
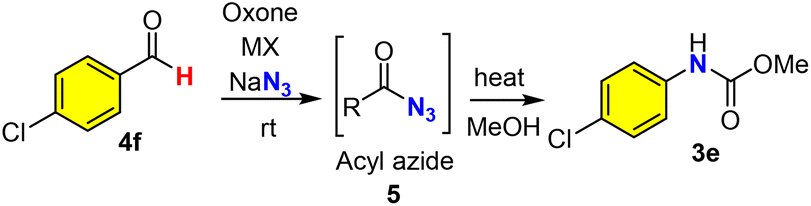
Unified and green oxidation of amides and aldehydes for the Hofmann and Curtius rearrangements†
Liyan
Song
 *a,
Yufei
Meng
a,
Tongchao
Zhao
a,
Lifang
Liu
a,
Xiaohong
Pan
a,
Binbin
Huang
a,
Hongliang
Yao
b,
Ran
Lin
*a and
Rongbiao
Tong
*bc
*a,
Yufei
Meng
a,
Tongchao
Zhao
a,
Lifang
Liu
a,
Xiaohong
Pan
a,
Binbin
Huang
a,
Hongliang
Yao
b,
Ran
Lin
*a and
Rongbiao
Tong
*bc
aKey Laboratory of Biopesticide and Chemical Biology (Ministry of Education), College of Plant Protection, Fujian Agriculture and Forestry University, Fuzhou, Fujian, China. E-mail: songliyan@fafu.edu.cn; linran@fafu.edu.cn
bGuangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Institute of Zoology, Guangdong Academy of Sciences, Guangzhou, Guangdong, China
cDepartment of Chemistry, The Hong Kong University of Science and Technology, Clearwater Bay, Kowloon, Hong Kong, China. E-mail: rtong@ust.hk; Fax: +(852)23581594; Tel: +(852) 23587357
First published on 27th November 2023
Abstract
The Hofmann and Curtius rearrangements have been widely used in organic synthesis and developed for the industrial production (5–100 kg) of pharmaceutically relevant amines/amides. However, the existing use of a stoichiometric organic oxidant [(diacetoxyiodo)benzene or trichloroisocyanuric acid for the Hofmann rearrangement] for amides or an activating reagent (diphenylphosphoryl azide for the Curtius rearrangement) for carboxylic acids is environmentally unfriendly and economically less attractive. Herein, we report the first green oxidation of amides and aldehydes with oxone and halide (and NaN3) to generate N-halo amides and acyl azides, respectively, both of which rearrange into the common isocyanate intermediates and subsequently produce stable carbamates or ureas (the Hofmann and Curtius rearrangements) when trapped with alcohols or amines. This unified green approach is highly efficient as demonstrated by more than 30 examples for each rearrangement. Importantly, this approach generates inorganic nontoxic K2SO4 as the only byproduct, which is advantageous over the existing methods that produced stoichiometric, toxic, and organic iodobenzene, and chloro-isocyanuric acid, or diphenylphosphoric acid. Notably, three urea-based drugs and eight chiral urea catalysts were efficiently synthesized from corresponding aldehydes by this green oxidative Curtius rearrangement. This green oxidative approach for the Hofmann and Curtius rearrangements is expected to find wide applications in organic synthesis and process chemistry.
Introduction
The Hofmann and Curtius rearrangements1–3 have found wide applications in organic synthesis because they offer a convenient and reliable way to convert carboxylic acid derivatives (amides and acyl azides) into synthetically valuable amines or protected amines (carbamates and ureas) through a common isocyanate intermediate as a result of formal insertion of nitrogen into C–C bond (Scheme 1a). Notably, both the Hofmann and Curtius rearrangements have been used for (continuous flow) industrial production of pharmaceutically relevant amines/carbamates:4 (a) the Hofmann rearrangement using (diacetoxyiodo)benzene as the oxidant has been applied to the industrial degradation (100 kg quantities) of asparagine derivatives;5 (b) a continuous flow Hofmann rearrangement using trichloroisocyanuric acid (TCCA) has been developed for the massive production of highly medicinally valuable 2-benzoxazolinone (23 g h−1) from inexpensive salicylamide;6 and (c) a continuous flow Curtius rearrangement4,7,8 typically using diphenylphosphoryl azide (DPPA)9,10 as an activating reagent for carboxylic acids has been rigorously studied for the large-scale preparation of the medicine-relevant carbamate intermediates including the carbamate (5.97-kg scale) of AZD7648 (a selective inhibitor of DNA-dependent protein kinase for anticancer treatment)11 and p-methoxybenzyl carbamate (0.8 kg h−1, 40 kg scale) for the synthesis of a CCR1 antagonist.12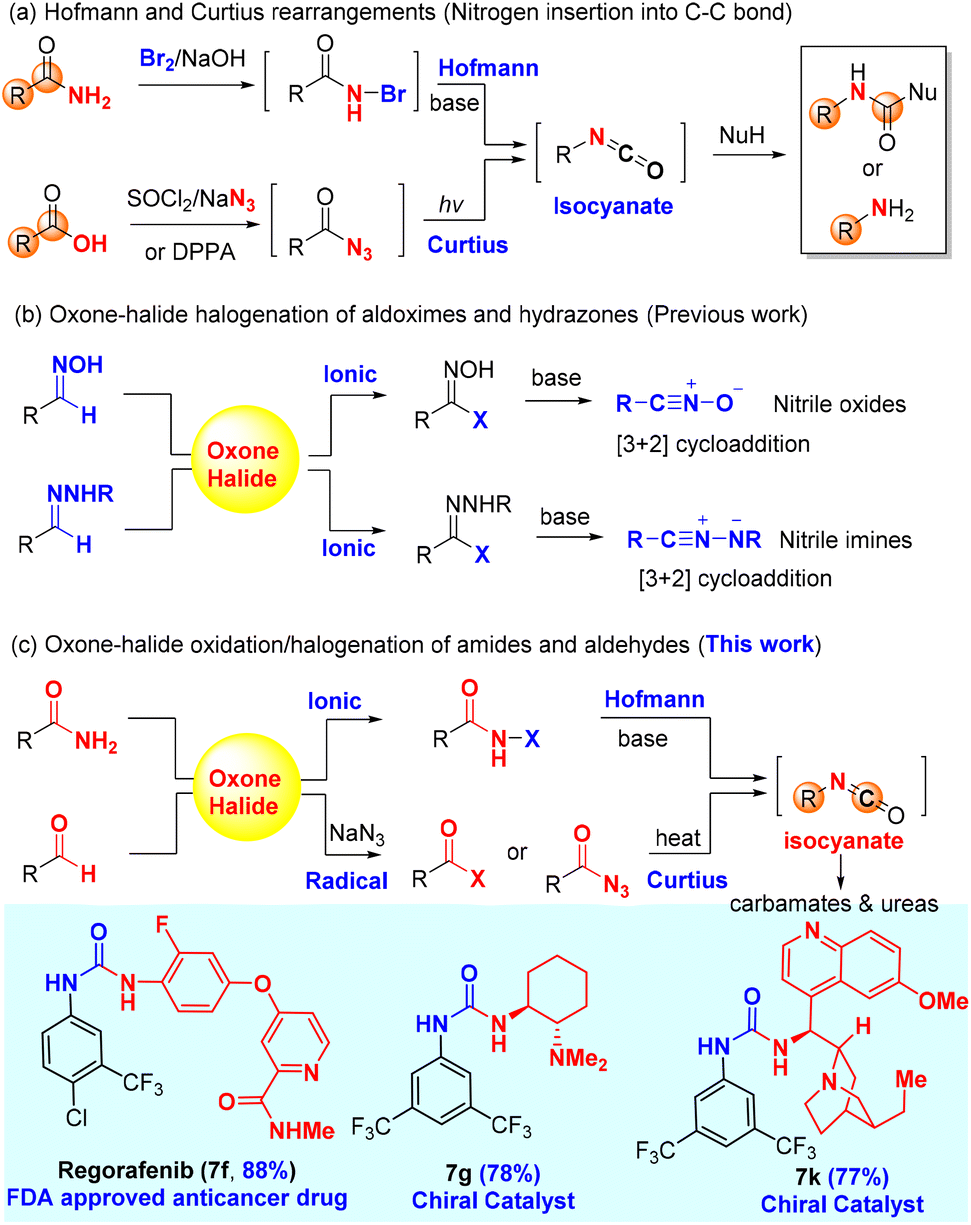 | ||
| Scheme 1 The Hofmann and Curtius rearrangements and oxone–halide oxidation (previous work and this work). | ||
An in situ generation of the unstable rearrangement substrates, N-halo amides and acyl azides, is generally conducive to the overall efficiency of both the Hofmann and Curtius rearrangements, which can be readily achieved by the halogenation of primary amides and the azidation of carboxylic acid derivatives (i.e., acid chloride), respectively. The halogenation of amides is conventionally carried out with reactive brominating species (i.e., bromine13 and N-bromo succinimide14) or hypervalent iodine(III)15 [PhI(OAc)2 and PhI(OCOCF3)2], while the classical azidation of carboxylic acids employs either SOCl2/NaN3 (acyl chloride intermediate) or DPPA. The major limitation of these standard procedures is ungreen and non-sustainable due to the use of hazardous chemicals (bromine and thionyl chloride) and/or producing stoichiometric toxic organic waste (hypervalent iodine, TCCA, and DPPA), which would become a big hurdle of practical applications in organic synthesis.
Our research group has a long-standing interest in developing alternative green protocols for important organic reactions.16–25 The combination of environmentally friendly oxone (2KHSO5·KHSO4·K2SO4)26,27 and halide (alkali halides such as NaCl and KBr) has demonstrated practical synthetic utility as an alternative green protocol to other halogenating reagents28,29 (i.e., halogen, N-halo succinimides, N-halo amides/imides, N-halo sulfonamides, etc.) in organic synthesis. Oxone–halide is not only used for the classical halogenation of arenes, alkenes, alkynes, and ketones (α-halogenation),17,21,26,27,30,31 but also employed in various oxidation reactions to provide non-halogenated products.20,32–36 The latter application is very interesting because the halide serves as a catalyst or mediator. For instance, our laboratory reported the oxone–halide system for the halogenation of aldoximes and hydrazones to form the unstable intermediates (hydroximoyl chloride and hydrazonyl bromide, respectively), which upon the treatment of bases generated in in situ reactive nitrile oxides and nitrile imines, respectively, for 1,3-dipolar cycloaddition with alkenes and alkynes (Scheme 1b).20,36 Inspired by this work, we hypothesized that the halogenation of amides and aldehydes with oxone–halide might generate N-halo amides and acyl azides (in the presence of sodium azide), which would undergo the Hofmann and Curtius rearrangements through the common isocyanate intermediate to provide carbamates/ureas (Scheme 1c). If successful, this would represent a unified green protocol for the Hofmann and Curtius rearrangements with great potential of practical large-scale production.
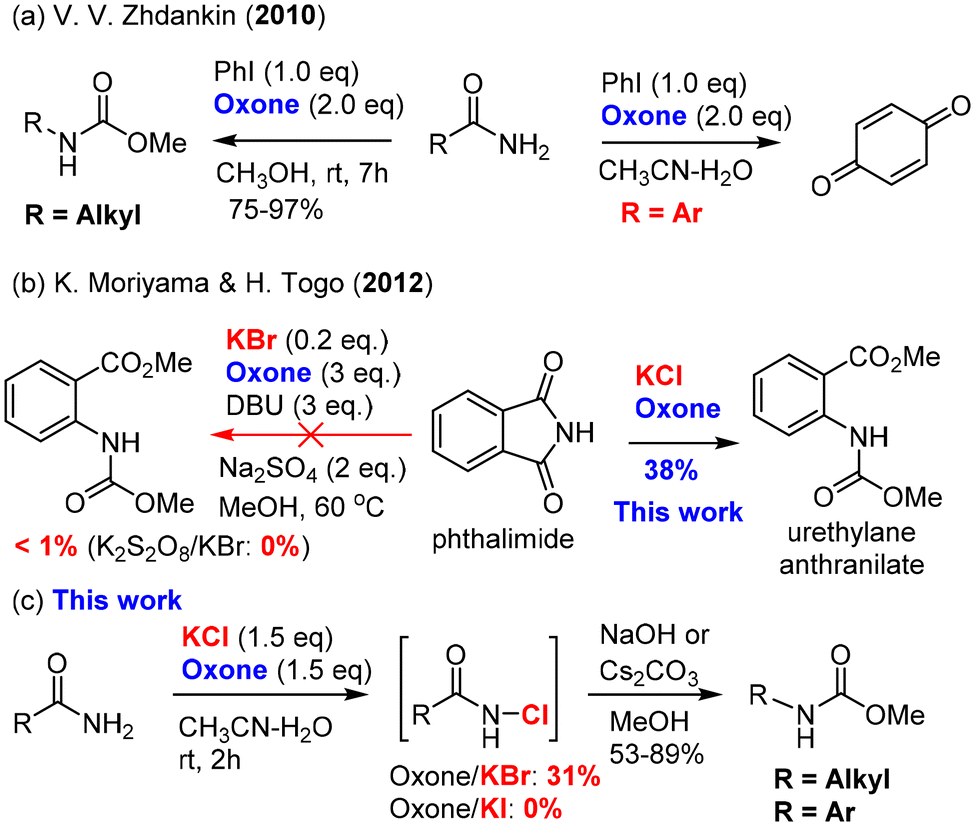
In this work, we report for the first time the chlorination of primary amides with oxone–KCl at room temperature to generate the desired intermediate N-chloro amides, which undergo a one-pot base-promoted Hofmann rearrangement to provide carbamates/ureas in good to excellent yields. Importantly, both aromatic and alkyl primary amides are suitable for this oxone–KCl system for the Hofmann rearrangement, which addresses the major limitation of Zhdankin's oxone–ArI system37,38 incompatible with aromatic primary amides (Scheme 2a). Secondly, we describe the development of an unprecedented oxidative azidation of aldehydes with oxone–KBr–NaN3, which gives expedited access to the reactive acyl azides for the Curtius rearrangement. Controlled experiments reveal that the oxidation of aldehydes to acyl bromide/azide might proceed in a radical mechanism, which is rarely reported for the oxidation with either oxone itself or the oxone–halide system. This work offers a practical advantage by using non-hazardous, inorganic, and inexpensive ACS-grade reagents (oxone: <20 US$ per kg; KCl: <10 US$ per kg; and KBr: <20 US$ per kg) for transforming readily available primary amides and aldehydes into drug molecules and a small library of high-value chiral urea catalysts used for asymmetric catalysis (Scheme 1c).
 | ||
| Scheme 2 The Hofmann rearrangement with oxone as a terminal oxidant. | ||
Results and discussion
Oxone–halide halogenation of primary amides for the Hofmann rearrangement
Since the classical procedure for the Hofmann rearrangement using an alkaline solution of bromine could be unsatisfactory and unreliable,13 many modifications of this protocol have been made including two major categories of reagents: (a) hypervalent iodine reagents15 [PhI(OCOCF3), PhI(OAc)2, PhIO–HCO2H, and PhI(OTs)OH] and N-bromosuccinimide (NBS) base (NBS–DBU,39 NBS–AgOAc,40 and NBS–KOH14). Recent efforts have led to the development of several remarkable catalytic processes such as bromide-catalyzed electrochemistry41–44 and ArI-catalyzed oxidation.37,45 For unknown reasons (economy or safety?), large-scale applications of these catalytic procedures have not been reported. The development of new and green protocols for the Hofmann rearrangement continues to be an important research field.Our hypothesis of using oxone as a terminal oxidant for the Hofmann rearrangement was first challenged by two precedented examples: Zhdankin's oxone/PhI37,38 was not applicable to aromatic amides (Scheme 2a), while Moriyama and Togo46 discovered that oxone/KBr (and K2S2O8/KBr) was ineffective for phthalimide (Scheme 2b). It was believed that aniline derivatives from the Hofmann rearrangement of aromatic amides were further oxidized by oxone to produce 1,4-benzoquinone in the oxone/PhI system. In the case of the ineffectiveness of phthalimide, we suspected that nitrogen was deactivated by two carbonyls and not electron-rich enough for the halogenation by BrOH or Br2 generated mainly by oxone/KBr. Our preliminary study showed that oxone/KI were completely ineffective to induce the Hofmann rearrangement of primary amides (Scheme 2c). Fortunately, we found that oxone/KCl was effective at room temperature when a base was added in a one-pot manner upon completion of the halogenation. Notably, both aromatic and alkyl primary amides were suitable for this oxidative Hofmann rearrangement and most importantly phthalimide was a suitable substrate for the Hofmann rearrangement to provide highly desirable urethylane anthranilate (Scheme 2b).
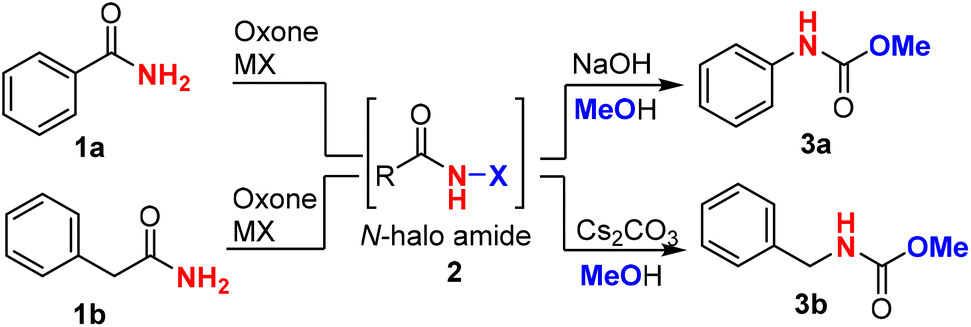
Firstly, we chose benzamide 1a as a representative aromatic amide and phenylacetamide 1b as a representative alkyl amide to optimize the reaction conditions (Table 1). We found that KI was not an effective halide for the halogenation of amide with oxone, while KBr as the halide source resulted in a lower yield (entries 1 and 2). To our delight, oxone–KCl was effective at halogenating the benzamide (1a) in a mixed solvent system (MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 10:1) and produced carbamate 3a in 90% yield when NaOH (1.5 equiv.) and methanol (1.5 mL) were added in a one-pot two-step manner (entry 3). The progress of the first-step reaction should be monitored constantly using thin layer chromatography (TLC) to avoid overoxidation of benzamide, which would lead to reduced yields. Surprisingly, NaCl and LiCl were inferior to KCl (entries 4 and 5), which might be due to the different abilities of forming metal ion hydrates and thus resulted in different oxidation potentials of the counter ion (Cl−).47 We further evaluated the different solvents, including protic and nonprotic solvents (entries 6–8), and found that acetonitrile/water was the optimal combination of solvents, which was consistent with our previous results. Since a base was mechanistically needed for the generation of isocyanate intermediate in the Hofmann rearrangement, we investigated other bases (NaOH, K2CO3, Cs2CO3, etc.) and found that NaOH was superior (entries 9–11). The amount of methanol could be reduced to 0.6 mL without decreasing the yield (entry 12). When phenylacetamide (1b) (aliphatic amide) was employed, similar conditions could deliver carbamate 3b with an excellent yield of 86% (entries 13–15). It was noted that cesium carbonate prevailed over NaOH, resulting in a cleaner reaction and slightly higher yield. Other protic or nonprotic solvents led to lower yields (entries 17–19). It was found that a smaller amount of methanol (0.6 mL) in the Hofmann rearrangement would significantly reduce the yield (entry 20). We have tried the Fenton–halide systems, including CeCl3, FeCl3, FeCl2, FeBr3etc., and found that the Fenton–halide system16,17,19,22–25 is not applicable to the Hofmann reaction. This might be due to iron/cerium coordinating strongly with primary amides and preventing their oxidation.
:H2O = 10:1) and produced carbamate 3a in 90% yield when NaOH (1.5 equiv.) and methanol (1.5 mL) were added in a one-pot two-step manner (entry 3). The progress of the first-step reaction should be monitored constantly using thin layer chromatography (TLC) to avoid overoxidation of benzamide, which would lead to reduced yields. Surprisingly, NaCl and LiCl were inferior to KCl (entries 4 and 5), which might be due to the different abilities of forming metal ion hydrates and thus resulted in different oxidation potentials of the counter ion (Cl−).47 We further evaluated the different solvents, including protic and nonprotic solvents (entries 6–8), and found that acetonitrile/water was the optimal combination of solvents, which was consistent with our previous results. Since a base was mechanistically needed for the generation of isocyanate intermediate in the Hofmann rearrangement, we investigated other bases (NaOH, K2CO3, Cs2CO3, etc.) and found that NaOH was superior (entries 9–11). The amount of methanol could be reduced to 0.6 mL without decreasing the yield (entry 12). When phenylacetamide (1b) (aliphatic amide) was employed, similar conditions could deliver carbamate 3b with an excellent yield of 86% (entries 13–15). It was noted that cesium carbonate prevailed over NaOH, resulting in a cleaner reaction and slightly higher yield. Other protic or nonprotic solvents led to lower yields (entries 17–19). It was found that a smaller amount of methanol (0.6 mL) in the Hofmann rearrangement would significantly reduce the yield (entry 20). We have tried the Fenton–halide systems, including CeCl3, FeCl3, FeCl2, FeBr3etc., and found that the Fenton–halide system16,17,19,22–25 is not applicable to the Hofmann reaction. This might be due to iron/cerium coordinating strongly with primary amides and preventing their oxidation.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Amide | MX | Base | Solv. | Yieldc (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
a Conditions: Halide and oxone were added to a solution of amide 1 (0.3 mmol) in the mixed solvent (1.1 mL, v/v solvent:H2O = 10:1) at room temperature and stirred for 2 h, then MeOH (1.5 mL) and a base (0.45 mmol) were added. The progress of the reaction was monitored using TLC.
b 0.6 mL MeOH was used.
c Isolated yield.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1a | KI | NaOH | MeCN | 0 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 1a | KBr | NaOH | MeCN | 31 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 1a | KCl | NaOH | MeCN | 90 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 1a | NaCl | NaOH | MeCN | 73(3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 1a | LiCl | NaOH | MeCN | 82(3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 1a | KCl | NaOH | THF | 11 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 1a | KCl | NaOH | DCM | 85 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 1a | KCl | NaOH | MeOH | 45 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 1a | KCl | LiOH | MeCN | 67 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1a | KCl | Cs2CO3 | MeCN | 71 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | 1a | KCl | K2CO3 | MeCN | 46 (3a) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | 1a | KCl | NaOH | MeCN | 89 ( 3a ) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | 1b | NaCl | NaOH | MeCN | 82 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | 1b | LiCl | NaOH | MeCN | 83 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 15 | 1b | KCl | NaOH | MeCN | 86 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 16 | 1b | KCl | Cs 2 CO 3 | MeCN | 88 ( 3b ) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 17 | 1b | KCl | Cs2CO3 | THF | 40 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 18 | 1b | KCl | Cs2CO3 | CH2Cl2 | 70 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 19 | 1b | KCl | Cs2CO3 | MeOH | 66 (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 20 | 1b | KCl | Cs2CO3 | MeCN | 55b (3b) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
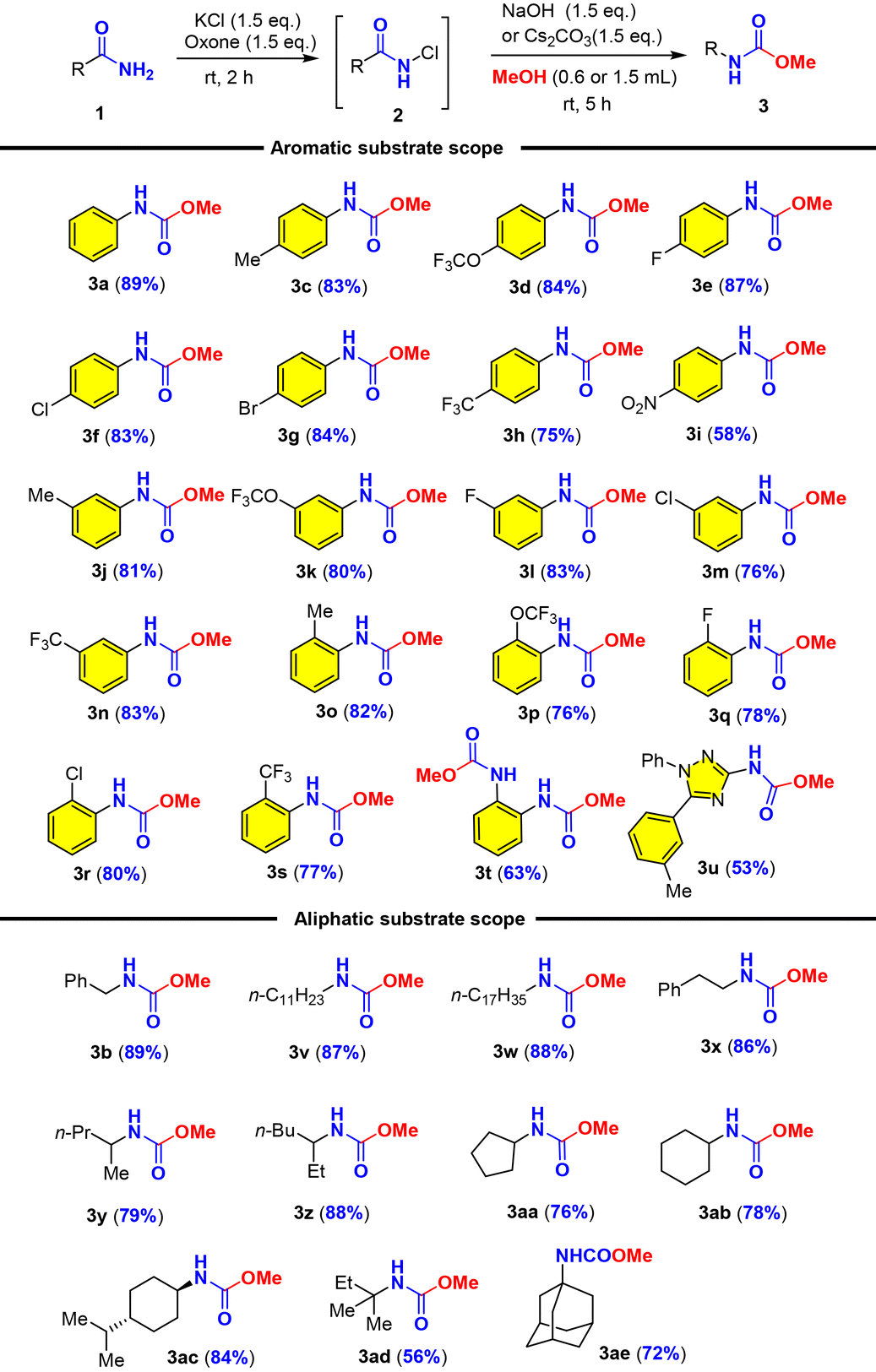
With the optimal conditions in hand, we set out to examine the amide scope including aromatic and aliphatic amides (Table 2). We first examined the effect of para-substituted aromatic amides and found that electron-donating and mild electron-withdrawing substituents (Me, OCF3, F, Cl, Br, and CF3) were well tolerated with good to excellent yields of carbamates (3c–3h), while strong electron-withdrawing substituents such as the nitro group (NO2) considerably reduced the yield (3i). It should be noted that electron-rich arenes tend to undergo halogenation with oxone–halide or further oxidation into quinones as recognized by Zhdankin et al. It was therefore significant to obtain electron-rich p-trifluoromethoxyl phenyl carbamate 3d in 84% yield without overoxidation. The effect of ortho- or meta-substituents (Me, OCF3, F, Cl, and CF3) on the arenes was also examined and they exerted insignificant steric/electronic effects on the reaction (3j–3s). It should be noted that phthalimide could be used for a double oxidative Hofmann rearrangement under our conditions to provide the corresponding dicarbamate in 63% yield (3t). Phthalimide that did not undergo the Hofmann rearrangement under oxone–KBr–DBU (Moriyama and Togo)46 could deliver 38% yield of urethylane anthranilate (Scheme 2b). Most noteworthy was that heterocyclic amide (1,2,4-triazole derived amide) was also suitable for this oxidative Hofmann rearrangement to deliver triazole carbamate with good 53% yield (3u). As illustrated by phenylacetamide (1b), aliphatic amides were further examined as competent substrates for our oxone–KCl oxidative Hofmann rearrangement. All alkyl amides reacted smoothly with oxone–KCl to generate the N-chloro amides and then the base-promoted Hofmann rearrangement provided carbamates (3v–3ae). The long-chain fatty amide lauric amide and stearamide showed good reactivity towards oxone–KCl, providing the desired carbamate products in 87% and 88% yields, respectively (3v–3w). Hydrocinnamide also afforded corresponding carbamates (3x) in 86% yield. α-Substituted and cyclic alkylcarboxamides were applicable for our oxone–KCl oxidative Hofmann rearrangement to generate secondary and tertiary carbamates (3y–3ae) in good to excellent yields regardless of the possible steric hindrance. It was particularly noteworthy that high fidelity of configuration retention was observed for chiral carboxamide (3ac).
|
a Conditions: KCl (0.45 mmol) and oxone (0.45 mmol) were added to a solution of primary amide (1, 0.3 mmol) in MeCN/H2O [10:1 (v/v), 1.1 mL] at room temperature and stirred for 2 h, and then MeOH/NaOH (0.6 mL/0.45 mmol) or MeOH/Cs2CO3 (1.5 mL/0.45 mmol) were added. The progress of the reaction was monitored using TLC.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Oxone–halide mediated azidation of aldehydes for the Curtius rearrangement
As an alternative to the Hofmann rearrangement, the Curtius rearrangement generates isocyanate intermediates via thermal decomposition of acyl azides. While the classical methods for the preparation of the rather unstable acyl azide substrates from carboxylic acids48 remain dominant in academia and industrial applications (Scheme 3a), great interest in the direct use of aldehydes for the in situ generation of acyl azides via oxidative azidation (Scheme 3b) has led to the identification of several efficient oxidants49–52 such as manganese(IV),50 chromium(VI),51,52 hypervalent iodine53,54 and 3,3′,5,5′-tetra-tert-butyldiphenoquinone55 (TBDP). However, a green and sustainable method for the synthesis of acyl azides from either carboxylic acids or aldehydes remains unknown in the literature. We herein proposed the oxone–halide–azide system as a green and sustainable approach for the in situ generation of acyl azide (Scheme 3c). We believed that the reaction of oxone, halide, and azide might generate in situ halogen azide (ClN3, BrN3 and IN3),56,57 which then would oxidize aldehyde into acyl azide via a radical mechanism (Scheme 3c, path i). Alternatively, oxone–halide might oxidize (unprecedentedly) aldehyde into acyl halide, which underwent acyl substitution with azide to produce the acyl azide intermediate (Scheme 3c, path ii). The challenge lies on the poor solubility of these inorganic salts (oxone, halide and NaN3) in organic solvents and instability of acyl azides towards water that is usually required for oxone-mediated reactions.26,27 | ||
| Scheme 3 Synthetic methods for the generation of acyl azides for the Curtius rearrangement. | ||
To verify our hypothesis, we chose p-chlorobenzaldehyde 4f as our model compound to examine the oxidative azidation of aldehyde with the oxone–halide–azide system for the Curtius rearrangement (Table 3). As anticipated, the addition of water to improve the dissolution of the three salts (oxone, MX and NaN3) caused an explosive release of nitrogen gas from the reaction system without detecting any carbamate products. After careful screening of various solvents (CHCl3, CH2Cl2, CCl4, hexane, THF, t-BuOH, MeCN, EtOAc and PhCF3) without using water as a co-solvent (entries 1–9), we found that only halogenated solvents (CHCl3, CH2Cl2, CCl4 and PhCF3) or non-polar hexane could afford the desired acyl azide intermediate and then the carbamate product. The reaction in such non-polar organic solvent appeared to be a solid–liquid biphasic system. The yield of CCl4 was slightly higher than that of PhCF3 (entries 3 and 9). However, as CCl4 is a highly hazardous solvent,58 we selected PhCF3 as a green and practical solvent59,60 for our study along with CCl4 for comparison. Notably, KBr was essential (entry 9) and outperformed the corresponding KCl and KI (entries 10 and 11). The better performance of KBr over KCl might be due to the faster and more efficient oxidation of KBr with oxone than the corresponding oxidation of KCl. Slightly higher loadings of oxone and NaN3 would improve the yield (entries 12 and 13), while a higher concentration was found to be detrimental to the reaction in terms of yield (entry 14). Some Fenton–halide conditions (H2O2/FeBr3 and H2O2/CeCl3) would result in the desired acyl azide in low yields (20–30%), which could be rationalized by the presence of water in the solution, which could hydrolyze the reactive acyl azide intermediate.
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | MX | Oxone (equiv.) | NaN3 (equiv.) | Solvent | Yieldc (%) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
a Conditions: Halide, oxone and NaN3 were added to a solution of 4f (0.3 mmol) in the solvent (3 mL, ACS grade and used as received) at room temperature. After stirring for 24 h, the reaction mixture was filtered and the filtrate was concentrated. The residue was dissolved in dry toluene/MeOH [10:1 (v/v), 3.3 mL] and stirred at 100 °C for 2 h. The progress of the reaction was monitored using TLC.
b 1.5 mL PhCF3 was used.
c Isolated yield. Note: reviewers suggested that CCl4 should be replaced by a green solvent and PhCF3 was identified as a green alternative to CCl4 with comparable yields.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | KBr | 1.5 | 2.0 | CHCl3 | 68 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | KBr | 1.5 | 2.0 | CH2Cl2 | 57 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | KBr | 1.5 | 2.0 | CCl4 | 76 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | KBr | 1.5 | 2.0 | Hexane | 46 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | KBr | 1.5 | 2.0 | THF | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | KBr | 1.5 | 2.0 | tBuOH | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | KBr | 1.5 | 2.0 | MeCN | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | KBr | 1.5 | 2.0 | EtOAc | 0 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | KBr | 1.5 | 2.0 | PhCF3 | 71 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | KCl | 1.5 | 2.0 | PhCF3 | 42 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | KI | 1.5 | 2.0 | PhCF3 | 16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 | KBr | 1.8 | 2.5 | PhCF3 | 82 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 13 | KBr | 1.8 | 2.2 | PhCF3 | 75 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 14 | KBr | 1.8 | 2.5 | PhCF3 | 66b | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
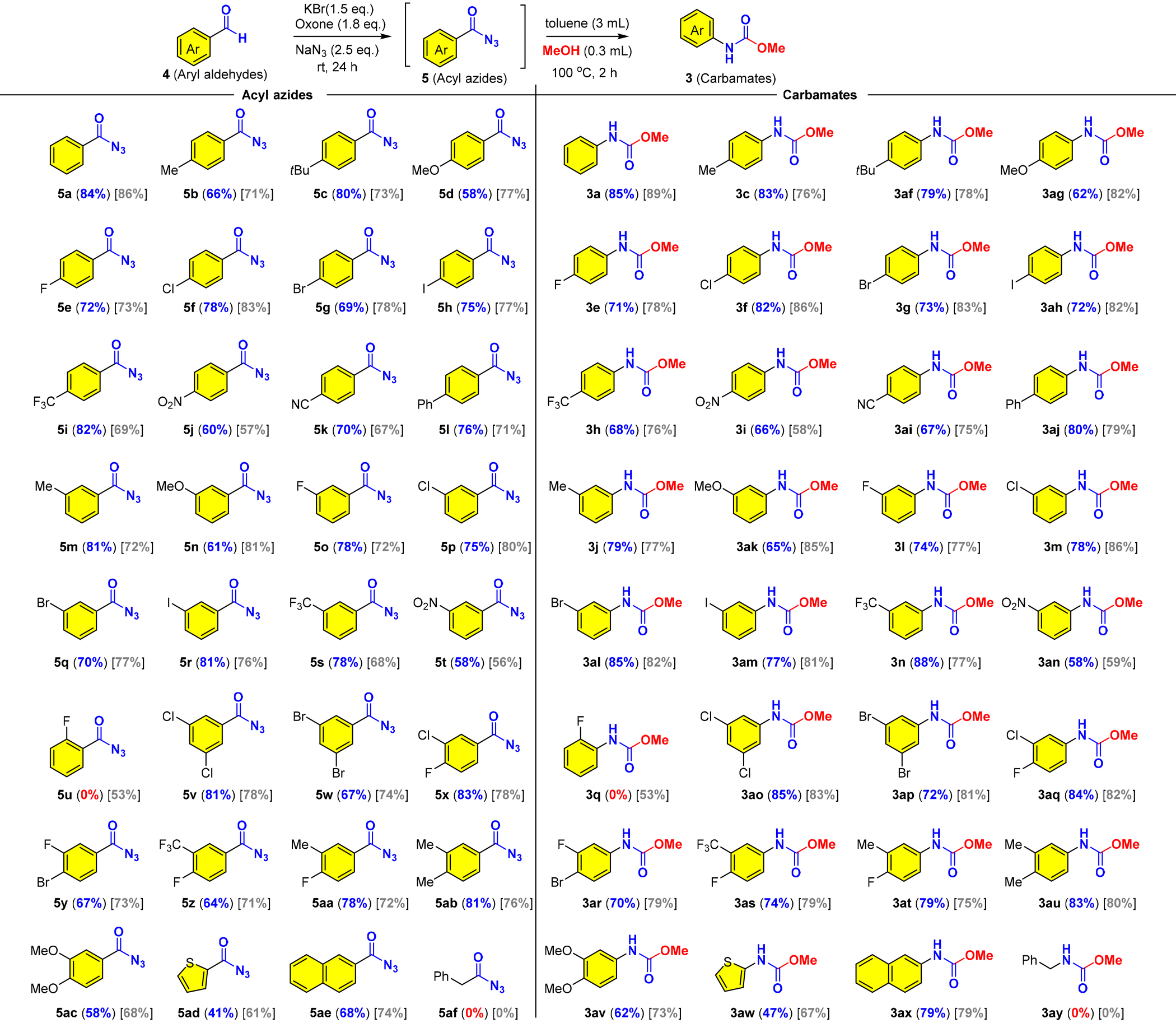
Next, we set out to examine the substrate scope and limitations of the reaction (Table 4). We found that aromatic aldehydes with different electronic properties could be transformed into the corresponding carbamates through acyl azides while aliphatic aldehydes failed in the oxidative azidation (5af/3ay). Notably, the aromatic acyl azides (5a–5ae) are stable for isolation and characterization with 1H NMR and 13C NMR and therefore presented together with related carbamates as shown in Table 4. We then investigated the effects of para-, meta-, and ortho-substituents (Me, Ph, t-Bu, OMe, F, Cl, Br, I, CF3, NO2, and CN) on benzaldehyde and found that most mild electron-donating and mild electron-withdrawing groups had no significant influence on the reaction, while a strong electron-donating substituent (OMe) and an electron-withdrawing substituent (NO2) on the benzaldehyde lowered the reaction yields to 58–66% (5d, 5j, 5n, 5t, 3i, 3ag, 3ak and 3an). For the substrates with meta substituents (5m–5t), no significant effect from the steric hindrance was observed and the reactions proceeded smoothly as para-substituted substrates (5b–5l). It was well known that ortho-substituted benzoyl azides were more reactive toward the Curtius rearrangement than those lacking an ortho-substituent.61 It was believed that the ortho-substituted aroyl azide generated in the reaction would quickly rearrange into the corresponding isocyanate and then was partially converted into ArNH2, which then reacted with isocyanate (ArNCO) to produce urea instead of the predicted carbamate. As a result, no desired product was obtained under our optimized conditions. In sharp contrast, only one example of ortho-substituted carbamate was attained in moderate yield (5u and 3q) in CCl4. This limitation was not observed in the oxidative Hofmann rearrangement (Table 2). Then we studied the meta/para di-substituted benzaldehydes bearing mild electron-withdrawing (Cl, Br, F, and CF3) or electron-donating (Me) groups. Gratifyingly, good yields of the desired products (acyl azides and carbamates) were obtained regardless of the electronic property (5v–5ab and 3ao–3au). The presence of two halides (5v, 5w, 5x, 5y; 3ao, 3ap, 3aq, and 3ar) offered a great chemical space for further elaborations (cf., transition metal catalyzed cross coupling reactions) in organic synthesis and drug discovery. Noteworthy was that naphthalene (5ae/3ax) could survive through the oxidative conditions, while the electron-abundant arene (5ac/3av) and thiophene (5ad/3aw) were obtained in lower yields. We found that PhCF3 provided comparable yields as compared to CCl4, except for the electron-abundant arenes (5d, 5j, 5n, 5t, 5ac, 3i, 3ag, 3ak, 3an and 3av), thiophene (5ad/3aw) and the ortho-substituted substrate (4u).
|
a Conditions: KBr (0.45 mmol), oxone (0.54 mmol) and NaN3 (0.75 mmol or 0.66 mmol) were added to a solution of 4 (0.3 mmol) in PhCF3 or CCl4 (analytical grade, 3 mL) at room temperature, and stirred for 24; the filtrate was concentrated and the residue was dissolved in dry toluene/MeOH [10:1 (v/v), 3.3 mL] and heated at 100 °C for 2 h. The progress of the reaction was monitored using TLC. The yield of carbamates was an isolated yield over two steps (from aldehyde) without isolation/purification of acyl azide intermediates. The yields in blue color in parentheses “()” were obtained in PhCF3, while the yields in gray color in square brackets “[]” were obtained in CCl4.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Synthetic utility of the oxone–halide protocols for the Hofmann and Curtius rearrangements
One of the key merits of this oxone–halide protocol is the green and efficient transformation of low-cost and commercially available aldehydes/amides into high-value pharmaceutically relevant amines/carbamates/ureas as exemplified by the following prices (Sigma-Aldrich) of compounds: 3,4-dimethoxybenzaldehyde (1.1 US$ per g) vs. 3,4-dimethoxyaniline (8.0 US$ per g); 2-naphthaldehyde (4.62 US$ per g) vs. 2-naphthylamine (664 US$ per g), and 2-thiophenecarboxaldehyde (0.55 US$ per g) vs. 2-aminothiophene (not commercially available). To further expand the utility of this new and green approach for synthesis of amine derivatives from amides and aldehydes, we explored the addition of other nucleophiles to trap the isocyanate intermediates for the synthesis of unsymmetrically substituted ureas/carbamates, drug molecules, and chiral catalysts as shown in Scheme 4. Oxone–KCl chlorination of benzamide afforded N-chloroamide 2a (Scheme 4a). Primary amines were used as both base and trapping nucleophile for the Hofmann rearrangement to provide asymmetrically substituted ureas (6a–6c). The addition of aqueous ammonia to N-chloroamide 2 smoothly triggered the Hofmann rearrangement at room temperature to provide N-phenyl urea 6a in 82% yield. Similarly, aqueous methyl amine and cyclohexylamine were also a competent base and nucleophile for the Hofmann rearrangement to yield urea products 6b and 6c in excellent yields. When N-chloroamide 2a was treated with sodium hydroxide to generate an isocyanate intermediate, benzamide could serve as a potential nucleophile to isocyanate to synthesize the corresponding benzoyl urea 6d. Similarly, a homoallyl alcohol was used to trap isocyanate and this afforded carbamate 6e in 85% yield, which was comparable with the addition of methanol. In addition to access to ureas and carbamates, our green oxone–halide method permitted low-cost synthesis of valuable ureas, including approved antitumor drugs and chiral urea catalysts from inexpensive aromatic aldehydes (Scheme 4b). We chose oxidative azidation of substituted benzaldehydes for the Curtius rearrangement to illustrate this application. Oxone–KBr mediated azidation of aromatic aldehydes (4) occurred at room temperature to generate the acyl azide, which upon heating at 100 °C resulted in the formation of isocyanates. The isocyanates could react with various amines to provide ureas. In order to avoid possible pyrolysis of chiral amines, the reaction of isocyanates with amines was carried out at room temperature by adding amines to the in situ generated isocyanates. Ureas 7a and 7b were produced by adding aqueous ammonia and aniline to the related N-(4-chloro)phenyl isocyanate. Interestingly, amide 7c was obtained by trapping the isocyanate with benzoic acid as a nucleophile. While the detailed mechanism remained elusive, it was believed that benzoyl carbamate was formed initially followed by the exclusion of carbon dioxide as reported by Mahajan et al.62 Vacor (7d), an old rodenticide, was identified as a novel antitumor agent by depleting NAD.63 Sorafenib (7e) and regorafenib (7f) were approved by the FDA for the treatment of carcinoma, possessing non-selective kinase inhibitor activities.64 These three antitumor agents (7d–7f) could be conveniently prepared using our methodology in excellent yields (84–85%). Chiral ureas are powerful organocatalysts that have been widely used in asymmetric catalysis. However, their preparation65–69 is generally limited to the availability of expensive isocyanates. To showcase the utility of our oxidative azidation of aldehydes for the Curtius rearrangement, we synthesized two small series of chiral urea catalysts namely trans-1,2-diamino cyclohexanes (7g–7j) and cinchona-derived bifunctional ureas (7k–7n) in excellent yields. Two gram-scale reactions were established to afford chiral ligands 7i and 7j with comparable yields (75% and 85%), demonstrating the practical application of the method. In principle, we could rapidly synthesize hundreds of chiral urea catalysts either from readily commercially available substituted benzaldehydes (4) listed in Table 4 or from primary amides listed in Table 2, which might be useful for the identification and optimization of chiral urea-based catalysts in the development of new asymmetric catalytic methodologies. | ||
| Scheme 4 The oxone–halide mediated oxidative Hofmann and Curtius rearrangements with other nucleophiles (not methanol). For the Curtius reaction, the yields in blue color in parentheses were obtained in PhCF3, while the yields in gray color in square brackets were obtained in CCl4. | ||
Mechanistic insights into the oxidation of amides/aldehydes with oxone–halide
The mechanism of the oxidative Hofmann rearrangement of amides was well-elucidated in the literature via the in situ generation of N-haloamide and isocyanate intermediates, which were consistent with our observation of N-chloroamide via TLC and the use of base for the rearrangement (Scheme 5f). Moreover, both benzamide (1a) and phenylacetamide (1b) were stable towards oxone if KCl was absent (Scheme 5a), which suggested the key role of chloride for the oxidation of amides into N-chloro amide. Notably, this represents the first example of the halogenative oxidation of amides with oxone–KCl, while the oxidative halogenation of amines into N-chloroamines was reported in 2018 by Madabhushi et al.70 In order to fully elucidate the mechanism, intermediate 2a (N-chloro benzamide) was prepared and characterized with 1H NMR and 13C NMR (see the ESI†). Next, we turned our attention to the mechanistic investigation of oxone–halide mediated azidation. Aromatic aldehyde (4f) was stable for at least 12 h in the presence of oxone (Scheme 5a). The radical pathway of our hypothesis (Scheme 5g) was supported by the results of our controlled experiments: the radical scavenger (1,4-benzoquinone, 1,1-diphenylethylene, and TEMPO) completely halted the oxidation reaction (Scheme 5b). Fresh preparation of bromo azide (BrN3) from oxone, KBr and NaN3 was performed in the PhCF3 solution by following Kappe's procedure56 and used for subsequent azidation reactions, which could provide benzoyl azide in 60% yield. This finding revealed that the true azidating species might be BrN3, which oxidizes the aldehyde into acyl azide in a radical pathway71 (Scheme 5g). On the other hand, we discovered that p-chlorobenzoyl bromide (8) could be prepared from p-cholorobenzaldehyde by oxone and KBr (Scheme 5d) and reacted smoothly with NaN3 to afford acyl azide 5f.72–74 To reconcile the seemingly contradictory results, we designed ortho-allyl benzaldehyde to investigate the possible presence of acyl radical, which if it exists might be trapped by the tethered alkene (Scheme 5e). Under our optimized conditions, we found that the oxidation reaction of o-allyl benzaldehyde (9) could afford the expected cyclization product 10, which might be derived from the generation of acyl radical and subsequent 5-exo radical cyclization/HX (HBr or HN3) elimination process. Taking all these experimental results into consideration, we believed that a radical pathway was well supported, while it was uncertain which of the radical pathways generated the acyl azide (Scheme 5g): (i) the in situ generation of hypobromous acid from oxone and bromide and then the reaction with an aldehyde in a radical mechanism to generate acyl bromide, which reacted with sodium azide as supported in Scheme 5d; or (ii) the in situ generation of bromine azide and then the reaction with an aldehyde through a radical mechanism to generate acyl azide directly without involving the acyl bromide intermediate and substitution. While the differentiation of these two pathways was difficult to confirm, they shared the common intermediate acyl radical, which was generated from the reaction of aldehyde with either the hypobromous acid or bromine azide. When the key acyl azide was formed, the subsequent Curtius rearrangement could be achieved by heating to generate the isocyanate intermediate, which reacted with alcohols or amines to provide the carbamates or ureas. | ||
| Scheme 5 Control experiments and proposed mechanisms of the oxidation of amides and aldehydes with oxone–halide. The corresponding reactions carried out in CCl4 resulted in comparable yields. | ||
Conclusion
In summary, we have developed the first green and efficient approach for the oxidation of amides and aldehydes with oxone–halide, leading to a sustainable and flexible access to N-halo amides and acyl azides, which undergo the Hofmann or Curtius rearrangement to provide synthetically and pharmaceutically valuable amines or protected amines (carbamates and ureas) through a common isocyanate intermediate. Our oxone–KCl system for the oxidative Hofmann rearrangement of primary amides not only addresses the limitation of Zhdankin's oxone–PhI method that is not applicable to primary aromatic amides but also overcomes the ineffectiveness of Moriyama/Togo's oxone–KBr protocol for phthalimide. It is the first example of N-halogenation of amides with oxone–halide. Our oxone–KBr–NaN3 system offers a green and sustainable approach for the in situ generation of water-sensitive acyl azides from aromatic aldehydes, enabling the oxidative Curtius rearrangement of aromatic aldehydes through formal insertion of nitrogen (from NaN3) between the aryl and carbonyl groups. We demonstrated the efficacy of our oxone–KBr–NaN3 system for the oxidative Curtius rearrangement with 30 examples and uncovered the rare radical mechanism of oxone–halide responsible for the in situ generation of acyl azides from aldehydes. We have showcased the synthetic utility by facile and green synthesis of three antitumor drugs and eight chiral urea organocatalysts. One of the key merits of this oxone–halide protocol is the green and efficient transformation of low-cost and commercially available aldehydes/amides into synthetically valuable and pharmaceutically relevant amines/carbamates/ureas. We expect that this green and benign oxone–halide system will substitute other halogenating reagents (NBS, NCS, t-BuOCl, Br2, etc.) and find wide applications for the oxidation and halogenation reactions in organic synthesis and process chemistry.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was financially supported by the National Key R&D Program of China (2022 YFD1400700), Research Grants Council of Hong Kong (C6026-19G, C6022-22W, 16306920, 16300921, 16308922). The authors also acknowledged the financial support from the Natural Science Foundation of Fujian Province of China (2020J01526), the Foundation of Education Department of Fujian Province of China (JAT200096), the GDAS Special Project of Science and Technology Development (2020GDASYL-20200202001 and 2022GDASZH-2022010110), and the Research and Development Projects in Key Fields in Guangdong Province (2020B1111580001 and 2021TQ060920).References
- P. Debnath, Curr. Org. Chem., 2019, 23, 2402–2435 CrossRef CAS.
- A. K. Ghosh, A. Sarkar and M. Brindisi, Org. Biomol. Chem., 2018, 16, 2006–2027 RSC.
- A. K. Ghosh, M. Brindisi and A. Sarkar, ChemMedChem, 2018, 13, 2351–2373 CrossRef CAS PubMed.
- A. I. Alfano, S. Pelliccia, G. Rossino, O. Chianese, V. Summa, S. Collina and M. Brindisi, ACS Med. Chem. Lett., 2023, 14, 326–337 CrossRef CAS.
- L.-H. Zhang, J. C. Chung, T. D. Costello, I. Valvis, P. Ma, S. Kauffman and R. Ward, J. Org. Chem., 1997, 62, 2466–2470 CrossRef CAS PubMed.
- G. Gambacorta and I. R. Baxendale, Org. Process Res. Dev., 2022, 26, 422–430 CrossRef CAS.
- T. A. Phung Hai, L. J. S. De Backer, N. D. P. Cosford and M. D. Burkart, Org. Process Res. Dev., 2020, 24, 2342–2346 CrossRef CAS.
- C. R. Sagandira and P. Watts, Eur. J. Org. Chem., 2017, 6554–6565 CrossRef CAS.
- M. Baumann, A. Leslie, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2021, 25, 452–456 CrossRef CAS.
- M. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin, C. D. Smith and J. P. Tierney, Org. Biomol. Chem., 2008, 6, 1577–1586 RSC.
- C. J. Mallia, N. G. McCreanor, D. H. Legg, C. R. Stewart, S. Coppock, I. W. Ashworth, J. Le Bars, A. Clarke, G. Clemens, H. Fisk, H. Benson, S. Oke, T. Churchill, M. Hoyle, L. Timms, K. Vare, M. Sims and S. Knight, Org. Process Res. Dev., 2022, 26, 3312–3322 CrossRef CAS.
- M. A. Marsini, F. G. Buono, J. C. Lorenz, B.-S. Yang, J. T. Reeves, K. Sidhu, M. Sarvestani, Z. Tan, Y. Zhang, N. Li, H. Lee, J. Brazzillo, L. J. Nummy, J. C. Chung, I. K. Luvaga, B. A. Narayanan, X. Wei, J. J. Song, F. Roschangar, N. K. Yee and C. H. Senanayake, Green Chem., 2017, 19, 1454–1461 RSC.
- E. S. Wallis and J. F. Lane, in Organic Reactions, 1946, vol. 3, pp. 267–306 Search PubMed.
- C. H. Senanayake, L. E. Fredenburgh, R. A. Reamer, R. D. Larsen, T. R. Verhoeven and P. J. Reider, J. Am. Chem. Soc., 1994, 116, 7947–7948 CrossRef CAS.
- R. M. Moriarty, C. J. Chany II, R. K. Vaid, O. Prakash and S. M. Tuladhar, J. Org. Chem., 1993, 58, 2478–2482 CrossRef CAS.
- P. Tian and R. Tong, Green Chem., 2023, 25, 1345–1350 RSC.
- L. Song, Y. Zhou, H. Liang, H. Li, Y. Lai, H. Yao, R. Lin and R. Tong, J. Org. Chem., 2023, 88, 504–512 CrossRef CAS PubMed.
- Y. Zheng, Y. T. Cheung, L. Liang, H. Qiu, L. Zhang, A. Tsang, Q. Chen and R. Tong, Chem. Sci., 2022, 13, 10479–10485 RSC.
- G. Zhao, Y. Wang, C. Wang, H. Lei, B. Yi and R. Tong, Green Chem., 2022, 24, 4041–4049 RSC.
- L. Song, Y. Lai, H. Li, J. Ding, H. Yao, Q. Su, B. Huang, M.-A. Ouyang and R. Tong, J. Org. Chem., 2022, 87, 10550–10554 CrossRef CAS.
- L. Liang, L.-D. Guo and R. Tong, Acc. Chem. Res., 2022, 55, 2326–2340 CrossRef CAS.
- C. He, F. Ma, W. Zhang and R. Tong, Org. Lett., 2022, 24, 3499–3503 CrossRef CAS.
- G. Zhao, E. Wang and R. Tong, ACS Sustainable Chem. Eng., 2021, 9, 6118–6125 CrossRef CAS.
- G. Zhao, L. Liang, E. Wang and R. Tong, ACS Catal., 2021, 11, 3740–3748 CrossRef CAS.
- G. Zhao, L. Liang, E. Wang, S. Lou, R. Qi and R. Tong, Green Chem., 2021, 23, 2300–2307 RSC.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS.
- S. Alvi, V. Jayant and R. Ali, ChemistrySelect, 2022, 7, e202200704 CrossRef CAS.
- I. Saikia, A. J. Borah and P. Phukan, Chem. Rev., 2016, 116, 6837–7042 CrossRef CAS.
- W. M. Gołebiewski and M. Gucma, Synthesis, 2007, 3599–3619 CrossRef.
- J. Ren and R. Tong, Org. Biomol. Chem., 2013, 11, 4312–4315 RSC.
- J. Xu and R. Tong, Green Chem., 2017, 19, 2952–2956 RSC.
- J. Xu, L. Liang, H. Zheng, Y. R. Chi and R. Tong, Nat. Commun., 2019, 10, 4754 CrossRef PubMed.
- Z. Li and R. Tong, J. Org. Chem., 2016, 81, 4847–4855 CrossRef CAS.
- G. Zhao and R. Tong, Green Chem., 2019, 21, 64–68 RSC.
- G. Zhao and R. Tong, Tetrahedron, 2019, 75, 1669–1675 CrossRef CAS.
- G. Zhao, L. Liang, C. H. E. Wen and R. Tong, Org. Lett., 2019, 21, 315–319 CrossRef CAS.
- A. Yoshimura, K. R. Middleton, M. W. Luedtke, C. J. Zhu and V. V. Zhdankin, J. Org. Chem., 2012, 77, 11399–11404 CrossRef CAS.
- A. A. Zagulyaeva, C. T. Banek, M. S. Yusubov and V. V. Zhdankin, Org. Lett., 2010, 12, 4644–4647 CrossRef CAS PubMed.
- X. Huang, M. Seid and J. W. Keillor, J. Org. Chem., 1997, 62, 7495–7496 CrossRef CAS.
- S.-S. Jew, H. Geun Park, H.-J. Park, M.-S. Park and Y.-S. Cho, Tetrahedron Lett., 1990, 31, 1559–1562 CrossRef CAS.
- S. Tatsuya, M. Yoshihiro, Y. Shin-ichiro and K. Shigenori, Chem. Lett., 1982, 11, 565–568 CrossRef.
- Y. Matsumura, T. Maki and Y. Satoh, Tetrahedron Lett., 1997, 38, 8879–8882 CrossRef CAS.
- Y. Matsumura, Y. Satoh, K. Shirai, O. Onomura and T. Maki, J. Chem. Soc., Perkin Trans. 1, 1999, 2057–2060, 10.1039/A904126E.
- L. Li, M. Xue, X. Yan, W. Liu, K. Xu and S. Zhang, Org. Biomol. Chem., 2018, 16, 4615–4618 RSC.
- K. Miyamoto, J. Yamashita, S. Narita, Y. Sakai, K. Hirano, T. Saito, C. Wang, M. Ochiai and M. Uchiyama, Chem. Commun., 2017, 53, 9781–9784 RSC.
- K. Moriyama, K. Ishida and H. Togo, Chem. Commun., 2012, 48, 8574–8576 RSC.
- H. Y. Chen and E. Ruckenstein, J. Phys. Chem. B, 2015, 119, 12671–12676 CrossRef CAS PubMed.
- D. Joseph and S. Lee, Org. Lett., 2022, 24, 6186–6191 CrossRef CAS PubMed.
- N. D. Arote and K. G. Akamanchi, Tetrahedron Lett., 2007, 48, 5661–5664 CrossRef CAS.
- S. S. Elmorsy, Tetrahedron Lett., 1995, 36, 1341–1342 CrossRef CAS.
- J. G. Lee and K. H. Kwak, Tetrahedron Lett., 1992, 33, 3165–3166 CrossRef CAS.
- P. S. Reddy, P. Yadagiri, S. Lumin, D.-S. Shin and J. R. Falck, Synth. Commun., 1988, 18, 545–551 CrossRef CAS.
- Y. Shinomoto, A. Yoshimura, H. Shimizu, M. Yamazaki, V. V. Zhdankin and A. Saito, Org. Lett., 2015, 17, 5212–5215 CrossRef CAS.
- D. S. Bose and A. V. N. Reddy, Tetrahedron Lett., 2003, 44, 3543–3545 CrossRef CAS.
- S. De Sarkar and A. Studer, Org. Lett., 2010, 12, 1992–1995 CrossRef CAS.
- D. Cantillo, B. Gutmann and C. Oliver Kappe, Org. Biomol. Chem., 2016, 14, 853–857 RSC.
- R. A. Valiulin, S. Mamidyala and M. G. Finn, J. Org. Chem., 2015, 80, 2740–2755 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. J. Maul, P. J. Ostrowski, G. A. Ublacker, B. Linclau and D. P. Curran, Modern Solvents in Organic Synthesis, 1999, vol. 206, pp. 79–105 Search PubMed.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. V. Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC.
- F. M. Miloserdov and V. V. Grushin, Angew. Chem., Int. Ed., 2012, 51, 3668–3672 CrossRef CAS.
- A. Kurnar, N. Kumar, R. Sharma, G. Bhargava and D. Mahajan, J. Org. Chem., 2019, 84, 11323–11334 CrossRef.
- D. Buonvicino, F. Mazzola, F. Zamporlini, F. Resta, G. Ranieri, E. Camaioni, M. Muzzi, R. Zecchi, G. Pieraccini, C. Dolle, M. Calamante, G. Bartolucci, M. Ziegler, B. Stecca, N. Raffaelli and A. Chiarugi, Cell Chem. Biol., 2018, 25, 471–482 CrossRef CAS PubMed.
- R. Ronchetti, G. Moroni, A. Carotti, A. Gioiello and E. Camaioni, RSC Med. Chem., 2021, 12, 1046–1064 RSC.
- T. Okino, Y. Hoashi, T. Furukawa, X. N. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS PubMed.
- T. Azuma, A. Murata, Y. Kobayashi, T. Inokuma and Y. Takemoto, Org. Lett., 2014, 16, 4256–4259 CrossRef CAS.
- S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS PubMed.
- M. S. Manna and S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS.
- S. C. Mallojjala, R. Sarkar, R. W. Karugu, M. S. Manna, S. Ray, S. Mukherjee and J. S. Hirschi, J. Am. Chem. Soc., 2022, 144, 17399–17406 CrossRef.
- V. Sriramoju, S. Kurva and S. Madabhushi, Synth. Commun., 2018, 48, 699–704 CrossRef CAS.
- V. K. Yadav, V. P. Srivastava and L. D. S. Yadav, Tetrahedron Lett., 2016, 57, 2502–2505 CrossRef CAS.
- M. Swetha, P. V. Ramana and S. G. Shirodkar, Org. Prep. Proced. Int., 2011, 43, 348–353 CrossRef CAS.
- H. Y. Zeng, Q. Tian and H. W. Shao, Green Chem. Lett. Rev., 2011, 4, 281–287 CrossRef CAS.
- P. Bellotti, J. Brocus, F. El Orf, M. Selkti, B. Konig, P. Belmont and E. Brachet, J. Org. Chem., 2019, 84, 6278–6285 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04355j |
| This journal is © The Royal Society of Chemistry 2024 |