 5725–5759 CrossRef CAS.
- T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS PubMed.
- J. C. Mejuto and J. Simal-Gandara, Int. J. Mol. Sci., 2022, 23(24), 15730 CrossRef PubMed.
- J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1981, 1137–1139 RSC.
- J. Costante-Crassous, T. J. Marrone, J. M. Briggs, J. A. McCammon and A. Collet, J. Am. Chem. Soc., 1997, 119, 3818–3823 CrossRef CAS.
- A. Bouchet, T. Brotin, M. Linares, H. Ågren, D. Cavagnat and T. Buffeteau, J. Org. Chem., 2011, 76, 4178–4181 CrossRef CAS PubMed.
- O. Baydoun, T. Buffeteau and T. Brotin, Chirality, 2021, 33, 562–596 CrossRef CAS PubMed.
- T. Brotin, R. Montserret, A. Bouchet, D. Cavagnat, M. Linares and T. Buffeteau, J. Org. Chem., 2012, 77, 1198–1201 CrossRef CAS PubMed.
- T. Brotin, P. Berthault, D. Pitrat and J.-C. Mulatier, J. Org. Chem., 2020, 85, 9622–9630 CrossRef CAS PubMed.
- S. Kancherla and J. H. Hansen, Eur. J. Org. Chem., 2023, e202301050 Search PubMed.
- T. Traoré, L. Delacour, S. Garcia-Argote, P. Berthault, J.-C. Cintrat and B. Rousseau, Org. Lett., 2010, 12, 960–962 CrossRef PubMed.
- L. K. S. von Krbek, C. A. Schalley and P. Thordarson, Chem. Soc. Rev., 2017, 46, 2622–2637 RSC.
- M. Bastos, O. Abian, C. M. Johnson, F. Ferreira-da-Silva, S. Vega, A. Jimenez-Alesanco, D. Ortega-Alarcon and A. Velazquez-Campoy, Nat. Rev. Methods Primers, 2023, 3, 17 CrossRef CAS.
- C. Sgarlata, J. S. Mugridge, M. D. Pluth, B. E. F. Tiedemann, V. Zito, G. Arena and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 1005–1009 CrossRef CAS PubMed.
- O. H. Lloyd Williams and N. J. Rijs, Front. Chem., 2021, 9, 407 Search PubMed.
- T. Brotin, D. Cavagnat, P. Berthault, R. Montserret and T. Buffeteau, J. Phys. Chem. B, 2012, 116, 10905–10914 CrossRef CAS PubMed.
- L.-L. Chapellet, J.-P. Dognon, M. Jean, N. Vanthuyne, P. Berthault, T. Buffeteau and T. Brotin, ChemistrySelect, 2017, 2, 5292–5300 CrossRef CAS.
- T. Brotin, S. Goncalves, P. Berthault, D. Cavagnat and T. Buffeteau, J. Phys. Chem. B, 2013, 117, 12593–12601 CrossRef CAS PubMed.
- A. Bouchet, T. Brotin, M. Linares, D. Cavagnat and T. Buffeteau, J. Org. Chem., 2011, 76, 7816–7825 CrossRef CAS PubMed.
- B. F. Abrahams, N. J. FitzGerald, T. A. Hudson, R. Robson and T. Waters, Angew. Chem., Int. Ed., 2009, 48, 3129–3132 CrossRef CAS PubMed.
- H. Zhang, M. Grabenauer, M. T. Bowers and D. V. Dearden, J. Phys. Chem. A, 2009, 113, 1508–1517 CrossRef CAS PubMed.
- L. Cera and C. A. Schalley, Chem. Soc. Rev., 2014, 43, 1800–1812 RSC.
- H. P. Ryan, Z. S. Fishman, J. T. Pawlik, A. Grommet, M. Musial, F. Rizzuto, J. C. Booth, C. J. Long, K. Schwarz, N. D. Orloff, J. R. Nitschke and A. C. Stelson, J. Am. Chem. Soc., 2023, 145, 19533–19541 CrossRef CAS PubMed.
- N. Geue, R. E. P. Winpenny and P. E. Barran, Chem. Soc. Rev., 2022, 51, 8–27 RSC.
- G. Carroy, V. Lemaur, J. De Winter, L. Isaacs, E. De Pauw, J. Cornil and P. Gerbaux, Phys. Chem. Chem. Phys., 2016, 18, 12557–12568 RSC.
- P. B. Armentrout, Int. J. Mass Spectrom., 1999, 193, 227–240 CrossRef CAS.
- M. B. More, D. Ray and P. B. Armentrout, J. Am. Chem. Soc., 1999, 121, 417–423 CrossRef CAS.
- S. E. Anderson, D. J. Bodzin, T. S. Haddad, J. A. Boatz, J. M. Mabry, C. Mitchell and M. T. Bowers, Chem. Mater., 2008, 20, 4299–4309 CrossRef CAS.
- S. Tainaka, T. Ujihira, M. Kubo, M. Kida, D. Shimoyama, S. Muramatsu, M. Abe, T. Haino, T. Ebata, F. Misaizu, K. Ohshimo and Y. Inokuchi, J. Phys. Chem. A, 2020, 124, 9980–9990 CrossRef CAS PubMed.
- L. Tapia, Y. Pérez, J. Solà, S. V. Luis, I. Alfonso and C. Vicent, Analyst, 2022, 147, 5546–5556 RSC.
- C. Przybylski and V. Bonnet, Carbohydr. Polym., 2022, 297, 120019 CrossRef CAS PubMed.
- J. L. Casas-Hinestroza, M. Bueno, E. Ibáñez and A. Cifuentes, Anal. Chim. Acta, 2019, 1081, 32–50 CrossRef CAS PubMed.
- K. A. Servage, J. A. Silveira, K. L. Fort and D. H. Russell, Acc. Chem. Res., 2016, 49, 1421–1428 CrossRef CAS PubMed.
- E. Kalenius, M. Groessl and K. Rissanen, Nat. Rev. Chem., 2019, 3, 4–14 CrossRef CAS.
- J. Shen and D. V. Dearden, Isr. J. Chem., 2018, 58, 225–229 CrossRef CAS.
- J. Lee, S.-S. Lee, S. Lee and H. B. Oh, Molecules, 2020, 25(18), 4048 CrossRef CAS PubMed.
- H. V. Schröder, J. M. Wollschläger and C. A. Schalley, Chem. Commun., 2017, 53, 9218–9221 RSC.
- T. Brotin and J.-P. Dutasta, Eur. J. Org. Chem., 2003, 2003, 973–984 CrossRef.
- J. B. Maglic and R. Lavendomme, J. Appl. Crystallogr., 2022, 55, 1033–1044 CrossRef CAS PubMed.
- N. Geue, T. S. Bennett, A.-A.-M. Arama, L. A. I. Ramakers, G. F. S. Whitehead, G. A. Timco, P. B. Armentrout, E. J. L. McInnes, N. A. Burton, R. E. P. Winpenny and P. E. Barran, J. Am. Chem. Soc., 2022, 144, 22528–22539 CrossRef CAS PubMed.
- C. Larriba-Andaluz and C. J. Hogan Jr., J. Chem. Phys., 2014, 141, 194107 CrossRef PubMed.
- H. Ouyang, C. Larriba-Andaluz, D. R. Oberreit and C. J. Hogan, J. Am. Soc. Mass Spectrom., 2013, 24, 1833–1847 CrossRef CAS PubMed.
- C. Larriba and C. J. Hogan, J. Comput. Phys., 2013, 251, 344–363 CrossRef CAS.
- C. Larriba and C. J. Hogan Jr., J. Phys. Chem. A, 2013, 117, 3887–3901 CrossRef CAS PubMed.
- N. Mehta, B. F. Abrahams and L. Goerigk, Chem. – Asian J., 2020, 15, 1301–1314 CrossRef CAS PubMed.
- K. Giles, J. Ujma, J. Wildgoose, S. Pringle, K. Richardson, D. Langridge and M. Green, Anal. Chem., 2019, 91, 8564–8573 CrossRef CAS PubMed.
- A. Bouchet, T. Brotin, D. Cavagnat and T. Buffeteau, Chem. – Eur. J., 2010, 16, 4507–4518 CrossRef CAS PubMed.
- R. E. Ireland and D. M. Walba, Org. Synth., 1977, 56, 44 CrossRef CAS.
- L.-L. Chapellet, J. R. Cochrane, E. Mari, C. Boutin, P. Berthault and T. Brotin, J. Org. Chem., 2015, 80, 6143–6151 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- O. Taratula, P. A. Hill, N. S. Khan, P. J. Carroll and I. J. Dmochowski, Nat. Commun., 2010, 1, 148 CrossRef PubMed.
- A. I. Joseph, S. H. Lapidus, C. M. Kane and K. T. Holman, Angew. Chem., Int. Ed., 2015, 54, 1471–1475 CrossRef CAS PubMed.
- M. F. Bush, I. D. G. Campuzano and C. V. Robinson, Anal. Chem., 2012, 84, 7124–7130 CrossRef CAS PubMed.
- D. K. Eggers, S. Fu, D. V. Ngo, E. H. Vuong and T. Brotin, J. Phys. Chem. B, 2020, 124, 6585–6591 CrossRef CAS PubMed.
- G. Seeber, G. J. T. Cooper, G. N. Newton, M. H. Rosnes, D.-L. Long, B. M. Kariuki, P. Kögerler and L. Cronin, Chem. Sci., 2010, 1, 62–67 RSC.
- K. Ohshimo, X. He, R. Ito, K. Tsunoda, S. Tainaka and F. Misaizu, EPJ Tech. Instrum., 2023, 10, 11 CrossRef.
- K. A. Servage, J. A. Silveira, K. L. Fort, D. E. Clemmer and D. H. Russell, J. Phys. Chem. Lett., 2015, 6, 4947–4951 CrossRef CAS PubMed.
- R. Pollet, J.-P. Dognon and P. Berthault, ChemPhysChem, 2024, 25, e202300509 CrossRef CAS PubMed.
- R. Pollet, J.-P. Dognon and P. Berthault, Phys. Chem. Chem. Phys., 2024, 26, 23561–23565 RSC.
- O. Baydoun, T. Buffeteau, N. Daugey, M. Jean, N. Vanthuyne, L.-L. Chapellet, N. De Rycke and T. Brotin, Chirality, 2019, 31, 481–491 CrossRef CAS PubMed.
- T. Brotin, N. Daugey, N. Vanthuyne, E. Jeanneau, L. Ducasse and T. Buffeteau, J. Phys. Chem. B, 2015, 119, 8631–8639 CrossRef CAS PubMed.
- V. Sidey, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 626–633 CrossRef CAS PubMed.
- J. Canceill, M. Cesario, A. Collet, J. Guilhem, L. Lacombe, B. Lozach and C. Pascard, Angew. Chem., Int. Ed. Engl., 1989, 28, 1246–1248 CrossRef.
- S. Kolboe, J. Chem. Theory Comput., 2014, 10, 3123–3128 CrossRef CAS PubMed.
- F. L. Thorp-Greenwood, M. J. Howard, L. T. Kuhn and M. J. Hardie, Chem. – Eur. J., 2019, 25, 3536–3540 CrossRef CAS PubMed.
- S. Mecozzi and R. Julius Jr., Chem. – Eur. J., 1998, 4, 1016–1022 CrossRef CAS.
- Z. Hall, A. Politis, M. F. Bush, L. J. Smith and C. V. Robinson, J. Am. Chem. Soc., 2012, 134, 3429–3438 CrossRef CAS PubMed.
- T.-C. Lee, E. Kalenius, A. I. Lazar, K. I. Assaf, N. Kuhnert, C. H. Grün, J. Jänis, O. A. Scherman and W. M. Nau, Nat. Chem., 2013, 5, 376–382 CrossRef CAS PubMed.
- G. Carroy, C. Daxhelet, V. Lemaur, J. De Winter, E. De Pauw, J. Cornil and P. Gerbaux, Chem. – Eur. J., 2016, 22, 4528–4534 CrossRef CAS PubMed.
- S. Moorthy, H. Lambert, N. Mohan, T. Schwarzlose, W. M. Nau, E. Kalenius and T.-C. Lee, Angew. Chem., Int. Ed., 2023, 62, e202303491 CrossRef CAS PubMed.
- I. Czerwinska, A. Kulesza, C. Choi, F. Chirot, A.-L. Simon, J. Far, C. Kune, E. de Pauw and P. Dugourd, Phys. Chem. Chem. Phys., 2016, 18, 32331–32336 RSC.
- O. H. Lloyd Williams, O. Rusli, L. Ezzedinloo, T. M. Dodgen, J. K. Clegg and N. J. Rijs, Angew. Chem., Int. Ed., 2023, e202313892 Search PubMed.
- S. C. North, K. R. Jorgensen, J. Pricetolstoy and A. K. Wilson, Front. Chem., 2023, 11, 1152500 CrossRef CAS PubMed.
- D. Ongari, P. G. Boyd, O. Kadioglu, A. K. Mace, S. Keskin and B. Smit, J. Chem. Theory Comput., 2019, 15, 382–401 CrossRef CAS PubMed.
- H. A. Fogarty, P. Berthault, T. Brotin, G. Huber, H. Desvaux and J.-P. Dutasta, J. Am. Chem. Soc., 2007, 129, 10332–10333 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic and analytical methods, and supplementary figures as mentioned in the text. Additional literature citations. See DOI: https://doi.org/10.1039/d4dt01824a |
| This journal is © The Royal Society of Chemistry 2024 |