
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Accessing five- and seven-membered phosphorus-based heterocycles via cycloaddition reactions of azophosphines†
Ethan D. E.
Calder
 ,
Louise
Male
and
Andrew R.
Jupp
*
,
Louise
Male
and
Andrew R.
Jupp
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: a.jupp@bham.ac.uk
First published on 9th August 2024
Abstract
Heterocycles containing both phosphorus and nitrogen have seen increasing use in recent years in luminescent materials, coordination chemistry and as building blocks for inorganic polymers, yet their chemistry is currently dominated by five- and six-membered derivatives. Seven-membered P/N heterocycles are comparatively scarce and lack general, high yielding syntheses. Here, we explore the synthesis and characterisation of 1,2,5-diazaphosphepines from azophosphines. The mechanism has been probed in detail with both computational and experimental studies supporting a stepwise mechanism to form a five-membered ring, and subsequent ring expansion to the diazaphosphepine. Regioselective synthesis of five- and seven-membered rings is possible using asymmetric alkynes. The Lewis acidic borane B(C6F5)3 could either catalyse the formation of the seven-membered ring (iPr derivative) or trap out a key intermediate via a frustrated Lewis pair (FLP) mechanism (tBu derivative).
Introduction
The incorporation of phosphorus into heterocyclic systems can lead to significant changes in their properties and reactivity. Phospholes, first synthesised in 1959,1 have emerged as highly valuable compounds in numerous fields ranging from photophysical applications to coordination chemistry, and often display aromaticity.2–5 Utilising phosphorus alongside its lighter group 15 congener, nitrogen, can bestow further reactivity in the heterocycle, particularly when featuring a direct PV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond. This arises due to the highly polarised nature of the PVN bond, which yields significant single-bond character and a more basic nitrogen centre. Such phosphorus and nitrogen containing heterocycles have subsequently been used in areas including luminescent materials6 and as building blocks for inorganic polymers.7
N bond. This arises due to the highly polarised nature of the PVN bond, which yields significant single-bond character and a more basic nitrogen centre. Such phosphorus and nitrogen containing heterocycles have subsequently been used in areas including luminescent materials6 and as building blocks for inorganic polymers.7
The majority of currently reported phosphorus/nitrogen containing heterocycles are based on either five- or six-membered rings.8 Seven-membered rings containing both phosphorus and nitrogen and comparatively scarce. The most common approach for preparing seven-membered P/N rings relies on the condensation of amines with halophosphine derivatives, with concomitant release of HCl, which necessarily affords products with direct P–N bonds.9–11 There are fewer examples of P/N heterocycles that contain solely P–C bonds, yet heterocycles of this type recently have been applied as ligands for Ni in electrocatalysts or as chelating ligands for lanthanide ions,12–14 and in medicinal applications (Fig. 1A).15 Azaphosphepines are unsaturated seven-membered P/N heterocycles, and convenient and high-yielding synthetic routes to such heterocycles remain limited in scope. The most pertinent examples of azaphosphepine synthesis for this current study are ring-expansions via P–N cleavage of an appropriate five-membered ring precursor. In 1990, Ried et al. reported the ring-expansion of a five- to seven-membered ring via the cleavage of a P–N bond with electronically activated alkynes.16 The products were characterised crystallographically and spectroscopically, although the mechanism of reaction was not discussed. In 2019, Lozovskiy et al. also reported intramolecular ring-expansion by P–N bond cleavage in a five-membered ring to yield a seven-membered ring.17 The only prior example of 1,2,5-diazaphosphepine chalcogenides was reported by Barkallah et al., where 2,2′-(phenylphosphoryl)bis(cyclopentan-1-one) (or the sulfur and selenium analogues) was reacted with hydrazine and acetic acid (Fig. 1B), although the products were not structurally characterised.18
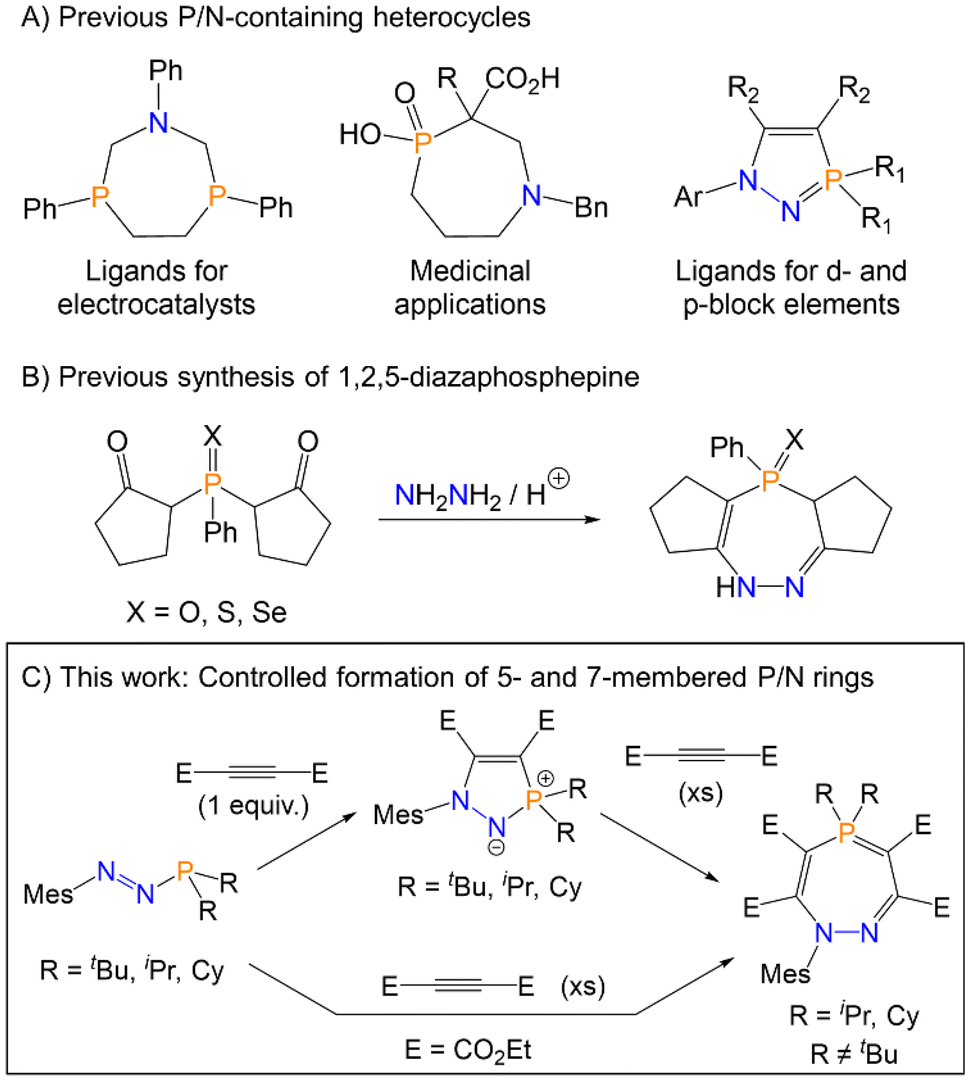 | ||
| Fig. 1 (A) Applications of previously reported P/N-containing heterocycles. (B) Only previous example of a 1,2,5-diazaphosphepine. (C) This work. | ||
Azophosphines are phosphorus-containing analogues of triazenes with the chemical formula ArNN–PR2, with a trivalent phosphorus centre that can be exploited for further reactivity. A limited range of azophosphines were synthesised in the 1970s and 1980s, but no further reactivity was explored.19–21 This class of compounds was ignored until 2021, when Cummins and co-workers synthesised MesN2PA (Mes = mesityl, A = anthracene).22 This molecule was shown to undergo a range of cycloaddition reactions with unsaturated substrates, including [Na(dioxane)2.5][PCO] and AdCP (Ad = adamantyl), with loss of the labile anthracene moiety. The following year, the same group isolated azophosphines of the form ArN2PR2 (R = iPr, Cy, Ph, NMe2), and demonstrated the cycloaddition reactions of these species with cyclooctyne, which yielded five-membered N-heterocyclic iminophosphoranes.23 One example of a 1,3-dipolar cycloaddition of the azophosphine (p-Me)C6H4N2PPh2 with the electronically activated alkyne dimethyl acetylenedicarboxylate (C(CO2Me))2 was also reported. Very recently, we reported a general synthetic route to azophosphines which allowed for tolerance of bulky P-substituents, and demonstrated the use of these systems as ligands in Ru complexes via the phosphorus and nitrogen centres.24
Motivated by these results, we reasoned that azophosphines may be able to serve as precursors to a wider range of phosphorus and nitrogen containing heterocycles beyond those currently reported. In this manuscript, we report the synthesis and characterisation of five- and seven-membered phosphorus- and nitrogen-containing rings from azophosphines. The seven-membered rings are the first crystallographically characterised 1,2,5-diazaphosphepines, and the mechanism of this transformation has been probed both computationally and experimentally. The interactions of these species with the Lewis acidic borane B(C6F5)3 has also been explored, which was shown to catalyse the formation of the seven-membered heterocycle for the iPr-derivative and result in the trapping of a key intermediate for the tBu-derivative.
Results & discussion
Synthesis and characterisation of heterocycles
We recently reported a new synthesis of azophosphines via the reaction of arenediazonium salts ([ArN2][BF4]; Ar = Mes or (p-NMe2C6H4)) with deprotonated secondary phosphine-boranes (HPR2·BH3), although the P-substituent was limited to R = tBu and Ph.24 The same synthetic procedure was used here, and was shown to tolerate smaller alkyl P-substituents. The secondary phosphine-boranes HPR2·BH3 (R = iPr, Cy (cyclohexyl)) were synthesised from the corresponding dialkylchlorophosphines by a modified literature procedure (Scheme 1A).25 Deprotonation of these substrates with sec-BuLi at −78 °C, followed by reaction with mesitylenediazonium tetrafluoroborate ([MesN2][BF4]) allowed for the formation of the target azophosphine-boranes (MesN2PR2·BH3) 1·BH3 (R = iPr) and 2·BH3 (R = Cy) as a purple oil or purple solid, respectively, in moderate yields following purification (1·BH3 = 41%, 2·BH3 = 44%; Scheme 1B). The borane protecting group could then be fully removed via reaction with pyrrolidine to yield the free azophosphines (MesN2PR2) 1 (R = iPr) and 2 (R = Cy) in good yields (1 = 78%, 2 = 71%; Scheme 1), both as red oils.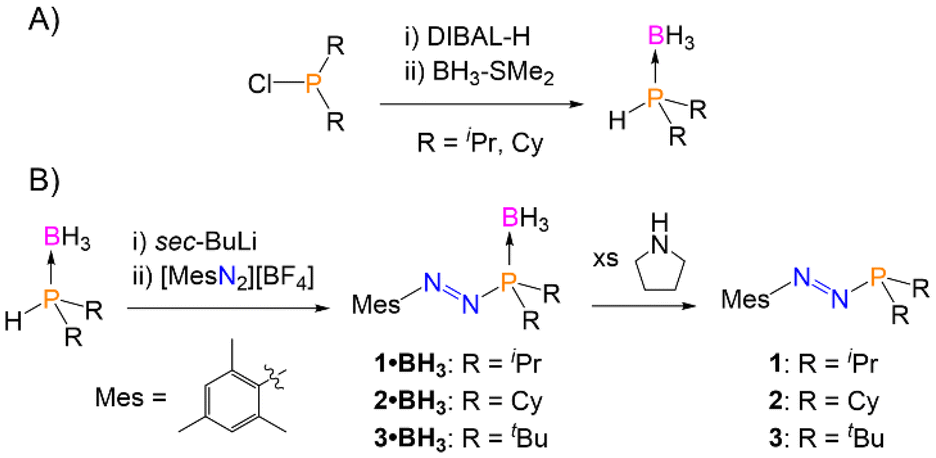 | ||
| Scheme 1 (A) Synthesis of phosphine-borane precursors. (B) Synthesis of azophosphine-boranes and free azophosphines. | ||
With azophosphines 1 and 2 in hand, as well as the previously reported azophosphine 3 (MesN2PtBu2), we next investigated the cycloaddition reactions of these systems. No reactivity was observed with the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond of diphenylacetylene, nor the CN bond of benzonitrile. However, the reaction of azophosphines 1–3 with one equivalent of the electron-poor alkyne diethyl acetylenedicarboxylate (C(CO2Et))2 in toluene gave an immediate colour change from red (1, 2) or purple (3) to pale yellow, and formation of the five-membered heterocycles (R = iPr (4), Cy (5), tBu (6); Scheme 2). Reaction monitoring by 31P{1H} NMR spectroscopy showed quantitative conversion from azophosphine to product; removal of the solvent in vacuo yielded analytically pure compounds 4–6 in good isolated yields (80–85%).
C bond of diphenylacetylene, nor the CN bond of benzonitrile. However, the reaction of azophosphines 1–3 with one equivalent of the electron-poor alkyne diethyl acetylenedicarboxylate (C(CO2Et))2 in toluene gave an immediate colour change from red (1, 2) or purple (3) to pale yellow, and formation of the five-membered heterocycles (R = iPr (4), Cy (5), tBu (6); Scheme 2). Reaction monitoring by 31P{1H} NMR spectroscopy showed quantitative conversion from azophosphine to product; removal of the solvent in vacuo yielded analytically pure compounds 4–6 in good isolated yields (80–85%).
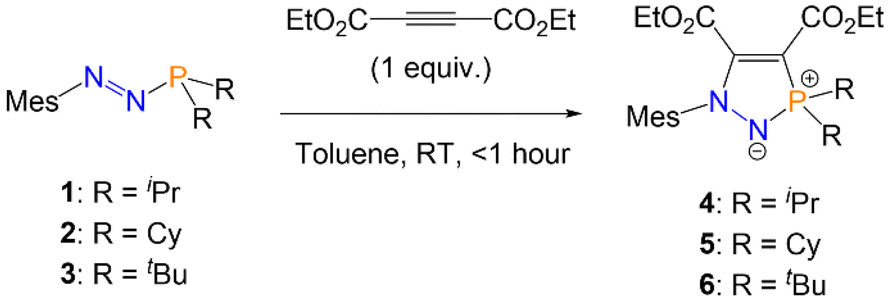 | ||
| Scheme 2 Synthesis of five-membered heterocycles 4–6via azophosphines. | ||
Single crystals of 4 were grown by slow evaporation of a hexane solution, and of 6 by slow evaporation of a THF–hexane solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10, v/v), allowing for characterisation of the solid state structures by single-crystal X-ray diffraction (SXRD) (Fig. 2A and B). Both 4 and 6 possess planar five-membered cores, as shown in the side-on views in Fig. 2C and D. 6 displays an N–N bond length of 1.4115(19) Å, indicative of an N–N single bond,26 and significantly elongated compared to the NN double bond [1.226(6) Å] observed in the previously reported solid state structure for its azophosphine-borane precursor 3·BH3.24 The P–N bond length of 6 exists somewhere between a single and double bond [1.6461(14) Å], but clearly shortened compared to the P–N bond of 3·BH3 [1.769(5) Å]. 4 displays similar N–N [1.4096(17) Å] and N–P [1.6442(14) Å] bond lengths to 6. The planarity and bond lengths of both 4 and 6 are in good agreement with the N-heterocyclic iminophosphoranes reported by Cummins, which also displayed planar heterocyclic cores and comparable N–N and P–N bond distances.23 To corroborate these findings, density functional theory (DFT) and natural bond orbital (NBO) analyses were carried out (see ESI† for details). The optimised structures are in good agreement with the observed solid-state structures, with similarly planar heterocyclic cores and N–N and P–N bond metrics. Natural population analysis (NPA) showed a highly electron-rich N2 nitrogen centre (−0.859 for 4, −0.874 for 6) and electron-deficient phosphorus centre (+1.809 for 4, +1.841 for 6), with calculated Wiberg bond indices for the P–N bonds of 1.07 (4) and 1.05 (6). These data are suggestive of more single bond than double bond character in the P–N bond.
:10, v/v), allowing for characterisation of the solid state structures by single-crystal X-ray diffraction (SXRD) (Fig. 2A and B). Both 4 and 6 possess planar five-membered cores, as shown in the side-on views in Fig. 2C and D. 6 displays an N–N bond length of 1.4115(19) Å, indicative of an N–N single bond,26 and significantly elongated compared to the NN double bond [1.226(6) Å] observed in the previously reported solid state structure for its azophosphine-borane precursor 3·BH3.24 The P–N bond length of 6 exists somewhere between a single and double bond [1.6461(14) Å], but clearly shortened compared to the P–N bond of 3·BH3 [1.769(5) Å]. 4 displays similar N–N [1.4096(17) Å] and N–P [1.6442(14) Å] bond lengths to 6. The planarity and bond lengths of both 4 and 6 are in good agreement with the N-heterocyclic iminophosphoranes reported by Cummins, which also displayed planar heterocyclic cores and comparable N–N and P–N bond distances.23 To corroborate these findings, density functional theory (DFT) and natural bond orbital (NBO) analyses were carried out (see ESI† for details). The optimised structures are in good agreement with the observed solid-state structures, with similarly planar heterocyclic cores and N–N and P–N bond metrics. Natural population analysis (NPA) showed a highly electron-rich N2 nitrogen centre (−0.859 for 4, −0.874 for 6) and electron-deficient phosphorus centre (+1.809 for 4, +1.841 for 6), with calculated Wiberg bond indices for the P–N bonds of 1.07 (4) and 1.05 (6). These data are suggestive of more single bond than double bond character in the P–N bond.
 | ||
| Fig. 2 Single crystal structures of 4 and 6. Top-down view shown in (A) and (B); side-on view for (C) and (D). Selected bond distances (Å) and torsion angles (°): 4: N1–N2 1.4096(17), N2–P1 1.6442(14), C10–C11 1.403(2), N1–C1 1.442(2), P1–C18 1.8326(16), P1–C21 1.8244(17), C10–C12 1.503(2), C11–C13 1.436(2), N1–N2–P1–C11 −2.37(11), N1–C10–C11–P1 −0.34(17); 6: N1–N2 1.4115(19), N2–P1 1.6461(14), C10–C11 1.401(2), N1–C1 1.442(2), P1–C18 1.8670(18), P1–C22 1.8655(17), C10–C12 1.503(2), C11–C13 1.437(2), N1–N2–P1–C11 −5.77(11), N1–C10–C11–P1 −2.50(18). 4 crystallised with two product molecules in the asymmetric unit; only one is shown here for clarity and bond metrics are statistically similar for both independent molecules so only one set is given above. Thermal ellipsoids were drawn at the 50% probability level at 100 K.27 | ||
Given the partial single-bond character of the P–N bond in five-membered rings 4–6, we hypothesised that this bond could be cleaved by certain substrates to yield larger phosphorus-containing heterocycles, by analogy with the aforementioned ring expansion chemistry.16,17 Reactions of a P–N bond within five-membered rings has previously been reported with ketones, ketenes, and isocyanates, although in all cases either acyclic or bicyclic structures were observed instead of ring expansion.28–30 Cummins also reported one example of P–N bond cleavage in an N-heterocyclic iminophosphorane with Ph3SiH, although this resulted in loss of the heterocyclic structure and formation of a Ph3Si–N(R)–H moiety.23 Five-membered heterocycle 4 (R = iPr) was thus reacted with an excess (4 equiv.; Scheme 3) of (C(CO2Et))2 in toluene. Monitoring by 31P{1H} NMR spectroscopy showed a new species forming, product 7, with quantitative conversion within 48 hours at room temperature; the solution also became a visibly brighter yellow colour. 7 could also be formed directly from azophosphine 1, by reacting 1 with the same excess of (C(CO2Et))2. Monitoring this latter reaction by 31P{1H} NMR spectroscopy showed 4 initially forming at 74.9 ppm, before giving way to 7 at 60.4 ppm. Analogous reactivity was observed with azophosphine 2 (MesN2PCy2), which yielded seven-membered heterocycle 8via5. Recrystallisation and removal of the solvent in vacuo yielded analytically pure compounds 7 and 8 in excellent isolated yields (7 = 88%, 8 = 92%), with no further work-up necessary. However, when the sterically more hindered azophosphine 3 (MesN2PtBu2) was used, no seven-membered ring could be detected by 31P{1H} NMR spectroscopy. Even with a large excess of alkyne (10 equiv.) and heating to 60 °C, only formation of five-membered ring 6 was observed (Scheme 3); heating above 60 °C gave an intractable mixture of products.
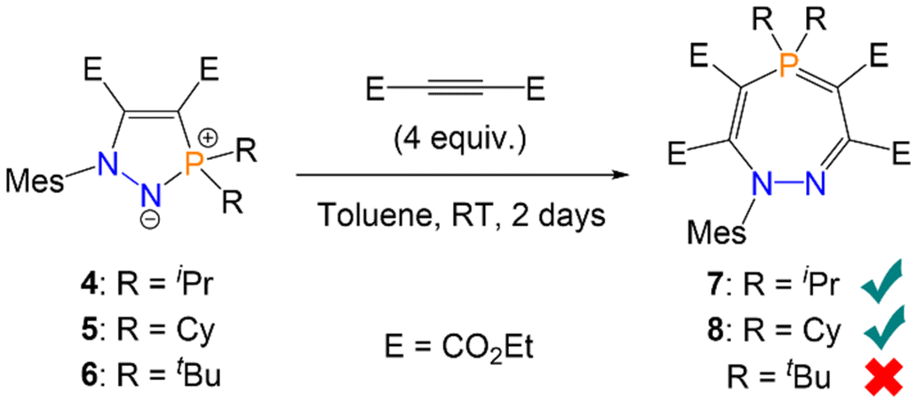 | ||
| Scheme 3 Synthesis of seven-membered heterocycles 7 and 8via azophosphines. 6 does not react with excess alkyne to form the analogous seven-membered ring. | ||
Single crystals of 7 and 8 were produced by slow diffusion of n-hexane into a concentrated THF solutions of the product at −35 °C, for which single-crystal X-ray diffraction revealed the unusual seven-membered ring structure featuring cleavage of the P–N bond by a second equivalent of alkyne (Fig. 3). In contrast to the planar geometries of five-membered rings 4 and 6, the seven-membered rings of 7 and 8 feature non-planar, puckered cores (Fig. 3C and D). An analysis of the bond metrics shows that four major resonance structures can be considered for these seven-membered rings (α, β, γ, and δ in Fig. 3E). The metric data for 7 and 8 are analogous and so 7 will be discussed here as a representative example. The P1–C13 bond [1.7465(14) Å] is significantly shorter than P1–C11 [1.7851(14) Å], highlighting the contribution from resonance α. The negative charge in β can also be delocalised into the proximal ester group as shown in γ [C13–C17 = 1.4316(19) Å, C17–O7 = 1.2235(18) Å], or onto N2 as shown in δ [C12–C13 = 1.438(2) Å]. The crystallographic data are corroborated by density functional theory (DFT) and natural bond orbital (NBO) analyses (see ESI† for details) for 7.
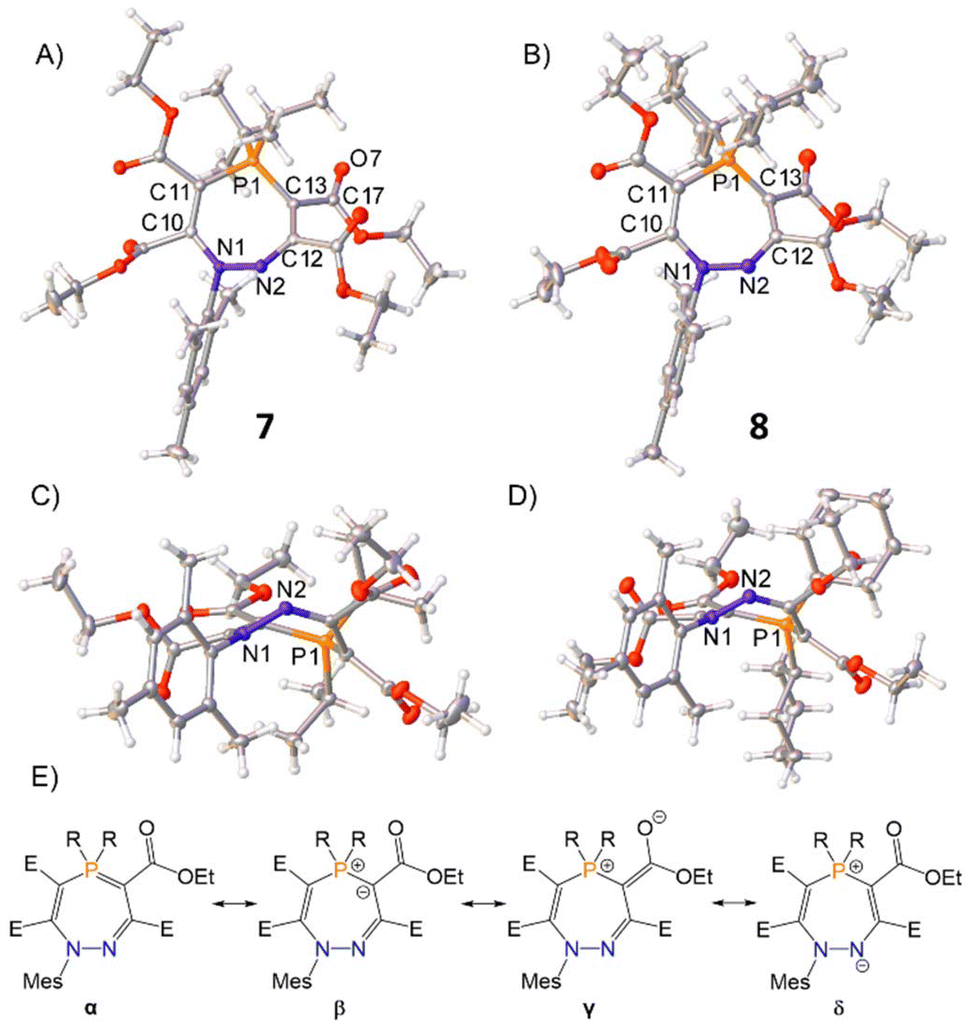 | ||
| Fig. 3 Single crystal structures of 7 and 8. Top-down view shown in (A) and (B); side-on view for (C) and (D). Selected bond distances (Å): 7 N1–N2 1.4172(16), N2–C12 1.2908(18), C12–C13 1.438(2), C13–P1 1.7465(14), C11–P1 1.7851(14), N1–C1 1.4472(17), C10–C14 1.5364(18), C11–C15 1.4659(19), C12–C16 1.5321(19), C13–C17 1.4313(19), P1–C26 1.8381(14), P1–C29 1.8344(14); 8 N1–N2 1.4177(18), N2–C12 1.2926(19), C12–C13 1.443(2); C13–P1 1.7407(16), C11–P1 1.7892(15), N1–C1 1.4487(18), C10–C14 1.531(2), C11–C15 1.480(2), C12–C16 1.530(2), C13–C17 1.434(2), P1–C26 1.8239(14), P1–C32 1.8321(15). Thermal ellipsoids were drawn at the 50% probability level at 100 K.27 (E) Possible resonance structures for seven-membered heterocycles 7 and 8 (E = CO2Et). | ||
Mechanistic insight
To determine the minimum energy pathway for the formation of five-membered rings 4 and 6, DFT calculations were carried out (see ESI† for details), using the xylyl group instead of mesityl for the calculations as a simplified model. A recent computational study by Zhang and Su exploring the cycloaddition of structurally related azophosphines with cyclooctyne found a concerted reaction pathway, with an activation barrier of 22.0 kcal mol−1.31 Cummins also calculated the reaction of their MesN2PA unit with cyclooctyne to be thermodynamically favourable via a concerted pathway, with subsequent loss of the anthracene unit.22 Our calculations showed a stepwise mechanism to instead be preferred for the cycloadditions of azophosphines 1 and 3 with diethyl acetylenedicarboxylate (Scheme 4). Focusing on the pathway for 3 to 6, initial attack of the phosphorus lone pair (HOMO) of 3 to the alkyne yields the zwitterionic intermediate I1, with a corresponding activation barrier of 20.4 kcal mol−1 (TS1) corresponding to P–C bond formation. I1 adopts a partially allenic, partially bent structure (Scheme 4). This is supported by a C1–C2 bond length of 1.329 Å, consistent with a CC double bond, and an elongated C2–C3 bond length of 1.401 Å, with a C1–C2–C3 bond angle of 138.3°. The HOMO of I1 has contributions from both the electron-rich carbon and oxygen centres, while the LUMO is mainly centred on the NN π* orbital. Subsequent HOMO to LUMO ring-closing proceeds over a small activation barrier of 3.3 kcal mol−1 (TS2). Product 6 is favoured by 50.1 kcal mol−1 relative to the starting materials. An identical pathway was found for the formation of 4 from azophosphine 1, with similar activation barriers calculated. The preference for a stepwise pathway in our system compared to the previously computed concerted pathways may be rationalised by the stabilisation of the zwitterionic intermediate I1 by the electron-withdrawing ester groups, and the fact that our study is on P,P-dialkyl azophosphines that are more nucleophilic than the previously studied P,P-diphenyl systems.22,31 A preference for stepwise over concerted reaction pathways has previously been shown for cycloaddition reactions of the analogous alkyne dimethyl acetylenedicarboxylate with dienylisobenzofurans.32
 | ||
| Scheme 4 Computed pathways (Gibbs free energy, kcal mol−1) for the cycloadditions of 1 and 3 with (C(CO2Et))2 to form 4 and 6 at the ωB97XD/def2TZVP//ωB97XD(toluene)//def2QZVP level of theory using the xylyl group instead of mesityl for a slightly simplified model. For full computational details see ESI.† | ||
We also sought to determine the minimum energy pathway for the formation of seven-membered ring 7, and rationalise the why the analogous heterocycle with R = tBu substituents did not form. Our calculations suggest that the first step, corresponding to nucleophilic attack by the dicoordinate nitrogen centre of 4/6 to the alkyne (TS3), is the rate-limiting step of the reaction (Scheme 5). This proceeds over an energy barrier of 18.1 kcal mol−1 for 4 (R = iPr), but the corresponding energy barrier for 6 (R = tBu) is significantly higher, at 24.7 kcal mol−1. This barrier is accessible for 4 at room temperature, but was inaccessible for 6 in our experiments. Attempts to overcome this barrier by heating the reaction led to an intractable mixture of products. Seven-membered rings 7 and 8 form at room temperature but decompose above 50 °C, which may indicate that if the energy barrier for R = tBu substituents can be overcome at elevated temperatures, the product subsequently decomposes and cannot be isolated. After this first step, the subsequent intermediate, I2, adopts a partially allenic and partially bent structure (analogous to I1, Scheme 4), although in this case the bent structure is stabilised to a greater extent as the carbanion has a weak interaction with the proximal electron-poor phosphorus centre. We were also able to locate a further intermediate (I3), which was only present for R = iPr substituents, in which the P–N interatomic distance is significantly longer (2.145 in I3vs. 1.756 in I2) and P–C bond shorter (1.842 in I3vs. 2.012 in I2). After extensive searching, we were unable to locate any transition states between I2 and the products, although relaxed potential energy surface scans indicated that these steps would be <5 kcal mol−1 for R = iPr and <7 kcal mol−1 for R = tBu and are thus clearly not rate-limiting (see ESI† for further details).
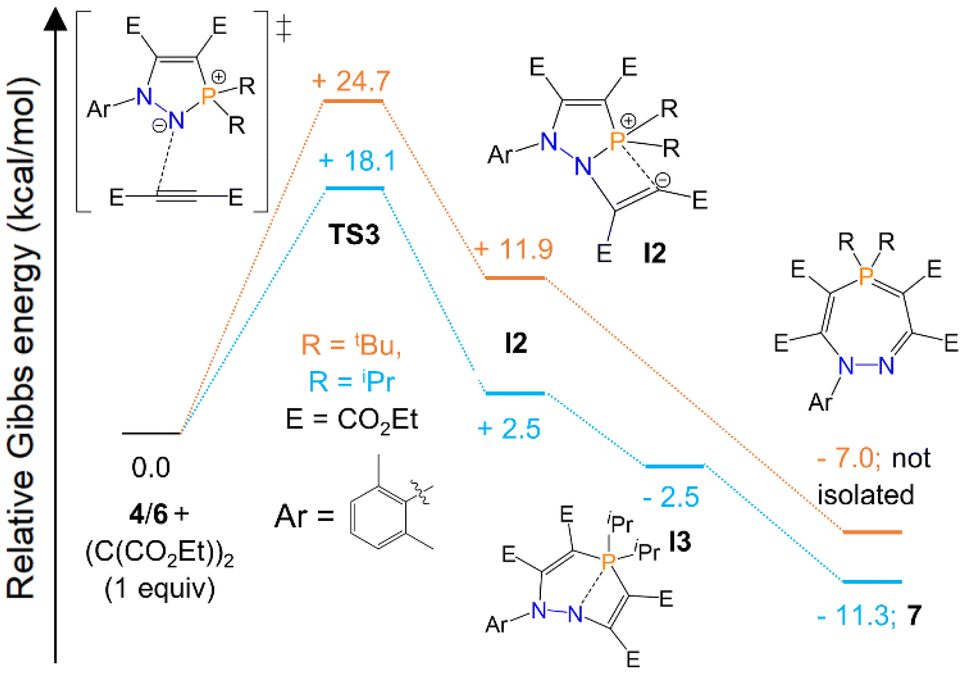 | ||
| Scheme 5 Computed pathways (Gibbs free energy, kcal mol−1) for the reaction of 4 and 6 with (C(CO2Et))2 at the ωB97XD/def2TZVP//ωB97XD(toluene)//def2QZVP level of theory using the xylyl group instead of mesityl for a slightly simplified model. For full computational details see ESI.† | ||
Regioselective formation of heterocycles
The stabilisation of I1 (Scheme 4) by electron delocalisation into the ester group prompted us to probe the regioselectivity of these reactions; namely, whether the use of an asymmetric alkyne would yield one, or several, regioisomers. Azophosphine 3 (MesN2PtBu2), was thus reacted with an excess (20 equiv.) of ethyl 2-butynoate (H3CCC(CO2Et)) in toluene. After 4 days, only one major product was observed by 31P{1H} NMR spectroscopy, which after work-up was observed to be product 9 (Scheme 6A). This regioisomer could be confirmed in solution by 2D-NOESY NMR spectroscopy, as a nOe (nuclear Overhauser effect) could be observed between the tBu groups and the methyl protons bound to the ring (–CCCH3), but not between the tBu groups and any of the protons on the ester groups. Single crystals of 9 were grown via slow evaporation of a concentrated hexane solution, and SXRD confirmed the product as the expected regioisomer (Fig. 4A). The bond metric data for 9 are similar to those previously discussed for 4 and 6.
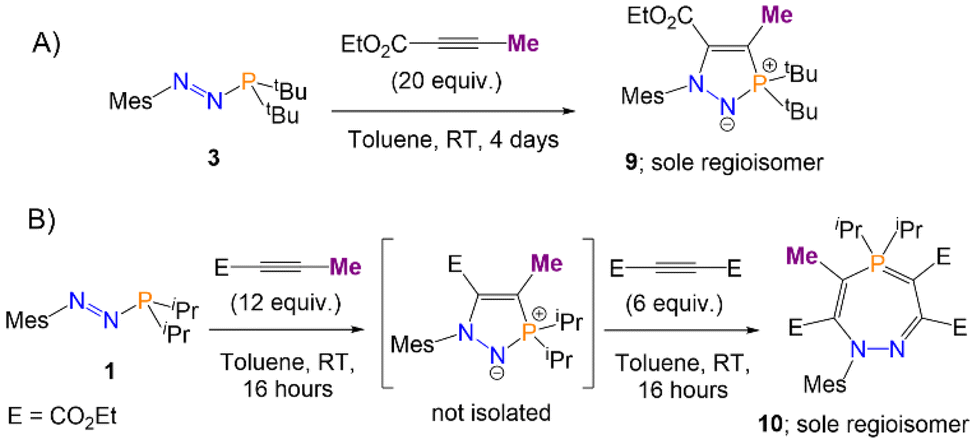 | ||
| Scheme 6 Regioselective synthesis of heterocycles 9 (A) and 10 (B) via the reaction of azophosphines with ethyl 2-butynoate. | ||
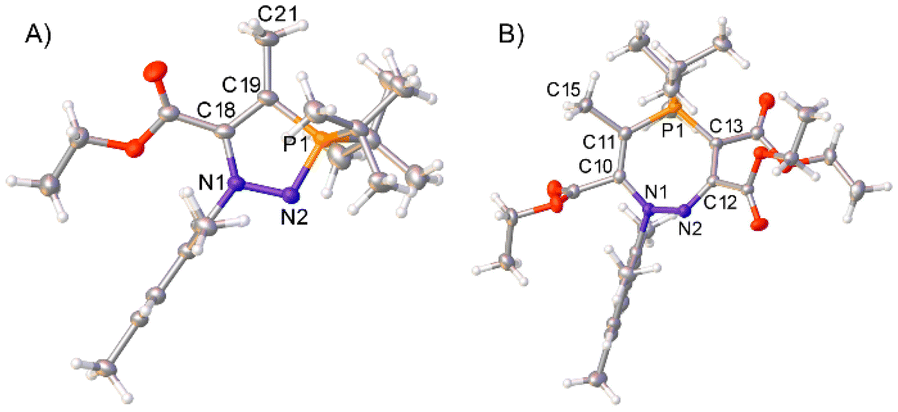 | ||
| Fig. 4 Single crystal structures of 9 (A) and 10 (B). Selected bond distances (Å): 9 N1–N2 1.4117(18), N2–P1 1.6382(13), C18–C19 1.390(2), N1–C1 1.4349(18), P1–C10 1.8790(17), P1–C14 1.8618(18), C18–C20 1.485(2), C19–C21 1.508(2), 10 N1–N2 1.4103(18), N2–C12 1.286(2), C12–C13 1.452(2), C13–P1 1.7268(17), C11–P1 1.7737(17), N1–C1 1.444(2), C10–C14 1.519(2), C11–C15 1.529(2), C12–C16 1.526(2), C13–C17 1.430(2), P1–C24 1.8264(18), P1–C27 1.8316(17). 9 crystallised with two product molecules in the asymmetric unit; only one is shown here for clarity. Thermal ellipsoids were drawn at the 50% probability level at 100 K for 9, and 120 K for 10.27 | ||
The analogous reaction with azophosphine 1 (MesN2PiPr2) was attempted, as the smaller P-substituents could potentially enable access to the corresponding seven-membered ring. Azophosphine 1 was reacted with an excess (12 equiv.) of ethyl 2-butynoate overnight in toluene (Scheme 6B). Complete consumption of the azophosphine was observed by 31P{1H} NMR spectroscopy, and formation of a new species at 72.4 ppm, consistent with formation of a five-membered heterocycle, although we were unable to cleanly isolate this species. There was no evidence of any formation of the seven-membered ring, despite the excess of the alkyne used, which we postulate is due to the reduced electrophilicity of ethyl 2-butynoate compared to diethyl acetylenecarboxylate (i.e. the analogous barrier that would correspond to TS3 from Scheme 5 in this system is too high to be accessible). However, subsequent addition of an excess (6 equiv.) of the more reactive diethyl acetylenedicarboxylate ((C(CO2Et))2) to this compound yielded species 10 as the sole product. Single crystals of 10 were grown via slow evaporation of a hexane solution, for which SXRD confirmed the product as the expected regioisomer of the seven-membered ring (Fig. 4B); this structural assignment was again supported by 2D-NOESY NMR spectroscopy. The formation of products 9 and 10 demonstrate the regioselective synthesis of five- and seven-membered rings from azophosphines.
Catalysis and trapping of intermediate
The formation of the seven-membered rings is relatively sluggish, and an excess of the alkyne was often used to drive the reaction to completion in a reasonable timeframe (four equivalents of alkyne required to form 7 in two days). Although the alkyne is relatively volatile and can be recycled for future experiments, we sought to improve the efficiency of the reaction. The rate-determining step for the formation of the seven-membered ring from the five-membered precursor is the attack of the electrophilic alkyne by the nitrogen centre (TS3 in Scheme 5). We therefore rationalised that a Lewis acidic borane could bind to the alkyne and activate it (thereby lowering the energy of TS3), with the caveat that the borane would have to be sterically bulky enough to preclude binding to the nucleophilic nitrogen centre on the five-membered ring and quenching its reactivity. We opted to use tris(pentafluorophenyl)borane (BCF), B(C6F5)3, which is highly Lewis acidic and relatively bulky, and is widely employed as a main-group Lewis acid catalyst and as a component of frustrated Lewis pair catalysts.33–39Five-membered heterocycle 4 (R = iPr) was thus combined with one equivalent of (C(CO2Et))2 and one equivalent of B(C6F5)3 in toluene (Scheme 7). The reaction resulted in the rapid formation (<1 hour) of 7·BCF, in which B(C6F5)3 is bound to an ester group in the corresponding seven-membered ring 7. This adduct was characterised by SXRD (Fig. 5A) and multinuclear NMR spectroscopy, with 7·BCF having a subtly different 31P{1H} NMR chemical shift to 7 (60.0 ppm (7·BCF), 60.4 ppm (7)). The fact that the borane is bound to this particular ester group of the four available is consistent with the electron-rich nature of this oxygen centre due to the aforementioned resonance structure γ (Fig. 3E). The bound borane increases the contribution from resonance γ relative to the free 7; this can be seen in the longer P1–C13 bond distance in 7·BCF (1.7791(15) Å vs. 1.7465(14) Å in 7). 7·BCF could be readily converted to spectroscopically pure 7 by addition of the nucleophilic amine 4-dimethylaminopyridine (DMAP; see ESI, Section S4.3† for details). The same reaction was then monitored with a substoichiometric amount of B(C6F5)3 to assess whether the borane could be used catalytically; the rate of reaction of 4 with one equivalent of (C(CO2Et))2 with 20 mol% of B(C6F5)3 was compared to the same reaction without B(C6F5)3 (Fig. 5B). B(C6F5)3 was indeed observed to have a catalytic effect, with the reaction containing 20 mol% B(C6F5)3 reaching 82% conversion in under two days, and full conversion within five days; whereas the reaction without B(C6F5)3 had reached only 21% and 43% conversion at these same timepoints, respectively.
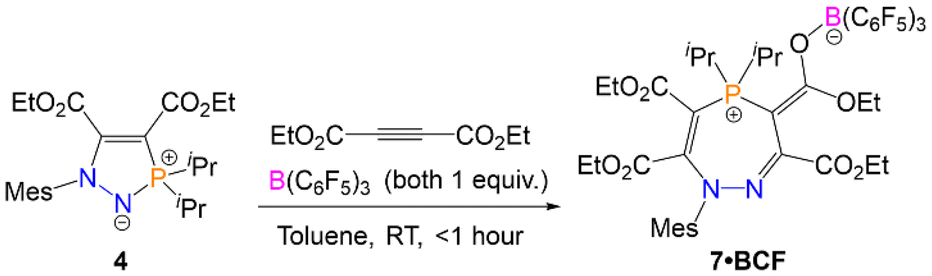 | ||
| Scheme 7 Synthesis of 7·BCFvia the reaction of 4 with (C(CO2Et))2 and B(C6F5)3. | ||
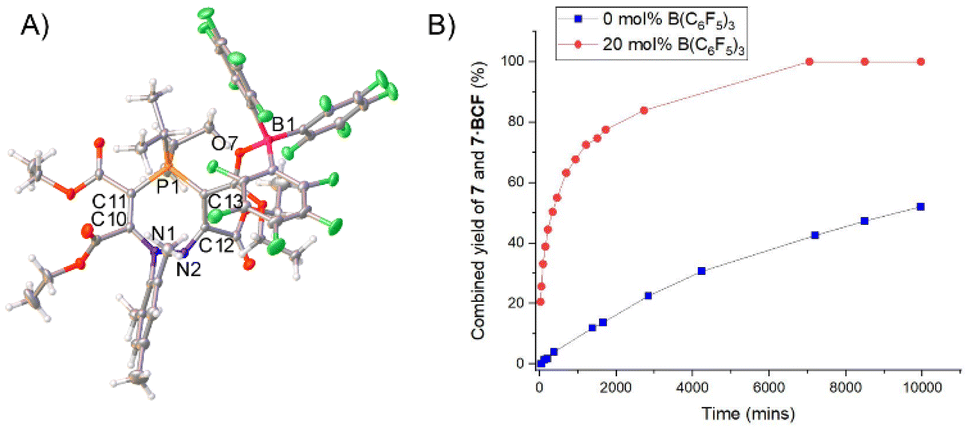 | ||
| Fig. 5 (A) Single crystal structure of 7·BCF. Selected bond distances (Å): P1–C13 1.7791(15), P1–C11 1.7833(16), C12–C13 1.465(2), N2–C12 1.281(2), N1–N2 1.4068(18), O7–B1 1.538(2), N1–C1 1.4571(19), C10–C14 1.530(2), C11–C15 1.471(2), C12–C16 1.533(2), C13–C17 1.383(2), P1–C26 1.8336(16), P1–C29 1.8434(17). Thermal ellipsoids were drawn at the 50% probability level at 120 K.27 (B) Graph showing conversion of 4 into 7 and 7·BCF in the absence (blue squares) and presence of 20 mol% (red circles) B(C6F5)3 in toluene at RT. | ||
It was established previously that the reaction of the tBu-derivative five-membered ring, 6, does not react further with the alkyne (C(CO2Et))2 to give the seven-membered ring. When this reaction was repeated with the addition of one equivalent of B(C6F5)3, complete conversion to a new product 11 was evidenced by a new chemical shift in the 31P{1H} NMR spectrum at 85.5 ppm. This chemical shift is significantly more downfield than the 31P{1H} NMR shifts for seven-membered rings 7 and 8. Slow evaporation of an n-hexane solution of 11 yielded crystals suitable for SXRD, which revealed that the borane had trapped out the intermediate I2 from Scheme 5 by binding to the electron-rich carbonyl group in the second equivalent of the alkyne (Scheme 8). Similarly to 4, the inclusion of B(C6F5)3 presumably lowers the energy barrier to the first step by activating the alkyne to attack from 6, allowing 11 to form. However, the binding of the borane would also raise the energy of the second step (attack of the nucleophilic carbon to the phosphorus centre) as the carbon is less nucleophilic. In this case with R = tBu substituents, the energy barrier for the subsequent steps is presumably raised to such an extent that this now become rate-limiting and inaccessible, preventing formation of the analogous seven-membered ring, and stopping the reaction at 11.
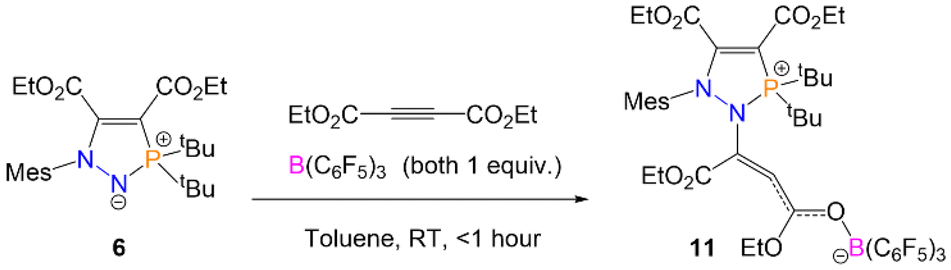 | ||
| Scheme 8 Reaction of heterocycle 6 with (C(CO2Et))2 and B(C6F5)3 to form 11. | ||
In good agreement with the structure of I2 (and I1) predicted by DFT, the single crystal structure of 11 features a partially allenic, partially bent structure, although the bound borane increases the contribution from the allenic structure (Fig. 6). The N2–C26 [1.440(6) Å] and C26–C27 [1.302(6) Å] bond lengths are consistent with an N–C single bond and CC double bond, while the C27–C28 [1.384(6) Å], being somewhere between single and double bond character.26,40 The C26–C27–C28 bond angle is 131.9(5)°, consistent with a geometry somewhere between that of an allene and alkene. In the solution phase, C27 also possesses a very downfield shift (205.9 ppm) in the 13C{1H} NMR spectrum of 11, in line with other reported allenes.41,42 DFT and NBO analyses of the structure of 11 (see ESI† for details) is in agreement with the observed crystallographic data. The N2–C26 and C26–C27 bonds possess WBI values of 0.96 and 1.82 respectively, while the WBI for C27–C28 (1.36) is indicative of its partial single- and double-bond character. The HOMO of 11 is located on the lone pair of C27, which resides in an sp3 orbital.
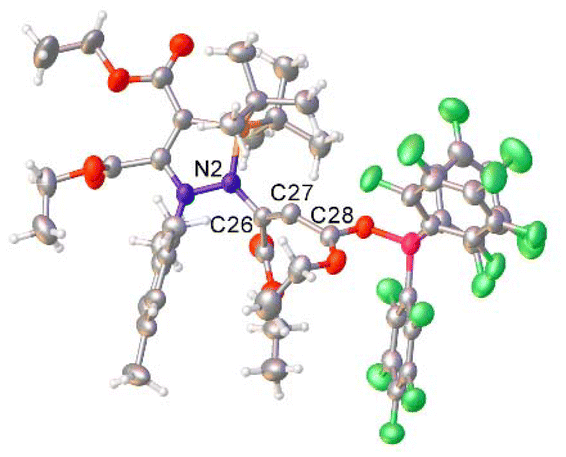 | ||
| Fig. 6 Single crystal structure of 11. Selected bond distances (Å) and angles (°): N2–C26 1.440(6), C26–C27 1.302(6), C27–C28 1.384(6), C26–C27–C28: 131.9(5). 11 crystallised with two product molecules and one hexane molecule in the asymmetric unit; for clarity, only one product molecule is shown here, and the hexane molecule has been omitted. Thermal ellipsoids were drawn at the 50% probability level at 100 K.27 | ||
11 can also be viewed as an example of frustrated Lewis pair (FLP) capture of diethyl acetylenedicarboxylate, in which 6 is acting as the Lewis base and B(C6F5)3 acting as the Lewis acid. While capture of alkynes with FLPs is common in the literature, including in FLP-catalysed hydrogenations, non-terminal alkynes predominantly undergo 1,2-addition with FLPs to form an alkene-linked zwitterion.43 The preference here for 1,4-addition is presumably due to the sterically bulky alkyne, and bulky nitrogen centre on 6 disfavouring 1,2-addition, as well as the ability to form a strong boron–oxygen bond via 1,4-addition. The structurally similar alkyne dimethyl acetylenedicarboxylate has indeed been shown to undergo unusual reactivity with FLPs, including 1,4-addition.44
Conclusions
In conclusion, we have described a general route to seven-membered P/N-containing heterocycles via azophosphines. This occurs through initial formation of the corresponding five-membered ring, before P–N bond cleavage yields the analogous seven-membered ring. The stepwise, rather than concerted, nature of these reactions was confirmed by DFT studies, and asymmetric alkynes could be employed for the regioselective synthesis of new heterocycles. The incorporation of B(C6F5)3 revealed both catalytic activity, and allowed for trapping of a reaction intermediate via a frustrated Lewis pair-type mechanism. We are continuing to explore the reactivity of the five-membered and seven-membered heterocycles with small molecules for future applications.Author contributions
Ethan Calder: investigation, formal analysis, visualisation, writing – original draft preparation. Louise Male: formal analysis (SXRD). Andrew Jupp: conceptualisation, funding acquisition, project administration, resources, supervision, visualisation, writing – reviewing and editing.Data Availability
The data associated with this manuscript are available at https://doi.org/10.25500/edata.bham.00001119.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Royal Society (URF\R1\201636), the EPSRC (EP/W036908/1), and the University of Birmingham for funding. The computations described in this paper were performed using the University of Birmingham's BlueBEAR HPC service, see https://www.birmingham.ac.uk/bear for more details.Notes and references
- F. C. Leavitt, T. A. Manuel and F. Johnson, J. Am. Chem. Soc., 1959, 81, 3163–3164 CrossRef CAS.
- L. Weber, Angew. Chem., Int. Ed., 2002, 41, 563–572 CrossRef CAS.
- M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- M. P. Duffy, W. Delaunay, P. A. Bouit and M. Hassler, Chem. Soc. Rev., 2016, 45, 5296–5310 RSC.
- N. Asok, J. R. Gaffen and T. Baumgartner, Acc. Chem. Res., 2023, 56, 536–547 CrossRef CAS PubMed.
- J. A. W. Sklorz and C. Müller, Eur. J. Inorg. Chem., 2016, 595–606 CrossRef CAS.
- J. Bedard and S. S. Chitnis, Chem. Mater., 2023, 35, 8338–8352 CrossRef CAS.
- J. N. McNeill, J. P. Bard, D. W. Johnson and M. M. Haley, Chem. Soc. Rev., 2023, 52, 8599–8634 RSC.
- S. E. Denmark and T. Wynn, J. Am. Chem. Soc., 2001, 123, 6199–6200 CrossRef CAS PubMed.
- T. Takeda and M. Terada, J. Am. Chem. Soc., 2013, 135, 15306–15309 CrossRef CAS PubMed.
- X. Gao, J. Han and L. Wang, Org. Lett., 2015, 17, 4596–4599 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- A. A. Karasik, A. S. Balueva, E. I. Moussina, R. N. Naumov, A. B. Dobrynin, D. B. Krivolapov, I. A. Litvinov and O. G. Sinyashin, Heteroat. Chem., 2008, 19, 125–132 CrossRef CAS.
- D. M. Weekes, M. G. Jaraquemada-Peláez, T. I. Kostelnik, B. O. Patrick and C. Orvig, Inorg. Chem., 2017, 56, 10155–10161 CrossRef CAS PubMed.
- A.-P. Schaffner, P. Sansilvestri-Morel, N. Despaux, E. Ruano, T. Persigand, A. Rupin, P. Mennecier, M.-O. Vallez, E. Raimbaud, P. Desos and P. Gloanec, J. Med. Chem., 2021, 64, 3897–3910 CrossRef CAS PubMed.
- W. Ried, M. Fulde and J. W. Bats, Helv. Chim. Acta, 1990, 73, 1888–1893 CrossRef CAS.
- S. V. Lozovskiy, A. Y. Ivanov and A. V. Vasilyev, Beilstein J. Org. Chem., 2019, 15, 1491–1504 CrossRef CAS PubMed.
- S. Barkallah, M. Boukraa, H. M'Rabet and H. Zantour, Synth. Commun., 1999, 29, 1911–1920 CrossRef CAS.
- J. Kroner, W. Schneid, N. Wiberg, B. Wrackmeyer and G. Ziegleder, J. Chem. Soc., Faraday Trans. 2, 1978, 74, 1909–1919 RSC.
- N. Wiberg, Adv. Organomet. Chem., 1984, 23, 131–191 CrossRef CAS.
- O. A. Attanasi, P. Filippone, P. Guerra and F. Serra-Zanetti, Synth. Commun., 1987, 17, 555–561 CrossRef CAS.
- M. Y. Riu, W. J. Transue, J. M. Rall and C. C. Cummins, J. Am. Chem. Soc., 2021, 143, 7635–7640 CrossRef CAS PubMed.
- K. Tanaka, M. Y. Riu, B. Valladares and C. C. Cummins, Inorg. Chem., 2022, 61, 13662–13666 CrossRef CAS PubMed.
- E. J. Jordan, E. D. E. Calder, H. V. Adcock, L. Male, M. Nieger, J. C. Slootweg and A. R. Jupp, Chem. – Eur. J., 2024, e202401358, DOI:10.1002/chem.202401358.
- C. Busacca, T. Bartholomeyzik, S. Cheekoori, R. Raju, M. Eriksson, S. Kapadia, A. Saha, X. Zeng and C. Senanayake, Synlett, 2009, 287–291 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2008, 15, 186–197 CrossRef PubMed.
- Deposition numbers 2356186 (4), 2356185 (6), 2356190 (7), 2356192 (7·BCF), 2356188 (8), 2356189 (9), 2356187 (10), and 2356191 (11) contain the supplementary crystallographic data for this paper.†.
- A. Schmidpeter and T. von Criegern, J. Chem. Soc., Chem. Commun., 1978, 470–471 RSC.
- A. Schmidpeter and T. von Criegern, Chem. Ber., 1978, 111, 3747–3749 CrossRef CAS.
- W. S. Sheldrick, D. Schomburg, A. Schmidpeter and T. von Criegern, Chem. Ber., 1980, 113, 55–69 CrossRef CAS.
- Z.-F. Zhang and M.-D. Su, Dalton Trans., 2023, 52, 4796–4807 RSC.
- Y. Chen, S. Ye, L. Jiao, Y. Liang, D. K. Sinha-Mahapatra, J. W. Herndon and Z. X. Yu, J. Am. Chem. Soc., 2007, 129, 10773–10784 CrossRef CAS PubMed.
- J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627–8643 CrossRef CAS PubMed.
- G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs I: Uncovering and Understanding, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- G. Erker and D. W. Stephan, Frustrated Lewis Pairs II: Expanding the Scope, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- J. C. Slootweg and A. R. Jupp, Frustrated Lewis Pairs, Springer, Cham, 2021 Search PubMed.
- A. R. Jupp and D. W. Stephan, Trends Chem., 2019, 1, 35–48 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- B. M. Trost, A. B. Pinkerton and M. Seidel, J. Am. Chem. Soc., 2001, 123, 12466–12476 CrossRef CAS PubMed.
- A. S. K. Hashmi, R. Döpp, C. Lothschütz, M. Rudolph, D. Riedel and F. Rominger, Adv. Synth. Catal., 2010, 352, 1307–1314 CrossRef CAS.
- J. Guo, M. Yan and D. W. Stephan, Org. Chem. Front., 2024, 11, 2375–2396 RSC.
- L. Wang, J. Li, D. Deng, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Dalton Trans., 2019, 48, 11921–11926 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2356185 (6), 2356186 (4), 2356187 (10), 2356188 (8), 2356189 (9), 2356190 (7), 2356191 (11) and 2356192 (7·BCF). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt02248c |
| This journal is © The Royal Society of Chemistry 2024 |