
An asymmetrically coordinated ZnCoFe hetero-trimetallic atom catalyst enhances the electrocatalytic oxygen reaction†
Changli
Chen‡
ab,
Jing
Chai‡
c,
Mengru
Sun‡
b,
Tianqi
Guo
*d,
Jie
Lin
e,
Yurong
Zhou
d,
Zhiyi
Sun
b,
Fang
Zhang
f,
Liang
Zhang
 *c,
Wenxing
Chen
*b and
Yujing
Li
*b
*c,
Wenxing
Chen
*b and
Yujing
Li
*b
aSchool of Chemistry and Chemical Engineering, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250353, P. R. China
bEnergy & Catalysis Center, School of Materials Science and Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: wxchen@bit.edu.cn; yjli@bit.edu.cn
cCenter for Combustion Energy, School of Vehicle and Mobility, State Key Laboratory of Intelligent Green Vehicle and Mobility, Tsinghua University, Beijing 100084, China. E-mail: zhangbright@tsinghua.edu.cn
dInternational Iberian Nanotechnology Laboratory (INL), Braga 4715-330, Portugal. E-mail: tianqi.guo@inl.int
eNingbo Institute of Materials Technology and Engineering, Chinese Academy of Science, Ningbo 315201, China
fAnalysis and Testing Center, Beijing Institute of Technology, Beijing 100081, P. R. China
First published on 15th February 2024
Abstract
Synthesizing heterometal atomic sites with asymmetric coordination structures is of great significance for improving the electrocatalytic performance of atomically dispersed catalysts, yet it is also a challenge. Herein, an unusual ZnCoFe hetero-trimetallic atom site is elaborately developed with the nitrogen-coordinated Co and Zn atoms adjacent to the sulfur/nitrogen dual-coordinated Fe atoms (ZnN3CoN3FeN2S) anchored in sulfur/nitrogen-doped carbon via a simple two-step wet chemistry strategy based on a metal–organic framework (MOF) and a post-coordination process. The ZnCoFe-TAC/SNC shows the smallest ΔE of 0.676 V, indicating an outstanding bifunctional catalytic activity. Furthermore, a ZnCoFe-TAC/SNC-based Zn–air battery displays high peak power density (304 mW cm−2) and specific capacity (760 mA h g−1). The in situ XAS results show that Co is the main active site and Fe is a co-catalytic site. Zn acts as an “electron regulator” to regulate the electron structures around the catalytic sites. Density functional theory (DFT) calculations further reveal the synergetic effect of the interactions among Zn, Co, and Fe metal atoms on the catalytic performance. This work provides a universal insight into the controllable synthesis of trimetallic atom catalysts and a proposal for regulating the performance in energy conversion and catalytic applications.
Broader contextSingle-atom catalysts represented by non-noble metal-based materials show comparable activities with expensive Pt or Ir/Rh-based catalysts in the oxygen reduction reaction and the oxygen evolution reaction. However, achieving an optimum charge redistribution is difficult because of the high free energy of the metal single atoms, which limits the content of the metal atoms. In this study, we developed an asymmetrically coordinated hetero-trimetallic atom catalyst (TAC) consisting of ZnN3CoN3FeN2S active sites by a simple two-step wet chemistry strategy. The experimental and theoretical results suggest the electronic synergy of Zn, Co and Fe, optimizing the adsorption/desorption of oxygenated intermediates and thereby accelerating the reaction kinetics. As a result, ZnCoFe-TAC/SNC exhibits competitive ORR/OER activity with a superior ultrasmall ΔE of 0.676 V. Moreover, rechargeable Zn–air batteries based on ZnCoFe-TAC/SNC catalysts present a high power density, high specific discharge capacity and long lifespans. This work provides a universal insight into the controllable synthesis of trimetallic atom catalysts (TACs) and a proposal for regulating the performance in energy conversion and catalytic applications. |
Introduction
The sluggish kinetics of the cathode oxygen reduction reaction (ORR) and the anode oxygen evolution reaction (OER) greatly hinder the wide application of energy conversion devices.1–6 Although some non-noble metal (Fe, Co, Ni, etc.) catalysts show comparable catalytic performances to noble metal (Pt, Ir, Ru, Pd, etc.) catalysts, non-precious metal single-atom catalysts (SACs) still show great limitations, owing to the difficulty in breaking the scaling relationship.7,8 The asymmetric coordination structures show great potential in regulating the reaction intermediate absorption on the active sites.9–18 The multi-metal catalysts, owing to the interactions among various mental atoms, can optimize the adsorption energies of oxygen species by modulating the 3d electronic structure, which promotes the oxygen reaction performances.13,19 However, achieving an optimum charge redistribution is difficult because of the high free energy of the metal single atoms, which limits the content of the metal atoms. The excessive metal atoms favor aggregation, leading to weakened activity. Besides, heteroatom (S, N, P, etc.) doping is commonly used to modulate charge redistribution of the metal center through metal–heteroatoms.20–25 Together with metal coupling and heteroatom doping, it can more effectively modulate charge redistribution through the metal–metal and metal–heteroatom electron coupling effects.Although asymmetric coordination of the single-atom (SA) site is beneficial to optimizing the electronic structure of the catalyst and further achieving highly efficient catalysis, the controllable design of such asymmetric coordination atomic site remains a great challenge.26–28 For instance, Ma et al. designed an asymmetrically coordinated Co–N4P SA site by introducing the asymmetric P atoms into symmetrically coordinated Co–N4 SA sites, which boosts the catalytic activity.26 The Yang group developed a N,O-asymmetrically coordinated Co–N2O2 site to enhance the electrochemical performance of the lithium metal anode.29 Chen et al. reported a rational design strategy to obtain the single Cu atoms coordinated with S and N atoms, exhibiting excellent ORR activity.30 The asymmetrically coordinated atomic sites provide more regulatory possibilities for SA catalysts. However, the problem of low metal loading and the metal aggregation effect remains unsolved. In contrast to SA catalysts, the dual-metal atomic site, consisting of two adjacent metal atoms, shows high metal atom loading and atom utilization. Fu's group found that Cu/Zn–NC catalyst with Cu–N4 and Zn–N4 sites exhibits excellent ORR activity.31 During the ORR process, the Zn–N4 sites can prevent the aggregation of Cu–N4 sites. As our previous work reported, a hetero Co–Fe dual-atomic catalyst, wherein the Co atom is coordinated with two nitrogen atoms and one sulfur atom and the Fe atom is only coordinated with three nitrogen atoms, shows high OER activity.32
Inspiringly, the hetero-trimetallic atom catalyst (TAC) as a fresh and boom catalyst shows superior catalytic activity owing to their unique active site structure.33–35 Firstly, each metal of the TAC serves as a different adsorption site, avoiding the competitive adsorption between various intermediates, which is conducive to optimizing the catalytic path. Secondly, heterogeneous trimetallic atom sites integrate the electronic properties of each metal element and optimize the charge distribution around the active site, which can break the limitations in activity for both the active site structure and the intermediates. Nevertheless, synthesizing TACs at the single-atom level remains a huge challenge due to the metal atoms’ different physical and chemical properties. Although the synthesis of the TACs can be learned from dual metal atom catalysts (DACs), a stable heterometallic atom site with exact configurations is challenging to fabricate. Therefore, it is important to design a strategy to obtain an asymmetrically coordinated multi-heterometal atomic catalyst with a controllable, stable, and high-efficiency active site.
Herein, we successfully designed a novel asymmetrically coordinated hetero-trimetallic SA catalyst (ZnCoFe-TAC/SNC) through a simple two-step wet chemistry strategy based on forming the metal–organic framework materials and the post-coordination process. The unique post-coordination approach can not only maintain the N-coordinated Zn/Co/Fe atom sites but also modulate the asymmetrical electron structure abounding the ternary heterometal atom sites via S-coordinated Fe atoms. The atomic resolution aberration-corrected electron microscopy confirms the existence of the ternary atomic sites, and the X-ray absorption spectroscopy (XAS) revealed the configuration of the symmetrically coordinated heterometal atomic sites, where the nitrogen coordinated single Co and Zn atoms adjacent to the sulfur/nitrogen dual-coordinated Fe atoms (ZnN3CoN3FeN2S). The in situ XAS results show that Co is the main active site, and Fe is a co-catalytic site. Zn acts as an “electron regulator” to regulate the electron structures around the catalytic sites. According to the DFT calculations, the Co atom is the active site because it is the most suitable binding of oxygen intermediates. Besides, the strong OH* adsorption on the Zn site can enhance the OER activity, and the Fe atoms of the ZnN3CoN3FeN2S site can reduce the d-band center of the Co atom, which weakens the Co–O binding energy, improving the catalytic performance of materials. Accordingly, the as-synthesized ZnCoFe-TAC/SNC exhibited extraordinary ORR and OER performances, outperforming those of the other monometallic or bimetallic SA catalysts and benchmark Pt/C catalysts. Moreover, Zn–air battery tests demonstrate that ZnCoFe-TAC/SNC exhibits competitive performance compared with Pt/C.
Results and discussion
Synthesis and morphology characterization
The synthetic procedure for the ZnCoFe-TAC/SNC is shown in Fig. 1(a). Firstly, the ZnCo-ZIF precursor is self-assembled with Co2+, Zn2+, and 2-methyl imidazole ligand, as shown in the experimental part of the ESI.† Subsequently, the Fe3+/C4H8N2S polymer polymerized by iron precursors and allylthiourea wraps the ZnCo-ZIF precursor to obtain the ZnCo-ZIF@Fe3+/C4H8N2S precursor. The ZnCoFe-TAC/SNC catalyst with the asymmetrically coordinated ternary heterometal atom site anchored in the sulfur/nitrogen-rich hollow carbon shell is then obtained after the pyrolysis treatment under a N2 atmosphere at 1000 °C. For comparison, the Zn–NC, ZnCo-DAC/NC, and ZnCoFe-TAC/NC catalysts are prepared using a similar procedure. The macroscopic color and morphology of the obtained catalysts are displayed in Fig. S1 (ESI†). The ZnCoFe-TAC/SNC shows a uniform hollow dodecahedral structure with an average size of 200–300 nm (Fig. 1(b)). The average sizes of the Zn–NC, ZnCo-DAC/NC, and ZnCoFe-TAC/NC are similar to those of the ZnCoFe-TAC/SNC (Fig. S2, ESI†). In Fig. 1(c) and Fig. S3 (ESI†), neither metal nor metallic oxide nanoparticles are detected, which confirms that the Zn, Co, or Fe atoms exist in the atomically dispersed state. Additionally, ZnCoFe-TAC/SNC shows the most severe facial collapse and the thinnest carbon walls among the catalysts, which is attributed to the effect of Fe3+ on the dissociation and recombination of carbon under high temperatures. Such unique structures facilitate the exposure of the active sites and the mass transfer process.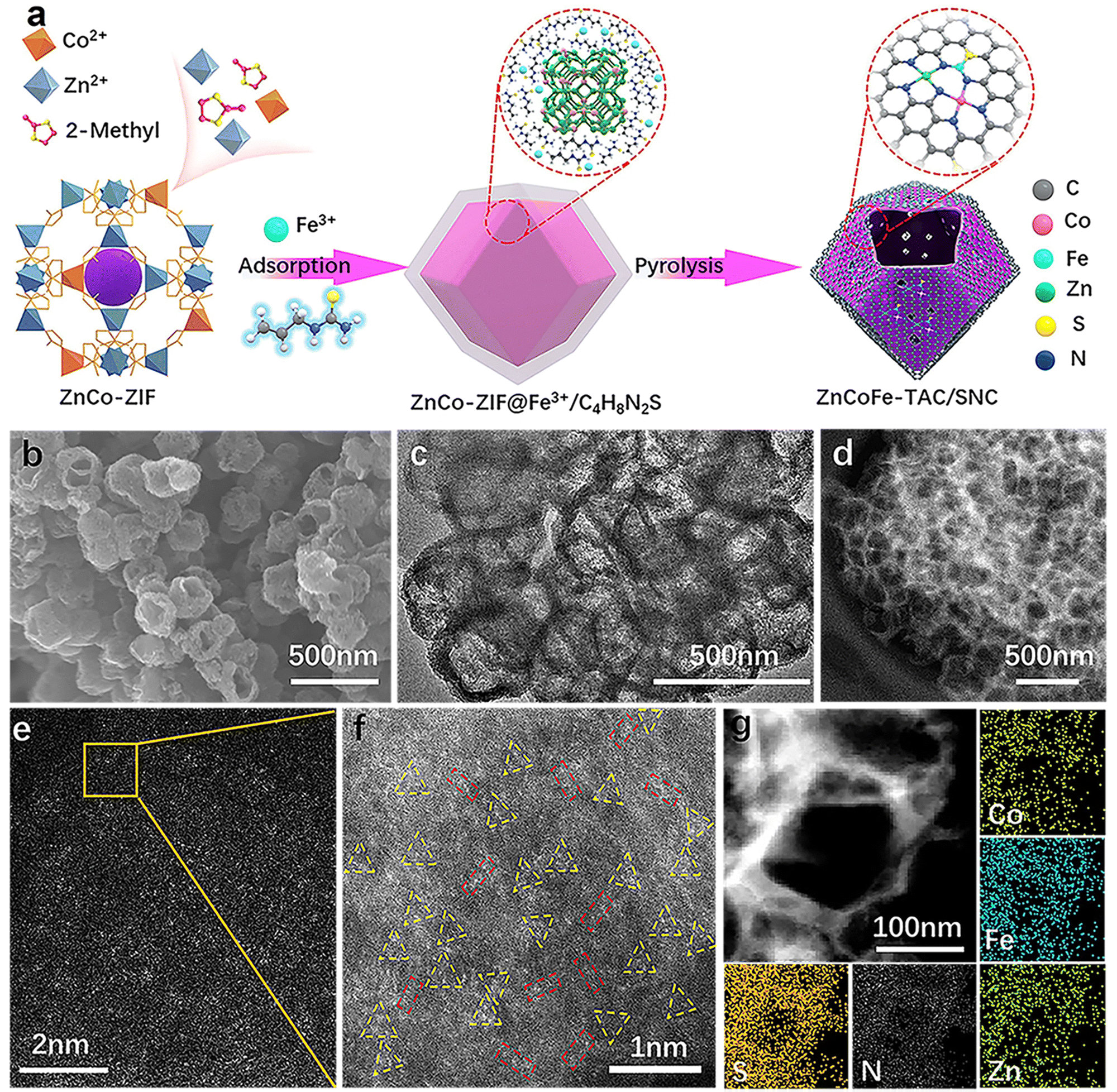 | ||
| Fig. 1 Morphology and component analysis of ZnCoFe-TAC/SNC. (a) Schematic synthesis of ZnCoFe-TAC/SNC; (b) SEM image of ZnCoFe-TAC/SNC; (c) TEM image of ZnCoFe-TAC/SNC; (d) HAADF-STEM image of ZnCoFe-TAC/SNC; (e) HAADF-STEM image and (f) the enlarged image of ZnCoFe-TAC/SNC catalyst; the red rectangle contains bimetallic atoms, and the yellow triangle contains trimetallic atoms. (g) Element-mapping (Fe: blue, Co: cyan, N: white, S: orange, Zn: green). | ||
Zeolitic imidazolate framework-8 (ZIF-8) is considered one of the best templates for preparing nitrogen-doped carbon catalysts because of their simple synthesis method and excellent porous structures.36,37 Other metals can be injected to form the multi-metal components-based ZIF materials. After the high-temperature treatment, abundant carbon defect structures and stable metal–N active sites are formed. The ZnCo-ZIF is synthesized and directly pyrolyzed. Very few Zn atoms remain in the material due to the evaporation of the Zn atoms during the pyrolysis process (Table S1, ESI†). Considering that the ZnCo-ZIF surface is rich in positive charges, we designed an electrostatic adsorption strategy to wrap a protective layer of allylthiourea outside of ZnCo-ZIF. This protective layer plays three critical roles in the final ZnN3CoN3FeN2S active sites: blocking the evaporation of Zn atoms, doping the S atoms into the ZIF material, and providing the N/S coordination sites for the post-adsorbed Fe3+. In this case, the evaporated Zn atoms escape from the framework to the surface and are immediately intercepted by the allylthiourea protective layer. The N-coordinated Zn and Co atoms gradually approach N/S-coordinated Fe atoms in the allylthiourea layer, which provides a limited environment for forming the tri-metal atom site. Additionally, the final and most critical driving force to ensure the formation of the tri-metal atom site is the high oxidation of Fe3+. The highly oxidizing Fe3+ can promote the dissociation and recombination of carbon under high temperatures, which is beneficial to the reconstruction of the metal active sites.
Noteworthily, another indirect effect of the allylthiourea on the catalyst is to promote the adsorption of Fe3+. The adsorbed allylthiourea can neutralize the positive charge of the ZnCo-ZIF surface, which reduces the adsorption resistance of Fe3+. The high Fe3+ adsorbed on the surface not only increases the incorporation of iron elements (Table S1, ESI†) but also strengthens the decomposition and reconstruction of carbon materials (Fig. 1(b)–(d)), which is conducive to the formation of hollow structures. To prevent the formation of Fe–Fe, Co–Co diatoms, or metal clusters, the feeding of the Fe and Co precursors, especially the Fe precursors, is strictly controlled at an appropriate level. A too-low addition is not conducive to forming the tri-metal atom site, and a too-high addition makes it easy to form metal clusters.
The X-ray diffraction (XRD) patterns in Fig. S4 (ESI†) show only two peaks at ∼24.5° and 43.5°, corresponding to the (002) and (100) crystallographic planes of carbon, which is attributed to the amorphous carbon matrix. No metal or metallic oxide nanoparticle signals are detected, which further confirms the single atom state of the metal atoms in the obtained catalysts. Aberration-corrected high-angle annular dark-field scanning TEM (HAADF-STEM) images further reveal information on the structure and the state of the metal. As shown in Fig. 1(d)–(f), the absence of the metal nanoparticles further indicates the atomic dispersion of Zn, Co, and Fe. Notably, the triangular trimetallic atom site (highlighted by a yellow triangle) and linear bimetallic atom site (highlighted by a red rectangle) could be observed, enlightening the speculation of an unusual active site. The ratio of the bimetallic and trimetallic atom sites has been analyzed, as shown in Fig. S5 (ESI†). The percentage of trimetallic atom sites is approximately 70%, the dominant metallic sites in ZnCoFe-TAC/SNC.
Besides, it is important to emphasize that the atomic site in the axial position of the trimetallic atom site is easily mislabeled as the bimetallic atom site. Therefore, some of the bimetallic atom sites marked in Fig. 1(f) may be unheeded trimetallic atom sites. Fig. 1(g) shows that the Zn, Co, Fe, C, N, and S elements distribute uniformly along the hollow pore wall of ZnCoFe-TAC/SNC. In contrast, the elements of other obtained catalysts are distributed evenly throughout the whole dodecahedron (Fig. S6–S8, ESI†). The Zn, Co, and Fe contents tested by the inductively coupled plasma optical emission spectrometer (ICP-OES) are values of 0.81, 0.73, and 0.75 wt%, respectively, close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 (Table S1, ESI†). The porous structures of the catalysts were validated by the nitrogen adsorption–desorption analysis, as shown in Fig. S9 (ESI†). The isotherms of ZnCoFe-TAC/SNC display a steep increase in adsorption volume at a low-pressure region and an obvious hysteresis loop at a higher pressure, suggesting a typical mesoporous structure. ZnCoFe-TAC/SNC exhibits a high specific surface area of 933 m2 g−1 and a pore volume of 0.51 m3 g−1, respectively. Noticeably, ZnCoFe-TAC/SNC shows lots of distinctive macro-pores, which are beneficial to mass transport during the catalytic process.
:1:1 (Table S1, ESI†). The porous structures of the catalysts were validated by the nitrogen adsorption–desorption analysis, as shown in Fig. S9 (ESI†). The isotherms of ZnCoFe-TAC/SNC display a steep increase in adsorption volume at a low-pressure region and an obvious hysteresis loop at a higher pressure, suggesting a typical mesoporous structure. ZnCoFe-TAC/SNC exhibits a high specific surface area of 933 m2 g−1 and a pore volume of 0.51 m3 g−1, respectively. Noticeably, ZnCoFe-TAC/SNC shows lots of distinctive macro-pores, which are beneficial to mass transport during the catalytic process.
Electronic interactions and atomic coordination structure analysis
The N/S/Zn/Co/Fe coordination structure information was investigated by X-ray photoelectron spectroscopy (XPS) and X-ray absorption fine spectroscopy (XAFS). The XPS survey spectrum of ZnCoFe-TAC/SNC displayed in Fig. S10 (ESI†) confirms the coexistence of C, O, N, S, Zn, Co, and Fe elements. The atom contents of each element of ZnCoFe-TAC/SNC derived from the XPS survey spectrum are listed in Table S3 (ESI†). In Fig. S11 and S12 (ESI†), the N 1s XPS spectra can be deconvoluted into four N species, and the dominant N species are graphitic N and pyrrolic N, followed by the pyridinic-N/M–N including the Zn–N, Co–N and Fe–N bonds.31 The Zn 2p XPS spectra in Fig. S13 (ESI†) exhibit two main signal peaks at 1020.8 eV and 1044.1 eV, which are due to the high valence of the Zn atom.38 ZnCoFe-TAC/NC shows a positive shift of binding energy for the Zn 2p XPS signal compared with that of Zn/NC and ZnCo-DAC/NC, indicating that the electrons transition from Zn atoms to Fe atoms. The binding energy of the Zn 2p XPS signal of ZnCoFe-TAC/SNC is more positive than that of ZnCoFe-TAC/NC, resulting from the electron-regulation function of S atoms. Besides, the binding energy of Co 2p XPS signal in ZnCoFe-TAC/SNC is located between that in ZnCoFe-TAC/NC and ZnCo-DAC/NC due to the electron-regulation function by Fe and S atoms (Fig. S14, ESI†). Fig. S15 (ESI†) shows the Fe 2p XPS spectrum of ZnCoFe-TAC/SNC, indicating the existence of the Fe. The S 2p XPS spectra indicate the existence of the Fe–S bond, and the thiophenic-S is the dominant S species (Fig. S16, ESI†). X-ray absorption near-edge structure (XANES) spectra are measured to further evaluate the state of the C, N, and S elements in the ZnCoFe-TAC/SNC catalyst. In Fig. 2(a), the two features of C located at 286.8 and 293.3 eV can be assigned to the π* C-ring, attributing to the C–N bonds and σ* C-ring ascribing to the C–C bonds, respectively.32 The N K-edge XANES spectrum was also conducted to monitor the nitrogen chemical configurations (Fig. 2(b)). The peaks of a, c, and d are attributed to the pyridinic N, pyrrolic N, and C–N bonds, respectively. The peak of b could be assigned to M–N.39 Meanwhile, the S L-edge XANES spectrum displays three peaks in the 162.7–167.1 eV region, arising from the reduced S species (Fig. 2(c)). | ||
Fig. 2 Atomic coordination structure characterization of catalysts by EXAFS. (a) C (a is π*(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) ring, b is σ*(CC) ring), (b) N (a is pyridinic-N, b is metal (M)–N, c is pyrrolic-N, d is σ*-N), (c) S K-edge NEXAFS spectra; (d) Fourier-transformed EXAFS signals of ZnCoFe-TAC/SNC and the references (solid lines); (e) Fe, (f) Co and (g) Zn K-edge EXAFS analysis of ZnCoFe-TAC/SNC in R spaces; (h) the structure of the active site (ZnN3CoN3FeN2S) derived from the EXAFS results. C) ring, b is σ*(CC) ring), (b) N (a is pyridinic-N, b is metal (M)–N, c is pyrrolic-N, d is σ*-N), (c) S K-edge NEXAFS spectra; (d) Fourier-transformed EXAFS signals of ZnCoFe-TAC/SNC and the references (solid lines); (e) Fe, (f) Co and (g) Zn K-edge EXAFS analysis of ZnCoFe-TAC/SNC in R spaces; (h) the structure of the active site (ZnN3CoN3FeN2S) derived from the EXAFS results. | ||
The coordination structure and valence of Fe, Co, and Zn elements are investigated using the XAFS measurements. Fig. S17 (ESI†) shows that the Zn, Co, and Fe atoms in ZnCoFe-TAC/SNC carry partially positive charges. In Fig. 3(d), no signal peaks of metal–metal (Fe–Fe at 2.2 Å, Co–Co at 2.2 Å, and Zn–Zn at 2.3 Å) bonds are observed in the Fourier transform extended X-ray absorption fine structure (FT-EXAFS) spectra of metal K-edge in ZnCoFe-TAC/SNC. It suggests that the Fe, Co, and Zn atoms are dispersed on the carbon matrix in a single atomic state, further confirmed by the wavelet-transform images (Fig. S18, ESI†). The Fe K-edge FT-EXAFS spectra of ZnCoFe-TAC/SNC show that two peaks are located at 1.6 Å and 1.8 Å, which are attributed to the Fe–N and Fe–S coordination in the structures. A broad and distorted peak is observed on the Co K-edge FT-EXAFS spectra of ZnCoFe-TAC/SNC, corresponding to the Co–N1 and Co–N2 coordination structures in ZnCoFe-TAC/SNC. The Zn K-edge FT-EXAFS spectra indicate the existence of the Zn–N1 and Zn–N2 in ZnCoFe-TAC/SNC. The local coordination structures of Fe, Co, and Zn atoms in ZnCoFe-TAC/SNC are further identified by the quantitative EXAFS fitting curves (Fig. 3(e)–(g), and Fig. S19, ESI†). As shown in Fig. 3(e), the fitting curve is consistent with the experimental curve of the Fe K-edge in ZnCoFe-TAC/SNC, consisting of Fe–N and Fe–S. The fitting result of the Co K-edge shows two N coordination structures corresponding to Co–N1 and Co–N2 (Fig. 2(f)). Zn K-edge fitting curves show similar coordination structures with that of Co K-edge, including Zn–N1 and Zn–N2 (Fig. 2(g)). Herein, according to the XAFS fitting data (Table S4, ESI†), the N1 can be assigned to the N atom that bridges two metal atoms, which shows short metal-bond length due to the electronic coupling between two metals. The N2 belongs to the N atom that coordinates with only one metal atom, leading to the large metal–N bond length. To sum up, the fitting tri-atom site is shown in Fig. 2(h), i.e., one site contains Zn, Co, Fe, N, and S atoms simultaneously.
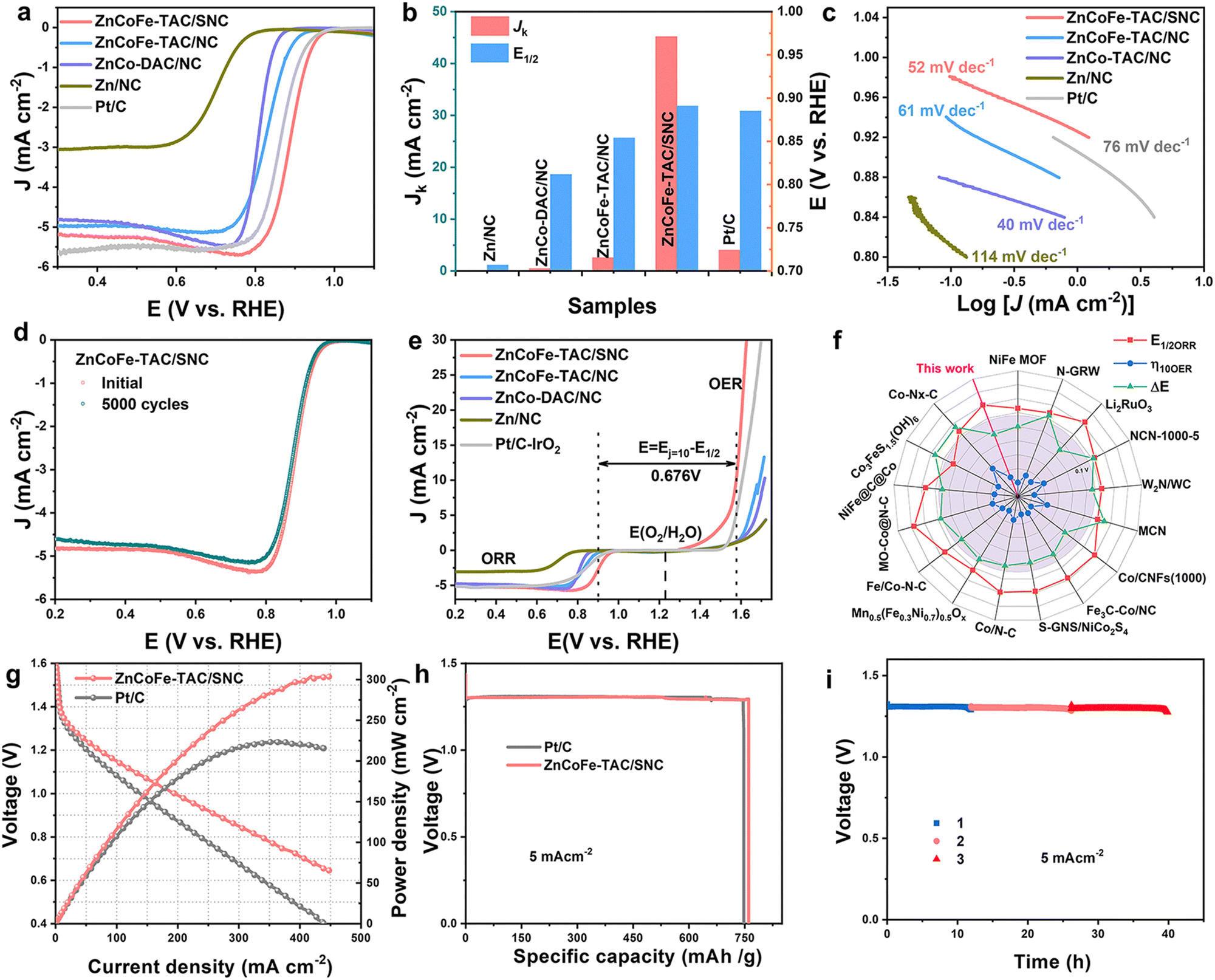 | ||
| Fig. 3 The electrocatalytic performances for ORR, OER, and Zn–air batteries. (a) LSV curves of ZnCoFe-TAC/SNC and references; (b) the comparisons of Jk at 0.85 V and E1/2 for ZnCoFe-TAC/SNC and references; (c) the Tafel plots of ZnCoFe-TAC/SNC and references; (d) the LSV curves before and after ADT; (e) the overall polarization curves of the ORR and OER; (f) the comparison of the catalytic activities in this work with the reported catalysts; (g) the discharge polarization and power density curves of of ZnCoFe-TAC/SNC in Zn–air batteries; (h) the specific capacity curves of ZnCoFe-TAC/SNC and Pt/C-based Zn–air batteries; (i) durability test curves of ZnCoFe-TAC/SNC-based Zn–air batteries (piece 1 is the test curve in the initial electrolyte solution, piece 2 is the test curve in the first-replaced electrolyte solution, piece 3 is the test curve in the second-replaced electrolyte solution). | ||
The electrocatalytic performances
The ORR performances of the obtained catalysts are evaluated in 0.1 M KOH. In Fig. 3(a), the ZnCoFe-TAC/SNC displays excellent catalytic activity. The half-wave potential (E1/2) of the ZnCoFe-TAC/SNC is 0.901 V (vs. RHE, the same standard below), which is higher than that of Pt/C (E1/2, 0.885 V). The kinetic current density (Jk) of the ZnCoFe-TAC/SNC is 45.17 mA cm−2 at 0.85 V, 11 times as high as that of Pt/C. Additionally, the Tafel slope of the ZnCoFe-TAC/SNC is 52 mV dec−1, which is smaller than that of Pt/C (76 mV dec−1), indicating the favorable ORR catalytic kinetic. As shown in Fig. S20 (ESI†), the average number of transferred electrons (n) is 3.97, suggesting the 4e− ORR process. No significant decay after 5000 cycles (Fig. 3(d) and Fig. S21, ESI†) indicates that ZnCoFe-TAC/SNC displays excellent catalytic stability. The favorable catalytic stability of ZnCoFe-TAC/SNC is further verified by the i–t curves (Fig. S22, ESI†). Besides, ZnCoFe-TAC/SNC also exhibits outstanding tolerance to methanol crossover (Fig. S23, ESI†). In Fig. S24 (ESI†), all the elements remain in ZnCoFe-TAC/SNC after the stability test. Significantly, no obvious aggregation or degradation occurred at the atomically dispersed metal sites (Fig. S25, ESI†), resulting in high catalytic stability. The potential difference (ΔE = EOER,j=10 − EORR,1/2) is usually used to assess the ORR/OER bifunctional catalytic performance. In Fig. 3(e), the ΔE of the ZnCoFe-TAC/SNC is 0.676 V, which is smaller than that of the Pt/C + RuO2. Compared with the reported catalysts, the ZnCoFe-TAC/SNC also shows comparable electrocatalytic performances (Fig. 3(f) and Table S5, ESI†).20,40–54 To further evaluate the bifunctionally catalytic activity of ZnCoFe-TAC/SNC in a practical application,30,55,56 a homemade rechargeable ZnCoFe-TAC/SNC-based Zn–air battery (Fig. S26, ESI†) was assembled. In Fig. S27 (ESI†), the ZnCoFe-TAC/SNC-based Zn–air battery delivers 1.40 V of open circuit voltage. As shown in Fig. 4(g), the ZnCoFe-TAC/SNC-based Zn–air battery shows a high maximum power density of 304 mW cm−2, outperforming Pt/C (223 mW cm−2). The specific capacity of ZnCoFe-TAC/SNC-based Zn–air battery is 760 mA h g−1 at the discharge of 5 mA cm−2. Moreover, the ZnCoFe-TAC/SNC-based battery can be used stably for up to 40 h with a slight decrease in discharge voltage (Fig. 4(i)), indicating excellent durability. | ||
| Fig. 4 In situ XANES measurements for ZnCoFe-TAC/SNC under electrochemical conditions. The normalized XANES spectra of (a) Zn, (c) Co, and (e) Fe K-edge: the insets are the enlarged image at the near-edge region; the first-derivative XANES curves of (b) Zn, (d) Co and (f) Fe K-edge; (g) the changing trend of the oxidation state at Zn, Co and Fe K-edge for ORR derived from (a)–(f); (h) the changing trend of the oxidation state at Zn, Co and Fe K-edge for OER derived from Fig. S29 (ESI†). | ||
In situ XAS analysis
To explore the conversions of the three metal atoms in the active site, the in situ XAS spectra (Fig. S28, ESI†) were conducted in standard fluorescence excitation mode during the potentials relevant to the ORR.57–59 In Fig. 4(a), the Zn absorption edge gradually migrates to a lower energy at the applied potential of 1.0 V, resulting in the lower oxidation state (Fig. 4(b) and (g)), which is attributed to the electron transition from the adjacent Co and Fe atoms to Zn atoms. In contrast, for the Co K-edge XANES (Fig. 4(c)), the absorption edge shifts to the higher energy at the applied potential of 1.0 V, leading to the higher oxidation state (Fig. 4(d) and (g)), which is attributed to the adsorption of the oxygen species on the Co sites. When a oxygen reduction potential of 0.85 V is applied, the energy of the absorption edge decreases due to the electrons transfer from Zn atoms to Co sites during the ORR process. Accordingly, the Co atom shows a lower oxidation state at 0.85 V than that of 1.0 V, and Zn shows a higher oxidation state at 0.85 V than that of 1.0 V (Fig. 4(h)). Interestingly, the absorption edge of Fe XANES changes similarly to that of the Co atom, suggesting that the Fe atom, as a co-catalytic site, plays a role in the oxygen reduction process. Briefly, the Co and Fe sites are synergistic catalytic active centers that control the adsorption and desorption behavior of intermediates. The Zn atom promotes catalytic activity by regulating the distribution of electrons around the active centers.The dynamic structure of the ZnCoFe tri-metal site for OER was also clarified by the in situ XAS spectra.60–62 As shown in Fig. S29a (ESI†), the Zn absorption edge shifts to lower energy under the applied potentials, indicating lower oxidation (Fig. S29b, ESI† and Fig. 4(i)), attributed to the electron transition from the nearby Co and Fe atoms. Conversely, the absorption edge of Co XANES moves to the higher energy (Fig. S29c, ESI†), resulting from the higher oxidation state induced by the adsorbed OH− under the electrochemical conditions (Fig. S29d, ESI† and Fig. 4(i)). The high oxidation state Co species possess a high reactivity towards OER. In Fig. S29e and f (ESI†), the adjacent Fe absorption edge also shifts to the positive direction, which shows a similar change trend to the Co site. As the analysis of the above, the Co and Fe go through the step of increasing oxidation state under OER condition (Fig. 4(h)), owing to the adsorption of the oxygen species, which is in favor of the improvement of the OER performance. Zn atoms play a role in regulating electron distribution to optimize surface oxygen adsorption energy. In summary, in the ORR and OER conditions, the Co serves as the main active site, the Fe as the the co-catalytic sites, and the Zn is the “electron regulator”. Co and Fe sites constate the synergetic catalytic center, and the Zn atom regulates the electron distribution around the catalytic center.
DFT analysis
According to the experimental results, a ternary atomic structure ZnCoFe was constructed. As depicted in Fig. 5(a) and Fig. S29 (ESI†), Co and Zn coordinate with four nitrogen atoms, while Fe coordinates with two nitrogen atoms and one sulfur atom. We first constructed the Pourbaix diagram by calculating different intermediate adsorption states on the ZnCoFe (Fig. 5(b)). The surface resting state of the ZnCoFe under the reaction condition has all metal sites covered by O*, and thus were considered for computing the OER and ORR free energy profiles on the ZnCoFe system. Compared with Fe and Zn sites in the ZnCoFe system, the Co site performed the highest catalytic activity. Hence, the Co site was selected as the active site, and the corresponding single and dual atomic structures CoN4 and CoZn were also studied as references. Furthermore, for a comprehensive understanding, structures such as FeN4, ZnN4, CoFe, and CoCoFe were also constructed. Additional details can be found in Fig. S30 and S31 (ESI†).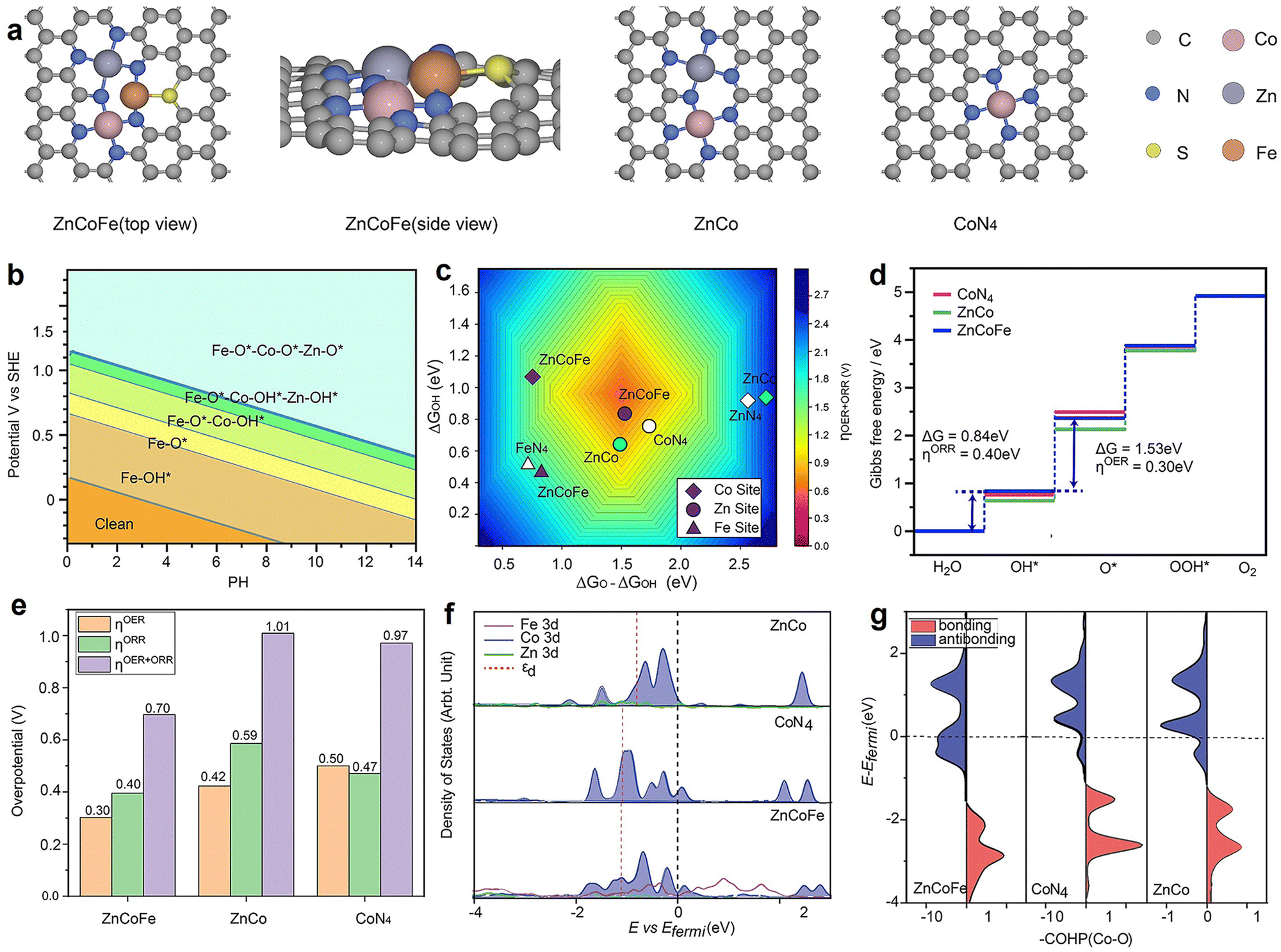 | ||
| Fig. 5 The theoretical calculation for the ZnCoFe. (a) The configurations of ZnCoFe (top view), ZnCoFe (side view), CoZn, and CoN4. Grey, blue, yellow, brown, pink, and silver balls represent the C, N, S, Fe, Co, and Zn atoms; (b) the Pourbaix diagram of CoZnFe; (c) calculated total theoretical overpotential (ηtotal) volcano plot with ΔGO* − ΔGOH* and ΔGOH* as descriptors; (d) the free energy diagrams of ZnCoFe, CoN4, and CoZn. The pink, blue, and green crosses represent ZnCoFe, CoN4, and CoZn, respectively; (e) the histogram of overpotential. Orange, green, and purple represent the overpotential of OER, ORR, and OER + ORR; (f) the diagram of the projected density of states. Among this program, the black dotted line represents the E-Fermi level, and the red dotted line represents the d-band center; (g) crystal orbital Hamiltonian population (COHP). The red area represents the bonding component, the blue area represents the antibonding area, and the black dotted line represents the E-Fermi level. | ||
Following the conventional four-electron transfer mechanism and the linear scaling between ΔGOH* and ΔGOOH* (Fig. S32–S34, ESI†), Fig. 5(c) plots the total theoretical overpotential volcano as a function of ΔGO* − ΔGOH* and ΔGOH*. The red central region represents the top of the volcano, with the lowest overpotential, and hence, the highest ORR and OER activity. In general, the catalytic activity of the Co sites stood out prominently, particularly notable in their concentrated presence in the central region. In contrast, the Fe and Zn sites were located in the lower left region and the right half of the Fig. 5(c), respectively, owing to their stronger or weaker adsorption to oxygen intermediates. The observed variation in activity can be attributed primarily to the adsorption strength of OH*, with the trend being CoZn < CoN4 < ZnCoFe. The presence of Zn strengthens the adsorption of OH* at the Co site, leading to a lower OER overpotential (CoZn 0.42 V and CoN4 0.50 V) but suppressed ORR performance (CoZn 0.59 V and CoN4 0.47 V). Meanwhile, the synergetic effect from Fe effectively optimizes the surface adsorption to a moderate range for the ΔGO* − ΔGOH* value, leading to a significant reduction in both ORR and OER overpotentials. Consequently, the total overpotential (ηtotal) is effectively reduced from 1.01 V to 0.70 V, which aligns well with the electrochemical test results shown in Fig. 3(g).
To further understand the effect of Fe atom doping on the electronic structure, the partial density of states (PDOS) and crystal orbital Hamilton population (COHP) analyses were performed. Fig. 5(f) shows that the d-band center of the Co atom undergoes a significant reduction, shifting from −0.80 eV to −1.11 eV upon the introduction of the Fe dopant, implying a weakened adsorption capacity of ZnCoFe for intermediates and improvement of catalytic activity. The nature of the Co–O interaction was explored through crystal orbital Hamilton population (COHP) analysis, the 3d orbital of Co and 2p orbital of O were selected, and the results are shown in Fig. 5(g). The incorporation of Fe resulted in a downward shift of bonding and antibonding states, weakening Co–O interaction. The −ICOHP values serve as indicators of bonding capabilities, and the observed trend is as follows: CoZn (1.16) > CoN4 (1.04) > ZnCoFe (0.83). In brief, the consistency between PDOS analysis and COHP analysis aligns with the observed trend in adsorption energy. The introduction of Fe proves effective in enhancing the catalytic performance of materials, attributed to its influence on altering the electronic structure and bonding properties of the materials.
Conclusions
In summary, an asymmetric hetero-trimetallic SA site catalyst (ZnCoFe-TAC/SNC), in which the nitrogen-coordinated Co and Zn atoms adjacent to the sulfur/nitrogen dual-coordinated Fe atoms (ZnN3CoN3FeN2S) anchored in S/N-doped carbon, is successfully designed. The novel asymmetric hetero-trimetallic atom active site is optimized through both the metal–non-metal (Zn–N, Co–N, Fe–N, and Fe–S) bond and the electron cloud metal–metal interactions. The resulting ZnCoFe-TAC/SNC displays an excellent ORR performance with E1/2 of 0.901 V and Jk of 45.17 mV cm−2 at 0.85 V in alkaline media. Besides, the ZnCoFe-TAC/SNC shows the smallest ΔE of 0.676 V, indicating an outstanding ORR/OER bifunctional catalytic activity. The ZnCoFe-TAC/SNC-based Zn–air battery displays a comparable maximum power density of 304 mW cm−2. The in situ XAS results show that Co is the main active site, and Fe is a co-catalytic site. Zn acts as an “electron regulator” to regulate the electron structures around the catalytic sites. DFT results further indicate the synergetic effect of the interactions among the three metal atoms on the catalytic performance. The design philosophy of the novel asymmetric hetero-trimetallic SA site catalyst will inspire the rational design and fabrication of the oxygen catalyst.Author contributions
C. C., M. S., W. C., and Y. L. conceived the idea, designed the study and wrote the paper. C. C. and M. S. synthesized and characterized the samples, performed experiments. W. C. and C. C. carried out the XAFS characterization and data analysis. J. C. and L. Z. performed the DFT calculations. Z. S. carried out the Zn–air battery measurements. F. Z. helped with the spherical aberration electron microscopy test and discussion. T. G., J. L., Y. Z., and Z. L. helped with the modification of the paper. All authors read the manuscript and contributed to the discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2023QB142), the National Natural Science Foundation of China (52171199, 22375019, 22373055, and 12374390), the Ningbo 3315 Innovative Teams Program (grant no. 2019A-14-C), the member of Youth Innovation Promotion Association Foundation of CAS, China (2023310), the Beijing Natural Science Foundation (2212018), the Talent Research Project of Qilu University of Technology (Shandong Academy of Sciences) (2023RCKY090), the Science, the Education and Industry Integration Pilot Project Plan of Qilu University of Technology (Shandong Academy of Sciences) (2023PX097), the State Key Laboratory of Intelligent Green Vehicle and Mobility (ZZ2023-063) and the Tsinghua University Initiative Scientific Research Program.References
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- S. Ott, A. Orfanidi, H. Schmies, B. Anke, H. N. Nong, J. Hubner, U. Gernert, M. Gliech, M. Lerch and P. Strasser, Nat. Mater., 2020, 19, 77–85 CrossRef CAS PubMed.
- W.-Y. Huo, S.-Q. Wang, W.-H. Zhu, Z.-L. Zhang, F. Fang, Z.-H. Xie and J.-Q. Jiang, Tungsten, 2021, 3, 161–180 CrossRef.
- X.-X. Li, X.-C. Liu, C. Liu, J.-M. Zeng and X.-P. Qi, Tungsten, 2023, 5, 100–108 CrossRef.
- C. Y. Zheng, X. Zhang, Z. Zhou and Z. P. Hu, eScience, 2022, 2, 219–226 CrossRef.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 46, 2060–2086 RSC.
- Y. Wang, Q. Wang, J. Wu, X. Zhao, Y. Xiong, F. Luo and Y. Lei, Nano Energy, 2022, 103, 107815 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J.-N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- L. Xie, S. Zhou, J. Liu, B. Qiu, T. Liu, Q. Liang, X. Zheng, B. Li, J. Zeng, M. Yan, Y. He, X. Zhang, H. Zeng, D. Ma, P. Chen, K. Liang, L. Jiang, Y. Wang, D. Zhao and B. Kong, J. Am. Chem. Soc., 2021, 143, 6922–6932 CrossRef CAS PubMed.
- S. Gupta, L. Qiao, S. Zhao, H. Xu, Y. Lin, S. V. Devaguptapu, X. Wang, M. T. Swihart and G. J. Wu, Adv. Energy Mater., 2016, 6, 1601198 CrossRef.
- X. Hao, Z. Jiang, B. Zhang, X. Tian, C. Song, L. Wang, T. Maiyalagan, X. Hao and Z.-J. Jiang, Adv. Sci., 2021, 8, 2004572 CrossRef CAS.
- H. B. Zhang, W. Zhou, T. Chen, B. Y. Guan, Z. Li and X. W. Lou, Energy Environ. Sci., 2018, 11, 1980–1984 RSC.
- A. Zitolo, N. Ranjbar-Sahraie, T. Mineva, J. Li, Q. Jia, S. Stamatin, G. F. Harrington, S. M. Lyth, P. Krtil, S. Mukerjee, E. Fonda and F. Jaouen, Nat. Commun., 2017, 8, 957 CrossRef.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS.
- Q. Lv, W. Si, J. He, L. Sun, C. Zhang, N. Wang, Z. Yang, X. Li, X. Wang, W. Deng, Y. Long, C. Huang and Y. Li, Nat. Commun., 2018, 9, 3376 CrossRef PubMed.
- W. Guo, X. Gao, M. Zhu, C. Xu, X. Zhu, X. Zhao, R. Sun, Z. Xue, J. Song, L. Tian, J. Xu, W. Chen, Y. Lin, Y. Li, H. Zhou and Y. Wu, Energy Environ. Sci., 2023, 16, 148–156 RSC.
- H. Wang, R. Liu, Y. Li, X. Lü, Q. Wang, S. Zhao, K. Yuan, Z. Cui, X. Li, S. Xin, M. L. Ru Zhang and Z. Lin, Joule, 2018, 2, 337–348 CrossRef CAS.
- T. Zhou, W. Xu, N. Zhang, Z. Du, C. Zhong, W. Yan, H. Ju, W. Chu, H. Jiang, C. Wu and Y. Xie, Adv. Mater., 2019, 31, 1807468 CrossRef PubMed.
- Q. Wang, D. Sun-Waterhouse, T. Zhang and G. Waterhouse, SmartMat, 2021, 2, 154–175 CrossRef CAS.
- J. Zhang, G. Shi, J. Liu, J. Chen and L. Dai, Angew. Chem., Int. Ed., 2016, 128, 2270–2274 CrossRef.
- W. Zhang, Z. Wu, H. Jiang and S. Yu, J. Am. Chem. Soc., 2014, 136, 14385–14388 CrossRef CAS PubMed.
- H. Shen, E. Gracia-Espino, J. Ma, K. Zang, J. Luo, L. Wang, S. Gao, X. Mamat, G. Hu, T. Wagberg and S. Guo, Angew. Chem., Int. Ed., 2017, 56, 13800–13804 CrossRef CAS PubMed.
- Y. Zhou, M. Liu, C. Zou, Z. Liang, P. D. Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- H. Liu, Q. Lei, R. Miao, M. Sun, C. Qin, L. Zhang, G. Ye, Y. Yao, B. Huang and Z. Ma, Adv. Funct. Mater., 2022, 32, 2207408 CrossRef CAS.
- D. Chen, L.-H. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed.
- W. Xu, H. Tang, H. Gu, H. Xi, P. Wu, B. Liang, Q. Liu and W. Chen, J. Mater. Chem. A, 2022, 10, 14732–14746 RSC.
- Y. Li, D. Fang, X. L. Li, D. Yan, S. Xi, T. C. Li, C. Lin, S. Huang, J. Qiu, X. Xu and H. Y. Yang, Energy Environ. Mater., 2023, 6, e12449 CrossRef CAS.
- H. Shang, X. Zhou, J. Dong, A. Li, X. Zhao, Q. Liu, Y. Lin, J. Pei, Z. Li, Z. Jiang, D. Zhou, L. Zheng, Y. Wang, J. Zhou, Z. Yang, R. Cao, R. Sarangi, T. Sun, X. Yang, X. Zheng, W. Yan, Z. Zhuang, J. Li, W. Chen, D. Wang, J. Zhang and Y. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- C. Chen, M. Sun, F. Zhang, H. Li, M. Sun, P. Fang, T. Song, W. Chen, J. Dong, B. Rosen, P. Chen, B. Huang and Y. Li, Energy Environ. Sci., 2023, 16, 1685–1696 RSC.
- A. Yang, K. Su, W. Lei, Y. Tang and X. Qiu, Adv. Energy Mater., 2022, 13, 2203150 CrossRef.
- R. Li, W. Fan, P. Rao, J. Luo, J. Li, P. Deng, D. Wu, W. Huang, C. Jia, Z. Liu, Z. Miao and X. Tian, ACS Nano, 2023, 17, 18128–18138 CrossRef CAS PubMed.
- J. Wang, W. Zang, S. Xi, M. Kosari, S. J. Pennycook and H. C. Zeng, J. Mater. Chem. A, 2020, 8, 17266–17275 RSC.
- N. Yuan, Y.-R. Deng, S.-H. Wang, L. Gao, J.-L. Yang, N.-C. Zou, B.-X. Liu, J.-Q. Zhang, R.-P. Liu and L. Zhang, Tungsten, 2022, 4, 269–283 CrossRef.
- J. Pei, L. Yang, J. Lin, Z. Zhang, Z. Sun, D. Wang and W. Chen, Angew. Chem., Int. Ed., 2024, 63, e202316123 CrossRef CAS PubMed.
- J. Wang, H. Li, S. Liu, Y. Hu, J. Zhang, M. Xia, Y. Hou, J. Tse, J. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 181–185 CrossRef CAS PubMed.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS.
- W. Cheng, X. Zhao, H. Su, F. Tang, W. Che, H. Zhang and Q. Liu, Nat. Energy, 2019, 4, 115–122 CrossRef CAS.
- H. B. Yang, J. Miao, S.-F. Hung, J. Chen, H. B. Tao, X. Wang, L. Zhang, R. Chen, J. Gao, H. M. Chen, L. Dai and B. Liu, Sci. Adv., 2016, 2, e1501122 CrossRef PubMed.
- H. Jang, W. Jin, G. Nam, Y. Yoo, J. S. Jeon, J. Park, M. G. Kim and J. Cho, Energy Environ. Sci., 2020, 13, 2167–2177 RSC.
- H. Jiang, J. Gu, X. Zheng, M. Liu, X. Qiu, L. Wang, W. Li, Z. Chen, X. Ji and J. Li, Energy Environ. Sci., 2019, 12, 322–333 RSC.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2020, 32, 1905679 CrossRef CAS PubMed.
- J. Yan, Y. Wang, Y. Zhang, S. Xia, J. Yu and B. Ding, Adv. Mater., 2021, 33, 2007525 CrossRef CAS PubMed.
- Z. Yang, C. Zhao, Y. Qu, H. Zhou, F. Zhou, J. Wang, Y. Wu and Y. Li, Adv. Mater., 2019, 31, 1808043 CrossRef PubMed.
- C. Yang, S. Zai, Y. Zhou, L. Du and Q. Jiang, Adv. Funct. Mater., 2019, 29, 1901949 CrossRef.
- W. Liu, J. Zhang, Z. Bai, G. Jiang, M. Li, K. Feng, L. Yang, Y. Ding, T. Yu, Z. Chen and A. Yu, Adv. Funct. Mater., 2018, 28, 1706675 CrossRef.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- J. Yang, X. Wang, B. Li, L. Ma, L. Shi, Y. Xiong and H. Xu, Adv. Funct. Mater., 2017, 27, 1606497 CrossRef.
- B. Chen, X. He, F. Yin, H. Wang, D.-J. Liu, R. Shi, J. Chen and H. Yin, Adv. Funct. Mater., 2017, 27, 1700795 CrossRef.
- X. Chen, J. Pu, X. Hu, Y. Yao, Y. Dou, J. Jiang and W. Zhang, Small, 2022, 18, 2200578 CrossRef CAS PubMed.
- H.-F. Wang, C. Tang, B. Wang, B.-Q. Li and Q. Zhang, Adv. Mater., 2017, 29, 1702327 CrossRef PubMed.
- B.-Q. Li, C.-X. Zhao, S. Chen, J.-N. Liu, X. Chen, L. Song and Q. Zhang, Adv. Mater., 2019, 31, 1900592 CrossRef PubMed.
- Z. Jiang, W. Sun, H. Shang, W. Chen, T. Sun, H. Li, J. Dong, J. Zhou, Z. Li, Y. Wang, R. Cao, R. Sarangi, Z. Yang, D. Wang, J. Zhang and Y. Li, Energy Environ. Sci., 2019, 12, 3508–3514 RSC.
- H. Shang, W. Sun, R. Sui, J. Pei, L. Zheng, J. Dong, Z. Jiang, D. Zhou, Z. Zhuang, W. Chen, J. Zhang, D. Wang and Y. Li, Nano Lett., 2020, 20, 5443–5450 CrossRef CAS PubMed.
- B. Wu, T. Sun, Y. You, H. Meng, D. M. Morales, M. Lounasvuori, A. B. Askari, L. Jiang, F. Zeng, B. Hu, X. Zhang, R. Tai, Z. J. Xu, T. Petit and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202219188 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 8525–8534 CrossRef CAS PubMed.
- W. Zhou, H. Su, W. Cheng, Y. Li, J. Jiang, M. Liu, F. Yu, W. Wang, S. Wei and Q. Liu, Nat. Commun., 2022, 13, 6414 CrossRef CAS PubMed.
- K. Zhu, X. Zhu and W. Yang, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef CAS PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- S. Zuo, Z.-P. Wu, H. Zhang and X. W. Lou, Adv. Energy Mater., 2022, 12, 2103383 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00134f |
| ‡ C. C., J. C., and M. S. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |