
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActivation of uranyl peroxides by ionizing radiation prior to uranyl carbonate formation†
Zoe C.
Emory
 a,
Jay A.
LaVerne
bc and
Peter C.
Burns
*ad
a,
Jay A.
LaVerne
bc and
Peter C.
Burns
*ad
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: pburns@nd.edu
bRadiation Laboratory, University of Notre Dame, Notre Dame, Indiana 46556, USA
cDepartment of Physics and Astronomy, University of Notre Dame, Notre Dame, Indiana 46556, USA
dDepartment of Civil and Environmental Engineering and Earth Sciences, University of Notre Dame, Notre Dame, Indiana 46556, USA
First published on 26th September 2024
Abstract
The solid form of the uranyl peroxide cage (UPC) cluster LiU28 (Li28[(UO2)28(O2)42]) was irradiated by 5 MeV He2+ ions to achieve doses up to 42 MGy. An intermediate compound formed that reacts with atmospheric CO2 to form uranyl carbonates. The role of water in the UPC to uranyl carbonate transformation was studied by flowing either dry or hydrated Ar over samples during He2+ irradiation, and by storing samples in dry and humid environments before and after irradiation. Raman, infrared, and X-ray photoelectron spectroscopies and electrospray ionization mass spectrometry were used to characterize solid Li–U28 salts before and after He2+ irradiation. The highest yield of uranyl carbonates occurred when hydrated Ar gas was flowed across the sample during He2+ irradiation. Electron paramagnetic resonance spectroscopy provided evidence of hydroxyl and superoxide radicals in both unirradiated and γ-irradiated Li–U28.
Introduction
Uranyl peroxides are currently attracting attention in diverse areas such as direct air capture of CO2,1 stabilization of hydroxyl and superoxide radicals,2,3 the nuclear fuel cycle,4–8 studies of gelation,9 and environmental impact.10 The response of uranyl peroxide materials to ionizing radiation is important for nuclear waste storage and the environment.11–13 Studtite, [(UO2)(O2)(H2O)2](H2O)2, and its lower hydrate metastudtite, are the only known peroxide-bearing minerals. Studtite is used to recover uranium during in situ leach mining processes, precipitates on spent nuclear fuel immersed in water in laboratory studies, and has formed on Chernobyl's “lava” that resulted from the core-melt accident.12,14,15Uranyl carbonates are an important group of minerals, some of which form in nuclear waste environments, in close association with studtite.15–17 Recently, a uranyl superoxide was observed to convert to a uranyl peroxo-carbonate species in atmospheric conditions.18,19 Another study reported that the uranyl peroxide Na4(UO2)(O2)3·9H2O converts to Na4(UO2)(CO3)3 after exposure to ionizing radiation.5 Although nuclear waste occurs in various environments, all experience ionizing radiation.15–17,20 The radiolytic facilitated formation and degradation of uranyl peroxides has been explored.21–23 Studies of radiation-induced solid-state transformations of Ca2[UO2(O2)3]·9H2O (CaUT) and Li4[(UO2)(O2)3]·10H2O (LiUT) revealed that terminal peroxide ligands in CaUT are replaced by hydroxyl groups through an interaction with lattice waters, whereas LiUT transforms to a uranyl oxyhydrate phase.23
An extensive family of uranyl peroxide cage clusters self-assemble in alkaline aqueous solutions containing uranyl and hydrogen peroxide.24 One of these, Li–U24 (Li24[(UO2)(O2)(OH)]24) formed when an aqueous solution containing the Li–uranyl tri-peroxide monomer (LiUT) was γ-irradiated, likely due to production and recombination of hydroxyl radicals to form hydrogen peroxide from water radiolysis.21 Upon further γ-irradiation, Li–U24 degrades to a uranyl oxide hydroxide hydrate phase. The behavior of other uranyl peroxides including studtite, U60 (Li44K16[(UO2)(O2)(OH)]60), U60Ox30 (Li12K48[{(UO2)(O2)}60(C2O4)30]), and U24Pp12 (Li24Na24[(UO2)24(O2)24(P2O7)12]) has been studied under He2+ ion irradiation.22 These were done using dry solid material that was subsequently characterized by powder X-ray diffraction (PXRD) and Raman spectroscopy. He2+ irradiation induced X-ray amorphization and a visible color change of the materials.
Li–U28 (Li28[(UO2)28(O2)42]) is a fullerene-topology cage cluster comprised of 28 uranyl ions bridged through peroxide ligands (Fig. 1). Properties of Li–U28 have been investigated previously in part due to its relative simplicity and its readily reproducible synthesis that yields pure material.25–27 Due to the extensive characterization of Li–U28, it serves as a model system for this study. This investigation includes He2+ irradiation of Li–U28 under dry or hydrated argon flow, with three types of sample storage (ambient, desiccated, and 75% relative humidity (RH)). The material was characterized before and after He2+ irradiation by Raman, infrared, and X-ray photoelectron spectroscopies. We identify the formation of uranyl carbonate species, likely resulting from a reactive radiation induced intermediate. The influence of water content on the formation of uranyl carbonates was observed through spectroscopic characterization. We also identify the presence and γ-radiolytic production of oxygen radicals in Li–U28 with electron paramagnetic spectroscopy.
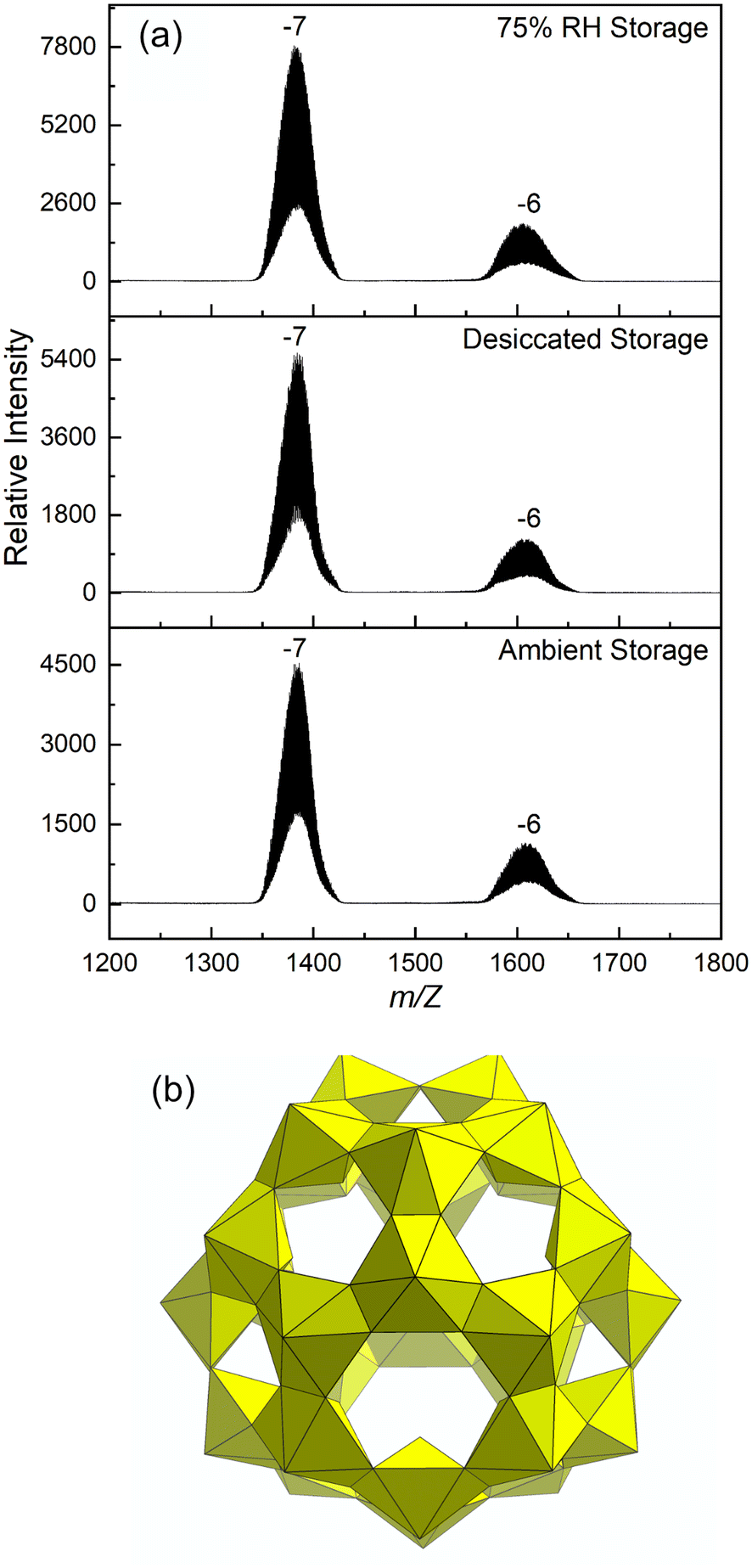 | ||
| Fig. 1 (a) ESI-MS spectra of Li–U28 dissolved in water after storage prior to irradiation. (b) Polyhedral representation of the UPC Li–U28. | ||
Experimental methods
Caution: Although depleted uranium was used in this study, it is a radioactive alpha emitter, and its daughters emit beta radiation. Depleted uranium should only be handled by trained individuals in appropriate facilities.Synthesis
Li–U28 was synthesized by combining an aqueous solution of uranyl nitrate hexahydrate (0.5 M, 10 mL) and hydrogen peroxide (30%, 10 mL) in a Falcon centrifuge tube, yielding studtite, [(UO2)(O2)(H2O)2](H2O)2, as a precipitate. LiOH monohydrate (2.38 M, 6 mL) was added, and the solution was stirred until the precipitate fully dissolved, resulting in a transparent yellow solution. Using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of methanol to reaction mixture, Li–U28 was crystallized by slow methanol diffusion. Four vials, each of which contained 0.75 g of reaction solution, were placed in a parafilm-covered beaker containing 3.00 mL of methanol. Crystals with dimensions of ∼100 μm formed within three days and were harvested by vacuum filtration. The identity of the Li–U28 crystals was confirmed by single crystal X-ray diffraction while the crystals were cooled in a nitrogen gas stream (Table S1†). Loss of lattice water during exposure to the ambient atmosphere quickly reduces long range order, rendering further crystallographic analysis impractical. After drying the material, Raman spectroscopy and electrospray ionization mass spectrometry (ESI-MS) confirmed the persistence of the Li–U28 cluster (see below).
:1 ratio of methanol to reaction mixture, Li–U28 was crystallized by slow methanol diffusion. Four vials, each of which contained 0.75 g of reaction solution, were placed in a parafilm-covered beaker containing 3.00 mL of methanol. Crystals with dimensions of ∼100 μm formed within three days and were harvested by vacuum filtration. The identity of the Li–U28 crystals was confirmed by single crystal X-ray diffraction while the crystals were cooled in a nitrogen gas stream (Table S1†). Loss of lattice water during exposure to the ambient atmosphere quickly reduces long range order, rendering further crystallographic analysis impractical. After drying the material, Raman spectroscopy and electrospray ionization mass spectrometry (ESI-MS) confirmed the persistence of the Li–U28 cluster (see below).
Irradiation
The Li–U28 material was adhered to carbon tape on an aluminum stub for He2+ irradiation. Ion radiolysis was performed using the 9S accelerator at the Nuclear Science Laboratory at the University of Notre Dame to produce 5 MeV He2+ ions, which mimic most α-particles. This method achieves high local doses in the MGy range with a beam current of approximately 25 nA and a beam diameter of 0.635 cm. The sample area is significantly larger than the beam. After irradiation, pristine material remains for comparison to the portion of the sample that was impacted by the beam. It is straightforward to visually discern pristine and irradiated material owing to a change in sample color resulting from He2+ irradiation.The SRIM/TRIM software was used to estimate the beam penetration depth of approximately 30 μm using material composition, density, and beam energy.28 Dose was accounted for by combining the incident energy with the integrated beam current. The Li–U28 average particle size, approximately 170 μm, exceeds the beam penetration depth, leaving the bulk of the Li–U28 material unirradiated. Samples were irradiated to doses of 8, 13, 21, and 42 MGy. To minimize indirect effects from the atmosphere, argon (Ar) gas (either dry or hydrated) flowed over the surface of the sample during He2+ irradiation. Hydrated Ar flow was used to prevent dehydration of the samples during He2+ irradiation, which occurs with the dry Ar flow.
Samples of solid Li–U28 were γ-irradiated up to 20 kGy using a sealed Shepherd 109-68R 60Co γ-source located at the University of Notre Dame Radiation Laboratory for EPR experiments. γ-Irradiation was used to induce similar radiolysis products that likely form during He2+ irradiation. Samples were irradiated in quartz glass tubes sealed under vacuum. Fricke dosimetry was used to calculate a dose rate of 50 Gy min−1 and was adjusted accounting for natural decay at the time of the experiment. No conversion was made for differences in electron density due to the uncertainties involved in photon absorption cross sections.
Vibrational spectroscopy
Fourier transform infrared (FTIR) spectra were collected using an attenuated total reflectance (ATR) objective on a Bruker Lumos FT-IR. All spectra were collected from the He2+ irradiated material on Al stubs with a spectral acquisition range of 600–3998 cm−1 and a resolution of 3.0 cm−1. Spectra were collected for unirradiated material, as well as for unirradiated portions of each irradiated sample that were exposed to gas flow conditions but not the He2+ beam, in addition to the irradiated portions.Raman spectra were collected using a Renishaw inVia Raman microscope outfitted with a 785 nm laser using a thermoelectrically-cooled CCD detector. Across all samples, exposure times and accumulations were varied for optimal signal over an extended scan range of 100–2000 cm−1 with the power of the 300 mW laser at 0.01–0.1%. Cosmic ray removal was done for all Raman spectra. Spectra were collected across multiple spots for each sample.
X-ray photoelectron spectroscopy (XPS)
XPS was performed on a PHI VersaProbe II X-ray photoelectron spectrometer equipped with a monochromatic Al Kα X-ray source. Survey scans were collected over the binding range of 0–1486 eV using a pass energy of 187.85 eV. High resolution scans were taken of uranium, oxygen, and carbon binding envelopes using a pass energy of 23.5 eV. The surface was then sputtered for 30 s using an Ar ion gun, and another high-resolution scan was collected. Spectra were calibrated to the adventitious carbon (C–C) peak at 284.8 eV. Data analysis was completed using the PHI Multipak software.Electrospray ionization mass spectrometry (ESI-MS)
ESI-MS was used to confirm cluster identity and purity prior to irradiation experiments. Salts containing Li–U28 were dissolved in 18 MΩ ultrapure water for a U concentration of ∼200 ppm and the pH was adjusted to ∼9.6 using tetraethylammonium hydroxide (1%), yielding a pale-yellow solution. Data were collected in negative ion mode using a Bruker Compact Quadrupole Time-of-Flight (QTOF) ESI-MS at an electrospray voltage of −3.5 to −4.0 kV. The solution containing dissolved Li–U28 was injected into the instrument at a rate of ∼6 μL per minute with a collection time of 15 minutes, using N2 nebulizer gas. Collision induced dissociation tandem MS measurements were done following published methods for cluster identification.25Electron paramagnetic resonance (EPR)
EPR spectra were collected on a Bruker EMX spectrometer for γ-irradiated Li–U28. Five scans were collected and averaged at room temperature in the X-band frequency range (∼9.8 GHz) on solid Li–U28 sealed under vacuum in quartz glass tubes. The magnetic field was centered at 3500.00 G, with a sweep width of 1000 G. To investigate the decay of radical species after γ-irradiation to 20 kGy, the samples were maintained in the sealed tubes for 24 hours and then the EPR spectra were re-collected. After 24 hours and collection of the EPR spectra, one sample was opened and exposed to air for an additional day while the other sample remained sealed, and EPR spectra were taken again for each of these samples.Sample storage
Samples of Li–U28 labeled ‘ambient storage’ were maintained on the bench top in a plastic sample holder before and after He2+ irradiation. To control for fluctuating atmospheric humidity in subsequent experiments, samples were contained in perfluroalkoxy (PFA) jars with either a saturated salt solution, ∼75% relative humidity (RH), or a desiccant to reduce humidity before and after He2+ irradiation. To achieve 75% RH, a slurry of NaCl and 20.0 mL of water was used.29 Samples were elevated above the salt solution or desiccant. Samples were exposed to the ambient atmosphere during characterization.Results and discussion
Powder X-ray diffraction is not effective for establishing the identity and purity of the material under study because the solid form of U28 is prone to dehydrating with loss of crystallinity. To confirm that harvested material contains the U28 cluster, solid material was dissolved in water that was then injected into an electrospray ionization mass spectrometer. Previous studies have established that ESI-MS gives broad peaks at high m/z when uranyl peroxide cage clusters are present.25 The spectrum for dissolved U28 collected during the current study contains the expected broad m/z envelopes that indicate the U28 cluster [(UO2)28(O2)42]28− travels to the detector with various combinations of Li+, H+ and H2O with charge states of −7 and −6 (Fig. 1).He2+ irradiation of samples under flowing dry or hydrated Ar
Samples of Li–U28 that were He2+ irradiated at the onset of the current investigation had been stored under ambient atmospheric conditions on the benchtop. Samples were irradiated to 8 to 42 MGy under flowing dry or hydrated Ar gas. Raman spectra were collected as soon as possible after irradiation, typically within two hours (Fig. 2a) The Raman microscope used is in a different building than the accelerator and samples were exposed to the ambient atmosphere after irradiation prior to collection of Raman spectra.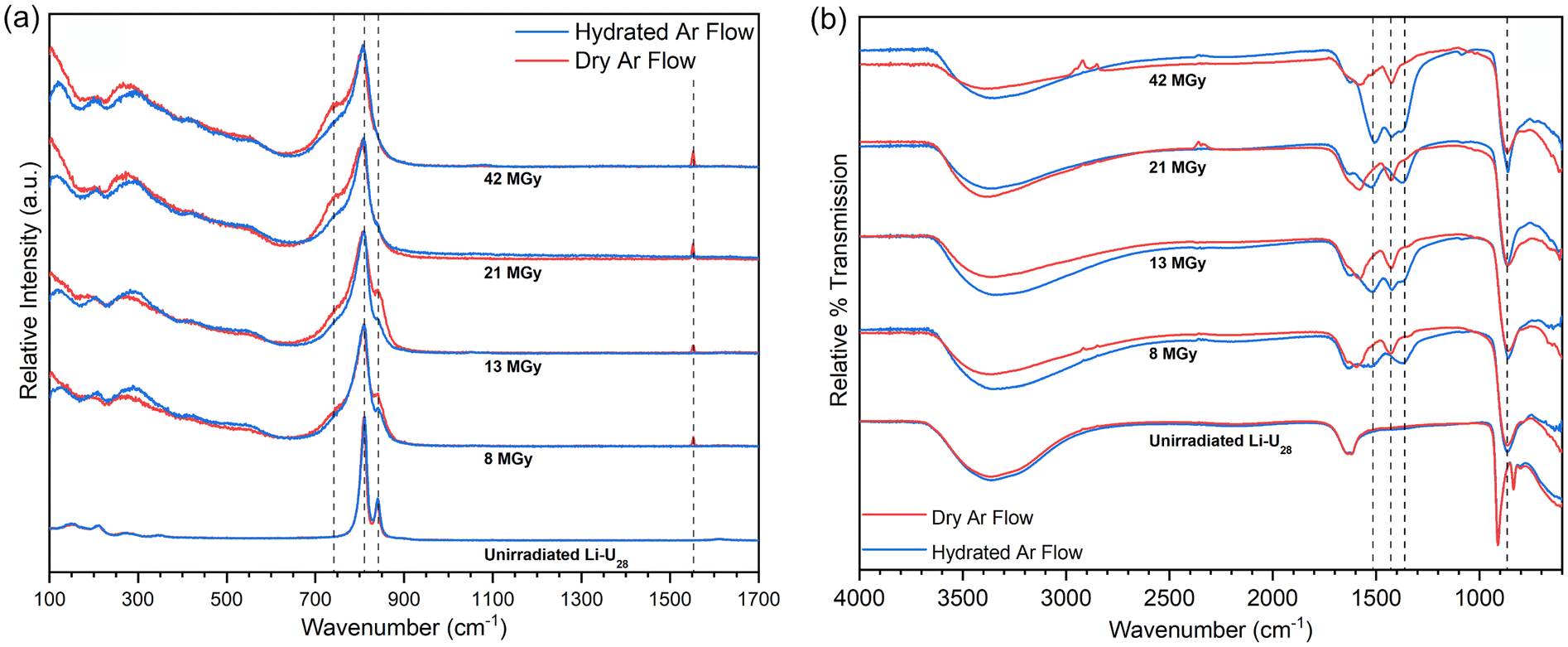 | ||
| Fig. 2 (a) Normalized Raman spectra of He2+ irradiated Li–U28. (b) Normalized FTIR spectra of He2+ irradiated Li–U28. Dashed lines between 1600–1300 cm−1 correspond to locations of carbonate binding motifs whereas the dashed line at 860 cm−1 highlights the shifted uranyl asymmetric stretch. For (a) and (b), stack plots show increase in dose, and red and blue traces indicate dry or hydrated Ar gas flow during irradiation, respectively. | ||
Raman spectra were collected for samples that had been subjected to progressively higher doses of radiation (Fig. 2a) The peroxo (O–O) symmetric stretch at 840 cm−1 gradually decreases with increasing dose, and is eventually lost, indicating breakdown of the peroxide bridges present in U28. Peroxide is an essential component of U28 and it is unlikely that U28 clusters remain following peroxide breakdown. The uranyl (U![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) symmetric stretch at 809 cm−1 broadens with increasing dose but maintains its position, indicating persistence of the uranyl ion. Upon further He2+ irradiation, a shoulder appears around 743 cm−1 that continues to grow with increasing dose. Carbonate (CO32−) in-plane bending (ν4) modes occur in the range 700–800 cm−1 and may be responsible for the ingrowth of the shoulder at 743 cm−1.30–32 The expected ν1 vibrational mode of carbonate is present but weak and is highlighted in Fig. S11.† Raman spectra were collected as extended scans ultimately impacting relative intensities of the present spectral features. Data collection was optimized for observing a large spectral window and details of the uranyl symmetric stretch, which greatly overshadows the carbonate symmetric stretch. IR spectra for irradiated material also contain modes assigned to carbonate vibrations (see below). It is likely that the irradiated material rapidly reacts with the ambient atmosphere to form uranyl carbonate complexes by capturing CO2, which has been observed in other reactive uranyl phases and irradiation studies.1,5,18 Carbonate out of plane bending modes (ν2), if present, overlap with the intense uranyl symmetric stretch.30
O) symmetric stretch at 809 cm−1 broadens with increasing dose but maintains its position, indicating persistence of the uranyl ion. Upon further He2+ irradiation, a shoulder appears around 743 cm−1 that continues to grow with increasing dose. Carbonate (CO32−) in-plane bending (ν4) modes occur in the range 700–800 cm−1 and may be responsible for the ingrowth of the shoulder at 743 cm−1.30–32 The expected ν1 vibrational mode of carbonate is present but weak and is highlighted in Fig. S11.† Raman spectra were collected as extended scans ultimately impacting relative intensities of the present spectral features. Data collection was optimized for observing a large spectral window and details of the uranyl symmetric stretch, which greatly overshadows the carbonate symmetric stretch. IR spectra for irradiated material also contain modes assigned to carbonate vibrations (see below). It is likely that the irradiated material rapidly reacts with the ambient atmosphere to form uranyl carbonate complexes by capturing CO2, which has been observed in other reactive uranyl phases and irradiation studies.1,5,18 Carbonate out of plane bending modes (ν2), if present, overlap with the intense uranyl symmetric stretch.30
The sharp peak at 1553 cm−1 in the Raman spectrum for Li–U28 He2+ irradiated under dry Ar gas is absent in the spectrum for material irradiated under a hydrated Ar gas flow. This peak is assigned to O2 gas trapped in the solid or possibly adsorbed on the surface.33 The O2 may arise from decomposition of peroxide or recombination of radiolytic radical species. The O2 is liberated upon addition of water, as confirmed visually and by gas chromatography (Fig. S3†). The absence of the signal at 1553 cm−1 for material irradiated under flowing hydrated Ar further establishes the role of water in O2 gas release.
FTIR spectra were collected the day after He2+ irradiation of the samples (Fig. 2b). The spectra of the unirradiated Li–U28 control samples contain major features including the –OH stretching mode between 3700–3000 cm−1, the water bending mode at 1632 cm−1, and the uranyl antisymmetric stretch (ν3) at 910 cm−1. FTIR spectra collected for samples that were irradiated all contain carbonate ν3 peaks between 1600–1300 cm−1. The uranyl ν3 stretch of the irradiated materials has shifted to 862 cm−1 in each case. This is attributed to weakening of the uranyl bonds caused by equatorial coordination by CO32−.30,34 The broadening of the uranyl ν3 stretch following irradiation is consistent with the presence of multiple bonding environments. In general, peaks in the carbonate ν3 region are more intense in FTIR spectra collected for samples He2+ irradiated under hydrated Ar than those irradiated under dry Ar, suggesting that water influences formation of the intermediate phase(s) and perhaps radical species that facilitate the capture of carbon dioxide from the atmosphere. Symmetry reduction increases the number of carbonate ν3 peaks, as would the occurrence of additional symmetrically distinct (UO2)2+ or (CO3)2−.30
FTIR spectra of Li–U28 He2+ irradiated under dry Ar gas contain peaks at 1574 and 1426 cm−1. Spectra for samples irradiated under hydrated Ar gas contain the same peaks, as well as peaks at 1515 and 1370 cm−1. Bidentate carbonate coordination to a metal center causes a splitting of the ν3 mode (Δν3) of ∼150 cm−1.35–37 Δν3 between 1574 and 1426 cm−1 is 148 cm−1, and between 1515 and 1370 cm−1 is 145 cm−1, both of which are consistent with bidentate coordination of carbonate to uranyl. He2+ irradiation under hydrated Ar followed by contact with the atmosphere yields two structurally distinct bidentate carbonate groups, in contrast to irradiation under dry Ar, which only produces one. Spectroscopic peak assignments are summarized in Table 1.
| ν 1 | ν 2 | ν 3 | ν 4 | |
|---|---|---|---|---|
| Unirradiated Li–U28 | ||||
| (UO2)2+ | 809 (R) | 910 (FTIR) | ||
| (O–O) | 840 (R) | 835 (FTIR) | ||
| He 2+ irradiated Li–U28 | ||||
| (UO2)2+ | 808 (R) | 204, 300 (R) | 862 (FTIR) | |
| (CO3)2− | 1089 (R) (FTIR) | 1370, 1426, 1515, 1574 (FTIR) | 743 (R) | |
Data collected from the first set of He2+ irradiations indicates that the solid form of Li–U28 undergoes significant changes during He2+ irradiation, and the resulting “damaged” material reacts with ambient air to rapidly capture CO2 as carbonate, particularly with the use of hydrated Ar. The breakdown of Li–U28 during irradiation likely results in several uranyl environments that are difficult to characterize, unlike crystalline Li–U28 that only contains uranyl ions coordinated by three bidentate peroxide groups. The uranyl and peroxide spectroscopic signals diagnostic of Li–U28 diminish markedly with increasing dose, with eventual complete loss of the signal at 840 cm−1 that corresponds to peroxide that is bridging two uranyl ions within the cage cluster. Breakdown of peroxide produced O2 gas that was trapped in the solid and later detected by spectroscopy. Peroxide breakdown doomed the U28 clusters as no uranyl peroxide cluster contain less than a 1:1 molar ratio of peroxide to uranyl (the ratio is 1.5 for pristine U28).
He2+ irradiation of samples stored in humid and desiccated environments
Initial He2+ irradiation experiments described above were done for samples of Li–U28 that had been stored under ambient conditions. To better understand the impact of water in the conversion of irradiated UPCs into uranyl carbonates, samples of Li–U28 were pre-treated in either a high humidity atmosphere or a desiccated atmosphere prior to He2+ irradiation to 42 MGy and storage in the same conditions afterward. Raman and infrared spectra for samples that had been stored in these two types of atmospheres prior to and post irradiation are in Fig. 3a and b, respectively. | ||
| Fig. 3 (a) Normalized Raman spectra of Li–U28 He2+ irradiated to 42 MGy under dry or hydrated argon gas flow with either 75% relative humidity (blue) or desiccated storage (red) conditions before and after irradiation. (b) Normalized FTIR spectra of Li–U28 He2+ irradiated to 42 MGy under dry or hydrated Ar gas flow with either 75% relative humidity (blue) or desiccated (red) storage conditions before and after irradiation. The dashed lines between 1600–1300 cm−1 correspond to positions of carbonate binding motifs and the dashed line at 863 cm−1 corresponds to the shifted uranyl asymmetric stretch. | ||
Overall, the Raman spectra of materials after He2+ irradiation are similar among the samples stored in desiccated or humid conditions, especially in the uranyl symmetric stretching region (Fig. 3a). The most notable differences occur in the low wavenumber regions. Samples irradiated under hydrated Ar flow produced almost identical spectra regardless of how they were stored prior to irradiation. However, irradiating samples under the dry Ar flow resulted in spectra that differed significantly depending on the sample storage, with less prominent features in the low wavenumber range for the spectrum collected for material that had been stored in a desiccated environment. Also, the corresponding spectrum contains a peak at 1553 cm−1 indicative of O2 gas in the sample, which is absent in the spectrum in which the sample was stored in a humid atmosphere. Evidently, the high humidity storage pre and post irradiation results in sufficient hydration of the sample to liberate oxygen generated during or after irradiation.
The low wavenumber signals in the Raman spectra provide some insights into chemical environments. The peak at 123 cm−1 may be due to lattice vibrations, indicating increased crystallinity despite irradiation, as some prior studies have observed.5,23 It is also possible that the peak at 123 cm−1 is due to in-plane bending of the equatorial ligands, which has been observed in other uranyl complexes.38 There is some ambiguity in the assignment for the peak between 200–250 cm−1. In uranyl chloride complexes, this has been attributed to the rocking vibrational mode (ν11) of the uranyl ion.39 Other literature suggests this peak could be bending (ν2) of the uranyl ion, which can become Raman active when the site symmetry of the uranyl ion is lowered.30,40 Those between 300–500 cm−1 are attributed to U–O vibrations in the equatorial positions of the uranyl bipyramids.40
FTIR spectra collected for samples stored in either desiccated or humid conditions emphasize the importance of hydrated versus dry Ar flow during He2+ irradiation (Fig. 3b). The spectrum of Li–U28 subjected to hydrated Ar during He2+ irradiation and humid storage contains intense peaks at 1429 and 1500 cm−1 with a Δν3 of 71 cm−1, likely indicating a monodentate bound carbonate.30 This might arise from an equatorial site of the uranyl bipyramid being occupied by excess water or a hydroxyl group. This spectrum also contains peaks corresponding to bidentate carbonate binding motifs (140 Δν3), which present as shoulders to the intense peak at 1429 cm−1. Conversely, the spectrum for the sample subjected to hydrated Ar during He2+ irradiation and desiccated storage contains intense peaks at 1522 and 1362 cm−1 with a 160 Δν3, indicative of only bidentate CO32− binding motifs. The excess water provided by both the high-humidity storage and hydrated gas flow during irradiation impacts how CO32− binds to UO22+ in the final products. Both spectra for samples irradiated under dry Ar flow contain analogous peaks to the spectra of the samples irradiated under the hydrated Ar flow with much less intensity regardless of the type of storage. The extent of alteration to a uranyl carbonate species depends on the amount of water associated with the material. Some transformation occurs in the dry Ar flow systems because Li–U28 has water inherently associated with the structure (Fig. 1a), as well as exposure to any atmospheric humidity during sample transfer and characterization, however, the dry Ar flow dries the surface while the hydrated Ar flow maintains surface water content during irradiation. Drying of the material exposed to the dry Ar flow during He2+ irradiation explains why the material stored in the humid environment does not alter nearly to the same extent as the samples exposed to hydrated Ar during irradiation. A weak ν1 symmetric carbonate stretch is visible in the FTIR spectra for the samples exposed to the hydrated gas flow condition at 1090 cm−1. In a symmetric carbonate group, this stretch should be Raman-only active, however, it can become IR-active when symmetry is lowered.40
The infrared spectra collected for the variously treated samples shown in Fig. 3b indicate that significant water is present in all cases, as indicated by the water bending mode and large envelope of peaks in the H bonding region. Storage of material in a desiccated environment without subsequent irradiation may have caused modest dehydration relative to the sample stored in a humid environment. The sample stored in a humid atmosphere is expected to contain much more surface water. Regardless, the intermediate compound produced by He2+ irradiation under flowing hydrated Ar was most effective in capturing CO2 from the atmosphere. CO2 readily dissolves in water to form H2CO3 that can further speciate to HCO3− and CO32−, providing a mechanism for how the irradiated material uptakes carbon that is dependent on the presence of surface water, rather than just structurally bound water. The reactivity of the intermediate exhibits a dependency on water content, particularly in the case of the hydrated Ar flow during He2+ irradiation. This dependency could suggest increased radical production during He2+ irradiation, as the UPC and associated water is undergoing radiolysis. We examined radical species generated by γ-irradiation in Li–U28 with EPR experiments (see below).
X-ray photoelectron spectroscopy (XPS)
XPS was used to characterize the C 1s, O 1s, U 4f7/2, and U 4f5/2 binding envelopes after He2+ irradiation for samples stored in a 75% RH or desiccated atmosphere. The binding energy was calibrated using the signal for adventitious carbon (C–C) at 284.7 eV (Fig. 4). The C 1s envelopes for unirradiated materials with humid and desiccated atmosphere storage conditions are largely comprised of the signal for adventitious carbon. In all cases after irradiation a carbonate (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(–O–)2) binding envelope is present at 289.7 eV.
C(–O–)2) binding envelope is present at 289.7 eV.
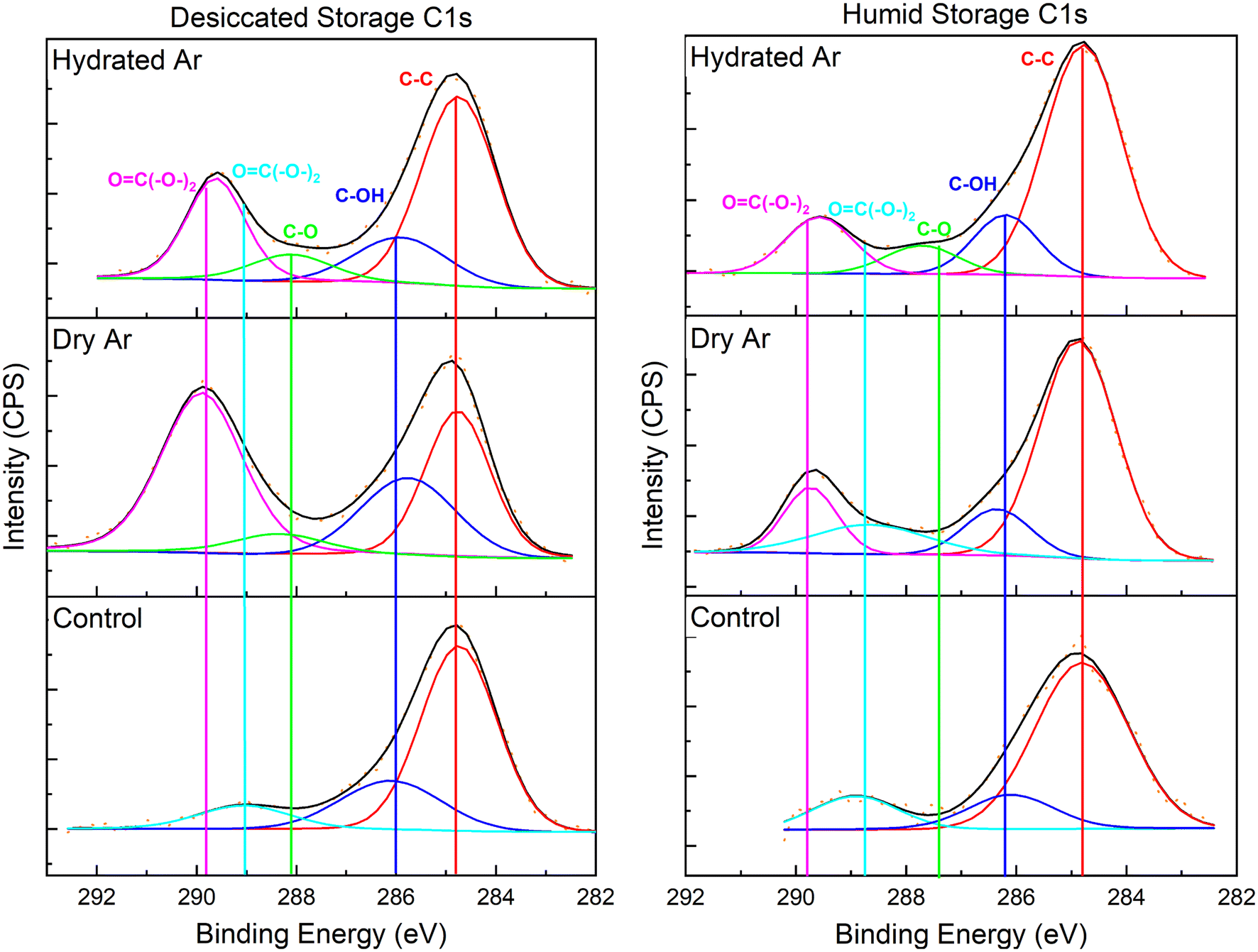 | ||
| Fig. 4 XPS spectra of the C 1s binding envelopes. Li–U28 stored in desiccated environment (left) Li–U28 stored in 75% RH environment (right). From top to bottom: Li–U28 He2+ irradiated to 42 MGy under hydrated Ar flow, Li–U28 irradiated to 42 MGy under dry Ar flow, unirradiated Li–U28 controls kept in respective storage environments. | ||
XPS spectra for the O 1s, U 4f7/2, and U 4f5/2 binding envelopes are in the ESI.† The O 1s envelope is impacted by both irradiation and storage environment. The signal at 529.7 eV assigned to equatorial U–O bonds linking uranyl polyhedra (U–O–U), decreases in intensity in spectra for the irradiated samples relative to spectra for unirradiated samples, however, it persists due to oxygen atoms still comprising the equatorial sites as (CO3)2−. At 531.6 eV, the carbonate (OC(–O–)2) bonding environment is notably larger after irradiation. Uranyl (OUO) and hydroxide (–OH) bonding environments are also present in all O 1s fits.
Electron paramagnetic resonance spectra
Radical oxygen species formed during He2+ irradiation of Li–U28 are potentially key in subsequent reactions that capture CO2 from the ambient atmosphere. The increased reactivity observed in more hydrated samples suggests that more radical oxygen species are forming during He2+ irradiation in these systems due to the additional water. Surprisingly, the EPR spectrum of unirradiated solid Li–U28 contains small peaks indicative of oxygen radicals with g-factors of 2.0167, 2.032, 2.0235, 2.0079. The EPR signals at g = 2.032, 2.0235, and 2.0079 can be attributed to superoxide (O2˙−) in the unirradiated Li–U28 sample.3,41,42 In samples of studtite, similar g-factors were observed, including g = 2.015, which was attributed to O2˙−; however, computations predicted the value to be around g = 2.023.3 Another group of researchers showed that U60 oxalate (a UPC with sixty uranyl ions bridged by peroxide and oxalate ligands) stabilizes ˙OH radical species for up to ten days in the solid state (g = 2.016), likely resulting from the breakdown of H2O2 in a Fenton-like process during synthesis.2 The principle component for the EPR spectra of unirradiated Li–U28 has a g-factor of 2.0167. This g-factor matches that of the hydroxyl radical (˙OH) identified in the U60 UPC as well as that of ˙OH in magnesium oxide.2,43 The presence of ˙OH in the unirradiated UPC could be explained by the radical being trapped in defect sites within the material, allowing it to persist despite the material being handled on the benchtop in air. We have also considered that the peak at 2.0167 could be due to an oxygen radical (O−), as these are longer lived radical species and have been observed in aluminum oxide and hydroxide compounds.44,45 Despite ambiguity in the identities of oxygen radicals, the EPR spectra reported herein are the first evidence that solid Li–U28 contains radical oxygen species.The EPR spectra of unirradiated Li–U28 and γ-irradiated Li–U28 (20 kGy) are shown in Fig. 5a. The unirradiated (black trace) and γ-irradiated (red trace) Li–U28 produce spectra with the same g-factor values for O2˙− and ˙OH or O− with much greater intensity in the case of the γ-irradiated sample. F-center defects from γ-irradiation of the quartz tube are in the EPR spectra with a g-factor of 2.0004 and as expected, this signal is absent in the spectrum of the unirradiated material. γ-Irradiation results in increased concentrations of both ˙OH or O− and O2˙− compared to the spectrum of the unirradiated material. The presence and formation of O2˙− in Li–U28 is particularly interesting in the context of this investigation because only one uranyl superoxide compound has been shown to transform into a uranyl carbonate bearing phase.18,19
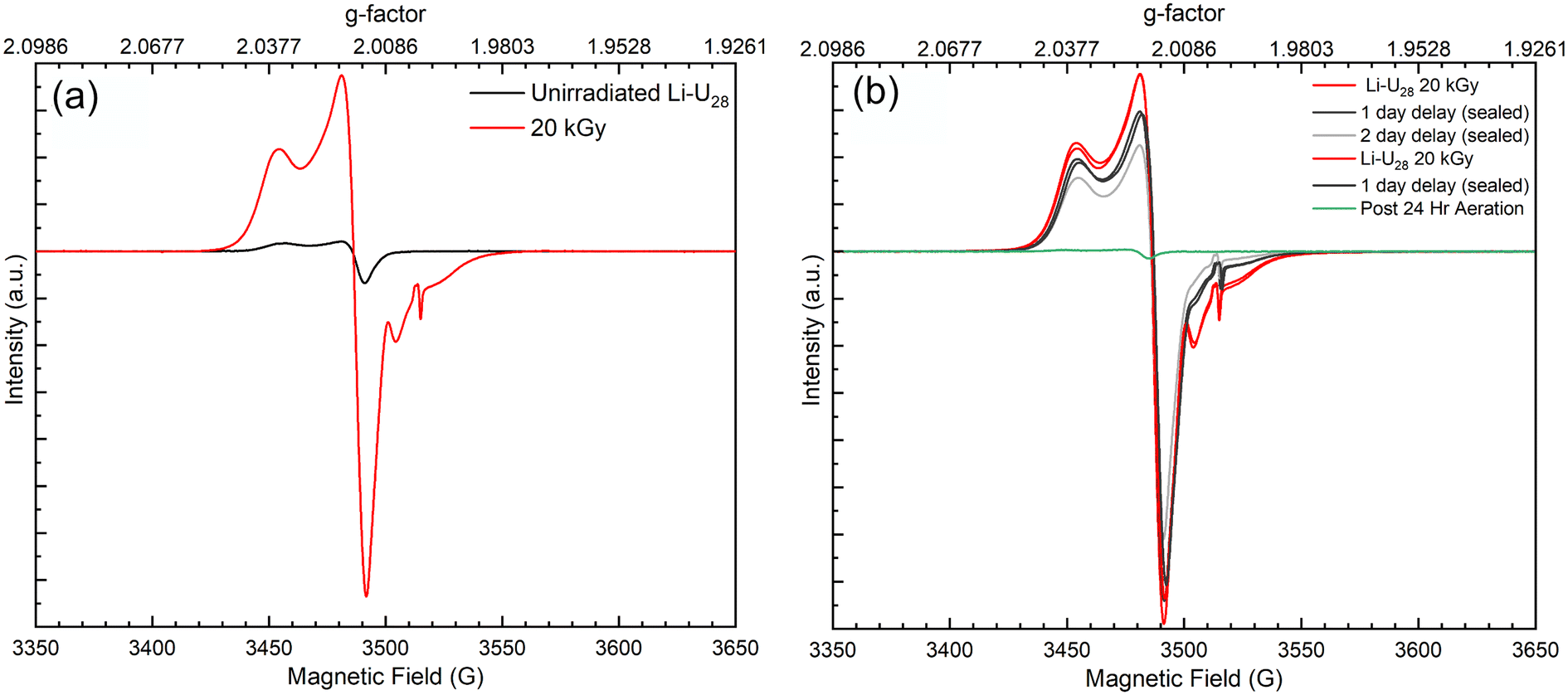 | ||
| Fig. 5 (a) EPR spectra of solid Li–U28 unirradiated (black) and 20 kGy γ-irradiated (red). (b) EPR spectra tracking radical decay over two days. Li–U28 20 kGy (red), Li–U28 one day after 20 kGy γ-irradiation sealed (black), Li–U28 two days after 20 kGy γ-irradiation sealed (gray), Li–U28 two days after γ-irradiation and 24 hours after exposure to air (green). | ||
The decay of radical species in γ-irradiated material was also examined using EPR spectroscopy. The two samples of Li–U28 that were γ-irradiated to 20 kGy remained sealed for 24 hours after which EPR spectra were taken again. After 24 hours and collection of the EPR spectra, one sample was opened and exposed to air for an additional day while the other sample remained sealed. The resulting spectra indicate a gradual decrease in intensity of radical species over two days when the sample is under vacuum, and an immediate quenching of radicals upon exposure to air. The radiation-induced radicals readily react with air, in contrast to the radical species stabilized in the unirradiated Li–U28. The reduction of radicals present in the aerated sample indicates that a majority of the γ-induced radical oxygen species are near the surface of the material, and therefore able to react with the atmosphere. This contrasts with other aeration experiments that conclude the reactive species can reside in the subsurface of various materials.40,42 Li–U28 synthesized and handled on the benchtop stabilizes oxygen radicals, as shown in the EPR of the unirradiated material, but once irradiated and exposed to the atmosphere again, this capacity is lost. These results indicate that small doses of ionizing radiation impact the material enough to prevent any subsurface trapping and increase the susceptibility of interacting with the atmosphere. The uranyl species present after He2+ irradiation induced Li–U28 breakdown are likely varied. However, it is evident from the EPR spectra that γ-irradiation creates oxide and hydroxide radicals in the material, which can be reactive toward atmospheric CO2 to form carbonates. Our experimental sets with added hydration during He2+ irradiation, likely form a greater amount of radical oxygen species that then react to form uranyl carbonates. Further EPR investigations varying water content are needed to confirm this.
Conclusions
Herein we expanded upon previous investigations on the behavior of various uranyl peroxides subjected to high doses of ionizing radiation.5,23 He2+ irradiation of Li–U28 produced a reactive intermediate that captured CO2 from the ambient atmosphere to form uranyl carbonate species. The peroxide units that bridge uranyl ions in Li–U28 degraded by 42 MGy, resulting in binding of CO32− ligands in a bidentate fashion in the equatorial plane of the uranyl ion shown by infrared spectroscopy. Li–U28 exposure to water pre and post He2+ irradiation by storage in 75% RH facilitated the alteration to a uranyl carbonate post-irradiation. The use of a hydrated Ar gas flow over the sample surface during He2+ irradiation was the most impactful factor in conversion of Li–U28 to a uranyl carbonate phase, regardless of the storage environment. The least amount of uranyl carbonate formed in samples stored in desiccated conditions and irradiated under a dry Ar gas, further demonstrating the importance of water in material alteration. Increased water content at the surface likely dissolves atmospheric CO2, followed by speciation to carbonate. EPR spectroscopy elucidated that unirradiated Li–U28 solid stabilizes hydroxyl or oxygen and superoxide radicals; however, this capacity is lost upon γ-irradiation. Irradiation induces formation of superoxide and hydroxyl or oxygen radicals, which likely play a role in uranyl peroxide degradation and subsequent carbonate formation. Further studies on the radiolytic mechanism and role of oxygen radicals in material alteration to form uranyl carbonate compounds are warranted.Data availability
All data collected for this study are available on Open Science Framework at https://osf.io/eyux9/?view_only=416407e619ff46efa01e38d4b463493e.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work of Z. C. E. and P. C. B. was funded by the University of Notre Dame and J. A. L. by DOE-BES. The authors acknowledge the Center for Sustainable Energy at Notre Dame (ND Energy) Materials Characterization Facility for the use of the XPS. The authors thank Prof. Michael Wiescher for making available the facilities of the Notre Dame Nuclear Science Laboratory, which is supported by the U.S. National Science Foundation through grant Phys-0758100, and Prof. Ian Carmichael for making available the facilities of the Notre Dame Radiation Laboratory, which is supported by DOE-BES through grant DE-FC02-04ER15533. This contribution is NDRL-5439 from the Notre Dame Radiation Laboratory.References
- A. Arteaga, T. Arino, G. C. Moore, J. L. Bustos, M. K. Horton, K. A. Persson, J. Li, W. F. Stickle, T. A. Kohlgruber, R. G. Surbella III and M. Nyman, The Role of Alkalis in Orchestrating Uranyl-Peroxide Reactivity Leading to Direct Air Capture of Carbon Dioxide, Chem. – Eur. J., 2024, 30, e202301687, DOI:10.1002/chem.202301687
.
- B. Lottes and K. P. Carter, Capture and Stabilization of the Hydroxyl Radical in a Uranyl Peroxide Cluster**, Chem. – Eur. J., 2023, 29(45), e202300749, DOI:10.1002/chem.202300749
- S. K. Scherrer, C. Gates, H. Rajapaksha, S. M. Greer, B. W. Stein and T. Z. Forbes, Superoxide Radicals in Uranyl Peroxide Solids: Lasting Signatures Identified by Electron Paramagnetic Resonance Spectroscopy, Angew. Chem., Int. Ed., 2024, 63, e202400379, DOI:10.1002/anie.202400379
- M. D. M. Badley, D. W. Shoesmith and J. J. Noël, Effect of Hydrogen on the Dissolution of Uranium Dioxide in Peroxide-Containing Environments, J. Electrochem. Soc., 2023, 170(9), 096506, DOI:10.1149/1945-7111/acf52b
- M. Fairley, G. E. Sigmon and J. A. LaVerne, Solid-State Transformation of Uranyl Peroxide Materials through High-Level Irradiation, Inorg. Chem., 2023, 62, 19780–19785, DOI:10.1021/acs.inorgchem.3c03373
- J. Li, L. Li and M. Jonsson, Formation and Stability of Studtite in Bicarbonate-Containing Waters, Ecotoxicol. Environ. Saf., 2023, 263, 115297, DOI:10.1016/j.ecoenv.2023.115297
- J. Li, X. Liu and M. Jonsson, UO2 Dissolution in Aqueous Halide Solutions Exposed to Ionizing Radiation, Appl. Surf. Sci., 2024, 646, 158955, DOI:10.1016/j.apsusc.2023.158955
- J. McGrady, Y. Kumagai, Y. Kitatsuji, A. Kirishima, D. Akiyama and M. Watanabe, UO2 Dissolution in Bicarbonate Solution with H2O2: The Effect of Temperature, RSC Adv., 2023, 13(40), 28021–28029, 10.1039/D2RA08131H
- X. Xu, Y. Yang, Y. Zhou, K. Xiao, J. E. S. Szymanowski, G. E. Sigmon, P. C. Burns and T. Liu, Critical Conditions Regulating the Gelation in Macroionic Cluster Solutions, Adv. Sci., 2024, 11, 2308902, DOI:10.1002/advs.202308902
- K. R. Smith, J. Ilavsky and A. E. Hixon, Crystallization of a Neptunyl Oxalate Hydrate from Solutions Containing NpV and the Uranyl Peroxide Nanocluster U60Ox30, Chem. – Eur. J., 2023, 29(19), e202203814, DOI:10.1002/chem.202203814
- C. R. Armstrong, M. Nyman, T. Shvareva, G. E. Sigmon, P. C. Burns and A. Navrotsky, Uranyl Peroxide Enhanced Nuclear Fuel Corrosion in Seawater, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(6), 1874–1877, DOI:10.1073/pnas.1119758109
- B. Hanson, B. Mcnamara, E. Buck, J. Friese, E. Jenson, K. Krupka and B. Arey, Corrosion of Commercial Spent Nuclear Fuel. 1. Formation of Studtite and Metastudtite, Radiochim. Acta, 2005, 93, 159–168, DOI:10.1524/ract.93.3.159.61613
- J. Li, A. C. Maier and M. Jonsson, Stability of Studtite in Aqueous Suspension: Impact of HCO3− and Ionizing Radiation on the Dynamics of Dissolution, ACS Appl. Energy Mater., 2020, 3(1), 352–357, DOI:10.1021/acsaem.9b01611
- S. O. Odoh, J. Shamblin, C. A. Colla, S. Hickam, H. L. Lobeck, R. A. K. Lopez, T. Olds, J. E. S. Szymanowski, G. E. Sigmon, J. Neuefeind, W. H. Casey, M. Lang, L. Gagliardi and P. C. Burns, Structure and Reactivity of X-Ray Amorphous Uranyl Peroxide, U2O7, Inorg. Chem., 2016, 55(7), 3541–3546, DOI:10.1021/acs.inorgchem.6b00017
- B. E. Burakov, E. E. Strykanova and E. B. Anderson, Secondary Uranium Minerals on the Surface of Chernobyl “Lava”, MRS Proc., 1996, 465, 1309, DOI:10.1557/PROC-465-1309
- K. J. Cantrell, K. M. Krupka, W. J. Deutsch and M. J. Lindberg, Residual Waste from Hanford Tanks 241-C-203 and 241-C-204. 2. Contaminant Release Model, Environ. Sci. Technol., 2006, 40(12), 3755–3761, DOI:10.1021/es0511568
- K. M. Krupka, H. T. Schaef, B. W. Arey, S. M. Heald, W. J. Deutsch, M. J. Lindberg and K. J. Cantrell, Residual Waste from Hanford Tanks 241-C-203 and 241-C-204. 1. Solids Characterization, Environ. Sci. Technol., 2006, 40(12), 3749–3754, DOI:10.1021/es051155f
- D. V. Kravchuk, N. N. Dahlen, S. J. Kruse, C. D. Malliakas, P. M. Shand and T. Z. Forbes, Isolation and Reactivity of Uranyl Superoxide, Angew. Chem., Int. Ed., 2021, 60(27), 15041–15048, DOI:10.1002/anie.202103039
- D. V. Kravchuk and T. Z. Forbes, Thermodynamics and Chemical Behavior of Uranyl Superoxide at Elevated Temperatures, ACS Mater. Au, 2022, 2(1), 33–44, DOI:10.1021/acsmaterialsau.1c00033
- B. Rosborg and L. Werme, The Swedish Nuclear Waste Program and the Long-Term Corrosion Behaviour of Copper, J. Nucl. Mater., 2008, 379(1), 142–153, DOI:10.1016/j.jnucmat.2008.06.025
- D. E. Felton, M. Fairley, A. Arteaga, M. Nyman, J. A. LaVerne and P. C. Burns, Gamma-Ray-Induced Formation of Uranyl Peroxide Cage Clusters, Inorg. Chem., 2022, 61(30), 11916–11922, DOI:10.1021/acs.inorgchem.2c01657
- M. Fairley, N. M. Myers, J. E. S. Szymanowski, G. E. Sigmon, P. C. Burns and J. A. LaVerne, Stability of Solid Uranyl Peroxides under Irradiation, Inorg. Chem., 2019, 58(20), 14112–14119, DOI:10.1021/acs.inorgchem.9b02132
- M. Fairley, D. E. Felton, G. E. Sigmon, J. E. S. Szymanowski, N. A. Poole, M. Nyman, P. C. Burns and J. A. LaVerne, Radiation-Induced Solid-State Transformations of Uranyl Peroxides, Inorg. Chem., 2022, 61(2), 882–889, DOI:10.1021/acs.inorgchem.1c02603
- P. C. Burns and M. Nyman, Captivation with Encapsulation: A Dozen Years of Exploring Uranyl Peroxide Capsules, Dalton Trans., 2018, 47(17), 5916–5927, 10.1039/C7DT04245K
- V. G. Rodriguez, H. J. Culbertson, G. E. Sigmon and P. C. Burns, Electrochemistry of Uranyl Peroxide Solutions during Electrospray Ionization, Inorg. Chem., 2023, 62, 4456–4466, DOI:10.1021/acs.inorgchem.2c03904
- H. Traustason, N. L. Bell, K. Caranto, D. C. Auld, D. T. Lockey, A. Kokot, J. E. S. Szymanowski, L. Cronin and P. C. Burns, Reactivity, Formation, and Solubility of Polyoxometalates Probed by Calorimetry, J. Am. Chem. Soc., 2020, 142(48), 20463–20469, DOI:10.1021/jacs.0c10133
- C. Falaise and M. Nyman, The Key Role of U28 in the Aqueous Self-Assembly of Uranyl Peroxide Nanocages, Chem. – Eur. J., 2016, 22(41), 14678–14687, DOI:10.1002/chem.201602130
- J. F. Ziegler, M. D. Ziegler and J. P. Biersack, SRIM – The Stopping and Range of Ions in Matter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268(11), 1818–1823, DOI:10.1016/j.nimb.2010.02.091
- L. Greenspan, Humidity Fixed Points of Binary Saturated Aqueous Solutions, J. Res. Natl. Bur. Stand., Sect. A, 1977, 81(1), 89–96, DOI:10.6028/jres.081A.011
- J. Čejka, J. Sejkora, J. Plášil, S. Bahfenne, S. J. Palmer and R. L. Frost, Raman Spectroscopic Study of the Uranyl Carbonate Mineral Čejkaite and Its Comparison with Synthetic Trigonal Na4[UO2(CO3)3], J. Raman Spectrosc., 2010, 41(4), 459–464, DOI:10.1002/jrs.2349
- R. J. P. Driscoll, D. Wolverson, J. M. Mitchels, J. M. Skelton, S. C. Parker, M. Molinari, I. Khan, D. Geeson and G. C. Allen, A Raman Spectroscopic Study of Uranyl Minerals from Cornwall, UK, RSC Adv., 2014, 4(103), 59137–59149, 10.1039/C4RA09361E
- R. L. Frost and J. Čejka, A Raman Spectroscopic Study of the Uranyl Carbonate Rutherfordine: Raman Spectroscopic Study of Rutherfordine, J. Raman Spectrosc., 2007, 38(11), 1488–1493, DOI:10.1002/jrs.1796
- A. Weber and E. A. McGinnis, The Raman Spectrum of Gaseous Oxygen, J. Mol. Spectrosc., 1960, 4(1–6), 195–200, DOI:10.1016/0022-2852(60)90081-3
- G. Lu, A. J. Haes and T. Z. Forbes, Detection and Identification of Solids, Surfaces, and Solutions of Uranium Using Vibrational Spectroscopy, Coord. Chem. Rev., 2018, 374, 314–344, DOI:10.1016/j.ccr.2018.07.010
- E. Koglin, H. J. Schenk and K. Schwochau, Vibrational and Low Temperature Optical Spectra of the Uranyl Tricarbonato Complex [UO2(CO3)3]4−, Spectrochim. Acta, Part A, 1979, 35(6), 641–647, DOI:10.1016/0584-8539(79)80121-X
- K. Coenen, F. Gallucci, B. Mezari, E. Hensen and M. van Sint Annaland, An In situ IR Study on the Adsorption of CO2 and H2O on Hydrotalcites, J. CO2 Util., 2018, 24, 228–239, DOI:10.1016/j.jcou.2018.01.008
- K. D. Dobson and A. J. McQuillan, An Infrared Spectroscopic Study of Carbonate Adsorption to Zirconium Dioxide Sol−Gel Films from Aqueous Solutions, Langmuir, 1997, 13(13), 3392–3396, DOI:10.1021/la962024i
- L. H. Jones, Systematics in the Vibrational Spectra of Uranyl Complexes, Spectrochim. Acta, 1958, 10(4), 395–403, DOI:10.1016/0371-1951(58)80107-1
- D. D. Schnaars and R. E. Wilson, Structural and Vibrational Properties of U(VI)O2Cl42− and Pu(VI)O2Cl42− Complexes, Inorg. Chem., 2013, 52(24), 14138–14147, DOI:10.1021/ic401991n
- J. Cejka, Infrared Spectroscopy and Thermal Analysis of the Uranyl Minerals, Rev. Mineral. Geochem., 1999, 38(1), 521–622 Search PubMed
- E. Giamello, M. Volante, B. Fubini, F. Geobaldo and C. Morterra, An EPR Study on the Formation of the Superoxide Radical Ion on Monoclinic Zirconia, Mater. Chem. Phys., 1991, 29(1), 379–386, DOI:10.1016/0254-0584(91)90032-P
- A. Amorelli, J. C. Evans and C. C. Rowlands, An Electron Spin Resonance Study of the Superoxide Radical Anion in Polycrystalline Magnesium Oxide and Titanium Dioxide Powders, J. Chem. Soc., Faraday Trans. 1, 1988, 84(5), 1723–1728, 10.1039/F19888401723
- E. Giamello, L. Calosso, B. Fubini and F. Geobaldo, Evidence of Stable Hydroxyl Radicals and Other Oxygen Radical Species Generated by Interaction of Hydrogen Peroxide with Magnesium Oxide, J. Phys. Chem., 1993, 97(21), 5735–5740, DOI:10.1021/j100123a045
- J. A. Kaddissy, S. Esnouf, D. Durand, D. Saffre, E. Foy and J.-P. Renault, Radiolytic Events in Nanostructured Aluminum Hydroxides, J. Phys. Chem. C, 2017, 121(11), 6365–6373, DOI:10.1021/acs.jpcc.6b13104
- J. Kuruc, Paramagnetic Centers by X-Ray-Irradiation of Aluminium Hydroxide, J. Radioanal. Nucl. Chem., 1991, 154(1), 61–72, DOI:10.1007/BF02163064
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt01841a |
| This journal is © The Royal Society of Chemistry 2024 |