
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Different routes of bismuth mineral transformation during pertechnetate and perrhenate uptake for subsurface remediation†
D.
Boglaienko
 *,
M. E.
Bowden
,
N. M.
Escobedo
,
Q. M.
Collins
,
A. R.
Lawter
,
T. G.
Levitskaia
* and
C. I.
Pearce
*
*,
M. E.
Bowden
,
N. M.
Escobedo
,
Q. M.
Collins
,
A. R.
Lawter
,
T. G.
Levitskaia
* and
C. I.
Pearce
*
Pacific Northwest National Laboratory, 902 Battelle Boulevard, Richland, WA 99354, USA. E-mail: daria.boglaienko@pnnl.gov; Tatiana.Levitskaia@pnnl.gov; carolyn.pearce@pnnl.gov
First published on 3rd October 2024
Abstract
We investigated basic bismuth subnitrate for removal of radioactive technetium-99 as pertechnetate (99TcO4−) from contaminated groundwater. This material removed 93% of the initial concentration of 99TcO4− within a week via formation of pH-dependent mineral phases that were identified here, but not reported previously. Perrhenate (ReO4−) removal was also studied because it is a widely used non-radiological analogue for 99TcO4−, considering their similar physicochemical properties. We found that removal of ReO4− was not identical to removal of 99TcO4− and led to formation of an additional transitional phase. This demonstrates that perrhenate and pertechnetate have different kinetics of contaminant removal as a result of variations in mineral transformation.
Water impactBasic bismuth subnitrate was studied to capture a contaminant 99TcO4− and its non-radioactive analogue ReO4− in a groundwater simulant. Removal of 99TcO4− without its release back within a two-month period occurred during bismuth mineral transformation. Application of the non-toxic bismuth minerals can be one of the efficient options for subsurface remediation but requires more in-depth investigations and insights. |
Introduction
Bismuth(III) (Bi3+, referred to as Bi) materials are non-toxic and versatile, and comprise a group of remediation materials with numerous advantages, e.g., environmentally friendly, high affinity for many contaminants, availability, cost effective, and easy to handle.1,2 In environmental research, Bi-based materials, mainly oxyhalides BiOX (X = Cl, Br, I), are predominantly studied for their photocatalytic properties for water disinfection, groundwater remediation, air purification, carbon dioxide reduction, energy production, and for electrochemical sensing of pollutants.2–6 However, there are synthesis challenges with BiOX-based materials, and lack of in-depth understanding of structure–property relationships.7Here, we investigated Bi-based materials for contaminant removal via mechanisms other than photocatalysis. Past nuclear waste disposal practices at the U.S. Department of Energy's Hanford Site (WA, USA) have driven the need for groundwater remediation. We have conducted a series of studies on sequestration of contaminants existing as anions in the subsurface, i.e. Cr(VI) as CrO42−, U as carbonate complexes of UO22+, 129I as IO3−, and 99TcO4− (further TcO4−), from contaminated groundwater with Bi-based materials.8–11 In these studies, we synthesized a disordered Bi oxyhydroxide (BOH), which was effective for uptake of all the abovementioned contaminants except pertechnetate, TcO4−. We found that pertechnetate can be removed with another Bi material, commercially available Bi subnitrate (BSN).9,10 The removal was pH-dependent and further studies were needed to elaborate removal mechanism and Bi mineral transformation of BSN in a groundwater simulant. Specifically, we aim to investigate which mineral phase is responsible for TcO4− uptake during BSN transformation and kinetics of TcO4− removal.
Bi aqueous chemistry is complex, with hydrolysis leading to olation and formation of polynuclear hydroxo species, and oxolation leading to oxo-bridged species through hydrolytic polymerization of olated cations. These oxo-hydroxo polynuclear species form Bi subnitrate during crystallization of dilute Bi3+ solutions in HNO3.12 If concentrated HNO3 is used, the pentahydrate Bi(NO3)3·5H2O is crystallized from Bi3+ solutions, and the basic salt BiO(NO3) precipitates upon dilution.13 A mixture of the basic compounds BiO(NO3), Bi(NO3)(OH)2, Bi2O2(OH)(NO3), and BiOOH, as well as a variety of Bi polynuclear species, were also reported in literature as a result of hydrolysis of Bi3+ aqueous solutions.14–16 Bi hexanuclear clusters are defined as [Bi6Ox(OH)8−x](10−x)+, i.e. [Bi6O5(OH)3](NO3)5·3H2O and [Bi6O4(OH)4](NO3)6·H2O,17 – both described first by Lazarini (1978 and 1979).15,18 They are represented by the two main complex ions [Bi6O5(OH)3]5+ and [Bi6O4(OH)4]6+ charge balanced by NO3− and OH− ligands through Bi–ONO2 bonds or OH–ONO2 hydrogen bonds,14,19 and their structural isomers, e.g., [Bi6O4(OH)4(NO3)5(H2O)](NO3), [Bi6O4(OH)4(NO3)6(H2O)2]·H2O.20 According to Christensen and Lebech (2012),21 the overall composition of a basic Bi nitrate can be described as a mixture of [Bi6O4(OH)4]6+ and [Bi6O5(OH)3]5+ with a general formula [Bi6O4(OH)4]0.5+x[Bi6O5(OH)3]0.5−x(NO3)5.5+x (ref. 21) or [Bi6O4.5(OH)3.5]2(NO3)11.22 In this mixture, the equilibrium between the clusters is pH-dependent:21
[Bi6O4(OH)4]6+ + H2O ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) [Bi6O5(OH)3]5+ + H3O+ [Bi6O5(OH)3]5+ + H3O+ | (1) |
For BSN, Liu et al. (2007)23 solved the structure that had been previously reported by Lazarini (1978),15 describing it as a dumbbell-like cluster consisting of two bridged [Bi6O5(OH)3]5+ polynuclear species: Bi12O10(OH)6(NO3)10·6H2O. Contaminant uptake by this Bi cluster in the BSN starting material was the foci of our work.
In our studies, we used groundwater simulants containing chloride (Cl−), sulfate (SO42−), bicarbonate/carbonate (HCO3−/CO32−), silicate (SiO32−), and nitrate (NO3−),9–11 to determine the effect of these common groundwater anions on the removal of contaminants of interest. We showed that the starting Bi polynuclear species transformed to different Bi mineral phases, and that the co-mingled effect of different anions in solution changed the Bi transformation paths. More detailed discussion is given in Escobedo et al. (in prep.).10
The layered crystalline structures of these Bi minerals exhibit electronic and spatial flexibility, due to the lone-pair electronic effect.24 This flexibility allows them to sequester various contaminants as negatively charged species. The layered structure is composed of Bi–O sheets ([Bi2O2]2+ in a fluorite-related structure) and two large groups, the Aurivillius and Sillén, cover numerous Bi minerals of various composition – e.g., BiPb2OCl, Bi2WO6, BiBa2Nb2O9, Bi4Ti3O12etc., − with alternating arrangement of anions of different nature between sheets of [Bi2O2]2+.1,25
Bismutite Bi2O2(CO3) and bismoclite BiOCl – layered Sillén related and Sillén structures, respectively – are two of the most common bismuth minerals found in the environment under ambient conditions. Bismutite is more thermodynamically stable (bismutite ΔGf = −916.2 kJ mol−1; bismoclite ΔGf = −322 kJ mol−1) and will dominate except under acidic conditions and the presence of chloride (eqn (2)).26
| Bi2O2CO3(s) + 2H+(aq) + 2Cl−(aq) = 2BiOCl(s) + CO2(g) + H2O | (2) |
Materials and methods
Caution! 99Tc is a β− emitter; energy 0.29 MeV (ref. 35) and should be handled in a suitably controlled facility. The experimental work with radioactive samples was conducted in a nuclear research facility by trained personnel in a fume hood designated for radiological contamination control.Materials
Bismuth(III) subnitrate, BSN, and bismuth(III) oxychloride, BiOCl, were procured from Sigma Aldrich. A cluster compound Bi6O4(OH)4(NO3)6 was synthesized following the procedure described in Henry et al. (2006).19 Briefly, 0.5 g Bi2O3 was dissolved in 2 mL concentrated HNO3 and added dropwise to a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water/ethanol solution (200 mL) with stirring. The precipitate was filtered, washed with deionized water (DI water), air-dried at room temperature, and characterized by PXRD. Groundwater simulant, here HSGW (Hanford simulated groundwater), was prepared using chemical reagents of analytical grade: silicic acid, H2SiO3·xH2O – Sigma Aldrich, potassium chloride, KCl – Fisher Chemical, magnesium carbonate, MgCO3 – Sigma Aldrich, sodium chloride, NaCl – Fisher Chemical, calcium sulfate dihydrate, CaSO4·2H2O – Acros Organics, calcium carbonate, CaCO3 – Baker. The in-house solution of ammonium pertechnetate, NH4TcO4, was prepared at the Radiochemical Processing Laboratory (Pacific Northwest National Laboratory), and sodium perrhenate, NaReO4, was acquired from Sigma Aldrich. Double deionized water of at least 18 MΩ cm was used to prepare all solutions.
:50 water/ethanol solution (200 mL) with stirring. The precipitate was filtered, washed with deionized water (DI water), air-dried at room temperature, and characterized by PXRD. Groundwater simulant, here HSGW (Hanford simulated groundwater), was prepared using chemical reagents of analytical grade: silicic acid, H2SiO3·xH2O – Sigma Aldrich, potassium chloride, KCl – Fisher Chemical, magnesium carbonate, MgCO3 – Sigma Aldrich, sodium chloride, NaCl – Fisher Chemical, calcium sulfate dihydrate, CaSO4·2H2O – Acros Organics, calcium carbonate, CaCO3 – Baker. The in-house solution of ammonium pertechnetate, NH4TcO4, was prepared at the Radiochemical Processing Laboratory (Pacific Northwest National Laboratory), and sodium perrhenate, NaReO4, was acquired from Sigma Aldrich. Double deionized water of at least 18 MΩ cm was used to prepare all solutions.
Batch kinetics experiments
The preparation procedure for HSGW was adopted from Truex et al., 2017 (i.e. Table 9).36 Final pH of the HSGW after filtering was 8.1 ± 0.1. The initial TcO4− and ReO4− stock solutions were prepared in DI water to have concentrations 40 mM KTcO4 and 0.1 M NaReO4. The same liquid to solid ratio was used in all batch experiments, i.e. 100 mL of liquid and 0.1 g of the solid bismuth material. The preparation of the samples was as follows: 100 mL of HSGW were added to a 250 mL Nalgene bottle, spiked with either TcO4− or ReO4− stock solution to have a target concentration 0.17 mM of each contaminant. After that, 0.1 g of bismuth material was mixed into the solution, samples were placed on an orbital shaking table at 200 rpm (MaxQ 2000, A class, J-Kem Scientific), and time was recorded as the starting point of the experimental series. Samples were taken every day for 3 days after the start of the experiment, followed by weekly sampling. Sacrificial samples were terminated at different times to examine changes to the Bi-based materials after exposure to the contaminants for up to almost two months. The control series had the same liquid to solid ratio of HSGW to BSN, excluding the contaminants; and the same concentrations of TcO4− and ReO4−, excluding BSN. Aliquots from the TcO4− series (50 or 100 μL) were filtered into 10 mL of liquid scintillation cocktail (Ultima Gold LLT, Perkin Elmer), and analyzed using a liquid scintillation counter (Tri-Carb 3100TR, PerkinElmer). The data were processed with correction for the background. Aliquots from the ReO4− series (1 mL) were collected for inductively coupled plasma mass spectrometry (ICP-MS). Both TcO4− and ReO4− aliquots were filtered using 0.45 μm pore size syringe filters (PTFE membrane, 25 mm, EZFlow). pH data were collected systematically for the ReO4− and control series.All experiments were conducted under aerobic conditions, at ambient temperature and pressure.
Powder X-ray Diffraction (PXRD)
The PXRD analysis with BSN starting material was conducted upon the termination of the experiments, at 59 days, when the samples were centrifuged, aqueous phase was decanted and the solid phase air dried. Additional samples were prepared and terminated for PXRD analysis at 24–28 and 45–49 days after the start of experiment. The PXRD patterns were collected using Cu Kα radiation (λ = 1.5418 Å) employing a Rigaku Miniflex II XRD unit. The Miniflex was operated at 30 kV and 15 mA and patterns were collected between 3 and 90° 2θ, scanning at 2° min−1 at 0.02° 2θ intervals. Identification of material phases was carried out using JADE® XRD pattern processing software (Materials Data Incorporated, CA) and reference patterns from the International Centre for Diffraction Data (ICDD) powder diffraction database.Results and discussion
In our earlier study we showed that the starting BSN material is the Bi12O10(OH)6(NO3)10·6H2O compound described by Lazarini (1978)15 and Liu et al. (2007),23 comprised of bridged hexanuclear [Bi6O5(OH)3]5+ clusters (Fig. S1, ESI†).9 It is helpful to distinguish between the dominant structural motifs in Bi compounds, and so we use the prefix “clus-” to denote compounds based on clusters, “lay-” for compounds based on layered [Bi2O2]2+ sheets, “dis-” for disordered compounds which give very broad PXRD peaks, and “unk-” for compounds where the structure is unknown. Hence, BSN is denoted as clus-Bi12O10(OH)6(NO3)10·6H2O.The clusters are charge balanced by anions, with the NO3− in the starting material, released and replaced by OH−, Cl−, SO42− and CO32−, depending on composition of aqueous media.10 In deionized water, there is decrease in pH (Fig. 1), as clusters undergo NO3− ligand exchange with OH− (eqn (3)). The pH decrease is less significant in aqueous media containing HCO3−/CO32− ions that participate in ligand exchange and buffer pH. Evidence of release of NO3− from BSN starting material in DI water with consequent pH drop was shown in our previous study.10
| [Bi6O4(OH)4](NO3)6 + (6 − x)H2O = [Bi6O4(OH)4](NO3)x(OH)(6−x) + (6 − x)H+ + (6 − x)NO3 | (3) |
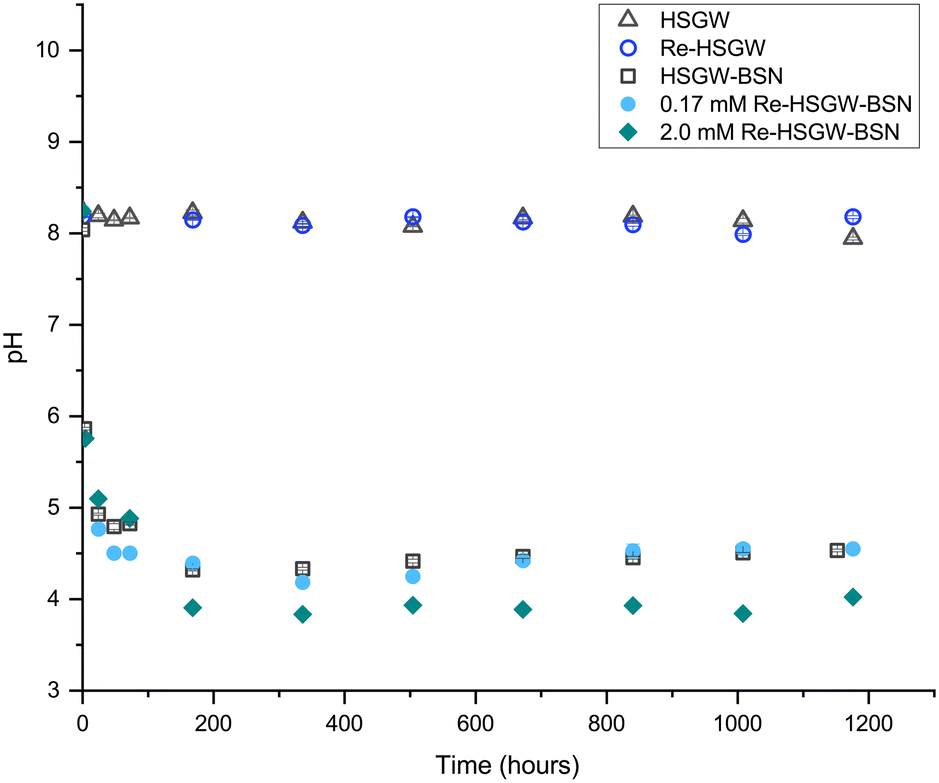 | ||
| Fig. 1 Change of pH with time in the 0.17 mM and 2.0 mM ReO4− series with 0.1 g BSN starting material in 100 mL HSGW simulant. Control series: HSGW – groundwater simulant only; Re-HSGW – 0.17 mM Re in the simulant; HSGW-BSN – 100 mL groundwater simulant only; Re-HSGW – 0.17 mM Re in the simulant; HSGW-BSN – 100 mL groundwater simulant and 0.1 g BSN starting material. Error bars within the symbols are ± 1 st. dev. of triplicate measurements. | ||
Fig. 1 shows changes of pH in the samples with BSN and 0.17 and 2.0 mM of ReO4− as compared to ReO4− in the groundwater simulant without BSN and to the groundwater simulant itself, where pH did not change by the end of the experiment. The decrease in pH caused by mineral transformation of BSN in HSGW simulant without ReO4− is shown for the control sample HSGW-BSN.
There were similar changes in pH trend, plateauing around 4.4, in the 0.17 mM ReO4− sample and HSGW-BSN control (BSN in the groundwater simulant without perrhenate) while pH decreased slightly below 4.0 in the 2.0 mM ReO4− sample.
This trend in pH for the 0.17 mM ReO4− samples agrees with pH measurements in our previous study for 0.17 mM TcO4− samples with BSN in HSGW at the same solution-to-solid ratio,10 where pH dropped to 6.7 after 1 day. After 60 days of the contact time, pH was 3.8, no intermediate pH measurements were performed. In that study, pH of the HSGW-BSN blank was 6.5 after 1 day and 3.5 after 60 days, which implies slightly higher buffering capacity of the batch of HSGW simulant prepared for these experiments, resulting in pH of 4.3 at the end of the experiment. The HSGW is saturated with respect to CaCO3, and the extent of saturation (thereby the buffering capacity) will vary depending on temperature, exposure to atmosphere etc. The overall similar trend between the studies shows that the pH data for the 0.17 mM ReO4− series is representative of the 0.17 mM TcO4− series.
NO3− anions in the BSN starting material undergo rapid ligand exchange with OH−, with pH decreasing below 6 after only three hours from the start of the experiment and plateauing after 168 hours (7 days) from the start of the experiment. Addition of 0.17 mM ReO4− to the HSGW simulant without BSN had no impact on the pH time profile. Consequently, we would expect contaminants to be removed by 7 days, or 168 hours, if the main mechanism for their removal is related to changes in BSN material caused by anion exchange. The removal kinetics for TcO4− (Fig. 2a) showed that 93% of the initial 0.17 mM TcO4− was removed from HSGW after 168 hours from the start of the experiment, but only 78% of the ReO4− was removed over the same time period (Fig. 2b). It took up to 672 hours for ReO4− removal to be below ICP-MS detection limit (0.027 mM of Re). The rate constant for TcO4− removal within the first 72 hours (1.2 ± 0.2) × 10−4 min−1 was slightly higher than the rate constant for ReO4− removal (0.7 ± 0.2) × 10−4 min−1 with an initial concentration of 0.17 mM and over the same time period (Fig. 2b).
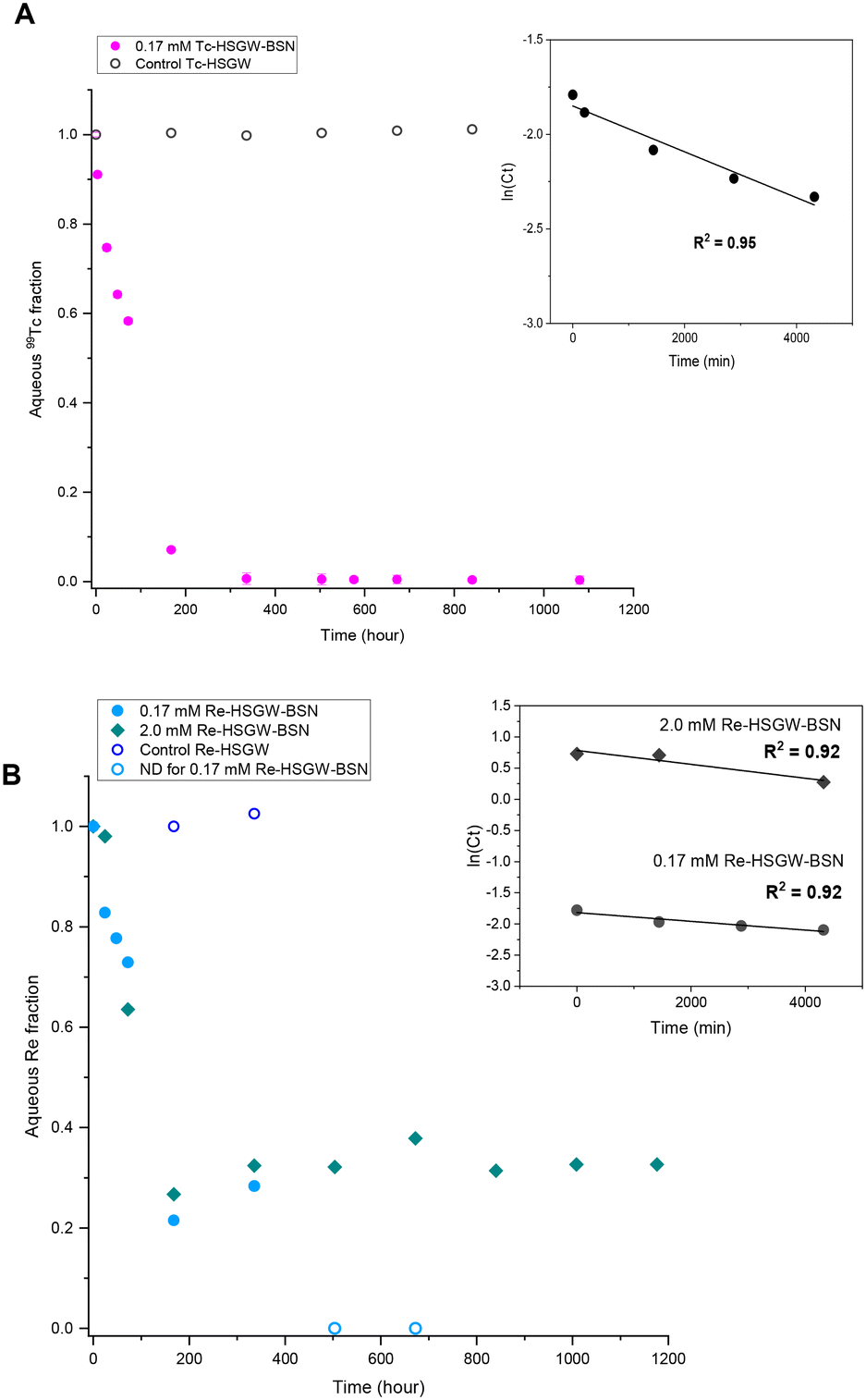 | ||
| Fig. 2 Kinetics of TcO4− and ReO4− removal with BSN starting material (0.1 g) in HSGW simulant (100 mL). A. Removal of 0.17 mM TcO4−. Control series: 0.17 mM TcO4− in HSGW simulant. B. Removal of 0.17 and 2.0 mM ReO4−. Control series: 0.17 mM ReO4− in HSGW simulant (only select time points were analyzed). Insets: First order linear models for 72 hours (4320 min). ND – not detected (ICP-MS EQL for Re was 0.027 mM). | ||
The rate constant for ReO4− removal from the 2.0 mM sample was (1.1 ± 0.3) × 10−4 min−1 for the first 72 hours. Concentrations of Re in solution at 72 hours were 0.13 mM and 1.3 mM for 0.17 mM and 2.0 mM ReO4− samples respectively. Later, after one week, it reached equilibrium, removing only, on average, 67% from the initial 2.0 mM Re concentration. Lower uptake of ReO4− in the 2.0 mM sample at the later time can be related to the different initial Bi to Re molar ratio, which in the 0.17 mM ReO4− sample was around 20.6, while in 2.0 mM ReO4− sample only 1.7. The equilibrium pH of this sample (3.91 ± 0.06) was lower compared to the 0.17 mM ReO4− sample (4.43 ± 0.08) revealing a greater extent of NO3− exchange for OH− in the Bi material during transformation.
The differences in the TcO4− and ReO4− removal kinetics with BSN imply different removal mechanisms, and we employed PXRD analysis to investigate the nature of the BSN mineral transformation in these experiments.
A summary of the PXRD patterns for TcO4− or ReO4− in contact with BSN material is given in Fig. 3, including the PXRD pattern for the blank, i.e. BSN in HSGW.
 | ||
| Fig. 3 Bi minerals after contact of the Bi subnitrate, or BSN, starting material with 0.17 mM TcO4− or 0.17 mM ReO4− in HSGW simulant for different number of days. Blank is BSN in HSGW simulant. Bi rhenium oxide Bi3ReO8 indicated as “o”; daubreeite related structure lay-BiO(OH,Cl) indicated as “d”; and the unknown phase unk-Bi(NO3)x(OH)yOz indicated as “x”. Numbers show peak positions of unidentified phases. Only the most clear and prominent peaks are marked. Disordered phase dis-BiOw(OH)x(NO3)y is evident as broad peaks in all patterns, except 2.0 mM Re 49 days. | ||
With both 0.17 mM ReO4− and TcO4−, the layered daubreeite related structure, lay-BiO(OH,Cl), and a disordered structure, with a general chemical formulae dis-BiOw(OH)x(NO3)y defined in our previous study,9 were present in all patterns. The disordered structure transformed into the unknown, i.e. not resolvable by PXRD analysis, unk-Bi(NO3)x(OH)yOz phase by 59 days (growing peaks in the region of 27–31° 2θ). Formation of this structure is consistent with our previous study, where unk-Bi(NO3)x(OH)yOz and lay-BiO(OH,Cl) formed after 60 days in the presence of TcO4−.10 The blank sample was less crystalline, composed of lay-BiO(OH,Cl) and dis-BiOw(OH)x(NO3)y structures. This sample is less crystalline compared to the blank sample in the previous study exposed to HSGW for 60 days.10 The blank samples were prepared identically for these two studies, but with different batches of the HSGW variable in carbonate saturation and buffering capacity. Differences in crystallinity and amount of the disordered phase in the blanks show that phase transformation of BSN starting material is sensitive to the presence of carbonate in a groundwater simulant. The disordered structure was described in more detail in Pearce et al.,9 where the broad diffraction pattern was like that for a metastable δ-polymorph of bismite (Bi2O3). The presence of 0.17 mM TcO4− and ReO4− in the HSGW enhanced the crystallinity of the solid phase, compared to the blank further enhanced in the 2.0 mM ReO4− sample (Fig. 3).
The experiments were conducted under aerobic conditions and no color change was observed. We assume that TcO4−, as well as ReO4−, is removed by non-specific adsorption and ion exchange with bismuth subnitrate and preserves its chemical form as pertechnetate, or perrhenate, anion.9 Despite expected similarities in TcO4− and ReO4− uptake by BSN, a different product, Bi rhenium oxide (Bi3ReO8), was formed in the presence of ReO4−. The relative amount of Bi3ReO8 to lay-BiO(OH,Cl) decreased with time in 0.17 mM ReO4− series and is more prominent in the 28 day Bi–Re sample than in the 49 day Bi–Re sample, disappearing in the 59 day Bi–Re sample. We hypothesize that this is due to the prolonged dissolution of Bi3ReO8. This hypothesis is supported by the Pourbaix diagram showing that ReO4−, not Bi3ReO8(s), is the predominant Re species at low pH (Fig. S2 (ref. 37) ESI†). The PXRD pattern for the 2.0 mM Bi–Re sample showed a more crystalline structure comprised of two clearly resolvable mineral phases, lay-BiO(OH,Cl) and Bi3ReO8 by 49 days (Fig. 3). Compared to 0.17 mM ReO4− sample at 49 days, the relative amount of Bi3ReO8 to lay-BiO(OH,Cl) was larger in the 2.0 mM ReO4− sample, suggesting that a larger fraction of the initial BSN material participated in formation of Bi3ReO8 in the 2.0 mM ReO4− sample. This is similar to the formation of lay-BiO(OH,Cl), which is controlled by the amount of Cl− in HSGW.
The presence of Bi3ReO8 in 0.17 mM Re sample and absence of Bi3TcO8 in 0.17 mM Tc sample is demonstrated in Fig. S3 ESI.† The formation of different products as a result of TcO4− and ReO4− uptake by the BSN starting material was an unexpected outcome of this study, as it was anticipated that these two oxyanions would behave in the same way, given their similar physical and chemical properties. However, it is known from the literature that ReO4− undergoes changes in coordination more readily than TcO4−, and it has a lower hydration energy (−330 kJ mol−1 for ReO4− and −251 kJ mol−1 for TcO4−); with both only weakly binding to positively charged binding sites (ligands with strong specific coulombic interactions).27 The difference in the oxidizing potentials of ReVII/ReIVvs. TcVII/TcIV is −0.51 vs. −0.74, meaning that TcVII is more prone to reduction than ReVII at the same conditions; and it was demonstrated in the study with steel coupons that TcO4− was predominantly reduced to TcIV and bound to the steel (iron) corrosion products, while ReO4− remained oxidized and did not bind to the corrosion products.38 It is also described that for Bi–Re–O and Bi–Tc–O systems, Bi3ReO8 and Bi2Tc2O7 are the predominant phases (studied under non-oxidizing conditions), respectively, where Re has oxidations state VII and Tc has oxidations state IV; Bi3TcO8 (TcVII) synthesis was only possible from the Bi2Tc2O7 (TcIV) with Bi2O3 in O2 upon heating.29 Formation of Bi2Tc2O7 (the precursor to Bi3TcO8) is not expected in this study as it would require reducing conditions (TcVII to TcIV).
Formation of the Bi3ReO8 phase resulted in slightly slower removal of ReO4− by BSN compared to the removal of TcO4− by BSN without formation of a Tc-bearing Bi phase, under the same experimental conditions (starting concentration of 0.17 mM). Formation of Bi3ReO8 may also explain the plateau at around 70% ReO4− removed for the higher starting concentration of 2.0 mM (Fig. 2b), as compared to almost complete removal with a starting concentration of 0.17 mM ReO4−. PXRD shows that formation of Bi3ReO8 is the initial ReO4− removal mechanism. At a starting ReO4− concentration of 0.17 mM, the initial molar ratio of Bi to Re was 20.6, which is sufficient to removal all of the ReO4− from solution by formation of the Bi3ReO8 phase. At a starting ReO4− concentration of 2.0 mM, the molar ratio of Bi to Re was at most 1.7 in the system, which is less than the ratio of 3 required to form the Bi3ReO8 phase. Therefore, there was insufficient Bi in the system to remove all the ReO4− from solution by formation of Bi3ReO8, and in this case 42% of ReO4− should have remained in solution if no uptake was caused by lay-BiO(OH,Cl). The fact that there was approximately 30% of the ReO4− suggests some surface interaction together with incorporation into the Bi solid phase.
After 59 days in the presence of 0.17 mM TcO4−, the dominant Bi mineral phases were unk-Bi(NO3)x(OH)yOz and the daubreeite-related structure lay-BiO(OH,Cl). We hypothesize that the unk-Bi(NO3)x(OH)yOz phase was responsible for Tc removal from the HSGW simulant. To test this hypothesis, bismoclite (lay-BiOCl or BiOCl; Fig. S4 ESI†), which is isostructural with the daubreeite-related structure lay-BiO(OH,Cl), was used as the starting Bi phase for TcO4− removal (Fig. S5a and b†). No TcO4− removal from HSGW was observed up to 59 days (Tc average fraction of 1.00 ± 0.01) and pH plateaued at 6.00 ± 0.05 after 24 days likely indicating completion of the phase transformation in this time. The PXRD patterns revealed that both the daubreeite-related structure lay-BiO(OH,Cl) and bismutite lay-Bi2O2(CO3) were present after 24 and 59 days, with the amount of bismutite, qualitative estimate, increasing over time (Fig. 5a). Thus, interlayer exchange of Cl− ions in bismoclite for OH− and CO32− ions in HSGW resulted in bismuth mineral transformation (eqn (2)), but the bismutite and daubreeite-related phases formed did not uptake TcO4−.
A final set of experiments was conducted to compare TcO4− removal by the commercial BSN with TcO4− removal by a synthesized clus-Bi6O4(OH)4(NO3)6 (Fig. S6 ESI†). It was more effective for TcO4− removal in the short-term, with 73% removed after 1 day (9.1 ± 0.1) × 10−4 min−1 (Fig. 4a), compared to 25% removed by BSN over the same time period. However, clus-Bi6O4(OH)4(NO3)6 took longer to reach near quantitative (99%) TcO4− removal (59 days compared to 14 days for BSN, Fig. 2a), implying a slow process of mineral incorporation of TcO4− (unidentified mineral phase). With clus-Bi6O4(OH)4(NO3)6, pH decreased more rapidly to 3.68 ± 0.08 after 34 hours (Fig. 4b), compared to 4.93 ± 0.01 after 24 hours for BSN. PXRD patterns revealed that both clus-Bi6O4(OH)4(NO3)6 and BSN formed unk-Bi(NO3)x(OH)yOz and lay-BiO(OH,Cl) after 59 days (Fig. 5b). However, the peaks corresponding to the unk-Bi(NO3)x(OH)yOz phase are clearly visible in the PXRD pattern after 1 day with clus-Bi6O4(OH)4(NO3)6, in both test and blank samples, but are not visible until after 59 days with BSN. This suggests that the readily available [Bi6O4(OH)4]6+ polycations in clus-Bi6O4(OH)4(NO3)6 rapidly transformed into the unk-Bi(NO3)x(OH)yOz phase, whereas BSN required an additional step to convert the [Bi6O5(OH)3]5+ polycations in its structure to [Bi6O4(OH)4]6+, followed by transformation to unk-Bi(NO3)x(OH)yOz.
 | ||
| Fig. 4 A. Removal of 0.17 mM TcO4− with clus-Bi6O4(OH)4(NO3)6 starting material (0.1 g) in HSGW simulant (100 mL). Control series: 0.17 mM TcO4− in HSGW simulant. Inset: First order linear model for up to 24 hours (1440 min); first order constant k = (9.1 ± 0.1) × 10−4 min−1. B. PH measurements for control series: HSGW only and HSGW with clus-Bi6O4(OH)4(NO3)6. | ||
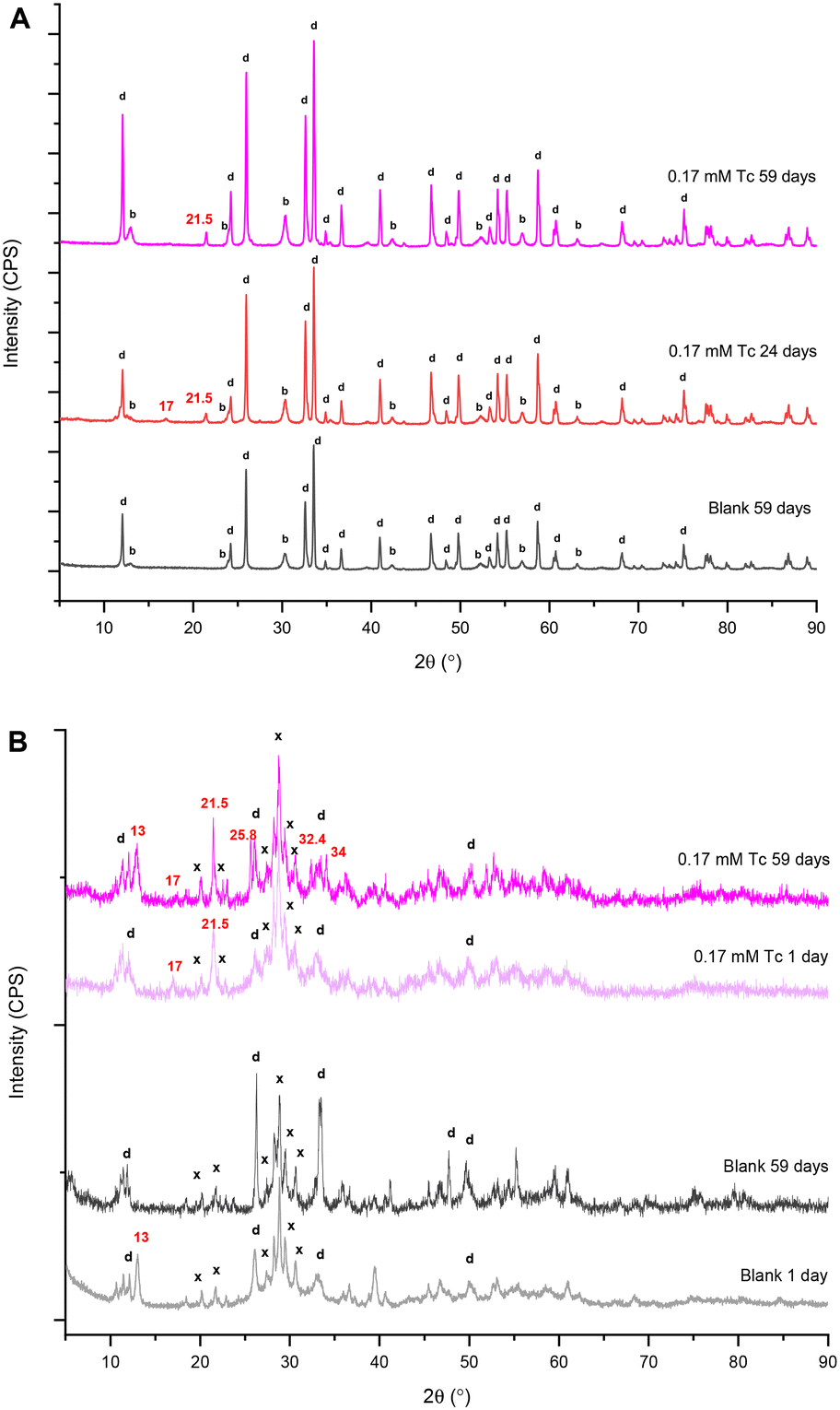 | ||
| Fig. 5 Bi minerals after contact with 0.17 mM TcO4− in HSGW simulant for different number of days. Blanks are the corresponding Bi materials in HSGW simulant. A. Bismoclite lay-BiOCl starting material. Daubreeite related structure lay-BiO(OH,Cl) indicated as “d”; bismutite lay-Bi2O2(CO3) indicated as “b”. B. clus-Bi6O4(OH)4(NO3)6 starting material. Daubreeite related structure lay-BiO(OH,Cl) indicated as “d”; and the unknown phase unk-Bi(NO3)x(OH)yOz indicated as “x”. Numbers show peak positions of unidentified phases. | ||
Formation of unk-Bi(NO3)x(OH)yOz also occurs in the absence of TcO4− through ligand exchange of BSN (clus-Bi12O10(OH)6(NO3)10·6H2O) and synthesized clus-Bi6O4(OH)4(NO3)6, which are present as [Bi6O4(OH)4]6+ polycations in acidic solution (eqn (1) and (3)). However, transformation of both clus-Bi6O4(OH)4(NO3)6 and BSN in the presence of TcO4− results in the formation of an additional unidentified phase (with peaks at approximately 25.8, 32.4, and 34° 2θ), which has also been observed in our previous studies.10 We hypothesize that mineral transformation into both this unidentified phase, and the unk-Bi(NO3)x(OH)yOz phase, is involved in the uptake of TcO4− (Fig. 3 and 5b), and further studies are needed to resolve the unknown Bi mineral phases. Peaks absent in the blanks, but present in the experiments with TcO4−, regardless of the starting Bi material (17 and 21.5° 2θ, Fig. 3 and 5a and b) also require additional evaluation.
Overall, removal of TcO4− and ReO4− from HSGW by the in situ transformation of clus-Bi12O10(OH)6(NO3)10·6H2O (BSN) and clus-Bi6O4(OH)4(NO3)6 starting materials is dependent on ligand exchange of Bi polynuclear oxo-hydroxo species, or clusters, removing OH− from solution and decreasing pH. Several research questions remain for future studies. For example, identification of the structure and formation mechanism of unk-Bi(NO3)x(OH)yOz; evaluation of long-term transformation of the unk-Bi(NO3)x(OH)yOz phase, possibly, into bismutite at higher pH and bismoclite as more thermodynamically stable products of Bi mineral transformation (eqn (2)), which may affect long term performance for TcO4− removal from groundwater in the subsurface.
Conclusions
Basic Bi subnitrate, BSN, starting material showed effective TcO4− removal from HSGW reaching 93% within 168 hours (7 days). Experiments were conducted up to almost two months and no release of removed TcO4− back to the aqueous phase was observed. BSN transformation by this time resulted in the “unknown” unk-Bi(NO3)x(OH)yOz phase and the mineral phase with the unidentified peaks, consistent with our previous studies,10 which were hypothesized to be responsible for TcO4− uptake. Formation of another mineral phase, daubreeite-related structure lay-BiO(OH,Cl), did not participate in TcO4− removal.The same starting material showed different removal kinetics for ReO4− from HSGW, the rate constants for 0.17 mM ReO4− removal vs. 0.17 mM TcO4− removal within the first 72 hours were (0.7 ± 0.2) × 10−4 min−1 and (1.2 ± 0.2) × 10−4 min−1, respectively. Along with unk-Bi(NO3)x(OH)yOz and lay-BiO(OH,Cl) phases formation of the intermediate Bi3ReO8 mineral phase was indicated by PXRD. Analogous mineral phase was not observed in the pertechnetate series of experiments, despite similarities in physical and chemical properties of these two species. Therefore, we conclude that BSN starting material is a promising efficient material for subsurface remediation, but more research is needed to investigate its long-term mineral transformation, characterization and investigation of the pH dependent formation of unk-Bi(NO3)x(OH)yOz and another unidentified phase, mechanisms of pertechnetate removal, as well as relevance to the conditions of the subsurface environment. Moreover, care should be taken when its non-radioactive analogue is being used for these and other kinds of studies.
Data availability
The data that support findings of this study are available from the corresponding author, D. Boglaienko, upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Pacific Northwest National Laboratory (PNNL) is operated by Battelle Memorial Institute for the U.S. Department of Energy (DOE) under Contract DE-AC05-76RL01830. We acknowledge the Deep Vadose Zone, Applied Field Research Initiative at PNNL. Funding for this work was provided by the DOE Richland Operations Office.Notes and references
- T. G. Levitskaia, N. P. Qafoku, M. E. Bowden, R. M. Asmussen, E. C. Buck, V. L. Freedman and C. I. Pearce, A Review of Bismuth (III)-Based Materials for Remediation of Contaminated Sites, ACS Earth Space Chem., 2022, 6, 883–908 Search PubMed.
- F. Franceschini, P. Jagdale, M. Bartoli and A. Tagliaferro, Perspectives on the use of bismuth based materials for sensing and removal of water pollutants, Curr. Opin. Environ. Sci. Health., 2022, 100345 Search PubMed.
- S. Nagendran, P. Sivasubramanian, J. H. Chang, S. Y. Shen, A. K. Nayak and M. Kumar, Fundamentals of environmental remediation techniques, in Bismuth-Based Materials for Environmental Remediation, IOP Publishing, 2022 Search PubMed.
- H. Chawla, A. Chandra, P. P. Ingole and S. Garg, Recent advancements in enhancement of photocatalytic activity using bismuth-based metal oxides Bi2MO6 (M = W, Mo, Cr) for environmental remediation and clean energy production, J. Ind. Eng. Chem., 2021, 95, 1–15 Search PubMed.
- Y. Liu, B. Yang, H. He, S. Yang, X. Duan and S. Wang, Bismuth-based complex oxides for photocatalytic applications in environmental remediation and water splitting: A review, Sci. Total Environ., 2022, 804, 150215 Search PubMed.
- J. Z. Hassan, A. Raza, U. Qumar and G. Li, Recent advances in engineering strategies of Bi-based photocatalysts for environmental remediation, Sustainable Mater. Technol., 2022, 00478 Search PubMed.
- Z. Wang, M. Chen, D. Huang, G. Zeng, P. Xu, C. Zhou, C. Lai, H. Wang, M. Cheng and W. Wang, Multiply structural optimized strategies for bismuth oxyhalide photocatalysis and their environmental application, Chem. Eng. J., 2019, 374, 1025–1045 Search PubMed.
- A. R. Lawter, T. G. Levitskaia, O. Qafoku, M. E. Bowden, F. C. Colon and N. P. Qafoku, Simultaneous immobilization of aqueous co-contaminants using a bismuth layered material, J. Environ. Radioact., 2021, 237, 106711 Search PubMed.
- C. I. Pearce, D. Boglaienko, A. R. Lawter, E. A. Cordova, K. J. Cantrell, M. E. Bowden, N. Lahiri, O. Qafoku, S. T. Mergelsberg, C. T. Resch, F. C. Colon, V. A. Garayburu-Caruso, N. D'Annunzio, M. Balasubramanian, S. A. Saslow, N. P. Qafoku, V. L. Freedman and T. G. Levitskaia, Mechanisms of interaction between bismuth-based materials and contaminants for subsurface remediation, in prep.
- N. M. Escobedo, S. A. Saslow, E. C. Cordova, A. R. Lawter, M. E. Bowden, O. Qafoku, C. T. Resch, N. Lahiri, N. D'Annunzio, D. Boglaienko, T. G. Levitskaia, C. I. Pearce, V. L. Freedman and R. D. Mackley, Part I: Structural transformation of bismuth-based materials in dynamic aqueous environments and implications for subsurface contaminant remediation, in prep.
- A. R. Lawter, N. P. Qafoku, E. C. Cordova, M. E. Bowden, O. Qafoku, F. C. Colon, N. D'Annunzio, D. Boglaienko, N. M. Escobedo, T. G. Levitskaia, C. I. Pearce and V. L. Freedman, Part II: Sediment interactions with layered bismuth materials and implications for subsurface contaminant remediation, in prep.
- D. T. Richens, The chemistry of aqua ions: synthesis, structure, and reactivity: a tour through the periodic table of the elements, Wiley, New York, 1997 Search PubMed.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2nd edn, 2005 Search PubMed.
- A. N. Christensen, M. A. Chevallier, J. Skibsted and B. B. Iversen, Synthesis and characterization of basic bismuth (III) nitrates, J. Chem. Soc., Dalton Trans., 2000, 265–270 Search PubMed.
- F. Lazarini, The crystal structure of a bismuth basic nitrate, [Bi6O5(OH)3](NO3)5·3 H2O, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3169–3173 Search PubMed.
- B. S. Brčić, D. Kolar, F. Lazarini and M. Malešič, Oxydation of bismuth with atmospheric oxygen in the presence of diluted nitric acid, Monatsh. Chem., 1973, 104, 365–375 Search PubMed.
- N. Henry, M. Evain, P. Deniard, S. Jobic, F. Abraham and O. Mentré, [Bi2O2] 2+ layers in Bi2O2 (OH)(NO3): synthesis and structure determination, Z. Naturforsch. B, 2005, 60, 322–327 Search PubMed.
- F. Lazarini, Bismuth Basic Nitrate [Bi6(H2O)(NO3)O4(OH)4](NO3)5, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 448–450 Search PubMed.
- N. Henry, O. Mentré, F. Abraham, E. J. MacLean and P. Roussel, Polycationic disorder in [Bi6O4(OH)4](NO3) 6: structure determination using synchrotron radiation and microcrystal X-ray diffraction, J. Solid State Chem., 2006, 179, 3087–3094 Search PubMed.
- L. Miersch, T. Ruffer, M. Schlesinger, H. Lang and M. Mehring, Hydrolysis studies on bismuth nitrate: synthesis and crystallization of four novel polynuclear basic bismuth nitrates, Inorg. Chem., 2012, 51, 9376–9384 Search PubMed.
- A. N. Christensen and B. Lebech, Investigation of the crystal structure of a basic bismuth (iii) nitrate with the composition [Bi6O4 (OH) 4] 0.54 (1)[Bi6O5 (OH) 3] 0.46 (1)(NO3) 5.54 (1), Dalton Trans., 2012, 41, 1971–1980 Search PubMed.
- N. Henry, M. Evain, P. Deniard, S. Jobic, O. Mentré and F. Abraham, [Bi6O4. 5 (OH) 3.5] 2 (NO3) 11: a new anhydrous bismuth basic nitrate. Synthesis and structure determination from twinned crystals, J. Solid State Chem., 2003, 176, 127–136 Search PubMed.
- B. Liu, W. W. Zhou, Z. Q. Zhou and X. Y. Zhang, Hydrolysis to the first dumbbell-like high-nuclearity bismuth-oxo cluster [Bi12 (μ3-OH) 4 (μ2-OH) 2 (μ3-O) 8 (μ4-O) 2 (NO3) 6] 4+: Synthesis, structure and spectroscopic characterizations, Inorg. Chem. Commun., 2007, 10, 1145–1148 Search PubMed.
- D. M. Driscoll, R. C. Shiery, N. D'Annunzio, D. Boglaienko, M. Balasubramanian, T. G. Levitskaia, C. I. Pearce, N. Govind, D. C. Cantu and J. L. Fulton, Water Defect Stabilizes the Bi3+ Lone-Pair Electronic State Leading to an Unusual Aqueous Hydration Structure, Inorg. Chem., 2022, 61, 14987–14996 Search PubMed.
- S. Sun and X. Yin, Progress and perspectives on aurivillius-type layered ferroelectric oxides in binary Bi4Ti3O12-BiFeO3 system for multifunctional applications, Crystals, 2020, 11, 23 Search PubMed.
- M. E. Clissold, Aspects of the supergene geochemistry of copper, nickel and bismuth, 2007 Search PubMed.
- E. A. Katayev, G. V. Kolesnikov and J. L. Sessler, Molecular recognition of pertechnetate and perrhenate, Chem. Soc. Rev., 2009, 38, 1572–1586 Search PubMed.
- C. Eiroa-Lledo, L. Lecrivain, T. G. Parker, D. E. Wall and N. A. Wall, Comparison of ReO4− and TcO4− in solvent extraction systems, Radiochim. Acta, 2020, 108, 443–449 Search PubMed.
- E. E. Rodriguez, F. Poineau, A. Llobet, K. Czerwinski, R. Seshadri and A. K. Cheetham, Preparation and crystal structures of bismuth technetates: a new metal oxide system, Inorg. Chem., 2008, 47, 6281–6288 Search PubMed.
- B. A. Lenell and Y. Arai, Perrhenate sorption kinetics in zerovalent iron in high pH and nitrate media, J. Hazard. Mater., 2017, 321, 335–343 Search PubMed.
- B. Wakoff and K. L. Nagy, Perrhenate uptake by iron and aluminum oxyhydroxides: An analogue for pertechnetate incorporation in Hanford waste tank sludges, Environ. Sci. Technol., 2004, 38, 1765–1771 Search PubMed.
- E. M. Pierce, W. W. Lukens, J. P. Fitts, C. M. Jantzen and G. Tang, Experimental determination of the speciation, partitioning, and release of perrhenate as a chemical surrogate for pertechnetate from a sodalite-bearing multiphase ceramic waste form, Appl. Geochem., 2014, 42, 47–59 Search PubMed.
- B. H. Hamilton, T. A. Wagler, M. P. Espe and C. J. Ziegler, Sequestering perrhenate with a borate-based coordination polymer: A model for pertechnetate separation, Inorg. Chem., 2005, 44, 4891–4893 Search PubMed.
- Q. H. Hu, Y. G. Wang, X. Gao, Y. Z. Shi, S. Lin, R. P. Liang and J. D. Qiu, Halogen microregulation in metal-organic frameworks for enhanced adsorption performance of ReO4-/TcO4, J. Hazard. Mater., 2023, 130744 Search PubMed.
- K. Schwochau, Technetium, Wiley-VCH, 2000 Search PubMed.
- M. J. Truex, J. E. Szecsody, N. Qafoku, C. E. Strickland, J. J. Moran, B. D. Lee, M. Snyder, A. R. Lawter, C. T. Resch and B. N. Gartman, Contaminant Attenuation and Transport Characterization of 200-DV-1 Operable Unit Sediment Samples, Pacific Northwest National Laboratory, Richland, WA (United States), 2017 Search PubMed.
- A. M. Patel, J. K. Nørskov, K. A. Persson and J. H. Montoya, Efficient Pourbaix diagrams of many-element compounds, Phys. Chem. Chem. Phys., 2019, 21, 25323–25327 Search PubMed.
- S. M. Heald, K. M. Krupka and C. F. Brown, Incorporation of pertechnetate and perrhenate into corroded steel surfaces studied by X-ray absorption fine structure spectroscopy, Radiochim. Acta, 2012, 100, 243–253 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns for BSN starting material, BSN after 0.17 mM Tc and Re uptake, pristine BiOCl, and clus-Bi6O4(OH)4(NO3)6 synthesized material; removal kinetics and pH profile for 0.17 mM Tc uptake with BiOCl; and a Pourbaix diagram. See DOI: https://doi.org/10.1039/d4ew00496e |
| This journal is © The Royal Society of Chemistry 2024 |