
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical depolymerisation of polylactic acid†
Sonja D.
Mürtz
 a,
Marcus S.
Lehnertz
a,
Justus
Kümper
a,
Eike
Häger
a,
Alexandra
Markus
a,
Tabea
Becker
b,
Sonja
Herres-Pawlis
bd and
Regina
Palkovits
*ac
a,
Marcus S.
Lehnertz
a,
Justus
Kümper
a,
Eike
Häger
a,
Alexandra
Markus
a,
Tabea
Becker
b,
Sonja
Herres-Pawlis
bd and
Regina
Palkovits
*ac
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen, Worringerweg 2, 52074 Aachen, Germany. E-mail: palkovits@itmc.rwth-aachen.de
bInstitute of Inorganic Chemistry, RWTH Aachen, Landoltweg 1a, 52074 Aachen, Germany
cInstitute for Sustainable Hydrogen Economy, Forschungszentrum Jülich, Am Brainenergy Park 4, 52428 Jülich, Germany
dBioeconomy Science Center (BioSC), Aachen, Germany
First published on 21st February 2024
Abstract
Herein, we present electrochemical depolymerisation as a promising new technique for chemical recycling of polylactic acid. Using platinum electrodes and a current density of 50 mA cm−2, a maximum yield of 87% lactic acid was obtained. Moreover, first mechanistic insights, the effect of the reaction conditions, and application to real waste streams are discussed in the following.
Plastics have revolutionized everyday life and are found in many products in various industries. They are lightweight, chemically stable, and easy to mold, making them useful in everything from healthcare and technology to construction and clothing.1 Therefore, production is increasing rapidly, 460 million tons were produced in 2019. However, only 9% of plastic waste was ultimately recycled demonstrating today's linear economy.2
Landfilling and uncontrolled dumpsites go hand in hand with immense pollution of the environment, particularly oceans.3,4 Microplastics are now found everywhere, even in the human body, posing a threat to the ecosystem and our health.5–7 It is therefore extremely important to reduce, reuse and recycle, as well as take responsibility throughout the entire life cycle of plastic products. In order to close the plastic loop and realize a holistic circular economy adhering to the principles of Green Chemistry,8 new approaches, methods, and resources are required for efficient and environmentally friendly recycling.9
In addition to contributing to pollution and waste, most plastics are also derived from fossil feedstocks. To defossilize the chemical sector, a shift to the use of biomass-based plastics is needed. Polylactic acid (PLA) has the highest market share among plastics made from renewable resources.10,11 It has a wide range of uses, including packaging materials12,13 up to biomedical applications.14 Life cycle analyses have shown that PLA has the potential to replace petroleum-based plastics such as PET.15,16 In addition, it is also biodegradable and thereby offers an intrinsic mistreatment protection in case of improper release into the enviroment.17,18 However, recycling is a more attractive option for addressing plastic waste, as it leads to recovery of the material value and preserves resources. Mechanical recycling is currently the most viable option, where plastics are ground and melted to be used in the manufacture of new products. However, single-variety polymer streams are required and the polymer quality decreases with the number of recycling runs.19 An alternative is chemical recycling breaking down plastic polymers into their monomers20,21 or new commodity chemicals according to the principle of open loop recycling.22,23 Exemplary for the chemical recycling is the depolymerisation of PLA to lactic acid (LA) shown in Scheme 1.
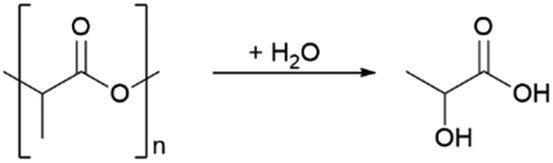 | ||
| Scheme 1 General reaction equation for depolymerisation of PLA to lactic acid. | ||
The driving force behind such a process is often elevated temperature, pressure, additives, or special solvents. Well-known methods for PLA are pyrolysis,24,25 acid or base hydrolysis,26–28 as well as chemo-catalysis with metal complexes29–33 or solid catalysts.34 A comparably high energy consumption, complex synthesis of the catalysts or high costs for downstream processes unify these methods. In addition to solvolytic, catalytic, and thermochemical approaches for the chemical recycling of polymers,35–37 it is also possible to break down polymers using electricity and a catalyst. However, electrochemical recycling is still relatively new and has received limited research. Shapoval and Pud published a series of papers on the electrochemical degradation of polytetrafluoroethylene (PTFE),38 polyethylene terephthalate (PET),39,40 polyvinyl chloride (PVC),41 and polycarbonate (PC).42 The carbon-chain of PTFE and PET was reported to break down during electrolysis when applied as a thin film on a cathode. This process, known as “surface electrochemical reactions and reducing degradation” (ECRD), may occur during the application of polymer coatings to cathodes or the use of polymer-modified electrodes.38,39 For the degradation of polycarbonates, a nucleophilic mechanism has been proposed leading to the production of bisphenol A and CO2.42
More recently, the focus has been on the oxidative depolymerisation of lignin. Butylated hydroxytoluene,43 carboxylic acids,44 as well as mixtures of vanillin45–47 with sinapic acid and guaiacol were produced in electrocatalytic processes. However, to the best of the authors’ knowledge, electrochemical recycling of bio-based plastics has not been reported yet.
In this study, we present a novel method for electrochemical recycling of PLA. Feasibility was demonstrated and underlined by mechanistic experiments. In addition, the effects of molecular weight, concentration, current density and electrode material were studied before using real waste such as cutlery and foil, all made of PLA. Beyond the scientific findings, we aim to expand the range of available recycling strategies and promote the use of waste streams as valuable feedstocks in a truly circular polymeric economy.
Feasibility and mechanistic insights
As a starting point for the electrochemical depolymerisation of PLA, a dioxane/water mixture (volumetric ratio: 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with lithium perchlorate as the electrolyte at 60 °C was identified as suitable reaction medium, enabling good solubility of the polymer, high conductivity, and potential for hydrolysis. More experimental details can be found in the ESI.† To demonstrate the practicality of the method from the outset, PLA from a commercially available cup (Mn = 48300 g mol−1) was used throughout the experiments. A galvanostatic electrolysis on platinum electrodes at a current density of 50 mA cm−2 led to a maximum yield of 87% using 1000 C (see Fig. 1a). In the blank experiments under the same conditions without using electricity no lactic acid was detected as no reaction took place. The yield is determined by high pressure liquid chromatography (HPLC) measurements and does not equal conversion because higher oligomers as well as gaseous products cannot be detected. The experiments had an estimated error range of ±3% obtained from a triple determination (more details in ESI†). Nuclear magnetic resonance (NMR) measurements show that decomposition of dioxane takes place as the only side reaction (see Fig. S3†). Hence, the selectivity is outstanding compared to other catalytic approaches where side products such as 1,2-propanediol or propionic acid occur.34 The yield increased linearly up to around 44% using 300 C, then the efficiency decreased as illustrated by the decreasing slope.
:1) with lithium perchlorate as the electrolyte at 60 °C was identified as suitable reaction medium, enabling good solubility of the polymer, high conductivity, and potential for hydrolysis. More experimental details can be found in the ESI.† To demonstrate the practicality of the method from the outset, PLA from a commercially available cup (Mn = 48300 g mol−1) was used throughout the experiments. A galvanostatic electrolysis on platinum electrodes at a current density of 50 mA cm−2 led to a maximum yield of 87% using 1000 C (see Fig. 1a). In the blank experiments under the same conditions without using electricity no lactic acid was detected as no reaction took place. The yield is determined by high pressure liquid chromatography (HPLC) measurements and does not equal conversion because higher oligomers as well as gaseous products cannot be detected. The experiments had an estimated error range of ±3% obtained from a triple determination (more details in ESI†). Nuclear magnetic resonance (NMR) measurements show that decomposition of dioxane takes place as the only side reaction (see Fig. S3†). Hence, the selectivity is outstanding compared to other catalytic approaches where side products such as 1,2-propanediol or propionic acid occur.34 The yield increased linearly up to around 44% using 300 C, then the efficiency decreased as illustrated by the decreasing slope.
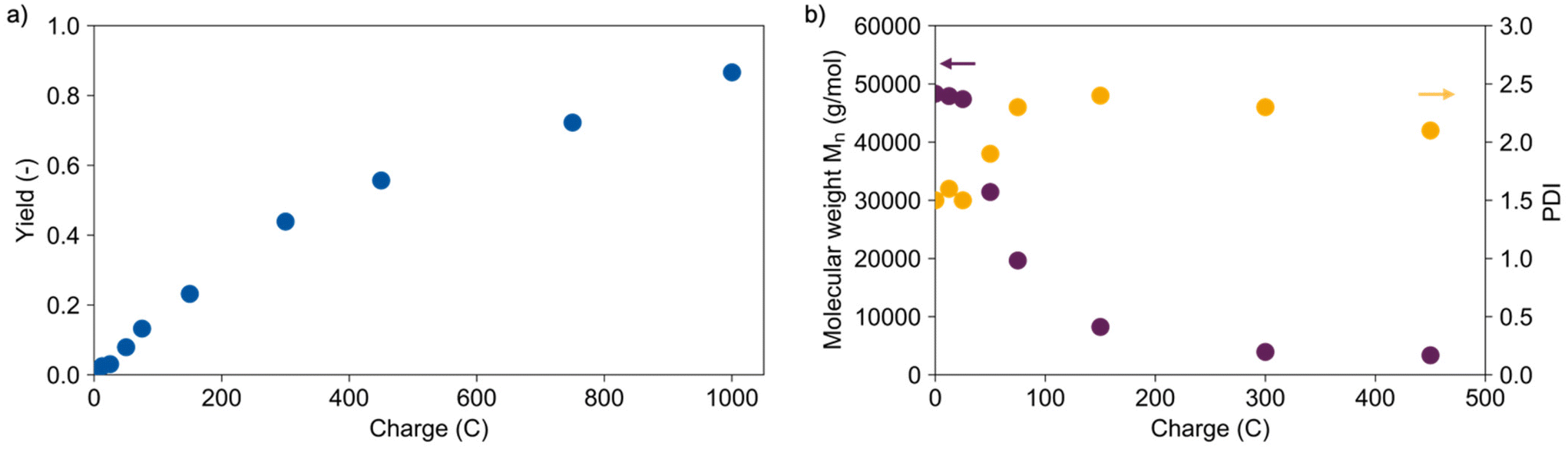 | ||
| Fig. 1 (a) Yield of lactic acid determined with HPLC over the transferred charge and (b) molecular weight and PDI of the precipitated polymer residues with increasing charge. Conditions: Pt cathode, Pt anode, 40 mA cm−2, dioxane/water (vol. 9:1), 0.25 M LiClO4, 60 °C, 5 mg mL−1 PLA (48300 g mol−1). | ||
The precipitated polymer residues were analysed with gel permeation chromatography (GPC) determining the number average molecular weights Mn and their distribution (see Fig. 1b). Short oligomers (Mn < 5000 g mol−1) are not included here. The molecular weight decreased exponentially with increasing transferred charge and yield, suggesting a random chain scission mechanism. This is supported by the fast increase of the polydispersity (PD) emphasizing a broad molecular weight distribution. Electrolysis in a divided cell showed the cathodic character of the depolymerisation. Two possible mechanisms for cathodic depolymerisation of PC were discussed by Pud et al.48,49 A single electron transfer was proposed to the carbonate group leading to decomposition into bisphenol A and CO2 as the anion-radical is extremely unstable.42,48 In parallel, degradation of PC is reported under the influence of negatively charged nucleophiles which are generated upon electrolysis of the supporting electrolyte (tetrabutylammonium perchlorate (TBAP) in DMF).49 Due to the similarities in polymer structure and reaction medium, similar molecular processes can be assumed for PLA in this reaction. Scheme 2 shows the two assumed depolymerisation pathways for PLA based on the mechanisms of PC.
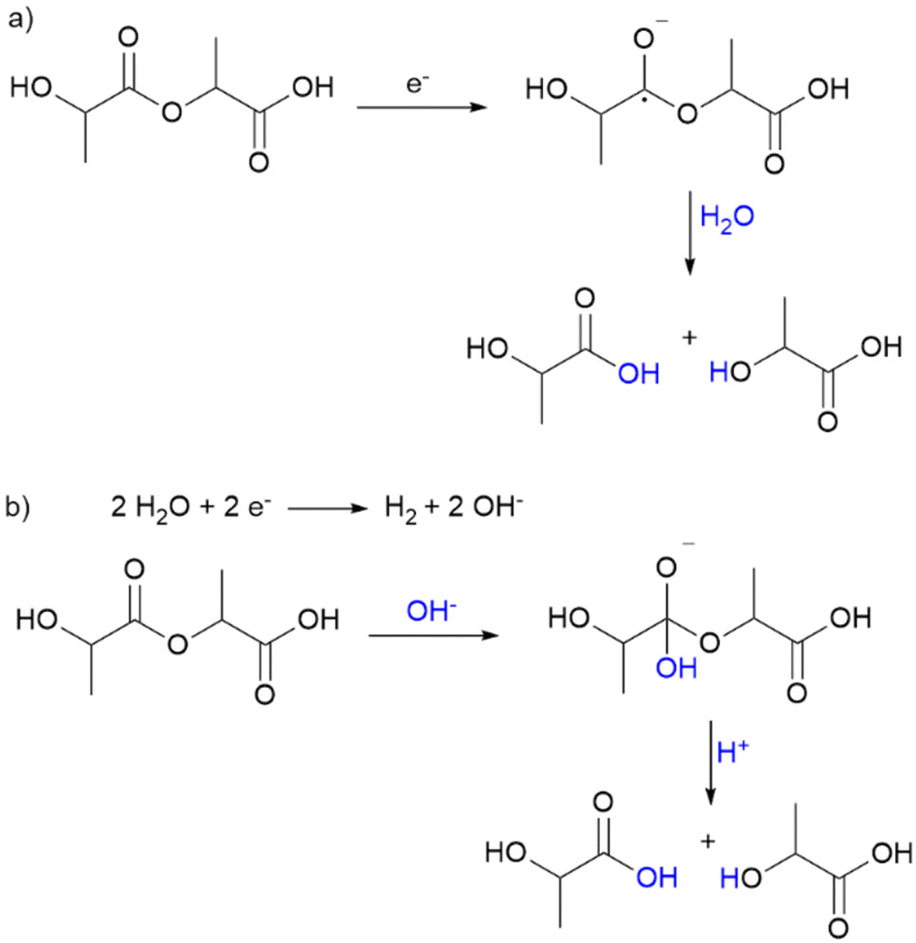 | ||
| Scheme 2 Proposed reaction pathways of electrochemical PLA depolymerisation according to (a) single electron transfer to carboxyl group and (b) hydroxyl ions from water splitting as nucleophiles. | ||
In the first pathway, a single electron is transferred from the cathode to the carboxyl group, resulting in cleavage of the C–O bond because the radical anion is unstable. In the second pathway, water splitting occurs as a competitive reaction, forming hydrogen and hydroxyl ions. These act as nucleophiles and lead to alkaline hydrolysis of the polymer. This hypothesis is supported by further experimental results (Fig. S4†), which show that lactic acid is not formed in a reaction solution without water but is formed in an alkaline solution without electricity. Preforming the electrolysis of PLA resulted in the same lactic acid formation as adding NaOH to the reaction mixture, but without performing electrolysis. The lactic acid yield was more than doubled when NaOH was added to the substrate mixture and electrolysis was performed, supporting the hypothesized mechanism of alkaline hydrolysis. Moreover, lactic acid is the only product which excludes other nucleophiles. Autocatalytic acid hydrolysis does not occur because the lactic acid yield does not increase further after interruption of charge transfer. However, further studies concerning the details of the depolymerisation mechanism of PLA are certainly required and will be addressed in future studies.
Influence of the reaction conditions
Reaction conditions were varied to gain a better understanding of the governing parameters and their impact on the depolymerisation. First, the PLA concentration was varied between 1.0 and 15 mg mL−1. The charge was adjusted accordingly to reach a comparable transferred charge per monomer unit, and a medium theoretical conversion was chosen for good comparability. For concentrations higher than 5 mg mL−1, a suspension was present due to the limited solubility of the polymer in the dioxane/water mixture. Fig. 2a shows an increase of the lactic acid yield from 22% for 1 mg mL−1 up to 36% for 5 mg mL−1 which suggests mass transfer limitations.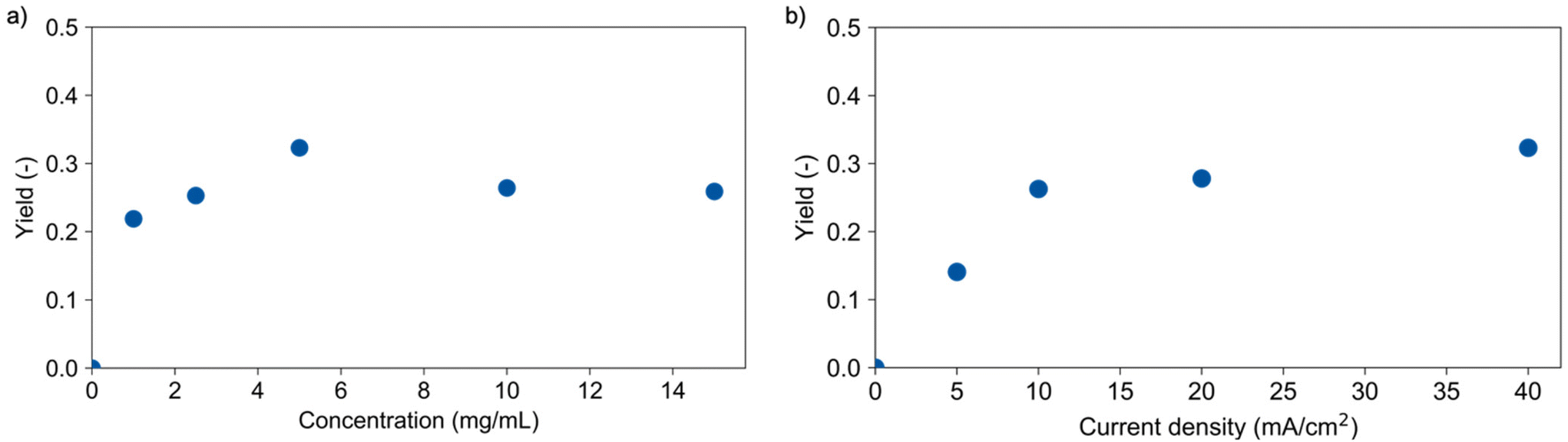 | ||
| Fig. 2 Yield of lactic acid determined with HPLC for (a) different concentrations (40 mA cm−2) and (b) different current densities (5 mg mL−1). Conditions: Pt cathode, Pt anode, dioxane/water (vol. 9:1), 0.25 M LiClO4, 60 °C, PLA (48300 g mol−1). | ||
Yields of about 26% for higher concentrations show that the electrolysis of a suspension is possible but is also accompanied by mass transfer challenges potentially causing the low influence of polymer concentration on yield. Polymer solubility, particle size as well as mixing and optimum electrode contact could be the limiting factors here.
Increasing the current density from 5 to 10 mA cm−2 led to a corresponding increase of the lactic acid yield from 14% to 28%, respectively. Here, the reaction is not limited by mass transfer and a linear increase of the reaction occurred. Further increase of the current density to 40 mA cm−2 led only to a slight increase to 36% lactic acid yield, again suggesting mass transfer limitations. This low correlation between yield and current densities suggests again the need for tailored reactor design. In this context, another important factor relates to the availability and costs of electrode materials. Titanium, graphite, and steel were tested as alternative cathode materials to replace expensive platinum. Fig. 3a shows the performance of these materials in the depolymerisation of PLA. A lactic acid yield of 32% was obtained using graphite and 28% with steel. The slightly lower performance of the graphite electrode compared to platinum (36%) may be due to its instability under the applied conditions, which is underlined by visible degradation of the electrode. Titanium yielded with 35% an equivalent amount of lactic acid as platinum and appeared therefore to be a cheap alternative cathode material for this depolymerisation reaction.
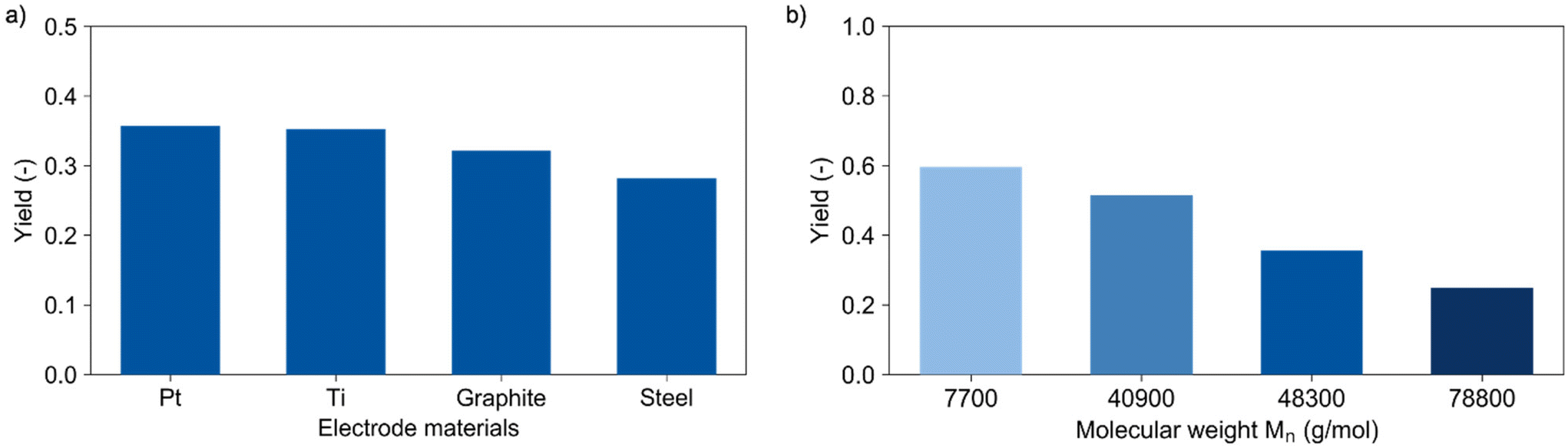 | ||
| Fig. 3 Yield of lactic acid determined with HPLC for (a) different cathode materials (Mn = 48300 g mol−1) and (b) different molecular weights (Pt cathode). Conditions: Pt anode, 40 mA cm−2, 150 C, dioxane/water (vol. 9:1), 0.25 M LiClO4, 60 °C, 5 mg mL−1 PLA. | ||
The strongest influence on the reaction was observed when varying the chain length of the polymer (Fig. 3b). Therefore, two samples with lower (Mn = 7700 and 40900 g mol−1) and one sample with higher molecular weight (Mn = 78800 g mol−1) compared to the plastic cup (Mn = 48300 g mol−1) were prepared as described in the ESI.† All materials were tested in a benchmark reaction under the same conditions without charge transfer for one hour. Formation of lactic acid was not observed for any of the samples. Using 150 C transferred to the solution within 50 min, the highest lactic acid yield of 60% was obtained for the polymer with the shortest chain-length. Extending the chain length by a factor of ten from a molecular weight of 7700 to 78800 g mol−1 resulted in approximate halving of the yield to 25%. This can be explained by a better accessibility of the short polymers due to a higher solubility and increased molecular mobility. However, one has to consider the current analytical limitations both for the experiments with varying PLA concentrations and the investigation concerning the impact of molecular weight. Indeed, the currently analytically available analysis of yield and molecular weight does not provide comprehensive insights concerning the number of chain scissions achieved under the different conditions, which, however, would be the required direct measure of reaction rate. Future studies will aim for appropriate reaction models.
Recycling of real waste
Regarding recycling of PLA waste, it must be considered that the materials may contain stabilizers, plasticizers, lubricants, and flame retardants. Therefore, real PLA waste materials were tested in the reaction. Fig. 4 demonstrates that electrolysis of real waste materials is indeed possible. Compared to the cup (Y(LA) = 35%, Mn = 48300 g mol−1), the yield of lactic acid from the cutlery (Mn = 41300 g mol−1) reached 30% and the one from the packaging foil (58300 g mol−1) 39%, respectively. The molecular weights of the residues after electrolysis obtained from GPC analyses were both 7000 g mol−1 similar to 8000 g mol−1 for the cup. This shows that additives such as colour pigments (cutlery) and plasticizers (packaging) may have a certain effect beyond the molecular weight correlation discussed previously. However, all results are in a comparable range concluding that the overall impact of additives on the reaction is low. This is an important finding for potential industrial applications. Nevertheless, long term studies concerning electrode stability in presence of the different compounds are certainly required towards technical feasibility of the presented technological approach.
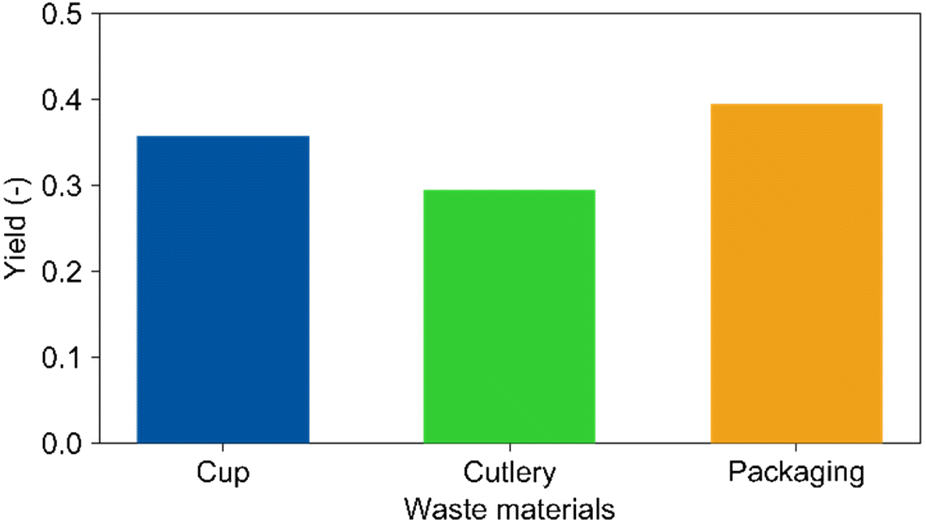 | ||
| Fig. 4 Yield of lactic acid determined with HPLC for the depolymerisation of real waste materials. Conditions: Pt cathode, Pt anode, 40 mA cm−2, 150 C, dioxane/water (vol. 9:1), 0.25 M LiClO4, 60 °C, 5 mg mL−1 PLA. | ||
Conclusions
In this study, we present the first approach of electrochemical depolymerisation of PLA. A maximum yield of lactic acid of 87% was obtained in the electrolysis on platinum electrodes in dioxane/water mixture (vol: 9:1) using a current density of 50 mA cm−2. First mechanistic insights were determined proposing a cathodic process with a random chain scission supported by GPC analyses. A combination of direct single electron-transfer to the polymer chain and formation of nucleophiles from the electrolyte attacking the carbonyl group was assumed based on literature and supported by experimental data. Moreover, the effect of the reaction conditions on the electrochemical depolymerisation of PLA was studied. Variation of the concentration showed that the reaction can be carried out using suspensions. In addition, titanium was found as a low-cost alternative to platinum as a cathode material, resulting in comparable yields. The highest impact was observed for the molecular weight. Extending the polymer chain by a factor of ten resulted in approximate halving of the yield. This was explained by a better accessibility of the short polymers due to a higher solubility and increased molecular mobility. Finally, the potential of this reaction to be applied to real waste materials such as plastic cutlery and packaging was demonstrated by obtaining similar yields regardless of the presence of additives like colour pigments, plasticizers, or stabilizers. Overall, this work introduces electrochemical depolymerisation of a renewable polymer as a promising new methodology to expand the options of chemical recycling and encourage the use of waste streams as valuable feedstocks in a closed or open loop circular polymer economy.
Data availability
Raw and meta data for this contribution are available at https://doi.org/10.5281/zenodo.10636333.Author contributions
Sonja Mürtz: Conceptualization, formal analysis, investigation, methodology, validation, visualization, writing – original draft. Marcus Lehnertz: Conceptualization, methodology, discussion, writing – review, and suggestions. Justus Kümper: Conceptualization, methodology, discussion, writing – review, and suggestions. Tabea Becker: Formal analysis, investigation, writing – review, and suggestions. Eike Häger: Investigation, validation. Alexandra Markus: Investigation, validation. Sonja Herres-Pawlis: Discussion, review. Regina Palkovits: Conceptualization, methodology, discussion, resources, supervision, funding acquisition, writing – review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Werner Siemens Foundation for funding in the frame of the WSS research prize. This work was also supported by the Cluster of Excellence Fuel Science Center (EXC 2186, ID: 390919832) funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities as well as the German Research Foundation within DFG PA 1689/17-1. The scientific activities of the Bioeconomy Science Center were financially supported by the Ministry of Culture and Science within the framework of the NRW Strategieprojekt BioSC (No. 313/323-400-002 13). The authors thank the analytical staff members Noah Avraham and Jens Heller for performing HPLC analysis.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Global Plastics Outlook, OECD, 2022 Search PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- M. Bergmann, S. Mützel, S. Primpke, M. B. Tekman, J. Trachsel and G. Gerdts, Sci. Adv., 2019, 5, eaax1157 CrossRef CAS PubMed.
- N. Ajith, S. Arumugam, S. Parthasarathy, S. Manupoori and S. Janakiraman, Environ. Sci. Pollut. Res., 2020, 27, 25970–25986 CrossRef PubMed.
- A. D. Vethaak and H. A. Leslie, Environ. Sci. Technol., 2016, 50, 6825–6826 CrossRef CAS PubMed.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, 1998 Search PubMed.
- V. G. Zuin and K. Kümmerer, Nat. Rev. Mater., 2022, 7, 76–78 CrossRef PubMed.
- Market – European Bioplastics e.V., https://www.european-bioplastics.org/market/, (accessed 2 January 2023).
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- A. J. R. Lasprilla, G. A. R. Martinez, B. H. Lunelli, A. L. Jardini and R. M. Filho, Biotechnol. Adv., 2012, 30, 321–328 CrossRef CAS PubMed.
- E. Rezvani Ghomi, F. Khosravi, A. Saedi Ardahaei, Y. Dai, R. E. Neisiany, F. Foroughi, M. Wu, O. Das and S. Ramakrishna, Polymers, 2021, 13, 1854 CrossRef PubMed.
- W. J. Groot and T. Borén, Int. J. Life Cycle Assess., 2010, 15, 970–984 CrossRef CAS.
- V. Rossi, N. Cleeve-Edwards, L. Lundquist, U. Schenker, C. Dubois, S. Humbert and O. Jolliet, J. Cleaner Prod., 2015, 86, 132–145 CrossRef.
- S. Spierling, C. Röttger, V. Venkatachalam, M. Mudersbach, C. Herrmann and H. J. Endres, Procedia CIRP, 2018, 69, 573–578 CrossRef.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- P. Mckeown and M. D. Jones, Sustainable Chem., 2020, 1, 1–22 CrossRef.
- A. R. Rahimi and J. M. Garciá, Nat. Rev. Chem., 2017, 1, 1–11 CrossRef.
- Y. Fan, C. Zhou and X. Zhu, Catal. Rev., 2009, 51, 293–324 CrossRef CAS.
- C. S. M. Pereira, V. M. T. M. Silva and A. E. Rodrigues, Green Chem., 2011, 13, 2658–2671 RSC.
- H. Zou, C. Yi, L. Wang, H. Liu and W. Xu, J. Therm. Anal. Calorim., 2009, 97, 929–935 CrossRef CAS.
- J. D. Badia, L. Santonja-Blasco, A. Martínez-Felipe and A. Ribes-Greus, Bioresour. Technol., 2012, 111, 468–475 CrossRef CAS PubMed.
- S. Lazzari, F. Codari, G. Storti, M. Morbidelli and D. Moscatelli, Polym. Degrad. Stab., 2014, 110, 80–90 CrossRef CAS.
- C. Y. Tham, Z. A. A. Hamid, Z. Ahmad and H. Ismail, Adv. Mater. Res., 2014, 970, 324–327 Search PubMed.
- T. Ivanova, I. Panaiotov, F. Boury, J. E. Proust, J. P. Benoit and R. Verger, Colloids Surf., B, 1997, 8, 217–225 CrossRef CAS.
- M. Fuchs, M. Walbeck, E. Jagla, A. Hoffmann and S. Herres-Pawlis, ChemPlusChem, 2022, 87, e202200029 CrossRef CAS PubMed.
- M. Fuchs, P. M. Schäfer, W. Wagner, I. Krumm, M. Walbeck, R. Dietrich, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2023, 16, e202300192 CrossRef CAS PubMed.
- T. O. Kindler, C. Alberti, E. Fedorenko, N. Santangelo and S. Enthaler, ChemistryOpen, 2020, 9, 401–404 CrossRef CAS PubMed.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, D. S. Reader, B. C. Dauphinais, S. P. McIlrath, A. A. Fischer, M. J. Carney and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC.
- M. S. Lehnertz, J. B. Mensah and R. Palkovits, Green Chem., 2022, 24, 3957–3963 RSC.
- A. L. Merchan, T. Fischöder, J. Hee, M. S. Lehnertz, O. Osterthun, S. Pielsticker, J. Schleier, T. Tiso, L. M. Blank, J. Klankermayer, R. Kneer, P. Quicker, G. Walther and R. Palkovits, Green Chem., 2022, 24, 9428–9449 RSC.
- A. Hermann, T. Becker, M. A. Schäfer, A. Hoffmann and S. Herres-Pawlis, ChemSusChem, 2022, 15, e202201075 CrossRef CAS PubMed.
- L. Burkart, A. Eith, A. Hoffmann and S. Herres-Pawlis, Chem. – Asian J., 2022, e202201195 Search PubMed.
- G. S. Shapoval, A. P. Tomilov and A. A. Pud, Teor. Eksp. Khim., 1984, 20, 247–249 CAS.
- A. A. Pud, Teor. Eksp. Khim., 1984, 20, 584–589 CAS.
- A. A. Pud, Polym. Degrad. Stab., 1990, 30, 181–193 CrossRef CAS.
- G. S. Shapoval, Èlektrohimiâ, 1994, 30, 686–689 CAS.
- A. A. Pud, S. P. Rogal'skii, G. S. Shapoval and V. G. Voloshchuk, Theor. Exp. Chem., 1998, 34, 292–296 CrossRef CAS.
- Y. M. Zhang, Y. Peng, X. L. Yin, Z. H. Liu and G. Li, J. Chem. Technol. Biotechnol., 2014, 89, 1954–1960 CrossRef CAS.
- D. Di Marino, T. Jestel, C. Marks, J. Viell, M. Blindert, S. M. A. Kriescher, A. C. Spiess and M. Wessling, ChemElectroChem, 2019, 6, 1434–1442 CrossRef CAS.
- M. Zirbes, D. Schmitt, N. Beiser, D. Pitton, T. Hoffmann and S. R. Waldvogel, ChemElectroChem, 2019, 6, 155–161 CrossRef CAS.
- D. Di Marino, D. Stöckmann, S. Kriescher, S. Stiefel and M. Wessling, Green Chem., 2016, 18, 6021–6028 RSC.
- N. Di Fidio, J. W. Timmermans, C. Antonetti, A. M. Raspolli Galletti, R. J. A. Gosselink, R. J. M. Bisselink and T. M. Slaghek, New J. Chem., 2021, 45, 9647–9657 RSC.
- A. A. Pud, S. P. Rogal'skii and G. S. Shapoval, Theor. Exp. Chem., 1998, 34, 348–351 CrossRef CAS.
- S. P. Rogal'skii, A. A. Pud, G. S. Shapoval, A. F. Mel'nik and M. T. Bryk, Teor. Eksp. Khim., 2004, 40, 29–33 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04234k |
| This journal is © The Royal Society of Chemistry 2024 |