
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing thermo-mechanochemistry of lignin enabled the production of high-quality low-cost carbon fiber†
Yixin
Luo
,
Moham Ed Abdur
Razzaq
,
Wangda
Qu
,
Abdulrahman A. B. A.
Mohammed
,
Alvina
Aui
,
Hamidreza
Zobeiri
,
Mark Mba
Wright
 ,
Xinwei
Wang
and
Xianglan
Bai
*
,
Xinwei
Wang
and
Xianglan
Bai
*
Department of Mechanical Engineering, Iowa State University, Ames, Iowa 50011, USA. E-mail: bxl9801@iastate.edu
First published on 7th February 2024
Abstract
Producing green carbon fiber from lignin is not only attractive to biorefineries for valorizing lignin but also contributes to the decarbonization of multiple industries. However, the difficulty of obtaining high-quality lignin-based carbon fibers at low cost remains the bottleneck and barrier to its commercial application. Previous approaches for chemically modifying lignin or blending with co-precursors were ineffective in delivering the carbon fiber with the desired quality. We discovered that introducing thermo-mechanochemistry of lignin to carbon fiber processing is a facile and green approach to overcome this challenge. Thermo-mechanochemistry can be introduced by properly integrating thermal heating and mechanical tension force applied to the spun fiber, which can manipulate the ordinary lignin chemistry to control the microstructure evolution from lignin. Under the influence of this newly known chemistry, lignin can transform into an oriented structure that can easily be graphitized at low temperatures. In this work, three precursors consisting of lignin or lignin blends were used to produce the carbon fibers with average tensile strength and modulus of 2.45 GPa and 236 GPa, 2.11 GPa and 215 GPa, 2.2 GPa and 225 GPa, respectively, using a surprisingly low carbonization temperature of only 700 °C. The preliminary techno-economic analysis suggests a low production cost. The discovery of the thermo-mechanochemistry of lignin will alter perceptions of lignin-based carbon fibers and provide opportunities to produce commercially viable low-cost green carbon fibers, advancing lignin valorization.
1. Introduction
Lignin is a byproduct abundantly available from paper industries and emerging biorefineries, with an estimated 225 million tons expected annually by 2030.1 Less than 2% of the paper industry-produced lignin is utilized for chemical production, with the rest being burned as boiler fuel. Accordingly, cost-effective lignin valorization at a market-size compatible strategy remains a significant challenge.1–3 Lignin-based carbon fiber (CF) offers a promising opportunity to valorize lignin.4–7 CF is a lightweight material with superior mechanical properties. It is popular in applications where high mechanical properties are available at light material weights, such as aerospace, automobile, wind turbine, construction, infrastructure, and sporting goods industries.4 For example, lightweight vehicles partly constructed with CF can improve fuel efficiency, contributing to decarbonization in the transportation sector.8 The CF composites market size continues to increase due to the unique advantages they can offer. According to market research, the CF and CF reinforced product market is projected to grow to $31.5 billion by 2025.9 Despite increasing demand, CF is mostly used in higher-end products due to its high costs. Over 60% of globally produced CFs are used in the aerospace and defense industries. Only 6.5% is used in the automotive industry, and 7% is used in wind turbines.10 About 82% of global CF is produced from polyacrylonitrile (PAN), whose price accounts for 51% of CF production cost.4–6 While commercial PAN-based CFs have tensile strength ranging from 3–7 GPa and a tensile modulus from 200 to 500 GPa, and the US Department of Energy (DOE) stated earlier that alternative precursor-based CFs with a tensile strength of 1.72 GPa and a modulus of 172 GPa, available at <$5–7 per lb could be widely used in automobile industries for the vehicle bodies and parts without compromising performance.11 Low-cost green CFs are also attractive to other industries (e.g., the construction industry, power industry, and mechanical engineering) for reducing costs and replacing non-renewable materials with renewable materials to lower carbon emissions.4Lignin has been extensively studied as an alternative precursor of CF because of its low costs, abundance, fixed carbon content, and absence of toxic gas emissions during its manufacturing process. However, the major bottleneck and barrier to developing commercially relevant lignin-based CFs are their poor tensile properties, far below the DOE target. So far, various lignins with different biomass origins isolated using different extraction methods have been studied. Other than directly spinning purified lignin, blending with other polymers or additives, or pre-modifying lignin using physical, chemical, and biological methods was also investigated.4–7,11–13 However, the resultant CFs mostly had tensile strengths below 1 GPa and tensile modulus below 100 GPa.6 A few previous CFs with relatively higher properties are the CF with a tensile strength of 1.07 GPa and modulus of 83 GPa produced using ultra-pure, experimental-grade lignin11 and the CF with a tensile strength of 1.39 GPa and modulus of 97 GPa obtained using solvent-fractionated and solvated kraft lignin that had ultra-high molecular weights.14 Previous studies indicated that the difficulties in producing quality CFs are mainly attributed to the intrinsic deficiencies of lignin as a precursor of CF.4–7 It was suggested that ideal precursors of CF must satisfy multiple requirements, such as high purity, high molecular weights, low polydispersity, and molecular orientation. Among them, the molecular linearity of a precursor is the most important factor in obtaining high-quality CFs.4,6,11 Usually, the tensile properties of a material are strongly correlated with the molecular orientation of the precursor.15 For example, PAN-based CFs are exceptionally strong due to the uniaxial orientation of polymer crystals along the fiber that could be graphitized into highly ordered structures. However, lignin is heterogeneous and intrinsically absent of molecular orientation.4–6 Lignin is a polyaromatic molecule in which three primary phenylpropane monomer units, coumaryl (H type), coniferyl (G type), and sinapyl alcohols (S type), are randomly crosslinked by C–O–C (β-O-4, α-O-4, and 4-O-5) or C–C (5–5, β-5 and β-1) bonds.16 Thus, lignin builds an amorphous three-dimensional network instead of the one-dimensional chain characteristic of most synthetic polymers. Furthermore, functional groups such as methoxyl, phenolic hydroxyl, aliphatic hydroxyl, benzyl alcohol, noncyclic benzyl ether, and carbonyl groups in lignin side chains contribute to the irregular, highly branched polymer structure. The lack of a defined molecular architecture severely limits the orientability of the drawn fibers, limiting their ability to graphitize upon carbonization.7 In previous studies, lignin with a higher content of aryl–ether linkage was prepared,17 or lignin was mixed with other well-defined linear long-chain polymers18–21 to increase precursor orientability. Unfortunately, these approaches still could not yield CFs with satisfactory tensile properties. Among the approaches, blending lignin with excessively high amounts of PAN and gel-spinning the solvent-doped mixed precursors at subzero temperatures followed by subsequent processing was most efficient in improving the CF properties.22 However, blending with PAN increases production costs and it is questionable if such CFs are still called lignin-based CFs. In summary, while lignin-based CFs were extensively investigated, the mechanical properties of previous CFs have been insufficient for use in structural applications. In this context, we previously used lignin bio-oil consisting of phenolic monomers and oligomers to synthesize acrylate polymers using a controlled radical polymerization technique.23,24 The lignin-derived linear thermoplastic polymer is further processed into CFs with a tensile strength of 1.7 GPa and tensile modulus of 182 GPa.24 These values are higher than previous results, emphasizing the importance of structural linearity in CF production.
While the previous studies mainly focused on modifying precursors, an overlooked aspect is the fiber fabrication process that transforms lignin into a CF structure. The fiber fabrication process of lignin-based CF is generally similar to that of PAN-based CF; both include fiber spinning, oxidative thermal stabilization, and carbonization. During the two-step thermal treatments, precursor fibers are chemically transformed to form carbonized materials. PAN is a linear one-dimensional long-chain polymer with repeated units. Since PAN does not melt before decomposing, PAN doped in a dissolving solvent is spun into a fiber form. The fiber is then rinsed in a water bath to remove the dissolving solvent. PAN has no fixed carbons and thus cannot be directly carbonized. Thus, spun fibers had to be first stabilized oxidatively to convert PAN into a new structure that can be carbonized. In the stabilization, PAN-spun fiber is heated in air or an oxidizing environment between 200–300 °C. When oxygen atoms enter its structure, the linear polymer chains of PAN are reorganized and cross-linked to form a nitrogen-containing cyclized ladder structure.4,25 Tension was also applied to reduce entangled polymer chains while oxidative transformation occurs. After stabilization, the fiber containing the oriented supramolecular structure is carbonized under the inert environment with temperatures above 1000 °C up to 1600 °C.25 During carbonization, the cyclized structure undergoes dehydrogenation and condensation to form turbostratic carbons oriented in the fiber direction. The turbostratic carbon structure, where many tetrahedral carbons crosslink the graphite-type carbon layers, is responsible for the high tensile strength of PAN-based CFs.25
The transformation of PAN during CF production was extensively studied and well understood.4 In comparison, much less is known about lignin transformation during its fiber fabrication. Lignin has completely different chemical structures and rheological properties than PAN. While it varies by individual lignin, lignin usually has much lower glass transition temperatures (Tg) than PAN. Thus, lignin with a sufficiently low Tg can be melt-spun at much lower costs than the solvent-based spinning of PAN fibers (e.g., about 1/10th of the cost).5 Unlike PAN, lignin contains oxygenated functional groups and thus can be carbonized. However, the lignin-spun fibers soften at elevated temperatures, causing the fiber to fuse or melt.11 Therefore, the fusible spun fibers must be converted to non-fusible fibers by stabilization. During stabilization, lignin-spun fibers were heated in the air at temperatures below 300 °C, where oxygen promotes crosslinking of lignin and thus increases the fiber Tg.11,26 Other than the fiber fusing, the as-spun fiber of lignin is also much weaker and significantly more brittle than the strong PAN-spun fiber. The weak spun fibers of lignin are attributed to low molecular weights, high dispersity, and lack of molecular orientation in lignin.6 Since as-spun fibers can break or melt, stabilization was usually performed without interruption by external mechanical force. Occasionally, minimal tension force was applied while the fibers softened to reduce fiber diameters.19,27 During the subsequent carbonization at 1000 °C or above, the stabilized fibers were further crosslinked and carbonized to remove non-carbon elements.6 Lignin-based CFs produced using the above method have amorphous carbon structures and randomly scattered nanocrystallites28 compared to the highly oriented turbostratic carbons found in PAN-based CFs, which explains the poor mechanical properties of lignin-based CFs.
As described above, the lack of repeated molecular units and orientation in lignin structures hindered the formation of graphene carbons and oriented structures desired for producing high-quality CFs. While lignin pretreatments or additives increased the production costs, such efforts were insufficient to overcome the tensile property issues of the CF. Lignin undergoes extensive chemical transformation and microstructural changes during the thermal treatments for producing CF. On the other hand, this also suggests that the fiber fabrication process can strongly affect the chemical reactions of lignin and microstructural evolution. However, this potential opportunity for altering lignin has been largely overlooked. Although chemical structures of raw lignins29 or oxidized lignin25 were analyzed in previous studies, such studies do not inform the chemical reactions nor the microstructural change dependent on the fiber processing conditions employed during actual CF production. In this work, we report a discovery of the thermo-mechanochemistry of lignin, which challenged the conventional understanding of lignin-based CF. Our findings show that a proper combination of thermal heating and mechanical tension force applied to lignin can introduce novel thermo-mechanochemistry, presenting us with an opportunity to manipulate the ordinary chemical reactions of lignin and control the microstructure evolution. Based on this approach, lignin can transform into high-quality CFs with graphitized structures and orientations without chemical treatments or additives but by applying heat and mechanical force. In the following, we will demonstrate CFs with unprecedented tensile properties produced solely from lignin and unexpectedly low carbonization temperatures for proof-of-concept. We will then track the changes in chemical and material structures of lignin at the different stages under different thermo-mechanical conditions to confirm the presence of thermo-mechanochemistry and elucidate its mechanisms for altering chemical reactions and controlling microstructural formation. Finally, we will present a cost analysis result to show the economic feasibility of lignin-based CFs.
2. Experimental
2.1. Lignin preparation
Softwood organosolv (OL) was provided by Attis, and softwood Indulin kraft lignin (KL) was obtained from Ingevity. As-received lignins were washed in 90 °C deionized water for 4 hours using a magnetic stirring bar to remove water-soluble impurities. The washed lignin was filtered and vacuum-dried overnight at 40 °C and kept in a sealed container before use.2.2. CF production method
Melt spinning of lignin precursors was performed using a bench-top twin-screw micro compounder (DACA Instruments, Santa Barbara, CA) with a nozzle size of 2 mm, and fibers were collected on a roller (DSM, Geleen, the Netherlands) at 100 m min−1. When OL was used, the lignin powder was directly injected into the extruder for fiber spinning. OL was melt-spinnable. However, KL cannot melt-spun because it did not soften before thermally decomposing. Thus, KL was blended with OL to prepare melt-spinnable precursors. The three precursors tested are 100% OL, 90/10 (w/w) OL/KL blend, and 80/20 (w/w) OL/KL blend. The blends containing higher than 20% of KL were difficult to melt spin. For preparing blended lignin precursors, OL and KL were first mixed in the extruder to make pellets. The mixing temperature was 185 °C for the 90/10 (w/w) OL/KL blend and 195 °C for the 80/20 (w/w) blend. The rotation speed was 150 rpm for both blends. The pellets were then re-injected into the extruder for fiber spinning. The extruder temperature and rotation speed were 155 °C and 100 rpm for OL, 195 °C and 100 rpm for 90/10 OL/KL, and 205 °C and 120 rpm for 80/20 OL/KL, respectively.Stabilization was performed using an Isotemp programable forced-draft furnace (Fisher Scientific; 12 × 6 inch dimension) using air. Before stabilization, the as-spun fiber (AF) bundle was cut into a length of 2 cm. Each end of the fiber bundle was held between two graphite strips using epoxy glue. The fibers and the graphite strip assembly were vertically hung inside the furnace. Tension was applied by attaching different amounts of weights to the bottom of the fiber assembly via a hook. The amount of tension stress applied to a single fiber was calculated by dividing the total gravity weight applied on the fibers (i.e., the sum of the mass of the fibers, the graphene strips, epoxy, hook, and the metal weights) by the cross-sectional area of the fiber bundles and the number of the fibers. The weights were adjusted multiple times during the stabilization. The cross-sectional areas of the fiber bundles were measured when the weights were adjusted to calculate the tensile stress of the fibers at the times of the adjustments. The heating and tension profiles employed during the stabilization are given in Fig. 1. In this work, the stabilized fibers (SF) produced under tension-applied conditions are indicated as “TSF”. For a comparison study, 100% OL-derived AF was also freely placed on an alumina boat and stabilized using the same heating profile given in Fig. 1 above for TSF but without applying tension. This non-tension applied SF is indicated as “NSF”.
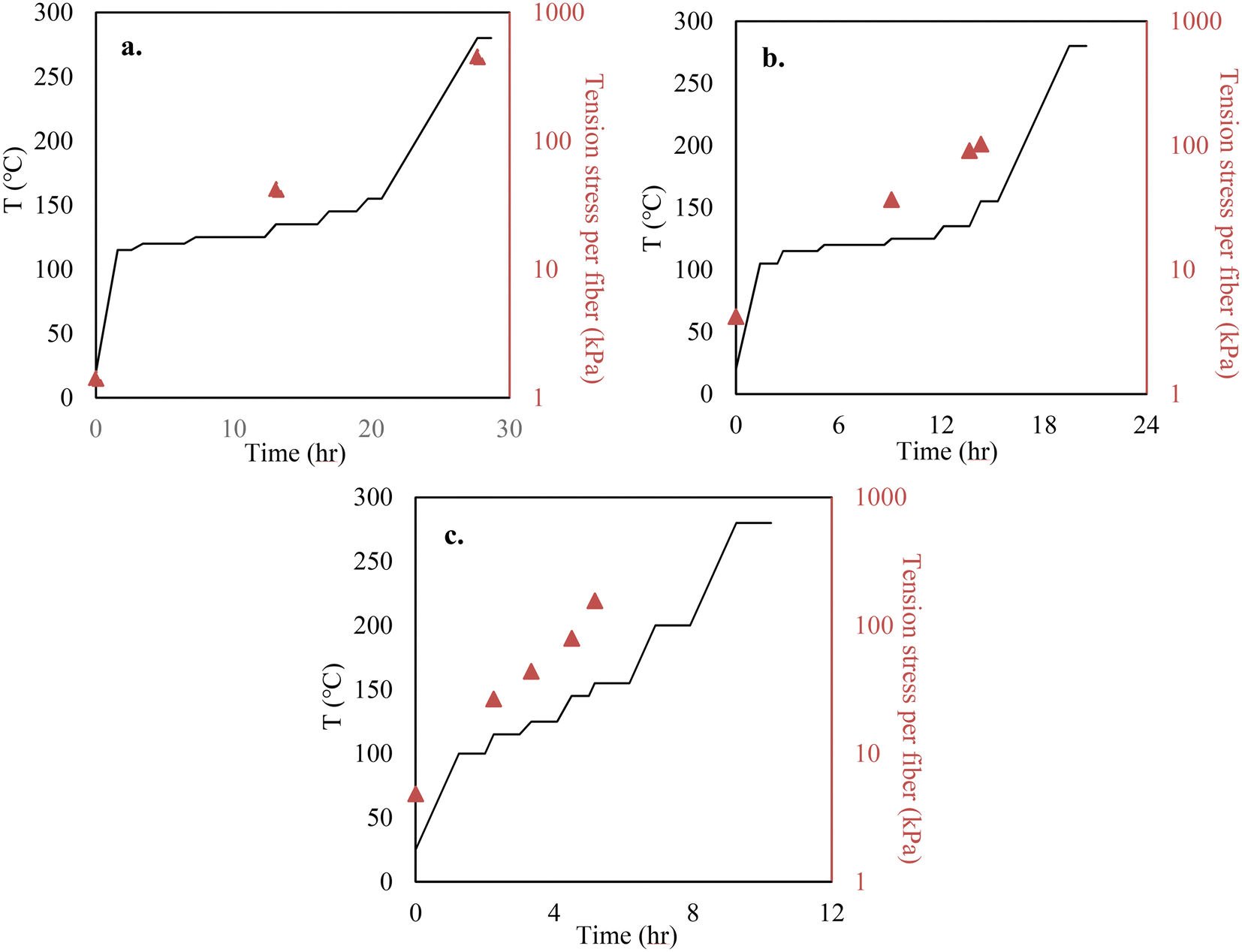 | ||
| Fig. 1 Heating profiles and tension adjustments of spun fibers during stabilization. (a) 100% OL; (b) 90/10 OL/KL; (c) 80/20 OL/KL. Orange triangles indicate when the weight loading attached to the fibers was adjusted. Weight loading remained constant until the next adjustment. The tension stress of fibers was measured only when the weight loading was adjusted. Due to decreasing fiber diameters during stabilization, tension stress per fiber is expected to increase even with the same weight loading. | ||
Carbonization of TSF and NSF was conducted in a tubular furnace (Lindberg Blue M, Thermo Scientific) purged by nitrogen gas. Before carbonization, the TSF bundle was placed onto a graphite sheet, and the ends of the fiber were fixed using a high-temperature glue (Ceramabond 503) to prevent fiber shrinkage. The fibers were carbonized from room temperature to 500 °C at 7 °C min−1, held at 500 °C for 5 min, from 500 °C to final temperatures at 2 °C min−1, and then kept for an hour. The final temperature was between 500 and 1000 °C for OL-based fibers, 700 and 800 °C for 90/10 OL/KL-based fibers, and 700 °C for 80/20 OL/KL lignin. The resulting CF prepared in the presence of tension during both stabilization and carbonization is indicated as “TCF”. For a comparison study, 100% OL-based NSF or TSF were also freely placed on an alumina boat for carbonization. The fibers were carbonized using the same heating profile used to produce TCF. The resultant CFs are indicated as “NCF” (i.e., the CF based on NSF) and “PCF” (the CF based on TSF), respectively. “PCF” indicates a partially-tension applied CF because tension was applied during stabilization but not carbonization. The carbonization temperatures were also included in fiber abbreviations. For example, TCFs obtained using the carbonization temperatures of 700, 800, 900, and 1000 °C are indicated as TCF700, TCF800, TCF900, and TCF1000, respectively. The fiber abbreviations and the corresponding processing conditions of the fibers are summarized in Fig. 2.
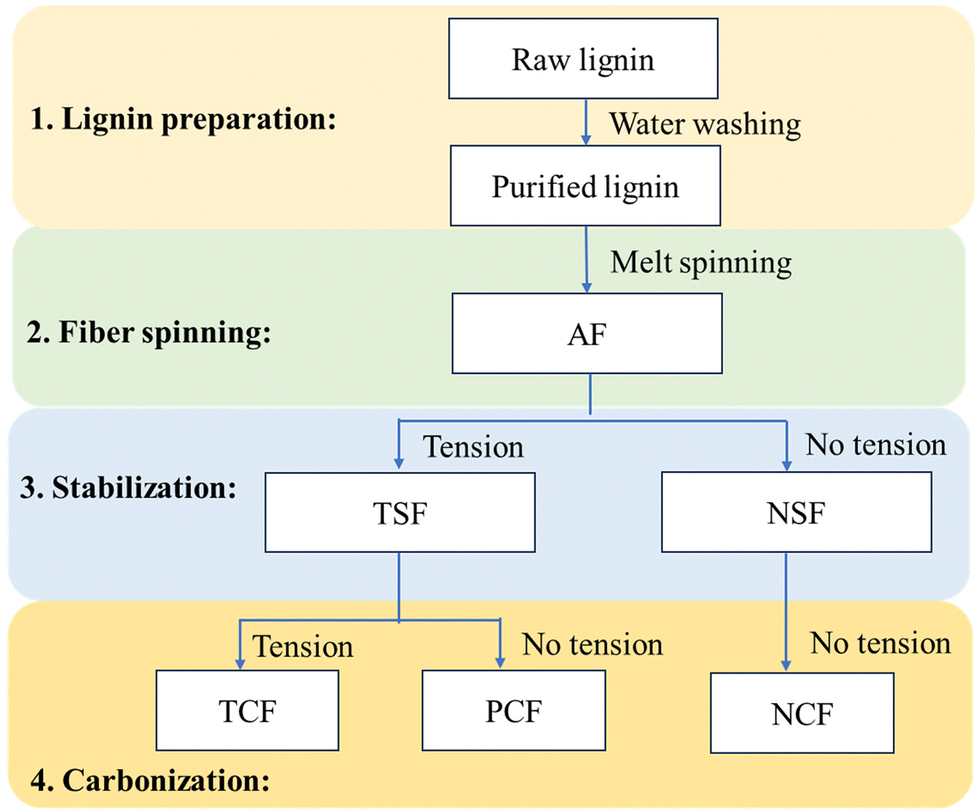 | ||
| Fig. 2 Preparations of stabilized fibers and carbon fibers under different tension conditions. | ||
2.3. Characterization methods
Gel Permeation Chromatography (GPC) analysis was conducted to determine the molecular weight distribution of lignin. A Dionex Ultimate 3000 series high-performance liquid chromatograph (HPLC) was equipped with a Shodex Refractive Index (RI) and Diode Array Detectors (DAD). Two Agilent PLgel 3 μm 100A0 300 × 7.5 mm (p/n PL1110–6320) columns were connected in series and maintained at 25 °C. An ultraviolet wavelength of 254 nm was used to detect the peaks. A GPC column was calibrated with six monodisperse polystyrene standards ranging from 162 to 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 120 g mol−1. Tetrahydrofuran (THF), with a flow rate of 1 mL min−1, was used as solvent and eluent in the column. The lignin samples were acetylated before the GPC analysis to increase their solubility using a previous method.30 Briefly, 100 mg of lignin was placed in a glass vial, and 3 mL of pyridine and 3 mL of acetic anhydride were added. The mixture was heated in an oil bath at 80 °C for three hours under continuous agitation. After the reaction, the solution was added to 500 mL of deionized water to precipitate the acetylated lignin. Precipitated lignin was filtered and washed with deionized water three times to remove the unreacted pyridine and acetic anhydride.
120 g mol−1. Tetrahydrofuran (THF), with a flow rate of 1 mL min−1, was used as solvent and eluent in the column. The lignin samples were acetylated before the GPC analysis to increase their solubility using a previous method.30 Briefly, 100 mg of lignin was placed in a glass vial, and 3 mL of pyridine and 3 mL of acetic anhydride were added. The mixture was heated in an oil bath at 80 °C for three hours under continuous agitation. After the reaction, the solution was added to 500 mL of deionized water to precipitate the acetylated lignin. Precipitated lignin was filtered and washed with deionized water three times to remove the unreacted pyridine and acetic anhydride.
Proximate analysis of lignin was performed using a thermal gravimetric analyzer (TGA/DSC 1 STARe system, Mettler Toledo). About 20 mg of vacuum-dried lignin was pyrolyzed at 10 °C min−1 to 900 °C using nitrogen with a flow rate of 100 mL min−1. After staying at 900 °C for 30 min, the air was introduced for combustion.
The glass transition temperature of lignin was determined using a DSC (Q2000, TA instruments). The sample was first rapidly heated to 200 °C and then cooled to 25 °C to eliminate thermal history. The sample was reheated to 200 °C at a heating rate of 10 °C min−1 to determine Tg.
Tensile properties of fibers were measured using a Discovery hybrid rheometer (DHR-2, TA Instruments) with dynamic mechanical analysis clamps by following an ASTM standard (ASTM C1557-03) based on single filament testing protocol. Data was acquired with a 0.001 s interval during the test. The fiber diameter was determined using a calibrated Leica LED inverted laboratory microscope with a 20× magnification lens with a resolution of 1 pixel. Each fiber diameter was the average of the measurements at five different locations along the fiber axial direction. The results of an average of 20 fibers were reported with a 95% confidence interval.
Scanning Electron Microscopy (SEM) analysis of CF was performed using FEI Inspect F50 Scanning Electron Microscope.
Transmission Electron Microscopy (TEM) analysis of CF was performed using a JEOL 2100 scanning TEM (Japan Electron Optics Laboratories) with a Gatan OneView 4K camera (Gatan, Inc.) and an operating voltage of 200 kV. ImageJ was used to determine crystallite size.
For preparing samples for heteronuclear-single-quantum-coherence nuclear magnetic resonance (HSQC-NMR) analysis, 100 mg of lignin or lignin-based fibers were dissolved in 1 mL of 4:1 (v/v) mixture of dimethyl sulfoxide (DMSO)-d6 and pyridine-d5 and sonicated for 2 hours. HSQC spectroscopies were obtained on a Bruker Biospin Advance 600 MHz spectrometer incorporated with a 5 mm cryogenically cooled z-gradient probe using the Bruker pulse sequence “hsqcetg-psisp.2”. The data processing was carried out using MestReNova v12.0.1 software.
Fourier Transform Infrared (FTIR) analysis was conducted using a Thermo Scientific Nicolet iS10 (Thermo Fisher Scientific Inc., Waltham, MA) with a Smart iTR accessory. The wave numbers of the FTIR analysis ranged from 750 to 4000 cm−1. Each sample was scanned 32 times at a resolution of 4 cm−1 and an interval of 1 cm−1.
Fast pyrolysis was performed using a Frontier micropyrolyzer system with an auto-shot sampler (Rx-3050 TR, Frontier Laboratories, Japan) and a single-stage furnace oven. Before pyrolysis, fibers were kept in a sealed container. During pyrolysis, approximately 0.5 mg of the sample in a deactivated stainless cup was dropped into a furnace preheated to 550 °C. Pyrolysis vapor products were directly swept into an online GC/MS. Helium was used as both the pyrolysis and carrier gas. An Agilent 7890B GC equipped with Agilent 5977A mass-selective detector (MSD), flame ionization detector (FID), and thermal conductivity analyzer (TCD) was used. The capillary column used in the GC was a ZB-1701 (60 m × 250 mm × 0.25 mm). The injection temperature at the GC was 250 °C. The oven temperature was kept at 40 °C for 3 min and then ramped to 280 °C at 3 °C min−1. Finally, the oven was held at 280 °C for another 4 minutes. The GC inlet temperature was maintained at 280 °C. The helium gas flow rate was 1 mL min−1, and the split ratio at the GC inlet was 20 to 1.
The elemental composition of fibers was determined using an elemental analyzer (Vario Micro Cube, Elementar, Germany). Carbon, hydrogen, nitrogen, and sulfur contents were measured, and oxygen content was determined by mass difference. The ash content of lignin was subtracted before calculating the elemental compositions.
Raman analysis of fibers was conducted using a confocal Raman system (Voyage, B&W Tek, Inc., Olympus BX51). A 532 nm Raman laser of 16 mW was focused on the fiber with a 503 lens. The measurement was conducted in two different views of the fibers. A 20 s integration time was used to obtain the spectrum, and Origin software was used to analyze the acquired Raman spectra with Gaussian curve fitting.
The crystallographic analysis of CF was conducted using an XRD (Siemens D500 diffractometer) using copper Kα radiation (λ = 1.5432 Å) with an accelerating voltage of 45 kV and current of 30 mA within the range of 2θ from 10° to 70° and a dwell time of 2 s. The fiber bundles were used, and equatorial scans were performed perpendicularly and parallel to the fiber axis.
A Xenocs Xuess 2.0 system with Cu (1.5406 Å) radiation was used to collect XPS data of CF. In the XPS spectra, C1 spectra were deconvoluted into four Gaussian peaks: –C–C–, –C–H– at 284.8 eV, –C–O– at 286.3 eV, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 288.0 eV, and –O–CO at 289.1 eV.
O at 288.0 eV, and –O–CO at 289.1 eV.
A Xenocs Xuess 2.0 system with Cu (1.5406 Å) radiation was used to collect Small-angle X-ray Scattering (SAXS) data.
2.4. Techno-economic analysis (TEA) modeling
The TEA was based on the study by Ellringman et al.12 and validated based on the U.S. DOE 2021 Carbon Fiber report.31 The base case facility produces 10000 metric tonnes (MT) of CF per year from lignin using a solvent-free melt-spinning process.
Annualized capital costs for the facility were estimated using the formula:
| Capital = DF × investment × area |
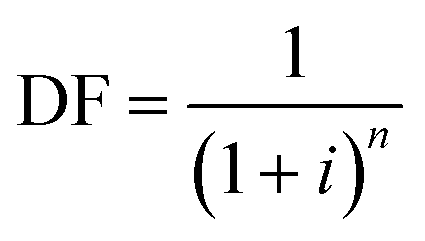
The investment factor is the cost per m2 of the facility, which is $1386 per m2 based on ref. 12. The area was calculated from the number of manufacturing lines required. A typical manufacturing line can process 1500 metric tonnes of CF per year, and each manufacturing line occupies 8650 m2. The manufacturing line cost was scaled up from 1.5 to 10 hours of stabilization time using a 0.7 economies-of-scale factor. Other capital-related costs include maintenance, insurance, and property tax, calculated as 3%, 0.5%, and 1% of the annual capital cost. CF capital cost can vary significantly due to several development factors, such as equipment type, stabilization time, and facility capacity. A DOE facility survey found that capital costs for commercial facilities varied between $110000 and $400000 when scaled to a common capacity of 10000 MT of CF per year.
Operating costs include material and utility costs. The main material is lignin. Lignin is available from different commercial sources, including pulp and paper mills and lignocellulosic biorefineries. Many factors, including the quality and condition of the lignin, and alternative markets, can influence the price of lignin. According to the DOE national laboratories, lignin prices range between $44 and $176 per MT. For this solvent-free process, the cost of chemicals is assumed to be negligible. Utility costs were estimated at 28.69 kW h kg−1 of CF with an electricity price of 0.0692. The electricity consumption was adjusted based on the CF conversion efficiency with a baseline of 0.476 corresponding to PAN to CF.
3. Results and discussion
3.1. TCF production using a thermo-mechanically controlled fabrication method
The mass recovery after water-washing was 95% for OL and 98% for KL. The characterizations of the purified lignins are given in Table S1 of the ESI.† The Tg of the lignin and lignin blends were 74 °C, 99 °C, and 106 °C for 100% OL, 90/10 (w/w) OL/KL blend, and 80/20 (w/w) OL/KL blend, increased with increased SKL content in the blends.Heating rates and tension applied to AF were controlled simultaneously to produce TSF. For all AFs, their viscosity decreased rapidly once the softening points of lignin were reached. Therefore, lower heating rates were used near their softening temperatures to prevent fiber fusing. Applying tension to the softened fiber at this stage can significantly reduce fiber diameters. However, AFs at the initial stage of the stabilization were very weak and could easily break if the tension force exceeded the limit the fibers could bear. The fibers became less fusible as the stabilization progressed. Thus, heating rates and tension loading were adjusted multiple times during the stabilization. Higher heating rates were used to reduce the stabilization time without causing fiber fusing. Additionally, the mechanical strength of the hardening fiber also increased as stabilization progressed. Thus, the weights attached to the fiber bundles were periodically added to ensure the fibers were stretched using as high a tension force as the fiber could bear without breaking. Increasing tension loading was also needed, otherwise, the fibers either stopped stretching too early or shrunk back if tension loading was insufficient.
The heating and tension profiles in Fig. 1 above differed for individual precursors. Higher heating rates were applied to a higher Tg precursor-based fiber containing more SKL because it was less fusible and hardened faster at lower stabilization temperatures than a lower Tg precursor-based fiber containing less or no SKL. This is because SKL has a stronger crosslinking tendency than OL. Since the fibers hardened faster for the high Tg precursor, the tension loading was increased sooner from lower stabilization temperatures to ensure the fibers were always under strong tension force and continued to stretch. It was important to tailor the tension conditions for individual precursor fibers. Different lignin precursors had different Tgs and reactivity due to their different chemical structures. Therefore, viscosity and crosslinking degree with increasing temperature varied for different lignins. These characteristics of different lignins also lead to individual precursor fibers undergoing stabilization to demonstrate different degrees of tolerance for tension stretching. To effectively stretch fibers, the fiber had to be pulled using as strong force as possible within the limit that the applied tension did not break the fiber. Since the precursor properties determine this limit, the tension profile must be tailored for individual precursors. For example, applying the tension profile suitable for an OL/KL blended precursor to 100% OL precursor can break the fibers because the OL precursor fiber was much weaker and hardened more slowly than the KL/OL blend-based fiber due to the lower reactivity of OL than KL. On the other hand, applying the tension profile suitable for 100% OL precursor fiber to the OL/KL blended precursor fiber led to a thicker fiber after stabilization because the tension force was insufficient to stretch the more rigid and faster-hardening fiber.
In this work, the fiber diameters were measured when the tension loading was adjusted. Thus, Fig. 1 above only reports the tension stresses of individual fibers at the adjustments. Due to decreasing fiber diameters during stabilization, the tension stresses applied to the fibers are expected to increase although the tension loading on the fiber bundles stays constant until the next adjustment. Compared to the methods used in previous studies where tension was either not applied or a fixed amount of tension was applied to stretch softened fibers, the tension and heating profiles were carefully integrated into each other in our method to ensure that the heated lignin-based fiber does not fuse while it was also stretched using near the maximum tension forces that its structure can bear. At the end of stabilization, not only did the length of the fiber stretch by around 10 times (Fig. S1†), but the stabilized fibers could also tolerate sufficiently high-tension loading, strikingly different from the weak AFs.
During the subsequent carbonization process, the TSF exhibited a strong tendency to shrink in the axial direction when the carbonization temperature exceeds ∼500 °C. Such a phenomenon was not seen with previously produced lignin-based CFs. The fiber shrinkage was prevented mechanically by placing the SFs on a graphite sheet base and fixing the two ends of the fiber bundles using a high-temperature adhesive. This way, external tension force created by the adhesive and graphite sheet can overcome fiber shrinkage. In previous methods, SFs were either carbonized to the final temperature of 1000 °C or above using a single heating rate or stepwise heating profiles with increasing heating rates at higher temperature regions.6 Our method differs from the previous methods as stepwise heating with a lower heating rate at higher temperatures was employed and most fibers were carbonized at final temperatures below 1000 °C. Higher heating rates can lead to greater fiber shrinkage to the TSF stabilized under tension stretching. As shown in Fig. S2,† the OL-based TSF shrunk so severely when it was carbonized using a single heating rate of 7 °C min−1 to 1000 °C, so that the shrinkage even caused the graphene sheet holder to bend along the fiber direction and broke fibers. The excessive shrinkage of the fibers was avoided in this study by lowering the heating rate at higher temperatures. Further details about the preparation and fabrication of the fibers can be found in the Experimental section.
The yields of TCFs were 48–50% for lignin and blended lignins. The microscopic images of representative CFs are shown in Fig. S3.† The fibers with diameters of a few micrometers had smooth surfaces and dense cores. The tensile properties of TCFs compared to previous CFs are listed in Table 1. The average tensile strength and modulus of the 100% OL-based TCF600 produced at a carbonization temperature of 600 °C were 1.06 GPa and 84 GPa and increased significantly to 2.45 GPa and 236 GPa for TCF700 by increasing the final carbonization temperature to 700 °C. On the other hand, the average fiber diameter decreased from 6.1 μm to 3.5 μm in the corresponding fibers. Interestingly, further increasing carbonization temperatures increased fiber diameters and decreased the tensile properties. The tensile strength and modulus of TCF800, TCF900, and TCF1000 were 2.12 GPa and 189 GPa, 1.81 GPa and 178 GPa, and 1.27 GPa and 130 GPa, respectively, and the average fiber diameters were 4.3, 4.4, and 5.9 μm. The increased CF diameters were due to the increased fiber shrinkage at higher carbonization temperatures. For 90/10 OL-based TCFs, the tensile properties were 2.11 GPa and 215 GPa for the carbonization temperature of 700 °C, 2.08 GPa and 194.1 GPa for 750 °C, and 1.94 GPa and 175.8 GPa for 800 °C. respectively. The tensile properties and fiber diameters of 90/10 OL/KL blend-based TCFs had the same temperature dependencies observed with 100% OL-based TCFs. Thus, 80/20 OL/KL blend-based fiber was carbonized using the optimal temperature of 700 °C. The resultant TCF700 had tensile properties of 2.2 GPa and 225 GPa. Representative strain-stress curves of the TCF700s are shown in Fig. S4.† Compared to previous studies, this work obtained CFs with unparalled tensile properties using unprecedently low carbonization temperatures.
| Precursors | Fiber spinning method | Carbonization temperature ( °C) | Tensile strength (MPa) | Tensile modulus (GPa) | Fiber diameter (μm) | Ref. |
|---|---|---|---|---|---|---|
| Lignin or lignin blends | ||||||
| Softwood organosolv lignin | Melt | 600 | 1060 | 84 | 6.1 | This work |
| Melt | 700 | 2456 | 236 | 3.5 | This work | |
| Melt | 800 | 2122 | 189 | 4.3 | This work | |
| Melt | 900 | 1809 | 178 | 4.4 | This work | |
| Melt | 1000 | 1272 | 130 | 5.9 | This work | |
| Softwood organosolv lignin and kraft lignin (90/10) | Melt | 700 | 2110 | 215 | 3.3 | This work |
| Melt | 750 | 2082 | 198 | 3.6 | This work | |
| Melt | 800 | 1941 | 176 | 3.8 | This work | |
| Softwood organosolv lignin and kraft lignin (80/20) | Melt | 700 | 2200 | 225 | 4.1 | This work |
| Hardwood kraft lignin | Melt | 1000 | 605 | 61 | 46 ± 8.0 | 47 |
| Softwood kraft lignin and hardwood kraft lignin (90/10) | Melt | 1000 | 300 | 30 | 20–90 | 48 |
| Alcell organosolv lignin | Melt | 1000 | 720 | 45.9 | 8.5 | 49 |
| Softwood kraft lignin | Melt | 1200 | 465 | 32 | 39–65 | 50 |
| Switchgrass organosolv lignin | Melt | 1000 | 587 | 35 | 16.2 ± 6.0 | 51 |
| Yellow poplar organosolv lignin and switchgrass organosolv lignin (85/15) | Melt | 1000 | 747 | 42 | 15.7 | 52 |
| Solvent fractionated ultra-high molecular weight softwood kraft lignin | Dry | 1000 | 1390 | 98 | 7 | 14 |
| Chemically modified lignin | ||||||
| Steam exploded hardwood lignin after hydrogenation | Melt | 1000 | 660 | 45 | 7.6 ± 2.7 | 53 |
| Phenolyzed steam exploded lignin | Melt | 1000 | 394 | 36 | — | 54 |
| Acetylated softwood kraft lignin | Dry | 1000 | 1040 | 52 | 6–8 | 27 |
| Pyrolysis oil of hardwood lignin | Melt | 1000 | 855 | 85 | 29–50 | 55 |
| Peracylated softwood kraft lignin | Melt | 1000 | 750 | 34 | 17 | 56 |
| Acetylated corn stover organosolv lignin | Melt | 1000 | 454 | 62 | 39.1 ± 5.4 | 57 |
| Oleic acid functionalized lignin | Melt | 1000 | 640 | 71 | 24.8 ± 4.3 | 58 |
| Acrylate thermoplastic polymer synthesized using pyrolysis oil of lignin | Melt | 1000 | 1700 | 182 | 5.1 | 24 |
| Lignin and additives | ||||||
| Hardwood kraft lignin and poly (ethylene oxide) (97/3) | Melt | 1000 | 458 | 59 | 33 ± 2 | 59 |
| Hardwood kraft lignin and polyethylene terephthalate (75/25) | Melt | 1000 | 703 | 94 | 34 ± 5 | 47 |
| Hardwood kraft lignin and syndiotactic polypropene (87.5/12.5) | Melt | 1000 | 437 | 54 | 44 ± 5 | 47 |
| Pyrolysis oil of lignin and 1% organoclay | Melt | 1000 | 438 | 32 | 47 ± 1 | 60 |
| Hardwood organosolv lignin and thermoplastic polyurethane (50/50) | Melt | 1400 | 1100 | 80 | 25 ± 3 | 21 |
| Hydroxyl-modified hardwood kraft lignin and thermoplastic polyurethane (50/50) | Melt | 1400 | 800 | 66 | 30 ± 1 | 21 |
| Pyrolysis oil of hardwood lignin and polyethylene terephthalate (95/5) | Melt | 1000 | 925 | 98 | 12.6 ± 2.1 | 18 |
| Kraft lignin, polyvinyl alcohol, and graphene oxide (66/29/5) | Wet | 1000 | 763 | 52 | — | 28 |
| Softwood kraft lignin and cellulose (70/30) | Wet | 1000 | 1070 | 76 | 7.6 ± 0.7 | 61 |
| Lignin and PAN | ||||||
| Softwood kraft lignin and PAN (35/65) | Wet | 1200 | 1680 | 201 | 16.8 | 62 |
| Annual plant soda lignin and PAN (30/70) | Gel (−50 °C) | 1100 | 1720 | 250 | 11.0 ± 1.1 | 63 |
| Annual plant soda lignin, PAN, carbon nanotube (30/70/3) | Gel (−50 °C) | 1100 | 1400 | 200 | 8.8 ± 0.3 | 63 |
| Lignosulfonate and acrylonitrile co-polymer (10/90) | Wet | 1400 | 649 | — | 19.2 | 64 |
| Softwood kraft lignin and PAN (50/50) | Wet | 1200 | 1200 | 105.7 | 7.0 ± 0.3 | 19 |
3.2. Comparison of CFs produced using different processing conditions
TCFs were produced solely from lignin without chemical treatments or additives. Instead of modifying precursors, we thermo-mechanically controlled the spun fibers undergoing stabilization and carbonization. To show that the high quality of the TCFs obtained in this work is not related to the lignin sources but due to the fiber fabrication method, OL-based CFs were also fabricated using the same heating profiles but without or partial tension control as described in the method section above.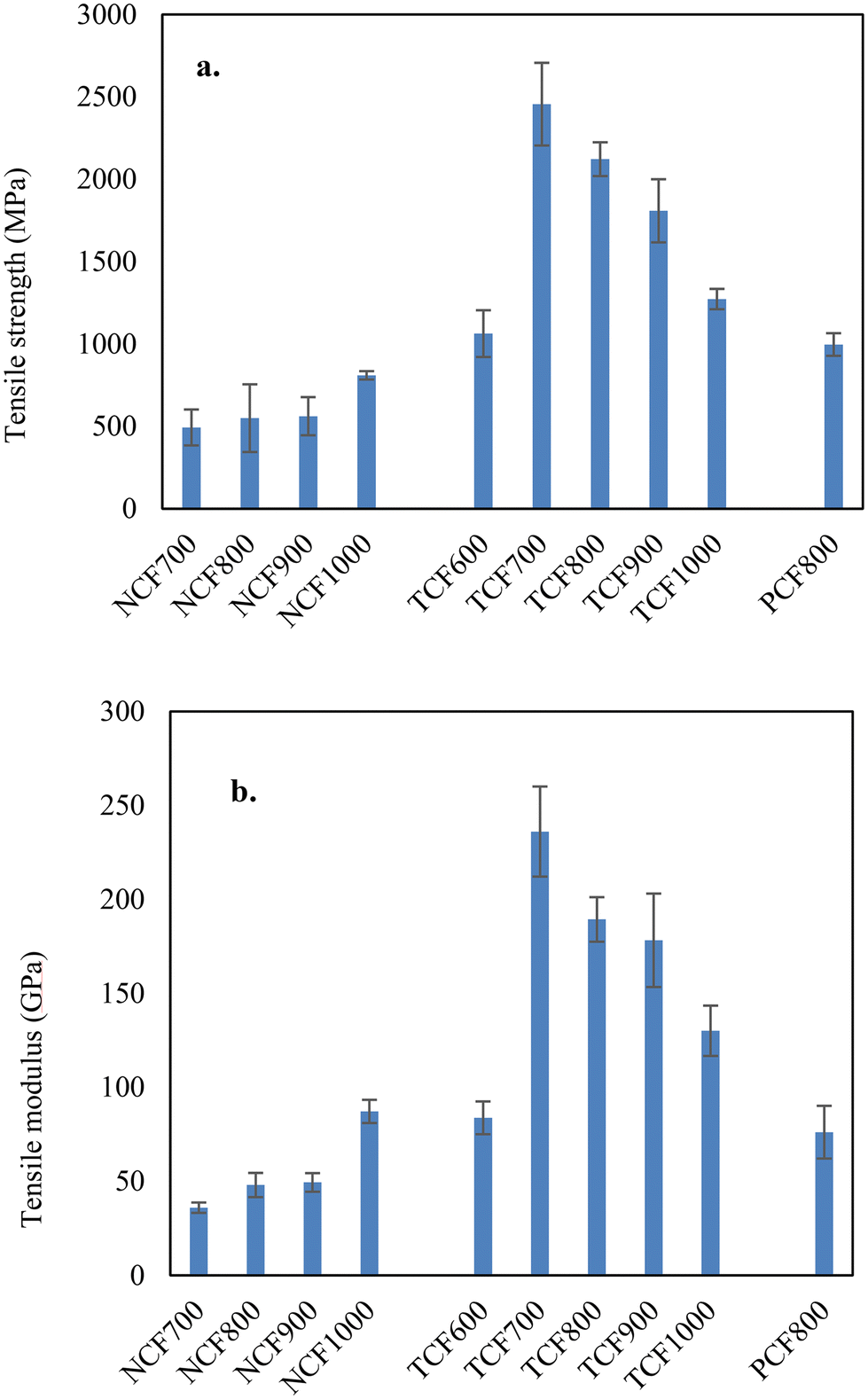 | ||
| Fig. 3 Tensile properties of CFs produced using 100% OL using different processing conditions. (a) Tensile strength; (b) tensile modulus. | ||
 | ||
| Fig. 4 TEM images of 100% OL-derived CFs. (a) NCF700; (b) NCF800; (c) NCF900; (d) NCF1000; (e) TCF700; (f) TCF800; (g) TCF900; (h) TCF1000. | ||
3.3. Analysis of SF produced under different processing conditions
The changes in the microstructures of lignin are the results of chemical reactions that took place during the CF production process. The formation of turbostratic structure from lignin at such low temperatures without catalysts has been impossible previously, suggesting the thermomechanical controlling altered the ordinary chemical reactions and microstructural evolution from lignin. To support this statement, the chemical reactions and the microstructures were tracked by analyzing OL-based fibers at different stages under different processing conditions. This section discusses the effects of thermomechanical controlling on lignin stabilization.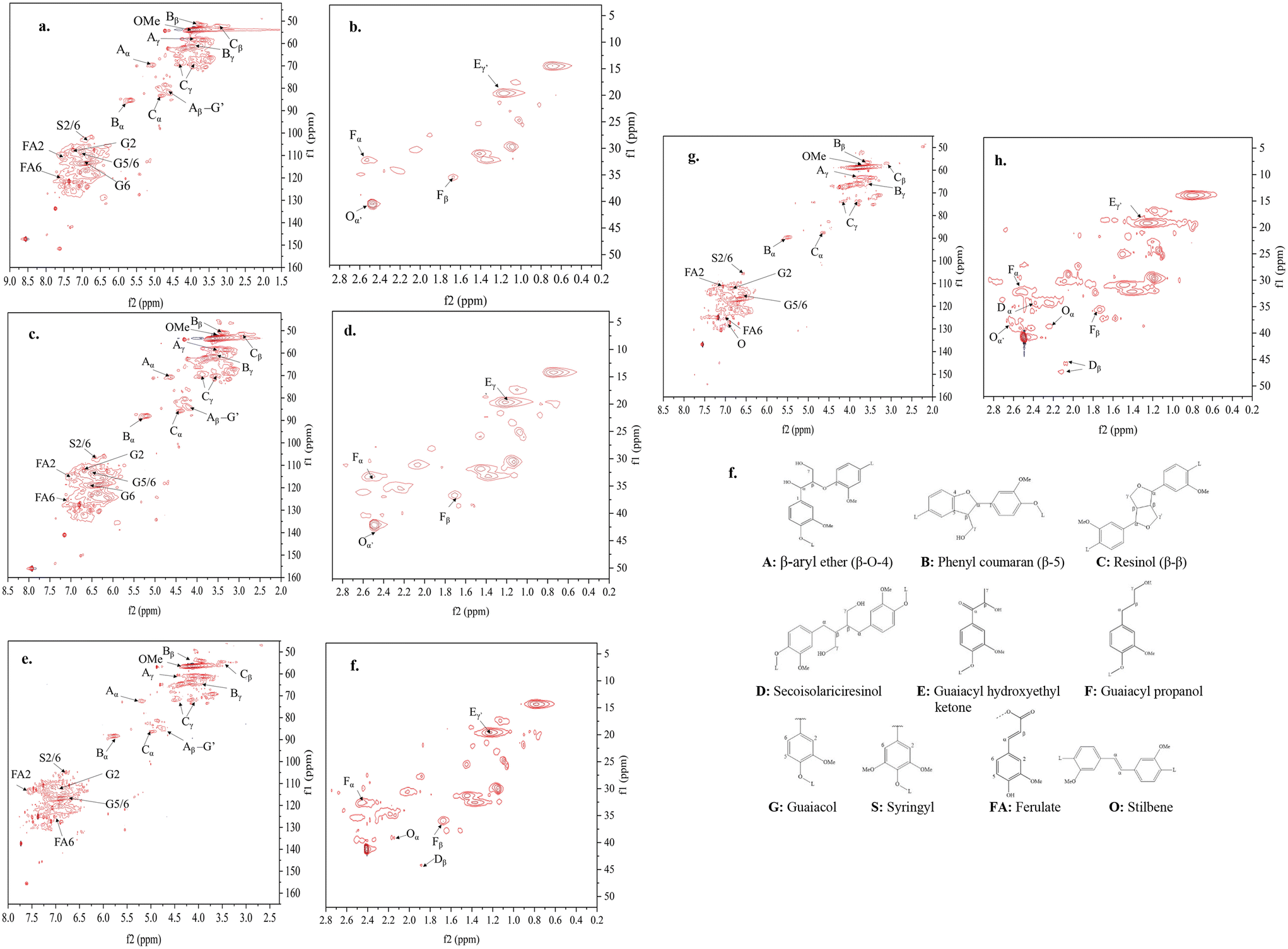 | ||
| Fig. 5 HSQC-NMR spectra of 100% OL-derived AF and SFs. (a & b) Lignin, (c & d) AF, (e & f) NSF stabilized up to 165 °C; (g & h) TSF stabilized up to 165 °C; (a, c, e, and g) are oxygen and aromatic-related regions. (b, d, f, and h) are non-oxygen aliphatic regions, and (i) the structures labeled in a–h. | ||
O groups is mainly due to oxidation at the aliphatic OH groups. Compared to NSF, TSF contained fewer amounts of CO and OH groups. On the other hand, TSF had more pronounced peaks of alkyl stretching (peaks 2 and 3) and aromatic ring vibrations (peaks 7 and 8) than NSF.
 | ||
| Fig. 6 (a) FTIR spectra of 100% OL-derived SFs: 1 (3100–3600 cm−1) for OH, 2 (2948 cm−1) & 3 (2850 cm−1) for alkyl C–H, 4 (1850 cm−1), 5 (1750 cm−1) & 6 (1700 cm−1) for CO, 7 (1600 cm−1) & 8 (1500 cm−1) for aromatic ring vibration, 9 (1232 cm−1) & 10 (1032 cm−1) for C–O stretch; Raman spectra of SFs measured from (b) cross-section view, (c) top view. | ||
O groups. Phenolic monomers were nearly absent when NSF was pyrolyzed, indicating a highly condensed structure. On the other hand, NSF produced significantly higher amounts of CO2 and CO than AF, attributed to the increased CO formation during oxidative stabilization. Unlike condensed NSF, several phenolic monomers with methyl and ether groups were observed from TSF pyrolysis, suggesting TSF structure is less condensed with fewer cross-linkages. TSF also produced fewer amounts of CO2 and CO than NSF, which agrees with the FTIR result that TSF contains fewer CO than NSF.
O. The characterization results of TSF compared to NSF showed that coupling thermochemistry with mechanochemistry introduced thermo-mechanochemistry to promote bond cleavages in the irregular lignin structure because the molecules are activated by both thermal energy and mechanical energy. Previous studies showed that mechanochemistry-induced bond rupturing causes chain fragmentation to degrade polymer structure.39 Thus, increasing interunit bond cleavages is supposed to reduce the mechanical strength of TSF. However, as described above, TSF had much higher tensile properties than NSF. Thus, the effect of thermo-mechanochemistry was more than deconstructing the heterogeneous polymer into smaller fragments. During the tension-applied stabilization, the partly cleaved lignin structures with reactive ends (i.e., radicals) are rearranged and repolymerized to form a new structure that is much stronger than the original lignin structure. The new structure is expected to consist of intra- or inter-molecular C–C or C–O linkages with high bond strengths aligned along the fiber axial direction because weaker bonds would not be able to survive the increasing temperature and the strong mechanical force simultaneously applied to the fibers. The weaker bonds of lignin (e.g., β-O-4) are likely eliminated and replaced by stronger bonds newly formed.
Mechanical stretching of the softening fiber also physically reduces fiber diameters during stabilization. The thinner fiber diameters of TSF can minimize heat transfer limitation during stabilization and promote uniform heat penetration toward the fiber core compared to NSF with thicker fiber diameters. Thus, the fiber surface and core structures are expected to be more uniform in TSF than in NSF. Although the extent of oxidation is supposed to be greater in TSF than in NSF, TSF has a lower carbonyl content and total oxygen content than NSF (elemental analysis results in Table S2†). The oxidation reactions in TSF likely promoted bond cleavages to release oxygenated volatile compounds and light gases rather than forming the end unit carbonyls. As indicated above, carbonyl and the residue hydroxyl groups were both lower in TSF than NSF. Thus, the aliphatic OHs were removed from lignin as volatile products rather than becoming CO. Since hydroxyl groups are the major active sites for lignin to crosslink, removing them reduced lignin's tendency to crosslink, which was evident in TSF pyrolysis results. The yield of TSF was much lower than that of NSF, which supports that the increased bond scissions and rearrangements in TSF promote lightweight byproducts.
Plausible reactions of the major lignin linkages under thermo-mechanochemistry are given in Fig. 7. During conventional thermal oxidation of lignin, oxygen promotes the abstraction of hydrogen from alcohol groups. The remaining ether CH2O radicals are stabilized to become terminal carbonyls. In comparison, the CH2O radicals are removed as formaldehyde in the presence of tension due to an additional C–C bond dissociation promoted by the mechanochemistry effect. As described in literature,39 external force can cause bond rotation, angle bending, and bond stretching to deform the original bond until the bond scission occurs. Deformed bonds have lower bond dissociation energies than free bonds. When tension is applied to stretch the bi-aromatic ring structures with interlinkages, the CH2O radicals present at the branches of the aromatic interlinkages can depart from their original positions in the molecules by responding to the force. When tension force is strong enough, the homolytic carbon–carbon bond session between the CH2O carbon and the adjacent alkyl carbon of the aromatic interlinkages will remove CH2O entirely as a volatile byproduct. After CH2O is removed, the remaining radical structures are stabilized to form linear aromatic structures with less branched interlinkages consisting of sp2 carbons. In lignin under tension, the CH2O radical can originate from hydrogen abstracted hydroxyls in β-O-4 or β-1 structures or ring-opening reactions of β–β or β-5 structures. Due to their orientability and sp2 hybridized amorphous carbons, the newly formed molecular structures after the CH2O removal can easily cyclize during continuous heat treatment by stabilization and carbonization. Beste33 previously studied the oxidation of lignin model compounds by performing a molecular dynamic simulation using a reactive force field method. The removal of aliphatic OH as formaldehyde was among the possible reactions. However, cracking was the major reaction under the high simulation temperatures (>2000 K) because most of the oxidation products were gases and other lightweight molecules. Therefore, the probability of these reactions occurring in thermally treated lignin at real stabilization temperatures is expected to be low. As observed in NSF characterization, OH groups were oxidized to become carbonyl groups after stabilization. Under tension, deconstructing and rearranging the irregular lignin structure to linearly stretched, orientable bi-aromatic ring structures and removing branched side chains with OHs to suppress random-directional crosslinking among molecules are expected to yield a structure with improved orientability and a higher content of graphitizable sp2 carbons in TSF. Such a new structure is difficult to obtain by thermally treating lignin. The uniform structure with strong covalent bonds in TSF is expected to reduce the risk of mechanical failures within the fiber when an external force is applied to the fiber.
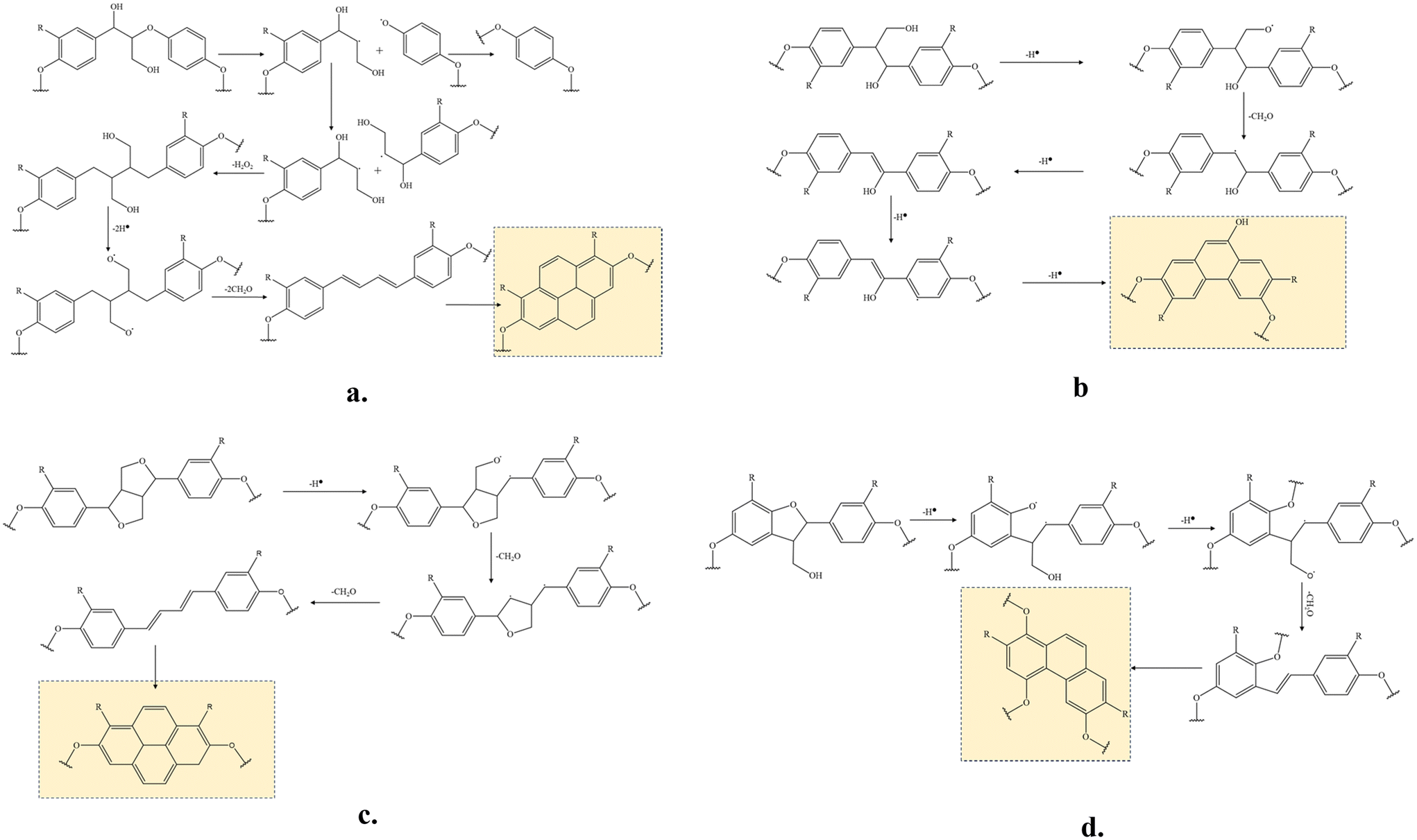 | ||
| Fig. 7 Plausible reactions occurring during tension-applied stabilization of lignin spun fiber. (a) β-O-4; (b) β-1; (c) β–β; (d) β-5. The structures in the colored boxes are after carbonization. | ||
3.4 Analysis of CFs produced using different processing conditions
In this section, TSF and NSF were carbonized to different final temperatures using the same heating profiles but with different conditions for mechanical control. Different CFs were analyzed for their chemical and microstructural transformations during carbonization.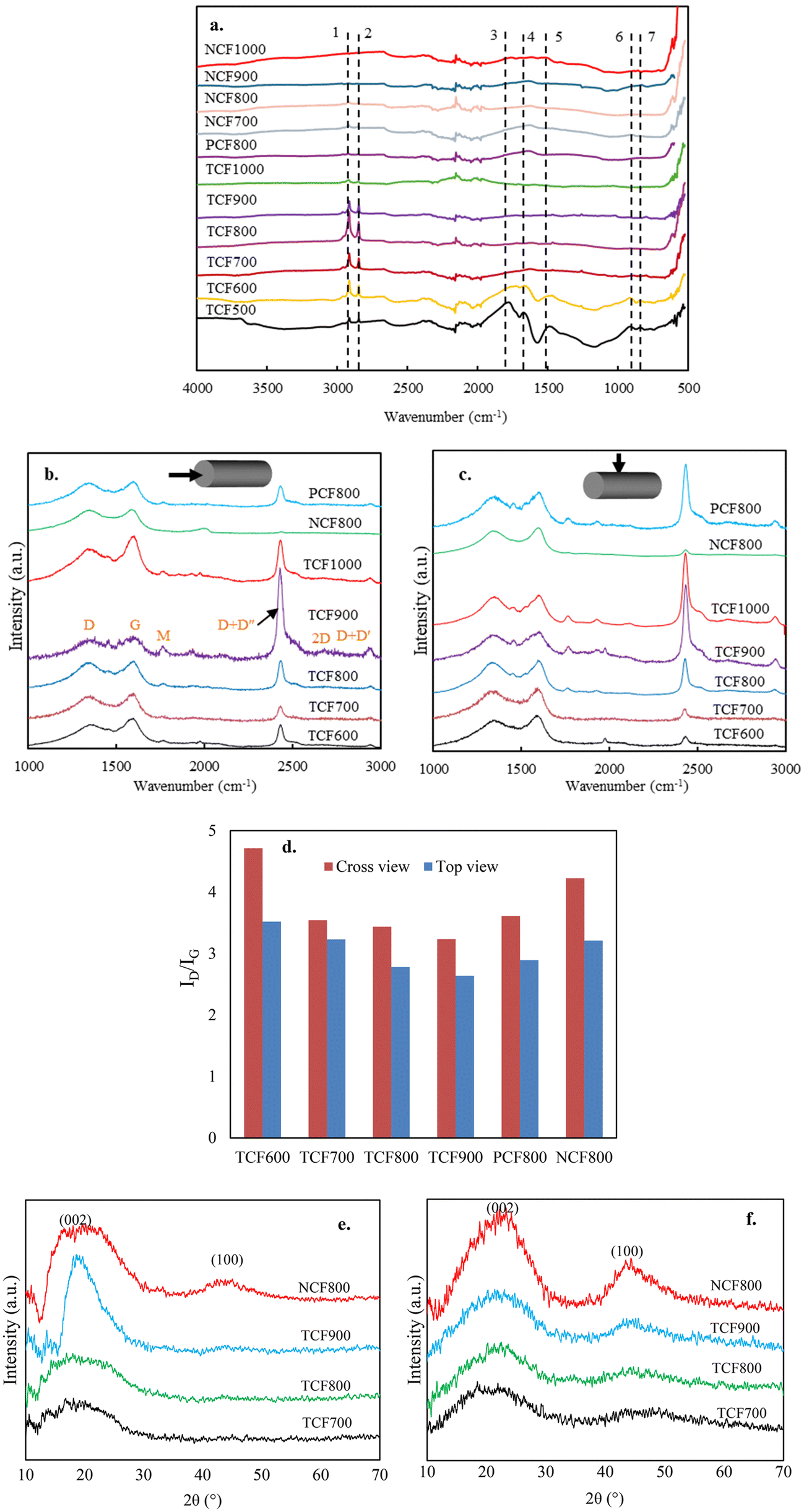 | ||
| Fig. 8 Characterization of 100% OL-derived CFs. (a) FTIR spectra: 1 (2914 cm−1) & 2 (2850 cm−1) for alkyl C–H stretch, 3 (1760 cm−1) & 4 (1650 cm−1) for CO, 5 (1500 cm−1), 6 (913 cm−1), and 7 (833 cm−1) for aromatic ring vibration. (b–d) Raman spectra: (b) cross-section view spectra; (c) top view spectra; (d) ID/IG ratios of CFs in different fiber directions after Gaussian fitting; (e & f) XRD equatorial scans measured at (e) parallel and (f) perpendicular to the fiber axis. | ||
O content decreased with increased temperatures to become negligible in TCF900. The –CO content was lowest in TCF700 and increased at the fibers with higher carbonization temperatures. When compared with NCF800, TCF800 contained significantly fewer amounts of oxygen-bonded carbons. For example, the –O–CO content was 22.3% in NCF800 whereas it was only 1.7% in TCF800. The XPS results correspond to the FTIR results of CFs, indicating that introducing thermo-mechanochemistry can reduce oxygen-bonded carbons in CF. Oxygen-bonded carbons can hinder the growth of crystallites during carbonization.43 Thus, the decreased oxygenated bonds are highly preferred for forming a graphene structure observed in TCFs.
| CFs | –C–C–/–C–H (%) | –C–O– (%) | –CO (%) |
–O–CO (%) |
|---|---|---|---|---|
| TCF700 | 81.20 | 3.02 | 8.66 | 7.12 |
| TCF800 | 71.58 | 8.67 | 18.30 | 1.45 |
| TCF900 | 84.49 | 2.96 | 12.55 | 0.0 |
| NCF800 | 57.40 | 9.15 | 11.18 | 22.27 |
| TCF700 | TCF800 | TCF900 | NCF800 | |
|---|---|---|---|---|
| d 002 (nm) | 0.3675 | 0.3572 | 0.3516 | 0.3697 |
| L c (nm) | 0.6155 | 0.8047 | 1.1003 | 0.9647 |
| L a (nm) | 1.0725 | 1.2029 | 1.3434 | 1.9109 |
While the thermo-mechanochemistry introduced during stabilization could manipulate and modify ordinary thermochemical reactions of lignin to transform the irregular molecular structure into a much-desired structure in TSF, introducing thermo-mechanochemistry during carbonization was important in ensuring the evolution of ordered microstructures from TSF. During carbonization, a strong tendency for fiber shrinkage along the fiber direction was only observed with TSF. When the stabilization of lignin occurs in the absence of thermo-mechanochemistry, various functional groups could freely interact in random directions of the fiber to form highly crosslinked and non-oriented structures. Thus, carbonizing NSF did not show noticeable fiber shrinkage. TSF exhibited the shrinking tendency, suggesting the oriented molecular structure of the stabilized fiber stretched along the fiber direction retains high chemical reactivity. TSF was less crosslinked and condensed than NSF, implying that the strong force applied to the fiber mechanically restricted the freedom of the molecular structure to prevent the intermolecular bonds from developing crosslinking in undesired fiber directions. If TSF is carbonized in the absence or with insufficient mechanical restrictions, the functional groups remaining in the no-longer restricted structure could freely react in different directions, causing the fiber to shrink. Allowing the fiber shrinkage during carbonization will lead to a microstructure with increased structural disorder and anisotropic, eventually lowering the mechanical properties in CF. In comparison, applying sufficient high force to restrict the fiber will ensure the oriented structure of TSF crosslinks only at the desired fiber direction as it carbonizes, which was critical in forming ordered microstructures and graphene carbons in TCF.
Until this study, it has been considered that the non-oriented amorphous carbons and low-tensile properties of CFs are unavoidable with lignin precursors. However, our results show that it is possible to overcome this challenging problem using a facile approach by controlling heat and mechanical force applied to lignin-derived fibers. Properly integrating thermal heating and mechanical stretching can introduce a novel thermo-mechanochemistry, which can manipulate ordinary chemical reactions to modify the molecular structure and control microstructural evolution, transforming lignin into an oriented structure with graphitized carbons. Our results suggest that the high tensile properties in lignin-based CF obtained in this work are due to the combined effects of structural ordering, isotropic structure, turbostratic carbons, and smaller pore sizes. TCF700 produced using the extremely low carbonization temperature had the highest tensile properties because the fiber could meet all these conditions, contributed by the newly introduced thermo-mechanochemistry. Unlike conventionally produced CFs, the tensile properties decreased at the TCFs produced at higher carbonization temperatures because the negative impacts caused by the increased anisotropic structure and larger pore size at these fibers were more prominent than the positive impact of the increased graphitization.
In this work, TSFs produced at low carbonization temperatures had both high tensile strength and modulus. Even for PAN-based CFs, carbonization temperatures above 1000 °C are usually required to obtain comparable tensile properties. PAN-based and lignin-based CF differ in their completely different molecular structures and reaction mechanisms for forming CF. PAN is a linear-chain polymer without ring structures. Thus, PAN must form individual benzene rings from scratch through extensive cyclization during stabilization and carbonization. In comparison, lignin intrinsically owns benzene rings in their basic units. By introducing thermo-mechanochemistry during stabilization, benzene rings are interconnected by stable aliphatic bonds to form an oriented structure susceptible to graphitization. Introducing thermo-mechanochemistry during the subsequent carbonization further promoted graphene structure by ensuring orientation and preventing undesired crosslinking. Since the benzene ring structure is already present in TSF, its graphitization can occur at a much lower temperature than the temperature required for PAN-based CFs, as confirmed by the TEM results above. Turbostratic structure and polyaromatic ring clusters in TCFs are connected via strong aliphatic linkages, increasing both the strength and rigidity of the material.
3.5. Techno-economic analysis of lignin-based CF
The above results show that CFs with mechanical properties exceeding the DOE targets could be produced using lignin alone based on the melt-spinning method. A preliminary techno-economic analysis (TEA) was conducted to determine the cost of melt-spun lignin-based CF. The cost estimation was based on a 10000 MT per year facility, with a lignin purchase price of $110 per MT and a 10-hour stabilization time as the base case (equivalent to the case for the 80%/20% OL/KL-based CF in this work). In Fig. 9, our current and projected lignin-based CF costs are compared to a literature-estimated cost of PAN-based CF.12 The results showed that the current production cost of lignin-based CF is estimated at $4.17 per lb, which is lower than that of the DOE automobile-grade CF. The impacts of techno-economic improvements on lignin-based cost projections were also studied. The cost can be reduced by lowering the lignin purchase price and increasing the plant capacity. Decreasing the lignin price to $50 per MT lowers the CF cost by $0.05 per lb to $4.12 per lb, and increasing the plant capacity by 20% decreases the cost by $0.19 per lb to $3.98 per lb. On the other hand, decreasing the stabilization time from 10 hours to 5 hours decreased costs by $1.25 per lb to $2.92 per lb. This work shows that the stabilization time is directly related to the precursor Tg. Extended stabilization hours were required in this work due to the low Tgs of the lignin precursors. We expect a substantial reduction in stabilization time when high Tg lignins are used.
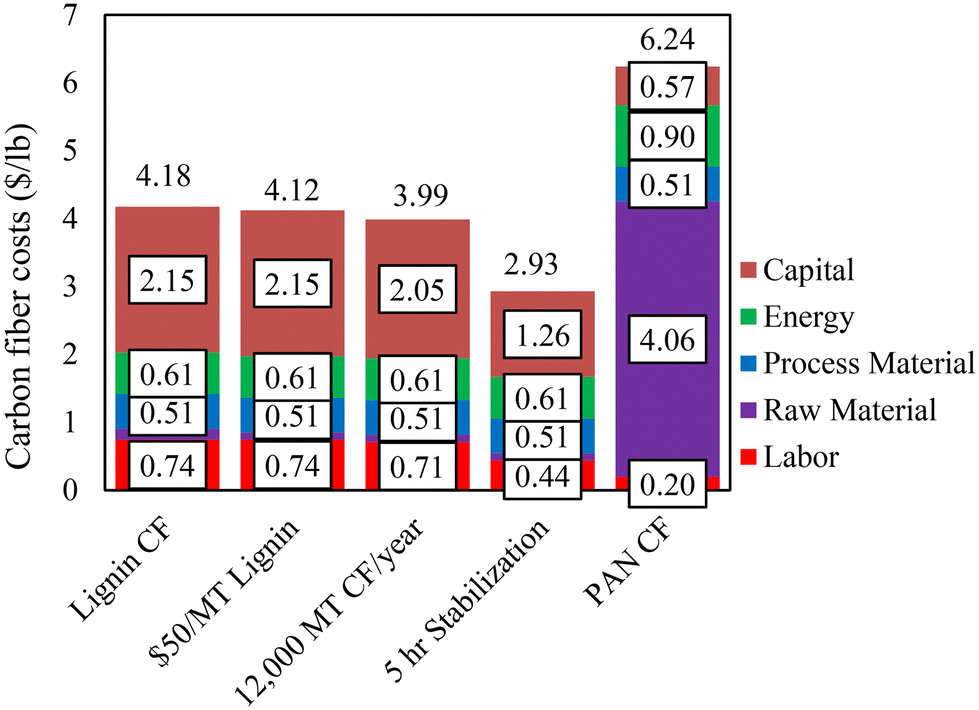 | ||
| Fig. 9 Comparison of current and projected production costs of lignin-based CF based on this work to literature-estimated PAN-based CF production cost (12). | ||
4. Conclusions
This work demonstrated the production of low-cost high-quality CFs using lignin alone without chemical treatment or additives. The property advancement is due to the introduction of thermo-mechanochemistry of lignin that is discovered in this work. This novel chemistry can be introduced by properly integrating thermal heating and mechanical stretching. The integration can transform lignin into oriented and graphene carbons by manipulating its chemical reactions and controlling the microstructure evolution. The proof-of-concept CFs demonstrated in the laboratory have smaller fiber diameters than commercial CFs, which are 5–8 μm. However, our results suggest that CF with higher tensile properties at industrial-relevant fiber diameters is expected when the fiber fabrication system with precision control is employed to fine-tune the heating and tension profiles. The discovery of the thermo-mechanochemistry of lignin and the improved understanding of lignin-based CFs reported in this work provide critical knowledge for developing a facile and green approach to enable high-quality CFs from broader lignin. Low-cost lignin-based CFs will simultaneously address the demands for low-cost green CFs in industries and the biorefinery bottleneck on lignin valorization.Author contributions
Conceptualization: X.B. Methodology: Y.L., M.R., A.M, W.Q., A.A., H.Z, Investigation: Y.L. M.R., W.Q. Writing – original draft: Y.L., M. R., A.A. writing – review & editing: X.B., X.W., M.M.W. funding acquisition: X.B. Supervision: X.B.Conflicts of interest
X. B, Y. L, and M. R along with Iowa State University Research Foundation have filed a US patent application no 18/495185.Acknowledgements
This work is supported by the United State Department of Agriculture through USDA-NIFA grants (2021-67021-34504 and 2023-67021-39598).References
- J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Lignin valorization: Improving lignin processing in the biorefinery, Science, 2014, 344, 6185 CrossRef PubMed
.
- S. Sethupathy, G. M. Morales, L. Gao, H. Wang, B. Yang, J. Jiang, J. Sun and D. Zhu, Lignin valorization: Status, challenges and opportunities, Bioresour. Technol., 2022, 347, 126696 CrossRef CAS PubMed
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, A concise review of current lignin production, applications, products and their environment impact, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Carbon fibers: Precursor systems, processing, structure, and properties, Angew. Chem., Int. Ed., 2014, 53, 5262–5298 CrossRef CAS PubMed
- S. Wang, J. Bai, M. T. Innocent, Q. Wang, H. Xiang, J. Tang and M. Zhu, Lignin-based carbon fibers: Formation, modification and potential applications, Green Energy Environ., 2021, 7, 578–605 CrossRef
- W. Qu, J. Yang, X. Sun, X. Bai, H. Jin and M. Zhang, Towards producing high-quality lignin-based carbon fibers: A review of crucial factors affecting lignin properties and conversion techniques, Int. J. Biol. Macromol., 2021, 189, 768–784 CrossRef CAS PubMed
- W. J. Sagues, A. Jain, D. Brown, S. Aggarwal, A. Suarez, M. Kollman, S. Park and D. S. Argyropoulos, Are lignin-derived carbon fibers graphitic enough?, Green Chem., 2019, 21, 4253–4265 RSC
- M. Pervaiz, S. Panthapulakkal, B. K. C. M. Sain and J. Tjong, Emerging Trends in Automotive Lightweighting through Novel Composite Materials, Mater. Sci. Appl., 2016, 07, 26–38 Search PubMed
- Mordor Intelligence™ Industry Reports. Automotive carbon fiber market-growth, trends, Covid-19 impact, and forecast (2023–2028), https://www.mordorintelligence.com/industry-reports/automotive-carbon-fiber-composites-market#:~:text=MarketOverview, the forecast period 2022–2027, (accessed 2023 Oct 5).
- Global Market Insights, Carbon Fibre Composites Market Size, Share and Industry Analysis Report by End-Use (Aerospace, Automotive, Wind Turbines, Sport & Leisure, Civil Engineering, Marine), and Matrix Material (Polymer [Thermosetting, Thermoplastics], Carbon, Ceramic, Metal. Report ID: GMI2180 07/2022., https://www.gminsights.com/industry-analysis/carbon-fibre-composites-market?gclid=CjwKCAjwq-WgBhBMEiwAzKSH6H6eBA4G-e9ctCOk2zLuCYrqhK45mwJjsvJ__N-qGfUIMIosUEwwTBoCYGMQAvD_BwE, (accessed 2023 Oct 5).
- D. A. Baker and T. G. Rials, Recent advances in low-cost carbon fiber manufacture from lignin, J. Appl. Polym. Sci., 2013, 130, 713–728 CrossRef CAS
- T. Ellringmann, C. Wilms, M. Warnecke, G. Seide and T. Gries, Carbon fiber production costing: A modular approach, Text. Res. J., 2016, 86, 178–190 CrossRef CAS
- J. Mikkilä, M. Trogen, K. A. Y. Koivu, J. Kontro, J. Kuuskeri, R. Maltari, Z. Dekere, M. Kemell, M. R. Mäkelä, P. A. Nousiainen, M. Hummel, J. Sipilä and K. Hildén, Fungal Treatment Modifies Kraft Lignin for Lignin- And Cellulose-Based Carbon Fiber Precursors, ACS Omega, 2020, 5, 6130–6140 CrossRef PubMed
- J. Jin, J. Ding, A. Klett, M. C. Thies and A. A. Ogale, Carbon Fibers Derived from Fractionated-Solvated Lignin Precursors for Enhanced Mechanical Performance, ACS Sustainable Chem. Eng., 2018, 6, 14135–14142 CrossRef CAS
- K. H. K. Chan, S. Y. Wong, X. Li, Y. Z. Zhang, P. C. Lim, C. T. Lim, M. Kotaki and C. B. He, Effect of molecular orientation on mechanical property of single electrospun fiber of poly[(R)-3-hydroxybutyrate-co-(R)-3-hydroxyvalerate], J. Phys. Chem. B, 2009, 113, 13179–13185 CrossRef CAS PubMed
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Liquid fuels, hydrogen and chemicals from lignin: A critical review, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS
- Q. Li, M. Li, H. S. Lin, C. Hu, P. Truong, T. Zhang, H. J. Sue, Y. Pu, A. J. Ragauskas and J. S. Yuan, Non-solvent fractionation of lignin enhances carbon fiber performance, ChemSusChem, 2019, 12, 3249–3256 CrossRef CAS PubMed
- W. Qu and X. Bai, Thermal treatment of pyrolytic lignin and polyethylene terephthalate toward carbon fiber production, J. Appl. Polym. Sci., 2020, 137, 1–10 CrossRef
- J. Jin and A. A. Ogale, Carbon fibers derived from wet-spinning of equi-component lignin/polyacrylonitrile blends, J. Appl. Polym. Sci., 2018, 135, 1–9 Search PubMed
- A. Bengtsson, J. Bengtsson, M. Sedin and E. Sjöholm, Carbon Fibers from Lignin-Cellulose Precursors: Effect of Stabilization Conditions, ACS Sustainable Chem. Eng., 2019, 7, 8440–8448 CrossRef CAS
- M. Culebras, A. Beaucamp, Y. Wang, M. M. Clauss, E. Frank and M. N. Collins, Biobased Structurally Compatible Polymer Blends Based on Lignin and Thermoplastic Elastomer Polyurethane as Carbon Fiber Precursors, ACS Sustainable Chem. Eng., 2018, 6, 8816–8825 CrossRef CAS
- H. C. Liu, J. Luo, H. Chang, A. A. B. Davijani, P. H. Wang and S. Kumar, Polyacrylonitrile sheath and polyacrylonitrile/lignin core bi-component carbon fiber, Carbon, 2019, 149, 165–172 CrossRef CAS
- W. Qu, Y. Huang, Y. Luo, S. Kalluru, E. Cochran, M. Forrester and X. Bai, Controlled Radical Polymerization of Crude Lignin Bio-oil Containing Multihydroxyl Molecules for Methacrylate Polymers and the Potential Applications, ACS Sustainable Chem. Eng., 2019, 7, 9050–9060 CrossRef CAS
- Y. Luo, W. Qu, E. Cochran and X. Bai, Enabling high-quality carbon fiber through transforming lignin into an orientable and melt-spinnable polymer, J. Cleaner Prod., 2021, 307, 127252 CrossRef CAS
- D. Choi, H. S. Kil and S. Lee, Fabrication of low-cost carbon fibers using economical precursors and advanced processing technologies, Carbon, 2019, 142, 610–649 CrossRef CAS
- J. L. Braun, K. M. Holtman and J. F. Kadla, Lignin-based carbon fibers: Oxidative thermostabilization of kraft lignin, Carbon, 2005, 43, 385–394 CrossRef CAS
- M. Zhang and A. A. Ogale, Carbon fibers from dry-spinning of acetylated softwood kraft lignin, Carbon, 2014, 69, 626–629 CrossRef CAS
- F. Torres-Canas, A. Bentaleb, M. Föllmer, J. Roman, W. Neri, I. Ly, A. Derré and P. Poulin, Improved structure and highly conductive lignin-carbon fibers through graphene oxide liquid crystal, Carbon, 2020, 163, 120–127 CrossRef CAS
- Q. Li, C. Hu, M. Li, P. Truong, J. Li, H. S. Lin, M. T. Naik, S. Xiang, B. E. Jackson, W. Kuo, W. Wu, Y. Pu, A. J. Ragauskas and J. S. Yuan, Enhancing the multi-functional properties of renewable lignin carbon fibersviadefining the structure-property relationship using different biomass feedstocks, Green Chem., 2021, 23, 3725–3739 RSC
- S. Zhou, Y. Xue, A. Sharma and X. Bai, Lignin Valorization through Thermochemical Conversion: Comparison of Hardwood, Softwood and Herbaceous Lignin, ACS Sustainable Chem. Eng., 2016, 4, 6608–6617 CrossRef CAS
- J. J. Adams, F. Nicola, F. Ray, G. Amit, A. Charlie, N. Amit and K. Logan, Consortium for Production of Affordable Carbon Fibers in the United States, 2021 DOI:10.2172/1829480
- D. J. Johnson, I. Tomizuka and O. Watanabe, The fine structure of lignin-based carbon fibres, Carbon, 1975, 13, 321–325 CrossRef CAS
- A. Beste, ReaxFF study of the oxidation of softwood lignin in view of carbon fiber production, Energy Fuels, 2014, 28, 7007–7013 CrossRef CAS
- M. H. Lahtinen, J. Mikkilä, K. S. Mikkonen and I. Kilpeläinen, Kraft Process - Formation of Secoisolariciresinol Structures and Incorporation of Fatty Acids in Kraft Lignin, J. Agric. Food Chem., 2021, 69, 5955–5965 CrossRef CAS PubMed
- C. Mattsson, S. I. Andersson, T. Belkheiri, L. E. Åmand, L. Olausson, L. Vamling and H. Theliander, Using 2D NMR to characterize the structure of the low and high molecular weight fractions of bio-oil obtained from LignoBoost™ kraft lignin depolymerized in subcritical water, Biomass Bioenergy, 2016, 95, 364–377 CrossRef CAS
- X. Li, J. Hayashi and C. Z. Li, FT-Raman spectroscopic study of the evolution of char structure during the pyrolysis of a Victorian brown coal, Fuel, 2006, 85, 1700–1707 CrossRef CAS
- T. Shimada, T. Sugai, C. Fantini, M. Souza, L. G. Cançado, A. Jorio, M. A. Pimenta, R. Saito, A. Gruneis, G. Dresselhaus, M. S. Dresselhaus, Y. Ohno, T. Mizutani and H. Shinohara, Origin of the 2450 cm-1 Raman bands in HOPG, single-wall and double-wall carbon nanotubes, Carbon, 2005, 43, 1049–1054 CrossRef CAS
- R. Wang, H. Zobeiri, H. Lin, W. Qu, X. Bai, C. Deng and X. Wang, Anisotropic thermal conductivities and structure in lignin-based microscale carbon fibers, Carbon, 2019, 147, 58–69 CrossRef CAS
- G. D. Bo, Mechanochemistry of the mechanical bond, Chem. Sci., 2017, 9(1), 15–21 Search PubMed
- J. Li, C. Nagamani and J. S. Moore, Polymer Mechanochemistry: From Destructive to Productive, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed
- E. Izak-Nau, D. Campagna, C. Baumann and R. Göstl, Polymer mechanochemistry-enabled pericyclic reactions, Polym. Chem., 2020, 11, 2274–2299 RSC
- S. Dabral, H. Wotruba, J. G. Hernández and C. Bolm, Mechanochemical Oxidation and Cleavage of Lignin β-O-4 Model Compounds and Lignin, ACS Sustainable Chem. Eng., 2018, 6, 3242–3254 CrossRef CAS
- J. D. Kim, J. S. Roh and M. S. Kim, Effect of carbonization temperature on crystalline structure and properties of isotropic pitch-based carbon fiber, Carbon Lett., 2017, 21, 51–60 CrossRef
- O. O. Sonibare, T. Haeger and S. F. Foley, Structural characterization of Nigerian coals by X-ray diffraction, Raman and FTIR spectroscopy, Energy, 2010, 35, 5347–5353 CrossRef CAS
- W. Dun, L. Guijian, S. Ruoyu and F. Xiang, Investigation of structural characteristics of thermally metamorphosed coal by FTIR spectroscopy and X-ray diffraction, Energy Fuels, 2013, 27, 5823–5830 CrossRef
- A. R. Chambers, J. S. Earl, C. A. Squires and M. A. Suhot, The effect of voids on the flexural fatigue performance of unidirectional carbon fibre composites developed for wind turbine applications, Int. J. Fatigue, 2006, 28, 1389–1398 CrossRef CAS
- S. Kubo and J. F. Kadla, Lignin-based carbon fibers: Effect of synthetic polymer blending on fiber properties, J. Polym. Environ., 2005, 13, 97–105 CrossRef CAS
- Y. Nordström, I. Norberg, E. Sjöholm and R. Drougge, A new softening agent for melt spinning of softwood kraft lignin, J. Appl. Polym. Sci., 2013, 129, 1274–1279 CrossRef
- D. A. Baker, F. S. Baker and N. C. Gallego, Thermal Engineering of Lignin for Low-cost Production of Carbon Fiber, in The Fiber Society 2009 Fall Confer-ence, University of Georgia, Athens, GA, USA, 2009, https://www.thefibersociety.org/Portals/0/PastConferences/2009_Fall_Abstracts.pdf, (accessed 2023 Oct 5).
- L. Salmén, E. Bergnor, A. M. Olsson, M. Åkerström and A. Uhlin, Extrusion of Softwood Kraft Lignins as Precursors for Carbon Fibres, BioResources, 2015, 10, 7544–7554 CrossRef
- N. Meek, D. Penumadu, O. Hosseinaei, D. Harper, S. Young and T. Rials, Synthesis and characterization of lignin carbon fiber and composites, Compos. Sci. Technol., 2016, 137, 60–68 CrossRef CAS
- O. Hosseinaei, D. P. Harper, J. J. Bozell and T. G. Rials, Improving processing and performance of pure lignin carbon fibers through hardwood and herbaceous lignin blends, Int. J. Mol. Sci., 2017, 18(7), 1410 CrossRef PubMed
- K. Sudo and K. Shimizu, A new carbon fiber from lignin, J. Appl. Polym. Sci., 1992, 44, 127–134 CrossRef CAS
- K. Sudo, K. Shimizu, N. Nakashima and A. Yokoyama, A new modification method of exploded lignin for the preparation of a carbon fiber precursor, J. Appl. Polym. Sci., 1993, 48, 1485–1491 CrossRef CAS
- W. Qu, Y. Xue, Y. Gao, M. Rover and X. Bai, Repolymerization of pyrolytic lignin for producing carbon fiber with improved properties, Biomass Bioenergy, 2016, 95, 19–26 CrossRef CAS
- L. M. Steudle, E. Frank, A. Ota, U. Hageroth, S. Henzler, W. Schuler, W. Neupert and M. R. Buchmeiser, Carbon Fibers Prepared from Melt Spun Peracylated Softwood Lignin: an Integrated Approach, Macromol. Mater. Eng., 2017, 302, 1–11 CrossRef
- W. Qu, J. Liu, Y. Xue, X. Wang and X. Bai, Potential of producing carbon fiber from biorefinery corn stover lignin with high ash content, J. Appl. Polym. Sci., 2018, 135, 1–11 Search PubMed
- D. Kang, Y. Lee, K. H. Park, J. S. Bae, S. M. Jo and S. S. Kim, Carbon Fibers Derived from Oleic Acid-Functionalized Lignin via Thermostabilization Accelerated by UV Irradiation, ACS Sustainable Chem. Eng., 2021, 9, 5204–5216 CrossRef CAS
- J. F. Kadla, S. Kubo, R. A. Venditti, R. D. Gilbert, A. L. Compere and W. Griffith, Lignin-based carbon fibers for composite fiber applications, Carbon, 2002, 40, 2913–2920 CrossRef CAS
- W. Qin and J. F. Kadla, Effect of organoclay reinforcement on lignin-based carbon fibers, Ind. Eng. Chem. Res., 2011, 50, 12548–12555 CrossRef CAS
- A. Bengtsson, P. Hecht, J. Sommertune, M. Ek, M. Sedin and E. Sjöholm, Carbon Fibers from Lignin-Cellulose Precursors: Effect of Carbonization Conditions, ACS Sustainable Chem. Eng., 2020, 8, 6826–6833 CrossRef CAS
-
P. J. Bissett and C. W. Herriott, Lignin/polyacrylonitrile-containing dopes, fibers and production methods, US Patent: US Patent; US 2012/0003471A1, 2010 Search PubMed
- H. C. Liu, A. T. Chien, B. A. Newcomb, Y. Liu and S. Kumar, Processing, Structure, and Properties of Lignin- and CNT-Incorporated Polyacrylonitrile-Based Carbon Fibers, ACS Sustainable Chem. Eng., 2015, 3, 1943–1954 CrossRef CAS
- Q. Ouyang, K. Xia, D. Liu, X. Jiang, H. Ma and Y. Chen, Fabrication of partially biobased carbon fibers from novel lignosulfonate–acrylonitrile copolymers, J. Mater. Sci., 2017, 52, 7439–7451 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04288j |
| This journal is © The Royal Society of Chemistry 2024 |