
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Emerging investigator series: preferential adsorption and coprecipitation of permafrost organic matter with poorly crystalline iron minerals†
Eva
Voggenreiter
 a,
Philippe
Schmitt-Kopplin
bc,
Laurel
ThomasArrigo
d,
Casey
Bryce
e,
Andreas
Kappler
af and
Prachi
Joshi
*a
a,
Philippe
Schmitt-Kopplin
bc,
Laurel
ThomasArrigo
d,
Casey
Bryce
e,
Andreas
Kappler
af and
Prachi
Joshi
*a
aGeomicrobiology, Department of Geosciences, University of Tübingen, Schnarrenbergstrasse 94-96, 72076 Tübingen, Germany. E-mail: prachi.joshi@uni-tuebingen.de
bAnalytical Biogeochemistry, Helmholtz Center Munich, Ingolstädter Landstrasse 1, 85764 Neuherberg, Germany
cAnalytical Food Chemistry, Technical University of Munich, Maximus-von-Imhof-Forum 2, 85354 Freising, Germany
dEnvironmental Chemistry, University of Neuchâtel, Avenue de Bellevaux 51, Neuchâtel, CH-2000, Switzerland
eSchool of Earth Sciences, University of Bristol, Wills Memorial Building, Queens Road, Bristol BS8 1RJ, UK
fCluster of Excellence: EXC 2124: Controlling Microbes to Fight Infection, Schnarrenbergstrasse 94-96, 72076 Tübingen, Germany
First published on 9th July 2024
Abstract
Future permafrost thaw will likely lead to substantial release of greenhouse gases due to thawing of previously unavailable organic carbon (OC). Accurate predictions of this release are limited by poor knowledge of the bioavailability of mobilized OC during thaw. Organic carbon bioavailability decreases due to adsorption to, or coprecipitation with, poorly crystalline ferric iron (Fe(III)) (oxyhydr)oxide minerals but the maximum binding extent and binding selectivity of permafrost OC to these minerals is unknown. We therefore utilized water-extractable organic matter (WEOM) from soils across a permafrost thaw gradient to quantify adsorption and coprecipitation processes with poorly crystalline Fe(III) (oxyhydr)oxides. We found that the maximum adsorption capacity of WEOM from intact and partly thawed permafrost soils was similar (204 and 226 mg C g−1 ferrihydrite, respectively) but decreased to 81 mg C g−1 ferrihydrite for WEOM from the fully thawed site. In comparison, coprecipitation of WEOM from intact and partly thawed soils with Fe immobilized up to 925 and 1532 mg C g−1 Fe respectively due to formation of precipitated Fe(III)–OC phases. Analysis of the OC composition before and after adsorption/coprecipitation revealed that high molecular weight, oxygen-rich, carboxylic- and aromatic-rich OC was preferentially bound to Fe(III) minerals relative to low molecular weight, aliphatic-rich compounds which may be more bioavailable. This selective binding effect was stronger after adsorption than coprecipitation. Our results suggest that OC binding by Fe(III) (oxyhydr)oxides sharply decreases under fully thawed conditions and that small, aliphatic OC molecules that may be readily bioavailable are less protected across all thaw stages.
Environmental significancePermafrost thaw leads to the release of organic carbon (OC), which may bind with ubiquitous ferric iron (Fe(III)) minerals. It is therefore important to understand the quantity and composition of Fe-bound OC in order to predict the (bio)availability of OC that is released. Here, we simulated adsorption and coprecipitation processes of OC along a permafrost thaw gradient with Fe(III) (oxyhydr)oxide minerals. We found that the quantity of Fe-bound OC decreases with permafrost thaw and observed preferential binding of less bioavailable OC by Fe(III) (oxyhydr)oxides. Our work therefore suggests that although these minerals bind OC in permafrost soil, the overall effect on OC protection against microbial degradation is not as strong as previously thought since binding selects for less bioavailable OC. |
Introduction
Permafrost soils are a major organic carbon (OC) stock, accounting for roughly one third of global soil OC (ca. 1300 Pg), yet only cover 15% of the global soil area.1,2 It has been estimated that permafrost thaw will cause the release of 67–237 Pg C as greenhouse gases by 2100 under business-as-usual climate scenarios2 due to microbial decomposition of OC that was previously not bioavailable. This microbially mediated release of OC as the greenhouse gases carbon dioxide (CO2) or methane (CH4) is still poorly constrained in timing and extent since it is influenced by a variety of biogeochemical factors.3–6 One of the key factors that controls the release of CO2 and CH4 is the bioavailability of OC for microbial decomposition. This OC bioavailability is lowered by its association with mineral surfaces in the soil, possibly leading to lower greenhouse gas emissions.7,8An array of different minerals can interact with OC in soil, such as phyllosilicates, metal (oxyhydr)oxides, as well as metal sulfides.8 Iron (Fe) (oxyhydr)oxides are especially known to bind a large quantity of OC in acidic soils9 due to their high surface area10 and thus play a larger role in permafrost peatland soils compared to other minerals, such as phyllosilicates.11 This binding can occur through (i) adsorption of OC onto existing Fe(III) (oxyhydr)oxides, or (ii) abiotic (or in certain cases, microbially mediated) precipitation of Fe(III) (oxyhydr)oxides by oxidation of dissolved Fe(II) in the presence of OC (referred to as “coprecipitation”).12,13 Adsorption takes place under both oxic and anoxic conditions while coprecipitation mainly occurs at redox interfaces when conditions transition from anoxic to oxic. Here, we refer to phases that are formed after either adsorption or coprecipitation as Fe–OC associations. The formation pathway of Fe–OC associations directly affects the properties of the Fe mineral. While adsorption of OC changes the surface properties of the mineral, coprecipitation with OC may affect Fe speciation and crystallinity of the resulting Fe–OC phase, which in turn may affect the reactivity of Fe.14–18
During the binding process, different OC compounds may be selectively bound while others remain in solution. Past studies using forest-floor extracts, plant litter, or humic reference materials suggested that the initial OC composition might determine which OC compounds are preferentially bound. Depending on the starting composition, it has been found that high molecular weight, oxygen-rich, carboxyl-rich, and aromatic compounds19–21 or in some cases carbohydrates22 could be preferentially bound to Fe(III) (oxyhydr)oxides. The composition of dissolved OC (DOC) after binding thus differs compared to its initial state, which has consequences for its bioavailability20 and its residence time in soils.23
Recently, several studies have shown that substantial fractions of OC may be bound to minerals (30–82%) in permafrost environments, with the dominant mineral fraction as Fe(III) (oxyhydr)oxides.12,24,25 The amount of Fe-bound OC is relatively consistent across different sites: ca. 10–20% of soil OC in intact permafrost soils of Scandinavia, Alaska, and the Tibetan Plateau.24,26–28 This suggests that part of the OC may remain unavailable due to binding with Fe minerals even with the increase in microbial activity due to permafrost thaw. However, our systematic understanding of the formation and composition of Fe–OC associations in permafrost soils and in subsequent thaw stages is limited. Along the thaw gradient, the soils undergo biogeochemical shifts due to changes in vegetation cover (higher abundance of sedges compared to shrubs and mosses),29,30 soil pH (acidic to circumneutral)31 and redox state (from oxic to anoxic).32 These changes affect the OC quantity and composition along the different thaw stages.29,33,34 For instance, bog and fen peat contain more tannin-, lignin- and protein-like compounds compared to palsa peat.29 Therefore, the amount of OC and the functional groups therein that react with Fe(III) (oxyhydr)oxides may differ over a thaw gradient. Extrapolating the results of adsorption and coprecipitation experiments from other ecosystems or OC reference compounds is difficult as the geochemical conditions (e.g., pH) and initial OC composition might differ substantially in permafrost systems.35,36 Systematic studies are thus necessary to understand the change in OC composition due to binding with Fe(III) (oxyhydr)oxides, independent of changes due to other biogeochemical factors during thaw. Finally, field-based studies37–39 only offer a snapshot of the net result of simultaneously occurring processes, making it difficult to disentangle effects of adsorption and coprecipitation alone. It is currently not known how much OC can potentially be bound by Fe(III) (oxyhydr)oxides via adsorption and coprecipitation along a permafrost thaw gradient, what impact these processes have on Fe mineralogy, and how they would influence preferential OC binding.
To address these knowledge gaps, the goal of this study was to systematically evaluate the quantity and selectivity of OC binding across permafrost thaw by simulating adsorption and coprecipitation, forming Fe–OC associations. The specific objectives were to (i) determine the maximum binding capacity of DOC from permafrost across different thaw stages to Fe(III) (oxyhydr)oxides by adsorption and coprecipitation, (ii) identify the speciation of Fe the formed Fe–OC associations, and (iii) evaluate changes in the chemical composition of OC due to binding to assess the potential changes in bioavailability. To achieve this, we utilized soils of a representative thawing permafrost peatland (Stordalen Mire, Sweden). We used in situ OC from soils of three distinct thaw stages, as previously defined by changes in vegetation and hydrology.30,40,41 The thaw stages were classified as (i) palsa underlain by permafrost, (ii) partly thawed, seasonally anoxic bog, and (iii) fully thawed, permanently anoxic fen areas. We chose to use water-extractable organic matter (WEOM) from the mineral-poor organic horizon as representative OC fractions for the experiments for two reasons: first, WEOM is the OC fraction which is most likely to enter the porewater and interact with Fe(III) (oxyhydr)oxides. Second, using WEOM allowed us to integrate all changes in OC composition due to vegetation, redox and pH condition shifts over the thaw gradient, thus being the most representative of environmental conditions.
Materials and methods
Field site description and sampling
Our field site, Stordalen Mire, is a subarctic permafrost peatland complex close to Abisko, Sweden (68 22′ N, 19 03′ E). It is located in the discontinuous permafrost zone and consists of several sub-habitats: (1) raised palsas underlain by intact permafrost and vegetated by dwarf shrubs, bryophytes, and lichens, (2) Sphagnum-rich bogs that experience seasonal water table fluctuations and have a higher active layer depth than palsas, and (3) fully inundated and anoxic fens, characterized by sedges (Eriophorum vaginatum, Carex rostrata).30,42 Along the thaw gradient, pH values of the porewater increase from acidic (pH 3.5 to 4.5) in palsa and bog soils to circumneutral (pH 5.0 to 6.5) in fen soils.31,43–45 The porewater generally has a low ionic strength; most dissolved species are in the μM-range.31In order to extract soil organic matter (see below) for the adsorption and coprecipitation experiments, we sampled the organic horizon of each thaw stage in July 2021 (Fig. S1†). The soil surface, covered with plant roots or young Sphagnum spp. layers, was removed and the soil below was collected by grab sampling, filled in plastic (LDPE) bags, and stored at 4 °C until further use. Soils in bog and fen areas were water-saturated and packed in the bags such that no headspace remained in order to limit oxygen exposure. Duplicate soil cores were collected per thaw stage at representative sites, based on the vegetation cover and pH values as described above, to compare solid phase OC composition to the WEOM. Cores were taken using a 50 cm long Humax corer as described previously.46
WEOM extraction
All glassware used in the extraction and further experiments was acid washed (1 M HCl), rinsed three times with double-deionized water (DDI, Millipore, >18 MΩ cm−1), and subsequently baked at 300 °C for 8 h before use. WEOM was extracted from the organic horizon of each permafrost thaw stage by mixing field-moist soil with DDI water for 24 h on an overhead shaker at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 weight/volume (w/v) ratio. Since we aimed to simulate conditions under which ferrihydrite would likely react with dissolved OC in permafrost soils, we carried out extractions for adsorption experiments under oxic conditions. For coprecipitation experiments, the extraction was done under anoxic conditions to simulate anoxic conditions in waterlogged soil layers. Afterwards, the suspensions were filled in glass serum bottles and centrifuged at 5250 rcf for 15 minutes. The supernatant was filtered sequentially through pre-rinsed 8 μm (Merck Millipore, MCE) and 0.22 μm (Merck Millipore, Steritop PES) filters. After filtration, extracts for the coprecipitation experiments were bubbled with N2 gas (99.999%) for 1 h to make them anoxic again after filtration. All extracts were stored in the dark at 4 °C until use (storage time was less than 24 h). Aliquots of all WEOM types were immediately freeze-dried after filtration, and the resulting solids were stored in the dark until analysis by Fourier-transform infrared spectroscopy (FTIR, details below). Initial DOC concentrations in the extracts of different soils and extraction conditions ranged from 9.8–23.5 mg C L−1 (Table S1†). They were quantified as described later in section “Setup of adsorption experiments”.
:10 weight/volume (w/v) ratio. Since we aimed to simulate conditions under which ferrihydrite would likely react with dissolved OC in permafrost soils, we carried out extractions for adsorption experiments under oxic conditions. For coprecipitation experiments, the extraction was done under anoxic conditions to simulate anoxic conditions in waterlogged soil layers. Afterwards, the suspensions were filled in glass serum bottles and centrifuged at 5250 rcf for 15 minutes. The supernatant was filtered sequentially through pre-rinsed 8 μm (Merck Millipore, MCE) and 0.22 μm (Merck Millipore, Steritop PES) filters. After filtration, extracts for the coprecipitation experiments were bubbled with N2 gas (99.999%) for 1 h to make them anoxic again after filtration. All extracts were stored in the dark at 4 °C until use (storage time was less than 24 h). Aliquots of all WEOM types were immediately freeze-dried after filtration, and the resulting solids were stored in the dark until analysis by Fourier-transform infrared spectroscopy (FTIR, details below). Initial DOC concentrations in the extracts of different soils and extraction conditions ranged from 9.8–23.5 mg C L−1 (Table S1†). They were quantified as described later in section “Setup of adsorption experiments”.
Ferrihydrite synthesis
Ferrihydrite was used in the adsorption experiments since it is a very common Fe(III) (oxyhydr)oxide mineral found in peatlands47,48 and has been shown to be present in soils of all thaw stages at Stordalen Mire.28 It is also typically the most relevant to OC binding in soils since it can bind the highest amounts of OC per g Fe due to its high specific surface area (>200 m2 g−1).49,50 It was synthesized as reported previously.51 Briefly, 40 g Fe(NO3)3·9H2O were dissolved in 500 mL DDI water and the pH was raised until 7.5 by addition of 1 M KOH under rapid stirring (800 rpm). The suspension was centrifuged (4250 rcf, 10 min) and washed with DDI water four times. Ferrihydrite was resuspended in 200 mL DDI water, stored at 4 °C in the dark, and used within 6 weeks after synthesis. Mineral identity of ferrihydrite was confirmed by 57Mössbauer spectroscopy and specific surface area was quantified after freeze-drying (further details in Text S1†).Setup of adsorption experiments
Adsorption experiments were carried out by adding aliquots of a ferrihydrite suspension (10.7 g L−1 Fh) to 20 mL of WEOM solution from palsa, bog, and fen soils inside closed 50 mL glass serum bottles. The WEOM was first adjusted to pH 4.5 (for palsa and bog WEOM) and pH 6 (for fen WEOM). For the adsorption isotherm of each WEOM type, the WEOM was diluted to different OC concentrations using a 3 mM NaCl solution as constant background ionic strength. The mass of added ferrihydrite was adjusted for each WEOM type, depending on its DOC concentration, to create the same initial molar C:Fe ratios across experiments (0.5 to 3.3, see Table S2†). After addition of ferrihydrite, the pH of the suspension was adjusted again to pH 4.5 (for palsa and bog WEOM) and pH 6 (for fen WEOM) using 0.1 M HCl. The pH was checked and re-adjusted 1 h later, if necessary. To quantify Fe concentrations in each replicate after adsorption, an aliquot (0.5 mL) of the suspension was acidified with 1 M HCl followed by total Fe quantification by the ferrozine assay.52 A control setup using 3 mM NaCl solution with no added WEOM but added ferrihydrite was used to determine background concentrations of DOC. No DOC originating from the ferrihydrite was detected.
Suspensions were then capped and shaken horizontally at 60 rpm for 24 h in the dark. They were filtered afterwards using pre-rinsed 0.22 μm filters (Merck Millipore, PVDF) inside filter cups (25 mm diameter, Millipore). The filtrate was used to quantify remaining DOC (as non-purgeable OC) after acidification with 2 M HCl by a TOC analyzer (multi N/C 2100S, Analytik Jena AG, Germany). The aromaticity of the filtrate was evaluated using specific ultraviolet spectroscopy at 254 nm (SUVA254, details below). The molecular composition of the WEOM before and after adsorption in reactors with an initial C:Fe ratio of ∼2 was analyzed by Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR-MS, details further below). This ratio was chosen since it was the highest common, initial ratio in all adsorption experiments. The solids of the same reactors (initial C:Fe ratio of ∼2) per WEOM type were further analyzed for OC functional groups by FTIR spectroscopy and Fe mineralogy by Fe K-edge X-ray absorption spectroscopy (XAS) and 57Fe Mössbauer spectroscopy. For solid phase analysis, the filter paper containing the ferrihydrite with adsorbed WEOM was washed twice with deionized water to remove loosely-bound ions. Then it was centrifuged (10055 rcf, 5 min) to release the particles from the paper surface. The supernatant was removed and samples were air-dried and stored at 4 °C until analysis.
Setup of coprecipitation experiments
Coprecipitation was simulated by abiotic oxidation of dissolved, anoxic Fe(II) in the presence of palsa and bog WEOM at different initial C:Fe ratios (0.35 to 1.2). Note that these ratios slightly differ from the ones used in the adsorption experiment since a higher Fe(II) addition was necessary to produce a sufficient amount of Fe precipitates. Coprecipitation was not simulated with fen WEOM since this thaw stage is permanently water-logged and anoxic53 with no redox fluctuations;38 thus abiotic coprecipitation due to Fe(II) oxidation is unlikely to play a major role.
Ferrous iron was added at a concentration of 1.5 mM to different dilutions of anoxic WEOM. A stock Fe(II) solution (0.3 M) was prepared by dissolving iron(II) chloride tetrahydrate (FeCl2·4H2O, Sigma Aldrich, USA) in anoxic DDI water, after which the solution was sterile-filtered (0.22 μm, Merck Millipore, PVDF) and stored in a N2-flushed, closed bottle in the dark. The added Fe(II) concentration is in the upper range of dissolved Fe(II) concentrations found at Stordalen Mire.46
The experiments were set up by adding the anoxic Fe(II) solution to 20 mL of anoxic WEOM solution inside an N2-flushed bottle. Afterwards, vials were opened and a cotton stopper was placed on top to ensure a constant O2 supply into the solution. The pH was measured at the start of the oxidation period and every 24 h thereafter, and adjusted back to pH 4.5 using 0.1 M NaOH if needed. The vials were shaken at 60 rpm on a horizontal shaker for 3 d in the dark. Afterwards, the same sampling procedure as described above for quantification of DOC, SUVA254, and determination of molecular OC composition by FT-ICR-MS and FTIR was carried out. Additionally, aliquots of the solutions before oxidation and the resulting suspensions after 3 d of oxidation as well as of the filtrate were acidified with 1 M HCl and the total Fe concentration was quantified using the ferrozine assay. The total Fe concentration in the filtrate was subtracted from the concentration in suspension after 3 d of oxidation to calculate the amount of Fe that had precipitated. To collect enough solid material for XAS analysis, 2.5 mM Fe(II) were added to 200 mL of WEOM in a sacrificial setup (initial molar C:Fe ratio = 0.6) and solids were collected after 3 d.
Fe K-edge X-ray absorption spectroscopy
Iron speciation in the Fe–OC associations was determined by Fe K-edge X-ray absorption spectroscopy at the SAMBA beamline at Synchrotron SOLEIL (Paris, France). Dried samples were homogenized with a mortar and pestle and pressed into pellets (7 mm diameter) with PVP (Polyvinylpyrrolidone K12, Carl Roth), before being sealed with Kapton® tape. At the beamline, transmission spectra were recorded in continuous scan mode at 20 K using a He(l) cryostat. An Si(220) monochromator was used and calibrated to the first derivative maximum of the K-edge absorption of an Fe(0) foil. Harmonic rejection was performed by two Si mirrors coated in Pd. Approximately 8–10 scans were collected per sample and merged to obtain the final spectra, used for analysis of Fe K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS). Iron oxidation state and speciation was determined through linear combination fit analysis (LCF) of XANES and EXAFS spectra, respectively, using Athena.54 The Fe binding environment was further determined by shell fitting using Artemis.54 Additional details of the XAS analyses are given in the ESI (Text S3).†57Fe Mössbauer spectroscopy
57Fe Mössbauer spectroscopy was used to identify changes in the Fe mineralogy after adsorption of WEOM (initial C:Fe ratio = 2) of each WEOM type to complement XAS measurements. The minerals were collected using filtration and the filter paper with particles was sealed with Kapton® tape. Details of spectral collection and analysis are given in the ESI (Text S1).† Spectra were recorded at 6 K. Analysis of coprecipitates was not possible due to limited sample mass.
Organic carbon analysis
Results and discussion
Quantity of organic carbon bound during adsorption and coprecipitation
The mass of OC bound to ferrihydrite via adsorption generally increased with increasing DOC concentration before reaching a threshold value (Fig. 1A). The maximum adsorption capacity, based on a Langmuir fit, of bog WEOM and palsa WEOM to ferrihydrite at pH 4.5 was 226 ± 3 and 204 ± 19 mg C g−1 Fh, while it was lower for fen WEOM at pH 6 (81 ± 4 mg C g−1 Fh). Our results for palsa and bog WEOM were comparable to previous studies using water extracts from forest floor litter (196–250 mg C g−1).19,22,62 The resulting maximum C:Fe ratios in ferrihydrite with adsorbed palsa and bog WEOM (Table S3†) were also comparable to permafrost OC bound on ferrihydrite-coated sand that were placed in the same thaw stages.38
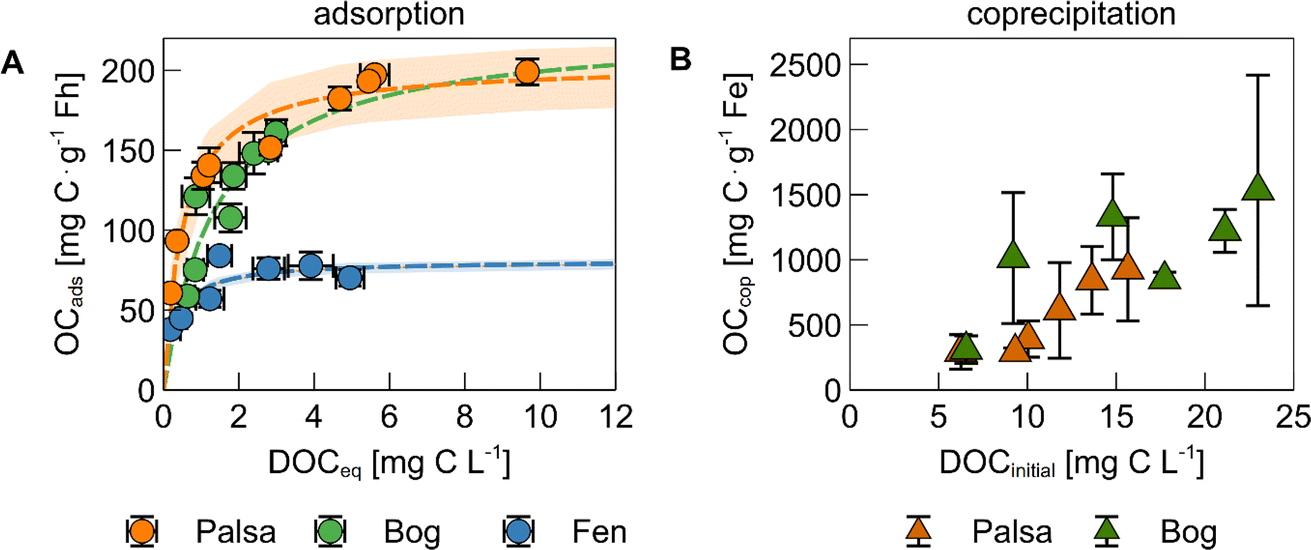 | ||
| Fig. 1 (A) Adsorption isotherm of water-extractable organic matter (WEOM) from different permafrost thaw stages on ferrihydrite (Fh). The mass of OC adsorbed (mg C g−1 Fh) is displayed against the equilibrium DOC concentration in solution after sorption. Data are denoted by circles and the Langmuir fit and 95% confidence interval is displayed as a dashed line and shaded area, respectively. The sorption experiments were performed at pH 4.5 for palsa, pH 4.5 for bog, and pH 6.0 for fen WEOM to represent environmentally relevant conditions. (B) Quantity of coprecipitated organic carbon (mg C g−1 Fe) as a function of the initial DOC (triangles). All data points and error bars represent the average and standard deviation of experimental triplicates. Note that the units and range of the y-axis are different for the two graphs. | ||
The lower maximum adsorption capacity in case of fen WEOM may be due to several factors: (i) pH and (ii) WEOM composition. At lower pH, a higher proportion of ferrihydrite surface sites are protonated, causing a more positive surface charge.63,64 As adsorption of OC to Fe(III) (oxyhydr)oxides is expected to be (mostly) electrostatic, negatively charged OC ions are therefore adsorbed to a higher degree to ferrihydrite at pH 4.5 than at pH 6.63 However, we speculate that this change in pH cannot explain the full magnitude of decrease in adsorption capacity. The net surface charge of ferrihydrite from pH 4.5 to pH 6 decreases by 38–50%,65,66 suggesting that the change in surface charge is too small to explain the discrepancy between the quantity of adsorbed bog and fen WEOM. Another possible reason for the differences in WEOM sorption at pH 4.5 and pH 6 is the difference in composition between bog and fen WEOM. Fen WEOM is more microbially processed due to a higher microbial diversity and higher plant litter input, resulting in a higher abundance of lipid-, amino-sugar, and carbohydrate-like compounds.31,33 In comparison, bog WEOM contains more aromatic-rich molecules.31,40,45 Differences in OM composition have been shown to result in changes in adsorption capacities,67–69 which is discussed in further detail below. The presence of bridging cations (Ca, Mg) could also increase the binding of OM to ferrihydrite,70 however, overall cation concentrations in WEOM from different thaw stages were low (<10 μM, Table S4†). Note that we performed experiments with fen WEOM only at pH 6 in order to best represent environmental conditions.
Coprecipitation of palsa and bog WEOM with iron generally led to higher amounts of Fe-bound OC than adsorption (Fig. 1B). The maximum masses of OC bound during coprecipitation with Fe were 925 ± 397 and 1530 ± 885 mg C g−1 Fe for palsa and bog WEOM, respectively. Resulting maximum C:Fe ratios were at least twice as high as previously reported for similar initial C:Fe ratios.19,22,71 One reason for this might be the use of higher pH values (pH 6 or 7) in other studies,18,19,22,71,72 which leads to a lower binding capacity of the precipitated Fe due to lower surface charge of the formed Fe(III) oxyhydroxides. Second, many studies used dissolved Fe(III) as a reactant14,19,22,73 during coprecipitation and increase the pH instead of oxidation of anoxic Fe(II) as in our study. The use of Fe(III) results in more precipitation of Fe relative to C at acidic pH values,74 due to the lower solubility of Fe(III) compared to Fe(II). This could lead to a lower content of bound OC, as illustrated by previously published values which are ca. 2 to 6 times lower at comparable initial C:Fe ratios and the same pH.73 We expect that the OC binding capacities due to coprecipitation measured here are more applicable to redox interfaces in permafrost peatlands than values of other studies19,22,71,73 as our experiments were done at environmentally relevant pH values and include Fe(II) oxidation.
After 3 days of oxidation, the percentage of oxidized and precipitated Fe was similar in experiments with palsa and bog WEOM (11 ± 5% and 10 ± 5%, respectively) and did not change based on the initial C:Fe ratio (Fig. S2†). Nierop et al. (2002)74 reported similarly low Fe precipitation when Fe(II) was added to OC of peat water extracts. Complexation of Fe(II) by DOC and the low pH could possibly hinder oxidation to Fe(III)75,76 and its subsequent precipitation. In contrast, coprecipitation led to removal of up to 44 ± 8% DOC from solution in palsa WEOM and up to 72 ± 1% in bog WEOM (Fig. S2†). For bog WEOM, the percentage of coprecipitated OC increased with the initial C:Fe ratio (Fig. S2†). This difference in share of coprecipitated OC is likely due to initial OC composition of palsa and bog WEOM (as discussed below). Coprecipitation in permafrost soils is thus able to remove much more OC from solution than adsorption, even with only a small percentage of Fe precipitating.
We acknowledge that there are also other minerals present along the thaw gradient which could bind OC, such as phyllosilicates or iron sulfide minerals.28 However, previous studies showed that both minerals bind less OC per g mineral (FeS: 55 mg C g−1 (ref. 77), phyllosilicates: 5–12 mg C g−1 (ref. 78)) at comparable initial C:Fe ratios than ferrihydrite by adsorption or during coprecipitation with dissolved Fe(II). Therefore, we expect that adsorption to, or coprecipitation with poorly crystalline Fe(III) (oxyhydr)oxides plays a larger role in permafrost environments.
Fe speciation in formed Fe–OC associations
To determine Fe speciation in the solid phase, we used Fe K-edge X-ray absorption spectroscopy. We first did linear combination fitting (LCF) of the XANES region in order to determine the Fe oxidation state. All Fe in ferrihydrite with adsorbed WEOM and in post-coprecipitation mineral phases was Fe(III) (Fig. S3 and Table S5†). Fitting results of EXAFS spectra showed that the post-adsorption phases consisted mostly of ferrihydrite and minor (<10%) fractions of lepidocrocite (Fig. 2). Additional Mössbauer spectra (Fig. S5†) of ferrihydrite with adsorbed WEOM at 6 K showed the presence of 2 sextets, with typical parameters (Table S7†) for poorly crystalline ferrihydrite.79,80 It is likely that the small fraction of lepidocrocite was not detected since parameters of lepidocrocite have been shown to overlap with those of ferrihydrite in Mössbauer spectra.81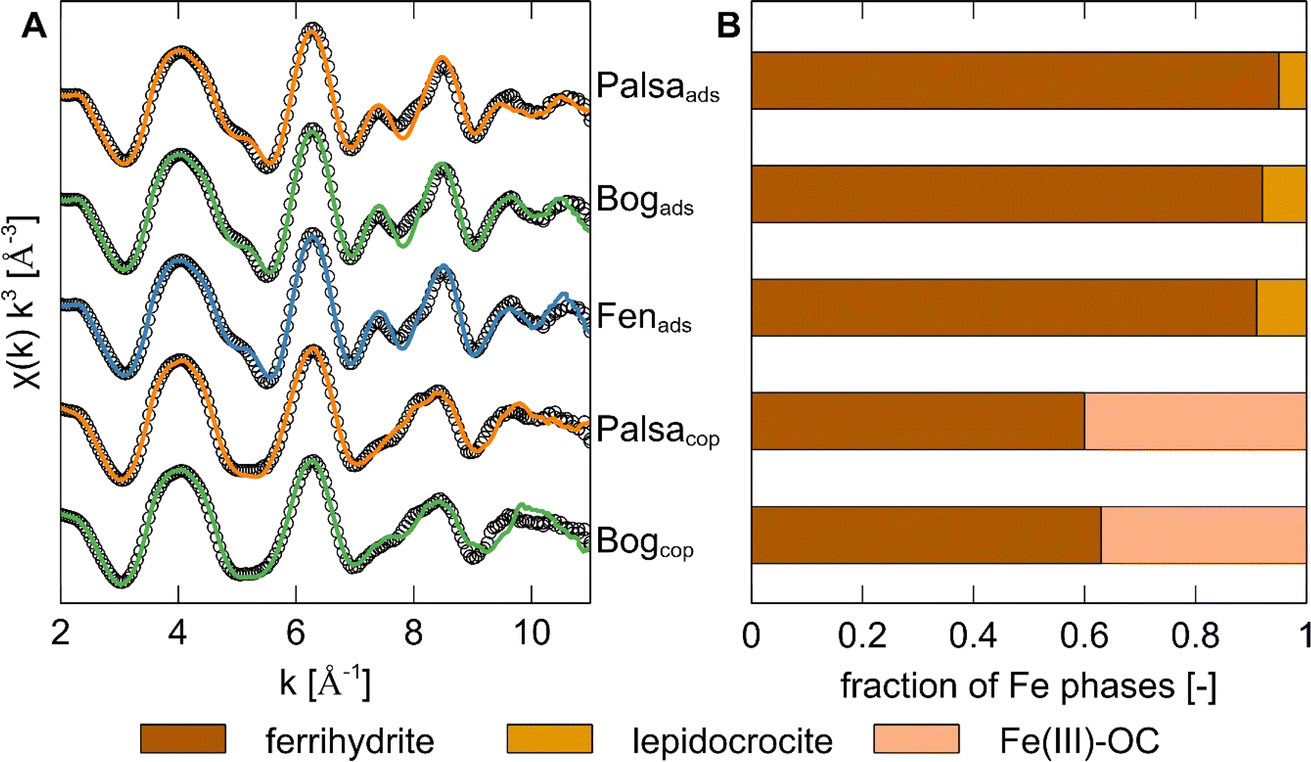 | ||
| Fig. 2 (A) Iron K-edge k3-weighted EXAFS spectra and linear combination fits (2 to 11 Å−1) of ferrihydrite with adsorbed WEOM (ads) from palsa, bog and fen, and Fe-OC coprecipitates (cop) synthesized with WEOM from palsa and bog. Experimental data is displayed as coloured lines and linear combination fits as black circles (see fit parameters in Table S6†). (B) Bar plot showing relative abundances of different Fe phases in the post-adsorption and post-coprecipitation mineral phases. The same labelling as for graph (A) applies here. Fe(III)–OC was fit using Fe(III)–citrate as a reference. | ||
In contrast, the Fe phase after coprecipitation with palsa and bog WEOM consisted of approximately 40% Fe(III) bound directly to OC (“Fe(III)–OC phases”) with the remaining fraction of Fe being ferrihydrite. This is consistent with observations by Schwertmann et al. (2005),82 who detected poorly soluble Fe(III)–OC phases when oxidizing Fe(II) in the presence of DOC. The detected Fe(III)–OC phase was best fit using Fe(III)–citrate (which contains carboxylic groups) as a reference, aligning with past studies of Fe–OC phases in non-permafrost peatlands.47,83 Since carboxylic groups are present in soils of all thaw stages40 and carboxyl-richness of natural OM has recently been proposed to be a controlling factor in OC binding to Fe(III) (oxyhydr)oxides,84 it is likely that carboxyl groups are significantly involved in coprecipitation with Fe in permafrost soils.
Shell fitting analysis of the Fourier-transformed Fe K-edge EXAFS region (Fig. S4, detailed results in Text S5†) further suggested that formed coprecipitates were likely more poorly crystalline and/or had a lower particle size than ferrihydrite with adsorbed WEOM.85,86 Within the coprecipitate set, the coprecipitates formed with palsa WEOM are likely more poorly crystalline than with bog WEOM, based on lower coordination numbers of both Fe–Fe paths (Table S7†). A higher proportion of carboxylic groups, which are present in palsa peat compared to subsequent thaw stages,40 might have led to this lower crystallinity.84 Thus, coprecipitation of OC in palsa and bog ecosystems likely causes formation of solid Fe(III)–OC phases and more poorly crystalline ferrihydrite, explaining the larger OC binding capacities of Fe–OC coprecipitates in contrast to adsorption by ferrihydrite.
Molecular fractionation of organic matter during adsorption and coprecipitation
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretches, respectively, and (iii) aromatic groups, visible as CC stretches in aromatic rings (1630 cm−1).19,87,88 Note that we could not evaluate the aliphatic C band caused by CH stretches of –CH2 groups (2840–2930 cm−1) because of an overlap of the band produced from O–H stretching in water87 (Fig. S6†). Relative peak heights of carboxylic and aromatic groups were higher than carbohydrates in palsa WEOM, whereas carbohydrates were relatively enriched in bog WEOM. This is in line with previous studies on changes of OC composition due to permafrost thaw at Stordalen Mire,29,40 in which carbohydrates often accumulate under the acidic and waterlogged conditions in bogs. Fen WEOM displayed weaker peaks of carbohydrate, carboxylic, and aromatic functional groups overall, compared to palsa and bog WEOM. We attribute this to higher overall microbial activity, leading to a depletion of labile carbohydrates in the water-extractable phase of the soil.31 Overall, the initial WEOM composition was comparable to that of the solid peat from representative soil cores (Fig. S7†).
O stretches, respectively, and (iii) aromatic groups, visible as CC stretches in aromatic rings (1630 cm−1).19,87,88 Note that we could not evaluate the aliphatic C band caused by CH stretches of –CH2 groups (2840–2930 cm−1) because of an overlap of the band produced from O–H stretching in water87 (Fig. S6†). Relative peak heights of carboxylic and aromatic groups were higher than carbohydrates in palsa WEOM, whereas carbohydrates were relatively enriched in bog WEOM. This is in line with previous studies on changes of OC composition due to permafrost thaw at Stordalen Mire,29,40 in which carbohydrates often accumulate under the acidic and waterlogged conditions in bogs. Fen WEOM displayed weaker peaks of carbohydrate, carboxylic, and aromatic functional groups overall, compared to palsa and bog WEOM. We attribute this to higher overall microbial activity, leading to a depletion of labile carbohydrates in the water-extractable phase of the soil.31 Overall, the initial WEOM composition was comparable to that of the solid peat from representative soil cores (Fig. S7†).
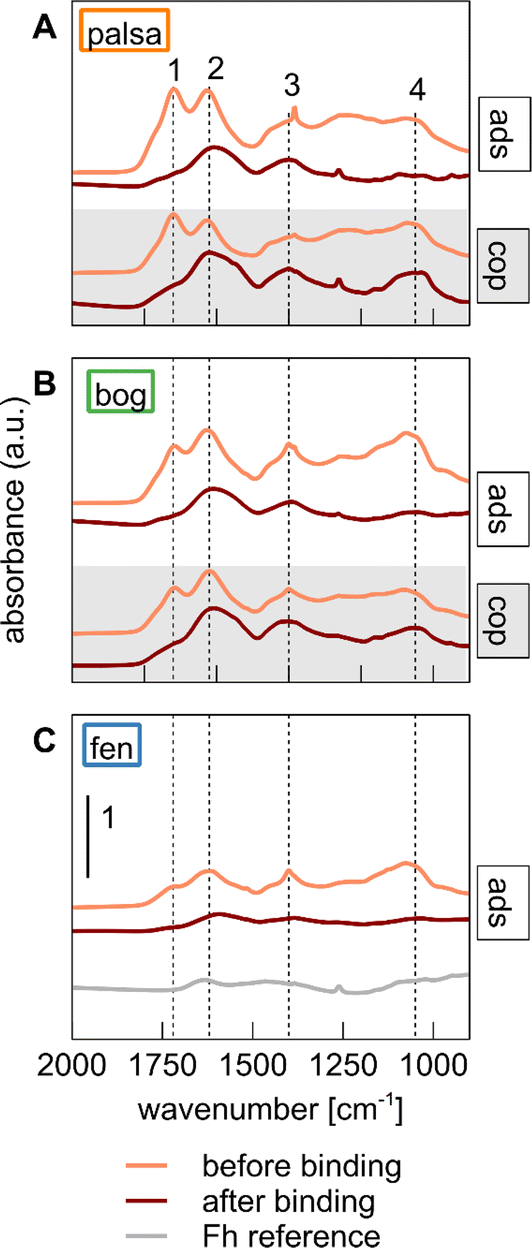 | ||
| Fig. 3 Normalized Fourier-transform infrared spectra of solids post-adsorption (ads) and post-coprecipitation (cop, both post-binding phases in maroon) in comparison to the original WEOM before binding (in orange) for (A) palsa, (B) bog, and (C) fen. The FTIR spectrum of ferrihydrite used for the adsorption experiments (grey) is given for reference. The scalebar in panel (C) applies for the other panels as well. Numbers and dashed lines stand for the following wavenumbers that are characteristic of the functional groups: 1 = 1720 cm−1 (CO stretch in COOH), 2 = 1630 cm−1 (aromatic CC stretch and/or asymmetric COO− stretch), 3 = 1400 cm−1 (symmetric COO− stretch), 4 = 1050 cm−1 (C–O stretch of carbohydrates).12,75,76 | ||
We observed preferential adsorption of certain organic functional groups to ferrihydrite for all WEOMs (Fig. 3). The magnitude of this effect was higher for palsa and bog WEOM than for fen WEOM. For example, the main OC functional groups bound on ferrihydrite from palsa WEOM were carboxylic (1400 and 1630 cm−1) and aromatic groups (1630 cm−1). In contrast, carbohydrates of palsa WEOM were adsorbed to a lower degree on ferrihydrite compared to carboxylic and aromatic groups. Preferential adsorption has been observed with plant litter or peat-derived OC in some previous studies with ferrihydrite and other Fe (oxyhydr)oxides.19,20,89,90 Coprecipitation led to less preferential binding of OC than adsorption for both palsa and bog WEOM, likely due to higher C:Fe ratios in the solid and more organic–organic interactions.21
To assess the change in OC preferential binding semi-quantitatively and compare changes between WEOM types, we calculated peak ratios (PR) of aromatic CC and COO− peaks to carbohydrate peaks before and after binding, respectively (Table S10†). These ratios, also termed humification indices in the context of organic matter decomposition,87 are used to show the selective decrease or increase of a particular functional group. Peak ratios generally increased after binding. Selective binding of aromatic groups relative to carbohydrates was highest for adsorption with palsa and bog WEOM (based on increases in PR values of 128 and 151%, respectively) while PR after adsorption of fen WEOM increased only by 17%. Similarly, the carboxylic to carbohydrate PR increased by 157 and 100% after adsorption of palsa and bog WEOM on ferrihydrite, while the PR for fen WEOM increased only by 25%. Selective binding may be higher for palsa and bog WEOM since they contain more oxidized OC moieties (e.g., carboxylic groups) than fen WEOM that have been shown to display a higher adsorption affinity to Fe(III) (oxyhydr)oxides.69,90 Coprecipitation of palsa WEOM with oxidized Fe(II) generally led to lower preferential binding of WEOM, with increases in PR by 50 and 33% for aromatic/carbohydrates and carboxylic/carbohydrates, respectively. Coprecipitation of bog WEOM induced almost no change in either PR. Taken together, OC binding to Fe(III) (oxyhydr)oxides in permafrost systems seems to be selective, favouring binding of carboxyl- and aromatic-rich OC moieties, especially during adsorption. However, this effect is lower under fully thawed conditions.
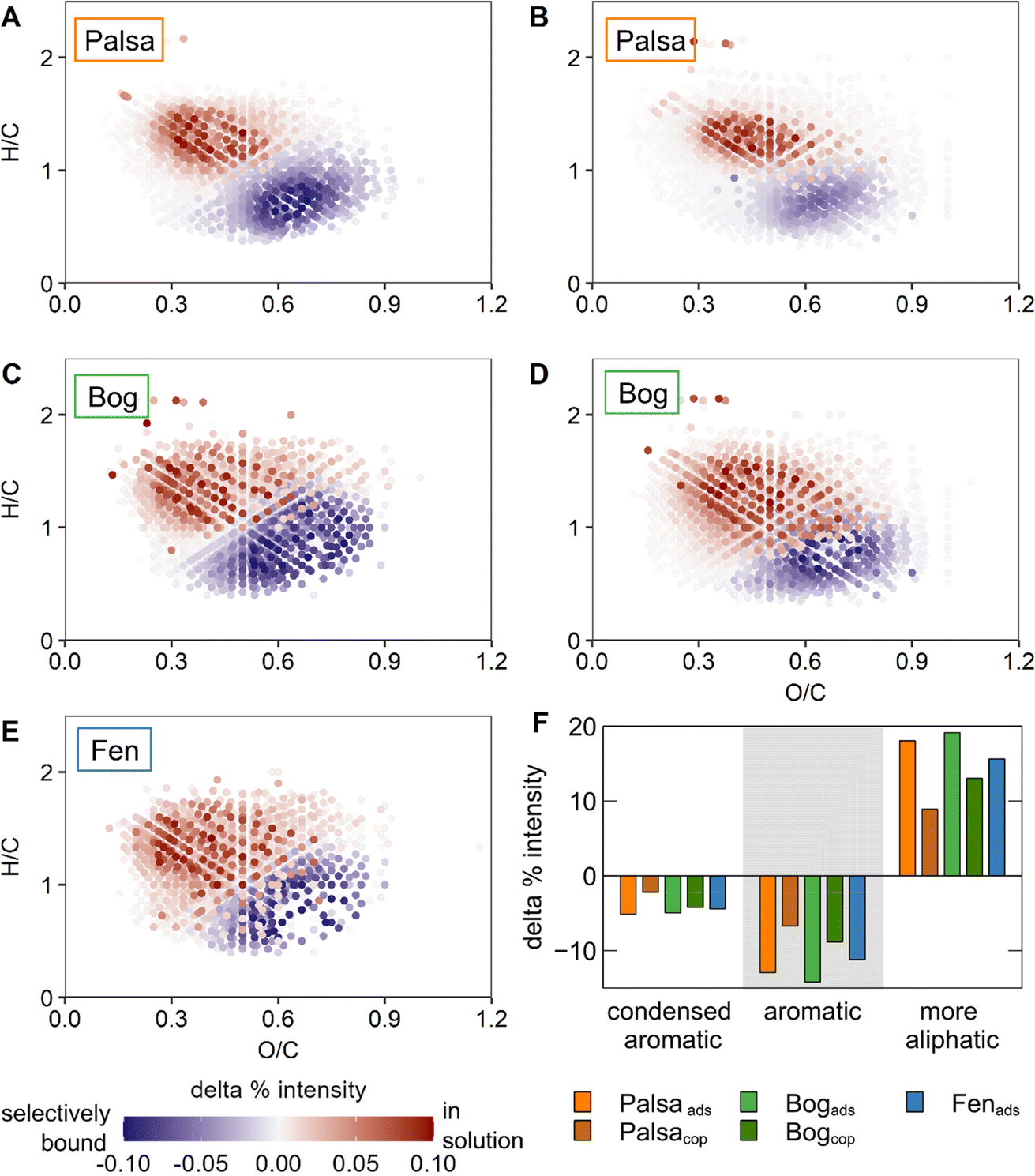 | ||
| Fig. 4 Van Krevelen diagrams showing changes in WEOM composition from palsa (A, B), bog (C, D) and fen (E) soils before and after adsorption (A, C, E) and coprecipitation (B, D). Organic compounds which are preferentially bound during adsorption or coprecipitation are displayed in blue and those retained in solution in red. (F) Change in molecular classes based on modified aromaticity index calculated for all experiments. Negative values indicate preferential adsorption while positive values mean compounds with formulae in those classes remained in solution. | ||
When comparing changes of different OC classes according to the modified aromaticity index, it was apparent that aromatic OC was preferentially adsorbed in all experiments (Fig. 4F). Adsorption induced a higher preferential binding of aromatic moieties than coprecipitation for both palsa and bog WEOM, consistent with the FTIR spectroscopy results. Comparing within the adsorption experiments, the relative change in aromatic groups is similar with OC from all thaw stages (11–14% increase after adsorption). In contrast, preferential binding of aromatic OC by coprecipitation is slightly higher in bog WEOM (11%) than palsa WEOM (7%), contradictory to the trends detected by FTIR. We assume these discrepancies stem from the insensitivity of FTIR spectroscopy to detect small changes in bound OC functional groups and possibly from the sample preparation step SPE, which only extracts a subset of OC molecules.56
To test changes in aromaticity over all initial C:Fe values, SUVA254 was measured. Results also indicated that aromaticity in palsa and bog WEOM after adsorption decreased compared to the initial WEOM (Fig. S10†). With decreasing DOC concentration in equilibrium, this preferential adsorption of aromatic groups in palsa and bog WEOM increased, as already reported elsewhere.19,90 However, fen WEOM showed increasing SUVA254 values with increasing DOC in equilibrium, meaning aromatic groups preferentially adsorbed with increasing DOC concentration. Since fen WEOM is rich in complex carbohydrates and amino acids,31 we speculate that those OC groups could dominate at lower WEOM concentrations and are thus bound to the mineral surface in a higher proportion than aromatic groups. Support for the dependence of preferential binding on initial composition is also given by Eusterhues et al. (2011)22 who found that different polysaccharides in a forest-floor extract preferentially adsorbed to ferrihydrite. SUVA254 values after coprecipitation of palsa WEOM also decreased with decreasing equilibrium DOC concentration, while those after coprecipitation with bog WEOM stayed constant (Fig. S10†). This points toward a higher preferential binding effect for palsa WEOM compared to bog WEOM.
Effect of preferential binding of organic matter on bioavailability towards microbial decomposition
We considered different parameters to judge the effect of selective binding of WEOM on changes to the bioavailability of dissolved OC. We considered structure, represented by aromaticity and double bond equivalents in the FT-ICR-MS data and partially by FTIR spectra and SUVA values as well as molecular weight based on FT-ICR-MS data. Based on these parameters, we propose that more bioavailable OC is likely relatively enriched in solution after adsorption and coprecipitation. The preferential binding of aromatic and high molecular weight compounds would leave smaller, more aliphatic compounds in solution that are considered to be more labile towards microbial decomposition. This is supported by the work of Li et al. (2021)20 who found that the extent of aerobic microbial degradation of ferrihydrite-bound OC, which was later again desorbed into the aqueous phase, was considerably lower than the original OC.We also considered NOSC as an indicator for bioavailability. This parameter differed from our proposed trend. The average NOSC of OC in solution decreased after binding, implying that compounds that were energetically favorable towards decomposition were bound in the (formed) Fe–OC associations. Based on theoretical definitions, the remaining dissolved low-NOSC compounds are more difficult to degrade for microorganisms.93 Possible reasons for this discrepancy are that NOSC is purely based on formula and not on structure or molecular weight of OC molecules. Bioavailability can also be regarded similar to energy availability, which is the ratio of invested to returned energy during OC decomposition.60 This emergent definition indicates that the total energy within an OC compound, which is proportional to its NOSC, may not be exactly related to what compound will be used energy efficiently by microorganisms.60 Further research should therefore focus on how compounds of similar NOSC but different structure are respired by microorganisms. As we see limitations in the interpretation of NOSC for bioavailability in this context, we consider the other indicators (supported by the bioavailability experiments by Li et al. (2021)20) as more reliable. Therefore, the changes in OC composition after binding indicate that less bioavailable OC is selectively bound to Fe(III) oxyhydroxides in thawing permafrost soils, leaving compounds that are degraded more easily in solution.
Conclusions
Our findings provide insights into the potential of Fe(III) (oxyhydr)oxides to bind porewater OC in (permafrost) peatlands. We found higher C:Fe ratios in Fe–OC associations formed by coprecipitation than by adsorption, even at relatively low initial C:Fe ratios. Given that very high C:Fe ratios (5–16 [g C g−1 Fe]) were observed in a variety of (permafrost) peatlands,26,28,94 coprecipitation as simulated in our study could be the major pathway for the formation of Fe–OC associations in palsa and bog ecosystems. It is feasible that the bound OC content is higher in a natural setting than in Fe–OC associations formed in batch experiments since DOC would be continuously supplied under natural conditions and reaction time scales are likely longer than 3 days. Coprecipitation thus likely plays a large role at trapping OC at redox interfaces, while adsorption, although binding less OC, is more relevant in upper, oxic soil layers.
Our results indicate that maximum adsorption capacities of OC drastically decreased from palsa and bog soils to fully thawed fen soils. This would mean that Fe(III) (oxyhydr)oxides provide less protection for OC in fully thawed, circumneutral permafrost conditions. Further, as more palsa soils transition to bogs or fens in the future, several processes could alter the binding of OC to poorly crystalline Fe(III) (oxyhydr)oxides. First, already existing Fe–OC coprecipitates could possibly further adsorb OC once they are formed. Second, under anoxic conditions in further thaw stages, the formed Fe–OC associations are likely to be microbially reduced and dissolved, releasing Fe2+ and OC into solution.46 Based on the high quantities of bound OC to Fe minerals in palsa and bog soils, this would lead to a substantial OC release during transitions between thaw stages. The previously Fe-bound OC could then be used for anaerobic respiration, increasing CO2 emissions and may ultimately also increase CH4 emissions by supplying more dissolved OC. It is also possible that the released OC is partly bound by phyllosilicate or iron sulfide minerals, which are present in the fen soil.28 Further, our observed preferential binding effects imply that Fe mineral protection of OC in permafrost soils is biased towards less bioavailable OC (albeit to a lower extent during coprecipitation than adsorption). This may affect OC cycling in primarily oxic soil horizons of permafrost soils since unbound, low-molecular weight compounds could partly be respired by microorganisms. Thus, Fe(III) (oxyhydr)oxides may limit DOC concentrations, especially in palsa and bog ecosystems, but may not substantially inhibit microbially mediated CO2 emissions.
Data availability
Data for this article, including the data presented in the main figures, are available at Zenodo at https://doi.org/10.5281/zenodo.12542760.Author contributions
Eva Voggenreiter: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft; Philippe Schmitt-Kopplin: methodology, formal analysis, writing – review and editing; Laurel ThomasArrigo: formal analysis, resources, writing – review and editing; Casey Bryce: conceptualization, writing – review and editing; Andreas Kappler: funding acquisition, conceptualization, supervision, writing – review and editing; Prachi Joshi: conceptualization, methodology, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the German Research foundation (DFG, KA 1736/66-1). A. K. acknowledges infrastructural support by the DFG under Germany's Excellence Strategy, cluster of Excellence EXC2124, project ID 390838134. Overall, we would like to thank the Swedish Polar Research Secretariat and SITES for the support of the work done at the Abisko Scientific Research Station. Special thanks go to Jennie Wikström and Erik Lundin for their help in the organization of the field campaign and sample shipment. We are also grateful to Ankita Chauhan, Katrin Wunsch, and Marie Mollenkopf for field assistance. We thank Hayley Green for assistance in laboratory analysis, as well as Lars Grimm and Tsz Ho Chiu for freeze-drying and BET measurements. We also thank Annette Flicker for her instructions during FTIR measurements and Thomas Borch for providing the XAS spectra of Fe(III)–OC references. We acknowledge SOLEIL (proposal no. 20210970) for the use of the synchrotron radiation facilities and thank Gautier Landrot (SAMBA) for his help during the synchrotron data collection. The TOC figure was made using https://www.BioRender.com.References
- G. Hugelius, J. Strauss, S. Zubrzycki, J. W. Harden, E. A. G. Schuur, C.-L. Ping, L. Schirrmeister, G. Grosse, G. J. Michaelson, C. D. Koven, J. A. O'Donnell, B. Elberling, U. Mishra, P. Camill, Z. Yu, J. Palmtag and P. Kuhry, Estimated stocks of circumpolar permafrost carbon with quantified uncertainty ranges and identified data gaps, Biogeosciences, 2014, 11, 6573–6593 CrossRef.
- E. A. G. Schuur, A. D. McGuire, C. Schädel, G. Grosse, J. W. Harden, D. J. Hayes, G. Hugelius, C. D. Koven, P. Kuhry, D. M. Lawrence, S. M. Natali, D. Olefeldt, V. E. Romanovsky, K. Schaefer, M. R. Turetsky, C. C. Treat and J. E. Vonk, Climate change and the permafrost carbon feedback, Nature, 2015, 520, 171–179 CrossRef CAS PubMed.
- D. Sihi, P. W. Inglett, S. Gerber and K. S. Inglett, Rate of warming affects temperature sensitivity of anaerobic peat decomposition and greenhouse gas production, Global Change Biol., 2018, 24, e259–e274 CrossRef PubMed.
- J. G. Ernakovich, L. M. Lynch, P. E. Brewer, F. J. Calderon and M. D. Wallenstein, Redox and temperature-sensitive changes in microbial communities and soil chemistry dictate greenhouse gas loss from thawed permafrost, Biogeochemistry, 2017, 134, 183–200 CrossRef CAS.
- J. Couwenberg, A. Thiele, F. Tanneberger, J. Augustin, S. Bärisch, D. Dubovik, N. Liashchynskaya, D. Michaelis, M. Minke, A. Skuratovich and H. Joosten, Assessing greenhouse gas emissions from peatlands using vegetation as a proxy, Hydrobiologia, 2011, 674, 67–89 CrossRef CAS.
- B. J. Woodcroft, C. M. Singleton, J. A. Boyd, P. N. Evans, J. B. Emerson, A. A. F. Zayed, R. D. Hoelzle, T. O. Lamberton, C. K. McCalley, S. B. Hodgkins, R. M. Wilson, S. O. Purvine, C. D. Nicora, C. Li, S. Frolking, J. P. Chanton, P. M. Crill, S. R. Saleska, V. I. Rich and G. W. Tyson, Genome-centric view of carbon processing in thawing permafrost, Nature, 2018, 560, 49–54 CrossRef CAS PubMed.
- R. C. Porras, C. E. Hicks Pries, M. S. Torn and P. S. Nico, Synthetic iron (hydr)oxide-glucose associations in subsurface soil: effects on decomposability of mineral associated carbon, Sci. Total Environ., 2018, 613–614, 342–351 CrossRef CAS PubMed.
- M. Kleber, I. C. Bourg, E. K. Coward, C. M. Hansel, S. C. B. Myneni and N. Nunan, Dynamic interactions at the mineral–organic matter interface, Nat. Rev. Earth Environ., 2021, 2(6), 402–421, DOI:10.1038/s43017-021-00162-y.
- C. Rasmussen, K. Heckman, W. R. Wieder, M. Keiluweit, C. R. Lawrence, A. A. Berhe, J. C. Blankinship, S. E. Crow, J. L. Druhan, C. E. Hicks Pries, E. Marin-Spiotta, A. F. Plante, C. Schädel, J. P. Schimel, C. A. Sierra, A. Thompson and R. Wagai, Beyond clay: towards an improved set of variables for predicting soil organic matter content, Biogeochemistry, 2018, 137, 297–306 CrossRef CAS.
- A. R. Saidy, R. J. Smernik, J. A. Baldock, K. Kaiser and J. Sanderman, The sorption of organic carbon onto differing clay minerals in the presence and absence of hydrous iron oxide, Geoderma, 2013, 209–210, 15–21 CrossRef CAS.
- E. Zhu, Z. Liu, S. Wang, Y. Wang, T. Liu and X. Feng, Organic Carbon and Lignin Protection by Metal Oxides Versus Silicate Clay: Comparative Study Based on Wetland and Upland Soils, J. Geophys. Res.: Biogeosci., 2023, 128(7) DOI:10.1029/2023JG007474.
- N. Gentsch, R. Mikutta, O. Shibistova, B. Wild, J. Schnecker, A. Richter, T. Urich, A. Gittel, H. Šantrůčková, J. Bárta, N. Lashchinskiy, C. W. Mueller, R. Fuß and G. Guggenberger, Properties and bioavailability of particulate and mineral-associated organic matter in Arctic permafrost soils, Lower Kolyma Region, Russia, Eurasian J. Soil Sci., 2015, 66, 722–734 CrossRef CAS.
- M. Kleber, K. Eusterhues, M. Keiluweit, C. Mikutta, R. Mikutta and P. S. Nico, Mineral–Organic Associations: Formation, Properties, and Relevance in Soil Environments, Elsevier, 2015, pp. 1–140 Search PubMed.
- K. Eusterhues, F. E. Wagner, W. Häusler, M. Hanzlik, H. Knicker, K. U. Totsche, I. Kögel-Knabner and U. Schwertmann, Characterization of ferrihydrite-soil organic matter coprecipitates by X-ray diffraction and Mössbauer spectroscopy, Environ. Sci. Technol., 2008, 42, 7891–7897 CrossRef CAS PubMed.
- M. Shimizu, J. Zhou, C. Schröder, M. Obst, A. Kappler and T. Borch, Dissimilatory reduction and transformation of ferrihydrite-humic acid coprecipitates, Environ. Sci. Technol., 2013, 47, 13375–13384 CrossRef CAS PubMed.
- K. Eusterhues, A. Hädrich, J. Neidhardt, K. Küsel, T. F. Keller, K. D. Jandt and K. U. Totsche, Reduction of ferrihydrite with adsorbed and coprecipitated organic matter: microbial reduction by Geobacter bremensis vs. abiotic reduction by Na-dithionite, Biogeosciences, 2014, 11, 4953–4966 CrossRef.
- C. Mikutta, X-ray absorption spectroscopy study on the effect of hydroxybenzoic acids on the formation and structure of ferrihydrite, Geochim. Cosmochim. Acta, 2011, 75, 5122–5139 CrossRef CAS.
- R. Angelico, A. Ceglie, J.-Z. He, Y.-R. Liu, G. Palumbo and C. Colombo, Particle size, charge and colloidal stability of humic acids coprecipitated with Ferrihydrite, Chemosphere, 2014, 99, 239–247 CrossRef CAS PubMed.
- C. Chen, J. J. Dynes, J. Wang and D. L. Sparks, Properties of Fe-organic matter associations via coprecipitation versus adsorption, Environ. Sci. Technol., 2014, 48, 13751–13759 CrossRef CAS PubMed.
- Y. Li, J. Lv, D. Cao and S. Zhang, Interfacial Molecular Fractionation Induces Preferential Protection of Biorefractory Organic Matter by Ferrihydrite, ACS Earth Space Chem., 2021, 5(5), 1094–1101, DOI:10.1021/acsearthspacechem.1c00019.
- X. Zhu, K. Wang, Z. Liu, J. Wang, E. Wu, W. Yu, X. Zhu, C. Chu and B. Chen, Probing Molecular-Level Dynamic Interactions of Dissolved Organic Matter with Iron Oxyhydroxides via a Coupled Microfluidic Reactor and an Online High-Resolution Mass Spectrometry System, Environ. Sci. Technol., 2023, 57, 2981–2991 CrossRef PubMed.
- K. Eusterhues, T. Rennert, H. Knicker, I. Kögel-Knabner, K. U. Totsche and U. Schwertmann, Fractionation of organic matter due to reaction with ferrihydrite: coprecipitation versus adsorption, Environ. Sci. Technol., 2011, 45, 527–533 CrossRef CAS PubMed.
- E. K. Coward, T. Ohno and A. F. Plante, Adsorption and Molecular Fractionation of Dissolved Organic Matter on Iron-Bearing Mineral Matrices of Varying Crystallinity, Environ. Sci. Technol., 2018, 52, 1036–1044 CrossRef CAS PubMed.
- F. Liu, S. Qin, K. Fang, L. Chen, Y. Peng, P. Smith and Y. Yang, Divergent changes in particulate and mineral-associated organic carbon upon permafrost thaw, Nat. Commun., 2022, 13, 5073 CrossRef CAS PubMed.
- J. Martens, C. W. Mueller, P. Joshi, C. Rosinger, M. Maisch, A. Kappler, M. Bonkowski, G. Schwamborn, L. Schirrmeister and J. Rethemeyer, Stabilization of mineral-associated organic carbon in Pleistocene permafrost, Nat. Commun., 2023, 14, 2120 CrossRef CAS PubMed.
- H. Joss, M. S. Patzner, M. Maisch, C. W. Mueller, A. Kappler and C. Bryce, Cryoturbation impacts iron-organic carbon associations along a permafrost soil chronosequence in northern Alaska, Geoderma, 2022, 413, 115738 CrossRef CAS.
- C. C. Mu, T. J. Zhang, Q. Zhao, H. Guo, W. Zhong, H. Su and Q. B. Wu, Soil organic carbon stabilization by iron in permafrost regions of the Qinghai-Tibet Plateau, Geophys. Res. Lett., 2016, 43(10), 286–294 Search PubMed.
- M. S. Patzner, C. W. Mueller, M. Malusova, M. Baur, V. Nikeleit, T. Scholten, C. Hoeschen, J. M. Byrne, T. Borch, A. Kappler and C. Bryce, Iron mineral dissolution releases iron and associated organic carbon during permafrost thaw, Nat. Commun., 2020, 11, 6329 CrossRef CAS PubMed.
- R. M. Wilson, M. A. Hough, B. A. Verbeke, S. B. Hodgkins, J. P. Chanton, S. D. Saleska, V. I. Rich and M. M. Tfaily, Plant organic matter inputs exert a strong control on soil organic matter decomposition in a thawing permafrost peatland, Sci. Total Environ., 2022, 152757 CrossRef CAS PubMed.
- N. Malmer, T. Johansson, M. Olsrud and T. R. Christensen, Vegetation, climatic changes and net carbon sequestration in a North-Scandinavian subarctic mire over 30 years, Global Change Biol., 2005, 11(11), 1895–1909, DOI:10.1111/j.1365-2486.2005.01042.x.
- R. AminiTabrizi, R. M. Wilson, J. D. Fudyma, S. B. Hodgkins, H. M. Heyman, V. I. Rich, S. R. Saleska, J. P. Chanton and M. M. Tfaily, Controls on Soil Organic Matter Degradation and Subsequent Greenhouse Gas Emissions Across a Permafrost Thaw Gradient in Northern Sweden, Front. Earth Sci., 2020, 8, 557961, DOI:10.3389/feart.2020.557961.
- C. R. Perryman, C. K. McCalley, A. Malhotra, M. F. Fahnestock, N. N. Kashi, J. G. Bryce, R. Giesler and R. K. Varner, Thaw Transitions and Redox Conditions Drive Methane Oxidation in a Permafrost Peatland, J. Geophys. Res.: Biogeosci., 2020, 125(3), e2019JG005526, DOI:10.1029/2019JG005526.
- M. Hough, S. McCabe, S. R. Vining, E. Pickering Pedersen, R. M. Wilson, R. Lawrence, K.-Y. Chang, G. Bohrer, W. J. Riley, P. M. Crill, R. K. Varner, S. J. Blazewicz, E. Dorrepaal, M. M. Tfaily, S. R. Saleska and V. I. Rich, Coupling plant litter quantity to a novel metric for litter quality explains C storage changes in a thawing permafrost peatland, Global Change Biol., 2022, 28, 950–968 CrossRef CAS PubMed.
- E. S. Kane, T. J. Veverica, M. M. Tfaily, E. A. Lilleskov, K. M. Meingast, R. K. Kolka, A. L. Daniels and R. A. Chimner, Reduction-Oxidation Potential and Dissolved Organic Matter Composition in Northern Peat Soil: Interactive Controls of Water Table Position and Plant Functional Groups, J. Geophys. Res.: Biogeosci., 2019, 124, 3600–3617 CrossRef CAS.
- Z. Guo, Y. Wang, Z. Wan, Y. Zuo, L. He, D. Li, F. Yuan, N. Wang, J. Liu, Y. Song, C. Song and X. Xu, Soil dissolved organic carbon in terrestrial ecosystems: Global budget, spatial distribution and controls, Global Ecol. Biogeogr., 2020, 29, 2159–2175 CrossRef.
- L. Heffernan, D. N. Kothawala and L. J. Tranvik, Review article: Terrestrial dissolved organic carbon in northern permafrost, Cryosphere, 2024, 18, 1443–1465 CrossRef.
- A. Monhonval, J. Strauss, E. Mauclet, C. Hirst, N. Bemelmans, G. Grosse, L. Schirrmeister, M. Fuchs and S. Opfergelt, Iron Redistribution Upon Thermokarst Processes in the Yedoma Domain, Front. Earth Sci., 2021, 9, 703339, DOI:10.3389/feart.2021.703339.
- M. S. Patzner, N. Kainz, E. Lundin, M. Barczok, C. Smith, E. Herndon, L. Kinsman-Costello, S. Fischer, D. Straub, S. Kleindienst, A. Kappler and C. Bryce, Seasonal Fluctuations in Iron Cycling in Thawing Permafrost Peatlands, Environ. Sci. Technol., 2022, 56, 4620–4631 CrossRef CAS PubMed.
- A. Monhonval, E. Mauclet, C. Hirst, N. Bemelmans, E. Eekman, E. A. Schuur and S. Opfergelt, Mineral organic carbon interactions in dry versus wet tundra soils, Geoderma, 2023, 436, 116552 CrossRef CAS.
- S. B. Hodgkins, M. M. Tfaily, C. K. McCalley, T. A. Logan, P. M. Crill, S. R. Saleska, V. I. Rich and J. P. Chanton, Changes in peat chemistry associated with permafrost thaw increase greenhouse gas production, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5819–5824 CrossRef CAS PubMed.
- M. E. Holmes, P. M. Crill, W. C. Burnett, C. K. McCalley, R. M. Wilson, S. Frolking, K.-Y. Chang, W. J. Riley, R. K. Varner, S. B. Hodgkins, A. P. McNichol, S. R. Saleska, V. I. Rich and J. P. Chanton, Carbon Accumulation, Flux, and Fate in Stordalen Mire, a Permafrost Peatland in Transition, Global Biogeochem. Cycles, 2022, 36(1), e2021GB007113, DOI:10.1029/2021GB007113.
- D. Olefeldt, N. T. Roulet, O. Bergeron, P. Crill, K. Bäckstrand and T. R. Christensen, Net carbon accumulation of a high-latitude permafrost palsa mire similar to permafrost-free peatlands, Geophys. Res. Lett., 2012, 39(3), 2011GL050355, DOI:10.1029/2011GL050355.
- M. Lupascu, J. L. Wadham, E. R. C. Hornibrook and R. D. Pancost, Temperature Sensitivity of Methane Production in the Permafrost Active Layer at Stordalen, Sweden: A Comparison with Non-permafrost Northern Wetlands, Arct. Antarct. Alp. Res., 2012, 44, 469–482 CrossRef.
- R. Mondav, B. J. Woodcroft, E.-H. Kim, C. K. McCalley, S. B. Hodgkins, P. M. Crill, J. Chanton, G. B. Hurst, N. C. VerBerkmoes, S. R. Saleska, P. Hugenholtz, V. I. Rich and G. W. Tyson, Discovery of a novel methanogen prevalent in thawing permafrost, Nat. Commun., 2014, 5, 3212 CrossRef PubMed.
- S. B. Hodgkins, M. M. Tfaily, D. C. Podgorski, C. K. McCalley, S. R. Saleska, P. M. Crill, V. I. Rich, J. P. Chanton and W. T. Cooper, Elemental composition and optical properties reveal changes in dissolved organic matter along a permafrost thaw chronosequence in a subarctic peatland, Geochim. Cosmochim. Acta, 2016, 187, 123–140 CrossRef CAS.
- M. S. Patzner, M. Logan, A. M. McKenna, R. B. Young, Z. Zhou, H. Joss, C. W. Mueller, C. Hoeschen, T. Scholten, D. Straub, S. Kleindienst, T. Borch, A. Kappler and C. Bryce, Microbial iron cycling during palsa hillslope collapse promotes greenhouse gas emissions before complete permafrost thaw, Commun. Earth Environ., 2022, 3(1), 76, DOI:10.1038/s43247-022-00407-8.
- A. Bhattacharyya, M. P. Schmidt, E. Stavitski and C. E. Martínez, Iron speciation in peats: Chemical and spectroscopic evidence for the co-occurrence of ferric and ferrous iron in organic complexes and mineral precipitates, Org. Geochem., 2018, 115, 124–137 CrossRef CAS.
- L. K. ThomasArrigo and R. Kretzschmar, Iron speciation changes and mobilization of colloids during redox cycling in Fe-rich, Icelandic peat soils, Geoderma, 2022, 428, 116217 CrossRef CAS.
- E. E. Roden and J. M. Zachara, Microbial Reduction of Crystalline Iron(III) Oxides: Influence of Oxide Surface Area and Potential for Cell Growth, Environ. Sci. Technol., 1996, 30, 1618–1628 CrossRef CAS.
- O. Larsen and D. Postma, Kinetics of reductive bulk dissolution of lepidocrocite, ferrihydrite, and goethite, Geochim. Cosmochim. Acta, 2001, 65, 1367–1379 CrossRef CAS.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory. Preparation and Characterization, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- L. L. Stookey, Ferrozine—a new spectrophotometric reagent for iron, Anal. Chem., 1970, 42, 779–781 CrossRef CAS.
- D. Olefeldt and N. T. Roulet, Effects of permafrost and hydrology on the composition and transport of dissolved organic carbon in a subarctic peatland complex, J. Geophys. Res., 2012, 117(G1), 2011JG001819, DOI:10.1029/2011JG001819.
- B. Ravel and M. Newville, ATHENA and ARTEMIS Interactive Graphical Data Analysisusing IFEFFIT, Phys. Scr., 2005, 2005, 1007 CrossRef.
- T. Dittmar, B. Koch, N. Hertkorn and G. Kattner, A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater, Limnol. Oceanogr.: Methods, 2008, 6, 230–235 CrossRef CAS.
- M. Chen, S. Kim, J.-E. Park, H.-J. Jung and J. Hur, Structural and compositional changes of dissolved organic matter upon solid-phase extraction tracked by multiple analytical tools, Anal. Bioanal. Chem., 2016, 408, 6249–6258 CrossRef CAS PubMed.
- B. P. Koch and T. Dittmar, From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter, Rapid Commun. Mass Spectrom., 2006, 20, 926–932 CrossRef CAS.
- D. E. LaRowe and P. van Cappellen, Degradation of natural organic matter: a thermodynamic analysis, Geochim. Cosmochim. Acta, 2011, 75, 2030–2042 CrossRef CAS.
- H. R. Naughton, M. Keiluweit, M. M. Tfaily, J. J. Dynes, T. Regier and S. Fendorf, Development of energetic and enzymatic limitations on microbial carbon cycling in soils, Biogeochemistry, 2021, 153, 191–213 CrossRef CAS.
- A. Gunina and Y. Kuzyakov, From energy to (soil organic) matter, Global Change Biol., 2022, 28, 2169–2182 CrossRef CAS PubMed.
- J. A. J. Dungait, D. W. Hopkins, A. S. Gregory and A. P. Whitmore, Soil organic matter turnover is governed by accessibility not recalcitrance, Global Change Biol., 2012, 18, 1781–1796 CrossRef.
- Z. Hu, A. M. McKenna, K. Wen, B. Zhang, H. Mao, L. Goual, X. Feng and M. Zhu, Controls of Mineral Solubility on Adsorption-Induced Molecular Fractionation of Dissolved Organic Matter Revealed by 21 T FT-ICR MS, Environ. Sci. Technol., 2024, 58, 2313–2322 CrossRef CAS PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides, Wiley-VCH, Weinheim, 2nd edn, 2003 Search PubMed.
- E. Tipping, The adsorption of aquatic humic substances by iron oxides, Geochim. Cosmochim. Acta, 1981, 45, 191–199 CrossRef CAS.
- A. Jain, K. P. Raven and R. H. Loeppert, Arsenite and Arsenate Adsorption on Ferrihydrite: Surface Charge Reduction and Net OH - Release Stoichiometry, Environ. Sci. Technol., 1999, 33, 1179–1184 CrossRef CAS.
- N. Bompoti, M. Chrysochoou and M. Machesky, Surface structure of ferrihydrite: Insights from modeling surface charge, Chem. Geol., 2017, 464, 34–45 CrossRef CAS.
- B. Gu, J. Schmitt, Z. Chen, L. Liang and J. F. McCarthy, Adsorption and desorption of natural organic matter on iron oxide: mechanisms and models, Environ. Sci. Technol., 1994, 28, 38–46 CrossRef CAS PubMed.
- B. Gu, J. Schmitt, Z. Chen, L. Liang and J. F. McCarthy, Adsorption and desorption of different organic matter fractions on iron oxide, Geochim. Cosmochim. Acta, 1995, 59, 219–229 CrossRef CAS.
- B. C. McAdams, J. Hudson, W. A. Arnold and Y.-P. Chin, Effects of aquatic dissolved organic matter redox state on adsorption to goethite, Aquat. Sci., 2023, 85(1), 19, DOI:10.1007/s00027-022-00912-0.
- T. D. Sowers, D. Adhikari, J. Wang, Y. Yang and D. L. Sparks, Spatial Associations and Chemical Composition of Organic Carbon Sequestered in Fe, Ca, and Organic Carbon Ternary Systems, Environ. Sci. Technol., 2018, 52, 6936–6944 CrossRef CAS PubMed.
- M. Sodano, C. Lerda, R. Nisticò, M. Martin, G. Magnacca, L. Celi and D. Said-Pullicino, Dissolved organic carbon retention by coprecipitation during the oxidation of ferrous iron, Geoderma, 2017, 307, 19–29 CrossRef CAS.
- A. R. Possinger, M. J. Zachman, J. J. Dynes, T. Z. Regier, L. F. Kourkoutis and J. Lehmann, Co-precipitation induces changes to iron and carbon chemistry and spatial distribution at the nanometer scale, Geochim. Cosmochim. Acta, 2021, 314, 1–15 CrossRef CAS.
- K.-Y. Chen, T.-Y. Chen, Y.-T. Chan, C.-Y. Cheng, Y.-M. Tzou, Y.-T. Liu and H.-Y. Teah, Stabilization of Natural Organic Matter by Short-Range-Order Iron Hydroxides, Environ. Sci. Technol., 2016, 50, 12612–12620 CrossRef CAS PubMed.
- K. G. J. Nierop, B. Jansen and J. M. Verstraten, Dissolved organic matter, aluminium and iron interactions: precipitation induced by metal/carbon ratio, pH and competition, Sci. Total Environ., 2002, 300, 201–211 CrossRef CAS PubMed.
- T. L. Theis and P. C. Singer, Complexation of iron(II) by organic matter and its effect on iron(II) oxygenation, Environ. Sci. Technol., 1974, 8, 569–573 CrossRef CAS.
- E. E. Daugherty, B. Gilbert, P. S. Nico and T. Borch, Complexation and Redox Buffering of Iron(II) by Dissolved Organic Matter, Environ. Sci. Technol., 2017, 51, 11096–11104 CrossRef CAS PubMed.
- H. Ma, P. Wang, A. Thompson, Q. Xie, M. Zhu, H. H. Teng, P. Fu, C. Liu and C. Chen, Secondary Mineral Formation and Carbon Dynamics during FeS Oxidation in the Presence of Dissolved Organic Matter, Environ. Sci. Technol., 2022, 56, 14120–14132 CrossRef CAS PubMed.
- R. Young, S. Avneri-Katz, A. McKenna, H. Chen, W. Bahureksa, T. Polubesova, B. Chefetz and T. Borch, Composition-Dependent Sorptive Fractionation of Anthropogenic Dissolved Organic Matter by Fe(III)-Montmorillonite, Soil Syst., 2018, 2, 14 CrossRef CAS.
- J. M. Byrne and A. Kappler, A revised analysis of ferrihydrite at liquid helium temperature using Mössbauer spectroscopy, Am. Mineral., 2022, 107, 1643–1651 CrossRef.
- E. Murad and J. Cashion, Mössbauer Spectroscopy of Environmental Materials and Their Industrial Utilization, Springer, New York, NY, 2011 Search PubMed.
- E. Murad and U. Schwertmann, The influence of crystallinity on the Mössbauer spectrum of lepidocrocite, Mineral. Mag., 1984, 48, 507–511 CrossRef CAS.
- U. Schwertmann, F. Wagner and H. Knicker, Ferrihydrite–Humic Associations, Soil Sci. Soc. Am. J., 2005, 69, 1009–1015 CrossRef CAS.
- L. K. ThomasArrigo, L. Notini, J. Shuster, T. Nydegger, S. Vontobel, S. Fischer, A. Kappler and R. Kretzschmar, Mineral characterization and composition of Fe-rich flocs from wetlands of Iceland: Implications for Fe, C and trace element export, Sci. Total Environ., 2022, 816, 151567 CrossRef CAS PubMed.
- L. Curti, O. W. Moore, P. Babakhani, K.-Q. Xiao, C. Woulds, A. W. Bray, B. J. Fisher, M. Kazemian, B. Kaulich and C. L. Peacock, Carboxyl-richness controls organic carbon preservation during coprecipitation with iron (oxyhydr)oxides in the natural environment, Commun. Earth Environ., 2021, 2, 1–13 CrossRef.
- C. Mikutta, J. Frommer, A. Voegelin, R. Kaegi and R. Kretzschmar, Effect of citrate on the local Fe coordination in ferrihydrite, arsenate binding, and ternary arsenate complex formation, Geochim. Cosmochim. Acta, 2010, 74, 5574–5592 CrossRef CAS.
- F. M. Michel, L. Ehm, S. M. Antao, P. L. Lee, P. J. Chupas, G. Liu, D. R. Strongin, M. A. A. Schoonen, B. L. Phillips and J. B. Parise, The structure of ferrihydrite, a nanocrystalline material, Science, 2007, 316, 1726–1729 CrossRef CAS PubMed.
- J. Niemeyer, Y. Chen and J.-M. Bollag, Characterization of Humic Acids, Composts, and Peat by Diffuse Reflectance Fourier-Transform Infrared Spectroscopy, Soil Sci. Soc. Am. J., 1992, 56, 135–140 CrossRef CAS.
- M. M. Tfaily, W. T. Cooper, J. E. Kostka, P. R. Chanton, C. W. Schadt, P. J. Hanson, C. M. Iversen and J. P. Chanton, Organic matter transformation in the peat column at Marcell Experimental Forest: Humification and vertical stratification, J. Geophys. Res.: Biogeosci., 2014, 119, 661–675 CrossRef CAS.
- K. Kaiser and G. Guggenberger, Sorptive stabilization of organic matter by microporous goethite: sorption into small pores vs. surface complexation, Eurasian J. Soil Sci., 2007, 58, 45–59 CrossRef CAS.
- J. Lv, S. Zhang, S. Wang, L. Luo, D. Cao and P. Christie, Molecular-Scale Investigation with ESI-FT-ICR-MS on Fractionation of Dissolved Organic Matter Induced by Adsorption on Iron Oxyhydroxides, Environ. Sci. Technol., 2016, 50, 2328–2336 CrossRef CAS PubMed.
- E. K. Coward, T. Ohno and D. L. Sparks, Direct Evidence for Temporal Molecular Fractionation of Dissolved Organic Matter at the Iron Oxyhydroxides Interface, Environ. Sci. Technol., 2019, 53, 642–650 CrossRef CAS PubMed.
- T. Riedel, D. Zak, H. Biester and T. Dittmar, Iron traps terrestrially derived dissolved organic matter at redox interfaces, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10101–10105 CrossRef CAS PubMed.
- K. Boye, V. Noël, M. M. Tfaily, S. E. Bone, K. H. Williams, J. R. Bargar and S. Fendorf, Thermodynamically controlled preservation of organic carbon in floodplains, Nat. Geosci., 2017, 10, 415–419 CrossRef CAS.
- X. Huang, X. Liu, J. Liu and H. Chen, Iron-bound organic carbon and their determinants in peatlands of China, Geoderma, 2021, 391, 114974 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4em00241e |
| This journal is © The Royal Society of Chemistry 2024 |