 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen-resistant [FeFe]hydrogenases: new biocatalysis tools for clean energy and cascade reactions
Francesca
Valetti
 *a,
Simone
Morra
b,
Lisa
Barbieri
ac,
Sabrina
Dezzani
ac,
Alessandro
Ratto†
a,
Gianluca
Catucci
a,
Sheila J.
Sadeghi
a and
Gianfranco
Gilardi
a
*a,
Simone
Morra
b,
Lisa
Barbieri
ac,
Sabrina
Dezzani
ac,
Alessandro
Ratto†
a,
Gianluca
Catucci
a,
Sheila J.
Sadeghi
a and
Gianfranco
Gilardi
a
aDepartment of Life Sciences and Systems Biology, University of Torino, Torino, Italy. E-mail: francesca.valetti@unito.it
bFaculty of Engineering, University of Nottingham, Nottingham, UK
cUniversity School for Advanced Studies IUSS Pavia, Italy
First published on 21st February 2024
Abstract
The use of enzymes to generate hydrogen, instead of using rare metal catalysts, is an exciting area of study in modern biochemistry and biotechnology, as well as biocatalysis driven by sustainable hydrogen. Thus far, the oxygen sensitivity of the fastest hydrogen-producing/exploiting enzymes, [FeFe]hydrogenases, has hindered their practical application, thereby restricting innovations mainly to their [NiFe]-based, albeit slower, counterparts. Recent exploration of the biodiversity of clostridial hydrogen-producing enzymes has yielded the isolation of representatives from a relatively understudied group. These enzymes possess an inherent defense mechanism against oxygen-induced damage. This discovery unveils fresh opportunities for applications such as electrode interfacing, biofuel cells, immobilization, and entrapment for enhanced stability in practical uses. Furthermore, it suggests potential combinations with cascade reactions for CO2 conversion or cofactor regeneration, like NADPH, facilitating product separation in biotechnological processes. This work provides an overview of this new class of biocatalysts, incorporating unpublished protein engineering strategies to further investigate the dynamic mechanism of oxygen protection and to address crucial details remaining elusive such as still unidentified switching hot-spots and their effects. Variants with improved kcat as well as chimeric versions with promising features to attain gain-of-function variants and applications in various biotechnological processes are also presented.
Introduction
One of the greatest challenges to current society is the design of cheap catalytic systems for efficient production and oxidation of carbon neutral energy carriers, such as molecular hydrogen. In this context a very promising perspective is the application of hydrogenases,1–5 the key enzymes involved in the metabolism of H2, able to catalyse the reversible reaction 2H+ + 2e− ↔ H2 at high turnover rates. Both [NiFe] and [FeFe]hydrogenases are highly appealing for exploitation: here the focus is on [FeFe] given their very high turnover frequency (kcat), reaching 104 catalytic cycles per second.The main factor that hinders the large-scale biotechnological application of [FeFe]hydrogenases is high sensitivity to oxygen, which is an irreversible inhibitor that destroys the active site of the enzyme. Recently, a new and game-changer [FeFe]hydrogenase, named CbA5H, has been characterised from Clostridium beijerinckii SM10, isolated from a pilot plant.6Clostridium beijerinckii is a Gram positive anaerobic bacterium, isolated from various sources and previously known for its capacity to produce H2 and other industrially-relevant biomolecules with high yields.7–9 The analysis of its genome highlighted the presence of six genes encoding for [FeFe]hydrogenases, one of which codes for CbA5H (CbHydA1, NCBI accession number: KX147468, Cbei_1773).6 CbA5H is a cytoplasmic soluble enzyme composed of 644 amino acids, with a molecular weight of 72 kDa and a modular structure typical of the M2c category.10 It belongs to the A5 subgroup,11 hence the name CbA5H. From the N-terminal to the C-terminal, this enzyme harbours a soluble-ligand-binding β-grasp motif (SLBB), a domain hosting two [4Fe-4S] clusters or bFd (homologous to bacterial ferredoxins), and the H-domain typical of all [FeFe]hydrogenases.12–15
This enzyme is the first of its kind due to its resilience towards O2: it can withstand the oxidative action of oxygen by switching to an inactive (Hinact) protected form in an aerobic environment.12 Conversely, in the absence of oxygen, it is able to return to an oxidised and fully active state (Hox). Even though it was not the first [FeFe]hydrogenase observed in which this particular Hinact state is formed and identified by a specific FTIR signature (it was reported16,17 for example in the enzyme DdH18 after aerobic purification from the producing Desulfovibrio species), it was the first one in which the transition from the Hox to the Hinact state was fully reversible and could be repeated several times without any activity loss or any damage observed to the H-cluster.12 Indeed, in DdH, once the Hinact form goes back to the Hox state, the former cannot be obtained again, and therefore the enzyme becomes O2-sensitive after the first exposure to air.16 Moreover, the Hinact state can be induced in some [FeFe]hydrogenases (DdH included) by adding sulfide under oxidizing conditions.19 Therefore, it was initially concluded that sulfide binds to the open coordination of the distal iron atom (Fed) of the H-cluster, protecting it from the attack of O2. However, in CbA5H the Hox → Hinact transition can be driven by mild organic oxidants, such as thionine or DCIP, but also by simple air exposure. CbA5H proved to be O2-resistant even without the need to add external sulfide,12–14 demonstrating an intrinsic property more likely influenced by the protein environment.
The capacity to overcome O2 attack is in fact possible thanks to the special role of cysteine 367, which coordinates with Fed avoiding the binding of O2 (acting as a “safety cap”).13 Cys367 is normally responsible for transferring protons to Fed, via the amine group of the azadithiolate ligand that connects the distal and proximal iron atoms. This residue is highly conserved among all [FeFe]hydrogenases,20 but it is normally located too far away from Fed to interact with it. The X-ray structure of CbA5H in the protected form, resulting from air exposure,13 shows some differences in comparison to anaerobically crystallised CpI: indeed CbA5H’s TSC-loop (T365-S366-C367) has a different conformation from the corresponding TSC-loop of CpI. This loop appears to be more flexible than in standard [FeFe]hydrogenases, enough to allow the sulfur atom of C367 to interact with Fed, lowering their distance from 5.9 Å (which is the reported value in CpI) to 3.1 Å. However, both the cysteine residue and the TSC-loop are strictly conserved among [FeFe]hydrogenases. Therefore, there needs to be another factor that contributes to the flexibility of the loop. In CbA5H, three residues have been identified, that probably play a role in making the TSC-loop more flexible, which are Leu364, Ala561 and Pro386.13 These residues do not significantly change the backbone structure from the one reported of CpI but have smaller and more flexible lateral chains which enable the mobility of the TSC-loop. A further increase in enzyme flexibility was also recently engineered.21
It is not clear though if the sequence features, which allow TSC-loop flexibility and are strictly required for this purpose (as demonstrated in loss-of-functions mutants),13 are also sufficient to grant the resiliency to oxygen of CbA5H. The details of the peculiar and immediate switch to Hinact and back to Hox allowed by this flexibility are still elusive,22 as well as the role of the modular structure and the unique SLBB domain23,24 present at the N-terminus of the protein. The crystal structure suggests the presence in the SLBB domain of a [4Fe4S] cluster bound to three cysteines and one histidine, but other ligands and/or redox partners might be interacting with this part of the protein.
In this work, a protein engineering approach has been used to investigate further the domains or residues involved in oxygen resistance of this peculiar [FeFe]hydrogenase, in the perspective of exploiting this feature for biotechnological applications. Mutated variants and chimeras obtained by shuffling domains with other hydrogenases are presented which add further information on the mechanism and lay the groundwork for robust hydrogen-driven catalysis. This can be achieved in combination with non-physiological partners or in chimeric multidomain proteins, as inspired by the modular “Lego approach” for protein engineering.25–30
Results and discussion
Site specific mutagenesis
In order to assess the fine-tuning role of the protein scaffold on the oxygen resistance and check if it is possible to implement the biotechnological exploitation of this robust enzyme, or even to confer oxygen resistance to other enzymes of the class, multiple amino acids substitutions were tested.The selected residues were identified within the inner (4 Å) and outer (8 Å) sphere of interaction of the diiron subcluster of the H-cluster with the protein moiety. Fig. 1 reports the corresponding regions (Fig. 1A: 4 Å, Fig. 1B: 8 Å) mapped on the reference structure of the Hox state of CpI.31 The highlighted residues were also mapped onto the sequence alignment (Fig. 1C) of the main studied enzymes of the [FeFe]hydrogenase class,32–36 with the same colour code (4 Å sphere positions are highlighted in green, 8 Å sphere in red). Many conserved residues are present, especially in the 4 Å sphere, but few fine differences can be spotted. Residue M393 of CbA5H is typically a glutamine in other well-characterized and oxygen sensitive hydrogenases and it is located in a region adjacent to the bridging CO portion of the H-cluster (check Fig. 2B for closer view). In the 8 Å coordination sphere, position 419 of CbA5H is also harbouring a chemically different amino acid if compared to most of the other enzymes of the group, since an alanine is present, whereas serine or threonine are the consensus conserved residues in oxygen sensitive enzymes (Fig. 1B). This implies the lack of possible H-bonding in CbA5H, while both serine and threonine in other more “standard” [FeFe]hydrogenases can grant this stabilising interaction. The longer distance to the H-cluster (Fig. 2B) is not hindering a possible effect if a long-range network of H-bonding interactions is involved.
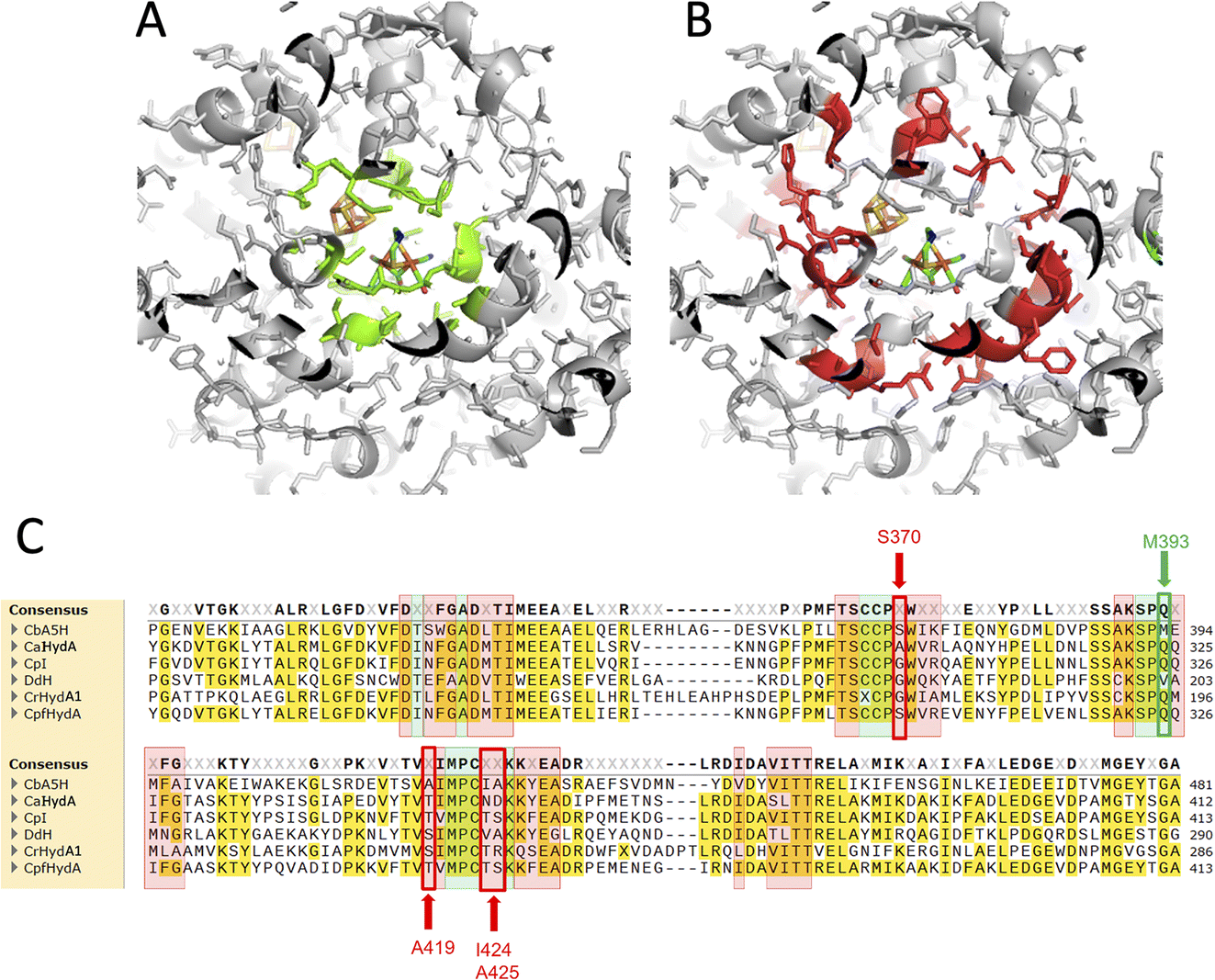 | ||
| Fig. 1 Protein regions interacting with H-cluster mapped on 3D structure and aligned sequences. (A) 4 Å sphere mapped on the crystal structure of CpI as a reference for Hox structure, highlighted in green. (B) 8 Å sphere mapped on the crystal structure of CpI. (C) 4 Å and 8 Å sphere residues mapped on the multiple alignment of CbA5H from C. beijerinckii, CaHydA from C. acetobutylicum, CpI from C. pasteurianum, DdH from D. vulgaris Hildenborough, CrHydA1 from C. reinhardtii, CpfHydA from C. perfringens. The same colour-code of A and B was used (green highlight: 4 Å sphere, red highlight: 8 Å). The alignment was performed using cobalt. | ||
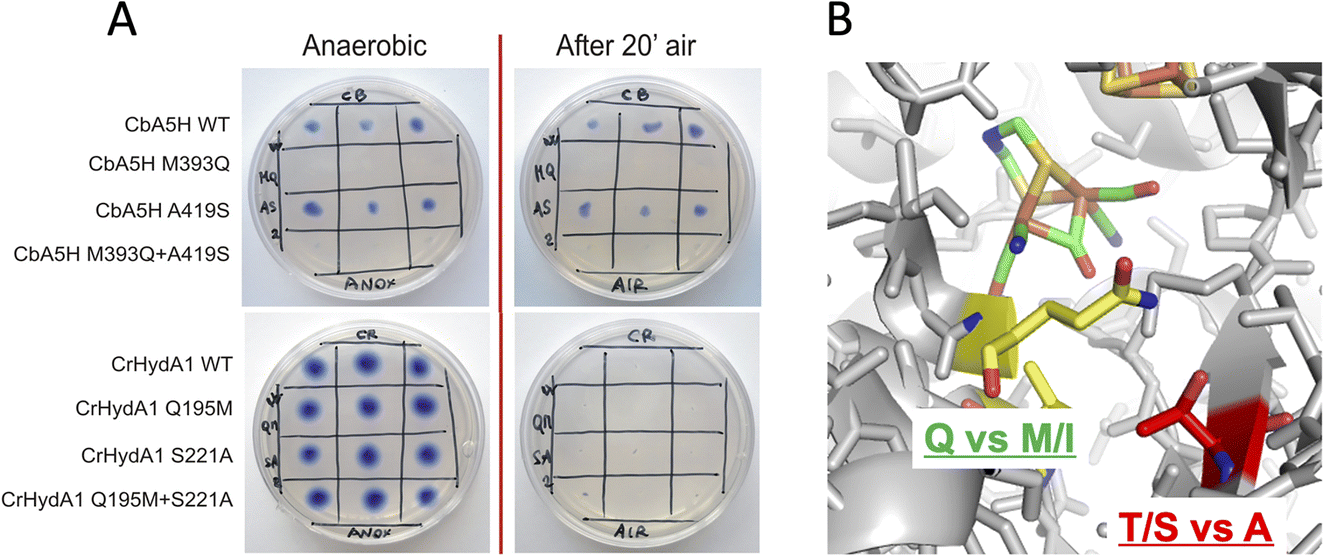 | ||
| Fig. 2 Mutations in the 2Fe subcluster vicinity, H-domain. (A) Results of the on-plate activity screening before and after air exposure on mutants M393Q, A419S of CbA5H and Q195M, S221A of CrHydA1. (B) Detail of the corresponding sites on the crystal structure of CpI as representative of the Hox state. | ||
For these reasons, both positions 393 and 419 were selected as mutagenesis targets to investigate a possible fine-tuning role in the peculiar behaviour of CbA5H. Results of the mutations were screened by a colorimetric on-plate method which can directly measure, with a semi-quantitative approach, the activity of the recombinant enzyme variants co-expressed in E. coli cells with the suitable maturases, as described in the Experimental section. The test can be performed before and after air exposure of the plate, granting information also on the capacity of the expressed enzymes to access the protected state typical of CbA5H. Fig. 2A shows the results on the CbA5H mutants M393Q and A419S. The typical behaviour of WT CbA5H (blue colour and halo indicative of activity both before and after air exposure) is achieved also by variant A419S. In contrast M393 is essential for activity in CbA5H, and the activity is abolished even before air exposure. Symmetrical mutations were performed on a typical oxygen sensitive [FeFe]hydrogenase, the HydA1 from Chlamydomonas reinhardtii (CrHydA1). Here the corresponding positions (195 and 221) were mutated to check if any stabilising effect to air degradation could be observed. Both variants Q195M and S221A, as well as a third variant with the combined presence of the two mutations behaved like the sensitive WT CrHydA1, since the blue colour and halo is present if the environment is always maintained anaerobic, but it disappears and cannot be restored if air exposure occurs, due to the typical irreversible damage of the sensitive and unprotected enzymes. Interestingly though, while position 393 in CbA5H is extremely sensitive to the replacement of M with Q, as shown by the loss of activity of the single M393Q and the combined M393Q/A419S mutants, the corresponding site in CrHydA1 is not affected, and Q195 can be replaced with methionine, even in the double mutant. This could not be linked specifically to oxygen resistance but, in view of a protein-engineering strategy turning sensitive enzymes to a CbA5H-like structure, it could be advantageous. Also, it can be worth further investigation to elucidate the reason of such effect in CbA5H.
A second set of mutations was performed on positions 424 and 425 of CbA5H, where the alignment suggests a strong difference in terms of polarity when comparing CbA5H to oxygen sensitive enzymes, except for DdH, in which a similarity can be highlighted. In fact, CbA5H harbours hydrophobic isoleucine and alanine (IA) while normally more polar residues are present (TR, TS, ND), with DdH containing instead similarly hydrophobic residues VA (Fig. 1C). These two amino acids are located in the vicinity of the [4Fe4S] cubane subcluster of the H-cluster (Fig. 3B) and changes in the protein environment have been demonstrated to affect potential and therefore properties of FeS clusters.37–40 The results of mutagenesis of positions 424–425 are reported in Fig. 3A.
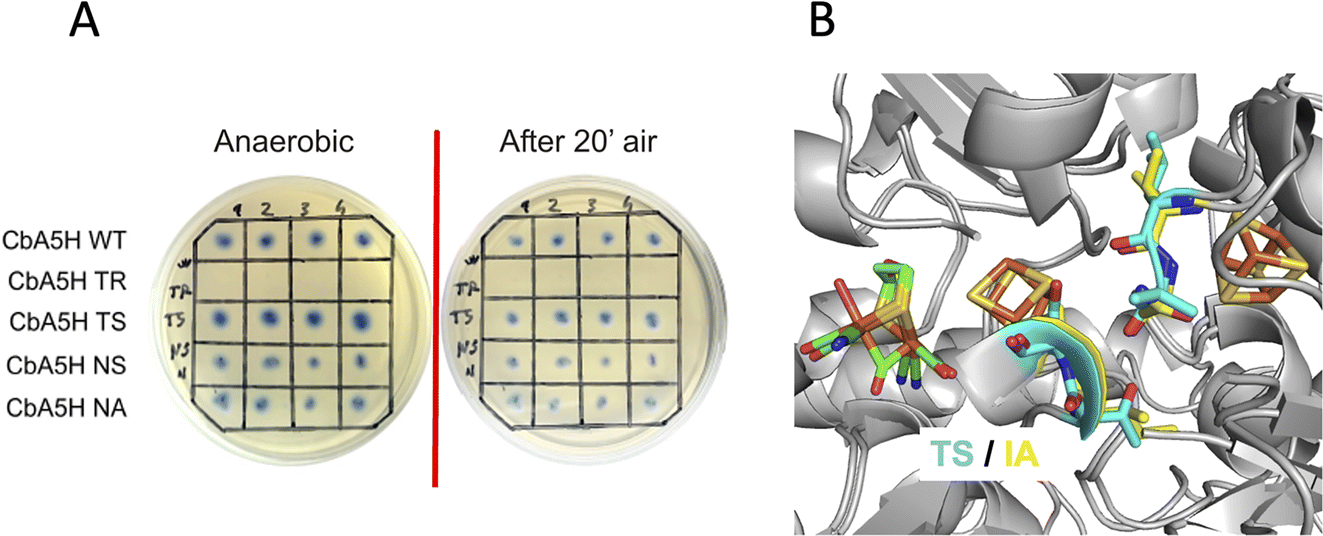 | ||
| Fig. 3 Mutations in the cubane subcluster vicinity, H-domain. (A) Results of the on-plate activity screening before and after air exposure on mutants of positions 424 and 425 of CbA5H and Q195M, S221A of CrHydA1. (B) Detail of the corresponding sites on the crystal structure of CpI (TS) and on structure of CbA5H obtained by homology modelling on the Hox structure (IA). Colour code as indicated. | ||
A disruptive effect is observed inserting TR, leading to an inactive CbA5H, but no apparent links to oxygen resistance are detected in the other mutated variants, which show a resistance similar to wild type in the on-plate screening. The variants were also expressed and purified and tested for hydrogen evolution activity (Table 1).
| Protein | H2 production activity kcat (s−1); mean ± SD |
|---|---|
| CbA5H NS | 2404 ± 424 |
| CbA5H TS | 5887 ± 691 |
| CbA5H NA | 2221 ± 361 |
| CbA5H A419S | 6757 ± 693 |
| CbA5H C236A | 9214 ± 400 |
| CbA5H WT batch 1 | 5449 ± 473 |
| CbA5H WT batch 2 | 4199 ± 544 |
The kcat is expressed as TOF (turnover frequency) already normalised for the protein concentration. Results are reported in Table 1, which also includes the activity of the purified variant A419S already discussed as for on-plate screening results, and of mutant C236A. This last was engineered to test the role of atypical cysteines present in CbA5H in the vicinity of the consensus sequences (Fig. 4C) required to coordinate cubane 1 and cubane 2 (also defined FSA and FSB) of the bFd domain of CbA5H. In particular C236 was observed, by modelling the structure of bFd of CbA5H (not visible in the crystal structure),13 to be possibly oriented towards cubane 1 (FSA) of this domain (Fig. 4B), suggesting a possible alternative site to the typical coordinating cysteines of the cluster or a cluster with unusual structure, which could be important for the peculiar features of CbA5H.41 The tested replacement with an alanine yielded the results reported in Fig. 4A: on-plate screening shows that the single replacement C236A is not affecting oxygen resistance of CbA5H and the variant has apparently a wild type-like behaviour, although a further investigation on the purified variant C236A suggests a subtle tuning effect.
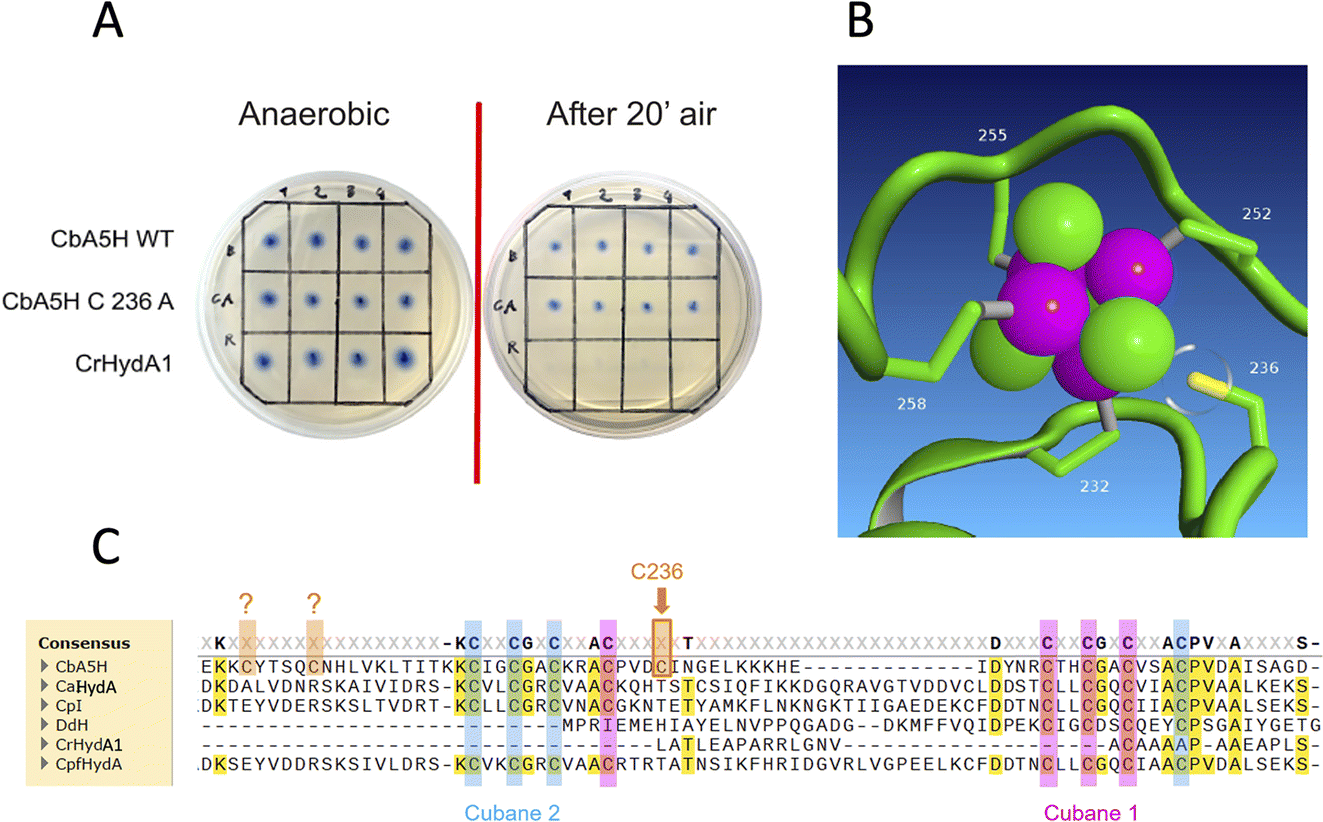 | ||
| Fig. 4 Mutation in the bFd domain. (A) Results of the on-plate activity screening before and after air exposure on mutant of position 236 of CbA5H. (B) Detail of the corresponding position of Cys236 and cubane 1 (FSA) on CbA5H structure obtained by homology modelling on the Hox CpI structure (CbA5H bFd domain could not be resolved in the crystal). (C) Alignment of CbA5H from C. beijerinckii, CaHydA from C. acetobutylicum, CpI from C. pasteurianum, DdH from D. vulgaris Hildenborough, CrHydA1 from C. reinhardtii, CpfHydA from C. perfringens with consensus sequences for cubane 1 (FSA) highlighted in pink and cubane 2 (FSB) highlighted in pale blue. The three extra cysteines of CbA5H (Cys236 is indicated) are highlighted in orange. DdH and CrHydA1 were inserted in the alignment even if their cluster arrangement is expected to be different. The alignment was performed using cobalt. | ||
In fact, by analysing the activity data obtained for the selected mutants, it is interesting to observe that the catalytic constant kcat of the wild type protein (values for two independent batches are reported, highlighting a relatively high variability) has values comparable only to that of the CbA5H TS mutant; the CbA5H NS and CbA5H NA mutants have a lower turnover than the wild type, while the point mutations CbA5H A419S and CbA5H C236A have a much higher turnover frequency. The data obtained therefore suggest that the mutations have generated subtle but significant changes in the protein environment. Although the mutations did not affect the mechanism of oxygen resistance, they could still have an important role in regulating the catalytic activity of the enzyme.
A further interesting single position, located in the proximity of the cubane of the H-cluster (identified within the 8 Å sphere represented in Fig. 1B and C) is serine 370 of CbA5H. In oxygen sensitive [FeFe]hydrogenases the corresponding residue is usually a glycine or an alanine, except for CpfHydA from C. perfringens, a strain normally present and isolated from the same environments in which C. beijerinckii is found,6 which could therefore represent an intermediate variant. Serine 370 is located in CbA5H in a position crucial for possible hydrogen-bonding in the vicinity of the cubane and the comparison of the crystal structure of CbA5H in the air exposed form to CpI in the Hox structure (Fig. 5A) shows that there is a shift in the backbone highlighting a possible role as switch between the two states. Also, recently a symmetrical mutation on CpI, where the corresponding position is numbered 302 (Fig. 5B), highlighted that the replacement of the canonical glycine with a serine can enhance the stability to oxygen exposure of CpI, possibly by a combined effect of the local blocking of O2 diffusion and the two newly formed H-bonds which trigger conformational changes onto S357 and might influence the flexibility of the protein, modifying the O2 diffusion channels.42
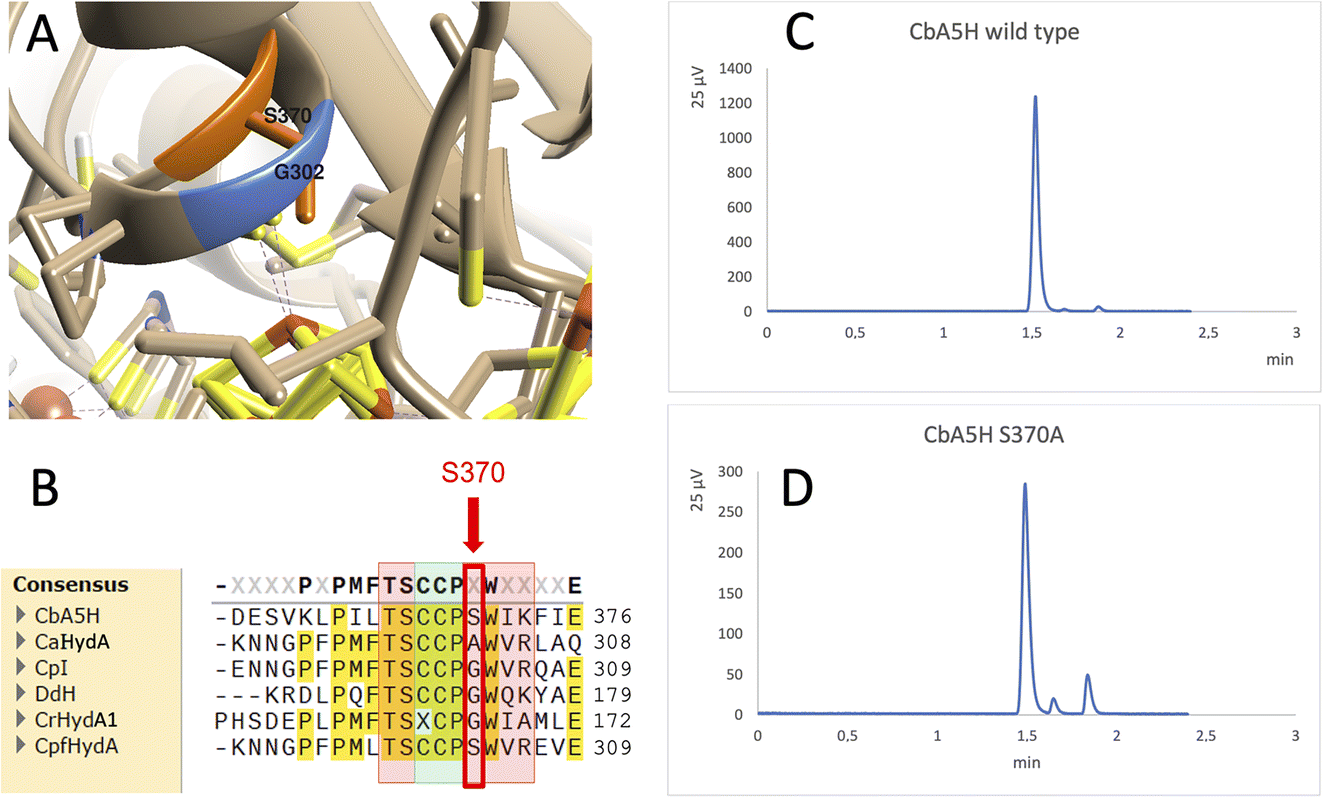 | ||
| Fig. 5 Mutation of Serine 370. (A) Comparison of the crystal structure of CbA5H with Serine 370 in the air exposed form to the corresponding glycine 302 of CpI in the Hox structure. (B) Corresponding alignment of the same region in CbA5H from C. beijerinckii, CaHydA from C. acetobutylicum, CpI from C. pasteurianum, DdH from D. vulgaris Hildenborough, CrHydA1 from C. reinhardtii, CpfHydA from C. perfringens. The colour-code is the same as indicated in Fig. 1. The alignment was performed using cobalt. (C) Gas chromatography signals of sampled headspace after 30′ of incubation at 37 °C of lysates expressing positive control WT CbA5H, with H2 peak at 1.5 minutes. (D) Gas chromatography signals of sampled headspace after 30′ of incubation at 37 °C of lysates (obtained as for WT) expressing mutant S370A of CbA5H, with H2 peak at 1.5 minutes. | ||
Preliminary tests were performed on a CbA5H S370A mutant, engineered and expressed on small scale. If compared to the wild type protein obtained with the same experimental set-up, the activity measured as H2 evolution by GC assay (Fig. 5C and D) is lowered to approximately 22% of the wild type, but further characterisation is needed to confirm an effect on the Hinact formation kinetics.
Domain shuffling
As a second strategy, different modular arrangement of the domains involved in electron transfer (ET) and possible key regulators in catalysis43,44 were engineered in truncated forms of CbA5H and in chimeras designed by combination with other oxygen sensitive [FeFe]hydrogenases such as CrHydA1 and CaHydA, producing variants in which the catalytic H-domain and the ET domains were shuffled between oxygen resistant and oxygen sensitive enzymes (Fig. 6).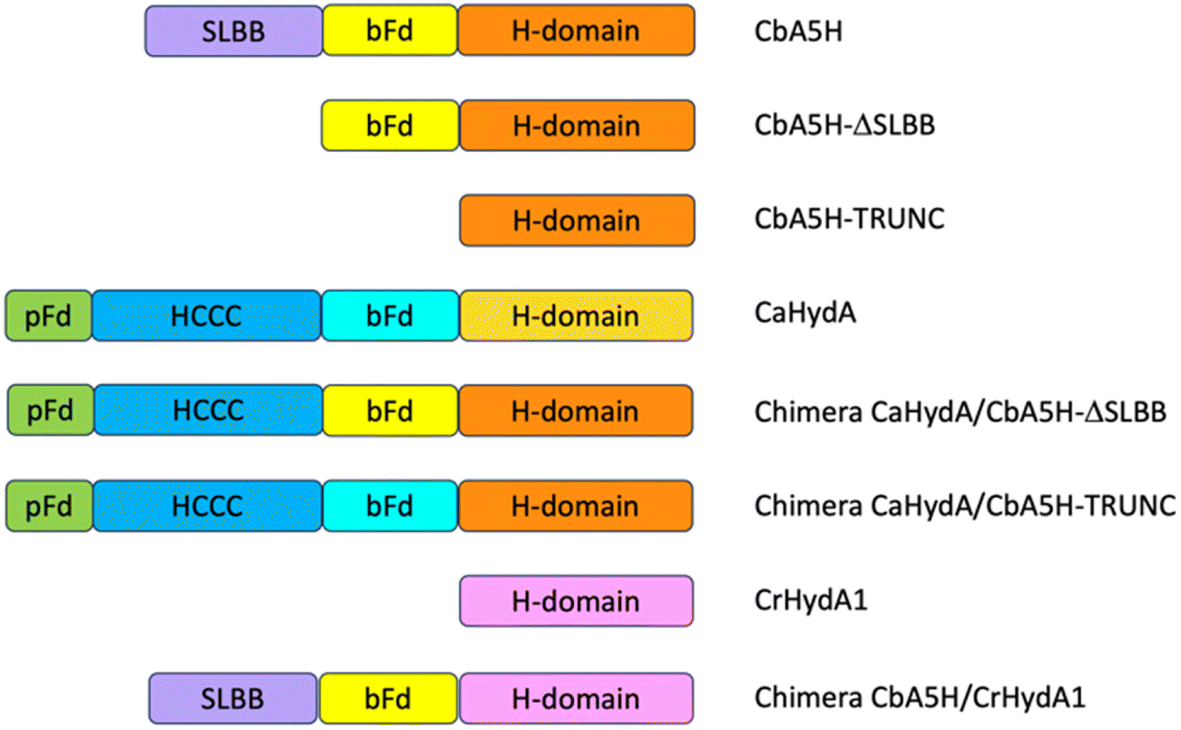 | ||
| Fig. 6 Strategy of domain swapping. In the figure the domains contained in CbA5H, CaHydA, CbA5H-Trunc and CbA5H-ΔSLBB proteins are schematically represented. In order to produce the CaHydA-CbA5H chimeras the accessory domains of CaHydA were transferred in the CbA5Hs truncated forms, following the diagram shown in the figure. Since the ΔSLBB mutant has an internal bFd domain, only the pFd and HCCC domains were transferred. The CbA5H-CrHydA Chimera was produced by linking the accessory domains of CbA5H to CrHydA which only contains the H-domain. bFd = bacterial ferredoxin-like 4F4S, HCCC = Cys3His [4Fe4S], pFd = plant ferredoxin-like [2F2S], SLBB = soluble ligand-binding β-grasp. | ||
Results of the effect of deleting domains is reported in Fig. 7A. Truncated forms of CbA5H (either removing SLBB alone or both SLBB and bFd) show no activity in the on-plate assay although both forms can be detected as expressed (Fig. 7B). Previously, truncated active versions were produced for [FeFe]hydrogenase MeHydA from Megasphaera elsdenii43 and for CaHydA from C. acetobutylicum.44 Nonetheless, in the latter case a dramatic reduction in catalytic activity was reported, therefore it is not surprising that the truncated forms of CbA5H do not show activity in the semi-quantitative on-plate assay.
 | ||
| Fig. 7 Truncated variants of CbA5H. (A) Results of the on-plate activity screening before and after air exposure on truncated form of CbA5H lacking SLBB only (CbA5H-ΔSLBB) or both SLBB and bFd domains (CbA5H-trunc). (B) Western blot analysis of lysates expressing the two truncated forms, showing the bands at 55 kDa (CbA5H-ΔSLBB) and at 40 kDa (CbA5H-trunc) in the induced cell lysates only. | ||
This lack of activity of the truncated forms hindered further investigation on the fine role of the atypical SLBB domain on the CbA5H oxygen resistance features. The other chimeras proposed in Fig. 6 were alternative strategies put in place to discriminate whether the SLBB domain, alone or in combination with the CbA5H bFd domain, is necessary for CbA5H to achieve the Hinact protected state and to explicate the recovery of activity upon air exposure, or if, conversely, SLBB can by itself grant protection to damage in oxygen sensitive [FeFe]hydrogenases.
The transfer of Clostridium acetobutylicum CaHydA accessory domains into the plasmids harboring the Trunc and ΔSLBB truncated sequences of CbA5H was successfully achieved, as confirmed in sequencing data, attesting the correct acquisition of the inserts in both samples. Fig. 8 reports preliminary results obtained for Chimeras CaHydA/CbA5H-ΔSLBB and CaHydA/CbA5H-TRUNC using cell lysates prepared in aerobic conditions and then tested, after reductive reactivation, in the GC H2 evolution assay. Compared to the negative control (Fig. 8A), in which non-induced cells were used, assays reported H2 production activity for the CaHydA/CbA5H-ΔSLBB expressing lysates (Fig. 8C), similarly to the positive control cells expressing CbA5H (Fig. 8B), although at lower levels, while in lysates which should express CaHydA/CbA5H-TRUNC Chimera no H2 production could be detected (Fig. 8D).
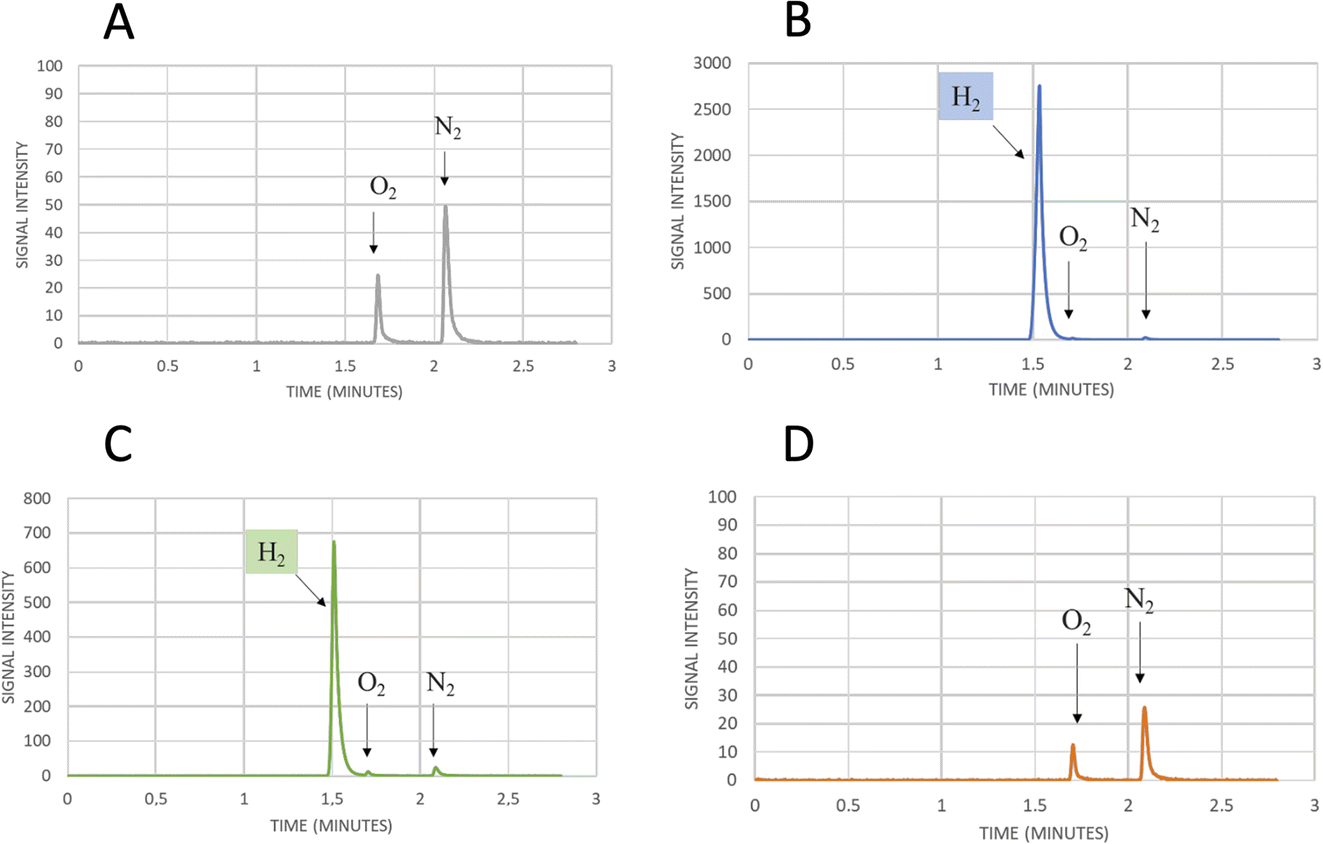 | ||
| Fig. 8 Gas chromatography signals of sampled headspace after 24 h of incubation at 37 °C of air exposed lysates from small-scale expression of (A) not induced cells as negative control, showing only the oxygen and nitrogen signals due to small air contamination in the inlet syringe, (B) cells expressing WT CbA5H as positive control, with the hydrogen production signal at 1.5 min, (C) cells expressing Chimera CaHydA/CbA5H-ΔSLBB, with the hydrogen production signal at 1.5 min, (D) cells expressing Chimera CaHydA/CbA5H-TRUNC, with no signal at 1.5 min and only oxygen and nitrogen signals as in the negative control. | ||
The results obtained allowed to highlight that the oxygen resistance is not severely affected by the absence of the SLBB domain, which could nonetheless have a role in allosteric regulation, since a significantly reduced activity was observed in the CaHydA/CbA5H-ΔSLBB chimera when compared to that of the wild type CbA5H. A dramatic impairment is instead caused by replacing the whole N-terminal part (including the bFd domain) of CbA5H with the corresponding N-terminal of CaHydA, although bFd is in principle a domain with higher similarity among the two [FeFe]hydrogenases than SLBB to the two domains (pFd and HCCC) transferred to replace the SLBB in the active CaHydA/CbA5H-ΔSLBB chimera. It is to be noted that the bFd domain of CbA5H, as discussed in the site specific mutagenesis part of this section, contains some peculiarity as for non-canonical sequences, i.e. the extra cysteines (like C236) in the cubane 1 and 2 (FSA and FSB) consensus regions. If not taken alone (given the high activity of the C236A mutant) these single changes could cumulatively make a difference in the whole structure and catalytic activity of CbA5H in combination with other unique features like the presence of SLBB.
As expected from previous data, granting to the H-domain of CbA5H a main role in driving the O2 resistance properties,13 it was confirmed (Fig. 9A) that CbA5H accessory domains (SLBB + bFd) are not sufficient to determine oxygen stability in the oxygen-sensitive CrHydA1. It is anyway very interesting to observe that the wild type (WT) CbA5H/CrHydA1 chimera is fully active in the on-plate assay if kept in an anaerobic environment. The CbA5H/CrHydA1 chimera was also used as a platform for mutagenesis to evaluate the combined effects of the two engineering strategies (site specific mutations and domain shuffling). Mutations were introduced on the previously identified positions (Fig. 1B): M393 and A419 of CbA5H, in the proximity of the 2Fe subcluster, replacing the corresponding Q195 and S221 of CrHydA1 (Chimera MA) and I424 and A425 of CbA5H, in the proximity of the cubane subcluster, replacing T226 and R227 of CrHydA1. The two sets of mutations were also combined to generate the chimera MAIA (Fig. 9A and B).
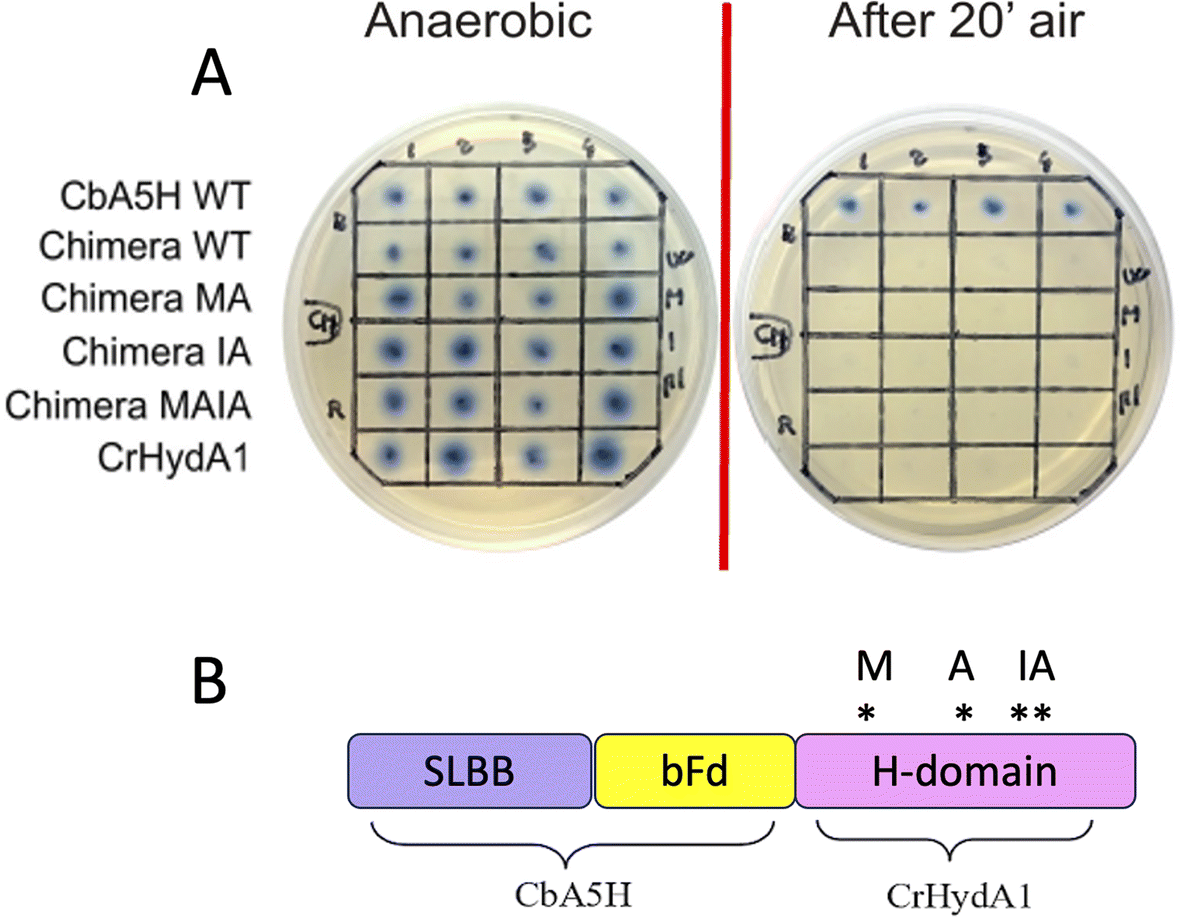 | ||
| Fig. 9 Chimeras CbA5H/CrHydA1 WT and mutants. (A) Results of the on-plate activity screening before and after air exposure on WT CbA5H/CrHydA1 Chimera and on mutated forms. (B) Domain arrangements of the chimera and graphical scheme of position of the mutations introduced. | ||
All mutated chimeras based on the CbA5H/CrHydA1 architecture were fully active in the on-plate screening in anaerobic conditions, but none gained any detectable resistance to air exposure in the on-plate assay. Minor improvement might be evaluated with a deeper analysis, but we expect further mutations to be necessary to attain a gain-of-function variant. The chimera CbA5H/CrHydA1 and its MAIA variant represent good platforms to introduce further changes, since they provide a “CbA5H-like” environment as for accessory ET domains and key positions, maintaining a good level of expression/activity and showing a high plasticity/tolerance to introduced mutations. In fact, it is to be noted that the mutation of R227 into A in the chimera did not affect the activity, while the symmetrical replacement in CbA5H (A425R in the TR variant, Fig. 3A) was the only one observed to completely abolish catalysis even in anaerobiosis. Also, CrHydA1 is an extremely well-studied model among [FeFe]hydrogenases,45–52 thus granting a “gold standard” comparison for data obtained on CbA5H/CrHydA1 chimeras.
Experimental
Materials
Culture media for bacterial growth and chemicals were analytical grade, purchased from Merck (Darmstadt, Germany) and Carlo Erba Reagents (Milan, Italy). Desthiobiotin, StrepTag resin and HABA were purchased from IBA Lifesciences GmbH (Göttingen, Germany).Mutagenesis and domain shuffling
Mutants were obtained by following an in-house adapted protocol20 based on the QuikChange strategy. Materials used were those of the QuikChange Lightning Kit (Agilent). The concentrations employed were those of the manufacturer’s protocol except for plasmid and primers which were doubled. Mutagenic primers were designed according to Agilent’s guidelines, following the criteria of melting temperature ≥55 °C and equal performances between forward and reverse primer, and purchased from Eurofins Genomics. DNA sequencings were outsourced to Eurofins Genomics, Germany, where they were carried out with the Sanger method. Samples were prepared according to the company’s instructions (for plasmid DNA: 15 μl at 50–100 ng μl−1) and shipped.On plate colorimetric screening
The screening procedure was previously developed in our group for site saturation mutagenesis.20,53 Briefly E. coli BL21 (DE3) competent cells were co-transformed with the desired hydrogenase plasmid and the maturation plasmid pFG, plated on LBA supplemented with antibiotics (ampicillin 150 μg ml−1 and streptomycin 50 μg ml−1) and incubated at 37 °C for 16–18 hours. After, 6 cm diameter plates were prepared containing 8 ml of M63 agar medium (12 g l−1 KH2PO4, 28 g l−1 K2HPO4, 8 g l−1 (NH4)2SO4, 5 g l−1 casamino acids, 120 mg l−1 anhydrous MgSO4, 1.5% w/v bacteriological grade agar, pH 7.3, 0.5 mg l−1 FeSO4·7H2O), supplemented with 206.5 μl of a mix solution containing 490 μl glucose 50% w/v, 490 μl ferric citrate 10 mM, 245 μl methyl viologen, 73.5 μl IPTG 1 M, 98 μl ampicillin 75 mg ml−1 and 49 μl streptomycin 50 mg ml−1.In each plate squares were drawn and appropriate E. coli BL21 (DE3) transformed colonies were transferred onto plates in triplicate (typically positive control, negative control, and samples to test for each plate); CbA5H-expressing colonies were used as positive control for oxygen resistance, while CrHydA1 WT-expressing colonies were used as negative control.
Plates were then placed in a sealed glass jar, with gas inlet and outlet, and fluxed with pure argon at 30 °C for 5 hours in order to remove the oxygen from the inside. Subsequently, the jar was incubated at 30 °C for further 20–21 hours to let the hydrogenases be expressed.
After this time, the enzymatic activity (hydrogen uptake) was detected by fluxing H2 for 60 minutes, during which the colourless [MV]2+ was reduced to [MV]+˙ yielding a blue spot around the colonies which harbour active hydrogenases. Oxygen stability was then determined by fluxing H2 for another 60 minutes after having exposed the plates to the air for 20 minutes to let the MV regress to colourless again. Colonies that redeveloped the blue spot were those whose hydrogenase activity was not compromised by air oxidation.
Protein expression and purification
CbA5H, mutants and chimeric variants were expressed as reported.12 Briefly, plasmid pCbA5H, containing the CbA5H gene12 or the mutated genes and the maturase gene hydE from Clostridium acetobutylicum,54 was transformed into E. coli Rosetta (DE3)/pFG (containing the plasmid pCaFG, with the maturase genes hydF and hydG from C. acetobutylicum).54Plates with streptomycin and ampicillin were used to grow the cells.
For the expression a single colony was used to inoculate 20 ml culture incubated overnight at 37 °C. The next day, the preculture was used to inoculate 1 l culture in 2 l total volume flasks. Each flask contained: 900 ml TB (12 g tryptone, 24 g yeast extract, 4 ml glycerol), 100 ml potassium phosphate solution (2.2 g KH2PO4, 9.4 g K2HPO4), 2 mM ferric ammonium citrate, 100 μg ml−1 ampicillin, 50 μg ml−1 streptomycin. The cultures were incubated aerobically at 37 °C, 200 rpm until OD600 = 0.4–0.6. When the culture reached the desired OD, it was supplemented with 0.5% glucose, 25 mM sodium fumarate, 2 mM cysteine and protein expression was induced with 1.5 mM IPTG. The cultures were transferred into 1 l bottles and kept under anaerobic conditions flushing argon for 24 h. The optimal post-induction temperature was 20 °C.55 Cells were harvested under anaerobic conditions and stored at −20 °C in airtight glass bottles.
Cell lysis was performed in Lysis Buffer (TrisHCl 100 mM, NaCl 150 mM, glycerol 5% v/v, Triton X-100 1% v/v, 1/4 tablet of COMPLETE protease inhibitor, lysozyme 1 mg ml−1, DNAse I 1 U ml−1 from Thermo Fisher, pH 8) as previously described,20,56 typically in glove box if not otherwise indicated.
Each purification was performed in anaerobic conditions, using Strep-Tactin Superflow high-capacity resin (IBA), following the manufacturer’s protocol, the protein was eluted with desthiobiotin in Buffer W (100 mM TrisHCl, 150 mM NaCl, pH 8.0), checked for purity via SDS-PAGE and finally the enzyme was concentrated and stored with Buffer W for further tests. Protein concentration was evaluated via the Bradford assay with bovine serum albumin as a standard. Typical yields were between 1 and 1.5 mg of pure protein per l culture.
Western blotting
All the hydrogenases and chimeras carried a Strep-tag® II (WSHPQFEK) against which the Strep-Tactin®-horseradish peroxidase conjugate (IBA Life Sciences) can be used for detection. Proteins separated by SDS-PAGE were electro-transferred onto a PVDF Transfer Membrane 0.45 μm (Thermo Scientific) with a Semiphor semi-dry transfer unit (Hoefer). The PVDF membrane was activated by dipping it in pure methanol for 5 minutes and after washing with deionized water the membrane was put in Towbin buffer (25 mM Tris base, 192 mM glycine, 20% v/v methanol, 0.1% w/v SDS, pH 8.3) for 5 minutes. The SDS-PAGE gel was prepared by soaking it in the Towbin buffer for 15 minutes. Inside the electro-transfer unit, a 6-layer system was assembled as described by the protocol: starting from the anode (+), 3 sheets of blotting paper (8.5 × 6 cm) imbued in Towbin buffer, the PVDF membrane, the SDS-PAGE gel and 3 more blotting paper sheets imbued in Towbin buffer were stacked on top of each other to finish the “sandwich”. The proteins were transferred during 60 minutes at 76 mA and at room temperature. After the run the membrane was incubated with 20 ml PBS-blocking buffer (PBS buffer with 3% BSA and 0.05% v/v Tween 20), overnight at 4 °C under gentle agitation.Subsequently the membrane was washed three times with 20 ml PBS-Tween buffer (PBS buffer with 0.1% v/v Tween 20) for 5 minutes each. After the last washing step, the membrane was incubated with gentle shaking for 1 hour with 10 ml PBS-Tween buffer and 2.5 μl Strep-Tactin® horseradish peroxidase conjugate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4000). After this step the membrane was washed two times with PBS-Tween buffer for 1 minute each and then two times with PBS-buffer for 1 minute each. From this point forth, IBA’s “Chromogenic detection with horse radish peroxidase (HRP)” protocol was followed, except for the substitution of 3% w/v 4-chloro-1-naphthol in methanol with 0.5 mg ml−1 3,3′-diaminobenzidine (DAB) in PBS buffer (4 mM KH2PO4, 16 mM Na2HPO4, 115 mM NaCl, pH 7.4) as chromogenic reagent.
:4000). After this step the membrane was washed two times with PBS-Tween buffer for 1 minute each and then two times with PBS-buffer for 1 minute each. From this point forth, IBA’s “Chromogenic detection with horse radish peroxidase (HRP)” protocol was followed, except for the substitution of 3% w/v 4-chloro-1-naphthol in methanol with 0.5 mg ml−1 3,3′-diaminobenzidine (DAB) in PBS buffer (4 mM KH2PO4, 16 mM Na2HPO4, 115 mM NaCl, pH 7.4) as chromogenic reagent.
GC H2 evolution activity assay
H2 evolution activity was evaluated by gas chromatography (GC) at 37 °C. The assay was conducted so as to recreate the ideal conditions for proteins re-activation. 20 ml glass vials were filled with 5 ml of Buffer W (pH 8) containing 1% v/v of methyl viologen 1 M, closed with rubber stoppers, sealed and saturated with argon (upside down) for 10 minutes. Subsequently 500 μl of 1 M sodium dithionite were added with a syringe and saturated with argon for another 10 minutes.Finally, a few μl of pure protein (adjusted depending on the concentration) were added and vials were incubated at 37 °C. H2 production was detected in time by analysing the headspace with an Agilent Technologies 7890A instrument equipped with purged packed inlet, Molesieve 5A column (30 m, ID 0.53 mm, film 25 mm) and thermal conductivity detector; argon was used as carrier gas.
Conclusions
The peculiarity that generates considerable interest in the protein CbA5H is its ability to induce a spontaneous and reversible highly stable state Hinact that can be converted into the active form simply through reductive treatment with dithionite or H2. A second intriguing feature is the presence of an SLBB domain, reported to be an adapter module, both in ancient prokaryotic organisms and in eukaryotes, for protein–protein and protein–ligand interaction in many redox-driven catalytic complexes.23,57–59Mutagenesis was here applied to gain insight into the dynamic mechanism of oxygen protection and to localise still unidentified switching hot-spots and their effects. Two selected mutants in the 2Fe-subcluster proximity did not determine oxygen stability change in CbA5H or CrHydA1 but highlighted a key position for CbA5H catalytic activity (M393) and granted a mutant (A419S) with increased kcat for H2 production. Changing the polarity environment of the cubane subcluster of CbA5H to increase hydrophilicity in positions 424–425 had variable results, ranging from no effect to a complete inactivation and in two cases lowering the kcat of the evolution reaction.
On a recently identified position in which oxygen sensitivity can be decreased in CpI,42 our data suggest that the mechanism of oxygen resistance in CbA5H is not affected by the absence of Serine 370, as the mutant has shown activity after exposure to an aerobic environment, so this excludes that the lateral chain of this residue can alone induce the transition to Hinact. Nonetheless, given the decreased activity of the S370A mutant, a role in the kinetics of switching and in general on catalysis cannot be excluded and the comparison with the data obtained on the symmetrical position in CpI suggests further experiments, for instance the replacement of S370 with glycine and a more detailed study by FTIR and electrochemistry to precisely dissect the replacement effects.
The production of chimeras by domain swapping clarified that SLBB is not strictly required for CbA5H to attain the O2 protected state. A robust and active hybrid chimera, with the SLBB and bFd domains of CbA5H at the N-terminal and the H-domain of CrHydA1 at the C-terminal, was built and further modified to drive the H-domain environment to a higher similarity to CbA5H. This will represent an excellent starting platform for protein engineering with the challenging aim of designing and producing a gain-of-function variant with O2 protection.
The availability of WT CbA5H and the design of derived chimeras, with alternative modular arrangements, open new perspectives of exploitation for applicative purposes. Robust devices, in which electrode interfacing of the enzyme, immobilization and entrapment for biofuel-cells attain enhanced stability, can be proposed and be relevant for real application.
Presently we are envisaging combination of CbA5H with other biocatalysts, either in soluble form or via the “Molecular-Lego approach”25–30 for cascade reactions, focusing on H2-driven CO2 conversion and on cofactors-regeneration (NADPH/NADH) with simplified product separation and improved sustainability.
Author contributions
Conceptualization: FV, SM, GC, GG, SJS. Data curation: SM, LB, SD, FV. Formal analysis: FV, SM, LB, SD, AR, GC. Funding acquisition: FV, SJS, GG. Investigation: SM, LB, SD, AR. Methodology: SM, FV, LB, SD, GC, SJS. Project administration: FV, SM, GG. Resources: FV, GG, SJS. Validation: SM, GC, FV, GG. Visualization: SM, LB, SD, AR, FV. Writing – original draft: FV, SM, LB, SD, GC. Writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding by 2022 PRIN project 2022JPPT55: “MORF: Molecular determinants of Oxygen Resistance in a unique [FeFe]hydrogenase”. SD and LB have been conducting this research during and with the support the Italian national inter-university PhD SDC course in Sustainable Development and Climate change (https://www.phd-sdc.it/) coordinated by IUSS Pavia and LB wishes to acknowledge funding by the European Union NextgenerationEU – PNRR. DM118/2023-M4C1-Inv. 3.4 – Transizioni digitali e ambientali.References
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- F. Wittkamp, M. Senger, S. T. Stripp and U.-P. Apfel, Chem. Commun., 2018, 54, 5934–5942 RSC.
- S. Morra, Front. Microbiol., 2022, 13, 853626 CrossRef PubMed.
- J. W. Sidabras and S. T. Stripp, JBIC, J. Biol. Inorg. Chem., 2023, 28, 355–378 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, P. Allegra, B. La Licata, F. Sagnelli, P. Zitella, G. Gilardi and F. Valetti, Int. J. Hydrogen Energy, 2014, 39, 9018–9027 CrossRef CAS.
- C.-M. Pan, Y.-T. Fan, P. Zhao and H.-W. Hou, Int. J. Hydrogen Energy, 2008, 33, 5383–5391 CrossRef CAS.
- X. Zhao, D. Xing, N. Fu, B. Liu and N. Ren, Bioresour. Technol., 2011, 102, 8432–8436 CrossRef CAS PubMed.
- P. Noparat, P. Prasertsan and S. O-Thong, Int. J. Hydrogen Energy, 2011, 36, 14086–14092 CrossRef CAS.
- M. Calusinska, T. Happe, B. Joris and A. Wilmotte, Microbiology, 2010, 156, 1575–1588 CrossRef CAS PubMed.
- C. Greening, A. Biswas, C. R. Carere, C. J. Jackson, M. C. Taylor, M. B. Stott, G. M. Cook and S. E. Morales, ISME J., 2016, 10, 761–777 CrossRef CAS PubMed.
- S. Morra, M. Arizzi, F. Valetti and G. Gilardi, Biochemistry, 2016, 55, 5897–5900 CrossRef CAS PubMed.
- M. Winkler, J. Duan, A. Rutz, C. Felbek, L. Scholtysek, O. Lampret, J. Jaenecke, U.-P. Apfel, G. Gilardi, F. Valetti, V. Fourmond, E. Hofmann, C. Léger and T. Happe, Nat. Commun., 2021, 12, 756 CrossRef CAS PubMed.
- P. S. Corrigan, J. L. Tirsch and A. Silakov, J. Am. Chem. Soc., 2020, 142, 12409–12419 CrossRef CAS PubMed.
- M. Heghmanns, A. Rutz, Y. Kutin, V. Engelbrecht, M. Winkler, T. Happe and M. Kasanmascheff, Chem. Sci., 2022, 13, 7289–7294 RSC.
- W. Roseboom, A. L. De Lacey, V. M. Fernandez, E. C. Hatchikian and S. P. J. Albracht, JBIC, J. Biol. Inorg. Chem., 2006, 11, 102–118 CrossRef CAS PubMed.
- A. L. De Lacey, V. M. Fernández, M. Rousset and R. Cammack, Chem. Rev., 2007, 107, 4304–4330 CrossRef CAS PubMed.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- P. Rodríguez-Maciá, E. J. Reijerse, M. van Gastel, S. DeBeer, W. Lubitz, O. Rüdiger and J. A. Birrell, J. Am. Chem. Soc., 2018, 140, 9346–9350 CrossRef PubMed.
- S. Morra, A. Giraudo, G. D. Nardo, P. W. King, G. Gilardi and F. Valetti, PLoS One, 2012, 7, e48400 CrossRef CAS PubMed.
- A. Rutz, C. K. Das, A. Fasano, J. Jaenecke, S. Yadav, U.-P. Apfel, V. Engelbrecht, V. Fourmond, C. Léger, L. V. Schäfer and T. Happe, ACS Catal., 2023, 13, 856–865 CrossRef CAS PubMed.
- P. S. Corrigan, S. H. Majer and A. Silakov, J. Am. Chem. Soc., 2023, 145, 11033–11044 CrossRef CAS PubMed.
- A. M. Burroughs, S. Balaji, L. M. Iyer and L. Aravind, Biol. Direct, 2007, 2, 4 CrossRef PubMed.
- A. M. Burroughs, S. Balaji, L. M. Iyer and L. Aravind, Biol. Direct, 2007, 2, 18 CrossRef PubMed.
- S. J. Sadeghi, Y. T. Meharenna, A. Fantuzzi, F. Valetti and G. Gilardi, Faraday Discuss., 2000, 116, 135–153 RSC.
- V. R. Dodhia, A. Fantuzzi and G. Gilardi, JBIC, J. Biol. Inorg. Chem., 2006, 11, 903–916 CrossRef CAS PubMed.
- K. J. McLean, H. M. Girvan and A. W. Munro, Expert Opin. Drug Metab. Toxicol., 2007, 3, 847–863 CrossRef CAS PubMed.
- S. J. Sadeghi and G. Gilardi, Biotechnol. Appl. Biochem., 2013, 60, 102–110 CrossRef CAS PubMed.
- G. Catucci, A. Ciaramella, G. Di Nardo, C. Zhang, S. Castrignanò and G. Gilardi, Int. J. Mol. Sci., 2022, 23, 3618 CrossRef CAS PubMed.
- D. Giuriato, D. Correddu, G. Catucci, G. Di Nardo, C. Bolchi, M. Pallavicini and G. Gilardi, Protein Sci., 2022, 31, e4501 CrossRef CAS PubMed.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS PubMed.
- T. Happe and J. D. Naber, Eur. J. Biochem., 1993, 214, 475–481 CrossRef CAS PubMed.
- G. Voordouw and S. Brenner, Eur. J. Biochem., 1985, 148, 515–520 CrossRef CAS PubMed.
- J. M. Kuchenreuther, C. S. Grady-Smith, A. S. Bingham, S. J. George, S. P. Cramer and J. R. Swartz, PLoS One, 2010, 5, e15491 CrossRef PubMed.
- M. F. Gorwa, C. Croux and P. Soucaille, J. Bacteriol., 1996, 178, 2668–2675 CrossRef CAS PubMed.
- S. Morra, B. Mongili, S. Maurelli, G. Gilardi and F. Valetti, Biotechnol. Appl. Biochem., 2016, 63, 305–311 CrossRef CAS PubMed.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 557–581 CrossRef CAS PubMed.
- P. Hosseinzadeh, N. M. Marshall, K. N. Chacón, Y. Yu, M. J. Nilges, S. Y. New, S. A. Tashkov, N. J. Blackburn and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 262–267 CrossRef CAS PubMed.
- M. Heghmanns, A. Günzel, D. Brandis, Y. Kutin, V. Engelbrecht, M. Winkler, T. Happe and M. Kasanmascheff, Biophys. Rep., 2021, 1, 100016 CAS.
- J. A. Zuris, D. A. Halim, A. R. Conlan, E. C. Abresch, R. Nechushtai, M. L. Paddock and P. A. Jennings, J. Am. Chem. Soc., 2010, 132, 13120–13122 CrossRef CAS PubMed.
- G. Caserta, L. Zuccarello, C. Barbosa, C. M. Silveira, E. Moe, S. Katz, P. Hildebrandt, I. Zebger and S. Todorovic, Coord. Chem. Rev., 2022, 452, 214287 CrossRef CAS.
- C. Brocks, C. K. Das, J. Duan, S. Yadav, U.-P. Apfel, S. Ghosh, E. Hofmann, M. Winkler, V. Engelbrecht, L. V. Schäfer and T. Happe, ChemSusChem, 2024, 17, e202301365 CrossRef CAS PubMed.
- G. Caserta, C. Papini, A. Adamska-Venkatesh, L. Pecqueur, C. Sommer, E. Reijerse, W. Lubitz, C. Gauquelin, I. Meynial-Salles, D. Pramanik, V. Artero, M. Atta, M. Del Barrio, B. Faivre, V. Fourmond, C. Léger and M. Fontecave, J. Am. Chem. Soc., 2018, 140, 5516–5526 CrossRef CAS PubMed.
- C. Gauquelin, C. Baffert, P. Richaud, E. Kamionka, E. Etienne, D. Guieysse, L. Girbal, V. Fourmond, I. André, B. Guigliarelli, C. Léger, P. Soucaille and I. Meynial-Salles, Biochim. Biophys. Acta, Bioenerg., 2018, 1859, 69–77 CrossRef CAS PubMed.
- S. Stripp, O. Sanganas, T. Happe and M. Haumann, Biochemistry, 2009, 48, 5042–5049 CrossRef CAS PubMed.
- P. Knörzer, A. Silakov, C. E. Foster, F. A. Armstrong, W. Lubitz and T. Happe, J. Biol. Chem., 2012, 287, 1489–1499 CrossRef PubMed.
- D. W. Mulder, M. W. Ratzloff, E. M. Shepard, A. S. Byer, S. M. Noone, J. W. Peters, J. B. Broderick and P. W. King, J. Am. Chem. Soc., 2013, 135, 6921–6929 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- C. Orain, L. Saujet, C. Gauquelin, P. Soucaille, I. Meynial-Salles, C. Baffert, V. Fourmond, H. Bottin and C. Léger, J. Am. Chem. Soc., 2015, 137, 12580–12587 CrossRef CAS PubMed.
- C. C. Pham, D. W. Mulder, V. Pelmenschikov, P. W. King, M. W. Ratzloff, H. Wang, N. Mishra, E. E. Alp, J. Zhao, M. Y. Hu, K. Tamasaku, Y. Yoda and S. P. Cramer, Angew. Chem., Int. Ed., 2018, 57, 10605–10609 CrossRef CAS PubMed.
- M. Senger, T. Kernmayr, M. Lorenzi, H. J. Redman and G. Berggren, Chem. Commun., 2022, 58, 7184–7187 RSC.
- E. C. Kisgeropoulos, V. S. Bharadwaj, D. W. Mulder and P. W. King, Front. Microbiol., 2022, 13 DOI:10.3389/fmicb.2022.903951.
- F. Valetti and G. Gilardi, Biomolecules, 2013, 3, 778–811 CrossRef PubMed.
- P. W. King, M. C. Posewitz, M. L. Ghirardi and M. Seibert, J. Bacteriol., 2006, 188, 2163–2172 CrossRef CAS PubMed.
- S. Morra, A. Cordara, G. Gilardi and F. Valetti, Protein Sci., 2015, 24, 2090–2094 CrossRef CAS PubMed.
- S. Morra, S. Maurelli, M. Chiesa, D. W. Mulder, M. W. Ratzloff, E. Giamello, P. W. King, G. Gilardi and F. Valetti, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 98–106 CrossRef CAS PubMed.
- M. V. Ordóñez, D. Nercessian and R. D. Conde, Extremophiles, 2012, 16, 437–446 CrossRef PubMed.
- N. A. Losey, S. Poudel, E. S. Boyd and M. J. McInerney, Front. Microbiol., 2020, 11 DOI:10.3389/fmicb.2020.01109.
- M. Gupta, R. Venkatramani and S. R. K. Ainavarapu, J. Phys. Chem. B, 2021, 125, 1009–1019 CrossRef CAS PubMed.
Footnote |
| † Presently at Department of Biochemistry, Louvain Institute of Biomolecular Science and Technology - LIBST, UCLouvain, Louvain-la-Neuve, Belgium. |
| This journal is © The Royal Society of Chemistry 2024 |