 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational study of the mechanism of a polyurethane esterase A (PueA) from Pseudomonas chlororaphis†
Katarzyna
Świderek
 *,
Sergio
Martí
,
Kemel
Arafet
and
Vicent
Moliner
*
*,
Sergio
Martí
,
Kemel
Arafet
and
Vicent
Moliner
*
BioComp Group, Institute of Advanced Materials (INAM), Universitat Jaume I, 12071 Castellón, Spain. E-mail: swiderek@uji.es; moliner@uji.es
First published on 18th March 2024
Abstract
The effective management of plastic waste has become a global imperative, given our reliance on a linear model in which plastics are manufactured, used once, and then discarded. This has led to the pervasive accumulation of plastic debris in landfills and environmental contamination. Recognizing this issue, numerous initiatives are underway to address the environmental repercussions associated with plastic disposal. In this study, we investigate the possible molecular mechanism of polyurethane esterase A (PueA), which has been previously identified as responsible for the degradation of a polyester polyurethane (PU) sample in Pseudomonas chlororaphis, as an effort to develop enzymatic biodegradation solutions. After generating the unsolved 3D structure of the protein by AlphaFold2 from its known genome, the enzymatic hydrolysis of the same model PU compound previously used in experiments has been explored employing QM/MM molecular dynamics simulations. This required a preliminary analysis of the 3D structure of the apo-enzyme, identifying the putative active site, and the search for the optimal protein–substrate binding site. Finally, the resulting free energy landscape indicates that wild-type PueA can degrade PU chains, although with low-level activity. The reaction takes place by a characteristic four-step path of the serine hydrolases, involving an acylation followed by a diacylation step. Energetics and structural analysis of the evolution of the active site along the reaction suggests that PueA can be considered a promising protein scaffold for further development to achieve efficient biodegradation of PU.
Introduction
Synthetic or semi-synthetic polymers have been developed in the last century to replace materials such as wood, glass and metals across many different applications due to their excellent and sometimes unique properties, coupled with low production costs. Global synthetic polymers consumption has quadrupled over the past 30 years, and nowadays plastics are the workhorse materials of the modern economy, outgrowing all human-made materials other than steel and cement.1 However, and precisely because the majority of them do not degrade, plastic waste management is a worldwide concern, as our current plastic economy is based on a linear model where the plastics are produced, used only once, and disposed of. Possible initiatives, such as incineration, accumulation in landfills, the synthesis of biodegradable polymers, or the use of biobased materials are not exempt from problems or side effects. Alternatively, inspired by recent observations that some microbes are adapting and evolving to use plastics as a source of nutrients, the use of enzymes capable of degrading synthetic polymers is offering a promising viable solution for the disposal of this kind of waste. In this regard, a lot of work on this direction has been carried out for the development of biocatalytic degradation of synthetic polymers, mainly focused on polyester-type plastics, such as polyethylene terephthalate (PET).2–5Computer simulations, and in particular those based on the combination of Quantum Mechanics and Molecular Mechanics potentials (QM/MM), have contributed to the understanding of the mechanism that governs the catalytic performance of hydrolases, such as those from Ideonella sakaiensis 201-F6, IsPETase and IsMHETase,6 or the metagenome-derived leaf-branch compost cutinase (LCC-ICCG variant),6 in the PET depolymerization. Further simulations of the structural changes of IsPETase induced by PET binding,7 whose results agree with recent results of Alper and co-workers,5 have provided information on the crucial polymer–enzyme binding step. On the other side, based on our previous computational studies showing the potential of the lipase B from Candida antarctica (CALB) as a protein scaffold to re-design new activities,8–12 together with experimental evidence that this highly promiscuous esterase can be used for the hydrolysis of aliphatic polyesters,13 we recently explored the hydrolysis of PET derived diesters and PET oligomers catalysed by CALB.14 The results generated by classical and QM/MM molecular dynamics (MD) simulations, combined with experimental Michaelis–Menten kinetics, suggested a selectivity switch of CALB for the formation of either mono(2-hydroxyethyl)terephthalate (MHET) or terephthalic acid (TPA) from bis-(hydroxyethyl)terephthalate (BHET) by modulating the pH conditions.14 Thus, the unknown role of the pH on the CALB regioselectivity toward the hydrolysis of bis-(hydroxyethyl) terephthalate (BHET) was revealed. In addition, this insight to perform pH-controlled biotransformation that selectively hydrolyses BHET to either its corresponding diacid or monoesters using both soluble and immobilized CALB was also exploited.14
The results derived from previous studies can constitute the bedrock to the understanding and controlling of the molecular mechanism that rules the performance of PET degrading enzymes. In this work, we focused on the enzymatic degradation of a different worldwide employed synthetic polymer: polyurethane (PU). Indeed, after polyesters, PUs make up the second largest class of hydrolysable plastics production and it is predicted a growth rate of 4.3% from 2022–2030.15,16 However PUs are difficult to recycle by mechanical or chemical methods due to their complex molecular composition containing different functional groups (aliphatic chains combining carbamate and ester groups) and sometimes, like thermoset PUs, showing three-dimensional cross-linking structures. Consequently, drastic recycling conditions are required when using chemical hydrolysis methods (high temperature, up to 450 °C, and pressure, up to 8 MPa)16 and the resulting monomers are of low purity or incapable of reacting in a new polymerization process. An additional difficulty in the use of efficient polyurethanases (PUases) is that the number of microbial and microbial consortia identified with certain activity in degrading PUs is much more limited than those degrading PET.17–23 Moreover, as mentioned by Badenhorst, Bornscheuer and co-workers,24 the PUases reported so far are misnamed polyester-hydrolases, like cutinases, acting on the polyester groups of polyester-polyurethanes while the urethane bonds in polyether-polyurethanes have remained inaccessible to biocatalytic hydrolysis. In any case, despite the studies devoted to understanding and designing PUs depolymerization enzymes, including the identification of the primary sequence for some of the proteins responsible for the PU degrading activity of microorganisms,25–28 no crystal structure has been yet solved. Consequently, computer simulations are hampered by the lack of highly accurate initial coordinates of the protein atoms. In fact, the only 3D structures reported so far are derived from comparative homology modelling techniques followed by classical molecular dynamics (MD) simulations using several lipases for multiple alignment, with the consequently associated uncertainty.29 In all, the current biodegradation of PU is still very limited and cannot meet the practical application of recycling.16,30
In the present study, we are investigating the molecular mechanism of polyurethane esterase A (PueA), which has been previously identified as responsible for the degradation of Impranil, a model compound that mimics polyester polyurethane (PU) chains, in Pseudomonas chlororaphis (Scheme 1).27 Because the 3D structure of PueA has not been yet solved, it has been generated by AlphaFold2 (ref. 31) from its known genome.27 Then, after a deep structural analysis of the structure, the docking of Impranil was carried out by an automatized software but also based on chemical intuition in order to identify the active site and the optimal protein–substrate binding. The enzymatic hydrolysis of the selected substrate has later been explored based on QM/MM molecular dynamics (MD) simulations. Free energy surfaces (FESs) have been computed thus allowing a detailed description of the complete chemical reaction. The present study must be considered as the starting point of a multidisciplinary and multistep project, which will imply the rational re-design of the wild-type enzyme to improve its potential initial activity. In this regard, it must be pointed out that, as mentioned by Wei, Bornscheuer and co-workers,4 improving the biocatalytic decomposition of plastic is undoubtedly a multifaceted problem that requires improvement not only of the catalytic efficiency but other relevant factors such as the accessibility of the enzyme binding to solid PU-based plastics, the affinity between the enzyme and the polymer chain and the thermostability of the protein.
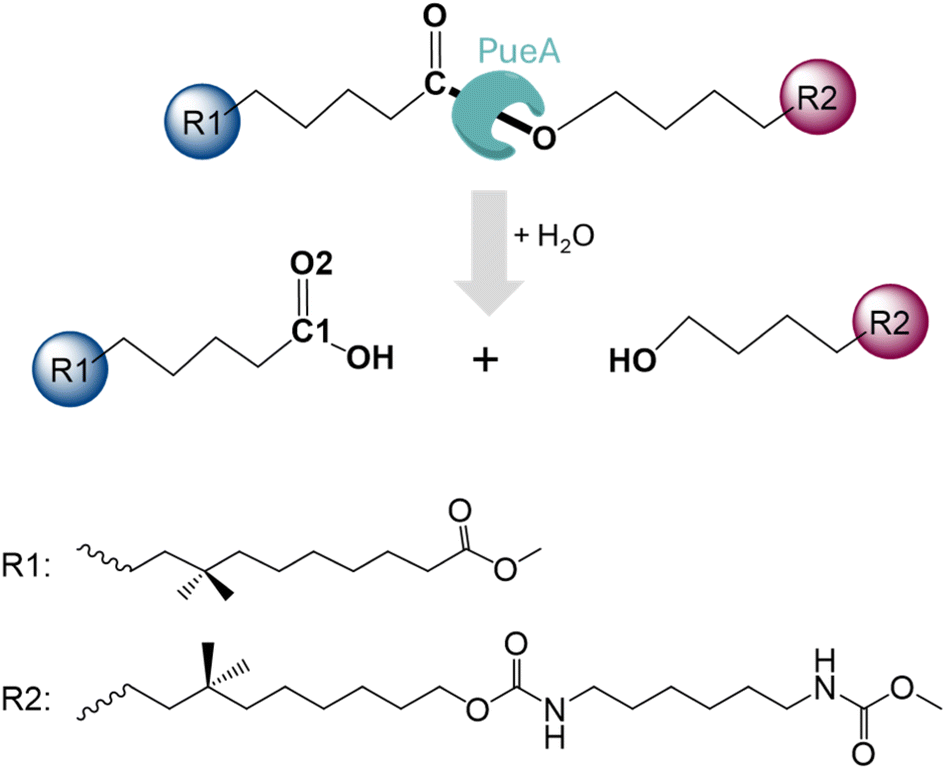 | ||
| Scheme 1 Hydrolysis of Impranil, as a model compound of PUs, catalyzed by PueA. | ||
Methods
Setting up the model
In the absence of structural data, the initial protein geometry was obtained using the AlphaFold2 program with the nucleotide sequence available in GenBank, (accession code #AF069748), as solved by Stern and Howard.27 A docking study of the Impranil molecule was carried out on this structure using AutoDock Vina,32 focusing on the position of the Ser-207 residue (within ±20 Å), since this is the only site where a triad composed of a serine, a histidine, and an aspartate was identified in the protein, in agreement with the fact that Ser207 was already proposed as the possible key catalytic residue.29 Charges and parameters from amber ff03 were used for the protein, whereas the Gasteiger charges33 and missing parameters for Impranil were automatically generated using Openbabel version 3.1.1 program.34 After inspecting the different poses (based on the scoring function and the geometry), a model was built where the ester group of Impranil was properly oriented toward Ser-207 and the adjacent His-313 thus forming a reactive conformation for the reaction to proceed (Fig. 1). In this structure, the substrate adopted a folded conformation inside the pocket of the protein, as described in next section. Subsequently, the missing parameters for the substrate were generated based on the General Amber Force Field (GAFF)35 and the atomic charges were computed at the AM1 method with bond charge corrections (AM1-BCC)36 using the Antechamber software37 (ESI Table S1†), which is available in the AmberTools package. The protonation state of titratable residues was determined at pH 7 using the PropKa ver. 3.0.3 empirical program,38 combined with a detailed inspection of the surroundings of each examined residue. A list of the computed pKa values is deposited in the ESI (Table S2†). The hydrogen atoms were added to the model using tleap software.37 Before proceeding with the next steps of system preparation, series of MD simulations with two alternative possible conformations of the substrate bound to the protein were carried out. These preliminary simulations were done in order to ensure the absence of Impranil refolding as predicted by the best pose of the docking studies (see Results section for details). Solvent water molecules were not yet added into the system to favor the protein:substrate interactions. The structure of the protein was first optimized, allowing side chains of amino acid residues especially those in the vicinity of substrate to adopt optimal positions, and consequently, 100 ns of classical molecular MD simulations with two constrained key distances defined between Oγ of Ser207 and C1 of the substrate, and Nε of His313 and O3 of the substrate (see Fig. 1). During these simulations done using the Namd-2 (ref. 39) program, only the Impranil molecule was allowed to move. Simulations confirmed the stability of the extended form of the substrate in the binding channel of the protein and therefore provided a starting model for the following steps of the system construction in which the overall negative charge of the system was neutralized by the addition of 30 sodium Na+ ions. Subsequently the system was soaked in a box of water molecules of 118 × 96 × 83 Å3. The protein and water molecules were described with AMBER ff03 (ref. 40) and TIP3P force fields,41 respectively. A cut-off for non-bonding interactions was set between 14.5 to 16 Å using a smooth switching function. The temperature during the simulations was controlled using the Langevin thermostat,42 and the pressure with the Nosé–Hoover–Langevin piston43 pressure control. In all simulations, periodic boundary conditions (PBC) were applied, as well as PME based long range electrostatic interactions. Then the position of all atoms was optimized using a preliminary minimization algorithm and gradual heating of the system to 298 K with 0.001 K temperature increments, followed by 500 ps of NPT equilibration and 100 ns of constrained NPT MD at 298 K with the SHAKE algorithm used to restrain all hydrogen bonds with a 2 fs time step. After constrained simulations, 500 ns of non-accelerated unbiased classical NPT MD simulations with a time step of 2 fs was carried out for two systems with two different orientations of Impranil (as described in detail in the Results section). Time evolution of the RMSD computed for the backbone atoms (C, O, Cα, and N) of protein and heavy atoms of Impranil were analyzed to confirm the stability of the initial reactant complex structures (ESI Fig. S1†).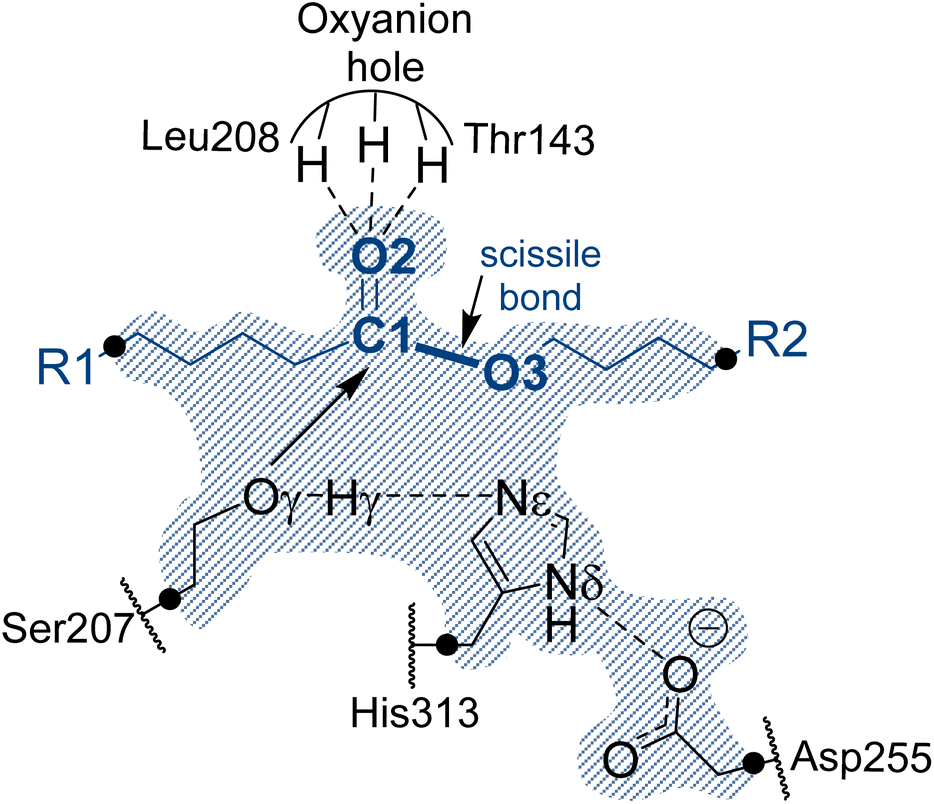 | ||
| Fig. 1 Schematic representation of the active site of PueA in complex with Impranil PU model compound in the non-covalent reactant complex, RC. Light blue shaded region contains atoms treated quantum mechanically. Dashed lines indicate hydrogen bond interactions. Quantum link atoms are represented as black dots. R1 and R2 as in Scheme 1. | ||
Free energy surfaces
A representative structure of the equilibrated system generated during the MD simulations was used as a starting point for the subsequent QM/MM reactivity studies, using the fDYNAMO library.44 The semiempirical AM1 (ref. 45) method and the M06-2X density functional46 were used to describe the QM sub-set of atoms, corresponding to the catalytic triad Ser207, His313 and Asp255, together with the substrate (Fig. 1). To saturate the valence of the QM-MM frontier atoms, 5 quantum link atoms were inserted (Fig. 1).The AMBER ff03 and TIP3P classical force fields were used to treat the protein and the solvent water molecules, respectively, as implemented in the fDynamo library.47 In each step of the proposed mechanism, the study of the corresponding associated potential energy surface was carried out using the AM1/MM hybrid Hamiltonian and applying a restraint of 5000 kJ mol−1 Å−2 to the different values of the distinguished reaction coordinates. Each PMF window consisted of 25 ps of which the first 5 ps were discarded when integrating the PMF. The force constants applied in the umbrella sampling (US) technique48 were 2500 kJ mol−1 Å−2, and the data generated were integrated in terms of the potential of mean forces (PMFs) using the weighted histogram analysis method (WHAM).49 Finally, since semiempirical Hamiltonians have been used to carry out the hybrid dynamics of the reactivity studies, the FESs were subsequently improved using spline under tension corrections, based on the original work of Truhlar and co-workers.50 As implemented in previous papers,51 a correction term is interpolated to any value along the reaction coordinates in the FES based on multiple structures derived from the AM1/MM simulations. A continuous energy function is used to obtain the corrected PMFs using the M06-2X functional with the standard 6-31+G(d,p) basis set. These calculations were carried out using the Gaussian09 (ref. 52) program combined with fDynamo. Finally, transition state (TS) structures selected from the stationary points of the FESs were optimized and characterized at the M06-2X/MM level and IRC paths were traced down to the precedent and posterior energy minima structures to confirm the reliability of the correcting protocol.
Results and discussion
Generation of the 3D structure of PueA and docking of Impranil
As described in the previous section, because the structure of PueA has not been solved, the 3D structure was generated with AphaFold2 based on the nucleotide sequence published by Stern and Howard.27 According to the results, the obtained structure, shown in Fig. 2, is a monomer composed of two domains, a C-domain containing 11 β-strands (in yellow) and an N-domain containing 8 α-helices (in purple), 617 residues in total. As stated before according to the crystal structure of a family I.3 lipase from Pseudomonas sp. MIS38,53 the N catalytic domain presents two lids (residues 46–74 and 146–167), in a closed conformation. Ser207 has been proposed as the reactive serine,29 which is located in the region of the protein with a high concentration of alpha helix conformations (N-domain). Interestingly, Ser207 is the only serine residue of the protein that is surrounded by an optimal dyad composed of histidine and an aspartate, confirming the existence of a potential reactive pocket to hydrolase an ester group following a molecular mechanism employed by serine hydrolyses. Thus, the active site presents, apart from Ser207, a well-positioned His313 that could activate the serine for the nucleophilic attack to the ester group of Impranil (see Fig. 2). An aspartate (Asp255) appears to be within a hydrogen bond distance of the Nδ atom of His313 suggesting a putative role in modulating histidine pKa, thus facilitating the different chemical steps of the catalytic process to take place. In all, the most promising structure of PueA, as generated by AlphaFold2, looks reasonable to advance to the next step in our study, which is exploring the binding of Impranil to generate a non-covalent reactant complex.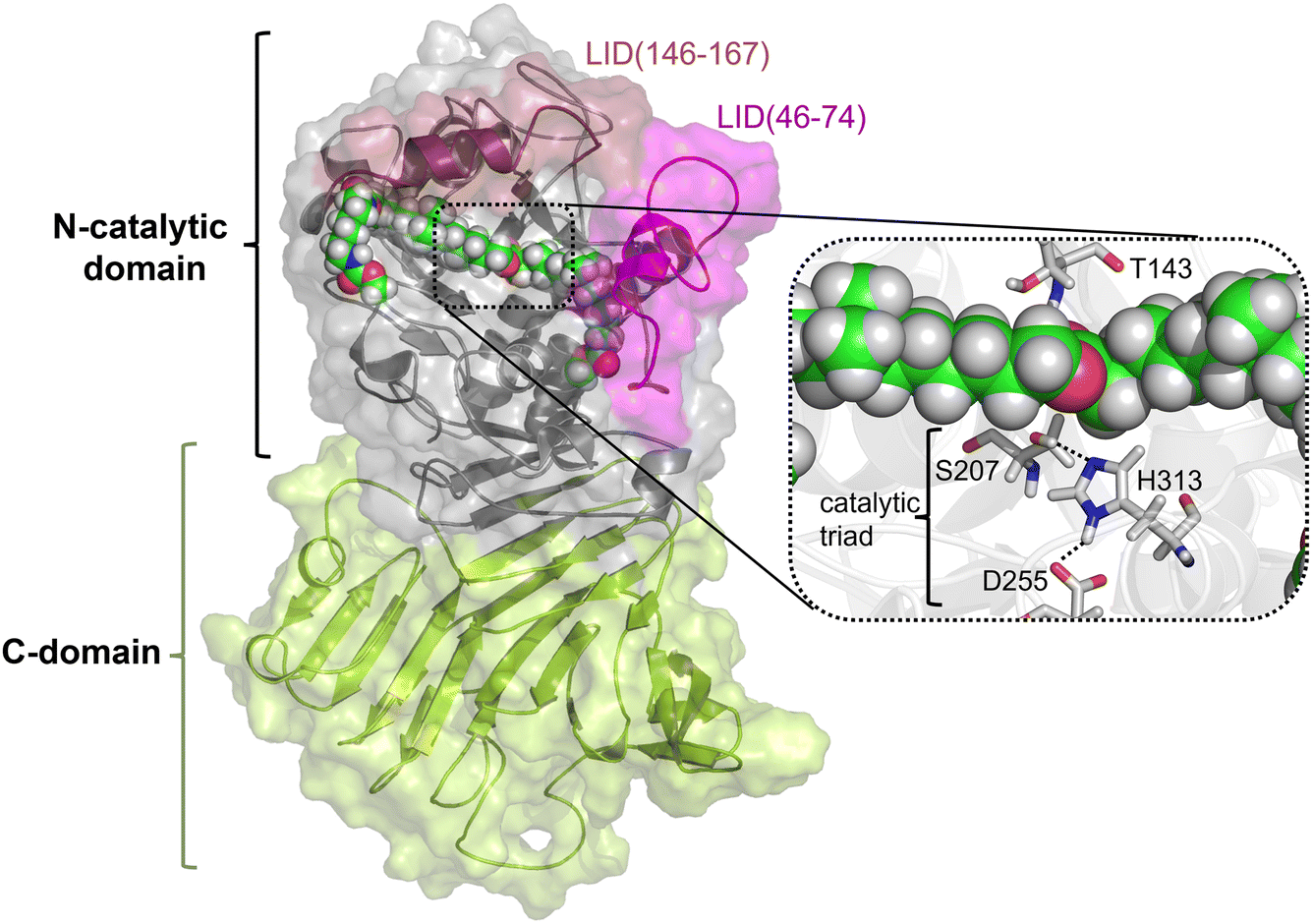 | ||
| Fig. 2 Structure of PueA generated by AlphaFold2 composed of a C-domain basically formed by β sheets (in yellow) and an N-catalytic domain with α helices (in grey with the indicated position of two LID structures in pink). The active site residues are depicted in liquorice while the Impranil, docked in the active site with an extended conformation, RC1, is represented as spheres. | ||
Substrate binding
The next step in setting up the full molecular model was to dock the selected substrate, Impranil, to the protein. After analysis of the different structures generated when running a blind docking between the protein and the substrate with AutoDock Vina, the best candidate (considering the scores of the different poses, complemented with a visual geometrical analysis) was selected as a possible protein:substrate reactant complex. The results show that the substrate adopts a folded conformation inside the active site that suggests the possibility of the chemical reactions taking place, according to the distances between the reactive Ser207 and the carbonyl carbon of the ester group of Impranil (ESI Fig. S2†). However, a deeper analysis of the structure of the substrate shows that this conformation of Impranil in the cavity of PueA is forcing not favourable intramolecular interactions in the aliphatic parts of Impranil (ESI Fig. S3A†). In addition, unconstrained MD simulations starting from this structure show how the substrate departs from the active site residues Ser207 and His 313 (ESI Fig. S3B†). Consequently, and keeping in mind the presence of a groove formed by hydrophobic residues in the surroundings of the selected active site of the enzyme, we manually extend the conformation of Impranil looking for a more stable and reliable protein:ligand initial reactant complex. Thus, two possible orientations of Impranil were prepared for further equilibrations, with the carbamate end group (R2 in Scheme 1) exposed to the solvent, RC1, or inside the cavity of the protein, RC2 (ESI Fig. S4†).As explained in the Methods section, 100 ns of MD simulations with constraints in the key substrate–enzyme distances, followed by 500 ns of free MD simulations were carried out to explore whether both extended conformations of Impranil were stable. Analysis of the time dependent evolution of the RMSD shows that the systems remain stable after 500 ns of MD simulation, when all the constraints were removed (Fig. S1†). The equilibrated structures (Fig. S4†) confirm how Impranil adopts extended poses on the surface of PueA, keeping stable the key inter-atomic distances in the active site, in both reactant complex (RC1 and RC2) conformations. Indeed, as shown in Fig. 3, the ester reactive group of Impranil adopts a promising pose in the active site of the enzyme for the chemical reaction to proceed in both RC1 and RC2: the hydroxyl group of Ser207 interacts with the Nε nitrogen atom of His313, while the Nδ nitrogen atom of His313 interacts with Asp255, as previously observed in the apoenzyme structure. This, together with the fact that the hydroxyl oxygen atom of Ser207 is at a short distance from the carbonyl carbon atom of the ester group of Impranil (Oγ–C1 distance), suggests a feasible nucleophilic attack of Ser207 to the substrate, after activation by His313. In addition, the structural analysis reveals the presence of an oxyanion hole formed by the N–H backbone groups of residues Thr143 and Leu208, which could be anchoring the substrate and could be also stabilizing the accumulation of negative charge on the carbonyl group of the substrate (O2) in putative tetrahedral intermediates that could appear along the reaction. Monitoring the end-to-end distances of Impranil confirms that, in both reactant complexes, the extended conformation is preserved along the unbiased MD simulations. As observed in Fig. 3, this distance oscillates around ca. 27 Å in RC1 and significantly more extended in the case of RC2, where this distance oscillates around 38 Å. See ESI Fig. S5 and S6† for a detailed monitoring of the time evolution of the discussed distances along the 500 ns of unbiased MD simulations of RC1 and RC2.
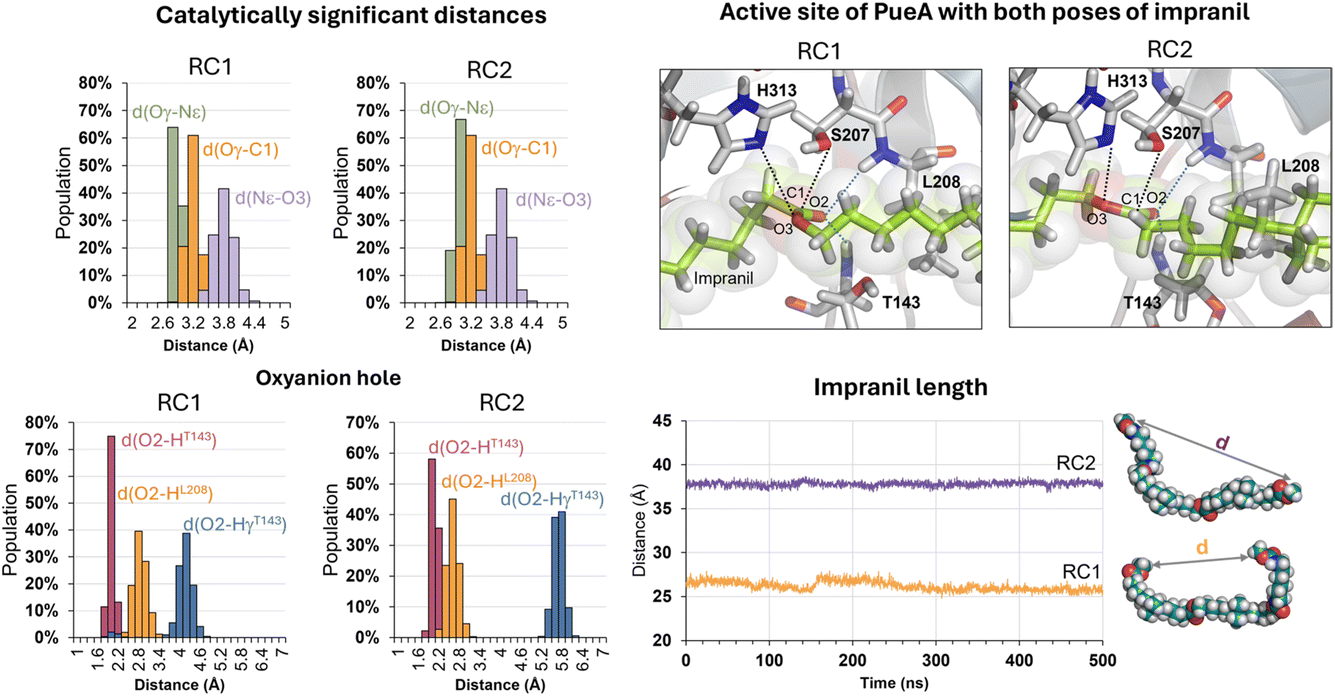 | ||
| Fig. 3 Analysis of key distances along the 500 ns of free MD simulation of the non-covalent protein:ligand reactants complex with the extended conformation of Impranil when the ester end-group was exposed to the solvent, RC2. Analysis was done for 2500 structures generated during 500 ns of MD simulation. | ||
Regarding other ligand–protein interactions responsible for the stability of this reactant-like complex, it is remarkable the complementarity between the aliphatic chains of Impranil and the groove formed by hydrophobic residues in the pocket of PueA, in both RC1 and RC2 conformations (Fig. 4A). In addition, some specific stabilizing interactions are established between the substrate and the protein, as revealed by the analysis of the average protein–ligand interaction energies, decomposed by residues (Fig. 4B). Interestingly, while several of these interactions appear to be preserved regardless the substrate poses, such as those established between Impranil and F36, G142, T143, D161, A164, A165, L166, L208, P239, L261, D262, W310, L311, H313, L314 and P315 residues, as indicated by yellow bars in Fig. 4B, others are specific to the particular orientation of Impranil in the pocket of PueA (grey bars in Fig. 4B). Thus, Impranil interacts with N34, G35, K169, A172, Y175, L211, S264, T265, F266, and L268 only in the case of RC1, while favorable interactions with Y40, G167, P168, S207, T240, G263, F288, D290, T316, R324, A343, N344 and R350 are established only in the RC2. Interestingly, the reactive Ser207 only shows a favorable interaction with the substrate in RC2. Another remarkable difference is observed with the interaction with Asp262: while in RC1 this is a destabilizing interaction, in RC it turns to be slightly favorable. This interaction is basically electrostatic in nature, which agrees with the fact that it is located 8.5 and 6.0 Å away from the substrate in RC2 and RC1, respectively (measured from the carboxylic group of D262 and the closest carbon atom of the backbone of Impranil). Finally, is also important to stress that RC2 shows a larger amount of stabilizing interactions than RC1 and, in addition, the interaction established with Asp262, which is unfavourable in RC1, is neutralized in RC2. A qualitative analysis of the protein:Impranil interactions in RC1, RC2, and also in the folded conformation directly derived from the docking with AutoDock Vina, can be also derived from diagrams of hydrogen bonding interaction and hydrophobic contacts (ESI Fig. S7†). The results show how RC2 presents the largest amount of hydrophobic contacts as well as more specific H-bond interactions between the protein and the polar groups of Impranil.
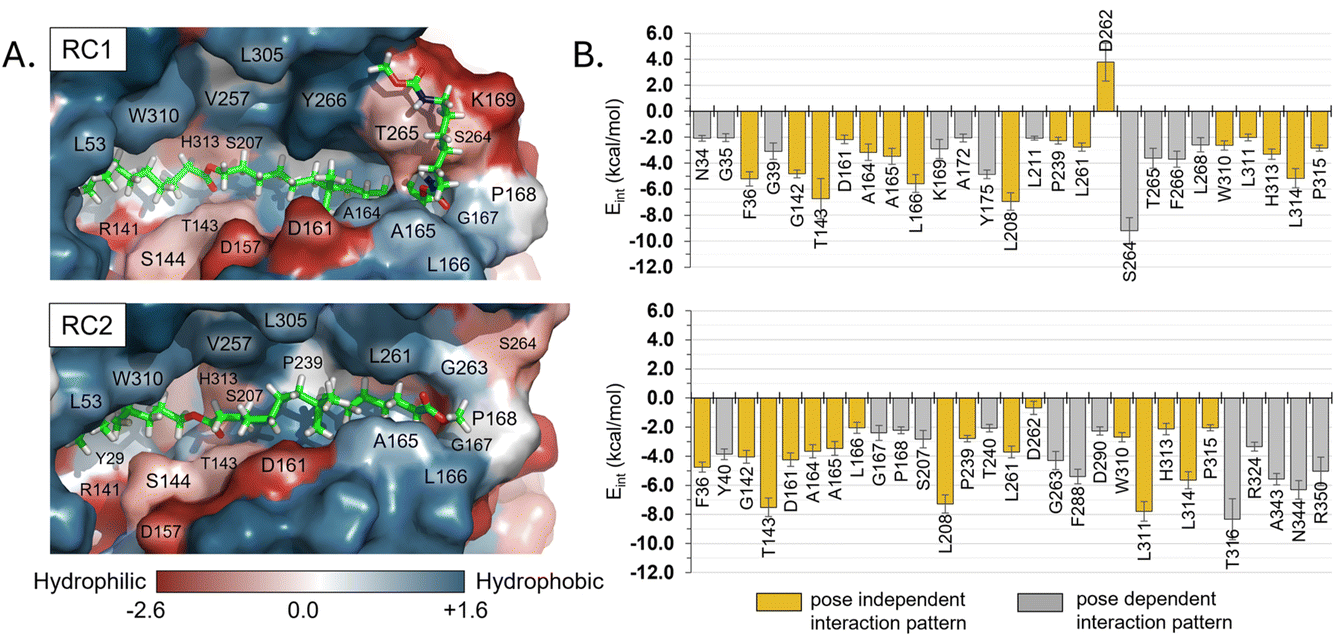 | ||
| Fig. 4 (A) Hydrophobicity map on the protein with the protein:Impranil reactant complexes, RC1 and RC2, generated based on the Eisenberg hydrophobicity scale.54 The increase of negative charges goes from blue (positive) to red (negative). (B) Protein:Impranil interaction energies computed as an average over 2500 snapshots from unbiased 500 ns MD simulations on reactant complexes, RC1 and RC2. Only values over 2 kcal mol−1 are displayed, except D262 on RC2, which is shown for comparative purposes. Bars in yellow are those whose interactions are present in both systems. | ||
Chemical reaction
The next step of our study consists of the exploration of the chemical steps of the hydrolysis of Impranil catalyzed by PueA. The free energy landscape was explored starting from the two possible orientations of the extended Impranil placed into the cavity of the protein, RC1 and RC2, considering that both of them appear to be stable and showing key interatomic distances between the reactive ester group and the residues of the active site compatible with the proposed reaction mechanism. Then, considering the kind of substrate and the reaction to be catalyzed by the enzyme PueA, we propose a molecular mechanism similar to those shown in other serine hydrolases, consisting of an acylation step followed by a diacylation or hydrolysis step (Fig. 5). The FESs for the acylation step obtained from the extended conformation RC1 are shown in ESI Fig. S8,† while those from the RC2 of Impranil are shown in Fig. 6. The corresponding free energy profiles derived from the FESs are presented in Fig. 7. As observed, when using RC1 as the starting structure, the activation free energy of the acylation is 29.8 kcal mol−1, defined by the decomposition of the first tetrahedral intermediate I1 to the acyl enzyme I2 intermediate. In contrast, when the reaction takes place from the RC2, the barrier is significantly lower, 19.9 kcal mol−1, and determined by the first step: the activation of the Ser207 that takes place concomitantly with the nucleophilic attack to the C1 atom of the Impranil. Interestingly, while the kinetics of this first step is basically equivalent in the two reactions (activation energies of 20.6 and 19.9 kcal mol−1, respectively), a dramatic difference is obtained in the second step of the acylation; the proton transfer from the protonated His313 to the ester oxygen atom of the Impranil (O2) required to release the first product of the reaction, the alcohol R2–OH. A geometrical analysis of the first tetrahedral intermediate I1 shows that after the formation of this intermediate, the Nε atom of His225 and the O1 atom of Impranil are in a much better orientation when the reaction was initiated from RC2 than from RC1. Thus, while the Nε–O3 distance is virtually the same in I1 coming from RC1 or RC2 (3.8 Å respectively, as shown in Fig. 3), the orientation of the substrate in the catalytic pocket in RC1 forces a conformation of the chain of Impranil where the –CH2– groups contiguous to the reactive ester group are pointing to the H atom of the Nε of His313, thus destabilizing I1 and TS2. Consequently, and in agreement with the more favorable interactions found in RC2 than in RC1 that suggest a more favorable binding of the former, it can be assumed that the only reactive reactant complex conformation is RC2, and the diacylation step was then explored only for this reaction path. The FESs of the diacylation steps are shown in Fig. 6, while the full free energy profile of the catalyzed decomposition of Impranil by PueA is depicted in Fig. 7, where details of the TS structures of the rate limiting steps of the acylation and deacylation TS-2 and TS-4, respectively, are also shown. As observed, the hydrolysis takes place in two steps, as proposed in the mechanism depicted in Fig. 5, with an activation free energy determined by the TS-4 (19.8 kcal mol−1). The reaction is slightly exergonic (−2 kcal mol−1), which is a good indication of the feasibility of the catalyzed process.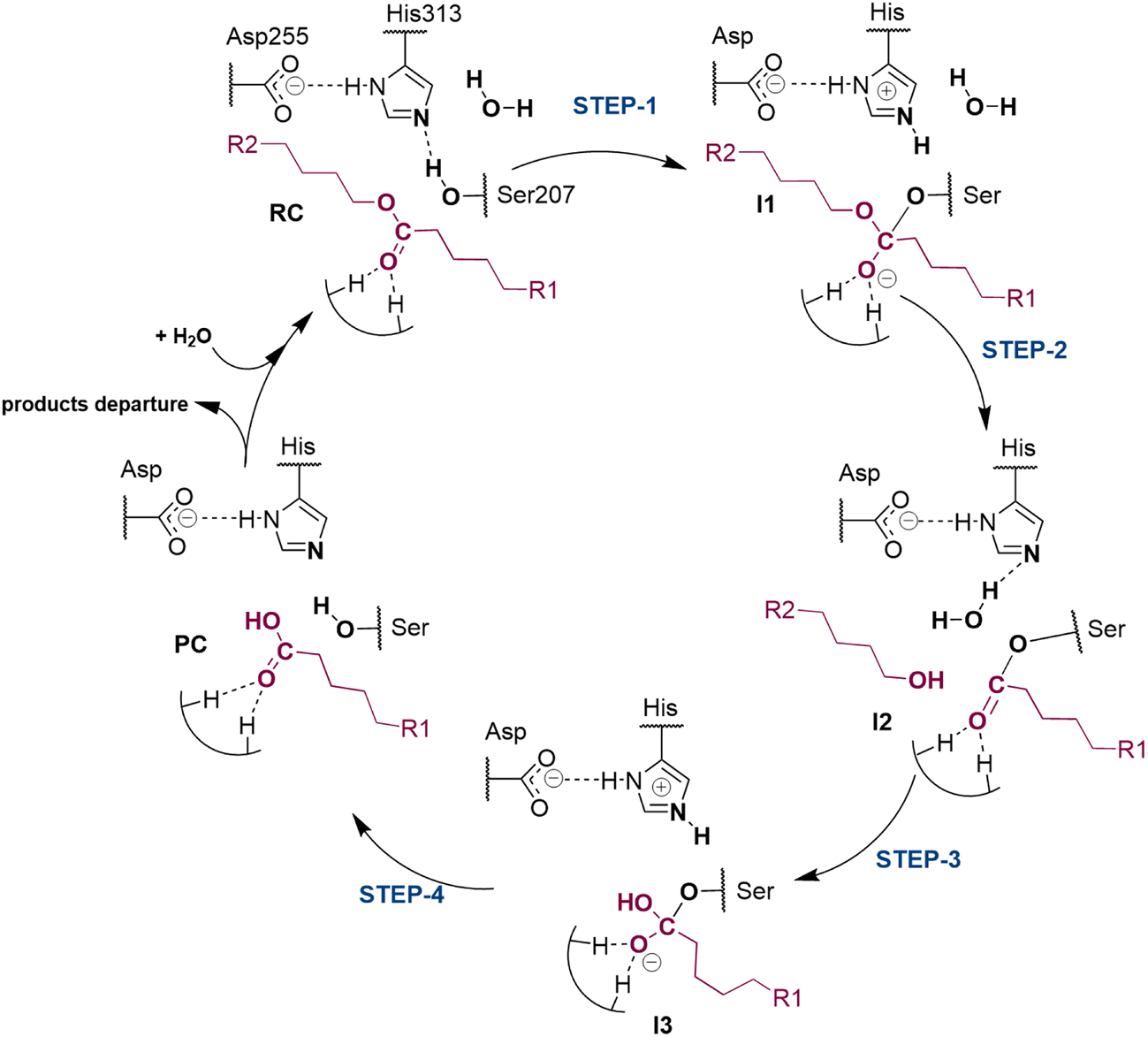 | ||
| Fig. 5 Proposed mechanism for hydrolysis of Impranil, as a model compound of polyester PUs, catalyzed by PueA. R1 and R2 as in Scheme 1. | ||
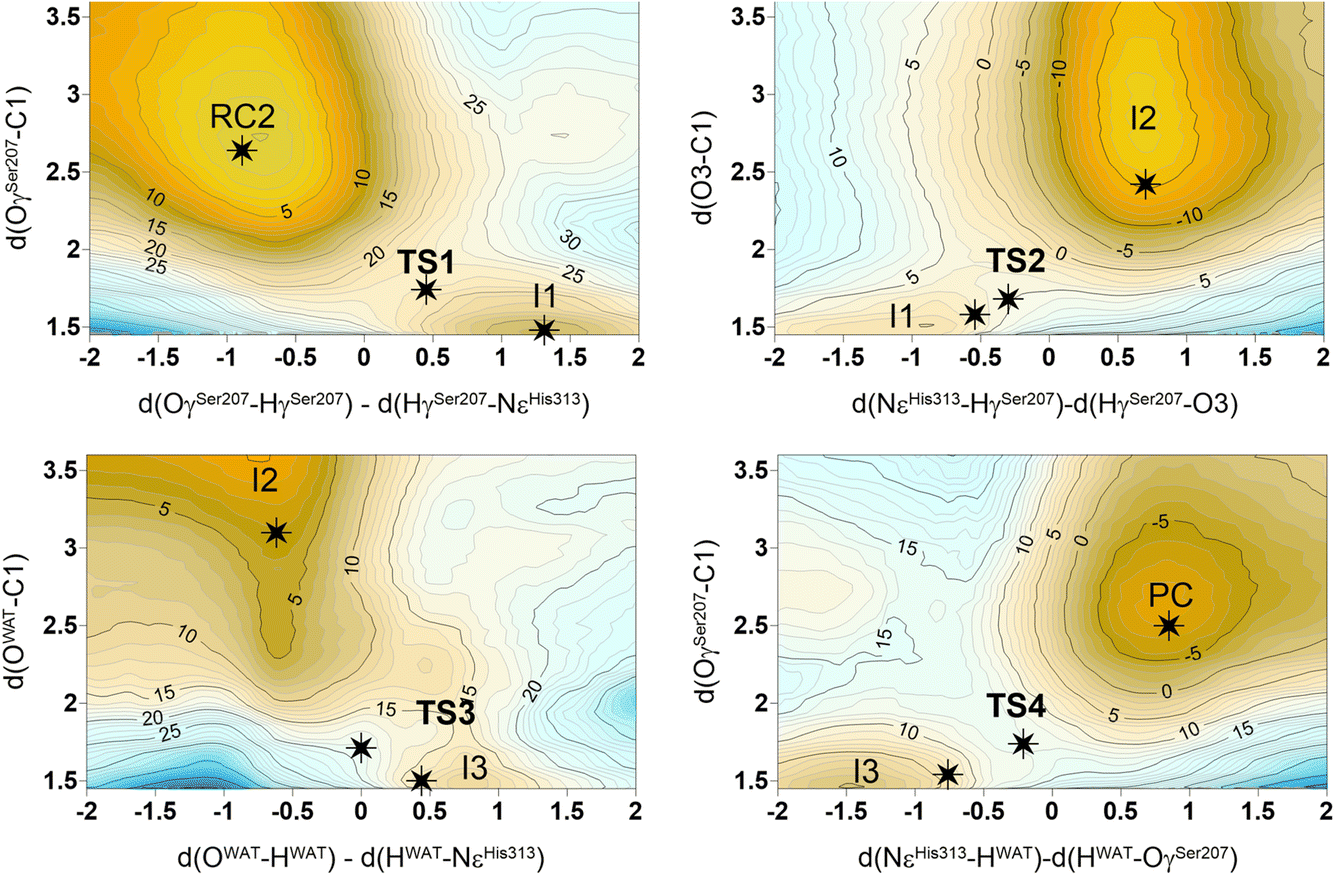 | ||
| Fig. 6 M06-2X:AM1/AMBER free energy surfaces of the acylation of Impranil catalyzed by PueA starting from RC2 conformations. Distances are given in Å and isoenergetic lines are in kcal mol−1. The black stars indicate the position of single stationary point structures fully optimized at M06-2X/AMBER level of theory. Structures of minima were generated from the M06-2X/AMBER IRC path computed from the located TS structures. | ||
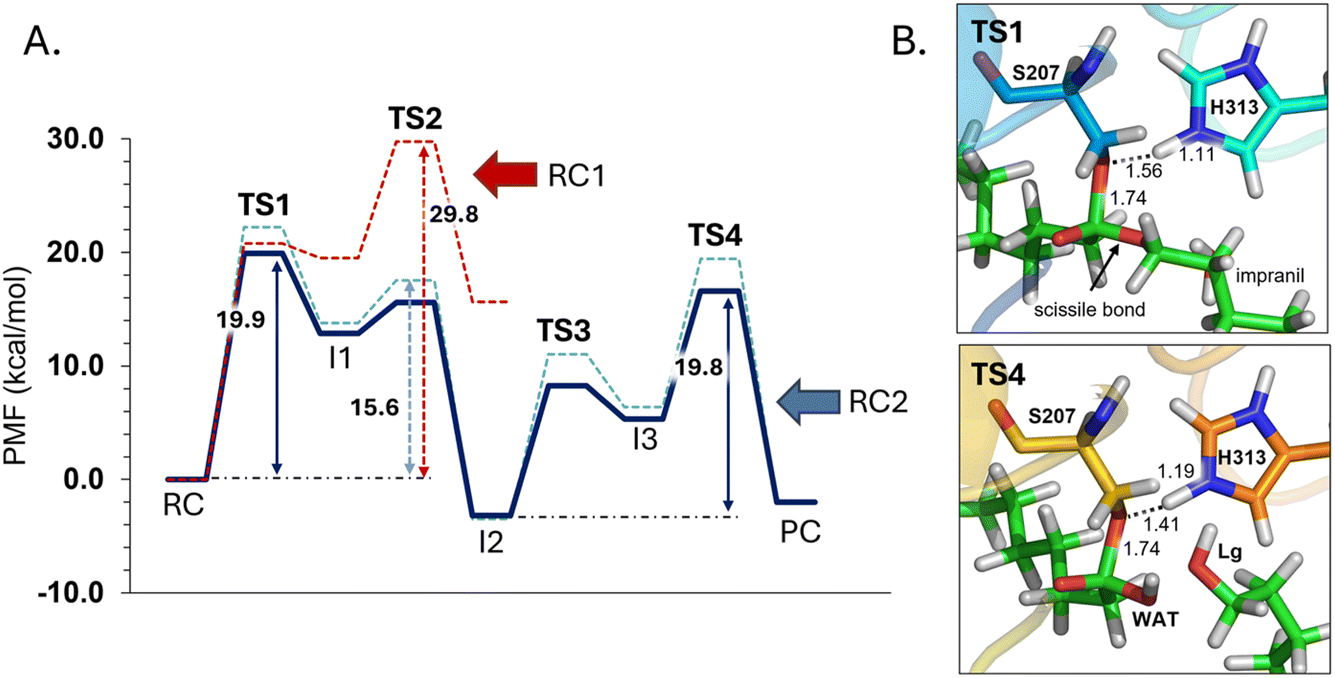 | ||
| Fig. 7 (A) M06-2X:AM1/AMBER free energy profile corresponding to acylation step of Impranil starting from RC1 (dashed red line) and the full depolymerization of the Impranil from the RC2 conformation (dashed blue line) derived from the corresponding FESs (Fig. S8† and 6, respectively). Dark blue line represents the free energy profile after zero-point vibrational corrections. (B) Detail of the M06-2X/AMBER optimized structures of the rate-limiting TS structures of the acylation and the deacylation process from the mechanism starting in RC2. Distances are given in Å. | ||
Conclusions
In the present work, we have investigated the hydrolysis of a model compound of polyester polyurethane, Impranil, catalyzed by polyurethane esterase A (PueA), which has been previously identified as the enzyme responsible for the degradation of this same sample by Pseudomonas chlororaphis.27 The unsolved 3D structure of the protein was generated by AlphaFold2 from the genome published by Stern and Howard.27 Analysis of the structure confirms the existence of a putative active site, composed of a Ser207-His313-Asp255 triad, in a solvent accessible pocket. This assembly of residues suggest that PueA could degrade this kind of substrate by means of a mechanism equivalent to those of other hydrolases,6,8–12 consisting of an acylation followed by a diacylation or hydrolysis step. No other Ser-His-Asp triad is identified in the generated 3D structure of PueA. Then, a docking of Impranil to the surface of PueA was carried out with Autodock Vina. The results showed how the putative reactive ester group of Impranil is positioned at good distance and orientation to the active site. However, the Impranil adopted a folded conformation inside the pocket, with intramolecular distances that suggest unfavourable interactions. In order to avoid this odd conformation, together with the fact that realistic substrates in future studies of PU biodegradation must be long polymeric chains, we manually extended the conformation of Impranil and searched for stable “unfolded” conformations. The results of unconstrained MD simulations confirm the stability of two possible orientations of the substrate in its extended form, depending on which terminal functional group (methyl carbamate or methyl ester) was exposed to the solvent or inside the cavity. Both possible non-covalent structures showed a promising orientation of the reactive ester group of Impranil in the surroundings of the reactive Ser207, His313 and Asp255. In addition, the carbonyl oxygen atom of the reactive ester group of Impranil occupied an oxyanion hole formed by the N–H backbone groups of residues Thr143 and Leu208, and the hydroxyl group of Thr143. However, analysis of protein:substrate interaction energies decomposed by residues reveals that the Impranil conformer with the carbamate end-group inside the cavity of the protein (RC2) better interacts with the protein than the inverse orientation (RC1).According to the free energy of activation for the acylation step, obtained by means of M06-2X:AM1/MM MD simulations, only one of the two possible orientations renders values that can be considered as compatible with a biocatalytic process (22 kcal mol−1). A deep analysis of the structures of the first tetrahedral intermediate, I1, confirmed that the high energy barriers obtained when the reaction was initiated with the Impranil conformer that exposes the carbamate end-group to the solvent (RC1) were associated with a non-reactive orientation of the protonated His313 and the ester oxygen atom of the substrate, which is the oxygen atom that should accept the proton to generate the first of the products (the alcohol compound). Consequently, the diacylation step was studied just for the most favourable conformation of the protein:substrate structure (RC2). The resulting free energy landscape confirms that the reaction can take place by a characteristic four-step path, involving an acylation followed by a diacylation step. The reaction would be kinetically controlled by the nucleophilic attack of the serine to the carbonyl carbon atom of the substrate, that takes place concomitantly with the activation of the serine by the nearby His313; first step of the acylation. The activation energy barrier appears to be slightly higher than those of known enzymatic processes. Thus, we predict a certain activity of wild-type PueA in degrading polyester PU samples, in agreement with the measurements of the growth rate of Bacillus subtilis vs. substrate concentration of Impranil reported by Stern and Howard that confirmed the capability of the microorganism to utilize this same sample of polyester polyurethane (Impranil) as its sole carbon and energy source.27 The obtained free energy of activation for the catalyzed process indicates that the polyurethanase activity of wild-type PueA is not high. Structural analysis of the evolution of the active site along the reaction progress together with the study of electrostatic effects generated by the protein can be performed in the future to improve the PUase efficiency of PueA.
Our results show that despite the studied activity of PueA has been just the polyester-hydrolase, because it is not attacking the carbamate group of the substrate but the ester group, this enzyme can be a promising protein scaffold for further developments in order to achieve an efficient biodegradation of PU, with future applications to address the environmental problems derived from generalized plastic consumption and disposal.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are very grateful to Prof. Fernando López-Gallego from the CIC biomaGUNE for discussions on the present study and reading the manuscript. This work was supported by the Spanish Ministerio de Ciencia, Innovación y Universidades (ref. PID2021-123332OB-C21), the Generalitat Valenciana (PROMETEO with ref. CIPROM/2021/079) and Universitat Jaume I (UJI-B2021-71). K. Ś. thanks the Ministerio de Ciencia e Innovación and Fondo Social Europeo for a Ramon y Cajal contract (ref. RYC2020-030596-I). The authors wish to thank the staff of the Servei d'Informàtica of the Universitat Jaume I, as well as the local computational resources funded by Generalitat Valenciana – European Regional Development Fund (REF: IDIFEDER/2021/02).References
- https://www.oecd.org/environment/plastic-pollution-is-growing-relentlessly-as-waste-management-and-recycling-fall-short.htm .
- S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, A bacterium that degrades and assimilates poly(ethylene terephthalate), Science, 2016, 351, 1196–1199, DOI:10.1126/science.aad6359.
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier and S. Gavalda, et al., An engineered PET depolymerase to break down and recycle plastic bottles, Nature, 2020, 580, 216–219, DOI:10.1038/s41586-020-2149-4.
- R. Wei, G. von Haugwitz, L. Pfaff, J. Mican, C. P. S. Badenhorst, W. Liu, G. Weber, H. P. Austin, D. Bednar, J. Damborsky and U. T. Bornscheuer, Mechanism-Based Design of Efficient PET Hydrolases, ACS Catal., 2022, 12, 3382–3396, DOI:10.1021/acscatal.1c05856.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Machine learning-aided engineering of hydrolases for PET depolymerization, Nature, 2022, 604, 662–667, DOI:10.1038/s41586-022-04599-z.
- S. Boneta, K. Arafet and V. Moliner, QM/MM Study of the Enzymatic Biodegradation Mechanism of Polyethylene Terephthalate, J. Chem. Inf. Model., 2021, 61, 3041–3051, DOI:10.1021/acs.jcim.1c00394.
- C. H. S. da Costa, et al., Assessment of the PETase Conformational Changes Induced by Poly(Ethylene Terephthalate) Binding, Proteins: Struct., Funct., Bioinf., 2021, 89, 1340–1352 CrossRef CAS PubMed.
- K. Świderek, S. Martí and V. Moliner, Theoretical Study of Primary Reaction of Pseudozyma antarctica Lipase B as the Starting Point to Understand Its Promiscuity, ACS Catal., 2014, 4, 426–434 CrossRef.
- M. À. Galmés, A. R. Nödling, L. Luk, K. Świderek and V. Moliner, Computational design of an amidase by combining the best electrostatic features of two promiscuous hydrolases, Chem. Sci., 2022, 13, 4779–4787 RSC.
- M. À. Galmés, E. García-Junceda, K. Świderek and V. Moliner, Exploring the Origin of Amidase Substrate Promiscuity in CALB by a Computational Approach, ACS Catal., 2020, 10, 1938–1946 CrossRef.
- M. À. Galmés, K. Świderek and V. Moliner, Computational Studies Suggest Promiscuous Candida antarctica Lipase B as an Environmentally Friendly Alternative for the Production of Epoxides, J. Chem. Inf. Model., 2021, 61, 3604–3614 CrossRef PubMed.
- M. À. Galmés, A. R. Nödling, L. Luk, K. Świderek and V. Moliner, Combined Theoretical and Experimental Study to Unravel the Differences in Promiscuous Amidase Activity of Two Nonhomologous Enzymes, ACS Catal., 2021, 11, 8635–8644 CrossRef PubMed.
- A. Kundys, E. Białecka-Florjańczyk, A. Fabiszewska and J. Małajo, Candida antarctica Lipase B as Catalyst for Cyclic Esters Synthesis, Their Polymerization and Degradation of Aliphatic Polyesters, J. Polym. Environ., 2018, 26, 396–407 CrossRef CAS.
- K. Świderek, S. Velasco-Lozano, M. À. Galmés, I. Olazabal, H. Sardon, F. López-Gallego and V. Moliner, Mechanistic studies of a lipase unveil effect of pH on hydrolysis products of small PET modules, Nat. Commun., 2023, 14, 3556, DOI:10.1038/s41467-023-39201-1.
- https://www.grandviewresearch.com/industry-analysis/polyurethane-pu-market .
- J. Liu, J. He, R. Xue, B. Xu, X. Qian, F. Xin, L. M. Blank, J. Zhou, R. Wei, W. Dong and M. Jiang, Biodegradation and up-cycling of polyurethanes: progress, challenges, and prospects, Biotechnol. Adv., 2021, 48, 107730, DOI:10.1016/j.biotechadv.2021.107730.
- J. R. Crabbe, J. R. Campbell, L. Thompson, S. L. Walz and W. W. Schultz, Biodegradation of a colloidal ester-based polyurethane by soil fungi, Int. Biodeterior. Biodegrad., 1994, 33, 103–113 CrossRef.
- T. Nakajima-Kambe, F. Onuma, N. Kimpara and T. Nakahara, Isolation and characterization of a bacterium which utilizes polyester polyurethane as a sole carbon and nitrogen source, FEMS Microbiol. Lett., 1995, 129, 39–42 CrossRef CAS PubMed.
- G. T. Howard and R. C. Blake, Growth of Pseudomonas fluorescens on a polyester-polyurethane and the purification and characterization of a polyurethanase-protease enzyme, Int. Biodeterior. Biodegrad., 1998, 42, 213–220 CrossRef CAS.
- G. T. Howard, C. Ruiz and N. P. Hilliard, Growth of Pseudomonas chlororaphis on a polyester-polyurethane and the purification and characterization of a polyurethanase-esterase enzyme, Int. Biodeterior. Biodegrad., 1999, 43, 7–12 CrossRef CAS.
- R. A. Pathirana and K. J. Seal, Studies on polyurethane deteriorating fungi. Part 1. Isolation and characterization of the test fungi employed, Int. Biodeterior., 1984, 20, 163–168 Search PubMed.
- L. Rowe and G. T. Howard, Growth of Bacillus subtilis on polyurethane and the purification and characterization of apolyurethanase-lipase enzyme, Int. Biodeterior. Biodegrad., 2002, 50, 33–40 CrossRef CAS.
- T. Tianyuan, T. Zhang, P. Liu, J. Bian, Y. Zheng, Y. Yuan, Q. Li, Q. Liang and Q. Qingsheng, Biodegradation of polyurethane by the microbial consortia enriched from landfill, Appl. Microbiol. Biotechnol., 2023, 107, 1983–1995 CrossRef PubMed.
- Y. Branson, S. Söltl, C. Buchmann, R. Wei, L. Schaffert, C. P. S. Badenhorst, L. Reisky, G. Jäger and U. T. Bornscheuer, Urethanases for the Enzymatic Hydrolysis of Low Molecular Weight Carbamates and the Recycling of Polyurethanes, Angew. Chem., Int. Ed., 2023, 62, e202216220 CrossRef CAS PubMed.
- N. Nomura, Y. Shigeno-Akutsu, T. Nakajima-Kambe and T. Nakahara, Cloning and sequence analysis of a polyurethane esterase of Comamonas acidovorans TB-35, J. Ferment. Bioeng., 1998, 86, 339–345 CrossRef CAS.
- C. Ruiz and G. T. Howard, Nucleotide sequencing of a polyurethanase gene (pulA) from Pseudomonas fluorescens, Int. Biodeterior. Biodegrad., 1999, 44, 127–131 CrossRef CAS.
- R. V. Stern and G. T. Howard, The polyester polyurethanase gene (PueA) from Pseudomonas chlororaphis encodes a lipase, FEMS Microbiol. Lett., 2000, 185, 163–168 CrossRef CAS PubMed.
- G. T. Howard, B. Crother and J. Vicknair, Cloning, nucleotide sequencing and characterization of a polyurethanase gene (pueB) from Pseudomonas chlororaphis, Int. Biodeterior. Biodegrad., 2001, 47, 141–149 CrossRef CAS.
- V. P. do Canto, C. E. Thompson and P. A. Netz, Polyurethanases: three-dimensional structures and molecular dynamics simulations of enzymes that degrade polyurethane, J. Mol. Graphics Modell., 2019, 89, 82–95 CrossRef CAS PubMed.
- A. Magnin, E. Pollet, V. Phalip and L. Averous, Evaluation of biological degradation of polyurethanes, Biotechnol. Adv., 2020, 39, 107457 CrossRef CAS PubMed.
- J. Jumper, R. Evans and A. Pritzel, et al., Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- (a) J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed; (b) O. Trott and A. J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- J. Gasteiger and M. Marsili, Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges, Tetrahedron, 1980, 36, 3219–3228 CrossRef CAS.
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, Open Babel: An open chemical toolbox, J. Cheminf., 2011, 3, 33 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and Testing of a General Amber Force Field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- A. Jakalian, D. B. Jack and C. I. Bayly, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation, J. Comput. Chem., 2002, 23, 1623–1641 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- (a) H. Li, A. D. Robertson and J. H. Jensen, Very fast empirical prediction and rationalization of protein pKa values, Proteins: Struct., Funct., Bioinf., 2005, 61, 704–721 CrossRef CAS PubMed; (b) M. H. M. Olsson, C. R. Søndergard, M. Rostkowski and J. H. J. Jensen, PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- C. James, et al., Scalable molecular dynamics on CPU and GPU architectures with NAMD, J. Chem. Phys., 2020, 153, 044130 CrossRef PubMed.
- Y. Zhang, H. Liu, S. Yang, R. Luo and H.-F. Chen, A Well-balanced Force Field ff03CMAP for Folded and Disordered Proteins, J. Chem. Theory Comput., 2019, 15, 6769–6780 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, Comparison of Simple Potential Functions for Simulating Liquid Water, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. S. Grest and K. Kremer, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3628–3631 CrossRef CAS PubMed.
- G. J. Martyna, D. J. Tobias and M. L. Klein, J. Chem. Phys., 1994, 101, 4177–4189 CrossRef CAS.
- M. J. Field, M. Albe, C. Bret, F. Proust-De Martin and A. Thomas, The Dynamo Library for Molecular Simulations Using Hybrid Quantum Mechanical and Molecular Mechanical Potentials, J. Comput. Chem., 2000, 21, 1088–1100 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, Development and Use of Quantum Mechanical Molecular Models. 76. AM1: A New General Purpose Quantum Mechanical Molecular Model, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Krzemińska, P. Paneth, V. Moliner and K. Świderek, Binding Isotope Effects as a Tool for Distinguishing Hydrophobic and Hydrophilic Binding Sites of HIV-1 RT, J. Phys. Chem. B, 2014, 119, 917–927 CrossRef PubMed.
- (a) B. Roux, The Calculation of the Potential of Mean Force Using Computer-Simulation, Comput. Phys. Commun., 1995, 91, 275–282 CrossRef CAS; (b) G. M. Torrie and J. P. Valleau, Non-Physical Sampling Distributions in Monte-Carlo Free-Energy Estimation – Umbrella Sampling, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. Swendsen and P. A. Kollman, The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- (a) J. C. Corchado, E. L. Coitiño, Y. Y. Chuang, P. L. Fast and D. G. Truhlar, Interpolated Variational Transition-State Theory by Mapping, J. Phys. Chem. A, 1998, 102, 2424–2438 CrossRef CAS; (b) K. A. Nguyen, I. Rossi and D. G. Truhlar, A Dual-Level Shepard Interpolation Method for Generating Potential Energy Surfaces for Dynamics Calculations, J. Chem. Phys., 1995, 103, 5522–5530 CrossRef; (c) Y. Y. Chuang, J. C. Corchado and D. G. Truhlar, Mapped Interpolation Scheme for Single-Point Energy Corrections in Reaction Rate Calculations and a Critical Evaluation of Dual-Level Reaction Path Dynamics Methods, J. Phys. Chem. A, 1999, 103, 1140–1149 CrossRef CAS.
- (a) J. J. Ruiz-Pernía, E. Silla, I. Tuñón, S. Martí and V. Moliner, Hybrid QM/MM Potentials of Mean Force with Interpolated Corrections, J. Phys. Chem. B, 2004, 108, 8427–8433 CrossRef; (b) J. J. Ruiz-Pernía, E. Silla, I. Tuñón and S. Martí, Hybrid Quantum Mechanics/Molecular Mechanics Simulations with Two-Dimensional Interpolated Corrections: Application to Enzymatic Processes, J. Phys. Chem. B, 2006, 110, 17663–17670 CrossRef PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- C. Angkawidjaja, D. You, H. Matsumura, K. Kuwahara, Y. Koga, K. Takano and S. Kanaya, Crystal structure of a family I.3 lipase from Pseudomonas sp. MIS38 in a closed conformation, FEBS Lett., 2007, 581, 5060–5064 CrossRef CAS PubMed.
- D. Eisenberg, E. Schwarz, M. Komaromy and R. Wall, Analysis of membrane and surface protein sequences with the hydrophobic moment plot, J. Mol. Biol., 1984, 179, 125–142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4fd00022f |
| This journal is © The Royal Society of Chemistry 2024 |