
Chemical and spectroscopic characterization of plutonium tetrafluoride
Jared S.
Kinyon
 a,
Eliel
Villa-Aleman
*a,
Elodia
Ciprian
b,
Amy E.
Hixon
b,
Bryan J.
Foley
a,
Jonathan H.
Christian
a,
Jason R.
Darvin
a,
Don D.
Dick
a,
Amanda J.
Casella
c,
Lucas E.
Sweet
c,
Edgar C.
Buck
c,
Forrest D.
Heller
c,
Aaron D.
Nicholas
c,
Cody A.
Nizinski
c and
Richard A.
Clark
c
a,
Eliel
Villa-Aleman
*a,
Elodia
Ciprian
b,
Amy E.
Hixon
b,
Bryan J.
Foley
a,
Jonathan H.
Christian
a,
Jason R.
Darvin
a,
Don D.
Dick
a,
Amanda J.
Casella
c,
Lucas E.
Sweet
c,
Edgar C.
Buck
c,
Forrest D.
Heller
c,
Aaron D.
Nicholas
c,
Cody A.
Nizinski
c and
Richard A.
Clark
c
aSavannah River National Laboratory, Aiken, SC 29808, USA. E-mail: Eliel.villa-aleman@srnl.doe.gov
bUniversity of Notre Dame, Notre Dame, IN 46556, USA
cPacific Northwest National Laboratory, P.O. Box 999, Richland, WA 99352, USA
First published on 21st October 2024
Abstract
Anhydrous plutonium tetrafluoride is an important intermediate in the production of metallic Pu. This historically important compound is also known to exist in at least two distinct, yet understudied hydrate forms, PuF4·xH2O(s) (0.5 ≤ x ≤ 2) and PuF4·2.5H2O(s). X-ray diffraction (XRD), thermogravimetric analysis (TGA), and scanning electron microscopy (SEM) are the most common tools used to characterize these materials, often in a context for studying structural and morphological changes that arise from aging or calcination. However, fundamental electronic and vibrational spectroscopic information is rather scarce. Here, we measured the visible and shortwave infrared (SWIR) diffuse reflectance, Fourier transform infrared (FTIR), fluorescence and Raman spectra of PuF4(s) and PuF4·xH2O(s) to obtain a better electronic and vibrational fingerprint. Our work provides clear indication of the polymeric structure of anhydrous PuF4, consistent with the Raman spectrum of UF4(s) and its hydrates. This is supplemented with XRD, TGA and SEM analysis. Findings in this study indicate that the spectra are modified by particle size, which in turn is influenced by synthetic technique.
Introduction
Since its discovery in 1940, plutonium (Pu) has held a unique position among the chemical elements. Though its destructive capabilities in nuclear weapons will live in infamy, plutonium and the chemical compounds that it forms are of fundamental importance for understanding the chemistry of the actinide series of elements. Despite this, chemical research on plutonium is relatively scarce compared to lighter actinides like uranium and thorium. This scarcity is largely due to a focus on the weaponization of Pu and is exacerbated by the health hazards and proliferation risks that are involved with handling and storing it.In the early days of plutonium research – not long after Pu was discovered – most studies were application-centric, with a clear focus on maximizing production of Pu metal to develop nuclear arsenals. During this process, plutonium tetrafluoride (PuF4(s)) was discovered to be one of the first plutonium-containing compounds to have utility, leading to its original development during World War II as an intermediate for Pu metal production. Most early studies of anhydrous PuF4 were metallurgical in nature and focused on optimizing fluorination conditions1 and engineering better metal recovery,2,3 with a clear aim to optimize production of Pu metal on an industrial scale.1–6 This narrow focus of weaponizing Pu provided few opportunities to study the fundamental properties of Pu and its chemical compounds. However, this changed after the collapse of the Soviet Union in 1991, which shifted Pu-based research away from weaponization and towards nuclear waste management and nonproliferation.
To date, the preponderance of Pu-based chemical research has focused on PuO2(s) due its prevalence in nuclear fuels7–12 and waste storage.13 By comparison, there is a dearth of modern studies pertaining to PuF4.14–22 This data gap is worsened by the structural instability of PuF4 and its hydrates. Many stockpiles of anhydrous PuF4 have degraded and become amorphous due to radiolytic decay, and most research facilities are no longer equipped to produce it, which is typically prepared by high temperature fluorination with highly corrosive HF gas.18 However, such work is important to understand periodic trends and improve modelling of actinide chemistry. As a general rule, periodic trends for actinide elements are difficult to establish theoretically, which is why experimental techniques such as Raman, infrared (IR), diffuse reflectance, nuclear magnetic resonance (NMR), electron paramagnetic resonance (EPR) and X-ray photoelectron spectroscopies (XPS) are quite useful. Practically, their non-destructive nature has made them well-suited for characterizing materials of importance to the nuclear fuel cycle, such as UF4 and its hydrates,23–31 PuO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14,32–41 and various forms of plutonium oxalate.14,37–40,42 Without foundational experimental data, computational studies of Pu can be quite cumbersome.43–49
14,32–41 and various forms of plutonium oxalate.14,37–40,42 Without foundational experimental data, computational studies of Pu can be quite cumbersome.43–49
In this manuscript, we utilize vibrational spectroscopy, X-ray diffraction, microscopy, and thermogravimetric analysis to provide an exquisite look at the structure and morphology of PuF4 and PuF4·xH2O, thus filling a longstanding knowledge gap, elucidating important trends in the actinide series of elements, and providing new data to improve the accuracy of actinide computational models.
Results and discussion
Structure and morphology
The diffraction pattern of anhydrous PuF4 (Fig. 1) is consistent with previous studies of PuF418,19 and isostructural to the anhydrous actinide fluorides NpF4 and UF4,16,50–52 both of which belong to the C2/c space group. Our pXRD data were initially fit to an NpF4 model53 and the unit cell parameters were refined to adjust for the slightly smaller Pu atoms. Refined unit cell parameters are not reported due to the uneven surface from our sample preparation. Our analysis resulted in a cell volume that was 14.82 Å3 smaller than NpF4, which is not unreasonable given that Pu4+ is smaller than Np4+.54 A broad feature around 15° 2θ was consistently measured in our PuF4 diffractograms but did not fit with our chosen structural model. This feature was not present in the diffraction pattern of the polyimide film that encased our sample. However, as our FTIR results show, this material is hygroscopic, which could explain the presence of this feature.
 | ||
| Fig. 1 The observed pXRD pattern of anhydrous PuF4 (black) is compared to a fit diffraction pattern refined from the known NpF4 structure (blue), while the difference plot is shown in red. | ||
In contrast, hydrated PuF4 is known to exist in at least two structurally distinct forms isostructural to the uranium variants—the simple cubic (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and orthorhombic (Pnma) geometries for PuF4·xH2O (0.5 ≤ x ≤ 2) and PuF4·2.5H2O, respectively.55–57 The 2.5 hydrate is generally stable at room temperature, but infrared measurements have shown that weakly bound water molecules are likely responsible for the variability of x in the cubic form.26 As shown in Fig. 2, our experimental diffractogram aligns with the published pattern reported by Dawson et al. (ICDD #00-034-0515) for the PuF4·xH2O structure.55 From a chemical perspective, the relatively simple diffractogram seems rather curious, as UF4·0.33H2O,27 UF4·1.33H2O58 and UF4·2.5H2O31,59,60 are all known to produce numerous diffraction bands that result from a complex structure.
m) and orthorhombic (Pnma) geometries for PuF4·xH2O (0.5 ≤ x ≤ 2) and PuF4·2.5H2O, respectively.55–57 The 2.5 hydrate is generally stable at room temperature, but infrared measurements have shown that weakly bound water molecules are likely responsible for the variability of x in the cubic form.26 As shown in Fig. 2, our experimental diffractogram aligns with the published pattern reported by Dawson et al. (ICDD #00-034-0515) for the PuF4·xH2O structure.55 From a chemical perspective, the relatively simple diffractogram seems rather curious, as UF4·0.33H2O,27 UF4·1.33H2O58 and UF4·2.5H2O31,59,60 are all known to produce numerous diffraction bands that result from a complex structure.
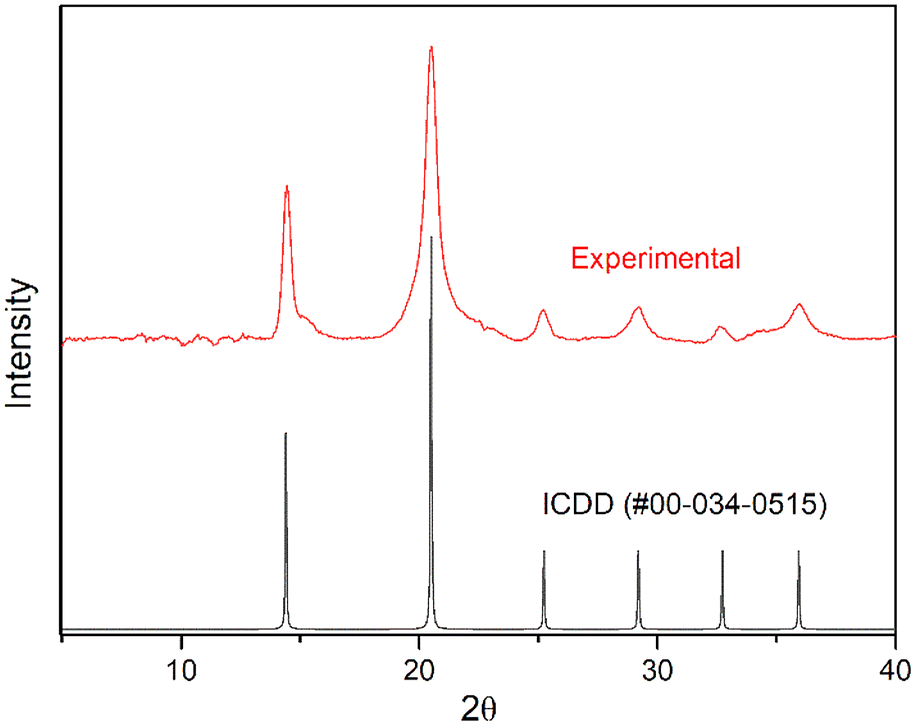 | ||
| Fig. 2 The experimental and ICDD (#00-034-0515) pXRD patterns of PuF4·xH2O. | ||
To supplement these results, SEM measurements were conducted on both hydrated and anhydrous PuF4. Micrographs of PuF4·xH2O (Fig. 3) reveal that our material contained agglomerates of rounded submicron particles, though the limited resolution hindered a quantitative description of size. The aggregated morphology indicates rapid precipitation from solution, which is supported by earlier solubility experiments.61 This was experimentally observed when our emerald-green Pu(NO3)4 solution promptly turned light pink upon the addition of concentrated HF. Synthetic efforts, including cooling reagents, reducing HF concentration, and reducing the rate at which HF was added to Pu(NO3)4, were attempted to reduce the rate of PuF4·xH2O precipitation and produce larger particles. Unfortunately, these attempts were unsuccessful.
 | ||
| Fig. 3 SEM-SE images of PuF4·xH2O (top) and SEM-CSE images of anhydrous PuF4 (bottom) at increasing magnification. | ||
In contrast, anhydrous PuF4 possessed larger particle sizes ranging from 5–10 μm in diameter. The particle surfaces were rough with pits and layered edges; such features have been observed after the calcination of Pu(IV) oxalate to PuO2.38 Elemental analysis of the material showed no evidence of discrete Pu or F rich regions. However, the anhydrous images (Fig. 3) are quite distinct from those reported on an aged powder produced at Los Alamos National Laboratory (LANL), which primarily consisted of crystalline aggregates 5–20 μm in length along their longest dimension.19 The differing morphologies likely originate from the precipitation and calcination steps, both of which have been shown to affect the size of PuO2 particulates.62–65
TGA was used to measure the mass loss of freshly synthesized PuF4·xH2O during heating (Fig. 4a). A total weight loss of 6.54 wt% was observed when heated to 800 °C under nitrogen. An initial weight loss of 5.4 wt% occurred from room temperature to 410 °C and is attributed to water removal, which could ostensibly originate from the lattice or surfaced-adsorbed molecules. For a pure lattice contribution, this would correspond with x = 1, which is consistent with pXRD results. Additionally, the dehydration is similar to what has been observed for anhydrous PuF4 with an 8% PuF4·1.6H2O crystalline phase.19 The remaining 1.1 wt% loss at 800 °C is assigned to the removal of weakly coordinated fluorine, which has been shown by both Wayne et al. and Dawson et al. to occur between 300 and 450 °C.19,55 Another possibility for the remaining mass loss could result from the reaction of PuF4 and PuO2 to produce O2 and PuF3.19,66
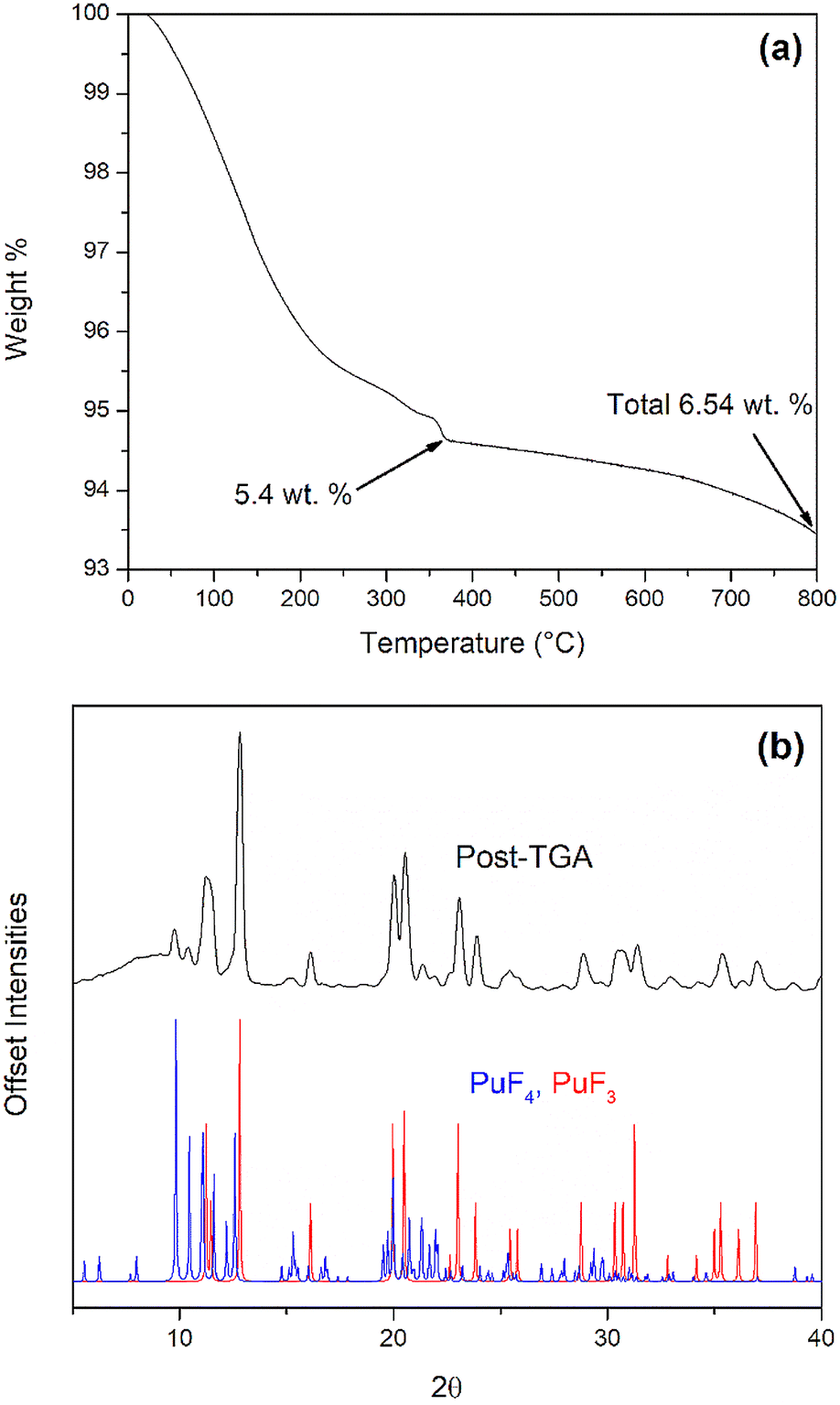 | ||
| Fig. 4 (a) Thermal decomposition of PuF4·xH2O under nitrogen heated to 800 °C at a ramp rate of 5 °C min−1. (b) ICDD patterns for PuF4 (#00-041-1218, blue) and PuF3 (#00-006-0327, black) in comparison to post-TGA PuF4·xH2O. | ||
The production of both PuF4 and PuF3 at high temperatures is further supported by pXRD results of the post-TGA PuF4·xH2O (Fig. 4b). This is an important observation, as previous literature work on this matter is unclear. For example, under vacuum at 10−4 Torr, Dawson et al. observed a PuF3 and PuF4 mixture when PuF4·xH2O is heated to 550 °C. For a second sample under vacuum Dawson et al. reported PuF3 and PuO2 when heating to 900 °C.56 The same products were produced for a hydrate when heated to 300 °C in an N2 atmosphere.55 It was suggested that dehydrated PuF4 could react with liberated water to produce PuO2, which in turn reacts with PuF4 to produce PuF3. In contrast, Wayne et al. did not observe the formation of PuF3; this is likely due to the use of open crucibles under an Ar atmosphere, which didn't allow for back-reaction with evolved gases.19 PuO2 has also been reported as a byproduct of heating PuF4·xH2O above 300 °C in vacuum or dry nitrogen,19,55 though its presence can usually be attributed to crystalline impurities inherent to the sample.18,19 No impurities were found in our sample; however, it is possible undetected impurities reacted with PuF4.
Raman spectroscopy
For the first time, Raman spectroscopy was used to elucidate the structures of PuF4 and PuF4·xH2O. Generally, it is challenging to measure high-quality spectra for metal fluorides because the intensity of Raman bands depend on bond polarizability, and metal–fluorine bonds are weakly polarizable. Fluorescence complicates matters, and has been shown to further obscure signals from UF4·0.33H2O, U3F12·H2O, and UF4·2.5H2O.24,27,31 We worked under the assumption that both PuF4 and its hydrate possess a complex polymeric structure, similar to UF4 and UF4·2.5H2O.50,59,67The Raman spectrum of PuF4·xH2O (Fig. 5a) primarily features a broad band centered around 400 cm−1 and several low intensity bands. The broad feature around 400 cm−1 likely results from the small particle morphology of PuF4·xH2O, as Raman spectra can be affected by phonon confinement68 leading to broad, asymmetric band shapes. Thus, our Raman measurements corroborate the SEM findings.
 | ||
| Fig. 5 (a) Raman spectra of PuF4·xH2O measured at 488 nm (blue) and 633 nm (red). (b) The 488 nm spectrum (blue) measured at 200 μW compared to another measured at 2 mW (black). Both plots are given a dual y-axis (intensities) for the convenient comparison of spectral features. | ||
Two challenges were encountered when measuring the Raman spectrum of PuF4·xH2O. First, naturally weak Raman scattering could not be enhanced by increasing laser power since the material was susceptible to decomposition and oxidation upon heating, even with long-term exposure to a low-powered laser. This is unsurprising, as submicron particles are known to undergo laser heating at powers as low as 1 mW. This phenomenon is show in Fig. 5b. At 2 mW, our samples of PuF4·xH2O had thermally decomposed to PuO2 nanoparticles, as evidenced by the observation of a broad (FWHM = 20 cm−1) band centered at 465 cm−1, with a weaker shoulder centered near 600 cm−1. Typically, PuO2 calcined at 450 °C displays a relatively sharp band at 476 cm−1 and a broad, weak band at 580 cm−1 which correspond to the T2g (1LO1) and 1LO2 bands of PuO2, respectively.32–35,69,70 In this case, the FWHM broadening and shift to lower frequencies can be explained by phonon confinement effects and crystallite dimensions of less than 10 nm. Consequently, very low laser powers (<250 μW) and long acquisition times were employed to avoid conversion of both PuF4 and PuF4·xH2O to PuO2.
The second challenge with measuring the Raman spectrum of PuF4·xH2O is wavelength-dependent fluorescence, which obscures vibrational modes. To determine which spectral features were Raman bands and which were fluorescence, the Raman spectrum of PuF4·xH2O was measured at 457, 488, 514, and 633 nm laser excitation wavelengths. Spectral features that were reproducible at different excitation wavelengths were labeled and assigned as Raman bands in Fig. 5a, while those shifted in position with respect to excitation wavelength were ascribed to fluorescence (Fig. 6a). Raman bands for PuF4·xH2O were located near 187, 240, 403 and 474 cm−1, which are in the same spectral region as UF4 metal–fluorine bands (70 cm−1 to 650 cm−1). These low intensity peaks were superimposed on a baseline rising towards the laser excitation wavelength; it is likely that this effect arises ostensibly not from fluorescence, but the significant number of low-frequency modes originating from F–F polymeric interactions and hydrogen bonding from water. Such observations have been made for UF4 and UF4·2.5H2O.24,31
 | ||
| Fig. 6 (a) Fluorescence spectra of PuF4·xH2O measured with 457 (black), 488 (red), and 514 (blue) nm excitation wavelengths, with respective powers of 444, 172 and 172 μW. (b) Unpublished fluorescence spectra of UF4 (black), UF4·0.33H2O (red) and UF4·2.5H2O (blue) taken at 325 nm. | ||
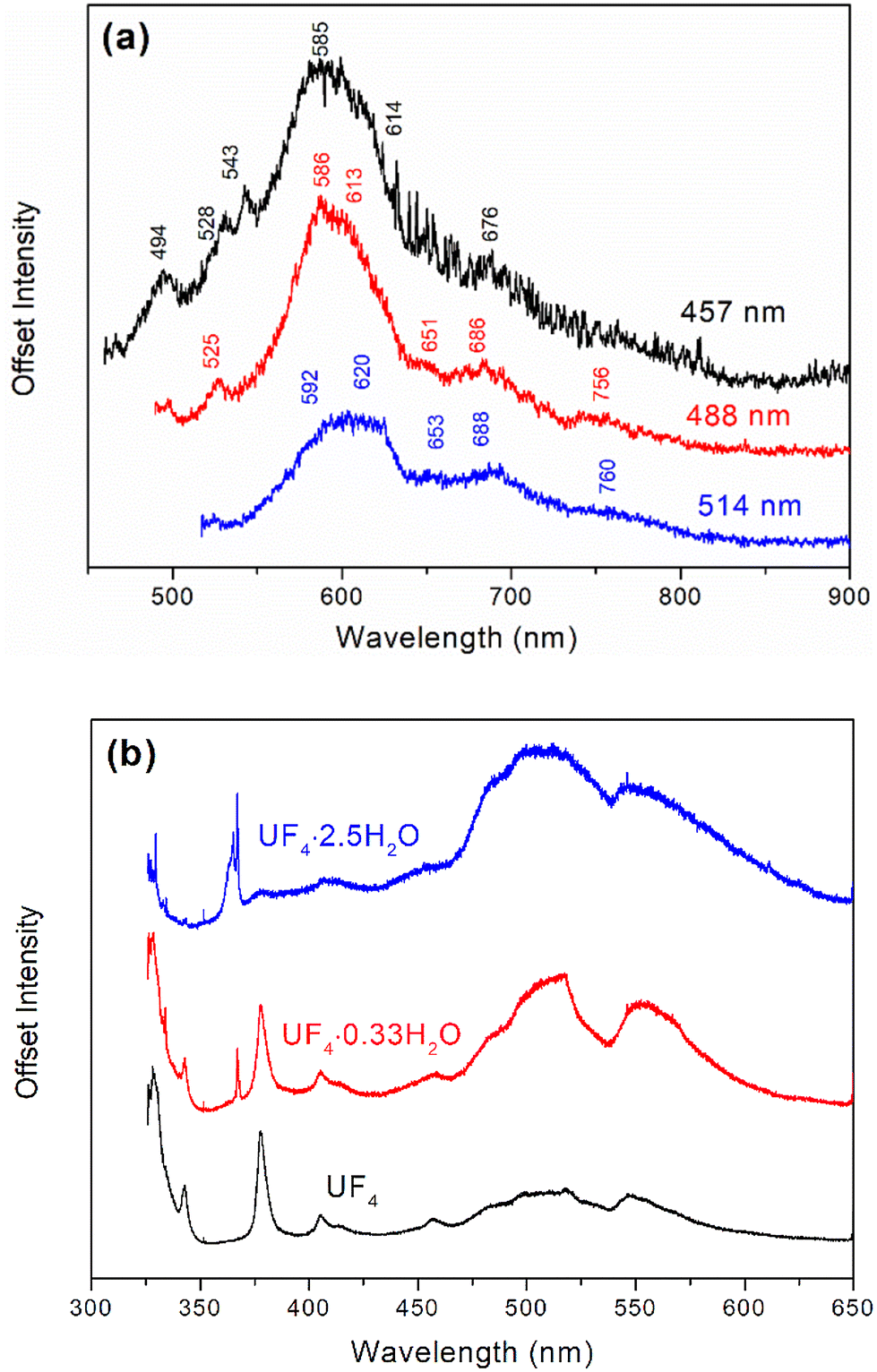
Fig. 6a and Table 1 show the fluorescence spectrum and peak positions, respectively, for PuF4·xH2O at 457, 488, and 514 nm. The large number of fluorescent features is quite interesting as fluorescent signatures are often used to rapidly assess chemical composition in nuclear forensics, such as for UF4 particulates.24,71 Fluorescence spectra collected at different wavelengths possessed a common peak and weak shoulder centered near 600 and 685 nm, respectively. The independence of overall band structure with excitation wavelength indicates that these peaks result from electronic transitions, as would be expected from the Vavilov rule. Larger energy transitions, observed at 457 and 488 nm, may have originated from otherwise inaccessible vibronic states.
| Fluorescent spectral bands | |||
|---|---|---|---|
| Excitation | 457 | 488 | 514 |
| Bands | 494 | ||
| 528 | 525 | ||
| 543 | |||
| 585 | 586 | 592 | |
| 614 | 613 | 620 | |
| 651 | 653 | ||
| 676 | 686 | 688 | |
| 756 | 760 | ||
Fluorescence generally indicates a preferred relaxation path for excited energy levels. As shown in Fig. 6b, UF4 and its hydrates have many identical features. This strongly suggests that the origin of the signal is based on the electronic states of U(IV), remaining largely uncoupled from its molecular structure. By analogy, we hypothesize that the fluorescence observed for PuF4·xH2O arises from different electronic transitions of Pu(IV). The relative strength of the fluorescence bands relative to the Raman could help in the detection and analysis of PuF4·xH2O.
The Raman spectrum of anhydrous PuF4 (Fig. 7) was markedly different than PuF4·xH2O and allowed for a more direct comparison with previously published spectra for UF4. As shown in Fig. 3, anhydrous PuF4 particles were much larger than PuF4·xH2O, which prevented phonon confinement and spectral broadening. Further, the larger particles of PuF4 were less susceptible to laser-induced heating during Raman measurements, enabling spectral acquisition using a laser power of to 500 μW, as opposed to 250 μW for PuF4·xH2O. However, long term exposure to laser power still results in the formation of PuO2, as it does for the hydrate. This work has identified at least 15 Raman bands between 50 cm−1 and 400 cm−1 and are listed in Table 2. As shown in Fig. 7b, the spectra of PuF4 and UF4 share many similarities. The numerous bands in the low frequency region are typical for a fluorine polymeric structure dominating a Raman spectrum24,27,28,31 and cannot be modeled on the basis of a tetrahedral monomer surrounding Pu.67
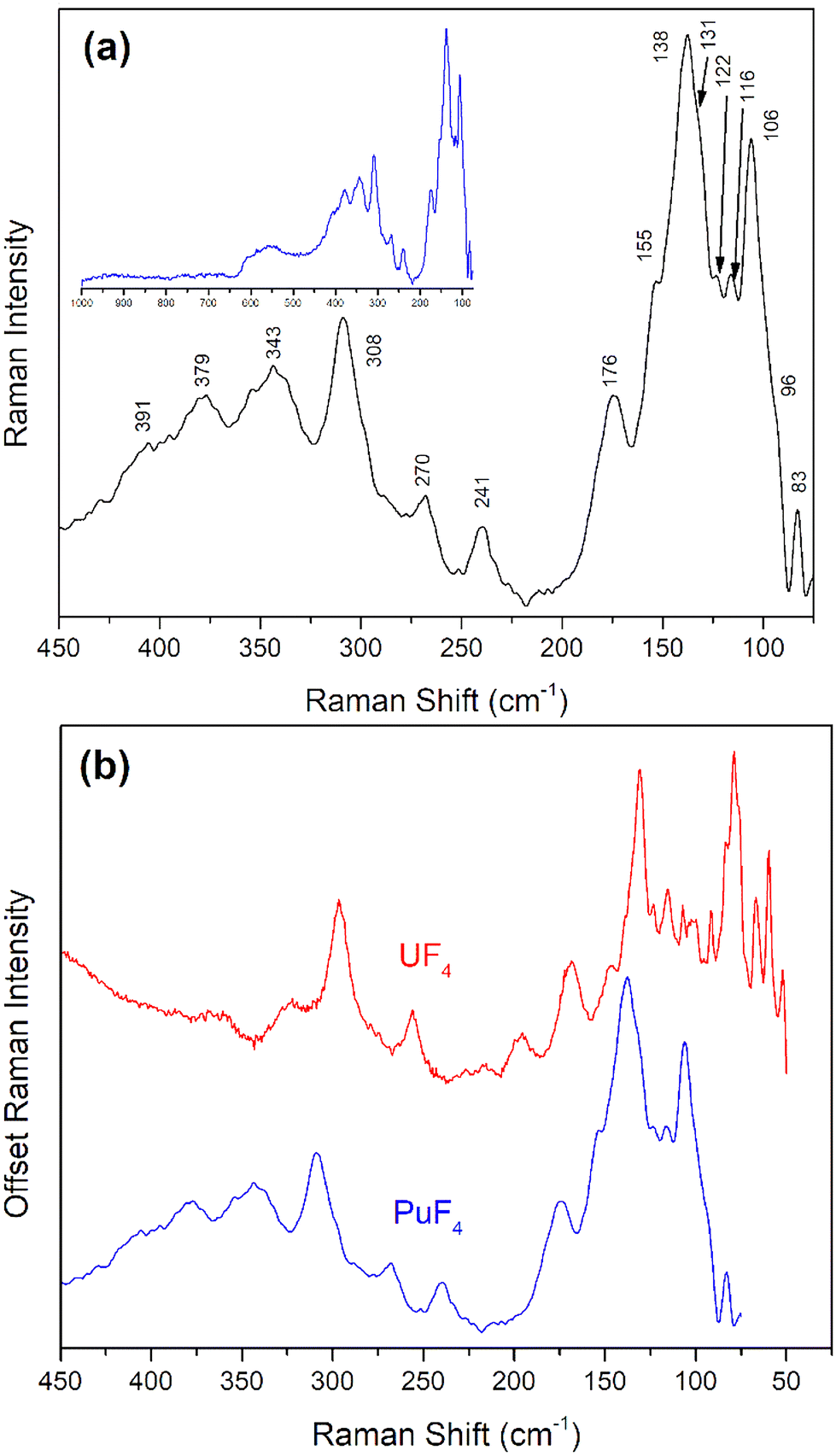 | ||
| Fig. 7 (a) Raman spectrum of anhydrous PuF4 measured with an excitation wavelength of 514 nm with bands labeled in the region of interest. The blue inset displays the full spectral range from 50–1000 cm−1. (b) A comparison of the anhydrous variants of UF4 (red)24 and PuF4 (blue) Raman spectra taken at 514 nm. Plot is given a dual y-axis for convenient comparison of spectral features. | ||
24 and UF4·0.33H2O.27 Save for the cubic PuF4·xH2O, the crystallographic structures are all similar and belong to C2/c
| Raman spectral bands | |||
|---|---|---|---|
| UF4 | PuF4 | UF4·0.33H2O | PuF4·xH2O |
| 50.9 | |||
| 59.4 | |||
| 66.8 | 61.7 | ||
| 78.9 | 82.7 | 76.6 | |
| 84.8 | |||
| 91 | 96.3 | ||
| 93.3 | |||
| 96.8 | |||
| 101.3 | 102 | ||
| 107.2 | 105.8 | 108 | |
| 115.9 | 116.2 | 118 | |
| 122.5 | 125 | ||
| 131.4 | 130.6 | 132 | |
| 138.1 | 141 | ||
| 148.5 | |||
| 155.1 | |||
| 170.4 | 175.6 | 175 | |
| 187 | |||
| 197.3 | |||
| 236 | |||
| 240.7 | 241 | ||
| 255.8 | |||
| 270.2 | |||
| 296.1 | |||
| 307.7 | 307 | ||
| 322.4 | |||
| 332 | |||
| 342.5 | |||
| 360.8 | 363 | ||
| 378.5 | 382 | ||
| 391 | |||
| 403 | |||
| 474 | |||
| 603.6 | |||
| 637 | |||
| 3532 | |||
| 3602 | |||
Infrared spectroscopy
FTIR was used to gain a more complete picture of the vibrational modes of the water molecules absorbed in PuF4 and PuF4·xH2O and are shown in Fig. 8, while the peak positions are listed in Table 3. These measurements are interesting since most infrared measurements on solid-state Pu(IV) samples are limited,38,42,72,73 with most of literature focusing on the analysis of aqueous solutions in the presence of species such as ClO4−, Cl−, NO3− and HNO3.74 | ||
| Fig. 8 Room-temperature FTIR spectra of (a) anhydrous PuF4 and (b) PuF4·xH2O taken at room temperature. | ||
Generally, actinide–fluoride bonds are not expected to possess IR active modes at frequencies greater than 500 cm−1. This is best reflected by the FTIR spectrum of anhydrous UF4, which has been shown to have no active vibrational mode between 500–4000 cm−1.31 This result is supported by DFT calculations, which predicted no IR-active modes for UF4 above 400 cm−1.23 Therefore, it is expected that the peaks for PuF4·xH2O and PuF4 displayed in Fig. 8 are related to water, OH stretches and low-frequency modes. The water band features in the anhydrous spectrum suggests either the presence of natural impurities (e.g., a small contribution of PuF4·xH2O within the anhydrous lattice) or an active sorption process, presumably from adsorbed water molecules or an isostructural hydrate with x < 0.5.
The sharp bands at 3593 cm−1 and 3527 cm−1 observed in the anhydrous spectrum of PuF4 (Fig. 8a) are superimposed on a broad band indicative of free OH experiencing different chemical environments within the crystal lattice,31 while the broad band itself suggests the presence of hydrogen bonding and polymeric fluorine bonding modes. The long tail from hydrogen/fluorine bonding has been observed in IR spectra of UO2F2-(H2O)x(HF)y75–78 and water with significant HF interactions.79 The asymmetry of the OH region likely results from broad, overlapping hydrogen-bonded OH peaks in slightly different chemical environments.31 Similar spectral features have been observed for UF4·0.33H2O27 and UF4·2.5H2O.31 Multiple peaks likely arise from OH groups weakly interacting with oxygen or fluorine neighbors.
At 1612 cm−1, there is another sharp peak overlayed on a weak asymmetric feature. The sharp band suggests an HOH bending mode in a specific configuration, while the weak broader asymmetrical component could be evidence of HOH bending in multiple chemical environments. A comparison to IR data from Khanaev et al.26 suggests that PuF4·0.33H2O may have been the hydrate in the sample prepared at PNNL and analyzed at SRNL. Though the means of H2O introduction to the sample remains unknown, it can be noted that PuF4 was synthesized in the arid environment in which PNNL is located. It is possible that anhydrous PuF4 absorbed most of the water once exposed to the more humid environment experienced at SRNL. Further research is warranted to examine whether the environment in which PuF4 is synthesized or handled can answer material provenance questions relevant to nuclear forensics.
The peaks for PuF4·xH2O (Fig. 8b) are related to water molecules in different chemical environments corresponding to OH stretches, H2O bending modes and low-frequency modes. FTIR data has been described in detail for PuF4·xH2O (x = 2.3, 0.9, 0.6 and 0.3) and closely matches the results from our material.26Table 3 compares our results to the x = 2.3 and x = 0.3 variants. The sharp shoulder peak at 3739 cm−1 is likely related to a free OH functional group; these usually have limited interactions with fluorine or water due to steric effects, resulting in shaper, higher-frequency bands with a FWHM < 50 cm−1 when compared with more strongly interacting OH groups. This contrasts with the broad, lower frequency peak at 3457 cm−1 seen in PuF4·xH2O, which is much more typical for a hydrogen-bonded OH group.27 Finally, an HOH bending mode common for hydrated lattices can be observed at 1654 cm−1.26,27 Comparisons with the UF4 hydrates strongly suggest that the water/PuF4 ratios are higher for PuF4·xH2O than for UF4·2.5H2O.31 Though this would contradict the TGA results, the hydrated PuF4 structure is based on submicron particulates and likely affected by surface properties. Considering this, it is very likely that this sample could have sorbed water between measurements.
Diffuse reflectance spectroscopy
Diffuse reflectance spectroscopy (DRS) is a useful tool for analyzing the local electronic structure of actinides, including plutonium tetrafluoride. While both Raman and infrared spectroscopy help elucidate local site symmetry, DRS is advantageous because of its sensitivity to electronic metal–ligand interactions. This allows it to provide detailed information based on the splitting of Laporte-forbidden f–f transitions due to the influence of the crystal field,14,15,80–82 making it highly sensitive to changes in the lattice symmetry resulting from alterations to oxidation state or structure.83 Its immunity to fluorescence, which often stymies the acquisition of Raman spectra, makes it an attractive technique.The room-temperature DRS spectra for both PuF4 and PuF4·xH2O are displayed in Fig. 9. These measurements are significant, as the available modern literature for diffuse reflectance on solid-state Pu complexes is limited, focusing instead on Pu-based coordination complexes arising from aqueous solvents,84–86 organic solvents,87–92 ionic liquids93 and solid-state melts.94 In the solid state, the primary interest in DRS has been to investigate the solubility and oxidative stability of Pu(IV) in solid matrices to assess their suitability for waste storage.95–98 Only recently have the oxalates and oxides been scrutinized with modern instrumentation.37,86
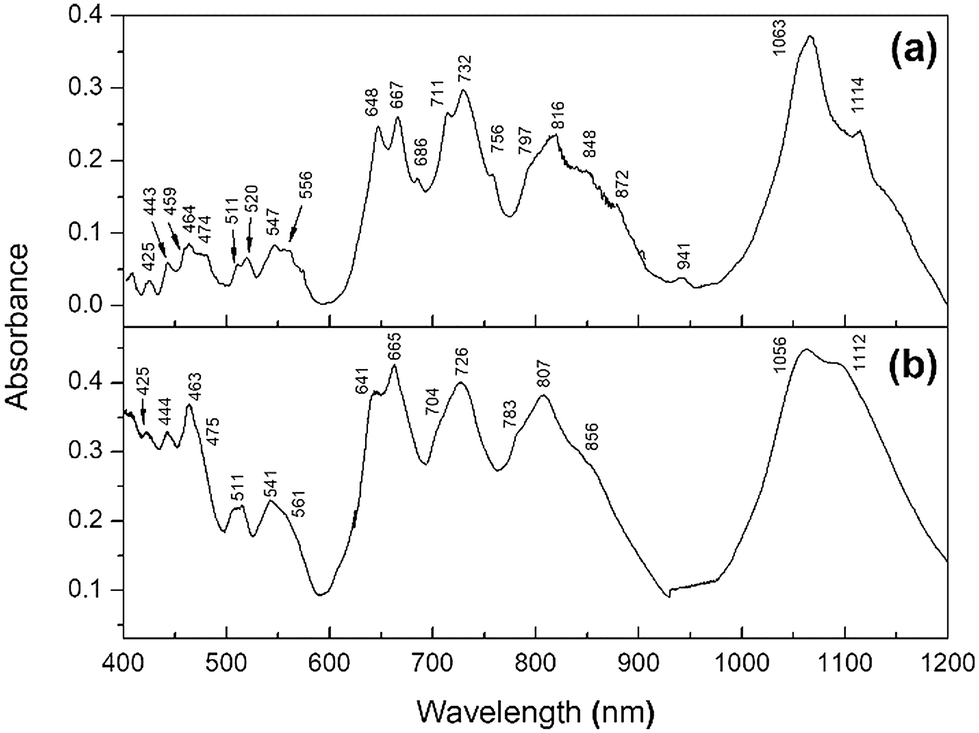 | ||
| Fig. 9 DRS spectra in the visible and shortwave infrared (SWIR) of (a) PuF4 and (b) PuF4·xH2O taken at room temperature. | ||
Each spectrum features a complex combination of several relatively broad and sharp features from 400–1200 nm. Despite their known differences in crystallographic symmetry, many of the spectral features and positions of PuF4·xH2O were shared with anhydrous PuF4, with the primary difference being in the broadness of the peaks, especially below 600 nm. The presence of water in the hydrate allows for greater overlap between the electronic bands resulting in peak broadening. The broadening of peaks observed in the hydrate could also be reflective of our SEM and XRD results; given that it is more amorphous than PuF4, and that DRS is sensitive to the degree of crystallinity, the DRS spectrum of the hydrate should be expected to possess broader peaks than the anhydrous variant. The relative intensity increases for the hydrate peaks, such as at 464 and 1114 nm are not currently understood, but could be explained by changes in the local symmetry, since the crystal-field plays an important role in transition probability.
Though not exhaustive, the DRS peaks are listed in Table 4 and compared to literature values. This includes the room-temperature DRS spectrum of plutonium tetrafluoride measured by Hobart et al. for a presumably anhydrous sample from 400–700 nm (ref. 14) and is shown in the inset of Fig. 10. The spectral features in the measured range match well, indicating no changes to the oxidation state of Pu(IV). The offset in peak position could be attributed to calibration issues. Given that the peak positions/intensities best match anhydrous PuF4 rather than PuF4·xH2O, our work clarifies that the original measurement was likely performed on an anhydrous sample. We also identified an additional peak at 686 nm and extended the range of the original measurement.
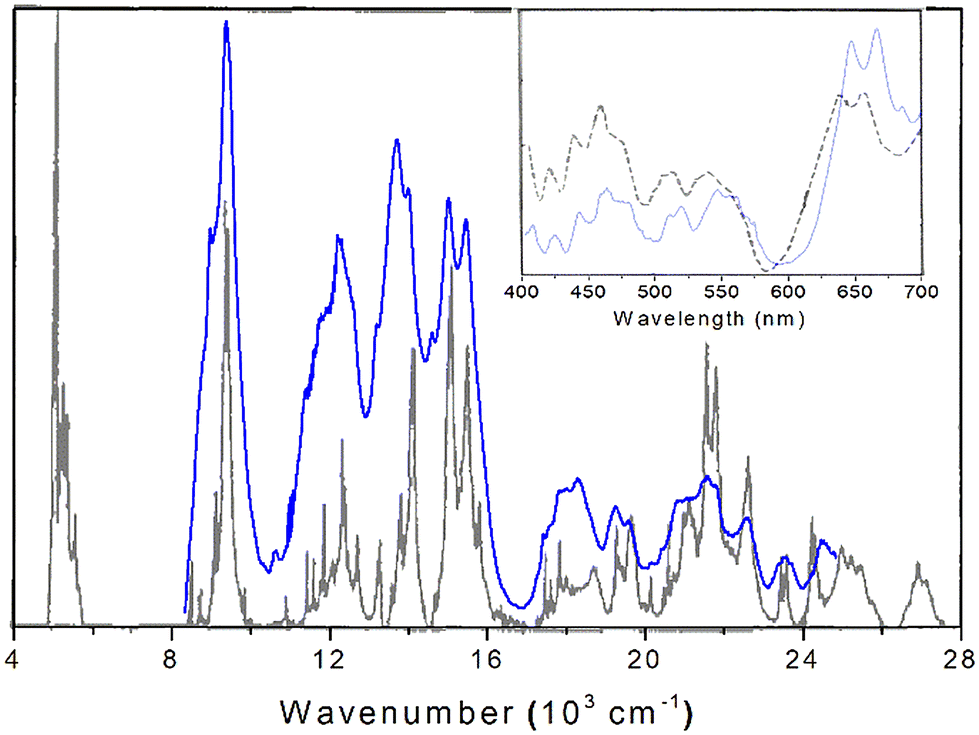 | ||
| Fig. 10 DRS spectra of anhydrous PuF4 at room temperature (blue), compared with its absorbance measured at 4 K (black, reproduced from ref. 15 with the permission of AIP Publishing). The inset depicts another comparison of our PuF4 DRS spectrum to an earlier one made at room temperature.14 | ||
| PuF414 |
PuF415 |
PuF4 | PuF4·xH2O |
|---|---|---|---|
| 421 | 426 | 425 | |
| 440 | 443 | 444 | |
| 459 | |||
| 460 | 464 | 464 | 463 |
| 470 | 473 | 474 | 475 |
| 506 | 509 | 511 | 511 |
| 513 | 519 | 520 | |
| 540 | 534 | 547 | 541 |
| 551 | 562 | 556 | 561 |
| 572 | 574 | ||
| 638 | 645 | 648 | 641 |
| 656 | 663 | 667 | 665 |
| 686 | |||
| 710 | 711 | 704 | |
| 724 | 732 | 726 | |
| 753 | 756 | ||
| 788 | 797 | 783 | |
| 808 | 816 | 807 | |
| 826 | |||
| 842 | 848 | 856 | |
| 860 | |||
| 873 | 872 | ||
| 921 | 941 | ||
| 1060 | 1063 | 1056 | |
| 1093 | |||
| 1114 | 1112 | ||
| 1170 |
Also worth mentioning is the experimental work of Carnall et al.15 measured the absorbance spectrum of anhydrous PuF4 pressed into a KBr pellet at 4 K from 360–2500 nm. Although band intensities might somewhat differ between absorbance and diffuse reflectance spectra, both provide the same information with regard to peak position and serves as a useful point of reference for the interpretation of our experimental results. Additionally, the low temperature of the measurement reduced spectral crowding and allowed for the identification of ground-state transitions. As seen in Fig. 10, there is good agreement in the absorption spectrum and our DRS results; peaks with similar central positions are listed in Table 4. Lower temperature measurements would be expected to cause further sharpening and splitting of the room temperature peaks.
Conclusions
The crystallinity, morphology and optical properties of anhydrous PuF4 and one of its hydrates, PuF4·xH2O, were studied with a variety of spectroscopic and microscopic techniques. Initial pXRD results showed a few peaks for PuF4·xH2O, as opposed to the more complex diffractogram expected for anhydrous PuF4 or the other hydrate, PuF4·2.5H2O. These results were augmented by SEM, which provided a clear difference in particulate dimensions prepared from the dry and wet methods. In particular, the wet method showed preferential submicron particulate formation of PuF4·xH2O resulting from rapid precipitation, in stark contrast to the large PuF4 agglomerates produced via dry synthesis. Additionally, TGA and post-XRD measurements clarified that the majority of mass lost from heating PuF4·xH2O occurs due to dehydration of the hydrate; an assumption of pure bonding and negligible adsorption would lead to an estimate of x = 1, which is consistent with pXRD results. The presence of PuF3 above 800 °C likely arises from the closed, non-oxidizing atmosphere of the TGA.For the first time, the Raman spectra of both PuF4 and PuF4·xH2O were recorded with several wavelengths. In particular, the spectrum of PuF4·xH2O was found to produce a poor signal, even when compared to UF4 and its hydrates. In addition to weak polarizability, broad bands and a rising baseline made interpretation quite difficult. A rising baseline likely arises from F–F agglomerates and hydrogen bonding from water, whereas peak broadness can be explained by phonon confinement of the small particulates. In contrast, anhydrous PuF4 had a much stronger response with sharper, well-defined Raman bands. Their spectral location closely mirrors UF4, representing a set of isostructural compounds (UF4, CeF4, etc.) where F–F vibrational modes dominate the Raman spectrum. The fluorescence spectra of PuF4·xH2O was also investigated and found to have bands analogous to those observed from UF4 and its hydrates, suggesting the presence of electronic transitions that could be used to identify PuF4 and its hydrates.
The water content was found to clearly impact the IR spectrum of both anhydrous PuF4 and PuF4·xH2O at higher energies, especially since no metal–fluoride peaks were expected below 500 cm−1. Bands corresponding to water bending or OH stretch modes in dry PuF4 suggest that water was sorbed during synthesis at PNNL or during handling in the humid environment at SRNL. Finally, DRS in the visible and shortwave infrared for both anhydrous PuF4 and PuF4·xH2O closely match earlier literature reports for PuF4. Save for peak broadening in PuF4·xH2O, the almost matching spectra indicate that excess water does not affect the electronic interactions between fluoride and Pu(IV) ions.
Experimental
Synthesis
All plutonium syntheses were performed inside Hazard Category II nuclear facilities that permit the safe handling of weapons-grade (WG) plutonium (>93% 239Pu). Anhydrous PuF4 was prepared at Pacific Northwest National Laboratory (PNNL) by first purifying WG Pu(IV) nitrate using anion exchange chromatography. The purified Pu was then precipitated as Pu(IV) oxalate through addition of oxalic acid. The oxalate was then calcined to PuO2 in air at 300 °C. The resulting PuO2 was then converted to PuF4 by reaction with anhydrous HF gas using the experimental set-up described by Casella et al.21 Briefly, PuO2 was heated to 300 °C in an air/O2 mixture. The atmosphere was then converted to an HF/O2 mixture, and the temperature was increased and held at 480 °C for 2 hours. The final product had a light pink color that was consistent with previous descriptions of PuF4. The product was subsequently analyzed by powder X-ray diffraction (pXRD) to determine purity and reaction yield.A hydrated sample of PuF4, henceforth referred to as PuF4·xH2O, was synthesized via low-temperature hydrofluorination of WG Pu. Specifically, 8.4 mL of 1 M HF was added to 21.45 mL of 0.098 M Pu(NO3)4(aq) to immediately yield a light pink solid that was dried overnight under a flow of argon. To limit the effects of radiolytic decay on chemical structure, all samples were analyzed readily after their production.
Powder X-ray diffraction
PuF4 was prepared by loading ∼1 g of powder into a 3D-printed sample holder with a μm thick polyimide film window. Powder X-ray diffraction data was collected using a Rigaku Ultima IV diffractometer equipped with a Cu sealed tube X-ray source and a 5° linear position sensitive detector on a 285 mm radius goniometer. Cu K-α X-rays were selected by use of a Ni filter. Diffraction data was collected from 5–159° 2θ in intervals of 0.02°. Data was analyzed using TOPAS version 6.99Diffractograms of PuF4·xH2O were collected with a Bruker Quazar single crystal X-ray diffractometer (SC-XRD) featuring monochromated Mo K-α radiation. The utilization of an SC-XRD as opposed to a powder X-ray diffractometer (pXRD) led to broader diffractograms, a feature primarily attributed to the lower resolution of the SC-XRD detector. The diffractometer had a 1024 × 1024 resolution and was positioned at a distance of 200 mm. Phi scans were conducted over 360° for a duration of up to 720 s, ranging from 4° to 54° 2θ, and diffractograms were obtained by integrating the resulting Debye rings. Diffractograms were obtained for freshly synthesized PuF4·xH2O both before and after subjection to thermogravimetric analysis (TGA). To address 2θ displacement, we employed lanthanum hexaboride (LaB6(s); Alfa Aesar, 99.5%) as an external standard. The International Centre for Diffraction Data (ICDD) database of crystal and powder X-ray diffraction100 was used for phase matching.
TGA
Thermogravimetric analysis (TGA) was used to provide information on PuF4·xH2O water content and probe chemical changes of hydrated PuF4 as a function of temperature. PuF4·xH2O (11.40 mg) was placed inside an alumina crucible and heated to 800 °C at a ramp rate of 5 °C min−1 and a sampling interval of 2 s pt−1. A balance flow rate of 50 mL min−1 and a sample flow rate of 80 mL min−1 was used during sample measurements. Increased flow rates were used to prevent corrosion of the instrument due to release of HF during heating.SEM
The imaging analysis for anhydrous PuF4 was performed using a Thermo-Fisher Inc., (Hillsboro, OR) Quattro Field Emission Gun (FEG) scanning electron microscope (SEM) equipped with a circular backscattered electron (CSE) detector and an iXRF Systems (Austin, TX) X-ray energy dispersive spectroscopy (EDS) detector. The system was modified to handle radioactive materials and the vacuum pumping system of the microscope was connected to a series of high efficiency filters to prevent the release of radioactivity to the environment. A beam energy of 20 keV was effective at revealing the light elements (F–K line) and the heavy element Pu–M line efficiently. The spatial resolution of the elemental mapping was unlikely to be better than 1 μm2.A JEOL JCM-6000 Plus Neoscope benchtop SEM was used to study the morphology of hydrated plutonium fluoride materials. Images were obtained using accelerating voltages ranging from 10 to 15 kV, with magnification reaching up to 5000× under secondary electron (SE) mode. To mitigate the hazards associated with handling dispersible plutonium, all sample materials were prepared within a negative pressure glovebox, and a minimal amount of plutonium powder was meticulously dispersed on carbon tape affixed to an aluminum stub. These stubs were tested for robustness and dispersibility then loaded out of the glovebox onto the benchtop.
Raman spectroscopy
Plutonium samples were contained within a double-walled containment cell containing BaF2 transmission windows for all vibrational spectroscopy and diffuse reflectance measurements. Raman and fluorescence spectra were acquired with a LabRAM HR800 (Horiba Jobin–Yvon) μ-Raman spectrometer equipped with an Andor iDus charge coupled device (CCD) detector (DU146A-LDC-DD). The detector had a 15 μm pixel resolution and a 2000 × 256 pixel array, and most experiments were conducted by binning the spectral array by a factor of two. The detector was maintained at a temperature of −92 °C with the aid of a water chiller and thermoelectric cooling. Excitation wavelengths of 457, 488, 514, and 633 nm were used, and spectra were processed using bandpass filters from Semrock Inc. The laser was focused onto the sample with a 50× objective and power at the sample ranged from 100–500 μW. Laser power was controlled by a half-wave plate and polarizer. Specular reflection from the sample was eliminated with ultra-steep, long pass edge filters produced by Semrock Inc., and an 1800 g mm−1 grating was used to disperse light onto the detector. Data acquisition periods ranged from 1–12 h. Labspec 5.78 software was used to control data acquisition parameters. For each integration time, at least two additional spectra were co-added to remove cosmic ray contributions. Fluorescence measurements were recorded using a 600 g mm−1 grating up to 900 nm with 457, 488, and 514 nm excitation for PuF4·xH2O samples. Acquisition periods for fluorescence measurements lasted approximately 5 minutes. Additional post-processing, including background subtraction, peak smoothing (10-point adjacent averaging), and peak fitting were conducted in OriginPro; all peaks were assumed to have a Lorentzian line shape.Infrared spectroscopy
Diffuse reflectance Fourier transform infrared spectroscopy (DRIFTS) was performed with a Continuum IR microscope coupled to a Nicolet 6700 spectrometer. A 15× Reflachromat objective from Thermo Fisher was used to focus light onto samples. A mercury cadmium telluride (MCT) detector, cooled with liquid nitrogen, was used to record the interferograms. Spectra were collected over a 800–7500 cm−1 spectral window with 4 cm−1 resolution. The BaF2 windows of the double-walled containment cell limited optical transmission to energies >800 cm−1. A data acquisition period consisted of the average of 2000 individual spectra, and multiple measurements were taken to ensure reproducibility. Baseline correction was performed with OriginPro software.Diffuse reflectance spectroscopy
An Olympus microscope coupled to an Andor Kymera 328i scanning spectrometer was used to perform diffuse reflectance spectroscopy (DRS) measurements. Illumination was provided with an Olympus quartz tungsten halogen (QTH) lamp with a spectral range of 400–2500 nm. Light was passed through a polarizer and focused onto the sample with an Olympus 20× objective. Reflected light polarized perpendicular to the incident light was collected with the objective at normal incidence, directed to a lens, focused into a broadband fiber optic with low OH content, and then directed into the Kymera 328i spectrometer. Three spectral regions were collected that used two different detectors and three different gratings. The first spectral range, 400–900 nm, was collected with an Andor Newton (DU920P-DU2) CCD detector and a 1200 g mm−1 grating blazed at 500 nm. The second spectral range, 600–1200 nm, was collected with and Andor iDus InGaAs-1.7 detector and 600 g mm−1 grating blazed at 1000 nm. The third spectral range, 930–1600 nm, was collected with an Andor iDus InGaAs-1.7 detector and a 600 g mm−1 grating blazed at 1200 nm. A longpass edge filter (900 nm) was used to block visible light that could result in second order reflections and produce false signals in the InGaAs detector. The spectral resolution was 1.0 nm. The system was calibrated, across the three spectral ranges, with the 404.7, 435.8, 576.96, 579.1, 696.5, 763.5, 811.5, 866.8, 912.3, 965.8, 1047.1, 1148.8, 1211.2, 1295.7, 1331.3, 1529.9 nm lines of a HgAr lamp. Spectralon placed in the double-walled cell was used to collect a reference spectrum, and Andor Solis software was used to collect absorption data. The three regions were then stitched together to construct the spectrum. Manual adjustments to the absorbance were unnecessary.Author contributions
Jared S. Kinyon: writing – original draft, writing – review and editing, visualization. Eliel Villa-Aleman: writing – original draft, writing – review and editing, data curation, conceptualization. Elodia Ciprian: writing – review and editing, data curation. Amy E. Hixon: writing – review and editing, resources. Bryan J. Foley: writing – review and editing, resources. Jonathan H. Christian: writing – review and editing, funding acquisition, project administration, conceptualization, resources. Jason R. Darvin: writing – review and editing, data curation, investigation. Don D. Dick: writing – review and editing, data curation. Amanda J. Casella: writing – review and editing, resources. Lucas E. Sweet: writing – review and editing, visualization, data curation, formal analysis. Edgar C. Buck: writing – review and editing, data curation, investigation. Forrest D. Heller: writing – review and editing. Aaron D. Nicholas: writing – review and editing. Cody A. Nizinski: writing – review and editing, data curation, investigation. Richard A. Clark: writing – review and editing.Data availability
Detailed datasets, including raw data and processed results, are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Office of Defense Nuclear Nonproliferation Research and Development within the U.S. Department of Energy's National Nuclear Security Administration for the synthesis of PuF4 and material characterization. This work was produced by Battelle Savannah River Alliance, LLC under Contract No. 89303321CEM000080 and/or a predecessor contract with the U.S. Department of Energy. Publisher acknowledges the U.S. Government license to provide public access under the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan). The United States Government retains and the publisher, by accepting this article for publication, acknowledges that the United States Government retains a non-exclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for United States Government purposes.References
- I. B. Johns and G. H. Moulton, Large-scale Preparation of the Anhydrous Fluorides of Plutonium. Report LA–193, Los Alamos Scientific Laboratory, Los Alamos, NM, 1944.
- R. D. Baker, Preparation of plutonium metal by the bomb method. Report LA–473, Los Alamos Scientific Laboratory, 1946.
- W. V. Conner, Process studies on the reduction of plutonium tetrafluoride to metal, Rocky Flats Div., Dow Chemical Co., Golden, Colo, 1966 Search PubMed.
- W. S. Moser and J. D. Navratil, Review of major plutonium pyrochemical technology, J. Less-Common Met., 1984, 100, 171–187 CrossRef CAS.
- I. B. Johns, Equilibrium Constants and Heats of Reaction for the Hydrofluorination of Uranium Dioxide and Plutonium Dioxide, United States, 1945 Search PubMed.
- D. L. Clark, et al., Plutonium, Springer, 2006 Search PubMed.
- M. Le Guellec, et al., Grain size analysis and characterization by Raman spectroscopy of a homogeneous sintered MOX fuel, J. Eur. Ceram. Soc., 2022, 42(16), 7553–7560 CrossRef CAS.
- Z. Talip, et al., Characterization of un-irradiated MIMAS MOX fuel by Raman spectroscopy and EPMA, J. Nucl. Mater., 2018, 499, 88–97 CrossRef CAS.
- C. Jegou, et al., Raman micro-spectroscopy of UOX and MOX spent nuclear fuel characterization and oxidation resistance of the high burn-up structure, J. Nucl. Mater., 2015, 458, 343–349 CrossRef CAS.
- C. G. Worley and G. J. Havrilla, Micro-X-ray Fluorescence Characterization of Mixed Oxide Fuel Surrogate Feed Material, Anal. Chem., 1998, 70(14), 2957–2963 CrossRef CAS.
- T. Abram and S. Ion, Generation-IV nuclear power: A review of the state of the science, Energy Policy, 2008, 36(12), 4323–4330 CrossRef.
- Status and Advances in MOX Fuel Technology, series=Technical Reports Series. 2003.
- L. Tandon, Radiolysis of Salts and Long-Term Storage Issues for Both Pure and Impure PuO2 Materials in Plutonium Storage Containers, United States, 2000 Search PubMed.
- D. E. Hobart, et al., Formation, characterization, and stability of plutonium(IV) colloid; A progress report, United States, 1989 Search PubMed.
- W. T. Carnall, et al., Analysis of the crystal–field spectra of the actinide tetrafluorides. I. UF4, NpF4, and PuF4, J. Chem. Phys., 1991, 95(10), 7194–7203 CrossRef CAS.
- S. Kern, et al., Temperature variation of the structural parameters in actinide tetrafluorides, J. Chem. Phys., 1994, 101(11), 9333–9337 CrossRef CAS.
- C. Capan, et al., Probing the Pu4+ magnetic moment in PuF4 with 19F NMR spectroscopy, Phys. Rev. B, 2016, 93(22), 224409 CrossRef.
- K. McCoy, et al., Radiation damage and annealing in plutonium tetrafluoride, J. Nucl. Mater., 2017, 496, 379–387 CrossRef CAS.
- D. M. Wayne, et al., X–ray diffraction, differential scanning calorimetry and evolved gas analysis of aged plutonium tetrafluoride (PuF4), J. Radioanal. Nucl. Chem., 2021, 329(2), 741–756 CrossRef CAS.
- E. D. Walter, et al., Measurement of local magnetic fields in actinide tetrafluorides, J. Chem. Phys., 2021, 154(21), 211101 CrossRef CAS PubMed.
- A. J. Casella, et al., In Stream Monitoring of Off-Gasses from Plutonium Fluorination, Actinide Res. Quart., 2019,(Second Quarter), 31–35 Search PubMed.
- L. E. Cox and J. D. Farr, 4f binding-energy shifts of the light-actinide dioxides and tetrafluorides, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39(15), 11142–11145 CrossRef CAS PubMed.
- A. Miskowiec, et al., Vibrational properties of uranium fluorides, Phys. B, 2019, 570, 194–205 CrossRef CAS.
- E. Villa-Aleman and M. S. Wellons, Characterization of uranium tetrafluoride (UF4) with Raman spectroscopy, J. Raman Spectrosc., 2016, 47(7), 865–870 CrossRef CAS.
- P. Gans, B. J. Hathaway and B. C. Smith, Diffuse reflectance spectra of some uranium(IV) complexes, Spectrochim. Acta, 1965, 21(9), 1589–1595 CrossRef CAS.
- E. I. Khanaev, E. G. Teterin and L. A. Luk'yanova, IR spectroscopic study of dehydration products of UF4 and PuF4 crystal hydrates in an anhydrous HF current, J. Appl. Spectrosc., 1967, 6(6), 533–538 CrossRef.
- M. A. DeVore, et al., Vibrational spectroscopy of uranium tetrafluoride hydrates, Vib. Spectrosc., 2021, 115, 103277 CrossRef CAS.
- B. J. Foley, et al., Probing the hydrolytic degradation of UF4 in humid air, Dalton Trans., 2022, 51(15), 6061–6067 RSC.
- S. N. Ghosh, W. Gordy and D. G. Hill, Paramagnetic Resonance in Uranium Salts, Phys. Rev., 1954, 96(1), 36–38 CrossRef CAS.
- A. Y. Teterin, et al., Electronic structure of solid uranium tetrafluoride UF4, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(4), 045101 CrossRef.
- J. H. Christian, et al., Characterizing the solid hydrolysis product, UF4(H2O)2.5, generated from neat water reactions with UF4 at room temperature, Dalton Trans., 2021, 50(7), 2462–2471 RSC.
- E. Villa-Aleman, et al., Raman microspectroscopy of PuO2 particulate aggregates, J. Nucl. Mater., 2019, 515, 140–149 CrossRef CAS.
- E. Villa-Aleman, et al., Raman spectroscopy: A tool to investigate alpha decay damage in a PuO2 crystal lattice and determining sample age since calcination, J. Raman Spectrosc., 2019, 50(6), 899–901 CrossRef CAS.
- E. Villa-Aleman, et al., Raman signatures from age-dating PuO2 since last calcination, J. Nucl. Mater., 2021, 551, 152969 CrossRef CAS.
- E. Villa-Aleman, et al., The electronic Raman scattering spectrum of PuO2, J. Raman Spectrosc., 2023, 54(3), 324–332 CrossRef CAS.
- E. Villa-Aleman, et al., Laser-induced annealing of aged PuO2, J. Raman Spectrosc., 2021, 52(8), 1486–1489 CrossRef CAS.
- E. Villa-Aleman, et al., Diffuse Reflectance Spectroscopy and Principal Component Analysis to Retrospectively Determine Production History of Plutonium Dioxide, Appl. Spectrosc., 2022, 77(5), 449–456 CrossRef PubMed.
- J. H. Christian, et al., Raman and infrared spectra of plutonium(IV) oxalate and its thermal degradation products, J. Nucl. Mater., 2022, 562, 153574 CrossRef CAS.
- J. H. Christian, et al., Probing the thermal decomposition of plutonium(III) oxalate with IR and Raman spectroscopy, X-ray diffraction, and electron microscopy, J. Nucl. Mater., 2023, 584, 154596 CrossRef CAS.
- N. Vigier, et al., Reaction mechanisms of the thermal conversion of Pu(IV) oxalate into plutonium oxide, J. Alloys Compd., 2007, 444–445, 594–597 CrossRef CAS.
- Y. A. Teterin, et al., Electronic structure and chemical bonding in PuO2, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(24), 245108 CrossRef.
- A. Karelin, et al., Thermal decomposition of Np(IV) and Pu(III, IV) oxalates, J. Radioanal. Nucl. Chem., 1990, 143(1), 241–252 CrossRef CAS.
- T. Vitova, et al., Exploring the electronic structure and speciation of aqueous and colloidal Pu with high energy resolution XANES and computations, Chem. Commun., 2018, 54(91), 12824–12827 RSC.
- P. S. Bagus, B. Schacherl and T. Vitova, Computational and Spectroscopic Tools for the Detection of Bond Covalency in Pu(IV) Materials, Inorg. Chem., 2021, 60(21), 16090–16102 CrossRef CAS PubMed.
- S. B. Isbill, et al., Computational insights into the lattice dynamics of Pu(IV) oxalates, J. Nucl. Mater., 2023, 573, 154106 CrossRef CAS.
- J. C. Wedal, F. Furche and W. J. Evans, Density Functional Theory Analysis of the Importance of Coordination Geometry for 5f36d1 versus 5f4 Electron Configurations in U(II) Complexes, Inorg. Chem., 2021, 60(21), 16316–16325 CrossRef CAS PubMed.
- C. J. South and L. E. Roy, Insights into the thermal decomposition of plutonium(IV) oxalate – a DFT study of the intermediate structures, J. Nucl. Mater., 2021, 549, 152864 CrossRef CAS.
- M. Pepper and B. E. Bursten, The electronic structure of actinide-containing molecules: a challenge to applied quantum chemistry, Chem. Rev., 1991, 91(5), 719–741 CrossRef CAS.
- J. C. Krupa, Optical excitations in lanthanide and actinide compounds, J. Alloys Compd., 1995, 225(1), 1–10 CrossRef CAS.
- A. C. Larson, R. B. Roof Jnr and D. T. Cromer, The crystal structure of UF4, Acta Crystallogr., 1964, 17(5), 555–558 CrossRef CAS.
- T. K. Keenan and L. B. Asprey, Lattice constants of actinide tetrafluorides including berkelium, Inorg. Chem., 1969, 8(2), 235–238 CrossRef CAS.
- L. B. Asprey and R. G. Haire, On the actinide tetrafluoride lattice parameters, Inorg. Nucl. Chem. Lett., 1973, 9(11), 1121–1128 CrossRef CAS.
- W. H. Zachariasen, Crystal chemical studies of the 5f-series of elements. XII. New compounds representing known structure types, Acta Crystallogr., 1949, 2(6), 388–390 CrossRef CAS.
- R. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767 CrossRef.
- J. K. Dawson, R. W. M. D'Eye and A. E. Truswell, The hydrated tetrafluorides of uranium and plutonium, J. Chem. Soc., 1954, 3922–3929 RSC.
- J. K. Dawson, et al., The preparation and some properties of plutonium fluorides, J. Chem. Soc., 1954, 558–564 RSC.
- W. H. Zachariasen, U. S. A. E. Commission and L. Argonne National, X-ray diffraction studies of fluorides of plutonium and neptunium: chemical identity and crystal structure, Atomic Energy Commission. MDDC, United States, 1396, Atomic Energy Commission, Oak Ridge, Tenn. 1947, 16 p Search PubMed.
- Y. V. Gagarinskii, et al., The crystal hydrate UF4·4/3 H2O, At. Energy, 1965, 18(1), 43–48 CrossRef.
- K. J. Pastoor, et al., Structural Characterization of Uranium Tetrafluoride Hydrate (UF4·2.5H2O), J. Phys. Chem. C, 2022, 126(31), 13256–13267 CrossRef CAS.
- S. V. Borisov and G. M. Zadneprovskii, The crystal structure of UF4·2.5H2O, At. Energy, 1971, 31(1), 761–763 CrossRef.
- C. J. Mandleberg and K. E. Francis, The solubility of PuF4 in Nitric acid solutions and the solubility product of PuF4, United Kingdom, 1952 Search PubMed.
- J. D. Moseley and R. O. Wing, Properties of plutonium dioxide, Rocky Flats Div., Dow Chemical Co., Denver, Colo, 1965 Search PubMed.
- X. Machuron-Mandard and C. Madic, Plutonium dioxide particle properties as a function of calcination temperature, J. Alloys Compd., 1996, 235(2), 216–224 CrossRef CAS.
- M. N. Myers, Thermal decomposition of plutonium(IV) oxalate and hydrofluorination of plutonium(IV) oxalate and oxide, General Electric Co. Hanford Atomic Products Operation, Richland, Wash, 1956 Search PubMed.
- P. K. Smith, et al., Effect of oxalate precipitation on PuO2 microstructures, United States, 1976, p. 11 Search PubMed.
- E. a. C. D. Y. Chudinov, At. Energ., 1970, 28, 151–153 CAS.
- M. Goldstein, R. J. Hughes and W. D. Unsworth, Vibrational spectra of some heavy metal teftafluorides in the solid state, Spectrochim. Acta, Part A, 1975, 31(5), 621–624 CrossRef.
- K. Roodenko, et al., Modified phonon confinement model for Raman spectroscopy of nanostructured materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(11), 115210 CrossRef.
- J. F. Corbey, et al., Quantitative Microstructural Characterization of Plutonium Oxalate Auto-Degradation and Evidence for PuO2 Nanocrystal Formation, Eur. J. Inorg. Chem., 2021, 2021(32), 3277–3291 CrossRef CAS.
- D. Hudry, et al., Ultra-Small Plutonium Oxide Nanocrystals: An Innovative Material in Plutonium Science, Chem. – Eur. J., 2014, 20(33), 10431–10438 CrossRef CAS PubMed.
- F. Pointurier, C. Lelong and O. Marie, Study of the chemical changes of μm-sized particles of uranium tetrafluoride (UF4) in environmental conditions by means of micro-Raman spectrometry, Vib. Spectrosc., 2020, 110, 103145 CrossRef CAS.
- L. M. Toth and H. A. Friedman, The IR spectrum of Pu(IV) polymer, J. Inorg. Nucl. Chem., 1978, 40(5), 807–810 CrossRef CAS.
- B. Arab-Chapelet, et al., Synthesis and characterization of mixed An(IV)An(III) oxalates (An(IV)=Th, Np, U or Pu and An(III)=Pu or Am), J. Nucl. Mater., 2008, 373(1), 259–268 CrossRef CAS.
- S. A. Bryan, et al., Spectroscopic monitoring of spent nuclear fuel reprocessing streams: an evaluation of spent fuel solutions via Raman, visible, and near-infrared spectroscopy, Radiochim. Acta, 2011, 99(9), 563–572 CrossRef CAS.
- D. P. Armstrong, W. D. Bostick and W. H. Fletcher, An FT-IR Study of the Atmospheric Hydrolysis of Uranium Hexafluoride, Appl. Spectrosc., 1991, 45(6), 1008–1016 CrossRef CAS.
- G. L. Wagner, et al., Morphologic and chemical characterization of products from hydrolysis of UF6, J. Fluorine Chem., 2015, 178, 107–114 CrossRef CAS.
- M. C. Kirkegaard, et al., Formation of a uranyl hydroxide hydrate via hydration of [(UO2F2)(H2O)]7·4H2O, Dalton Trans., 2019, 48(36), 13685–13698 RSC.
- K. Ohwada, Uranium-fluorine lattice vibration of uranyl fluoride, J. Inorg. Nucl. Chem., 1971, 33(6), 1615–1623 CrossRef CAS.
- R. K. Thomas and H. W. Thompson, Hydrogen bonding in the vapour phase between water and hydrogen fluoride: the infrared spectrum of the 1:1 complex, Proc. R. Soc. London, Ser. A, 1997, 344(1639), 579–592 Search PubMed.
- J. E. Lohr and G. Kortüm, Reflectance Spectroscopy: Principles, Methods, Applications, Springer Berlin Heidelberg, 2012 Search PubMed.
- J. C. Krupa and W. T. Carnall, Electronic structure of U4+, Np4+, and Pu4+ doped into ThSiO4 single crystal, J. Chem. Phys., 1993, 99(11), 8577–8584 CrossRef CAS.
- G. K. Liu, et al., Analysis of the crystal–field spectra of the actinide tetrafluorides. II. AmF4, CmF4, Cm4+:CeF4, and Bk4+:CeF4, J. Chem. Phys., 1994, 101(10), 8277–8289 CrossRef CAS.
- D. E. Hobart and K. Boland, Diffuse Reflectance Spectroscopy of Plutonium Solids, Actinide Res. Quart, 2011, 2, 37–41 Search PubMed.
- J. H. Matonic, B. L. Scott and M. P. Neu, High-Yield Synthesis and Single-Crystal X-ray Structure of a Plutonium(III) Aquo Complex: [Pu(H2O)9][CF3SO3]3, Inorg. Chem., 2001, 40(12), 2638–2639 CrossRef CAS PubMed.
- W. Runde, et al., Synthesis and structural characterization of a molecular plutonium(IV) compound constructed from dimeric building blocks, Chem. Commun., 2007,(17), 1728–1729 RSC.
- W. Runde, et al., Directed Synthesis of Crystalline Plutonium(III) and (IV) Oxalates: Accessing Redox-Controlled Separations in Acidic Solutions, Inorg. Chem., 2009, 48(13), 5967–5972 CrossRef CAS PubMed.
- S. D. Reilly, B. L. Scott and A. J. Gaunt, [N(n-Bu)4]2[Pu(NO3)6] and [N(n-Bu)4]2[PuCl6]: Starting Materials To Facilitate Nonaqueous Plutonium(IV) Chemistry, Inorg. Chem., 2012, 51(17), 9165–9167 CrossRef CAS PubMed.
- S. D. Reilly, et al., Synthesis and characterization of NpCl4(DME)2 and PuCl4(DME)2 neutral transuranic An(IV) starting materials, Dalton Trans., 2014, 43(4), 1498–1501 RSC.
- S. G. Minasian, et al., Synthesis and Structure of (Ph4P)2MCl6 (M = Ti, Zr, Hf, Th, U, Np, Pu), Inorg. Chem., 2012, 51(10), 5728–5736 CrossRef CAS PubMed.
- D. Rosario-Amorin, et al., Synthesis and Coordination Chemistry of Phosphine Oxide Decorated Dibenzofuran Platforms, Inorg. Chem., 2012, 51(12), 6667–6681 CrossRef CAS PubMed.
- S. D. Reilly, et al., Plutonium(IV) complexation by diglycolamide ligands—coordination chemistry insight into TODGA-based actinide separations, Chem. Commun., 2012, 48(78), 9732–9734 RSC.
- J. H. Matonic, et al., Synthesis and crystal structure of a ten-coordinate plutonium(IV) ion complexed by 2-[(diphenylphosphino)methyl]pyridine N,P-dioxide: [Pu(NO3)3{2-[(C6H5)2P(O)CH2]C5H4NO}2][Pu(NO3)6]0.5, J. Chem. Soc., Dalton Trans., 2002,(11), 2328–2332 RSC.
- S. I. Nikitenko and P. Moisy, Formation of Higher Chloride Complexes of Np(IV) and Pu(IV) in Water-Stable Room-Temperature Ionic Liquid [BuMeIm][Tf2N], Inorg. Chem., 2006, 45(3), 1235–1242 CrossRef CAS PubMed.
- R. F. Hess, et al., Synthesis and Structural Characterization of the First Quaternary Plutonium Thiophosphates: K3Pu(PS4)2 and APuP2S7 (A = K, Rb, Cs), J. Am. Chem. Soc., 2002, 124(7), 1327–1333 CrossRef CAS PubMed.
- Y. Zhang, E. R. Vance and T. McLeod, Diffuse reflectance spectroscopy of Np and Pu in zirconia and pyrochlore-structured Y2Ti2O7, J. Nucl. Mater., 2012, 420(1), 278–281 CrossRef CAS.
- Y. Zhang and E. R. Vance, Diffuse reflectance spectroscopy of tetravalent neptunium and plutonium ions in ThO2, J. Nucl. Mater., 2008, 374(1), 192–196 CrossRef CAS.
- D. J. Gregg, et al., The incorporation of plutonium in lanthanum zirconate pyrochlore, J. Nucl. Mater., 2013, 443(1), 444–451 CrossRef CAS.
- Y. Zhang and E. R. Vance, Plutonium in monazite and brabantite: Diffuse reflectance spectroscopy study, J. Nucl. Mater., 2008, 375(3), 311–314 CrossRef CAS.
- TOPAS, Bruker AXS software package Copyright 1999. 2016 Search PubMed.
- S. Gates-Rector and T. Blanton, The Powder Diffraction File: a quality materials characterization database, Powder Diffr., 2019, 34(4), 352–360 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |