DOI:
10.1039/D4DT01840K
(Communication)
Dalton Trans., 2024,
53, 18828-18833
A twist on a classic scaffold: rational design of a new bimetallic platform†
Received
25th June 2024
, Accepted 30th July 2024
First published on 31st July 2024
Abstract
Herein, we report the synthesis of a modular family of novel bimetallic tetraamidodiamine (tada) ligands, Li4-R-tada (R = Me3Si, tBuMe2Si, and iPr3Si). These silylamido ligands display two distinct binding pockets whose steric profiles can be easily tuned by choice of the substituents on silicon. We also show that salt metathesis is a convenient route to install these new ligands on the early transition metals titanium(IV) and vanadium(III).
Some of the most powerful transformations in nature are made possible through the action of catalysts featuring multimetallic active sites. For instance, soluble methane monooxygenase (sMMO), a diiron enzyme found in methanotropic bacteria, can convert methane to methanol under ambient conditions,1 a remarkable process that has yet to be replicated synthetically. Despite the key roles that these multimetallic species play, detailed studies on their mechanisms of action remain rare, in part because of the difficulties associated with studying such complex systems. Thus, it is of interest to examine simple, well-defined models of these active systems to gain valuable insights into how they function.2,3 These data can then be used to rationally design subsequent generations of improved catalysts.
In order to isolate the effects of metal–metal interactions on reactivity, we sought to prepare a new binucleating ligand scaffold that closely mirrored a well-studied, monometallic congener in which the primary coordination sphere would be conserved. We identified the tris(2-aminoethyl)amine (tren) framework as a promising candidate for this study. The triply-silylated tren derivative (Me3SiNHCH2CH2)3N was first reported by Verkade in 1989,4 where its original use was to stabilize Group 14 compounds. Several years later, Cummins and Schrock demonstrated that the trianionic form of above silylamine was an excellent ligand for early transition metals (see Scheme 1, left).5–7 The silyl groups of the metal complexes are oriented such that they create an accessible binding pocket at the axial coordination site opposite the tertiary amine donor atom. The substituents on the silyl also provide sufficient steric protection to prevent bimolecular decomposition while still allowing easy access to the transition metal for small molecule substrates. An appealing feature of this triamidoamine platform is that the substituents are highly variable, allowing for a wide degree of both steric and electronic tuning. We reasoned that a tetraamidodiamine (tada) analogue of such a ligand would be an appealing platform to enforce a bimetallic active site and allow for in-depth study of metal–metal interactions. Indeed, during the course of our studies reported here, Zhang, Hong, and Shi reported a binucleating, aryl-substituted tada ligand scaffold that could be applied towards dinitrogen activation with titanium (see Scheme 1, right).8 Interestingly, the authors reported successful C–H amination of the ligand backbone, which they propose occurs via a nitridyl intermediate.
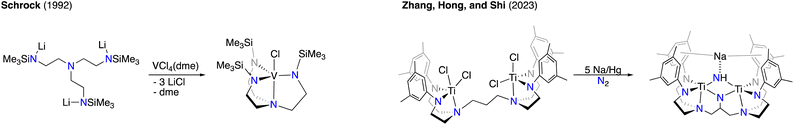 |
| | Scheme 1 Literature examples of triamidoamine and tetraamidodiamine complexes. | |
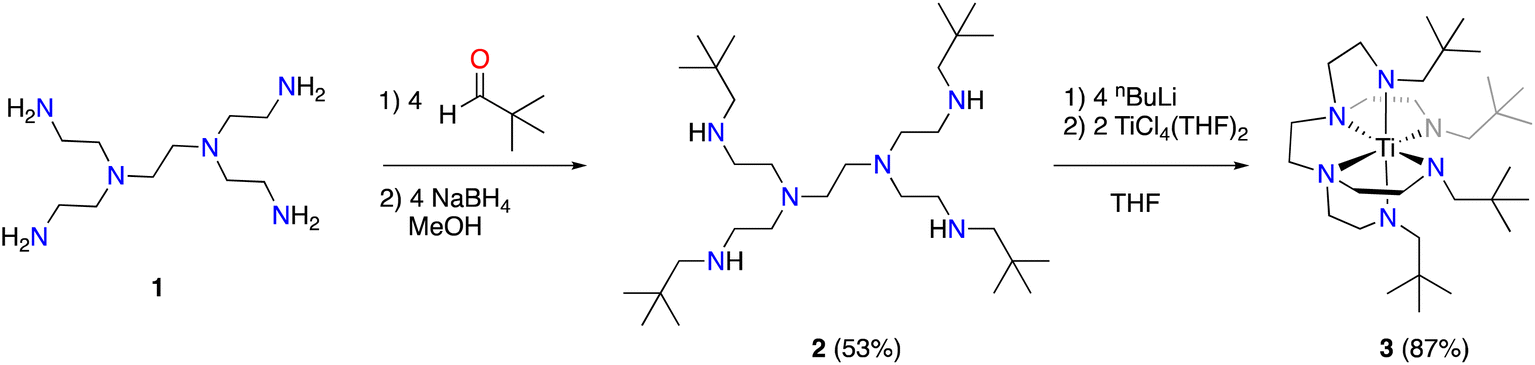
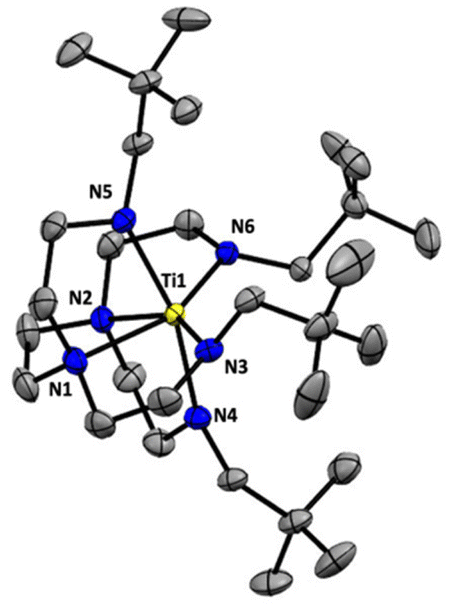
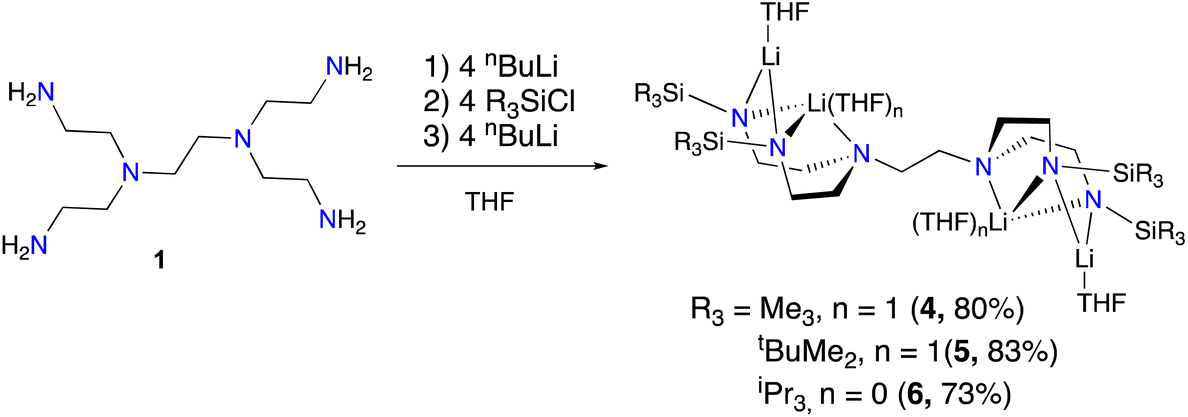
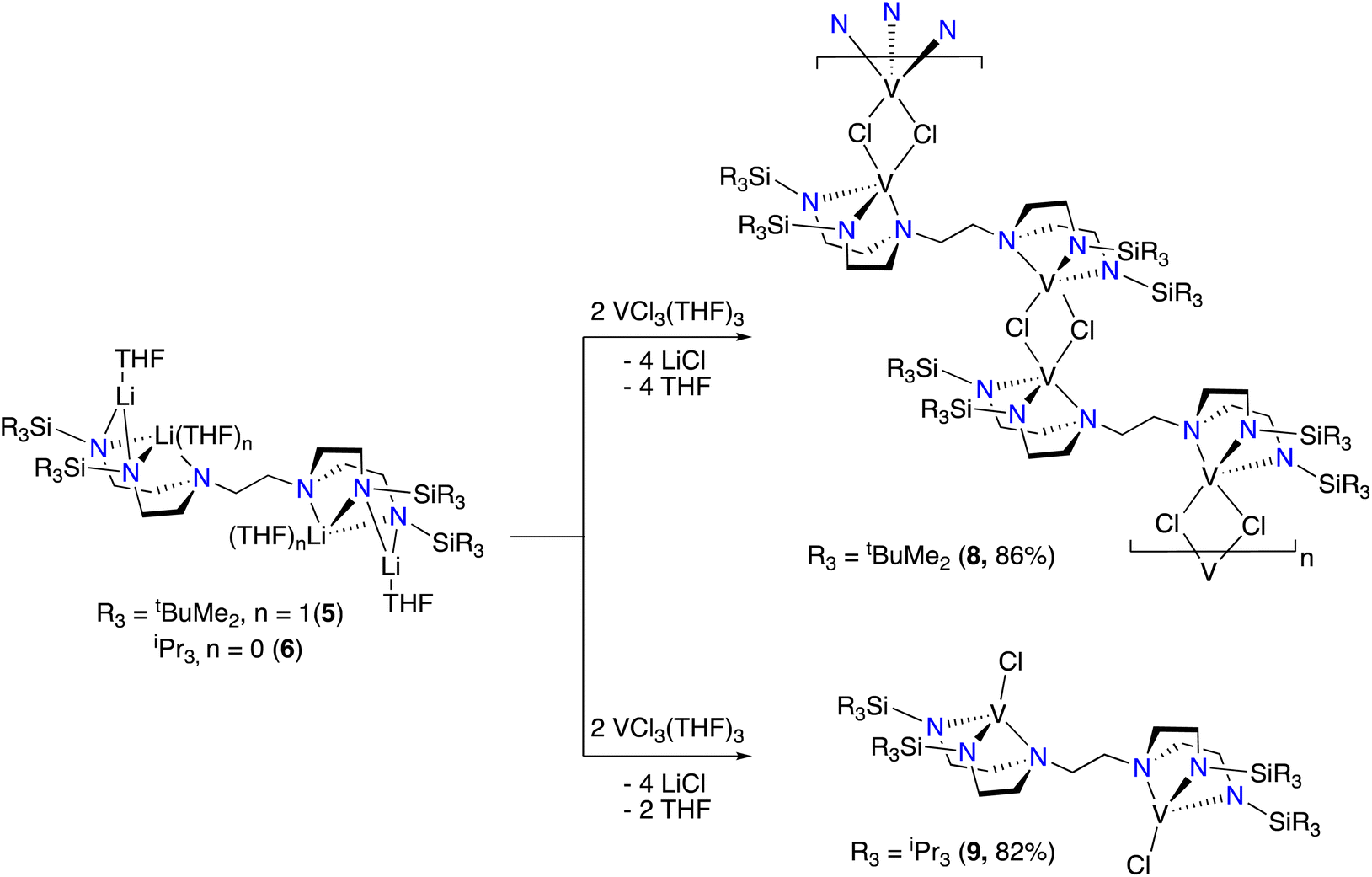
We selected hexaamine 1 (see Scheme 2) as a promising candidate upon which to construct our targeted binucleating ligand motif. Despite being known in the literature since 1952,9 only a few groups have made use of 1 as a ligand building block.10–13 Inspired by previous work from Verkade14 and Scheer,15 we found that the neopentyl-substituted ligand precursor H4-Np-tada (2, Np = neopentyl) could be prepared smoothly in a two-step, one-pot reductive amination procedure of 1 with 4 equivalents of pivaldehyde. Compound 2 was isolated as a colourless oil in moderate yield (53%) and was fully characterized by NMR spectroscopy and high-resolution mass spectrometry. With 2 in hand, we next sought to explore its coordination chemistry. Following the examples of Schrock6 and Zhang, Hong, and Shi,8 we sought to coordinate our new scaffold to early transition metals. Treatment of THF solutions of 2 with 4 equivalents of nBuLi resulted in a change from colourless to pale yellow. Subsequent addition of 2 equivalents of bright yellow TiCl4(THF)2 results in a rapid colour change to dark red-brown. After removal of the volatiles in vacuo, the dark brown residue was extracted with hexane, filtered, and dried again to yield an orange-brown powder. Analysis by 1H NMR spectroscopy revealed a single major product (Np-tada-Ti, compound 3), and recrystallization of the crude product could be achieved by cooling a saturated hexane solution to −35 °C overnight, resulting in the deposition of orange-red crystals suitable for X-ray diffraction. The solid-state structure of compound 3 is shown in Fig. 1. The complex is monometallic, with the titanium(IV) center adopting an octahedral geometry featuring both amine and all four amido donors from the ligand. We reasoned that larger substituents on the amido nitrogens would be required to open up the binding pocket to preferentially accommodate two metals. Historically, silylated triamidoamine ligands have been successfully utilized by several groups, including Schrock,5–7,16 Filippou,17,18 and Liddle.19,20 Following a modification of the procedure developed by Schrock, hexaamine 1 was suspended in THF and treated with 4 equivalents of nBuLi, followed by the addition of 4 equivalents of R3SiCl (R3 = Me3, tBuMe2, or iPr3, see Scheme 3). Following removal of the volatiles in vacuo and extraction with hexane, filtration through Celite yielded pale yellow solutions, which were subsequently treated with another 4 equivalents of nBuLi. After solvent removal and recrystallization, compounds R-tada-Li4 (R = Me3Si (4), R = tBuMe2Si (5), and R = iPr3Si (6)) could be isolated as colourless crystals in good yields. All three compounds were characterized by 1H and 13C NMR spectroscopy, as well as by diffraction studies. X-ray quality crystals of compounds 4 and 5 were grown from THF/hexane at −35 °C, while suitable crystals of 6 were grown by cooling a saturated hexane solution. The solid-state structures are shown in Fig. 2. All three of the complexes show two binding pockets that each contain external (κ2) and internal (κ3) Li centers. The structures of 4 and 5 are very similar, with each Li being bound by a single THF molecule. In contrast, compound 6 only features THF bound to the two κ2 Li centers (see Fig. 2). We attribute this to the increased steric hindrance of the bulky iPr3Si groups. The binding pockets of all three complexes can be considered as more open, dianionic analogues of the commonly used triazacyclononane (tacn) family of ligands.
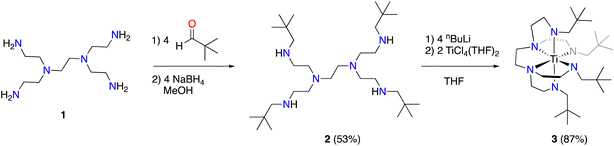 |
| | Scheme 2 Synthesis of compound 2 and complex 3. Isolated yields in parentheses. | |
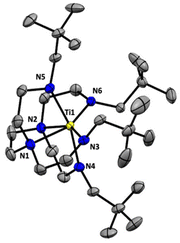 |
| | Fig. 1 Solid-state structure of 3 (50% probability ellipsoids). All hydrogen atoms omitted for clarity. | |
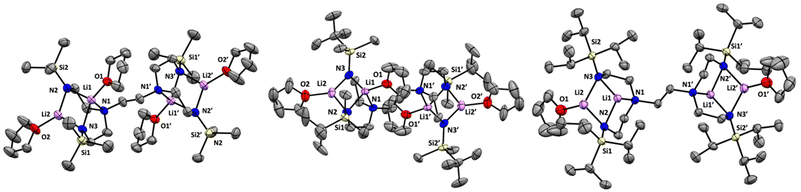 |
| | Fig. 2 Solid-state structures of 4 (left), 5 (middle), and 6 (right), all with 50% probability ellipsoids. All hydrogen atoms omitted for clarity. | |
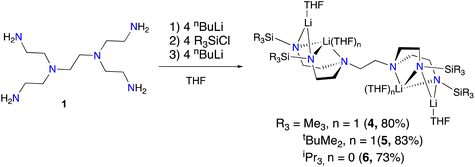 |
| | Scheme 3 Synthesis of tetralithium complexes 4, 5, and 6. Isolated yields in parentheses. | |
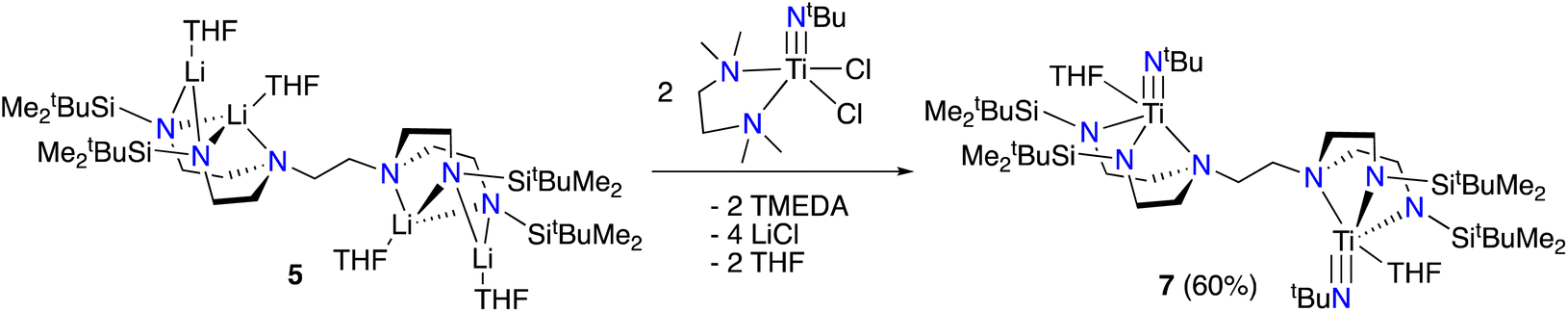
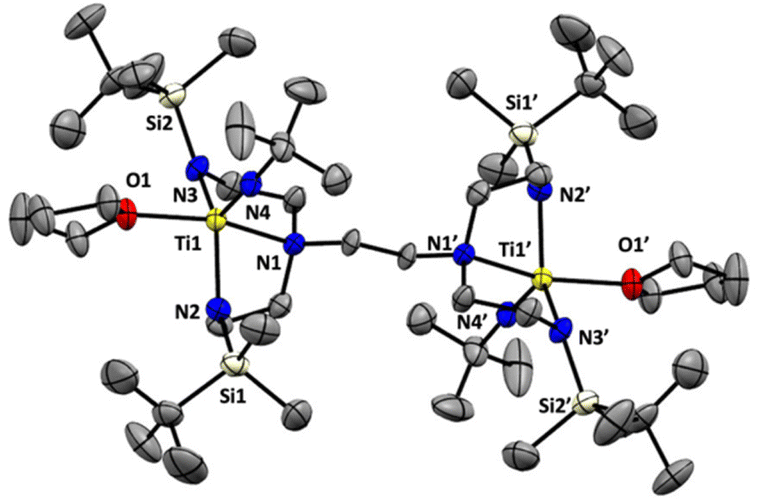
We next sought to explore the coordination chemistry of 4–6 with early transition metals. Unfortunately, while each lithiated ligand precursor did react with either TiCl4(THF)2 or TiCl3(THF)3, as indicated by rapid colour changes, characterization of the products has been hindered by the broadness of the peaks observed in the 1H NMR spectra, as well as the significant solubility of the products in organic solvents. Fortunately, switching to a different titanium(IV) precursor results in more tractable product formation. Addition of 5 to a yellow solution of [(tmeda)Ti(NtBu)Cl2]21 (tmeda = N,N,N′,N′-tetramethylethylenediamine) in THF along with gentle heating (50 °C) results in a colour change to orange-red. Analysis by 1H NMR spectroscopy reveals the formation of a new symmetric product, tBuMe2Si-tada-[Ti(NtBu)(THF)]2 (7, see Scheme 4) in 60% spectroscopic yield. Attempts to isolate compound 7 in pure form were hindered by its decomposition to multiple unidentified species during workup and recrystallization. Nevertheless, we were able to obtain orange-red crystals from these attempts which were suitable for diffraction analysis. The solid-state structure of 7 is shown in Fig. 3. Gratifyingly, the complex demonstrates successful incorporation of one metal into each of the binding pockets. Like the tetralithium complexes 4–6, the structure of 7 is centrosymmetric. The coordination geometry at titanium is a distorted square pyramid, with the imido moiety occupying the apical position. The last coordination site is filled by a THF ligand.
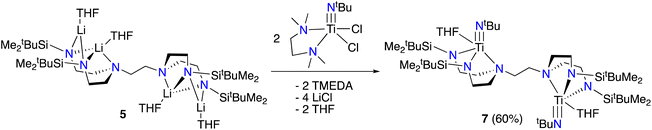 |
| | Scheme 4 Synthesis of complex 7. 1H NMR spectroscopic yield in parentheses. | |
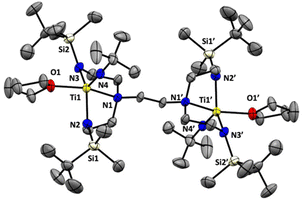 |
| | Fig. 3 Solid-state structure of complex 7 (50% probability ellipsoids). All hydrogen atoms omitted for clarity. | |
The structure of 7 reveals no metal–metal interactions, which is unsurprising given the d0–d0 configuration. To probe whether an increase in the d-electron count or the presence of potentially bridging chloride ligands would lead to observable metal–metal interactions in these complexes, we next turned our attention to vanadium(III). Stirring THF solutions of either 5 or 6 with 2 equivalents of VCl3(THF)3 resulted in colour changes from maroon to a dark red-brown or purple, respectively. Complexes tBuMe2Si-tada-[VCl]2 (8) and iPr3Si-tada-[VCl]2 (9, see Scheme 5) were both isolated from their respective reaction mixtures by extraction with hexane, and X-ray quality crystals of each were grown by cooling saturated hexane solutions to −35 °C overnight. The solid-state structures are shown in Fig. 4. Complex 8 crystallizes as an intermolecular coordination polymer, with the V–Cl moiety of one binding pocket coming together with that of another molecule of 8 to form a V2Cl2 diamond linkage, which daisy chains along the polymer backbone. Related geometries at vanadium(III) have been observed by Cloke22 and Gambarotta,23 The V–V distance across the V2Cl2 diamondoid in 8 is 3.7161(9) Å, slightly longer than the corresponding distance in the crystallographic data reported by Gambarotta.23 In contrast, the solid-state structure of 9 is monomeric, with each metal center adopting a distorted trigonal monopyramidal geometry. Like complex 7, the solid-state structure of 9 is centrosymmetric. While 8 shows a substantially different coordination environments from the vanadium(III) tren complex reported by Schrock,6 complex 9 displays a similar trigonal monopyramidal geometry We attribute the structural differences between 8 and 9 to the increased steric profile of the iPr3Si groups. To quantify the importance of steric effects in these tada systems, we turned to buried volume (Vbur).24,25 Not surprisingly, the monometallic complex 3, which is coordinatively saturated with the tada ligand and essentially binds both binding pockets, has a buried volume of 96.7%. The tada ligands of complexes 7 (Vbur = 73.7%), 8 (Vbur = 87.8%) and 9 (Vbur = 83.2%), on the other hand, display only marginally lower buried volumes despite consisting of a single binding pocket per metal center. Evans method analysis of 8 and 9 reveals solution-state effective magnetic moments of 3.4μB and 3.6μB per bimetallic complex, respectively. The observed values are higher than that observed for Cloke's V2Cl2 dimer,22,23 and may indicate a degree of antiferromagnetic coupling between the two (d2) vanadium(III) centers. Similar coupling has recently been observed with dititanium.26 Neither 8 nor 9 reacted with 2 equivalents of Et4NCl in THF at room temperature, as shown by UV/Vis spectroscopy. Further studies to coordinate these new multimetallic ligands to other transition metals, as well as exploring new oxidation states that may enable discrete metal–metal interactions, are ongoing in our laboratory.
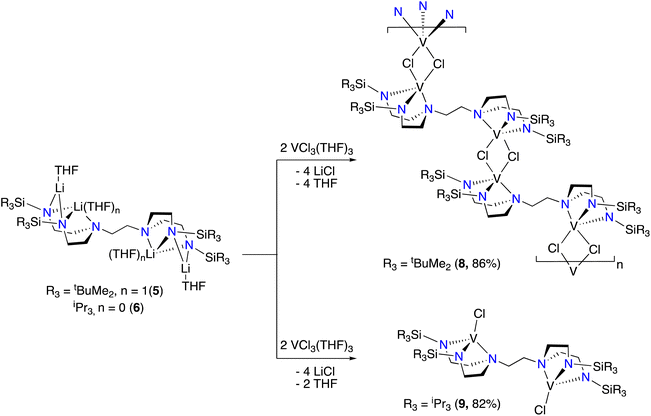 |
| | Scheme 5 Synthesis of complexes 8 and 9. Isolated yields in parentheses. | |
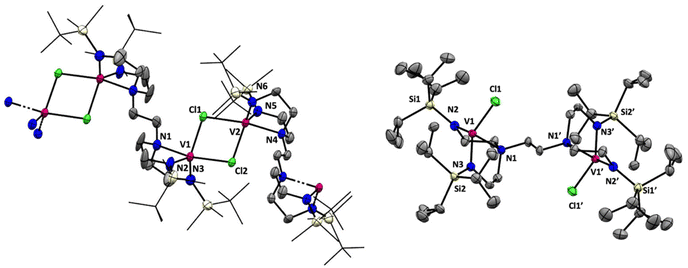 |
| | Fig. 4 Solid-state structures of complex 8 (left, 40% probability ellipsoids) and complex 9 (right, 50% probability ellipsoids). All hydrogen atoms omitted for clarity. The silyl substituents of 8 are in wireframe. | |
Conclusions
This report details the synthesis and structural characterization of three new lithiated tetraamidodiamine (tada) ligands. These silylamido ligands display two distinct binding pockets whose steric profiles can be easily tuned by choice of the substituents on silicon. Preliminary reactivity studies demonstrate successful coordination of these new ligands to titanium(IV) and vanadium(III) via salt metathesis.
Author contributions
The manuscript was written through contributions of all authors. All authors have agreed to the final version of the manuscript.
Data availability
The experimental data generated in this manuscript are included in the ESI,† including experimental protocols, 1H and 13C NMR spectroscopic data, mass spectrometry data, elemental analysis data, and X-ray Crystallographic data.
Crystallographic data for compounds 3–9 has been deposited at the CCDC under deposition numbers 2365242–2365248† and can be obtained from https://www.ccdc.cam.ac.uk/structures/?
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by start-up funds from Oregon State University. We acknowledge the support of the Oregon State University NMR Facility funded in part by the National Institutes of Health, HEI Grant 1S10OD018518, and by the M. J. Murdock Charitable Trust grant #2014162. We also acknowledge the M. J. Murdock Charitable Trust (grant SR-2017297) for acquisition of OSU's single crystal diffractometer. We thank Prof. Chong Fang for use of his UV/Vis spectrophotometer.
References
- R. G. Castillo, R. Banerjee, C. J. Allpress, G. T. Rohde, E. Bill, L. Que, J. D. Lipscomb and S. DeBeer, High-Energy-Resolution Fluorescence-Detected X-ray Absorption of the Q Intermediate of Soluble Methane Monooxygenase, J. Am. Chem. Soc., 2017, 139, 18024–18033 CrossRef CAS PubMed.
- Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Tuning metal-metal interactions for cooperative small molecule activation, Chem. Commun., 2021, 57, 2839–2853 RSC.
- I. G. Powers and C. Uyeda, ACS Catal., 2017, 7, 936–958 CrossRef CAS.
- D. Gudat and J. G. Verkade, New azasilatranes: synthesis and substitution reactions, Organometallics, 1989, 8, 2772–2779 CrossRef CAS.
- C. C. Cummins, R. R. Schrock and W. M. Davis, Synthesis of vanadium and titanium complexes of the type RM[(Me3SiNCH2CH2)3N] (R = Cl, alkyl) and the structure of ClV[(Me3SiNCH2CH2)3N], Organometallics, 1992, 11, 1452–1454 CrossRef CAS.
- C. C. Cummins, J. Lee, R. R. Schrock and W. D. Davis, Trigonal–Monopyramidal MIII Complexes of the Type [M(N3N)] (M = Ti, V, Cr, Mn, Fe; N3N = [(tBuMe2Si) CH2CH2]3N), Angew. Chem., Int. Ed. Engl., 1992, 31, 1501–1503 CrossRef.
- C. C. Cummins, R. R. Schrock and W. M. Davis, Synthesis of Terminal Vanadium(V) Imido, Oxo, Sulfido, Selenido, and Tellurido Complexes by Imido Group or Chalcogen Atom Transfer to Trigonal Monopyramidal V[N3N] (N3N = [(Me3SiNCH2CH2)3N3−), Inorg. Chem., 1994, 33, 1448–1457 CrossRef CAS.
- S.-J. Xie, R.-K. Wu, Y.-F. Huang, H.-L. Chen, S.-Q. Zhang, F. Liu, D.-D. Zhai, X. Hong and Z.-J. Shi, Direct Incorporation of Dinitrogen into an Aliphatic C–H Bond, J. Am. Chem. Soc., 2023, 145, 6773–6780 CrossRef CAS PubMed.
- W. Gauss, P. Moser and G. Schwarzenbach, N,N,N′,N′–Tetrakis–(β–aminoäthyl)–äthylendiamin, Helv. Chim. Acta, 1952, 35, 2359–2363 CrossRef CAS.
- B. K. Wagnon and S. C. Jackels, Synthesis, characterization, and aqueous proton relaxation enhancement of a manganese(II) heptaaza macrocyclic complex having pendant arms, Inorg. Chem., 1989, 28, 1923–1927 CrossRef CAS.
- P. V. Bernhardt, Diverse solid-state and solution structures within a series of hexaamine dicopper(II) complexes, Inorg. Chem., 2001, 40, 1086–1092 CrossRef CAS PubMed.
- P. V. Bernhardt and E. J. Hayes, Copper(II) complexes of mono-and di-nucleating hexaamines, J. Chem. Soc., Dalton Trans., 1998, 1037–1042 RSC.
- J. Xu, T. M. Corneillie, E. G. Moore, G. L. Law, N. G. Butlin and K. N. Raymond, Octadentate cages of Tb(III) 2-hydroxyisophthalamides: A new standard for luminescent lanthanide labels, J. Am. Chem. Soc., 2011, 133, 19900–19910 CrossRef CAS PubMed.
- P. B. Kisanga and J. G. Verkade, Synthesis of new proazaphosphatranes and their application in organic synthesis, Tetrahedron, 2001, 57, 467–475 CrossRef CAS.
- M. Scheer, J. Müller, G. Baum and M. Häser, Antimony as a symmetrically bridged ligand in a novel neutral complex, Chem. Commun., 1998, 2505–2506 RSC.
- C. C. Cummins and R. R. Schrock, Synthesis of an Iron(IV) Cyanide Complex That Contains the Triamido Amine Ligand [tBuMe2SiNCH2CH2)3N]3−, Inorg. Chem., 1994, 33, 395–396 CrossRef CAS.
- S. Schneider and A. C. Filippou, Triamidoamine complexes of chromium(III) and chromium(IV), Inorg. Chem., 2001, 40, 4674–4677 CrossRef CAS PubMed.
- A. C. Filippou, S. Schneider and G. Schnakenburg, A pair of remarkably stable mononuclear chromium(III) and chromium(IV) hydrides, Angew. Chem., Int. Ed., 2003, 42, 4486–4489 CrossRef CAS PubMed.
- L. R. Doyle, A. J. Wooles and S. T. Liddle, Bimetallic Cooperative Cleavage of Dinitrogen to Nitride and Tandem Frustrated Lewis Pair Hydrogenation to Ammonia, Angew. Chem., 2019, 131, 6746–6749 CrossRef.
- L. R. Doyle, A. J. Wooles, L. C. Jenkins, F. Tuna, E. J. L. McInnes and S. T. Liddle, Catalytic Dinitrogen Reduction to Ammonia at a Triamidoamine–Titanium Complex, Angew. Chem., 2018, 130, 6422–6426 CrossRef.
- T. S. Lewkebandara, P. H. Sheridan, M. J. Heeg, A. L. Rheingold and C. H. Winter, Terminal and Bridging Imido Complexes from Titanium Tetrachloride and Primary Amines. Implications for the Chemical Vapor Deposition of Titanium Nitride Films, Inorg. Chem., 1994, 33, 5879–5889 CrossRef CAS.
- G. P. Clancy, H. C. S. Clark, G. K. B. Clentsmith, F. G. N. Cloke and P. B. Hitchcock, Vanadium(III) complexes incorporating the silylamino(disilylamido) ligand [(Me3Si)N{CH2CH2N(SiMe3)}2]2− [N{N′′}2]2−. Synthesis and crystal structure of the dimeric, non-metallocene vanadium(III) hydride [{V(N{N′′}2)}2(μ-H)2], J. Chem. Soc., Dalton Trans., 1999, 3345–3347 RSC.
- K. Feghali, D. J. Harding, D. Reardon, S. Gambarotta, G. Yap and Q. Wang, Stability of metal-carbon bond versus metal reduction during ethylene polymerization promoted by a vanadium complex: The role of the aluminum cocatalyst, Organometallics, 2002, 21, 968–976 CrossRef CAS.
- L. Killian, R. L. M. Bienemann and D. L. J. Broere, Quantification of the Steric Properties of 1,8-Naphthyridine-Based Ligands in Dinuclear Complexes, Organometallics, 2023, 42, 27–37 CrossRef CAS PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Olivia, V. Scarano and L. Cavallo, Towards the Online Computer-Aided Design of Catalytic Pockets, Nat. Chem., 2019, 11, 872–879 CrossRef CAS PubMed.
- A. J. Cuellar De Lucio, I. C. Cai, R. J. Witzke, A. N. Desnoyer and T. D. Tilley, Synthesis, Characterization, and Reactivity of Low-Coordinate Titanium(III) Amido Complexes, Organometallics, 2022, 41, 1434–1444 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Lev N.
Zakharov
and
Addison N.
Desnoyer
,
Lev N.
Zakharov
and
Addison N.
Desnoyer