
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScalable electrified cementitious materials production and recycling†
Xiao Kun
Lu‡
 ab,
Wenxin
Zhang‡
ac,
Brianna N.
Ruggiero
b,
Linsey C.
Seitz
*b and
Jiaqi
Li
*ad
ab,
Wenxin
Zhang‡
ac,
Brianna N.
Ruggiero
b,
Linsey C.
Seitz
*b and
Jiaqi
Li
*ad
aAtmospheric, Earth, and Energy Division, Lawrence Livermore National Laboratory, 7000 East Avenue, Livermore, CA 94550, USA. E-mail: li88@llnl.gov
bDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: linsey.seitz@northwestern.edu
cDivision of Engineering and Applied Sciences, California Institute of Technology, 1200 E. California Blvd., Pasadena, CA 91125, USA
dDepartment of Civil and Environmental Engineering, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: licee@umich.edu
First published on 13th November 2024
Abstract
The production of Portland cement, the industry-standard cement, contributes ∼8% of global CO2 emissions through fossil-fuel heating and decomposition of limestone (the primary cement raw material). Decarbonization, e.g., via direct electrification, of this 200-year-old liming routine is extremely challenging at the industry scale. We propose a scalable electrochemical decarbonization approach to circumvent the limestone use by switching to carbon-free calcium silicates from abundant minerals and recycled concrete. Water electrolysis produces protons and hydroxides to drive a pH gradient that accelerates Ca2+ ion leaching from calcium silicates and captures atmospheric CO2 to form carbon-negative CaCO3, which serves as the feedstock for cement manufacturing or as the carbon-mineralized product for cement substitution with permanent carbon storage. Value-added co-products amorphous silica and green H2 further enhance cement performance and supplant fossil fuels for net-zero transition, respectively. The products readily meet present-day regulatory standards and demands, and the approach readily synergizes with business-as-usual cement manufacturing and concrete construction, which are important for upscaling and structural safety, promising ready reception by the public and industries. Blended Portland cement produced through our approach with carbon-negative CaCO3 and silica demonstrates enhanced resilience and achieves carbon neutrality or negativity when incorporating storage or circulation of CO2 from cement plant flue gas, respectively. This low-cost, electrochemical cement production approach using abundant ubiquitous raw materials enables electrification, transition to clean fuel, and decarbonization at a gigaton scale.
Broader contextThe cement industry is the second-largest industrial contributor to global greenhouse gas emissions. Portland cement is the largest regulated and ubiquitous commodity, second only to water. The main decarbonization challenge arises from the decomposition of limestone – the primary cement feedstock – which accounts for 60% of total CO2 emissions. No technology has yet decarbonized this 200-year-old liming routine for producing cement. Here, we develop a novel decarbonization approach to utilize abundant, carbon-free calcium silicates from rocks and wastes. By coupling enhanced weathering of calcium silicates with water electrolysis, we demonstrate the production of carbon neutral/negative calcium carbonate, green hydrogen, and amorphous silica suitable for direct integration with contemporary cement manufacturing. A zero-gap electrolyzer configuration was determined to improve hydrogen productivity and durability. Life cycle assessment and techno-economic analysis indicate this process can maintain profitability while being carbon neutral/negative. This approach could enable gigaton-scale annual decarbonization of the cement industry, meeting regulatory compliance with minimal capital investment. |
Introduction
Cement, the essential binder of concrete – the most used material worldwide, only second to water, is produced at over 4 Gigatonnes per year (Gt per y) and contributes to a significant 8% of global CO2 emissions.1,2 The annual global demand for cement is projected to increase by 50% by 2050 because urbanization and the surge in the renewable energy sector create significant demands for cement in the construction of building and infrastructure. Thus, it is urgent to incorporate carbon capture, utilization, and storage (CCUS) technologies to control and mitigate the carbon footprint of cement manufacturing.3CO2 emissions of Portland cement (the most common cement and a ubiquitous commodity) production mainly arise from (i) limestone (CaCO3) decomposition to CaO and CO2 (∼60% contribution) and (ii) fossil-fuel combustion for cement kiln heating for pyroprocessing (∼30% contribution).4 Despite extensive research, there has been no substantial breakthrough in decarbonization of this 200-year-old global industrial-standard liming routine, where the thermal efficiency of cement rotary kilns has been optimized over the 200 years to a ceiling. Most partial decarbonization strategies rely on partial Portland cement substitution by supplementary cementitious materials (SCMs). Common supplementary cementitious materials are vitreous silicates sourced from industrial byproducts, including fly ash from coal-fired power plants, which accounts for 50% of total share of the current supplementary cementitious materials market, and blast furnace slags from the iron/steel industry.5 However, these conventional supplies of supplementary cementitious materials are currently under shortage of over 0.5 Gt per y, while the shortage will be further exacerbated by the increasing cement demand and decommissioning of CO2-intensive traditional coal power plants and conventional steel manufacturing.6 Regardless, supplementary cementitious materials as a partial substitute for Portland cement are practically limited to <50% replacement level, therefore unable to achieve full cement decarbonization towards carbon-neutral/negative Portland cement.4 Besides, studies on incorporating CCUS and/or clean energy with cement production are burgeoning, but a lack of focus on scalability and long-term CO2-storage in concrete precludes technology transfer, especially since the 1450 °C heating of massive cement kilns is beyond the feasibility of direct electrification, which also lacks economic incentives due to long payback periods of cement plant remodeling.7 Recent studies suggest that many emerging cement decarbonization techniques relying on CO2 mineralization in concrete materials (e.g., CO2 curing of concrete blocks and direct utilization of cement carbonation products as cementitious materials) may not effectively offset the lifecycle carbon footprint of cement at scale.8,9 The lack of international standards for these alternative low-carbon cements also discourages the deployment of low-carbon technologies in the construction sector for the concerns over liabilities and safety.
Herein, we demonstrate a carbon-neutral-to-negative, economically attractive Portland cement production scheme that utilizes abundant carbon-free natural/recycled materials and renewable electricity while co-producing carbon-neutral-or-negative supplementary cementitious materials and clean fuels. The strategy couples water electrolysis and CO2 direct air capture with electrochemical generation of carbon-sequestered CaCO3 – which substitutes limestone as the primary cement manufacturing feedstock and neutralizes limestone decomposition-induced CO2 emissions – from non-carbonaceous precursors, which are highly available worldwide (e.g., basalt – half volume of the Earth's crust surface), and industry wastes (e.g., recycled concrete fines from construction and demolition waste).10 Our approach produces materials (Portland cement feedstock and supplementary cementitious materials) that are compatible with existing Portland cement manufacturing infrastructure and decarbonization technologies,11 avoiding regulatory limits on this safety-sensitive structural material. Portland cement developed through our approach complies with existing international and national standards, and its usage does not require new training for millions of materials and civil engineers or builders globally. Furthermore, the decreasing cost of renewable electricity, now below that of conventional fossil-fuel-powered electricity, together with potential carbon credit savings enabled by the present carbon-neutral-to-negative cement manufacturing scheme, provides further economic competitiveness beyond environmental benefits.11
Fig. 1 demonstrates the potential integration of our approach with cement manufacturing as well as CCUS processes and a circular CO2 pathway. Our room temperature electro-geochemical cell takes in electricity (possibly in the form of renewable energy) as well as captured atmospheric CO2 (or circulated concentrated CO2) and supports water electrolysis, which incurs a pH gradient enabling Ca2+ extraction from precursors calcium silicates to produce CaCO3, amorphous SiO2, and green H2 gas. By delivering the same primary feedstock as current Portland cement production, CaCO3, we provide a cost-efficient, scalable decarbonization and energy transition pathway to the cement industry, which has previously been reluctant to embrace emerging marginal decarbonization technologies due to their costly, long payback periods for retrofitting or remodeling modern cement plants.12
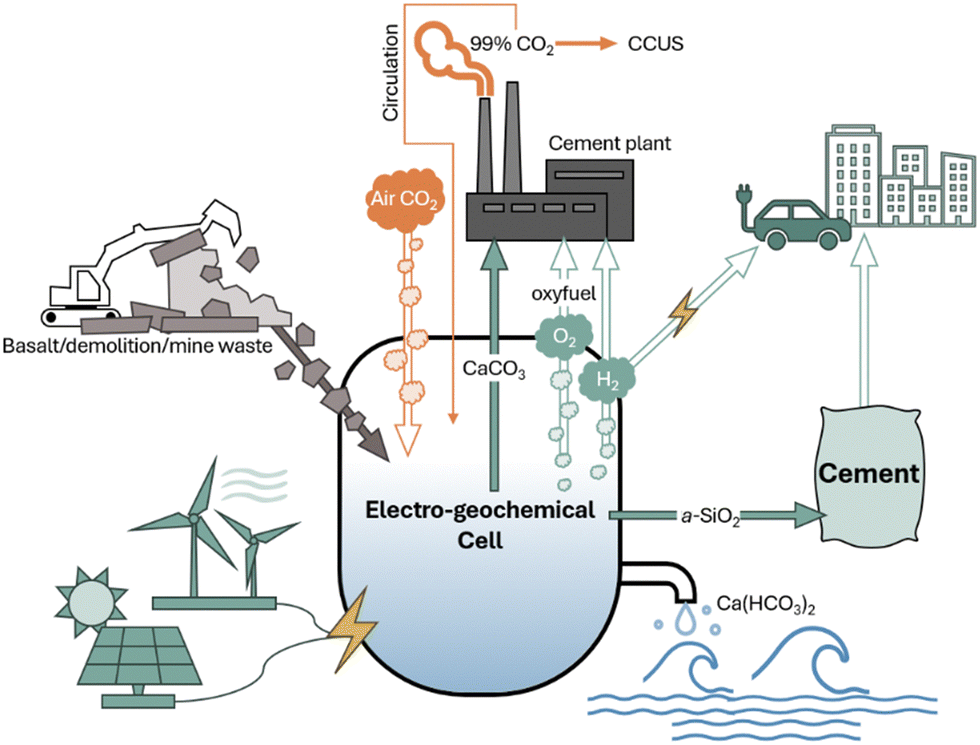 | ||
| Fig. 1 Scheme of the electro-geochemical cell powered by renewable electricity for converting Ca-bearing silicates (industrial/construction/mine wastes and rocks) to CaCO3 and high-value amorphous silica SCM for carbon neutral-to-negative Portland cement. Captured CO2 can transform into dissolved bicarbonate (e.g., Ca(HCO3)2) to mitigate ocean acidification. Water electrolysis generates H2 and O2 as a green energy carrier for zero-emission infrastructure and oxyfuel for CCUS, respectively. | ||
Our strategy offers the following benefits: (1) full carbon-neutralization of limestone decomposition through CO2 circulation or direct air-captured CO2 mineralization and storage at scale, (2) production of high-value supplementary cementitious materials (carbon-negative reactive CaCO3 and highly pure amorphous silica) to further decarbonize cement for carbon credit savings, enhance life-time concrete performance, and lower production and operational costs, (3) H2 generation for industrial heating, power generation, and chemical manufacturing, (4) O2 generation for oxy-fuel combustion aiding in sequestering concentrated CO2 flue gas at cement plants, and (5) generation of calcium (bi)carbonate water to mitigate ocean acidification.13–15 We studied this process in three electrochemical reactor configurations, evaluated the products and performances, and performed life cycle assessment and techno-economic analysis to probe the embodied carbon, energy use, and economic viability.
Results and discussion
Electrolytic dissolution of calcium silicates
| 2H2O + 2e− → 2OH− + H2(g) | (1) |
| 2H2O → 4H+ + O2(g) + 4e− | (2) |
| CaSiO3(s) + 2H+ → Ca2+ + SiO2(s) + H2O | (3) |
| Ca2+ + 2OH− + CO2(g) → CaCO3(s) + H2O | (4) |
 | ||
| Fig. 2 (A) Scheme of reactions occurring in the H-cell for weathering of CaSiO3 at room temperature; cation exchange membrane (CEM), anion exchange membrane (AEM), and bipolar membranes (BPM) are used as ion separators. (B) e-CaCO3-to-CaSiO3 product/precursor weight ratio post electrolysis. (C) Schematic of flow cell electrolyzer operation using ion exchange membranes and non-precious metal catalysts. (D) Current density of constant potential hold at 7 V. (E) Schematic of zero-gap electrolyzer and cascade cement recycling process. (F) Current density reached with zero-gap electrolyzer at 3.7 V for 12 distinct runs. | ||
The series of reactions occurring in the H-cell sums up to eqn (5).
| 2CaSiO3(s) + 2CO2(g) + 2H2O → 2CaCO3(s) + 2H2(g) + O2(g) + 2SiO2(s) | (5) |
At cell potentials ranging from 3 to 9 V, the current during electrolysis increases over initial operation due to increased electrical conductivity from H+ and OH− generation (Fig. S2A and B, ESI†) and decreases due to decreased electrolyte levels over longtime operation (Fig. S2, ESI†). The resulting mass ratio of e-CaCO3-to-CaSiO3 (i.e., calcium conversion rate) linearly scales with charge passed until the theoretical limit of ∼0.9 (Fig. 2B). Dissolved Ca2+ ion content in the anolyte increases linearly until ∼50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 C has passed, at which point the precursor no longer provides dissolvable Ca, and most of the Ca2+ ions have diffused to the cathodic compartment (Fig. S4B, ESI†).
000 C has passed, at which point the precursor no longer provides dissolvable Ca, and most of the Ca2+ ions have diffused to the cathodic compartment (Fig. S4B, ESI†).
For more efficient continuous operation, the precursor is placed in an agitated anolyte reservoir, so that continuous operation is achieved by replacing the precursor reservoir once the leaching of Ca2+ ions completes. Two consecutive batches of 2 g CaSiO3 yielded a total of 2.65 g e-CaCO3 (74% Ca converted) after 44 h of electrolysis. Although the flow-cell electrolyzers are operated continuously, membrane fouling due to CaCO3 precipitation on the cation exchange membrane (CEM) decreases current density by 30% from the first to the second cycle (Fig. S5 and S6, ESI†). Such fouling is common in state-of-the-art processes; therefore, for flow cell electrolyzers to be viable, engineering of membrane permeability for Ca2+ ions and mass transport to rapidly remove precipitates at the membrane is essential.19,20
As-produced e-HNO3 and e-NaOH were also utilized to recycle commercial ordinary Portland cement hardened paste (mainly consisting of hydrated calcium silicates), as a model for recycled concrete fines from construction and demolition waste. Ground paste was treated with e-HNO3 at pH < 1 and rapidly exhibited complete dissolution. Si in solution was recovered by adjusting the pH to 4.5 with e-NaOH (pH > 13) and filtered as a-SiO2 precipitates. Ca2+ ions in the solution carbon-mineralize to form e-CaCO3 with e-NaOH and air CO2, or solution with captured CO2 (e.g., 0.1 M Na2CO3). Performing dissolution and precipitation of cementitious materials allows for precise control over crystallization parameters23 and avoids heterogeneity in the electrochemical system, as real-world precursor feeds often contain impurities that are detrimental to membrane and electrode stability.24
Product characterization and performance
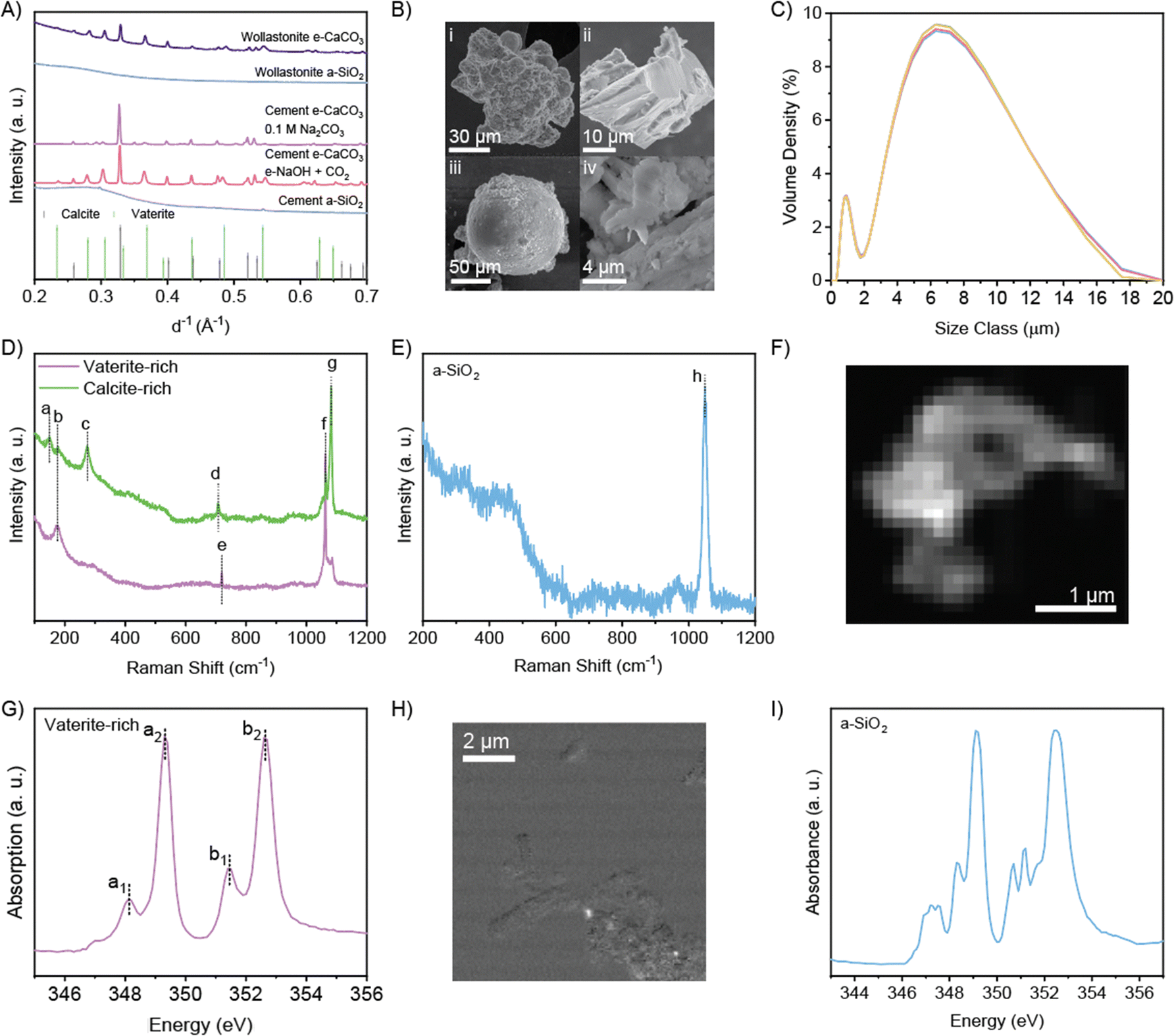 | ||
| Fig. 3 (A) XRD of e-CaCO3, a-SiO2, and references. (B) Scanning electron microscopy images of (i) aggregated vaterite, (ii) angular calcite, (iii) vaterite sphere, and (iv) a-SiO2. (C) Particle size distribution of a-SiO2 product after 1-day electrolysis at 5 V. Five measurements were recorded on the same sample mixture. (D) and (E) Raman spectrum of e-CaCO3 and a-SiO2 products. See Table S5 (ESI†) for assignment of peaks a–h. (F) and (G) STXM image and Ca L-edge NEXAFS spectrum of an e-CaCO3 particle. (H) and (I) STXM Ca mapping and Ca L-edge NEXAFS spectrum of a-SiO2 sample. | ||
As for the major decalcified coproduct, a-SiO2, XRD demonstrates that a-SiO2 from both precursors are amorphous according to the diffuse peak around d−1 of 0.25–0.3 Å−1 (Fig. 3A). Co-produced a-SiO2 from CaSiO3 (initial Si/Ca atomic ratio of 1) leaching retains the morphology of the precursor CaSiO3 and that from dissolution and re-precipitation of recycled cement paste (initial Si/Ca atomic ratio of 0.3) exhibits submicron sizes of the precipitates (Fig. 3Biv and Fig. S12–S14, ESI†). Energy dispersive X-ray spectroscopy (EDX) reveals high Si/Ca atomic ratios of 120 from leaching CaSiO3 and 24 from re-precipitation of recycled cement paste (Tables S6, S7 and Fig. S15, S16, ESI†), promising the superior quality of the present a-SiO2 as a siliceous supplementary cementitious material. The laser diffraction-based particle size characterization demonstrates that precursor CaSiO3 particles range ∼1–30 μm in size with a mean value of 7.2 μm (Fig. S17, ESI†), while the enhanced weathering product a-SiO2 particles range 0.2–20 μm in size with a mean value of 6.8 μm (Fig. 3C), primarily following the particle size distribution of the precursor with a small fraction of finer particles likely caused by fracturing upon Ca-leaching, agreeing with the measured specific area increase from 0.9 m2 g−1 of CaSiO3 to 1.7 m2 g−1 of decalcified a-SiO2. Again, both the particle size distribution and specific area of the electrochemical product fall closely to that of anhydrous Portland cement, suggesting their direct usage without the need for grinding or flowability adjustment via adding polymeric superplasticizers, whose manufacturing is intensive in cost, energy, and carbon emissions.26–29
Fig. 3D and E show the Raman spectra of e-CaCO3 and a-SiO2 with peak assignments given in Table S8 (ESI†). The two CaCO3 polymorphs were differentiated by the position of the peaks corresponding to in-plane bending and symmetric stretching of CO32−, i.e., peaks d and g at 708 and 1082 cm−1, respectively, as the signatures of the vaterite polymorph versus peaks e and f at 720 and 1063 cm−1, respectively, as the signatures of the calcite polymorph (Fig. 3D). Both calcite and vaterite-rich e-CaCO3 regions display peaks below 300 cm−1, which are assigned to the translational and rotational lattice modes. The Raman spectrum of a-SiO2 exhibits a prominent peak at 1049 cm−1, attributed to the symmetric stretching of Q3 (silicate tetrahedra connected at three corners into the network). This dominant peak reveals full dissolution of precursor CaSiO3, which contains solely Q2 (silicate tetrahedra connected at two corners as a chain), and that acid-stable a-SiO2 is left with Q3 structure that is the most resistant to acid leaching (Fig. 3E).30 This Q3 signal also suggests the silica product to be amorphous.
STXM-NEXAFS analysis further confirms the presence of the vaterite polymorph in e-CaCO3 produced (Fig. 3F and G). In the Ca L2,3-edge NEXAFS spectrum, an a1 peak (L3 pre-edge) position of 348.1 eV and a ΔE of 1.3 eV between the b2 peak (L2 edge) and b1 peak (L2 pre-edge) are characteristic of the vaterite phase (Fig. 3G).31 The absence of the calcite polymorph under STXM was due to the absorption saturation limit of the thicker calcite particles. The a-SiO2 resulting from precursor CaSiO3 contains trace amounts of Ca structures indicated by bright spots in Ca element mapping (Fig. 3H), which shows the spatially resolved difference in the absorbance level pre- versus on-Ca L3-edge, with the bright spot indicating large difference thus abundance of Ca (Fig. S18, ESI†). NEXAFS spectrum obtained from the Ca-remaining region on a-SiO2 exhibits multiple L2 and L3 peaks, suggesting disordered Ca coordination and low Ca content (Fig. 3I).
The cement hydration reaction is exothermic, where its main heat evolution peak (at ∼5 h for the ordinary Portland cement reference in Fig. 4A) signifies the rapid growth of calcium silicate hydrate (C–S–H, the primary binding phase and strength contributor of concrete) and of ettringite (the minor phase contributing to the setting of concrete).32,33 During this stage, the hydration products grow and interlock, allowing cement paste to harden and gain strength, which continues to increase asymptotically in the long term with continued hydration. In electrochemical-products-blended Portland cement, the main heat evolution peak is ∼1 h earlier with significantly increased area under the curve, meaning accelerated hydration reaction onset and greater heat flow compared to ordinary Portland cement (Fig. 4A). Both e-CaCO3 and a-SiO2 promote Portland cement hydration,34,35 accelerating the formation of C–S–H and the setting and strength development of cement. In detail, e-CaCO3 reacts with aluminates in Portland cement to form calcium carboaluminate hydrates for additional strength gain. e-CaCO3 also provides nucleation sites to facilitate C–S–H growth.36,37 a-SiO2 consumes portlandite (Ca(OH)2) – a chemically and mechanically vulnerable phase in hydrated Portland cement – to facilitate further C–S–H formation via the pozzolanic reaction.36,37
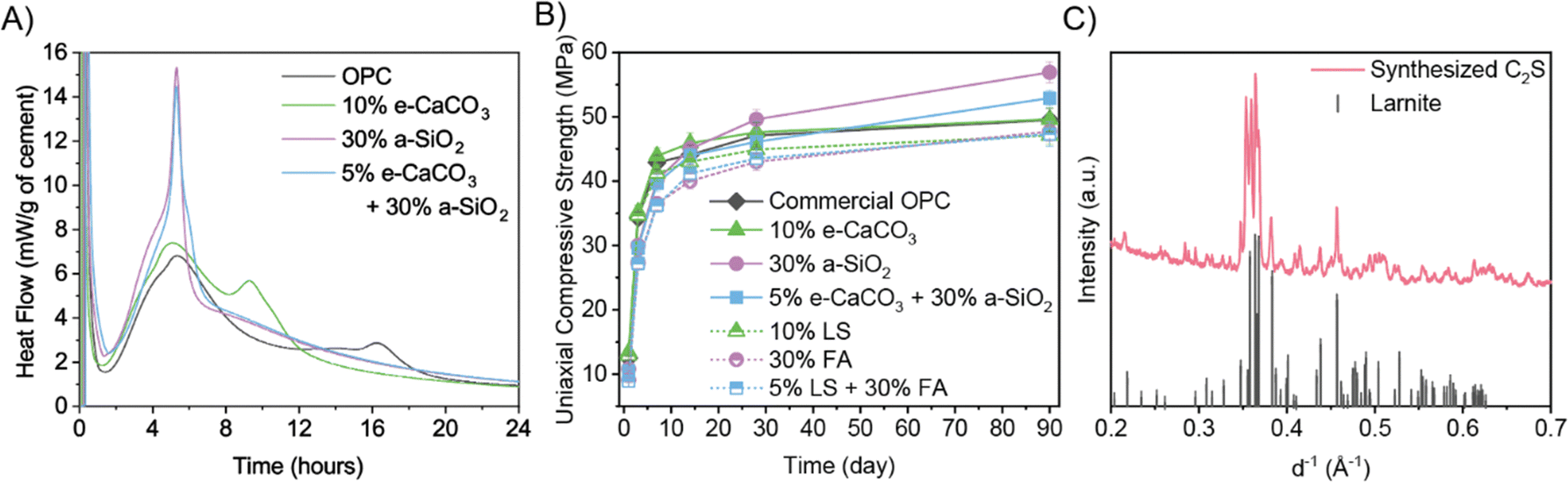 | ||
| Fig. 4 (A) Calorimetry of 5 g of (blended) cement paste. (B) Compressive strength of (blended) cement paste using electrochemical products (e-CaCO3 and a-SiO2) or conventional supplementary cementitious materials (limestone powder and fly ash) over 90 days. OPC = ordinary Portland cement; LS = limestone; FA = fly ash. (C) XRD of synthesized C2S and reference. | ||
Compared to ordinary Portland cement and conventional limestone powder-and-fly ash-blended Porland cements, the electrochemical-products-blended Portland cements achieve greater compressive strengths at all curing ages, with especially pronounced enhancement at late ages (Fig. 4B), explained by the higher reactivity of metastable vaterite in e-CaCO3 and the enhanced pozzolanic reactivity of a-SiO2. The enhancement effect of vaterite over calcite has also been found in other cement systems.38 Using the present electrochemical products overcomes the typical problem of low early-age strength of blended Portland cement due to the limited reactivity of common industrial byproduct supplementary cementitious materials, as manifested by the conventional blends compared to ordinary Portland cement in terms of their lower 28-day strengths, the most critical metric in practical construction applications. Moreover, while it is widely agreed that ordinary Portland cement gains the majority of compressive strength by 28 days,39 the electrochemical-products-blended Portland cement demonstrates 15% continued strength gain between 28 to 90 days at 30 wt% a-SiO2 substitution in contrast to just 6% gain for the ordinary Portland cement reference, benefiting from the pozzolanic reactivity of a-SiO2. The a-SiO2 blended cement typically shows higher durability (e.g., resistance to sulfate and acidic environments) due to the consumption of Ca(OH)2 and refined pore structure, meaning reduced repair/maintenance and longer lifetime, further decreasing the carbon footprint, especially when normalized by the service life of concrete structures.6
Using supplementary cementitious materials as Portland cement substitutes (typically up to 35 wt%) has been successfully applied in the cement industry for over 100 years, penetrating the market for our process to be scaled up.40 Electrochemically produced a-SiO2 is more chemically homogeneous than the highly heterogeneous conventional supplementary cementitious materials (e.g., fly ash and volcanic ash) and emerging supplementary cementitious materials (e.g., municipal solid waste incineration ash). These industrial waste-sourced supplementary cementitious materials experience varying pozzolanic reactivity due to intermixed inert impurities (e.g., mullite and quartz41,42) and are prone to cause cracking failures of concrete due to other detrimental impurities (e.g., aluminum fines).6 Thus, our a-SiO2 addresses the challenging quality control of blended Portland cement incorporating existing supplementary cementitious materials regarding mechanical and durability performances. Therefore, by superseding conventional supplementary cementitious materials with electrochemical products, our approach further contributes to the decarbonization and sustainability of built environment by enhancing the lifetime of concrete structures.
Life cycle assessment and techno-economic analysis
A cradle-to-gate life cycle assessment (LCA) compares the carbon footprint of industrial-standard Portland cement manufacturing (Fig. 5A and F) and the electrochemical Portland cement production schemes (Fig. 5B–E and G and H) under three treatment models for different destinations of concentrated CO2 flue gas from the cement kiln: “emission” model assuming CO2 release to the atmosphere, “circulation” model assuming circulating CO2 to feed the electrochemical reaction, and “CCS” assuming CO2 capture and storage. For each model, two or three scenarios are considered: “-conv” indicates a conventional cement manufacturing scheme business-as-usual; “FF” or “H2” indicate an electrochemical manufacturing scheme using fossil fuel or green H2 for cement kiln fueling, respectively. Thus, “FF” scenarios represent a low capital-intensive scheme using our electrochemical products to feed existing Portland cement plants; “H2” scenarios represent a moderately capital-intensive scheme additionally using our electrochemical co-product green H2 to fuel a cement kiln. Note S1 (ESI†) provides the energy consumption analysis, and Note S2 (ESI†) provides the full description of our LCA methodology and assumptions. Conventional ordinary Portland cement manufacturing (scenario “emission-conv”) incurs global warming potential (GWP) of 0.93 kg CO2-eq per kg ordinary Portland cement produced (Fig. 6Ai and Fig. S20 and Table S9, ESI†): 0.51 kg from limestone decomposition at the kiln, 0.39 kg from combustion of conventional fuels (mainly coal/coke) at the kiln, and 0.03 kg from electricity or transportation during other processes (e.g., quarrying, transporting, grinding, and in-plant conveying). By blending ordinary Portland cement with conventional supplementary cementitious materials (limestone powder and fly ash) up to 35 wt%, the total GWP is abated by up to 34%.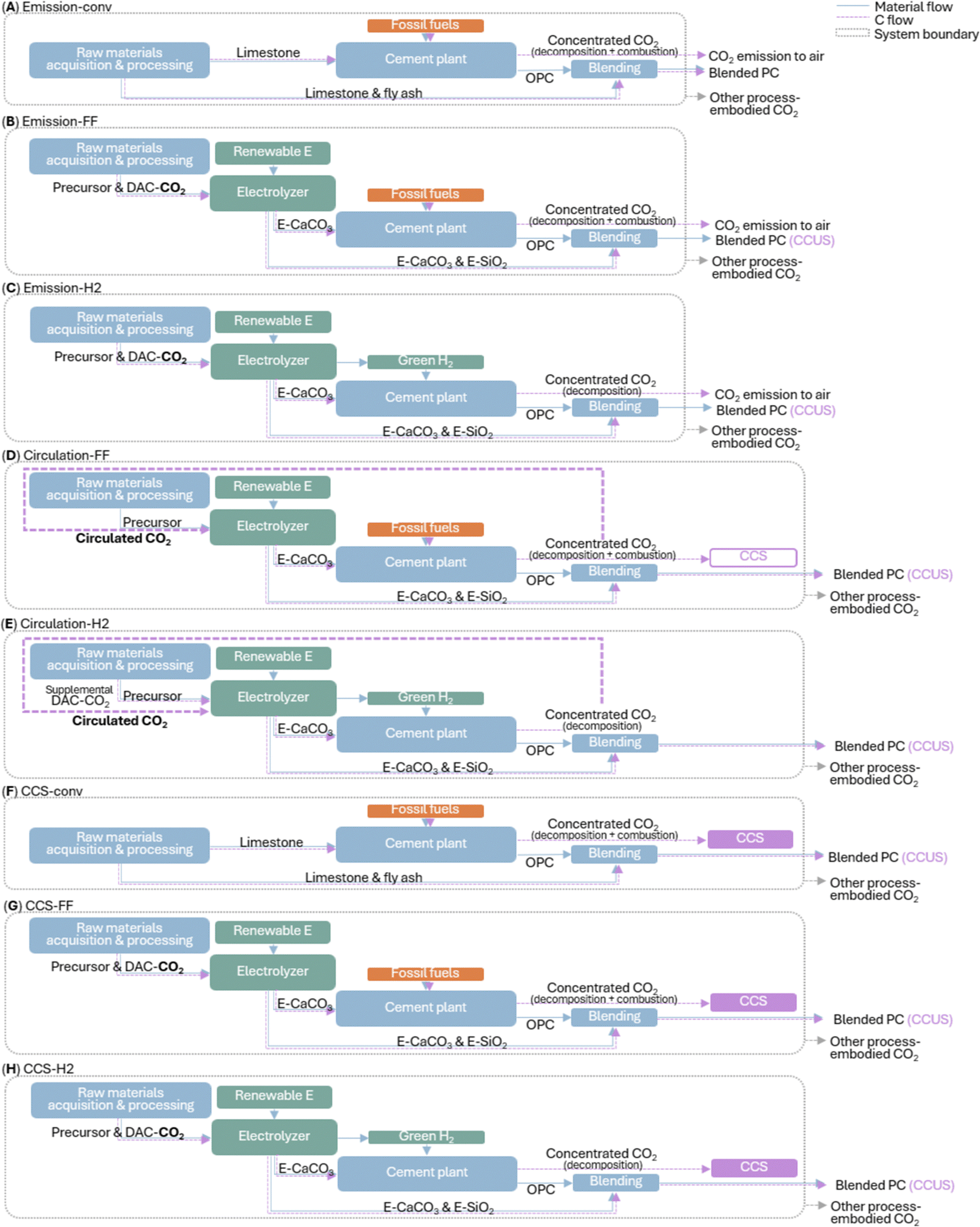 | ||
| Fig. 5 (A)–(H) Schematics of comparative LCA scenarios. In scenarios (B)–(E), (G) and (H), precursor refers to wollastonite or recycled cement paste. RE = renewable electricity; DAC = direct air capture; CC(U)S = carbon capture, (utilization,) and storage. Note that to clearly illustrate the electrochemical products and conventional equivalents’ flow in the manufacturing process, other cement production raw materials, e.g., clay, are not plotted in the diagrams but accounted for. | ||
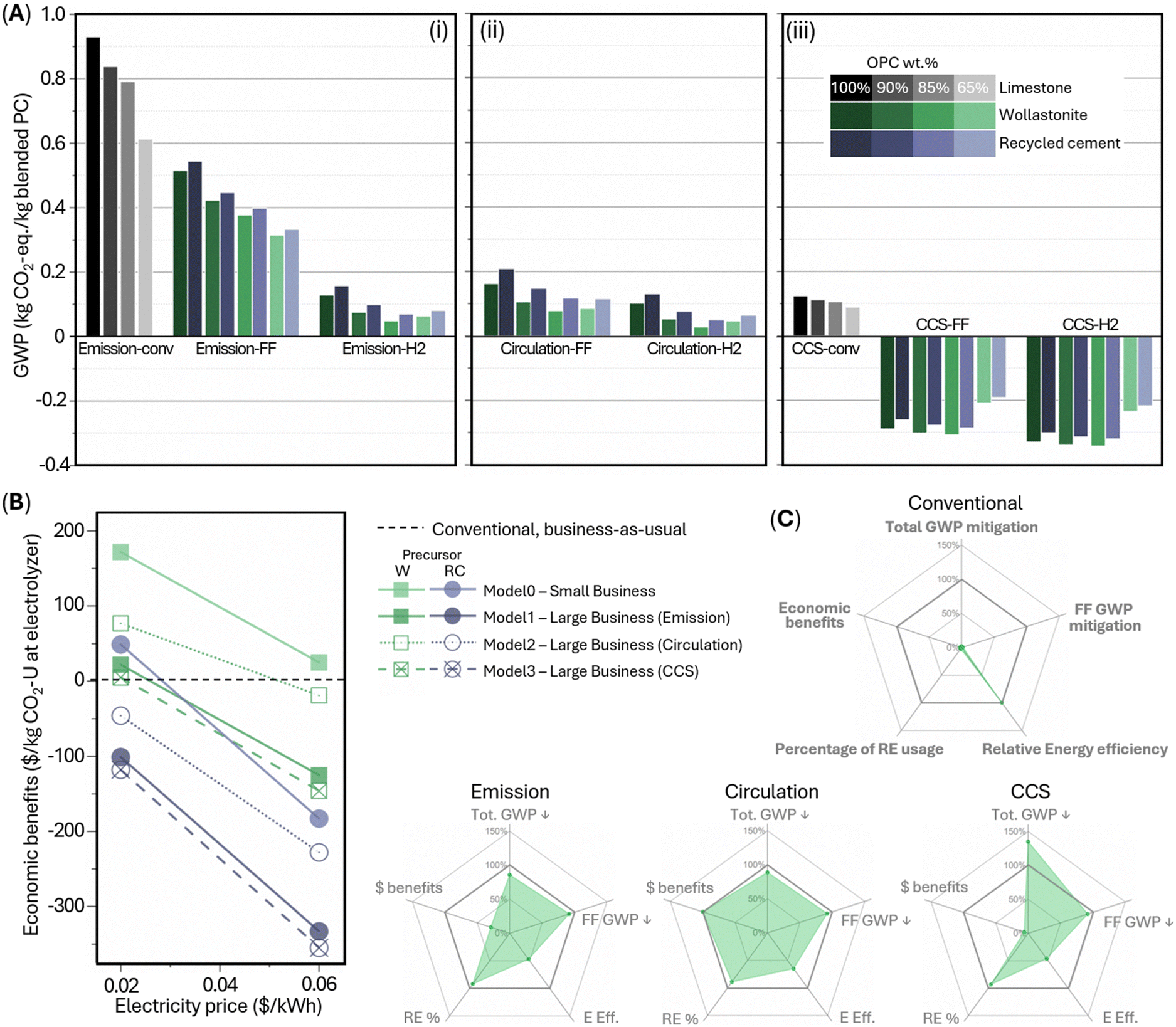 | ||
| Fig. 6 (A) Cumulative GWP of four blended cements (100 wt% ordinary Portland cement (OPC); 90 wt% OPC with 10 wt% CaCO3; 85 wt% OPC with 15 wt% CaCO3; 65 wt% OPC with 5 wt% CaCO3 and 30 wt% SiO2) under eight manufacturing scenarios following three CO2 treatment models (i) “emission”; (ii) “circulation”; (iii) (“CCS”); legends are shared between (i)–(iii) with grey for conventional manufacturing, green for wollastonite (CaSiO3) as the precursor, and purple for recycled cement paste as the precursor. (B) Cumulative economic benefits of ordinary Portland cement manufacturing under four TEA-models relative to conventional manufacturing (black dashed line); green for wollastonite, W, as the precursor and purple for recycled cement paste, RC, as the precursor. (C) Radar plots for comprehensive comparison of “conventional” and three wollastonite-based “large business” scenarios under different CO2 treatment models; RE = renewable energy; FF = fossil fuel; E = energy. | ||
In all electrochemical manufacturing scenarios, CO2-mineralized e-CaCO3 decarbonizes cement manufacturing in two-fold: (i) as the Portland cement feedstock, e-CaCO3 fully decarbonizes the decomposition-induced CO2 emissions; (ii) as a supplementary cementitious material (up to 15 wt% of Portland cement substitution), e-CaCO3 allows permanent CO2 storage and utilization in concrete, which alone, is a carbon-negative process. Besides, a-SiO2, the carbon-neutral electrochemical coproduct, is directly incorporated as a supplementary cementitious material up to 30 wt% of Portland cement substitution, or together with e-CaCO3 up to 35 wt%.
In scenario “emission-FF” with business-as-usual cement plant operation (Fig. 5B and 6Ai), wollastonite-based, electrochemical-products-blended Portland cement at 0–35 wt% substitution levels achieve 45–66% CO2 abatement. Switching the precursor from wollastonite to recycled cement paste increases GWP moderately, owing to the increased electrical energy demand to fully dissolve recycled cement paste compared to partial dissolution of wollastonite (5.40 vs. 2.86 MJ kg−1 e-CaCO3 produced). In scenario “emission-H2” (Fig. 5C and 6Ai), additionally, the co-product green H2 replaces fossil fuels for kiln heating, further offsetting the rest combustion-induced emissions, leading to approximately net zero emission of the cement kiln and total CO2 abatement by 86–95%. Such fuel switching is practical as the industry has adopted coal/coke as the primary fuel for current kilns for cost saving, superseding the dominance of natural gas as the primary fuel in the 1970s.43 Specifically, the 15 wt% e-CaCO3 blend achieves the lowest carbon intensity 0.049 kg CO2-eq per kg blended Portland cement produced, owing to the direct utilization of carbon-mineralized e-CaCO3 as a partial substitute for cement. Meanwhile, electrochemical production-associated processes, e.g., filtering of solid electrochemical products and pumping of CO2 and H2 gases, contribute marginally to the overall GWP.
Furthermore, under the “circulation” model (Fig. 5D, E and 6Aii), the flue gas with concentrated CO2, from e-CaCO3 decomposition (and fossil fuel combustion in “FF” scenarios), does not exit the system but is cycled as the electrochemical feedstock. Using the concentrated CO2 provides great energy benefits and CO2 abatement by avoiding the entropic penalty of gas separation in direct air capture (DAC) and low efficiency ∼10%, which invokes DAC energy demand of 2 MJ kg−1 e-CaCO3 produced. Note that the excess CO2 emissions from fossil fuel combustion in scenario “circulation-FF” is considered for CCS, while the net CO2 removal via CCUS in e-CaCO3 as a cement partial substitute in scenario “circulation-H2” is accounted for by introducing small amount of supplemental DAC-CO2 to keep a steady CO2 supply. Overall, by curtailing both the cement pyroprocessing CO2 emissions and energy-intensive DAC, wollastonite precursor-based “circulation” model leads to 83–92% GWP abatement without cement kiln modification (“FF”) or 89–97% with fuel switching, achieving a minimum of 0.029 kg CO2-eq per kg blended Portland cement produced.
Alternatively, under the “CCS” model (Fig. 5F–H and 6Aiii), the flue gas from cement pyroprocessing is not released or circulated but directly captured, transported, and geologically stored. Thus, the DAC-CO2 intake and mineralization at the electrolyzer lead to carbon-negative cement in all examined “CCS-FF” and “CCS-H2” scenarios, achieving as low as −0.342 kg CO2-eq per kg blended Portland cement produced (wollastonite-based scenario “CCS-H2” at 15 wt% e-CaCO3 as a partial cement substitute). On the other hand, scenario “CCS-conv” is associated with a higher GWP than “circulation-FF” and “circulation-H2,” suggesting the significant merit of a circular CO2 scheme, which enables almost net-zero cement manufacturing at low-to-moderate cement plant modifications and eliminates the need for the long-distance CO2 pipeline transport to geological CCS reservoirs that raise liability and infrastructure rollout issues.
Based on the life cycle inventory analysis of energy consumption, the wollastonite-based electrolyzer operation is associated with 2.9 MJ kg−1 e-CaCO3 produced, according to the thermodynamic energy requirement for the electrochemical production of 20 mol H+ required for leaching of Ca2+ per kg e-CaCO3 produced. This amounts to 4.5 MJ kg−1 ordinary Portland cement produced at a typical industrial electrolyzer efficiency of 75% (Notes S1 and S2, ESI†). This electrolyzer electrical energy consumption is moderately higher than the equivalent cement kiln thermal energy consumption of 3.0 MJ kg−1 ordinary Portland cement produced. Although the energy requirement in the conventional cement manufacturing scheme is lower than our electrochemistry-based counterparts, the inaccessibility to electrified heating in conventional manufacturing schemes always inevitably results in higher GWP. DAC and CCS derive additional demands of 2.3 and 0.4 MJ kg−1 ordinary Portland cement produced, respectively. Overall, scenario “circulation-H2” has the lowest energy consumption among all electrochemical manufacturing schemes, achieving a minimum of 3.96 MJ kg−1 blended Portland cement produced (Fig. S21, ESI†), comparable to the conventional cement manufacturing cradle-to-gate energy consumption of 3.84 MJ kg−1 ordinary Portland cement produced – but importantly, the former fully supplants the fossil fuel use by renewable energy (renewable electricity and green H2), accelerating cement manufacturing transition to clean electricity at the industry scale. Using recycled cement paste as the precursor instead, the electricity usage of the electrolyzer increases to 5.40 MJ kg−1 e-CaCO3 produced. Thus, the cradle-to-gate lifecycle energy consumption with recycled cement paste as the precursor increases by ∼3 MJ kg−1 blended Portland cement produced compared to wollastonite as the precursor under equivalent scenarios (Fig. S21, ESI†).
A techno-economic analysis (TEA) for ordinary Portland cement manufacturing via the present electrochemical scheme is conducted in relative to the “conventional” manufacturing (Fig. 6B and Table S5, ESI†). Note S3 (ESI†) contains a description of our TEA methodology and assumptions. TEA-model0 is a “small business” that operates only the electrochemical production with direct sale of all electrochemical products, e.g., to existing Portland cement plants. Maximum benefits of $172/t CO2 utilized at the electrolyzer, resulted from: sales of $170, $91, and $23 from a-SiO2, green H2, and e-CaCO3, respectively; carbon credit saving of $130; costs of $41, $48, and $23 from raw materials, electrolyzer operational cost, and DAC operation cost, respectively; and $27 and $100 from capital expenditure of electrolyzer and DAC, respectively.
TEA-model1, 2, and 3 consider a “large business” that runs both the electrochemical production and an existing Portland cement plant switched to green H2 kiln fueling ($15/t CO2 utilized from cement plant fossil fuels saving), consuming e-CaCO3 within the system boundary and treating green H2 surplus and a-SiO2 for sale. These models provide large corporations, who already own Portland cement plants, an economically attractive, near-term pathway towards clean energy transition and decarbonization without establishing remarkably capital-intensive new Portland cement plants. Among the three models for managing flue gas CO2 from cement pyroprocessing, “circulation” leads to the greatest economic benefits of maximumly $77/t CO2 utilized at the electrolyzer. Even though the “circulation” model does not allow for claiming carbon credits, its reduction of DAC operational and capital expenses is substantial. Future reduction of the DAC capital expenditure and increase of energy efficiency is expected to increase the economic benefits of “emission” and “CCS” models, but under the current assumptions, the margins are at least $55/t and $72/t CO2 utilized at the electrolyzer, respectively, lower than the economic benefit of the “circulation” model.
While scenarios involving wollastonite as the precursor are mostly economically beneficial at low to intermediate electricity prices, the scenarios involving recycled cement paste as the precursor are generally not as profitable as conventional cement manufacturing. However, the present TEA does not estimate the eliminated cost of waste disposal for recycled cement paste. Moreover, future opportunities with lower DAC and CCS costs and lower renewable electricity prices could increase economic competitiveness. Future studies are suggested to expand the system boundary to comprehensively evaluate the benefits of recycled cement paste versus wollastonite considering their different sources of industrial waste versus natural reserves.
Fig. 6C provides a comprehensive comparison between the models based on LCA and TEA using five metrics, where greater values and the area enclosed indicate model performances superior to the “conventional” reference. Total GWP mitigation and fossil fuel GWP mitigation evaluates the percentage of total and fossil fuel-induced GWP reduction relative to the “conventional” model, respectively, demonstrating that all three electrochemical production models are able to achieve >90% mitigation at >90% of renewable energy usage rate. Particularly, the “CCS” model is carbon-negative and reaches 135% total GWP mitigation. Relative energy efficiency evaluates the total energy demand of electrochemical cement manufacturing schemes relative to the “conventional” reference: values of ∼47% for “emission” and “CCS” models and 65% for “circulation” model result from the high energy demand for DAC and electrolyzer, despite fundamentally transforming cement manufacturing from fossil fuel-intensive to renewable energy-dominant. Lastly, the “circulation” model gains the greatest economic benefits relative to business-as-usual cement manufacturing, while “emission” and “CCS” models have intermediate to low profit margins, which could be improved with the maturity of carbon management and renewable electricity technologies in foreseeable future. In general, while “CCS” model achieves substantial carbon-negativity, “circulation” model is overall highly rated for the highest economic benefits, greater energy efficiency, and approximate carbon-neutrality.
Our sensitivity analysis primarily examines the influence of the renewable electricity's embodied carbon footprints on the total GWP of the electrochemical manufacturing scenarios. Fig. 7A lists the carbon footprint of renewable electricity resources up to 0.04 kg CO2-eq. per MJ of electricity generated as well as the projected carbon footprint of ∼0.022 kg CO2-eq. per MJ for Global 2050 electricity generation based on the forecasted 2050 mixed grid. The data points in Fig. 7A–D mark three representative cases of low, medium, and high renewable electricity carbon footprints from, respectively, land-based wind power, photovoltaic (PV) without and with Li-ion battery energy storage for improved stability and steady energy supply. Note that unlike alternative high-temperature electrolyzer requiring continuous electrical heating, the present electrolyzer can operate entirely at low temperatures (<100 °C), requiring significantly less strict operating environments. Therefore, the present electrochemical scheme is potentially more compatible with the use of waste heat from cement kilns and lower-carbon footprint, intermittent renewable electricity resources, relaxing the reliance on high-carbon footprint battery energy storage systems thus offering additional CO2 abatement compared to the alternative high-temperature electrochemical cement manufacturing techniques. In Fig. 7A–D, the slope is dictated by the average specific electricity consumption of blended Portland cement production, equaling 4.3–6.1 and 6.1–7.9 MJ kg−1 blended Portland cement produced for wollastonite-based electrolysis at high and low energy efficiency, respectively, or 5.3–7.0 and 7.6–9.5 MJ kg−1 blended Portland cement produced if recycled cement paste-based. Overall, the relative trends between the scenarios are consistent across the different electricity sources and precursors: “CCS-FF” and “CCS-H2” are mostly carbon-negative; “circulation-FF,” “circulation-H2,” and “emission-H2” achieve nearly carbon-neutral at low renewable electricity carbon footprint and maintain >50% CO2 abatement even at high renewable electricity carbon footprint; “emission-FF” leads up to ∼50% CO2 abatement but catch up conventional manufacturing (“emission-conv”) at higher renewable electricity carbon footprint due to the high electrical energy demand from DAC and electrolyzer operation. Nevertheless, the blended Portland cement produced in “emission-FF” at high renewable electricity carbon footprint would be still more industrially favorable due to the shortage of coal fly ash for conventional blended cement. Furthermore, the influence of electrolyzer energy efficiency on the total energy consumption is evaluated (Fig. S22, ESI†). At high industrial-electrolyzer energy efficiency (75%), relative energy efficiency of electrochemical cement manufacturing, averaged across the various blended cement designs, ranges 35–66% (wollastonite as the precursor) and 25–42% (recycled cement paste as the precursor), by normalizing to the total energy consumption of conventional cement manufacturing, nevertheless, enabling the cement industry to transition from fossil fuels to renewable resources. Lower efficiency (50% – low industrial-electrolyzer energy efficiency) causes decreases to 29–48% and 20–29%, respectively. On the other hand, improving electrolyzer energy efficiency towards 85% and 95% can achieve respectively 72% and 78% relative energy efficiency through wollastonite-based scenario “circulation-H2,” where the electrolyzer electricity demand becomes comparable to conventional cement kiln thermal energy use.
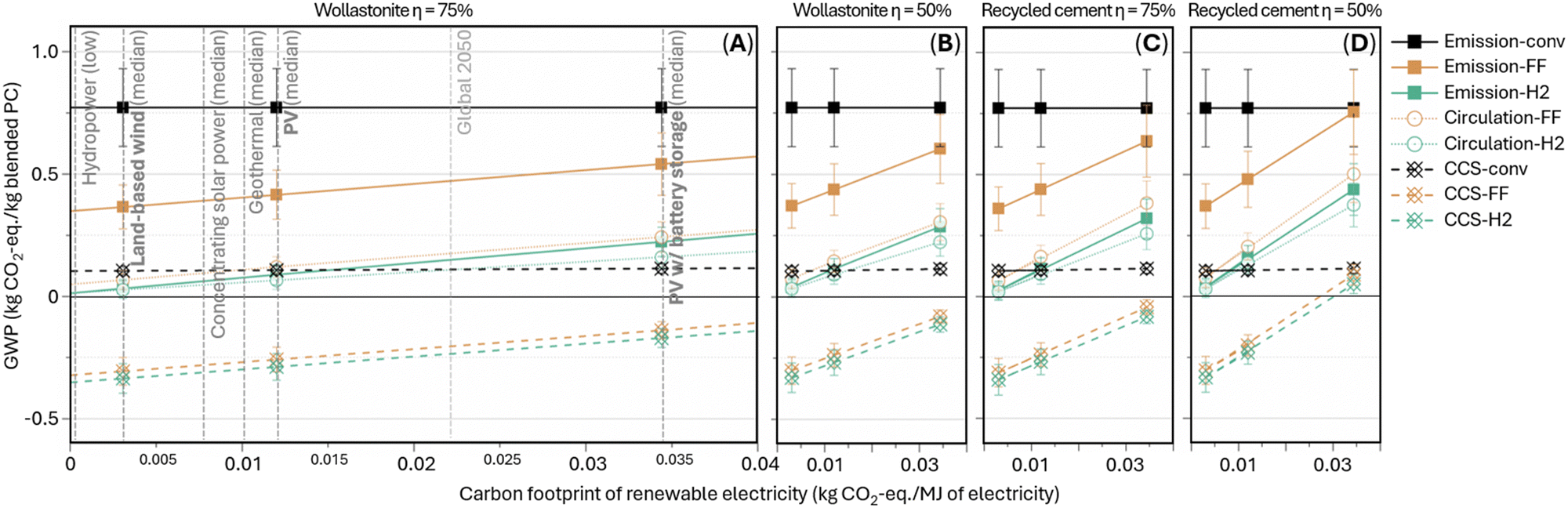 | ||
| Fig. 7 Sensitivity analysis – influence of renewable electricity carbon footprint and electrolyzer energy efficiency on total GWP of conventional and electrochemical cement manufacturing schemes. The data point value is averaged GWP of the four blend designs, and the error bar shows the min-to-max range of the four values.43,44 | ||
Discussions
Our TEA estimation is conservative as we consider the low-value fate of e-product CaCO3 for cement manufacturing at $10/t. If the carbon-mineralized CaCO3 is not sourced for liming but treated as a conventional CCS product, this carbon-negative electrochemical product can directly claim carbon credit at $57/t or $79/t CaCO3 in the U.S. at $130/t of direct atmospheric CO2 capture and utilization or at $180/t for direct atmosphere CO2 capture and storage. Although appearing even more profitable, this business model does not compete with the alternative conventional DAC with CCS techniques from the cost perspective due to its greater energy intensity and, more importantly, it sacrifices the substantial economic and decarbonization benefits to the cement/concrete industry and incurs extra concerns for landfill costs and impacts because end users, like concrete, with gigaton capability to the massively produced e-CaCO3 is rare. It is noteworthy that e-CaCO3 consumption (as well as the proposed circular CO2 scheme) within cement manufacturing potentially transforms the concrete industry from a gigaton carbon emitter to a gigaton CCUS enabler and furthermore largely alleviates the tremendous challenge and inertia behind large-scale underground carbon storage faced by conventional CCS technologies, not to mention their accompanying issues including long-distance transport, capital expenditure for new infrastructure, leakage/contamination liability, and more. Meanwhile, implementing the present electrochemical manufacturing scheme at scale can solve the industry-wide shortage of supplementary cementitious materials, particularly, coal fly ash, for conventional blended cements, facilitating net-zero transitions in both the energy and concrete sectors.A potential alternative solution is to harness e-CaCO3 for ocean acidification mitigation by enhancing alkalinity and promoting marine direct air capture and storage.45,46 Every t of e-CaCO3 ejected into oceans can capture up to 440 kg of air CO2 to Ca(HCO3)2.47 However, the location of marine electrochemical reactors is limited to onshore or offshore, imposing transportation optimization questions to co-produced a-SiO2 in order to be distributed to various regions (coastal areas benefit from barging with lower cost and CO2 intensity relative to rail and highway). Presently, this alternative electrolysis-based air CO2 capture scheme lacks optimal prototypes at pilot scale or higher, and the true economic and environmental benefits of this alternative electrolysis-based atmospheric CO2 capture scheme require further validation.
Besides, electrochemically produced O2 can power oxyfuel cement kilns as a superior source than air O2, which requires additional energy for N2 separation from air. Oxyfuel improves the thermal efficiency of combustion and negates the formation of NOx, toxic greenhouse gases. After initial fossil-fuel combustion, flue gas is recirculated to mix with pure O2 for subsequent combustion, allowing for more efficient CCUS from higher CO2 concentration (>90%)48 and integration with mature CO2 capture technologies, e.g., NOx-sensitive, amine-based CO2 sorption49 – transforming CO2-intensive cement products into gigatons of carbon sink. Moreover, the electrochemical process can separate Ca from Mg impurities (common in precursor minerals but forbidden in Portland cement) via precipitation pH difference, while precipitated Mg(OH)2 can directly capture and store CO2 from air.50
Prior to the present study, electrochemical production for cement manufacturing has focused on the electrified production of Ca(OH)2,16,19,21 which similarly avoids limestone decomposition-induced CO2 emissions but has the following drawbacks: (i) when using CaSiO3 as the precursor, the standard enthalpy of reaction is 113 kJ mol−1 CaSiO3 higher without introducing CO2 reactant to form CaCO3 (without CO2, Ca(OH)2 is formed instead), meaning greater energy demand; (ii) the utilization of Ca(OH)2 for Portland cement production requires modification of existing industrial cement plants, particularly to the preheater and precalciner prior to the cement kiln, which is unfavored by large cement manufacturing businesses, who prefer modification-free strategies due to capital investment concerns; (iii) Ca(OH)2 could absorb air CO2 during post-electrolysis processing and transportation, suggesting strict storage requirements for calcination at lower temperature in modified precalciners or compromised efficacy; (iv) the approach does not involve carbon-mineralized products for concrete carbon storage, thus infeasible to achieve carbon-neutral/negative. Therefore, the present electrochemical scheme via e-CaCO3 represents a more efficient, prompt, and preferable solution to cement decarbonization at the industry scale.
Indeed, clean energy sources with a low embodied carbon footprint and low prices are a prerequisite to the carbon-negativity or carbon-neutrality of the present electrochemical cement manufacturing scheme and positive economic benefit margins compared to business-as-usual conventional cement plants. While the large availability of low-carbon clean energy may have already been achieved in countries like Iceland, Norway, and Sweden, whose power grids are dominated by hydroelectric and geothermal sources, progress in decarbonizing electricity generation is still anticipated for the rest of the world toward approaching the Global 2050 goal, which would widely allow for the present green electrochemical manufacturing scheme with versatility. Ongoing research is dedicated to (1) upscaling the present laboratory gram-scale experiments into industrial-style systems, e.g., larger electrolyzers and stirred reactors and (2) examining the efficacy for multitudes of feedstock profiles including various industrial solid wastes, in order to better assess the efficiencies and challenges and evaluate the environmental impacts more comprehensively. Besides, due to experimental limitations in measuring the current CO2 capture and utilization processes, the present LCA and TEA use average values of energy demand and cost from references for a generic DAC process. Future studies are encouraged to carefully evaluate the direct capture and utilization of atmospheric CO2 under our electrochemical reaction scheme through experimental approaches to assess the energy consumption, efficiency, operational costs, and capital expenditure for more comprehensive LCA and TEA refinement. Additionally, it is crucial to compare these results with the utilization of circulated concentrated CO2, which demonstrates greater environmental and economic competitiveness in the present study.
Currently, the U.S. is estimated to generate ∼35–40 Mt y−1 waste hydrated cement paste (i.e., the reactive component in recycled concrete fines) from construction and demolition wastes generated at >600 Mt y−1.51–53 These waste fines have been commonly landfilled after concrete recycling and aggregates reclamation due to the high water demand of hydrated cement in the fines.51–53 Our work encourages research on recycled cement paste separation and treatment by providing a scalable pathway towards its valued use. At full scale in the U.S., the recycled cement paste may be converted to ∼21–24 Mt y−1 green ordinary Portland cement via the present strategy, accounting for ∼25% of annual U.S. cement production.51–53 Globally, at full capacity, our strategy can achieve CO2 abatement by 1.2 Gt per y without cement plant modification, 3 Gt per y with green H2 and CO2 circulation integrated, or 4.7 Gt per y with further CCS incorporated, equivalent to over 5% of total annual global CO2 emissions.54
Conclusion
We demonstrated an electrochemical approach potentially incorporating CO2 circulation as well as capture and storage for carbon-neutral/negative cement manufacturing that can be readily integrated to the existing cement industry and rapidly scaled up in the near term. Calcium silicates as naturally abundant rocks and industrial/municipal solid wastes undergo accelerated weathering and capture atmospheric CO2 to form carbon-negative CaCO3 to feed cement kilns, neutralizing the 200-year-old liming routine without modifying the conventional cement manufacturing process. The electrochemical products allow for direct cement substitution for long-term carbon storage and enhanced concrete lifetime; the co-produced green hydrogen provides an economically competitive solution to CO2 abatement for existing capital-intensive cement plants. These results elucidate a promising pathway for the fundamental decarbonization and clean energy transition of the cement industry, which can transform from a gigaton CO2 emitter to a gigaton-scale enabler for renewable energy, direct air carbon capture and storage, and green hydrogen.Author contributions
X. K. L. contributed to performing experiments, analyzing the data, writing and editing the manuscript. W. Z. contributed to performing experiments, analyzing the data, performing LCA and TEA, writing and editing of the manuscript. B. N. R. contributed to performing experiments, analyzing the data, editing the manuscript. L. C. S. contributed to editing the manuscript. J. L. contributed to conceiving the idea, performing experiments, writing and editing the manuscript.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work was performed under the auspices of the U.S. Department of Energy by Lawrence Livermore National Laboratory under Contract DE-AC52-07NA27344 with IM release number LLNL-JRNL-870523. The Advanced Light Source is supported by the Director, Office of Science, and Office of Basic Energy Sciences of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. This work made use of the EPIC facility of Northwestern University's NUANCE Center, which has received support from the SHyNE Resource (NSF ECCS-2025633), the IIN, and Northwestern's MRSEC program (NSF DMR-2308691). The U.S. Department of Energy Office of Science Graduate Student Research (SCGSR) program.References
- J. Lehne and F. Preston, Making Concrete Change: Innovation in Low-carbon Cement and Concrete, Chatham House, 2018 Search PubMed.
- I. H. Shah, S. A. Miller, D. Jiang and R. J. Myers, Nat. Commun., 2022, 13, 5758 CrossRef CAS PubMed.
- P. J. M. Monteiro, S. A. Miller and A. Horvath, Nat. Mater., 2017, 16, 698–699 CrossRef PubMed.
- S. A. Miller and R. J. Myers, Environ. Sci. Technol., 2020, 54, 677–686 CrossRef CAS PubMed.
- I. Diaz-Loya, M. Juenger, S. Seraj and R. Minkara, Cem. Concr. Compos., 2019, 101, 44–51 CrossRef CAS.
- M. C. G. Juenger, R. Snellings and S. A. Bernal, Cem. Concr. Res., 2019, 122, 257–273 CrossRef CAS.
- M. Wei, C. A. McMillan and S. de la Rue du Can, Curr. Sustainable/Renewable Energy Rep., 2019, 6, 140–148 CrossRef.
- E. Van Roijen, K. Sethares, A. Kendall and S. A. Miller, Nat. Commun., 2024, 15, 4848 CrossRef CAS PubMed.
- J. G. Driver, E. Bernard, P. Patrizio, P. S. Fennell, K. Scrivener and R. J. Myers, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2313475121 CrossRef CAS PubMed.
- C. Dessert, B. Dupré, J. Gaillardet, L. M. François and C. J. Allegre, Chem. Geol., 2003, 202, 257–273 CrossRef CAS.
- N. M. Haegel, R. Margolis, T. Buonassisi, D. Feldman, A. Froitzheim, R. Garabedian, M. Green, S. Glunz, H.-M. Henning, B. Holder, I. Kaizuka, B. Kroposki, K. Matsubara, S. Niki, K. Sakurai, R. A. Schindler, W. Tumas, E. R. Weber, G. Wilson, M. Woodhouse and S. Kurtz, Science, 2017, 356, 141 CrossRef CAS PubMed.
- IEA, Technology Roadmap – Low-Carbon Transition in the Cement Industry, 2018.
- J. Li, W. Zhang, C. Li and P. J. M. Monteiro, J. Cleaner Prod., 2019, 223, 662–679 CrossRef CAS.
- L. Zheng, T. P. Hills and P. Fennell, Faraday Discuss., 2016, 192, 113–124 RSC.
- B. Hönisch, A. Ridgwell, D. N. Schmidt, E. Thomas, S. J. Gibbs, A. Sluijs, R. Zeebe, L. Kump, R. C. Martindale and S. E. Greene, Science, 2012, 335, 1058–1063 CrossRef PubMed.
- L. D. Ellis, A. F. Badel, M. L. Chiang, R. J. Park and Y. M. Chiang, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12584–12591 CrossRef CAS PubMed.
- M. Stefanoni, U. M. Angst and B. Elsener, Nat. Mater., 2019, 18, 942–947 CrossRef CAS PubMed.
- R. Pärnamäe, S. Mareev, V. Nikonenko, S. Melnikov, N. Sheldeshov, V. Zabolotskii, H. V. M. Hamelers and M. Tedesco, J. Membr. Sci., 2021, 617, 118538 CrossRef.
- Z. Zhang, B. A. W. Mowbray, C. T. E. Parkyn, C. Waizenegger, A. S. R. Williams, E. W. Lees, S. Ren, Y. Kim, R. P. Jansonius and C. P. Berlinguette, Energy Environ. Sci., 2022, 15, 5129–5136 RSC.
- Z. Zhang, M. Bowbray, C. Parkyn, Y. Kim and C. Berlinguette, ChemRxiv, 2023, preprint DOI:10.26434/chemrxiv-2023-0zcw9.
- R. K. Miao, N. Wang, S.-F. Hung, W.-Y. Huang, J. Zhang, Y. Zhao, P. Ou, S. Wang, J. P. Edwards, C. Tian, J. Han, Y. Xu, M. Fan, J. E. Huang, Y. C. Xiao, A. H. Ip, H. Liang, E. H. Sargent and D. Sinton, ACS Energy Lett., 2023, 8, 4694–4701 CrossRef CAS.
- T. Burdyny and W. A. Smith, Energy Environ. Sci., 2019, 12, 1442–1453 RSC.
- J. M. Williams, D. Zhao, S. Moon, S. Kawashima, A.-H. A. Park and A. J. Moment, Cryst. Growth Des., 2023, 23, 8103–8115 CrossRef CAS.
- N. Li, S. S. Araya and S. K. Kær, J. Power Sources, 2019, 434, 226755 CrossRef CAS.
- K. Scrivener, R. Snellings and B. Lothenbach, A practical guide to microstructural analysis of cementitious materials, Crc Press, Boca Raton, 2016 Search PubMed.
- H. F. W. Taylor, Cement Chemistry, Thomas Telford, 1997 Search PubMed.
- P.-C. Aïtcin and R. J. Flatt, Science and technology of concrete admixtures, Woodhead Publishing, 2015 Search PubMed.
- S. A. Miller, A. Horvath, P. J. M. Monteiro and C. P. Ostertag, Environ. Res. Lett., 2015, 10, 131–143 Search PubMed.
- A. P. Gursel, H. Maryman and C. Ostertag, J. Cleaner Prod., 2016, 112, 823–836 CrossRef.
- G. Rim, A. K. Marchese, P. Stallworth, S. G. Greenbaum and A.-H. A. Park, Chem. Eng. J., 2020, 396, 125204 CrossRef CAS.
- R. T. DeVol, R. A. Metzler, L. Kabalah-Amitai, B. Pokroy, Y. Politi, A. Gal, L. Addadi, S. Weiner, A. Fernandez-Martinez, R. Demichelis, J. D. Gale, J. Ihli, F. C. Meldrum, A. Z. Blonsky, C. E. Killian, C. B. Salling, A. T. Young, M. A. Marcus, A. Scholl, A. Doran, C. Jenkins, H. A. Bechtel and P. U. P. A. Gilbert, J. Phys. Chem. B, 2014, 118, 8449–8457 CrossRef CAS PubMed.
- R. Snellings and K. L. Scrivener, Mater. Struct., 2016, 49, 3265–3279 CrossRef CAS.
- K. L. Scrivener, P. Juilland and P. J. M. Monteiro, Cem. Concr. Res., 2015, 78, 38–56 CrossRef CAS.
- B. Lothenbach, G. Le Saout, E. Gallucci and K. Scrivener, Cem. Concr. Res., 2008, 38, 848–860 CrossRef CAS.
- E. Berodier and K. Scrivener, J. Am. Ceram. Soc., 2014, 97, 3764–3773 CrossRef CAS.
- M. C. Juenger and R. Siddique, Cem. Concr. Res., 2015, 78, 71–80 CrossRef CAS.
- A. Palomo, P. Monteiro, P. Martauz, V. Bilek and A. Fernandez-Jimenez, Cem. Concr. Res., 2019, 124, 105829 CrossRef CAS.
- C. W. Hargis, A. Telesca and P. J. Monteiro, Cem. Concr. Res., 2014, 65, 15–20 CrossRef CAS.
- Y. Zhong, P. Wang, B. Zhang, Y. Wang, T. Liu, X. Li and Y. Niu, J. Build. Eng., 2023, 76, 107090 CrossRef.
- K. Celik, C. Meral, A. P. Gursel, P. K. Mehta, A. Horvath and P. J. M. Monteiro, Cem. Concr. Compos., 2015, 56, 59–72 CrossRef CAS.
- J. Li, K. Xu, G. Geng and H. E. Mason, Cem. Concr. Res., 2022, 161, 106947 CrossRef CAS.
- J. Li, Buildings, 2021, 11, 495 CrossRef.
- DOE, How Wind Can Help Us Breathe Easier, https://www.energy.gov/eere/wind/articles/how-wind-can-help-us-breathe-easier, (accessed 10/07/2024).
- S. Deutz and A. Bardow, Nat. Energy, 2021, 6, 203–213 CrossRef CAS.
- G. H. Rau, E. L. McLeod and O. Hoegh-Guldberg, Nat. Clim. Change, 2012, 2, 720–724 CrossRef.
- G. H. Rau, K. G. Knauss, W. H. Langer and K. Caldeira, Energy, 2007, 32, 1471–1477 CrossRef CAS.
- G. H. Rau, H. D. Willauer and Z. J. Ren, Nat. Clim. Change, 2018, 8, 621–625 CrossRef.
- C. E. Powell and G. G. Qiao, J. Membr. Sci., 2006, 279, 1–49 CrossRef CAS.
- S. A. Miller, V. M. John, S. A. Pacca and A. Horvath, Cem. Concr. Res., 2018, 114, 115–124 CrossRef CAS.
- I. Singh, R. Hay and K. Celik, Cem. Concr. Res., 2022, 152, 106673 CrossRef CAS.
- Cement, https://pubs.usgs.gov/periodicals/mcs2024/mcs2024-cement.pdf, 2024.
- Sustainable Management of Construction and Demolition Materials, https://www.epa.gov/smm/sustainable-management-construction-and-demolition-materials#:~:text=600%20million%20tons%20of%20C%26D,represents%20less%20than%2010%20percent, 2024.
- C. F. Dunant, S. Joseph, R. Prajapati and J. M. Allwood, Nature, 2024, 629, 1055–1061 CrossRef CAS PubMed.
- IEA, CO2 Emissions in 2022, 2022.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, LCA and TEA methods, electrochemistry data, XRD, SEM, TEM, STXM, Raman spectra, LCA and TEA results. See DOI: https://doi.org/10.1039/d4ee03529a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |