
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrthogonal assisted tandem reactions for the upgrading of bio-based aromatic alcohols using chitin derived mono and bimetallic catalysts†
Francesco
Zorzetto
 a,
Daniel
Ballesteros-Plata
b,
Alvise
Perosa
a,
Enrique
Rodríguez-Castellón
b,
Maurizio
Selva
*a and
Daily
Rodríguez-Padrón
*a
a,
Daniel
Ballesteros-Plata
b,
Alvise
Perosa
a,
Enrique
Rodríguez-Castellón
b,
Maurizio
Selva
*a and
Daily
Rodríguez-Padrón
*a
aDepartment of Molecular Science and Nanosystems, Ca’ Foscari University of Venice, Via Torino 155, 30175 Venezia, Italy. E-mail: selva@unive.it; daily.rodriguez@unive.it
bDepartment of Inorganic Chemistry, Facultad de Ciencias, Universidad de Málaga, Campus de Teatinos s/n, 29071 Málaga, Spain
First published on 26th March 2024
Abstract
The upgrading of a benzyl-type alcohols was explored via an orthogonal tandem sequence comprised of a first oxidative step producing the corresponding aldehydes, and a subsequent reductive amination to achieve both secondary and tertiary amines. To the scope, acetonitrile (ACN) was used as a solvent and a source/precursor of reactant amines, and different heterogeneous catalysts based on Rh and Mo, were designed as mono- and bi-metallic systems in the form of metal nanoparticles dispersed on a chitin-derived N-doped carbons. A parametric analysis carried out separately for the oxidation and the reductive amination allowed to choose the best performant catalyst for both the reactions of the tandem process. A one-pot two-step protocol was implemented accordingly: as an example, benzyl alcohol was quantitatively and selectively oxidised to benzaldehyde (>99%) which in turn, was converted to N-benzylethanamine (66%) or N-benzyl-N-ethylethanamine (60%) in the presence of [Rh(5%)-N/C–Mo(5%)]-N/C or [Rh(3%)-N/C–Mo(5%)]-N/C as catalysts, respectively. The tandem sequence proved successful also for other bio-based benzyl-type alcohols that afforded the corresponding secondary/tertiary amines in yields up to 53–93%. Overall, the study proved the viability of an innovative method aimed not only at process intensification for multistep synthesis, but also at the valorization of substrates (alcohols) and biopolymers (chitin) derived from biomass.
 Daily Rodríguez-Padrón | Daily Rodríguez-Padrón is a Marie-Curie Post-Doctoral researcher at Università Ca’ Foscari di Venezia, Italy (Marie Sklodowska-Curie Cofund Grant Agreement No. 945361). She earned her Bachelor's degree in Chemistry from the University of Havana, Cuba, in 2013, and completed her Ph.D. in the Department of Organic Chemistry at the University of Cordoba, Spain, in 2020. In April 2020, she joined KelAda Pharmachem Ltd (Dublin, Ireland) as a visiting postdoctoral researcher, contributing to the Horizon 2020 Marie Skłodowska-Curie Action (MSCA) RISE project titled GreenX4Drug. Dr Rodríguez-Padrón has undertaken research stays in esteemed universities, including the Universita degli Studi Mediterranea di Reggio Calabria and the Università degli Studi di Messina in Italy, as well as the PSL Research University, Chimie ParisTech CNRS, in France. She serves on the Editorial Board of Sustainable Chemistry and has been invited as a Guest Editor for various journals, including Current Opinion in Green and Sustainable Chemistry, Topics in Current Chemistry, and Nanomaterials. Dr Rodríguez-Padrón has been laureated with the Dan David Prize 2019 in the field of Combatting Climate Change from Tel-Aviv University in Israel and the Green Talent Award 2020 from the German Federal Ministry of Education and Research. Her research primarily focuses on mechanochemistry, biomass valorisation, heterogeneous catalysis, and sustainability. |
Introduction
Tandem reactions, especially in combination with renewables, offer a powerful tool to design strategies for the valorization of biomass derived molecules.1–3 Apart from simple bi-catalytic reactions, the nature of the catalysts and the mechanisms allow to classify different sequences as domino and tandem reactions, auto tandem, assisted tandem catalytic processes, and orthogonal tandem catalytic (OTC) trasformations.4,5 The last category, though it requires a challenging catalyst design, is perhaps the most attractive one for its intrinsic wide applicability based on the concept of two different catalysts operating side-by-side to perform two sequential catalytic cycles.6 OTC-reactions are further divided into orthogonal auto tandem catalysis and orthogonal assisted tandem catalysis depending on whether an input is provided after the first cycle is finished.As part of our long-standing interest on this subject,2–5 OTC assisted tandem catalysis caught our attention to devise protocols aimed at combining the upgrading of bio-based compounds with the use of catalysts synthesised from renewable sources. A one-pot two step sequence comprised of an initial oxidation of benzyl-type alcohols to the corresponding aldehydes followed by the reductive amination of the aldehydes to both secondary and tertiary alkylamines, was chosen for the scope of this paper (Scheme 1).
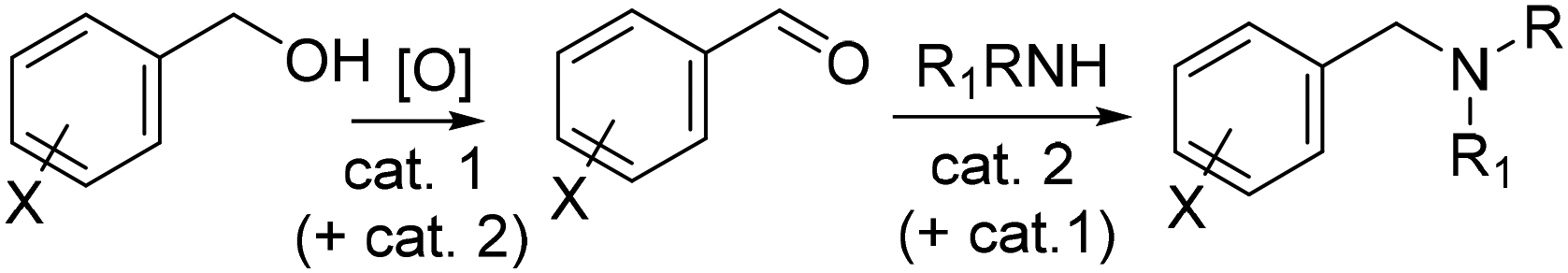 | ||
| Scheme 1 Orthogonal tandem catalytic (OTC) sequence in this work. | ||
Yet, the selection of catalysts (cat. 1 and cat. 2) and reaction conditions was neither obvious nor straightforward.
A literature inspection indicated that among the many catalytic systems available for the alcohols oxidation,7,8 emerging trends highlighted the use of supported nano-sized transition metals based on not endangered elements (Mo, Fe, Co),9 coupled to benign oxidants, such as molecular oxygen and air.10,11 Also, carbocatalysts, specifically carbon nanotubes (CNTs), have been recently proposed as sustainable materials for the conversion of benzyl alcohol to benzaldehyde, though a solid oxidant as peroxymonosulfate (PMS) was necessary.12
On the other hand, a variety of transition metal catalysts and conditions have been reported to convert carbonyl compounds to amines via protocols of reductive amination. This is a fundamental reaction in organic synthesis, covering about a quarter of all processes applied for the formation of C–N bonds in pharmaceutical and medicinal chemistry.13,14 The use of Rh as an active phase deserve attention in this context. Excellent results have been reported by immobilizing Rh nanoparticles (NPs) on spherical covalent organic frameworks (COFs) in which the nitrogen atoms of the framework/support served as anchoring sites to stabilize metal atoms, forming sufficiently strong coordinate bonds. These nanocatalysts allowed quantitative reductive amination processes (+30 examples) with selectivity often >90%, under mild conditions (2 MPa H2 and 90 °C).15 Other approaches described Rh-NPs supported on both N-doped carbons (achieved from mixtures of phenanthroline and activated carbons) and carbon nanofibers as highly effective catalysts for the conversion of aromatic aldehydes into primary and secondary amines with yields in the range of 90–99%.16 Moreover, aside from ammonia and amines, a range of nitrogen-containing derivatives with functional groups amenable to reduction have been described as reaction partners. These included nitriles, amides, nitro derivatives, urea, sulfonylhydrazines, azides, methyl carbamates, and isocyanates. Nitriles, in particular, were one of the most promising options from both environmental and large-scale production standpoints, because of their versatility to act in a double role as reaction solvents and precursors of amines obtained via in situ hydrogenation. This strategy was recently exemplified in a previous contribution by members of our group, that described a cascade reaction where a Pt/TiO2 nanocatalyst allowed the hydrogenation of acetonitrile to a mixture of primary and secondary amines that were used in the same reactor for the reductive amination of benzaldehyde (Scheme 2).17
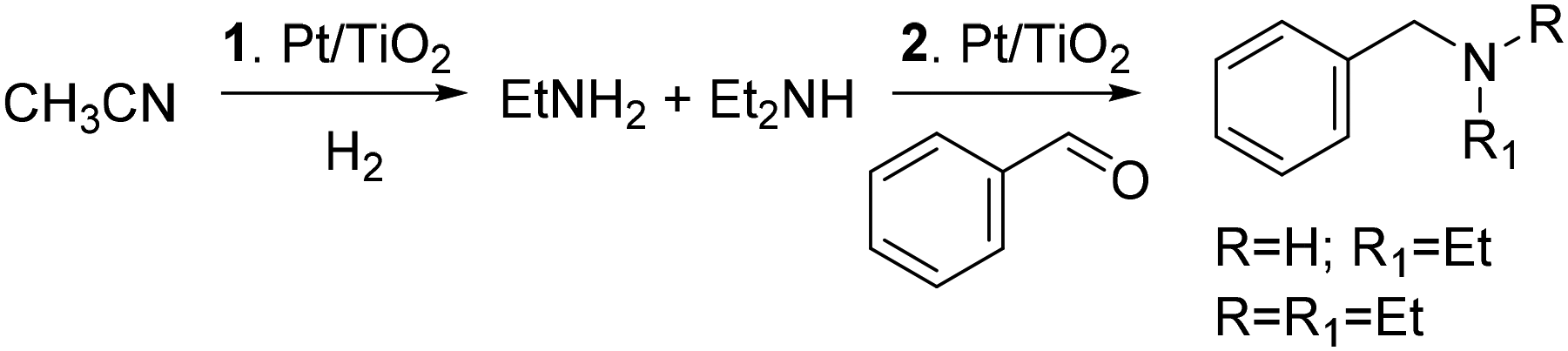 | ||
| Scheme 2 Pt/TiO2 nanocatalyst for a cascade reaction: 1: hydrogenation of acetonitrile; 2: reductive amination of benzaldehyde. | ||
Also, other TiO2-supported Rh, Ru, Pt and Pd nanoparticles proved effective for the acetonitrile-assisted reductive amination of furfural.18
From this analysis, the choice for the catalysts design in this paper fell on transition metal nanoparticles (MNPs) supported on N-doped carbons. These carbonaceous-based materials have in principle, two interesting attributes: (i) the N-sites may be active for base-catalyzed reactions, and (ii) if used as supports for MNPs, they allow a uniform dispersion of the active sites thanks to specific bonding interactions between the nanoparticles and N-functional groups, particularly pyridinic ones, distributed on the support surface.19 These interactions significantly prevent metal leaching, thereby leading to the synthesis of robust catalysts. The preparation of N-doped materials (N/C) has been described through a variety of carbonization processes carried out by combining different carbon and nitrogen sources,20 or alternatively, starting from N-containing polymers of both fossil and bio-based origin as polyvinylpyrrolidone,21 and chitosan,22 respectively. In this context, also the use of chitin, the second most abundant biopolymer on Earth, can be valorized for the sustainable synthesis of heterogeneous catalysts. Commercial chitin is available in dozen million tons from the extraction of biowaste of the fisheries sectors, mostly crustacean shells, but contrarily to chitosan, its applications are limited due to the low solubility in almost all media. Therefore, strategies to convert chitin into high-added value materials and composites are highly desirable in the circular economy vision.23–28 We recently contributed to this subject by designing a library of catalysts comprised of core–shell nanoparticles of metal/metal oxides based on Ni, Fe, Co and Mo supported on chitin-derived N-doped carbons.29 In the presence of air and acetonitrile as an oxidant and solvent, respectively, these systems proved effective for the oxidation of different benzyl-type alcohols; interestingly, supported Mo was the most performant catalyst to control the products distribution and achieve conversion and selectivity both above 90% towards the corresponding aldehydes as products of partial oxidation.
Inspired by these findings and considerations, we wish to report herein an innovative strategy for the tandem process outlined in Scheme 1 and further described in Scheme 3 which specifies the active metal phases and their involvement in the multiple steps of the overall sequence. Supported Mo was the oxidation catalyst by which alcohols dissolved in acetonitrile (ACN) were converted into aldehydes. ACN, solvent for the oxidation reaction, was the precursor of amines (as in Scheme 2) reactants for the step of reductive amination. A Rh-based system catalysed both the hydrogenation of acetonitrile and the reductive amination reaction.
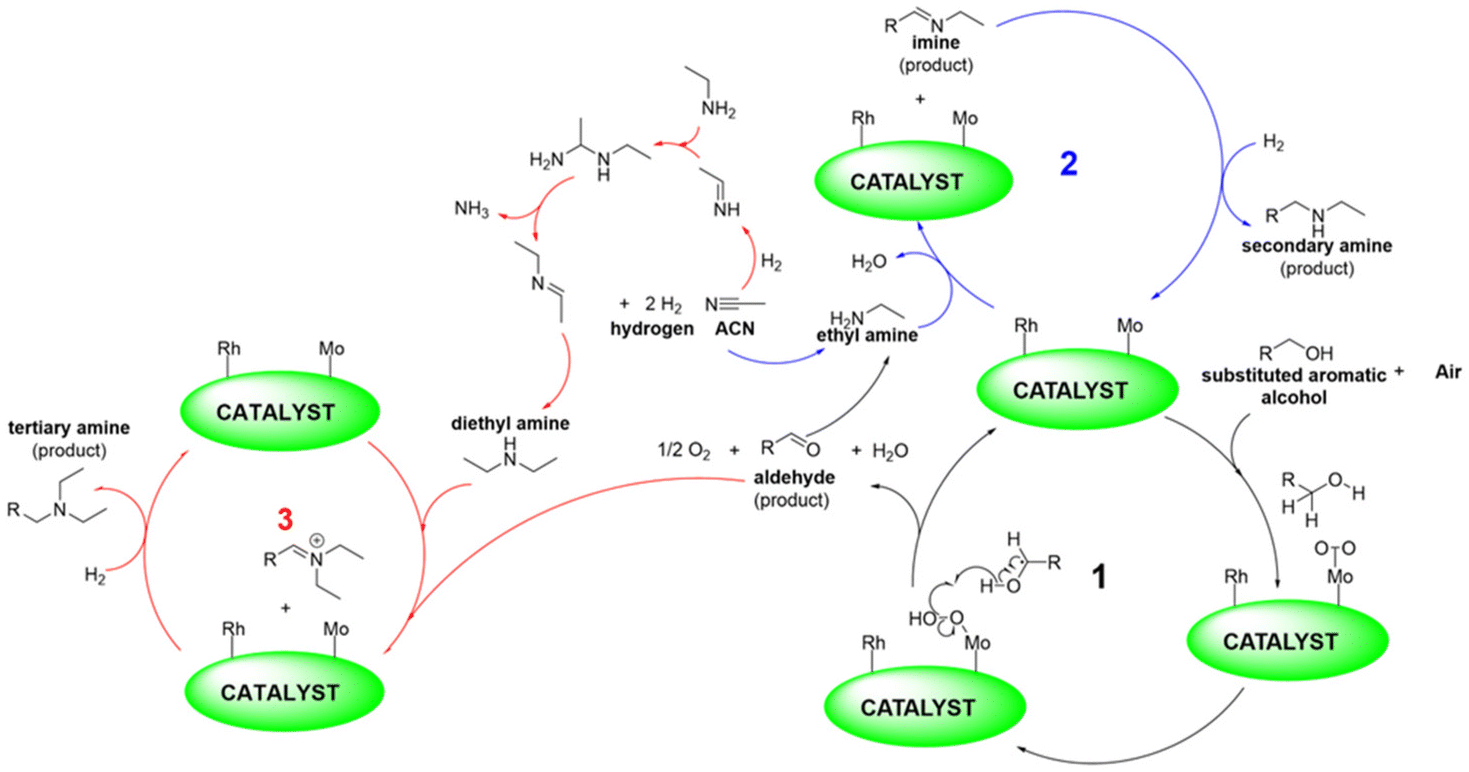 | ||
| Scheme 3 Tandem sequence comprised of oxidation of alcohols, hydrogenation of acetonitrile, and reductive amination of aldehydes. Further details are discussed in the ESI, Scheme S3.† | ||
This paper demonstrates that the catalysts achieved by either co-dispersion of Mo- and Rh-nanoparticles on N-doped carbons or by mechanical mixing of the corresponding mono-metallic systems, proved not only active for the desired reactions, but also stable and tolerant to the diverse (oxidative/reductive) environments experienced throughout the entire sequence. For example, in the presence of a mixture of [Rh(5%)-N/C–Mo(5%)]-N/C or [Rh(3%)-N/C–Mo(5%)]-N/C as catalysts and acetonitrile as solvent/amines precursor, benzyl alcohol dissolved in ACN was quantitatively and selectively oxidized to benzaldehyde (>99%) using air as an oxidant. Then, by switching from air to a H2 atmosphere, benzaldehyde was converted to a mixture of N-benzylethanamine or N-benzyl-N-ethylethanamine, whose proportions were increased up to 66% and 60%, respectively, by changing the Rh loading from 5 to 3 wt% in the used catalytic systems. The protocol was successfully extended to other biomass-derived alcohols.
For comparison, albeit this work was focused on molybdenum and rhodium-based materials, other metals (Pd, Ru) were also explored.
Results and discussion
Envisioning the design of a robust, versatile, and high-performance catalytic system tailored for OTC reactions, this investigation explored Mo, Pd, Ru, and Rh precursors. A solution-based methodology was followed by using EDTA as a chelating agent, 2-propanol as solvent, and chitin as a dual source of carbon and nitrogen (Fig. 1). After heating (80 °C, 9 h) with stirring, the suspension was filtered, and the residual solid was thermally treated (500 °C) under a N2 flow. Our previous studies,29 confirmed that this synthetic strategy was advantageous to achieve a better control over the morphology, size, and dispersion of the metal particles compared to conventional impregnation methods.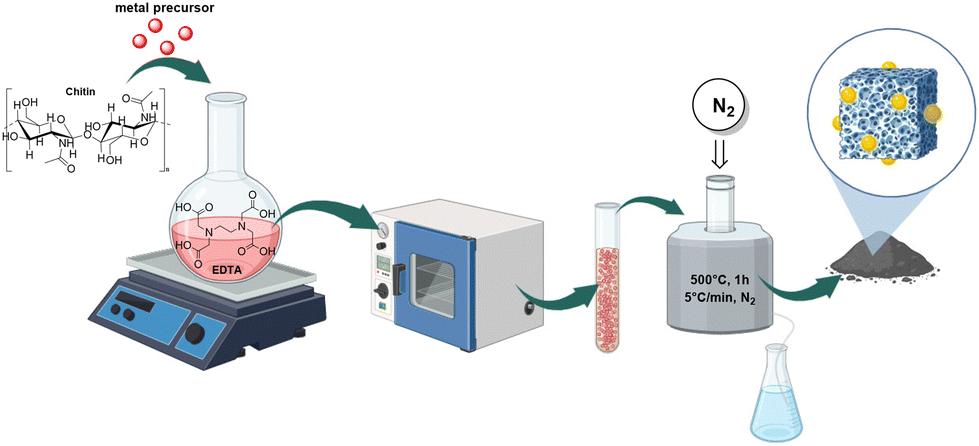 | ||
| Fig. 1 Schematic representation of the synthetic strategy used for the preparation of the catalytic samples. | ||
Catalysts characterization
All samples underwent comprehensive characterization through a multi-technique approach, encompassing X-ray Photoelectron Spectroscopy (XPS), X-ray powder diffraction (XRPD), N2-physisorption measurements, High-Resolution Transmission Electron Microscopy (HRTEM), and Inductively Coupled Plasma Mass Spectrometry (ICP-MS). The full characterization analysis of molybdenum nanoparticles supported on N-doped carbon materials was reported in a previous study of our group (details have been provided in the ESI†). A more comprehensive discussion is presented here for rhodium-based catalysts, along with their bimetallic (Mo/Rh) counterparts. Moreover, characterization results of other materials prepared using the same synthetic protocol, such as Pd and Ru-modified samples, also assessed in the reductive amination step with lower selectivity, are additionally presented in the ESI.†Monometallic catalysts
Rhodium nanoparticles supported on N-doped carbons were examined at various loadings of 5, 3, and 1 wt%. The corresponding samples were designated as Rh(5%)-N/C, Rh(3%)-N/C, and Rh(1%)-N/C, respectively. XRD patterns of the rhodium-based materials are shown in Fig. 2. | ||
| Fig. 2 XRD patterns of the Rh-based materials with loading of 5 wt% (black), 3 wt% (blue), and 1 wt% (green). | ||
A common feature of all samples, regardless the metal loadings, was the presence of a peak at around 25°. As for the Mo(5%)-N/C sample (see ESI, Fig. S1†), this peak was attributed to the (002) crystallographic plane, related to the parallel stacking of graphene-like sheets. Notably, the same signal was observed in the XRD diffractograms of all the other Ni, Co, Fe, Ru and Pd-based catalysts (shown in the ESI, Fig. S4 and S8†).
The broad peak shape was primarily due to the amorphous nature of the synthesized samples. The three least intense signals at 41, 48 and 70° were associated to the typical diffractions of the (111), (200), and (220) crystalline planes of metallic Rh.30 The intensity of these three peaks increased at the higher Rh loading, thereby confirming a correlation with the metal content (Fig. 2).
The textural properties of the Rh(5%)-N/C and Rh(1%)-N/C samples were investigated by N2-physisorption analysis. Fig. 3 compares the corresponding adsorption isotherms. Both samples exhibited mesoporous characteristics, featuring type-IV isotherms.
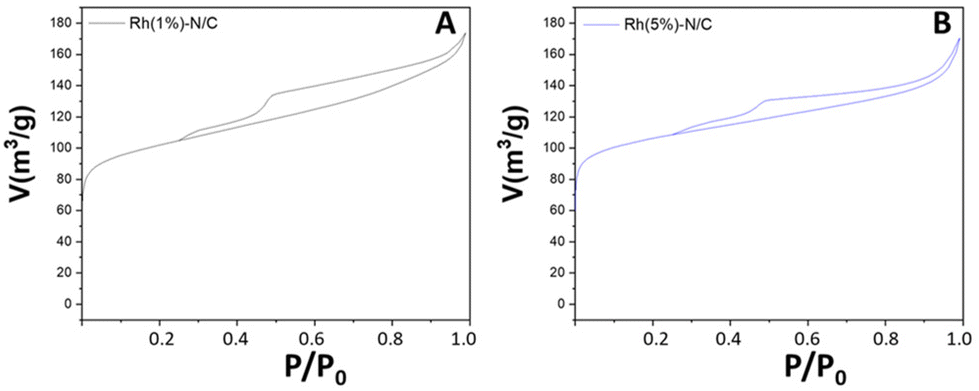 | ||
| Fig. 3 N2-physisorption isotherms of Rh(1%)-N/C (A) and Rh(5%)-N/C (B), and their comparison (C). | ||
The BET surface area, mean pore diameter, and pore volume of the Rh-N/C samples are shown in Table 1. The two samples exhibited outstanding features; interestingly, however, their surface area was nearly 40% larger than that of the molybdenum-based material (cfr. Table S1, ESI†). It was hypothesized that this difference was due to the different metal precursors used in each case, (NH4)MoO4 and RhCl3·H2O, respectively. The slight increase in the mean pore size diameter and the reduction in pore volume of the Rh(5%)-N/C sample compared to the Rh(1%)-N/C sample suggested that the higher metal loading brought about an occlusion of smaller pores within the carbonaceous matrix.
| Material sample | S BET [m2 g−1] | D BJH [nm] | V BJH [cm3 g−1] |
|---|---|---|---|
| a S BET: specific surface area was calculated by the Brunauer–Emmett–Teller (BET) equation. b D BJH: mean pore size dimeter was calculated by the Barret–Joyner–Halenda (BJH) equation. c V BJH: pore volumes were calculated by the Barret–Joyner–Halenda (BJH) equation. | |||
| Rh(1%)-N/C | 474 | 5.8 | 0.24 |
| Rh(5%)-N/C | 489 | 6.5 | 0.23 |
XPS analysis of a representative Rh-based sample, particularly Rh(5%)-N/C is presented in Fig. 4. Furthermore, the XPS results of the Rh(1%)-N/C and Rh(3%)-N/C samples are reported in Fig. S11 and S12,† respectively. Spectra confirmed the presence of Rh particles on the surface of the nitrogen-doped carbonaceous supports.
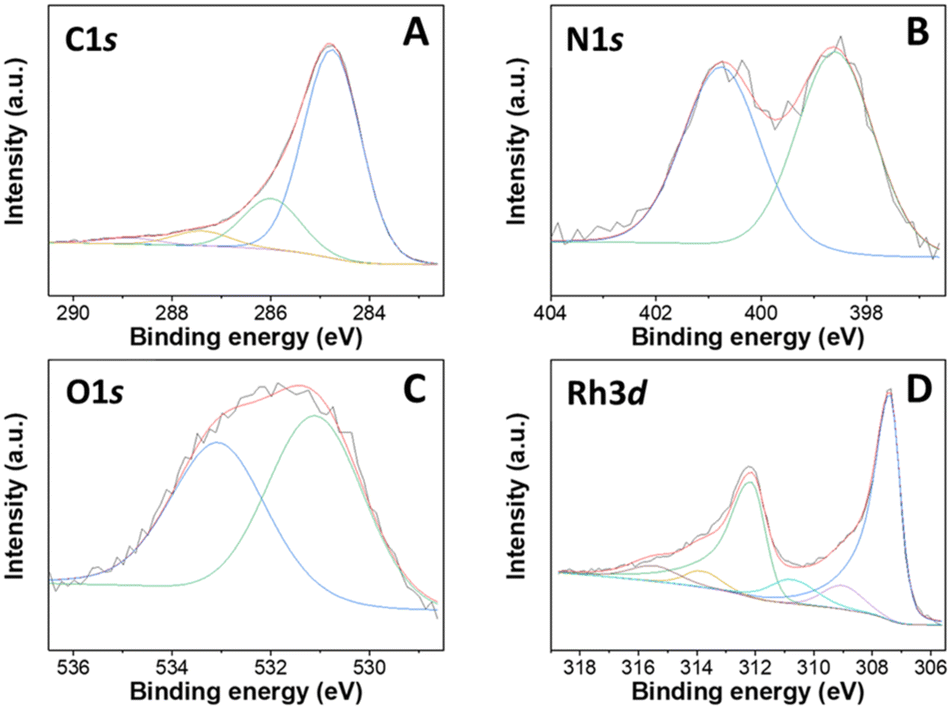 | ||
| Fig. 4 XPS spectra of the Rh(5%)-N/C sample, including C 1s (A), N 1s (B), O 1s (C) and Rh 3d (D) regions. | ||
The C 1s spectra of the Rh samples were deconvoluted into four contributions located at approximately 284.7, 286.1, 287.5, and 289.0 eV (Fig. 4A). Such contributions correspond to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (graphitic and aromatic carbon), C–OH or C–N/C–O, CO bonds, and carbonate, respectively. In addition, in the N 1s XPS regions, the core-level XPS spectrum revealed the presence of two main peaks at ca. 398.6 and 400.8 eV, associated to pyridinic (Np) and pyrrolic (Npyr) groups, respectively (Fig. 4B). Moreover, in the O 1s XPS region of the prepared materials, two primary contributions were observed at around 531.1 and 533.1 eV, attributed to lattice oxygen in metal oxides and adsorbed/bonded water, respectively (Fig. 4C).
C bonds (graphitic and aromatic carbon), C–OH or C–N/C–O, CO bonds, and carbonate, respectively. In addition, in the N 1s XPS regions, the core-level XPS spectrum revealed the presence of two main peaks at ca. 398.6 and 400.8 eV, associated to pyridinic (Np) and pyrrolic (Npyr) groups, respectively (Fig. 4B). Moreover, in the O 1s XPS region of the prepared materials, two primary contributions were observed at around 531.1 and 533.1 eV, attributed to lattice oxygen in metal oxides and adsorbed/bonded water, respectively (Fig. 4C).
In the Rh 3d region, peaks characteristic of both Rh0 and Rh3+ were detected at binding energies of 307 and 309 eV, respectively (Fig. 4D). More specifically, the peaks at 307 and 312 eV were assigned specifically to the doublet Rh 3d5/2 and Rh 3d3/2, distinctive of Rh0, and the peaks at 309 and 314 eV were respectively assigned to the doublet Rh 3d5/2 and Rh 3d3/2, of Rh3+.31 Furthermore, the presence of additional contributions at around 310.7 and 315.4 eV strongly indicated the formation of Rh–N bonds.
As previously commented, this metal–N interactions could likely enhance the stability of the samples preventing leaching phenomena during their use under liquid phase reaction conditions. Interestingly, a slight decrease in the pyridinic nitrogen content was noticed as the rhodium content in the samples increased (Fig. 5). This decrease was attributed to an increasing formation of nitrogen-metal bonds on the surface. XPS quantification analysis are reported in Table S2 in the ESI.†
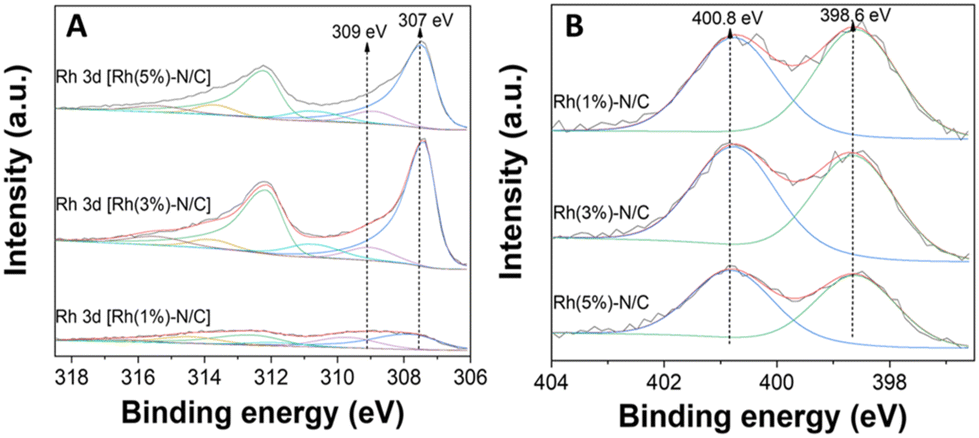 | ||
| Fig. 5 (A) Rh 3d regions of the Rh samples Rh(1%)-N/C, Rh(3%)-N/C, and Rh(5%)-N/C, (B) N 1s regions of the Rh samples Rh(1%)-N/C, Rh(3%)-N/C, and Rh(5%)-N/C. | ||
HRTEM and STEM images of the three Rh-based mono-metallic samples (Rh(5%)-N/C, Rh(3%)-N/C, and Rh(1%)-N/C) are displayed in Fig. 6. The presence of a well-defined laminar structure of the carbonaceous support was noticed in all samples. HRTEM images confirmed the presence of uniformly distributed Rhodium nanoparticles with an average diameter of (5.9 ± 2.0) nm for the Rh(5%)-N/C sample, (5.2 ± 2.0) nm for the Rh(3%)-N/C sample, and (3.9 ± 2.0) nm for the Rh(1%)-N/C sample. Additionally, micrographs and mapping analysis provided evidence of the formation of highly dispersed metallic entities throughout the sample (Fig. 6 and 7, respectively).
 | ||
| Fig. 6 HRTEM (A, B and C) and STEM (D, E and F) micrographs of the Rh-N/C samples with 5%, 3% and 1% of Rh respectively. | ||
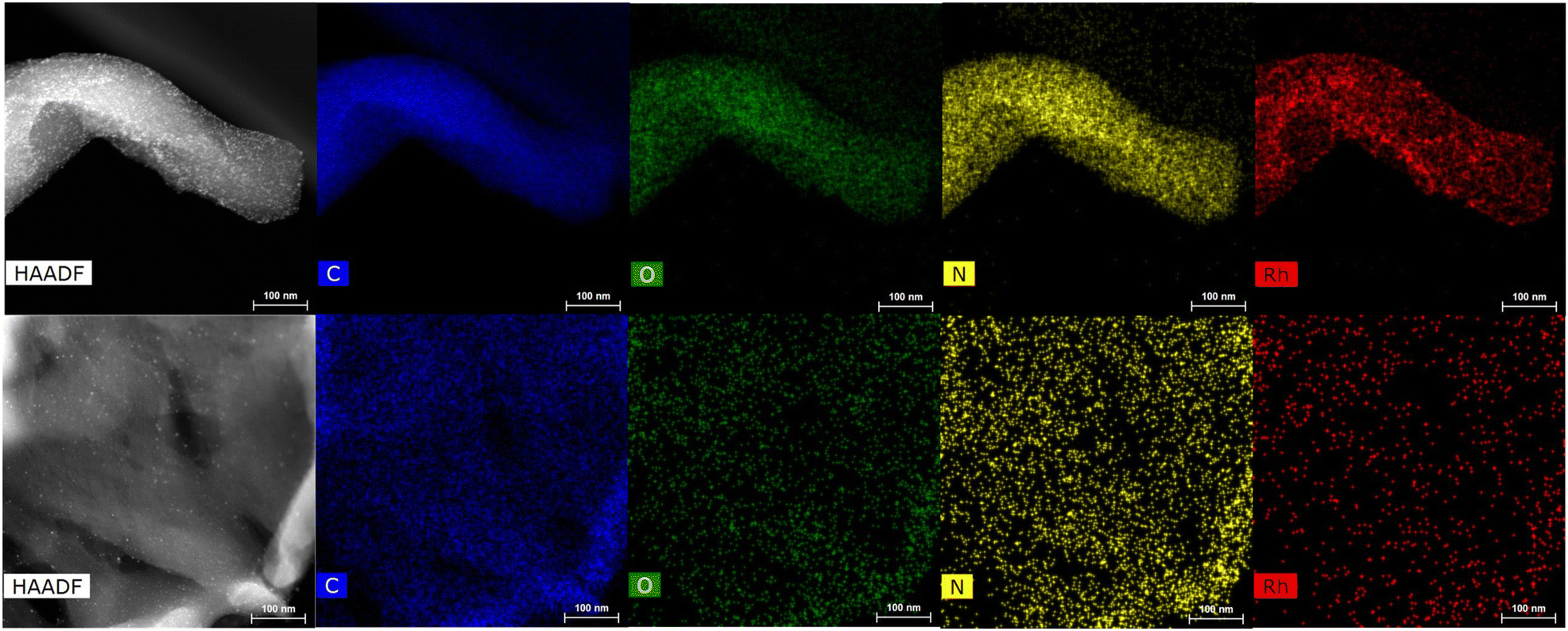 | ||
| Fig. 7 HRTEM in STEM mode-EDX micrographs of the Rh-N/C samples: Rh(3%)-N/C (top) and Rh(1%)-N/C (down). | ||
In addition to the monometallic samples, bimetallic counterparts were also prepared looking forward to developing OTC processes. Two bimetallic samples were prepared at a constant loading of molybdenum (5 wt%) and varying the Rh amount from 3 wt% to 5 wt%. These catalysts were labelled as [Rh(3%)-Mo(5%)]-N/C and [Rh(5%)-Mo(5%)]-N/C, respectively.
The [Rh(3%)-Mo(5%)]-N/C sample was chosen as the representative bimetallic sample for characterization in this work. It is reasonable to anticipate that bimetallic materials would display unique characteristics and behaviors compared to their monometallic counterparts. In this case, possible interactions between Rh and Mo metal centers could lead to unique physicochemical properties and different catalytic behaviors in the reactions under investigation. The discussion of these aspects will be presented in the following paragraphs.
The XRD pattern of the sample is shown in Fig. 8. Alike the monometallic materials, the peak at approximately 25° was associated to the (002) crystallographic plane related to the parallel stacking of graphene-like sheets.
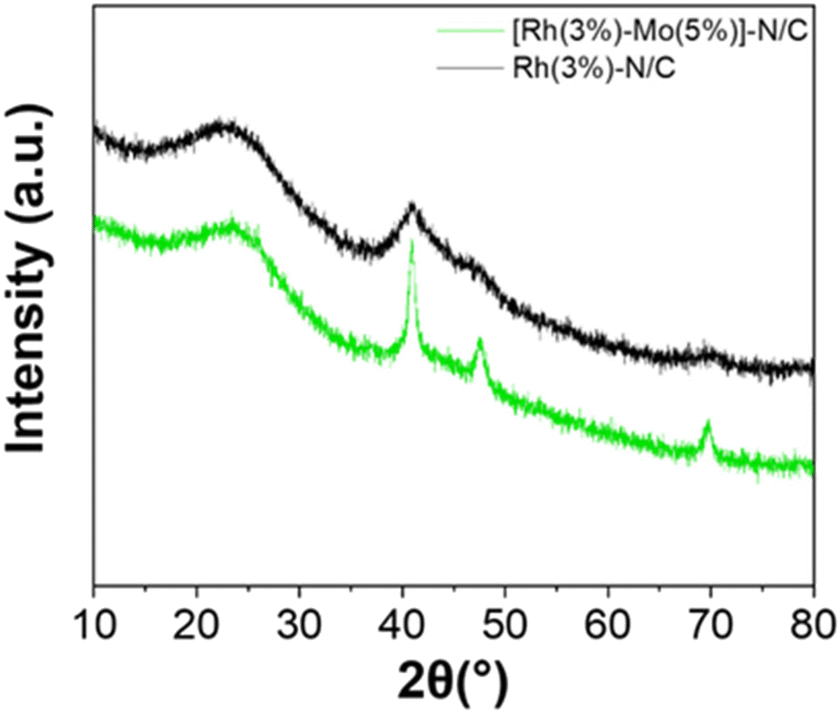 | ||
| Fig. 8 XRD patterns of the sample [Rh(3%)-Mo(5%)]-N/C (green) compared with the monometallic sample Rh(3%)-N/C (black). | ||
The three distinct and sharp peaks at 41, 47 and 70° were ascribed to the characteristic diffractions associated with the (111), (200) and (220) crystalline planes of metallic Rh. These results mirrored those observed for the monometallic Rh-based sample, albeit differences in signals intensity were discernible, with the bimetallic material exhibiting more well-defined peaks. Interestingly, no additional signals related to the presence of molybdenum entities were detected, thereby suggesting the formation of well-dispersed low-sized metal nanoparticles or even clusters.
The adsorption isotherm of [Rh(3%)-Mo(5%)]-N/C, as well as a comparison with the adsorption isotherms of the monometallic samples are shown in Fig. 9 and Fig. S13 (ESI).†
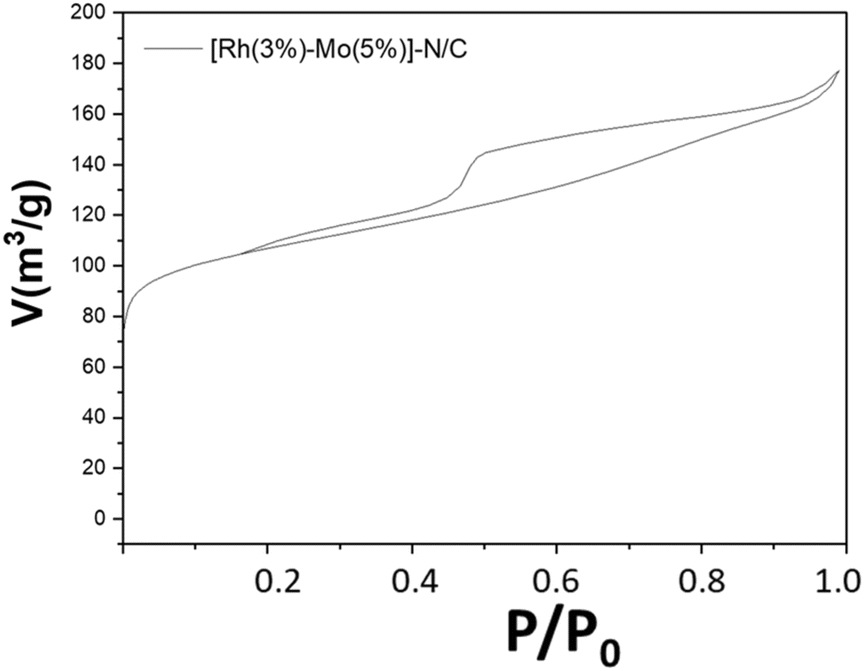 | ||
| Fig. 9 N2-physisorption isotherms at −196 °C of the prepared bimetallic [Rh(3%)-Mo(5%)]-N/C sample. | ||
Bimetallic catalysts
The bimetallic catalyst displayed a behavior analogue to that previously described for monometallic materials, exhibiting a type-IV isotherm reasonably associated with the presence of a mesoporous structure. Moreover, the surface area, mean pore diameter, and pore volume of [Rh(3%)-Mo(5%)]-N/C closely resembled those obtained for the monometallic rhodium catalyst (Table 1). This similarity suggested that the formation of the mesoporous architecture was mainly influenced by the rhodium precursor (Table 2).| Material sample | S BET [m2 g−1] | D BJH [nm] | V BJH [cm3 g−1] |
|---|---|---|---|
| a S BET: specific surface area was calculated by the Brunauer–Emmett–Teller (BET) equation. b D BJH: mean pore size dimeter was calculated by the Barret–Joyner–Halenda (BJH) equation. c V BJH: pore volumes were calculated by the Barret–Joyner–Halenda (BJH) equation. | |||
| [Rh(3%)-Mo(5%)]-N/C | 496 | 5.4 | 0.25 |
| Rh(1%)-N/C | 474 | 5.8 | 0.24 |
| Mo(5%)-N/C | 285 | 2.9 | 0.20 |
The bimetallic sample was also characterized by XPS analysis (see Fig. 10 and S14†). In analogy to the previously discussed XPS spectra of monometallic materials: (i) the C 1s region of [Rh(3%)-Mo(5%)]-N/C, showed four distinct contributions located at ca. 284.7, 285.9, 287.3, and 288.8 eV, which corresponded to C–C/CC bonds (graphitic Ng and aromatic carbon), C–OH or C–N/C–O, CO and carboxylate/carbonate bonds, respectively; (ii) the N 1s regions of the sample showed two main peaks at ca. 398.6 and 400.8 eV, due to pyridinic (Np) and pyrrolic (Npyr) groups, respectively; (iii) the O 1s XPS region showed two main contributions at approximately 531.0 and 533.1 eV, associated with lattice oxygen in metal oxides and adsorbed/bonded water, respectively.
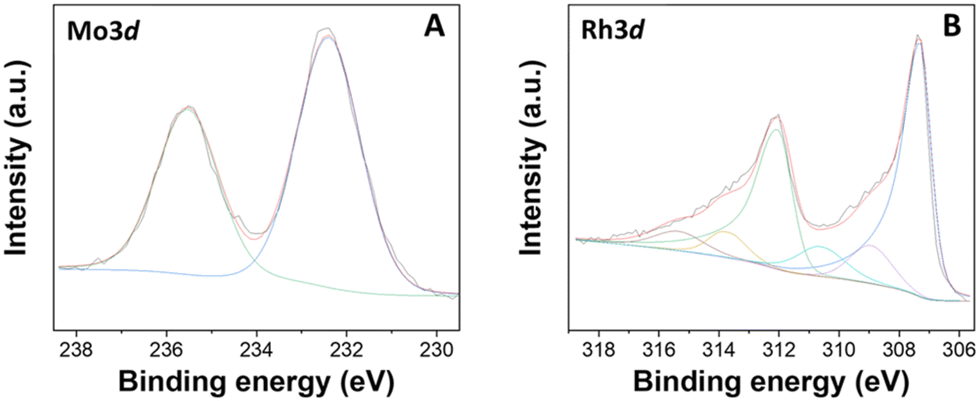 | ||
| Fig. 10 XPS spectra of the [Rh(3%)-Mo(5%)]-N/C sample, including (A), Mo 3d and (B) Rh 3d regions. | ||
The Mo 3d and Rh 3d regions were then inspected (Fig. 10). In the Mo 3d XPS region, two distinct contributions were identified at 232.4 and 235.5 eV, assigned to the doublet Mo 3d5/2–Mo 3d3/2, indicative of MoO3. Some remarkable differences were noticed from the comparison with the XPS spectrum of the monometallic Mo(5%)-N/C sample (Fig. S1, ESI†). Interestingly, the signals at binding energies of 230.4 and 233.5 eV, associated with the doublet Mo 3d5/2 and Mo 3d3/2 contributions of Mo(IV) in MoO2, previously observed in the monometallic material, were absent in the bimetallic sample. Moreover, no signals related to Mo–N bonds were detected for the [Rh(3%)-Mo(5%)]-N/C material. It was hypothesized that a stronger affinity between rhodium and nitrogen resulted in the exclusive formation of Rh–N bonds instead of Mo–N bonds.
By contrast, the Rh 3d regions of the bi- and the mono-metallic sample were substantially superimposable (Fig. 4 and 10), thereby indicating that the presence of molybdenum did not influence the chemical nature of the rhodium entities.
HRTEM images of the two Rh- and Mo-based bi-metallic samples ([Rh(5%)-Mo(5%)]-N/C and [Rh(3%)-Mo(5%)]-N/C) are displayed in Fig. 11. HRTEM images confirmed the presence of uniformly distributed rhodium nanoparticles. Rh particles in the [Rh(5%)-Mo(5%)]-N/C sample (Bi-M5) present similar dimensions to the mono-metallic Rh-based materials while, for the lower Rh loading of the [Rh(3%)-Mo(5%)]-N/C sample (Bi-M3), the particles displayed certain tendency to form agglomerates. On the other hand, Mo nanoparticles were found homogeneously dispersed in both bimetallic samples. It is pertinent to highlight that monometallic and bimetallic systems may exhibit distinct behaviors in terms of nanoparticle nucleation and formation. Deeper investigations are required to unveil the impact of molybdenum and higher rhodium concentrations on nanoparticles formation and dimensions.
 | ||
| Fig. 11 HRTEM in STEM mode-EDX micrographs of the bimetallic samples: [Rh(5%)-Mo(5%)]-N/C (top) and [Rh(3%)-Mo(5%)]-N/C (down). | ||
Catalytic activity
The synthesized catalytic systems, both the mono- and bi-metallic samples, were tested for the tandem sequence of Scheme 3 which was comprised of the oxidation of a benzyl-type alcohol to the corresponding aldehyde, the hydrogenation of acetonitrile and imines, and the reductive amination of the aldehyde.Moreover, under the investigated conditions, competitive transamination side-reactions occurred (described later in this section). Due to complexity of these multiple pathways, the approach used to study the tandem sequence was to explore the reaction steps separately in order to optimize the conditions independently for the single involved processes.
Selective oxidation of BnOH
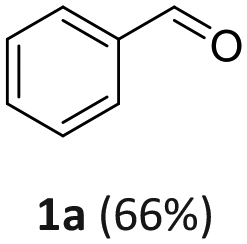
Benzyl alcohol was chosen as a model substrate to investigate the oxidation step. Air was used an oxidant for both safety and economic reasons. Initial experiments were carried out in an autoclave (a stainless-steel vessel) charged with a mixture of BnOH (1 mmol, 108 mg), acetonitrile as the solvent, and Mo(5%)-N/C as the catalyst.A parametric analysis of the reaction was performed by exploring different conditions, particularly by varying: (i) the amount of the catalyst, from 25 to 100 mg; (ii) the temperature, from 130 to 170 °C; (iii) the air pressure, from 1 to 20 atm; (iv) the reaction time, from 4 to 24 h, and the volume of acetonitrile (ACN), from 6 to 10 mL. At the end of each experiment, the reaction mixture was analyzed by NMR and GC/MS that confirmed the formation of benzaldehyde (1a) as the major, if not exclusive, product. In some cases, benzoic acid (1b) as a by-product of overoxidation of the alcohol was also detected.
Results are reported in Fig. S15 and 16 of the ESI.† The study not only demonstrated that the chosen catalyst was effective for the reaction, but it highlighted that the oxidation was most conveniently carried out at 170 °C and 1 bar, by dissolving BnOH in 10 mL of ACN. Under such conditions, in the presence of Mo(5%)-N/C (25 mg), the conversion of the alcohol was quantitative after 18 h, and benzaldehyde was obtained with a selectivity >99% (Scheme 4).
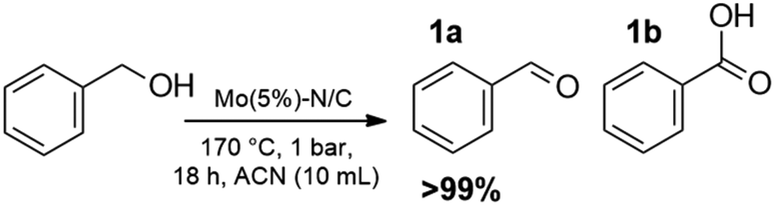 | ||
| Scheme 4 Selective oxidation of BnOH. | ||
Interestingly, the reaction took place without pressurizing the autoclave, with obvious advantages for both the safety and cost-efficiency of the protocol.27 In the autoclave reaction, autogenous pressure was generated.
The best conditions identified by the parametric analysis (170 °C, 1 bar, 18 h, VACN = 10 mL, cat. amount 25 mg) were then used to compare the performance of Mo(5%)-N/C to that of three bimetallic samples as Rh(3%)-Mo(5%)-N/C, Rh(5%)-Mo(5%)-N/C, and a mechanical mixture of Mo(5%)-N/C and Rh(5%)-N/C, respectively. The bimetallic samples were prepared with a quantity of molybdenum corresponding to the use of 25 mg of Mo(5%)-N/C. Further details are available in the Experimental section.
This study was aimed at assessing whether the presence of Rh-based systems which were essential for the hydrogenation and the reductive amination of the tandem sequence of Scheme 3, affected the activity of Mo for the oxidation of BnOH. The results are reported in Fig. 12.
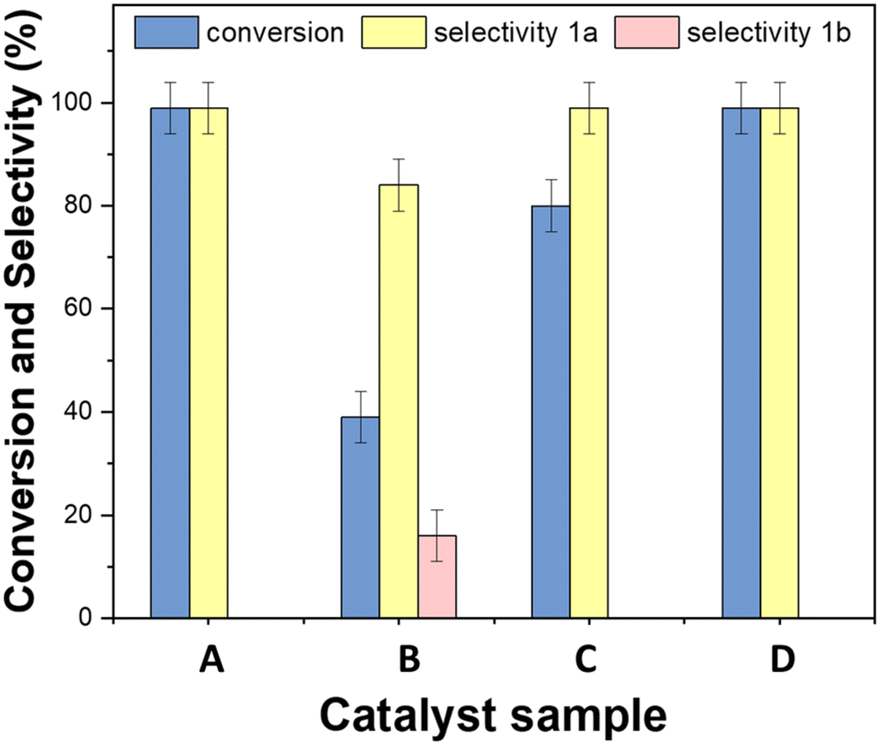 | ||
| Fig. 12 Comparison of the performance of different catalysts for the oxidation of BnOH (A) Mo(5%)-N/C, (B) [Rh(3%)-Mo(5%)]-N/C, (C) [Rh(5%)-Mo(5%)]-N/C, (D) mechanical mixture of Mo(5%)-N/C and Rh(5%)-N/C. Other conditions: 170 °C, 1 atm, 18 h, VACN = 10 mL. | ||
The mechanical mixture of Mo(5%)-N/C and Rh(5%)-N/C (D) yielded results comparable to those achieved by Mo(5%)-N/C (A) alone, with quantitative conversion and selectivity. Conversely, the use of bimetallic samples synthesized by co-impregnation of metal precursors, implied a reduction of the conversion to 39% and 78% for Rh(3%)-Mo(5%)-N/C (B) and Rh(5%)-Mo(5%)-N/C (C), respectively. Also, the benzaldehyde selectivity diminished to 85% in the case of Rh(3%)-Mo(5%)-N/C. As indicated by the XPS analysis, these results were consistent to the changes in the chemical nature of the molybdenum active sites (responsible for oxidation) induced by the presence of Rh species. The study led us to conclude that the most promising catalyst for the tandem sequence of Scheme 3, was the mechanical mixture of Mo(5%)-N/C and Rh(5%)-N/C that left unaltered the oxidation activity of Mo and at the same time, thanks to the presence of Rh, it was potentially suited for the other steps of hydrogenation and reductive amination.
Reductive amination of benzaldehyde
The reductive amination of benzaldehyde was explored using acetonitrile as a solvent/amine precursor in a H2 atmosphere. As anticipated in Scheme 3, this process was comprised of a cascade sequence which included the hydrogenation of acetonitrile followed by the reductive amination step. The products distribution was therefore determined by the multiple formation/occurrence of: (i) different amines from the hydrogenation reaction (Scheme 3 and S1†); (ii) competitive reductive aminations of benzaldehyde with different amines; (iii) alkylation/hydrogenation processes triggered by amines and imines species as carbon electrophiles.Mono-metallic catalysts
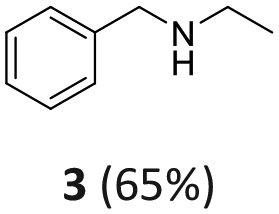
Benzaldehyde served as the model substrate for this study. Initial batch tests were conducted in an autoclave which was charged with a mixture of benzaldehyde (1 mmol, 106 mg), the catalyst (50 mg), and acetonitrile (ACN, 10 mL). The reactor was then pressurized to 30 bar with hydrogen and heated at 100 °C for 24 hours. Mono-metallic samples including Rh(5%)-N/C, Rh(3%)-N/C, and Rh(1%)-N/C with 5, 3, 1 wt% loading of Rh, respectively, were used as catalysts.The analysis of the reaction mixtures, carried out by NMR and GC/MS, confirmed the formation of three primary products: N-benzylethanimine (2), benzylethylamine (3), and benzyldiethylamine (4) (Scheme 5).
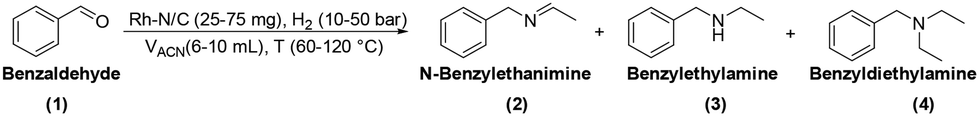 | ||
| Scheme 5 Three main products observed in the reductive amination of benzaldehyde using ACN as a solvent/amines precursor. | ||
According to the reductive amination mechanism, imine 2 was the first intermediate product formed by the nucleophilic attack of ethylamine (originated in situ by the direct hydrogenation of acetonitrile) to the carbonyl of benzaldehyde. Compound 3 was achieved by the hydrogenation of imine 2.
A hypothesis for the formation of product 4 was formulated based on the multiple transaminations occurring during the hydrogenation of acetonitrile (details are in Scheme S1†). Among other products, these reactions provided both diethylamine and ethanimine. Two different routes were postulated as shown in Scheme 6. The first one (A) was the catalytic reductive amination of benzaldehyde with diethylamine, a well-known process.32 The second option (B) involved a nucleophilic attack of 3 on ethanimine (as a carbon electrophile),33 that originated a diamino-intermediate species. Upon the release of ammonia and a final hydrogenation step, the tertiary amine 4 was obtained. In this case, 4 was not derived from a reductive amination, but from an N-alkylation/hydrogenation sequence.
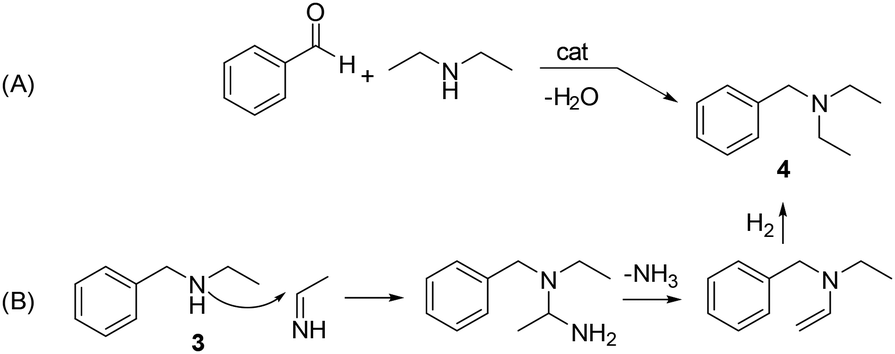 | ||
| Scheme 6 Hypotheses (A) and (B) for the formation of product 4. | ||
Other products (up to a 40% amount of the total observed products) were also detected during the experiments. These were categorized as “others” and their structures reported in the ESI,† suggested that their formation was plausibly triggered by a side reaction between benzaldehyde and ammonia, followed by a variety of other parallel and/or consecutive processes.
The results are reported in Fig. 13 which compares the conversion of benzaldehyde and the products distribution achieved with the use of the three investigated mono-metallic catalysts.
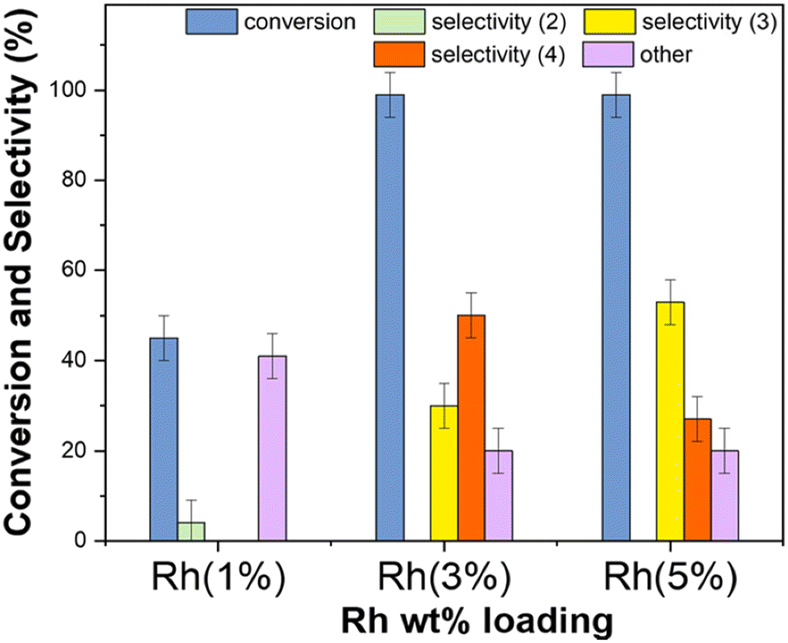 | ||
| Fig. 13 Reductive amination of benzaldehyde with ACN (10 mL) carried out at 100 °C, 30 bar of H2, and 24 h. Catalysts (50 mg) were mono-metallic Rh-based systems with 1, 3, and 5 wt% metal loading. The colored bars refer to conversion (blue), and amounts of products, 2 (green), 3 (yellow), 4 (orange), and others (violet). | ||
Rh(5%)-N/C and Rh(3%)-N/C allowed a quantitative conversion of benzaldehyde. The higher the Rh-loading, the greater the amount of the secondary amine 3: this product was obtained in 30% and 53% amounts with Rh(3%)-N/C and Rh(5%)-N/C, respectively. The tertiary amine 4 (50%) was instead, the predominant species when Rh(3%)-N/C was used. Overall, the total of products from the reductive amination remained undefined – ranging from a minimum of 30% (only 3) to a maximum of 81% (53 + 28 = 3 + 4) – because 4 could derive from different reaction pathways (Scheme 6).
The use of lower Rh-loading in the Rh(1%)-N/C brought about a remarkable drop in the conversion (43%) and even more significantly, the almost exclusive formation of undesired products “others” was achieved with only traces of the imine 2 (3%). An additional blank experiment (not shown in the figure) confirmed that negligible conversion (≤ 2%) was observed in the absence of any catalyst.
This behavior was rationalized by considering an inherent less activity of Rh(3%)-N/C compared to Rh(5%)-N/C, due to the lower Rh-metal loading. From the analysis of the hydrogenation of ACN, if the product ethylamine was obtained rapidly (Scheme S2, path A†), it could react further via transamination and hydrogenation processes to obtain both diethyl- and triethyl-amine (Scheme S2, paths B and C†). Once diethylamine was consumed, the residual triethylamine could no longer give the amination of benzaldehyde. GC-FID and GC-MS analyses of the mixtures collected after the reactions of Fig. 13, proved that in the presence of Rh(5%)-N/C, the concentration of triethylamine was higher than with Rh(3%)-N/C. This confirmed the hypothesis that the more active Rh(5%)-N/C catalyst, favored both the condensation of ethylamine with benzaldehyde to obtained the product 3, and the formation of triethylamine that hindered the reaction by which 4 was achieved. On the other hand, the tertiary amine 4 was produced by more than one mechanism (Scheme 6). Therefore, its amount in the reaction solution was not entirely dependent on the activity of the Rh-N/C catalyst.
Other metal catalysts as Pd(5%)-N/C and Ru(5%)-N/C were tested under the conditions of Fig. 13. However, in both cases, the reduction of the carbonyl group leading to benzyl alcohol occurred, instead of the reductive amination (for a comparative analysis, refer to Table S3 in the ESI,† which includes literature data).
Based on these findings, Rh(5%)-N/C was chosen to continue the investigation. A parametric analysis of the reductive amination of benzaldehyde was carried out by varying: (i) the amount of catalyst, from 25 to 75 mg; (ii) the temperature, from 60 to 120 °C; (ii) the hydrogen pressure, from 10 to 50 bar; (iii) the reaction time, from 4 to 24 h, and the volume of acetonitrile (ACN), from 6 to 10 mL. Results are reported in Fig. S17 in the ESI.† The study demonstrated that the kinetics of reductive amination took advantage from either the temperature or the availability of gaseous hydrogen in the liquid solution, which increased with pressure. Moreover, increasing the amount of solvent favored the reduction 2→3 by plausibly dissolving more hydrogen in the liquid phase and making the gas ready for the hydrogenation processes.
Overall, the parametric analysis led us to conclude that the reductive amination of benzaldehyde was most conveniently carried out under the conditions already highlighted in Fig. 13 [100 °C, 30 bar of H2, Rh(5%)-N/C (50 mg), 24 h and acetonitrile (10 mL) of as a solvent/reactant], by which the maximum amount of benzyl ethyl amine 3 was 53%. The tertiary amine 4, whether it was formed by reductive amination or by other mechanisms (Scheme 6), was obtained in a 28% amount.
Bi-metallic systems and mechanical mixtures of mono-metallic catalysts
The reductive amination of benzaldehyde was further investigated in the presence of bi-metallic systems and binary mechanical mixtures of mono-metallic catalysts. The experiments were carried out under the conditions of Fig. 14, using the following: (i) bi-metallic samples including [Rh(5%)-Mo(5%)]N/C (Bi-M5) and [Rh(3%)-Mo(5%)]N/C as (Bi-M3) with 5 and 3 wt% loading of Rh and 5 wt% loading of Mo, respectively; (ii) two mechanical mixtures indicated as MIX1 comprised of Rh(5%)-N/C + Mo(5%)-N/C, and MIX2 comprised of Rh(3%)-N/C + Mo(5%)-N/C, respectively, were used.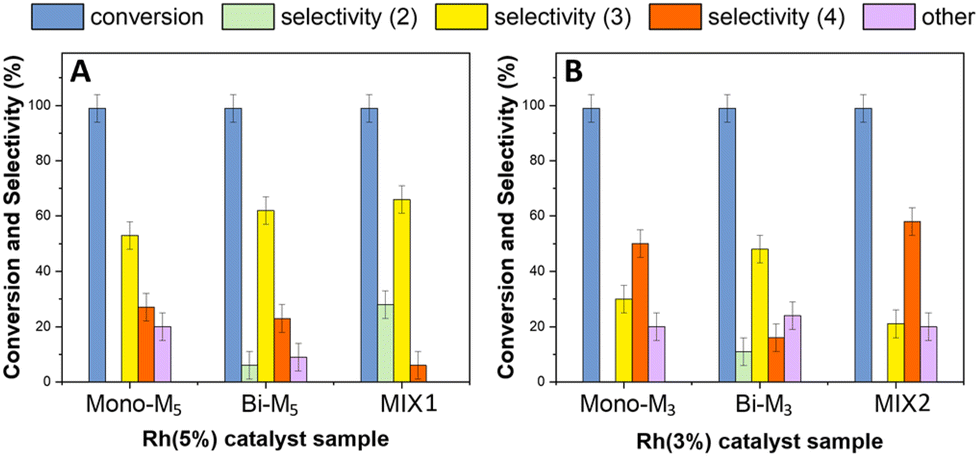 | ||
| Fig. 14 Reductive amination of benzaldehyde with ACN (10 mL) carried out at 100 °C, 30 bar of H2, and 24 h. (A) 5 wt% Rh-based catalysts either as mono-, bi-metallic (Bi-M5) samples or the mechanical mixture MIX1; (B) 50 bar of H2, 3 wt% Rh-based catalysts either as mono-, bi-metallic (Bi-M3) samples or the mechanical mixture MIX2. The colored bars refer to conversion (blue), and amounts of products 2 (green), 3 (yellow), 4 (orange), and others (violet). | ||
The amount of catalytic sample used in each reaction was calibrated to have the same quantity of Rh used in the tests with the monometallic systems. Further details are in the Experimental section.
In all cases, the analysis of the mixtures collected at the end of the tests confirmed the formation of same three main products N-benzylethanimine (2), benzylethylamine (3), and benzyldiethylamine (4), observed with the monometallic catalysts (cfr. Scheme 4), along with the presence of a mixture of derivatives (others).
The results are reported in Fig. 14 which shows the conversion of benzaldehyde and products distribution achieved with the use of: (i) the 5 wt% Rh-based catalysts either the bi-metallic (Bi-M5) samples or the mechanical mixture (MIX1) (left, Fig. 14A); the 3 wt% Rh-based catalysts either the bi-metallic (Bi-M3) samples or the mechanical mixture (MIX2) (right, Fig. 14B). For a more convenient comparison, this figure also reports the results obtained with the mono-metallic catalysts Rh(5%)-N/C (14A) and Rh(3%)-N/C (14B).
Compared to Rh(5%)-N/C, the use of bimetallic sample Bi-M5 and the MIX1 catalysts improved the formation of the secondary amine 3 up to 60 and 65%, respectively (Fig. 14A). MIX1 appeared the more perfomant system towards reductive amination since the two products 3 and 2 (the imine precursor of 3) reached an excellent total of 93%, with only 7% of the tertiary amine 4. The same derivatives 2 + 3 and 4 summed up 67% and 22%, respectively, with Bi-M5; moreover, a not negligible amount of others (11%) was detected in this case.
Rh(3%)-N/C and MIX2 showed a comparable behavior between each other and both favored the formation of the tertiary amine 4 (50–55%; Fig. 14B). By contrast, the bimetallic [Rh(3%)-Mo(5%)]-N/C sample (Bi-M3) was more selective towards the secondary amine 3 (48%). Albeit this was apparently inconsistent, a reason for this difference was inferred from the TEM analyses of Bi-M3, where metal clusters were noticed, as opposed to the presence of well-dispersed Rh-nanoparticles in both Rh(3%)-N/C and MIX2. In any case, however, a substantial amount of “others” (18–22%) was observed for all the three catalysts based on 3wt% of Rh.
The results of Fig. 14A and B confirmed the trend highlighted from the comparison of the monometallic samples: a higher Rh content was beneficial to improve the formation of the secondary amine 3 and its precursor 2, which were originated by the reductive amination of benzaldehyde. The 5wt% Rh-based bimetallic systems and particularly the mechanical mixture MIX1 proved even more effective than mono-metallic Rh(5%)-N/C, thereby suggesting that the presence of Mo altered the (hydrogenation) activity of Rh, plausibly by favoring a more uniform and homogeneous distribution of Rh-nanoparticles over the support.
Tandem reactions
The results of the independent investigation of the oxidation of BnOH and the reductive amination of benzaldehyde prompted us to study the tandem sequence of Scheme 3 by using the mechanically prepared mixture MIX1 as the preferred catalyst. Experimental conditions were selected according to those by which the highest selectivity and conversion of both steps were achieved. In particular: (1) for the oxidation reaction, T, P, and t were set to 170 °C, ambient (air) pressure, and 18 h, respectively; (2) for the reductive amination reaction, T, P, and t were set to 100 °C, 30 bar (H2 pressure), and 24 h, respectively.All tests were carried out in the presence of acetonitrile (10 mL) as a solvent/reagent and MIX1 in the form of a mixture of Rh(5%)-N/C (50 mg) and Mo(5%)-N/C (25 mg) as the catalyst. Under such conditions, once the first step (oxidation) was completed, the reactor (autoclave) was cooled to r.t., purged with N2, pressurized with H2, and finally heated to start both the hydrogenation of ACN and the reductive amination of benzaldehyde. The experiments proved that the tandem sequence proceeded as expected from the study of the two individual reactions (Fig. 12 and 14), even though the whole reaction mixture, catalyst and solvent were never treated/removed until the overall process was completed. The results are summarized in Scheme 7.
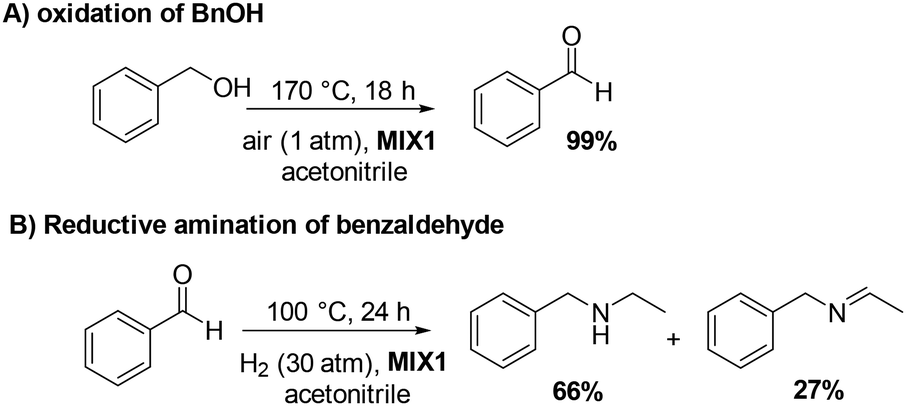 | ||
| Scheme 7 One-pot two-step tandem reaction catalyzed by MIX1: (A) oxidation of BnOH; (B) reductive amination of benzaldehyde. | ||
Both reactions proceeded to complete conversion. Not traces of BnOH or benzaldehyde were detected at the end of the sequence. BnOH was fully oxidized to benzaldehyde (99%) in a clean smooth process without any by-products. Benzaldehyde was then subjected to reductive amination. Albeit the control of the products distribution of this process was critical due to the presence of different amines and imines species, the product 3 and its precursor 2 were achieved in an amount of 66% and 27%, respectively, meaning that derivatives of reductive amination reached a 93% quantity (2 + 3). An additional experiment prolonged for up to 48 hours and aimed to enhance the yield of compound 3 by completing the hydrogenation of 2, was not successful: the amount of 3 was of only 69%, while the tertiary amine 4 (24%) was increased of almost the same quantity the intermediate imine 2 disappeared. This suggested that path B in Scheme 6 was likely the predominant reaction pathway for the formation of 4.
Also, further attempts carried out to improve the 2→3 conversion by enhancing the H2 pressure up to 50 bar, were not satisfactory because the tandem protocol proceeded with the major formation of 4 (60%).
Additional tests were carried out using MIX2 (a mixture of Rh(3%)-N/C (50 mg) and Mo(5%)-N/C (25 mg)) as the catalyst. The oxidation of BnOH was quantitative and fully selective to benzaldehyde under the conditions of Scheme 8 (MIX 2, 170 °C, 1 atm, 18 h). However, conditions for the second step were modified to optimize/improve the outcome of the reductive amination, resulting in a moderate-to-good selectivity (60%) towards the tertiary amine 4, at 100 °C, 50 bar (H2), and 4 h. These results are summarized in Scheme 8.
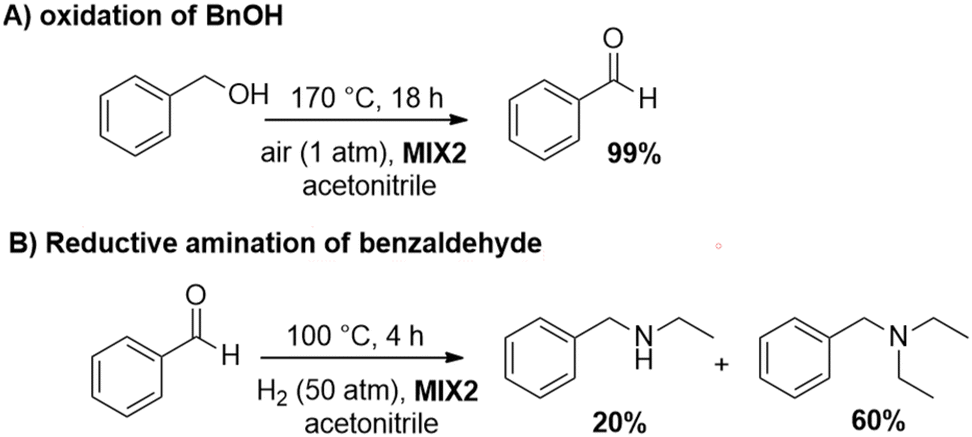 | ||
| Scheme 8 One-pot two-step tandem reaction catalyzed by MIX2: (A) oxidation of BnOH; (B) conversion of benzaldehyde to benzyldiethylamine (other products – not shown in path B, were detected and were the complement to 100% in the products distribution). | ||
Substrate scope
MIX1 [Rh(5%)-N/C (50 mg) and Mo(5%)-N/C (25 mg)] the best performing catalyst for the tandem reaction of Scheme 3 was explored to induce the same cascade reactions on other biomass-derived aromatic alcohols (or derivatives). The procedure and reaction conditions were those described in Scheme 7. The results are reported in Table 3.| Entry | Substrate | Oxidation (A) | Reductive amination (B) | ||
|---|---|---|---|---|---|
| Conversion | Major product (& selectivity) | Conversion | Major product (& selectivity) | ||
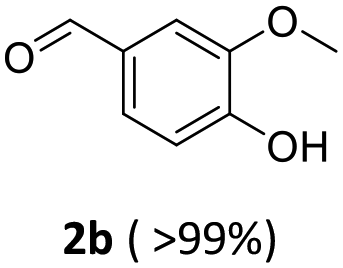
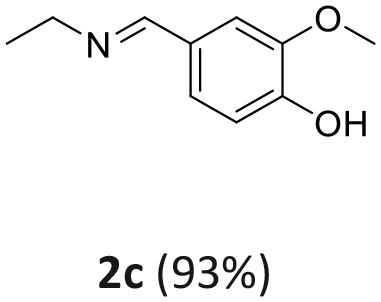
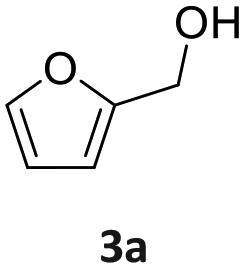
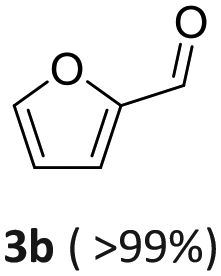
| 2a: vanillyl alcohol, 2b: vanillin, 2c: 4-(ethyliminomethyl)-2-methoxyphenol, 3a: furfuryl alcohol, 3b: furfural, 3c: N-ethyl-N-(2-furylmethyl)ethanamine, 4a: 4-bromo benzylic alcohol, 4b: 4-Br-benzaldehyde, 4c: N-ethyl-4-bromobenzylamine, 5a: phenylethyl alcohol, 1: benzaldehyde, 2: ethylbenzylamine, 6a: 2-aminobenzyl alcohol, 6b: imino quinoline. | |||||
| 1 |
|
90% |
|
> 99% |
|
| 2 |
|
> 99% |
|
95% |

|
| 3 |
|
80% |
|
99% |
|
| 4 |
|
> 99% |
|
> 99% |
|
| 5 |
|
> 99% |
|
n.d. | n.d. |
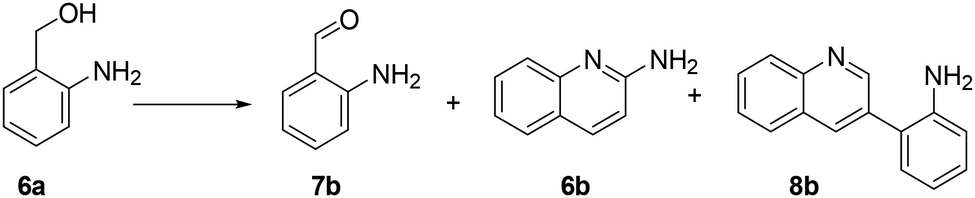 | ||
| Scheme 9 The reaction of 2-aminobenzyl alcohol (6a) under oxidative conditions (170 °C; 1 atm, 18 h, VACN = 10 mL). 2-amminobenzyl aldehyde iminoquinoline (6b), and 2-(3-quinolinyl) aniline (8b) were the observed products. | ||
Interestingly, an additional experiment conducted by replacing MIX1 with the Mo(5%)-N/C catalyst resulted in a significant improvement of the selectivity of aldehyde 7b (56%, Fig. S19,† right). This suggested that the presence of Rh was detrimental for the oxidation step. Based on this observation, the tandem sequence was carried out using Mo(5%)-N/C for the synthesis of the aldehyde 7b. Thereafter, once the oxidation was complete, Rh(5%)-N/C was introduced in the reactor to conduct the reductive amination step. The reaction was stopped until the formation of the imine product 6c which was observed with a selectivity higher than 99%, albeit a moderate conversion (36%) was reached (Scheme 10).
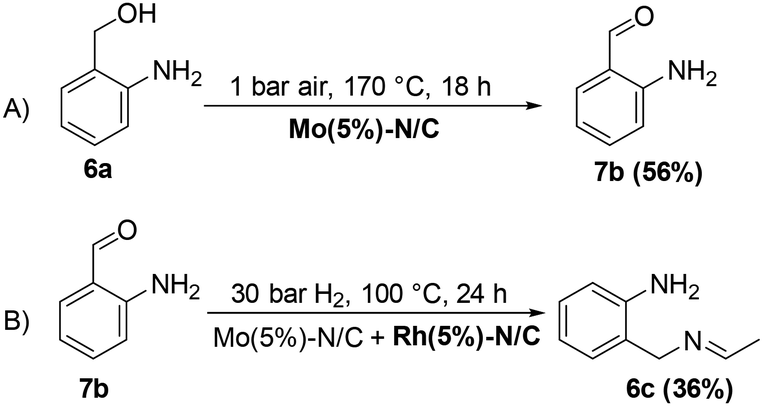 | ||
| Scheme 10 The reductive amination of 6a. | ||
Overall, the investigation of the substrate scope clearly highlighted that conditions for the tandem sequence needed a case-by-case study and a general protocol could not be defined. In other words, reaction parameters had to be set according to the reactivity of any given alcohol/substrate.
Catalyst reusability
The costs associated with the catalyst in a liquid-phase reaction can represent up to one-third of the overall process cost. Therefore, any loss of catalyst due to leaching or other factors is highly significant, underscoring the importance of its recovery and reuse.29 To assess the stability and reusability of both Mo(5%)-N/C and Rh(5%)-N/C catalytic samples, recycling experiments were designed under the investigated reaction conditions. After completing one reaction cycle, the catalysts were separated in each case, through filtration, rinsed with 30 mL of acetone and dried overnight.The recovered catalysts were suspended in fresh solutions of BnOH (1 mmol) in acetonitrile (10 mL), and new reactions (oxidation and reductive amination, respectively) were initiated. The recycling process was repeated for seven subsequent runs for both Mo(5%)-N/C in the oxidation of BnOH and Rh(5%)-N/C in the reductive amination of benzaldehyde. The entire series of reactions was performed twice to ensure reproducibility. The results are depicted in Fig. 15.
 | ||
| Fig. 15 Catalyst recycle. Left (A) Mo(5%)-N/C recycles in the oxidation of BnOH; right (B): Rh(5%)-N/C recycles in the reductive amination of benzaldehyde. | ||
Both catalytic samples maintained a steady conversion (>99%) over the six reuses in their respective reactions. The oxidation selectivity was constant at 99% towards benzaldehyde, while the selectivity towards amine 3 in the reductive amination was oscillating in a narrow range of 50–57%. Interestingly, from the second run, the selectivity towards tertiary amine 4 increased from 23% to 40–45%, respectively. However, in general, these results led us to conclude that the overall performance of Mo(5%)-N/C and Rh(5%)-N/C was not appreciably affected by the recycling process, except for the variation/increase in selectivity towards compound 4. To better assess the recyclability of the catalytic systems, additional studies were conducted at incomplete conversion. The reductive amination reaction was carried out using Rh(5%)-N/C at 80 °C, 10 bar of H2, and 24 hours. Results are reported in the ESI (Fig. S20†), confirming the good stability of the materials.
Post-recycle characterization
A post-characterization analysis of Rh(5%)-N/C was conducted to gain insights on the chemical, structural, and morphological features of the samples after the catalytic reactions and their reuse, under the previously reported conditions. The same multi-technique approach previously described for the fresh catalytic samples was used.As shown in Fig. 16A, N2-physisorption analysis revealed that the used material retained a mesoporous behaviour, since it exhibited typical Type IV isotherms with Type II adsorption hysteresis. However, a drastic reduction in the adsorbed volume was evident when compared to the fresh sample (as indicated in Table 4). Consequently, a reduction in surface area down to 44 m2 g−1 was also observed, which represents only 9% of the initial value of the fresh sample. The pore volume of 0.06 cm3 g−1 was almost 4 time lower than that of the fresh sample, while the mean pore diameter (5.5 nm) showed a lower value, in comparison with the fresh sample (6.5 nm). These results were explained by a collapse of the internal structure of the carbonaceous materials, resulting in smaller pores and lower surface area.
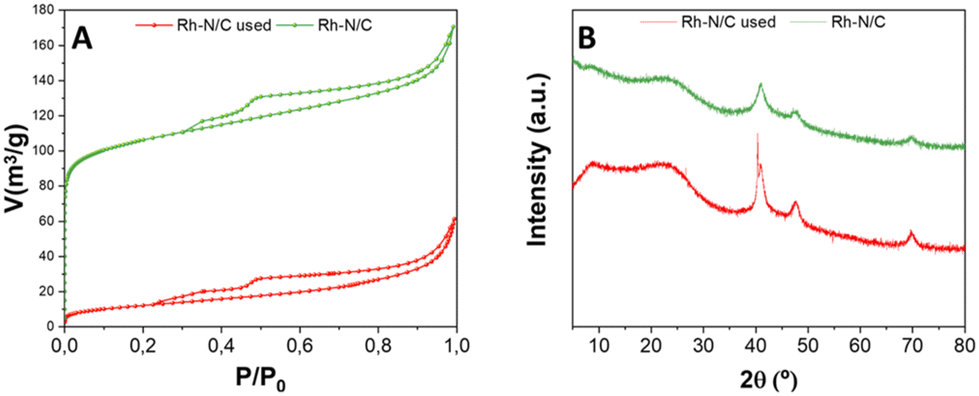 | ||
| Fig. 16 (A) Compared isotherms of used Rh-N/C and fresh Rh-N/C samples; (B) compared XRD spectra of used Rh-N/C and fresh Rh-N/C samples. | ||
| Material sample | S BET [m2 g−1] | D BJH [nm] | V BJH [cm3 g−1] |
|---|---|---|---|
| a S BET: specific surface area was calculated by the Brunauer–Emmett–Teller (BET) equation. b D BJH: mean pore size dimeter was calculated by the Barret–Joyner–Halenda (BJH) equation. c V BJH: pore volumes were calculated by the Barret–Joyner–Halenda (BJH) equation. | |||
| Fresh Rh(5%)-N/C | 489 | 6.5 | 0.23 |
| Used Rh(5%)-N/C | 44 | 5.5 | 0.06 |
The XRD patterns of the used Rh(5%)-N/C sample exhibited minor differences when compared to the XRD pattern of the fresh Rh(5%)-N/C sample (Fig. 16B). Particularly, three peaks were identified in the used sample, at 41, 48 and 70°, in accordance with the typical diffractions associated with the (111), (200), and (220) crystalline planes of metallic Rh, also found in the fresh sample. This confirms the presence of metallic Rh in the used catalyst. In addition to these observations, the sample maintained an amorphous structure, as indicated by the broad nature of the peaks.
The XPS analysis of the Rh-N/C used material is reported in Fig. 17. No significant differences were observed when comparing the XPS regions of the fresh Rh(5%)-N/C and used Rh(5%)-N/C samples. In the Rh 3d region of the used sample, peaks characteristic of both Rh0 and Rh3+ were detected with no shift in binding energy values from the fresh sample. Furthermore, the presence of additional contributions at around 310.7 and 315.4 eV, akin to the fresh sample, strongly indicated that the Rh–N bonds did not were affected, most likely contributing to the stability of the samples and the prevention of leaching during the catalytic transformations. This study highlighted that during its utilization/recyle, the Rh(5%)-N/C sample underwent textural, and structural modifications, but none of these changes had an adverse impact on its catalytic performance.
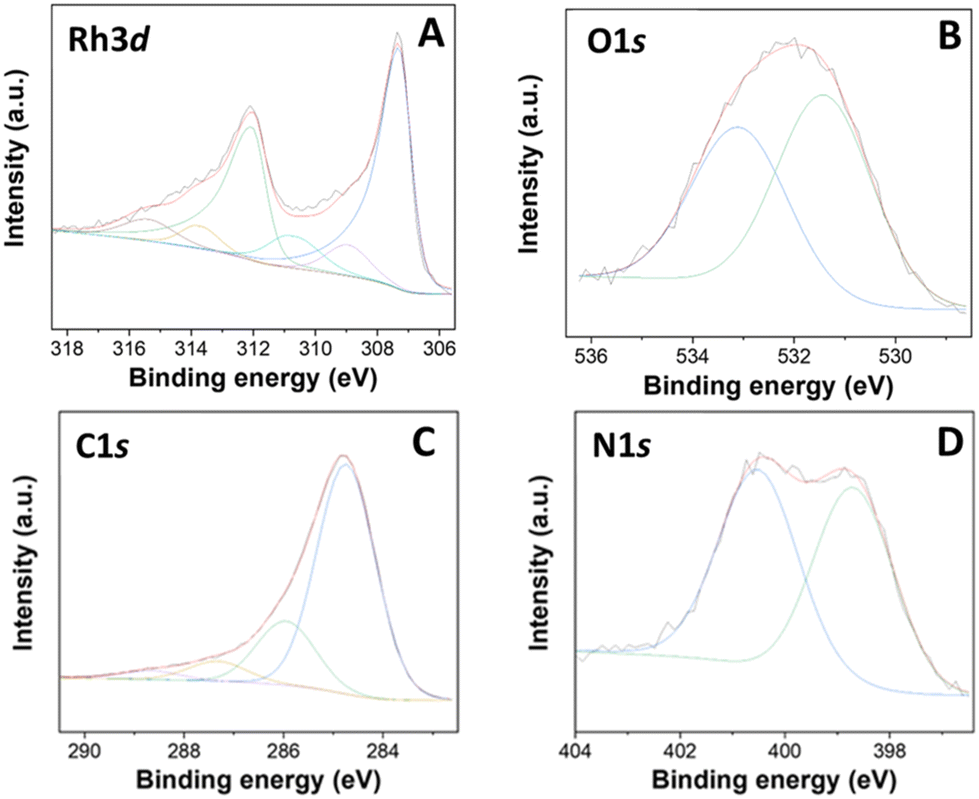 | ||
| Fig. 17 XPS results of (A) Rh 3d, (B) C 1s, (C) N 1s, (D) O 1s of the used Rh-N/C sample. | ||
HRTEM-EDX analyses and HRTEM micrographs (Fig. 18) of the re-used Rh(5%)-N/C sample did not exhibit significant changes as compared to the fresh sample, given that the mean particle diameter was calculated to be around (6.0 ± 2.0) nm, while it was (5.9 ± 2.0) nm for the fresh material. HRTEM-EDX results also demonstrated a homogeneous distribution of the active rhodium sites, maintaining a good dispersion. However, there were some agglomerated regions that likely contributed to the increase in the mean particle diameter.
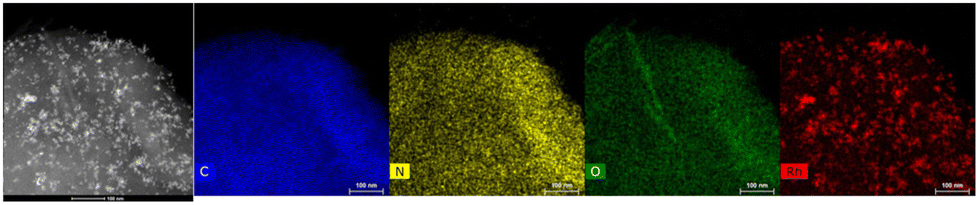 | ||
| Fig. 18 HRTEM in STEM mode-EDX micrographics of the Rh(5%)-N/C used sample. | ||
Mechanochemical perspectives
While solvents have a crucial role for chemical processes, they have a huge impact on health and safety, environment, regulatory compliance, and cost management. Proper risk assessment, safety measures, and sustainable practices are essential to address these challenges effectively. Even more, the design of solvent free protocols for organic transformations is a thrilling and challenging goal, which has recently spurred the scientific community to explore solventless mechanochemical protocols for the preparation of chemicals and materials.38–40Inspired by such premises, the final stage of this work was dedicated to evaluating the catalytic performance of the synthesized materials, particularly the Mo-N/C systems, through mechanochemically-assisted oxidation reactions. More specifically, a solvent free continuous-flow protocol was developed using a twin-screw mini extruder to produce vanillin by the selective oxidation of vanillyl alcohol. The choice of this reaction was dictated by the fact that both the starting material, vanillyl alcohol, and the desired product, vanillin, were solids with melting points significantly higher than room temperature (115 °C and 83 °C, respectively).41 This characteristic made them suitable candidates for a solvent-free mechanochemical approach.
During this investigation, three different oxidizing agents were considered, namely air, hydrogen peroxide, and urea hydroperoxide. Through all these experiments, the extrusion mixture was made of 2 g of substrate (vanillyl alcohol, 2a), 100 mg of Mo(5%)-N/C catalyst, and the oxidant in appropriate amounts (see Table S4, ESI†). The reaction conditions were set at 80 °C, 100 rpm, and 1 h of residence time. Notwithstanding its affordability, safety, and widespread availability, air use was unsuccessful (Scheme 11, A).
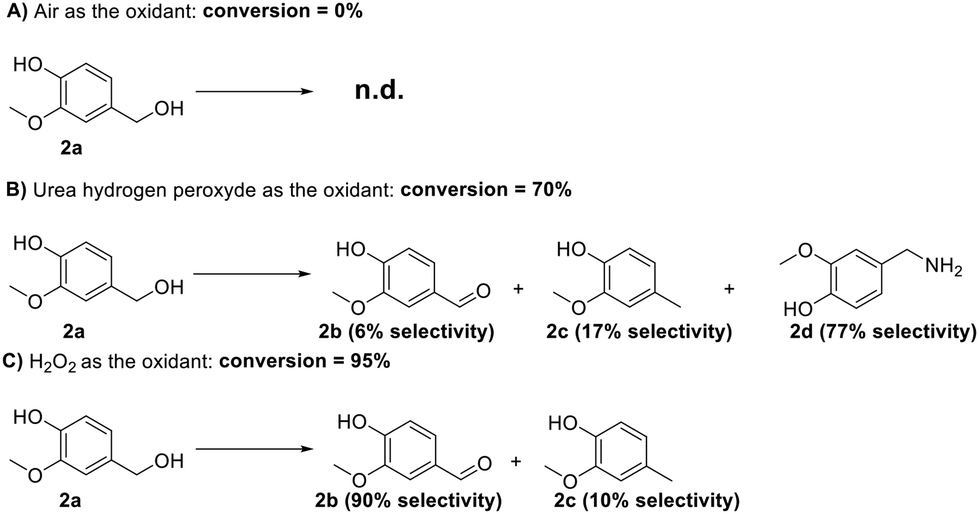 | ||
| Scheme 11 schematic representation of the oxidation of vanillic alcohol using (A) air, (B) urea peroxide, (C) hydrogen peroxide. 2b: vanillin, 2c: creosol, 2d vanillyl amine. | ||
Urea hydroperoxide was then selected based on a literature review of solid oxidizing agents with a relatively low melting point (up to 95 °C) and a good environmental acceptability when compared to other solid oxidants as, for instance, potassium permanganate. Urea hydroperoxide allowed a conversion of 70%, but the selectivity towards vanillin was of only 6%. The primary amine 2d emerged as the major product (77%, Scheme 11, B): albeit the mechanism of this reaction was not explored, the result was intriguing and potentially interesting to unlock new avenues for the synthesis of amine-containing compounds using a solventless mechanochemical approach.
Finally, hydrogen peroxide allowed an excellent reaction outcome with conversion and selectivity towards vanillin of 95% and 90%, respectively (approximately corresponding to an 86% yield) (Fig. 19). It should be noted here that when introduced in the liquid form, hydrogen peroxide allowed the formation of a slurry-like mixture with the reagent and catalyst under the conditions described above. Overall, hydrogen peroxide emerged as the optimal oxidizing agent for this reaction. Blank experiments in the absence of catalytic material were also conducted in this case, yielding a 20% conversion of vanillyl alcohol to vanillin, thereby confirming the crucial role of the catalyst. The encouraging preliminary findings achieved with hydrogen peroxide in this study will be a groundwork for more extensive investigations in our lab in the near future. Turnover Number (TON) and the Turnover Frequency (TOF) of this mechanochemical process were 474 and 474 h−1, respectively.42 Further studies in this regard will be performed to optimize the herein proposed mechanochemical approach.
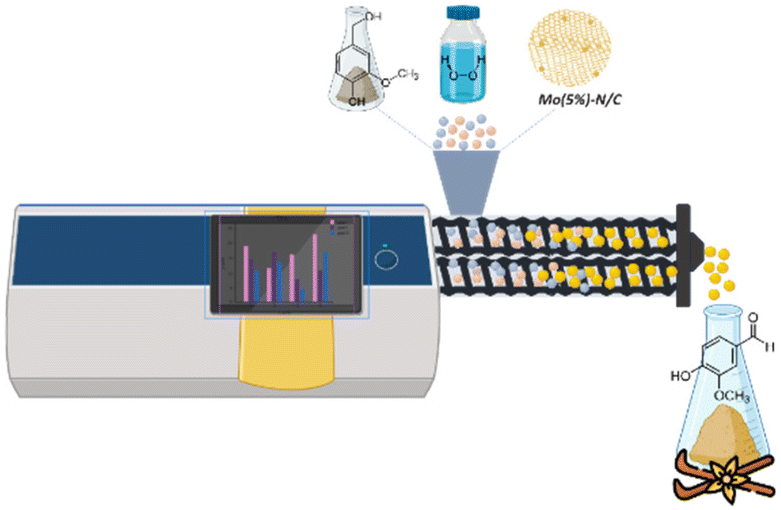 | ||
| Fig. 19 Mechanochemical synthesis of vanillin. | ||
CHEM21 toolkit: the application of Green Metrics
The final section of this work was dedicated to an assessment of the investigated protocols, employing the CHEM21 toolkit Zero Pass (see Table S3, ESI†).43 Specifically, the sustainability of the processes designed for the two sequential steps of the oxidation of benzyl alcohol to benzaldehyde and reductive amination of benzaldehyde have been evaluated, including also the mechanochemical approach for the production of vanillin from vanillyl alcohol.As it can be seen in Fig. 20A, the oxidation step presents excellent results for the metrics included in the Zero Pass of the CHEM21 toolkit (Table S5†). All values are proximate to 1, the maximum.
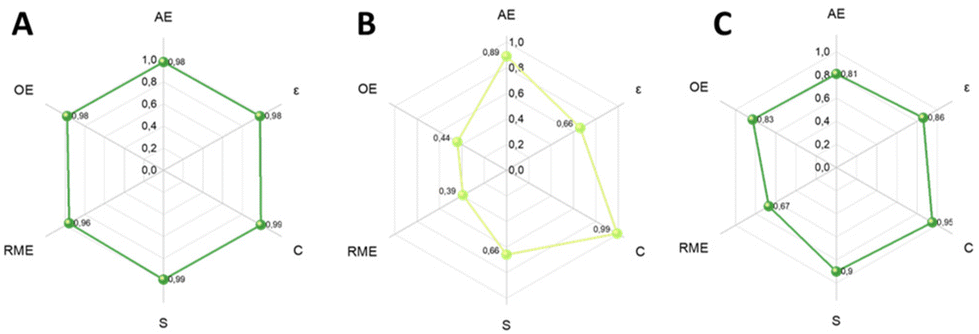 | ||
| Fig. 20 Hexagon radial chart comprising CHEM21 Zero Pass metrics applied to (A) oxidation reaction; (B) reductive amination reaction; (C) to the mechanochemical oxidation of vanillyl alcohol. | ||
Moreover, the Mass Intensity (MI) resulted to be 1.02, which is an excellent efficiency ratio. It is necessary to mention that ACN is solely a solvent in the oxidation step and should normally be considered in the calculation of MI. However, it is both a solvent and a reactant for the next reaction step. This complicates the assessment of the consumed mass of ACN, as it can be readily recycled and replenished during step B of the tandem reactions. If considered it would give a value of MI of 74.79 kg of required input material to yield 1 kg of product, which, however, does not truly represent this process.
The results of the metrics applied to the reductive amination step confirm that the major limiting factor of the reductive amination steps is the low yield (Fig. 20B). To calculate MI, ACN will first be considered as a recyclable solvent, leading to an MI of 5.10. On the other hand if the ACN mass was considered as a non-recoverable solvent, MI would be 92.81 kg kg−1.
Both yield and selectivity are green flags for the oxidation step (>89%), while in the reductive amination are red (<70%) although near to amber flag (70–89%) with 66% selectivity and yield for the secondary amine 3.
Finally, the green metrics of CHEM21 Zero Pass’ toolkit were applied to the mechanochemical extrusive oxidation reaction of vanillyl alcohol to vanillin and the hexagon graph shows excellent results (Fig. 20C). In this mechanochemical process no ACN or any other solvent was used, bringing the MI at 1.28 kg of mass needed to yield 1 kg of product, in contrast with the potential 74.79 kg in the oxidation process of BnOH to benzaldehyde in ACN. This was the best process in terms of greenness according to the CHEM21 Zero Pass toolkit. A summary is shown in Table 5.

|
Experimental
Materials and equipment
Benzyl alcohol, furfuryl alcohol, 1,5-bis(hydroxymethyl)furan, 1-phenylethanol, vanillyl alcohol, 1-pentanol, cyclopentanol, acetonitrile, 2-propanol, chitin, EDTA, CoCl2·6H2O, Fe(NO3)3·9H2O, Ni(Ac)2·4H2O, (NH4)MoO4, Pd(OAc)2, RuCl3·H2O and RhCl3·H2O were commercially available compounds sourced from Sigma-Aldrich. If not otherwise specified, reagents and solvents were employed without further purification. Air gas was purchased from SIAD, Italy. Extruding techniques were carried on using a mini-extruder (ZE 12 HMI extruder from Three Tec., Seon, Switzerland).GC-MS (EI, 70 eV) analyses were performed on a HP5-MS capillary column (L = 30 m, Ø = 0.32 mm, film = 0.25 mm). GC (flame ionization detector; FID) analyses were performed with an Elite-624 capillary column (L = 30 m, Ø = 0.32 mm, film = 1.8 mm). 1H, 13C NMR spectra were recorded in the Bruker Avance III HD 400 WB equipped with a 4 mm CP/MAS probe, at 400 and 101 MHz, respectively. Chemical shifts were reported downfield from tetramethylsilane (TMS) and MeOD, DMSO-d6 and CDCl3 were used as solvents.
Synthesis of supported metal nanoparticles on chitin derived N-doped materials
In a typical synthesis, the salt chosen as a metal precursor (based on Co, Fe, Ni, Mo, Pd, Ru, and Rh; see above) was dissolved in 2-propanol (60 mL), together with EDTA (1 g) (see Fig. 15). EDTA was utilized as a powerful metal complexing agent with the aim to achieve a dispersion of the metal active sites as uniform as possible over the support. The amount of the precursor salt was calibrated to achieve different metal loadings, from 1 up to 5 wt%. Subsequently, chitin (5 g) was added to the mixture, which was kept under stirring for 9 h at 80 °C under reflux. The suspension was filtered and the so obtained solid was dried at 100 °C overnight and finally, heated at 500 °C (heating rate was 5 °C min−1) under N2 flow (10 mL min−1) for 1 h. The catalytic samples achieved by this procedure were labelled as Ni–N/C, Fe–N/C, Co–N/C, Mo–N/C, Rh–N/C, Pd–N/C, Ru–N/C for the nickel, iron, cobalt, molybdenum, rhodium, palladium, and ruthenium catalysts, respectively. Also, bimetallic samples were synthesized using Mo and Rh metals: this metals combination was chosen as Mo and Rh separately exhibited the best catalytic activity for the investigated reactions. The synthesis of these materials was identical to that of their mono-metallic counterparts. The amount of metal precursor used for the materials preparation was always calculated assuming that, after calcination, the obtained catalyst mass would be ca. 30% of the total initial mass which comprises (i) metal salt precursor(s), (ii) 5 g of chitin, (ii) 1 g of EDTA as the complexing agent. Accordingly, 1 and 0.6 mmol of metal salt were used to produce 5 and 3 wt% respectively of metal on the final material. These values were unaltered when preparing the bimetallic samples, as the co-presence of Mo and Rh on the finished material did not bring significant changes in the preparation procedure.The resulting materials were ground to powder (particle size <200 μm) and stored in the oven (60 °C, 15 mbar) until further use. The yield of the obtained materials was ca. 25 ± 5%, based on the total weight of chitin and the metal precursor used.
Material characterization
Surface chemical composition analysis was conducted using XPS with a Physical Electronics VersaProbe II Scanning XPS Microprobe. The XPS system was equipped with monochromatic X-ray Al Kα radiation and operated at a vacuum level of 10−7 Pa. To reference the binding energies, the C 1s peak from adventitious carbon at 284.8 eV was used. High-resolution spectra were obtained with a concentric hemispherical analyzer using a constant energy pass of 29.35 eV and a 200 μm diameter analysis area. The analysis chamber maintained a pressure below 5 × 10−6 Pa throughout the measurements. Data acquisition and analysis were performed using PHI ACCESS ESCA-F V6 software. A Shirley-type background was subtracted from the signals, and Gauss-Lorentz curves were applied to accurately determine the binding energy of atomic levels for different elements in the recorded spectra.XRPD patterns were obtained in the laboratory using a PANalytical X'Pert Pro automated diffractometer located in the central research facilities (SCAI) at the University of Málaga. The measurements were performed in the Bragg–Brentano reflection configuration, employing a Ge(111) primary monochromator (Cu Kα1) and the X'Celerator detector. The data acquisition utilized a step size of 0.0167° (2θ). The XRPD patterns were collected within the 4 to 70° range in 2θ, with each step having an equivalent counting time of approximately 60 seconds.
N2 physisorption measurements were performed using a Micromeritics TriStar 3000 instrument. To prepare the samples, they were subjected to outgassing at 120 °C for a duration of 2 h. Subsequently, adsorption and desorption isotherms were recorded at a low temperature of −196 °C. Specific surface areas were determined employing the BET method, while pore volumes were calculated based on the adsorption isotherms. Pore size distributions were estimated utilizing the Barrett, Joyner, and Halenda (BJH) algorithm, which is integrated into the Micromeritics software.
HRTEM analysis was conducted using a TALOS F200x instrument operating in STEM mode (Scanning Transmission Electron Microscopy). This instrument was equipped with a High-Angle Annular Dark Field (HAADF) detector and operated at 200 kV with a 200 nA current. Microanalysis was carried out using an Energy-Dispersive X-ray (EDX) Super-X system, which featured four X-ray detectors and an X-FEG beam.
To assess metal leaching, ICP-MS was employed, utilizing an Elan DRC-e spectrometer manufactured by PerkinElmer SCIEX.
Typical oxidation reaction procedure and product analysis
Experiments were conducted within a 50 mL tubular reactor crafted from borosilicate glass (Pyrex). The reactor was charged with benzyl alcohol (1 mmol, 108 mg), along with the catalyst and acetonitrile. The quantities of the catalyst mass and acetonitrile volume were systematically varied during the optimization of reaction parameters. Subsequently, the reactor was positioned inside a jacketed stainless-steel autoclave, which was equipped with a manometer and two needle valves. This autoclave was pressurized to the desired air pressure and heated to the designated temperature. The reaction mixture was maintained under continuous magnetic stirring at a rate of 700 rpm. At the end of the reaction, the autoclave was gradually cooled to room temperature using an ice bath. It was subsequently gently purged. Upon completion of the reaction, a sample of the reaction mixture was withdrawn using a syringe and subjected to analysis by GC-FID to determine reaction conversion and selectivity. Lastly, the samples were evaporated using a rotary evaporator (60 °C, 15 mbar). The obtained dried sample was analyzed by GC-MS and NMR in CDCl3 solvent.Typical reductive amination reaction procedure and product analysis
The experiments were conducted within a 50 mL tubular reactor made of borosilicate glass (Pyrex). This reactor was charged with benzaldehyde (1 mmol, 106 mg), along with the catalyst and acetonitrile. The quantities of catalyst mass and acetonitrile volume were systematically varied during the optimization of reaction parameters.Subsequently, the reactor was placed inside a jacketed stainless-steel autoclave, which was equipped with a manometer and two needle valves. This autoclave was pressurized to the desired air pressure and heated to the desired temperature. The mixture was continuously stirred magnetically at a rate of 700 rpm. After the completion of the tests, the autoclave was cooled to room temperature using an ice bath, followed by gentle purge. Upon the conclusion of the reaction, a sample of the reaction mixture was withdrawn using a syringe and subjected to GC-FID analysis to determine reaction conversion and selectivity. Lastly, the samples were evaporated using a rotary evaporator (60 °C, 15 mbar). The obtained dried sample was analyzed by GC-MS and NMR in CDCl3 solvent.
Typical tandem reaction procedure and product analysis
Experiments were performed in a 50 mL tubular reactor of borosilicate glass (Pyrex) loaded with benzylic alcohol (1 mmol, 108 mg), the catalyst and acetonitrile. The quantity of mass of the catalyst and the volume of acetonitrile used, were studied during the optimization of reaction parameters. The reactor was then placed in a jacketed stainless-steel autoclave equipped with a manometer and two needle valves. The autoclave was pressurized with the desired air pressure and heated to the desired temperature. The mixture was kept under magnetic stirring at a rate of 700 rpm. Upon concluding the first step, the autoclave was allowed to cool to room temperature using an ice bath and was gently purged. Following this, the autoclave was charged with hydrogen at the desired pressure and heated to the optimized temperature for the second stage of the tandem process. After the reaction was completed, an aliquot of the reaction mixture was withdrawn by a syringe and analyzed by GC-FID to determine the reaction conversion and selectivity and rotary evaporated (60 °C, 15 mbar). The obtained dried sample was analyzed by GC-MS and NMR in CDCl3 solvent. Detailed characterization data can be found in the ESI.†Conclusions
This work presented the synthesis of heterogeneous metal catalysts supported on a biomass-derived N-doped carbon. These materials were prepared following a one-pot protocol whereby chitin was used as a sustainable source of carbon and nitrogen. Both non-noble and noble metals were considered along with their combinations, particularly bimetallic catalysts based on Mo and Rh.The catalytic behaviour of the designed materials was evaluated in a tandem sequence comprised of (A) the selective oxidation of biomass-derived alcohols to the respective aldehydes using air as a green oxidant, followed by (B) the reductive amination of aldehydes into alkylamines (or imines) using acetonitrile (ACN) as both solvent and reactant, and H2 as reducing agent. The designed mono-metallic catalytic materials and even more the mechanical mixture MIX1 based on Mo(5%)-N/C and Rh(5%)-N/C mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio, displayed excellent results in terms of conversion for both steps of the tandem reaction, that in most cases were quantitative. As for the selectivity, it was more than satisfactory for the oxidation reactions, while a complicated behaviour was encountered in the reductive amination, where the products distribution was also influenced by the in situ hydrogenation of acetonitrile, which leads to several parallel reactions, lowering the overall selectivity towards reductive amination products. Additionally, in both steps the heterogeneous catalytic system could be recycled for up to six repeated reactions, without any considerable loss of its performance, in terms of both conversion and selectivity. In particular, the oxidation step achieved green flags (recommended procedure) according to the CHEM21 Zero Pass toolkit, though a relatively high mass intensity was calculated (MI = 74.79 kg kg−1).
:2 ratio, displayed excellent results in terms of conversion for both steps of the tandem reaction, that in most cases were quantitative. As for the selectivity, it was more than satisfactory for the oxidation reactions, while a complicated behaviour was encountered in the reductive amination, where the products distribution was also influenced by the in situ hydrogenation of acetonitrile, which leads to several parallel reactions, lowering the overall selectivity towards reductive amination products. Additionally, in both steps the heterogeneous catalytic system could be recycled for up to six repeated reactions, without any considerable loss of its performance, in terms of both conversion and selectivity. In particular, the oxidation step achieved green flags (recommended procedure) according to the CHEM21 Zero Pass toolkit, though a relatively high mass intensity was calculated (MI = 74.79 kg kg−1).
Finally, the oxidation of vanillyl alcohol to vanillin was implemented by a solvent-free, continuous flow mechanochemical approach in a mini twin-screw extruder using H2O2 as a green oxidant. The results of this test were remarkable, obtaining 86% yield of vanillin (95% conversion and 90% selectivity) on a synthetic scale (2 g of substrate). This process was awarded a green flag according to the CHEM21 Zero Pass toolkit, providing an excellently low MI of 1.28 kg kg−1.
Author contributions
Conceptualization, D.R.-P. and M.S.; methodology, D.R.-P.; software, E.R.-C.; validation, A.P., M.S. and E.R.-C.; formal analysis, F.Z. and D.B.-P.; investigation, F.Z. and D.B.-P.; resources, A.P., M.S. and E.R.-C.; data curation, D.R.-P.; writing – original draft preparation, F.Z. and D.R.-P.; writing – review and editing, D.R.-P. and M.S.; visualization, A.P.; supervision, M.S. and E.R.-C.; project administration, A.P.; funding acquisition, M.S., A.P., E.R.-C. and D.R.-P. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Cofund Grant Agreement No. 945361. D. B. P. and E. R. C. thank to the Spanish Ministry of Science and Innovation, project PID2021-126235OB-C32 funded by MCIN/AEI/10.13039/501100011033 and FEDER funds, and project TED2021-130756B-C31 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe” by the European Union NextGenerationEU/PRTR.References
- A. Behr, A. J. Vorholt, K. A. Ostrowski and T. Seidensticker, Green Chem., 2014, 16, 982–1006 RSC.
- D. Rigo, G. Fiorani, A. Perosa and M. Selva, ACS Sustainable Chem. Eng., 2019, 7, 18810–18818 CrossRef CAS.
- D. Rigo, D. Polidoro, L. Marcuzzo, A. Perosa and M. Selva, ACS Sustainable Chem. Eng., 2023, 11, 12602–12613 CrossRef CAS.
- R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 1921–1941 RSC.
- R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 7609–7619 RSC.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed.
- C. M. Crombie, R. J. Lewis, R. L. Taylor, D. J. Morgan, T. E. Davies, A. Folli, D. M. Murphy, J. K. Edwards, J. Qi, H. Jiang, C. J. Kiely, X. Liu, M. S. Skjøth-Rasmussen and G. J. Hutchings, ACS Catal., 2021, 11, 2701–2714 CrossRef CAS.
- C. Parmeggiani, C. Matassini and F. Cardona, Green Chem., 2017, 19, 2030–2050 RSC.
- A. J. Hunt, A. S. Matharu, A. H. King and J. H. Clark, Green Chem., 2015, 17, 1949–1950 RSC.
- S. Najafishirtari, K. F. Ortega, M. Douthwaite, S. Pattisson, G. J. Hutchings, C. J. Bondue, K. Tschulik, D. Waffel, B. Peng, M. Deitermann, G. Wilma Busser, M. Muhler and M. Behrens, Chem. – Eur. J., 2021, 27, 16809–16833 CrossRef CAS PubMed.
- C. Jacquot, V. Middelkoop, A. Köckritz, A. Pohar, R. Bienert, S. Kellici, I. A. Bărăgău, B. Venezia, A. Gavriilidis, B. Likozar and A. M. Beale, Sustainable Mater. Technol., 2021, 30, e00329 CrossRef CAS.
- J. Li, M. Li, H. Sun, Z. Ao, S. Wang and S. Liu, ACS Catal., 2020, 10, 3516–3525 CrossRef CAS.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- N. Wang, J. Liu, L. Tang, X. Wei, C. Wang, X. Li and L. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 24966–24975 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2020, 120, 9583–9674 CrossRef CAS PubMed; F. Bucciol, E. Calcio Gaudino, A. Villa, M. C. Valsania, G. Cravotto and M. Manzoli, ChemPlusChem, 2023, 88, e202300017 CrossRef PubMed.
- C. Altuğ, M. J. Muñoz-Batista, D. Rodríguez-Padrón, A. M. Balu, A. A. Romero and R. Luque, Green Chem., 2019, 21, 300–306 RSC.
- D. Polidoro, C. Espro, N. Lazaro, O. Trentin, A. Perosa, S. M. Osman, D. Rodríguez-Padrón, R. Luque and M. Selva, Catal. Today, 2023, 423, 113890 CrossRef CAS.
- S. Mao, C. Wang and Y. Wang, J. Catal., 2019, 375, 456–465 CrossRef CAS.
- T. Horikawa, N. Sakao, T. Sekida, J. I. Hayashi, D. D. Do and M. Katoh, Carbon, 2012, 50, 1833–1842 CrossRef CAS.
- H. Xu, S. Zhang, J. Geng, G. Wang and H. Zhang, Inorg. Chem. Front., 2021, 8, 2829–2834 RSC.
- A. Samikannu, L. J. Konwar, P. Mäki-Arvela and J.-P. Mikkola, Appl. Catal., B, 2019, 241, 41–51 CrossRef CAS.
- M. Triunfo, E. Tafi and A. Guarnieri, et al. , Sci. Rep., 2022, 12, 6613 CrossRef CAS PubMed; M. Kostag and O. A. El Seoud, Carbohydr. Polym. Technol. Appl., 2021, 2, 100079 Search PubMed.
- L. D. Mthembu, D. Lokhat, R. Gupta and N. Deenadayalu, Waste Biomass Valorization, 2021, 12, 3179–3191 CrossRef CAS.
- H. Kargbo, J. S. Harris and A. N. Phan, Renewable Sustainable Energy Rev., 2021, 135, 110168 CrossRef CAS.
- R. Bosch, M. van de Pol and J. Philp, Nature, 2015, 523, 526–527 CrossRef CAS PubMed.
- C. Espro, E. Paone, F. Mauriello, R. Gotti, E. Uliassi, M. L. Bolognesi, D. Rodríguez-Padrón and R. Luque, Chem. Soc. Rev., 2021, 50, 11191–11207 RSC.
- T. Maschmeyer, R. Luque and M. Selva, Chem. Soc. Rev., 2020, 49, 4527–4563 RSC.
- D. Polidoro, D. Ballesteros-Plata, A. Perosa, E. Rodríguez-Castellón, D. Rodríguez-Padrón and M. Selva, Catal. Sci. Technol., 2023, 13, 2223–2238 RSC.
- Y. M. Bar-On, R. Phillips and R. Milo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 6506–6511 CrossRef CAS PubMed.
- J. Andraos, Org. Process Res. Dev., 2009, 13, 161–185 CrossRef CAS.
- A. W. Heinen, J. A. Peters and H. van Bekkum, Eur. J. Org. Chem., 2000, 2000, 2501–2506 CrossRef.
- Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445–450 CrossRef CAS PubMed.
- A. Fayeulle, E. Trudel, A. Damiens, A. Josse, N. Ben Hadj Youssef, P. Vigneron, M. Vayssade, C. Rossi and C. Ceballos, Sustainable Chem. Pharm., 2021, 22, 100471 CrossRef CAS.
- E. A. Hoff, G. X. De Hoe, C. M. Mulvaney, M. A. Hillmyer and C. A. Alabi, J. Am. Chem. Soc., 2020, 142, 6729–6736 CrossRef CAS PubMed.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed; H. Liu, M. Feng and X. Jiang, Chem. – Asian J., 2014, 9, 3360–3389 CrossRef PubMed.
- C. S. Cho, B. T. Kim, T.-J. Kim and S. C. Shim, Chem. Commun., 2001, 2576–2577 RSC.
- M. J. Muñoz-Batista, D. Rodriguez-Padron, A. R. Puente-Santiago and R. Luque, ACS Sustainable Chem. Eng., 2018, 6, 9530–9544 CrossRef.
- T. Friščić, D. G. Reid, I. Halasz, R. S. Stein, R. E. Dinnebier and M. J. Duer, Angew. Chem., Int. Ed., 2010, 49, 712–715 CrossRef PubMed.
- O. Trentin, D. Polidoro, A. Perosa, E. Rodríguez-Castellon, D. Rodríguez-Padrón and M. Selva, Chemistry, 2023, 5, 1760–1769 CrossRef CAS.
- PubChem, https://pubchem.ncbi.nlm.nih.gov/, (accessed September 28, 2023).
- H.-U. Blaser, Chem. Commun., 2003, 293–296 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc04848a |
| This journal is © The Royal Society of Chemistry 2024 |