
Photoredox streamlines electrocatalysis: photoelectrosynthesis of polycyclic pyrimidin-4-ones through carbocyclization of unactivated alkenes with malonates†
Minglin
Tao
a,
Qin
Feng
a,
Kaixing
Gong
a,
Xuege
Yang
a,
Lou
Shi
 *a,
Qiaowen
Chang
*b and
Deqiang
Liang
*a
*a,
Qiaowen
Chang
*b and
Deqiang
Liang
*a
aYunnan Key Laboratory of Metal–Organic Molecular Materials and Device, School of Chemistry and Chemical Engineering, Kunming University, Kunming 650214, China. E-mail: shil090@nenu.edu.cn; liangdq695@nenu.edu.cn
bState Key Laboratory of Advanced Technologies for Comprehensive Utilization of Platinum Metals, Kunming Institute of Precious Metals, Kunming 650106, China. E-mail: changqiaowen@126.com
First published on 28th February 2024
Abstract
Both polycyclic pyrimidin-4-one synthesis and the dehydrogenative coupling of malonates often require a redox agent, an elevated temperature, large amounts of transition-metal salts, and/or highly acidic/basic conditions, and the promising photoelectrocatalysis suffers from limited reaction patterns. We present herein a new photoelectrocatalytic mode and an electrolysis–photocatalysis–Brønsted base hybrid system for the synthesis of polycyclic pyrimidin-4-ones through dehydrogenative carbocyclization of unactivated alkenes with simple malonates under very mild and external-oxidant-free conditions. The reaction exhibits a good functional-group tolerance and is amenable for a gram-scale synthesis, and the sunlight could serve as the light source. Mechanistic studies suggest that the synergistic effect of light and electricity originates from the fast anchoring of an active electrochemical intermediate by the oxidative quenching photocatalytic cycle of Ir(ppy)3.
Introduction
Polycyclic pyrimidin-4-ones are found in the skeletons of many pharmaceuticals (Fig. 1a)1,2 and alkaloids (Fig. 1b),3–6 such as vibegron,1 risperidone,2 (−)-aglaroxin C,3 vasicinone,4 anisotin,5 and auranthine,6 and exhibit a broad range of important bioactivities. Tremendous efforts have been devoted to the construction of this structural motif, and conventionally the cycloaddition of 2-arylquinazolin-4-ones with one-7 or two-carbon synthons8 could afford 2,3-fused pyrimidin-4-ones (Scheme 1a). Alternatively, they could be prepared through the [4 + 2]-annulation of 1,4-dipoles such as isatoic anhydrides,9 isatins10–12 and 2-aminobenzoyls13–15 with 1,2-dipoles16 like 1,2,3,4-tetrahydroisoquinolines,11,13 isatins9,10 and pyridines (Scheme 1b).12,14 Both methodologies are rather appealing, yet generally proceed with a chemical oxidant and large amounts of transition-metal salts at a high temperature. Over the past decade, the alkene difunctionalization17 and cyclization18 have proven to be particularly fruitful lines of research, and it has been found recently that N-tethered alkenyl group quinazolin-4-ones19–23 and N-cyanobenzamides24 could undergo intriguing cyclizations to give fused quinazolin-4-ones, often at an elevated temperature and in the presence of a redox agent. On the other hand, the C–C bond formation is of fundamental interest in synthetic chemistry, and the dehydrogenative coupling of a C–H bond is most fascinating due to its atom- and step-economy.25 Malonates are readily available C–H feedstocks, and they could serve as nucleophiles to react with electrophiles usually using a stoichiometric excess of a base in the absence26 or presence of a noble-metal catalyst.27 Recent reports have revealed that a polarity-reversed malonate radical could be furnished upon the oxidation of the corresponding enolate, and such an electrophilic 1,3-dicarbonyl radical could be coupled with a wide range of acceptors28–31 as well as persistent radicals.32 Notwithstanding the extraordinary discoveries, these reactions frequently require as well high transition-metal loadings and/or highly acidic/basic conditions. Furthermore, it seems that the net-oxidative coupling of a malonate radical28–30 is more challenging than a redox-neutral one,31 because the former usually proceeds at an elevated temperature and is often restricted to fluoro-28 or alkyl-activated malonates.29 As a result, the development of green and mild protocols for the synthesis of polycyclic pyrimidin-4-ones and for the dehydrogenative coupling of simple malonates is still highly desirable.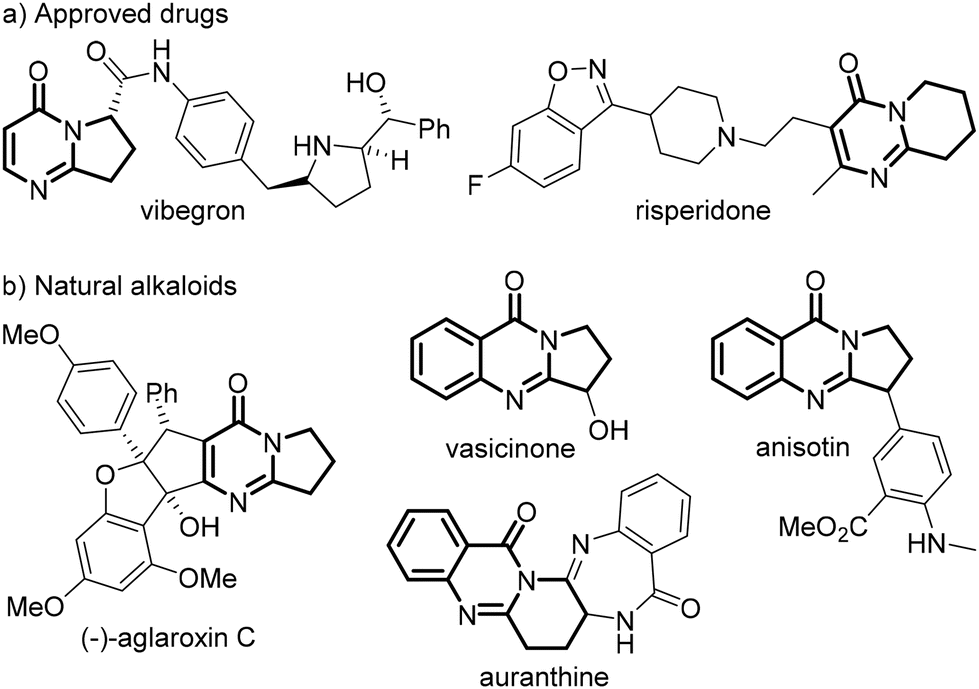 | ||
| Fig. 1 (a) Selected examples of pyrimidin-4-one pharmaceuticals; (b) Selected examples of pyrimidin-4-one alkaloids. | ||
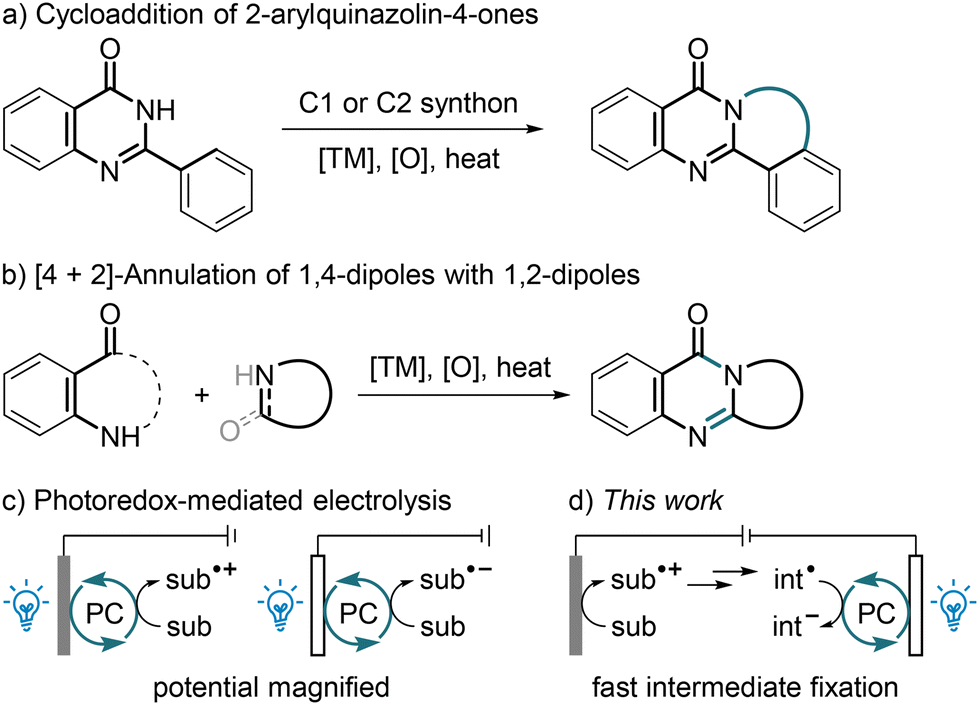 | ||
| Scheme 1 Synthesis of polycyclic pyrimidin-4-ones and electrophotocatalysis. | ||
Photocatalytic and electrochemical techniques have revolutionized modern organic synthesis over the past decade.33 In addition, enterprising pioneers, including Lambert34 and Xu,35 combined the two techniques to harness the powers of both light and electricity, spawning a new paradigm of electrophotocatalysis and pushing the boundaries of redox chemistry.36 Using a photocatalytic cycle as the electron shuttle between the reactants and an anode34,35,37–41 or cathode,42 a mild yet heterogeneous electrode potential might be turned into a homogeneous and highly biased excited-state potential, thus enabling the coupling of an inactive radical precursor, or enabling an improved reaction efficiency and/or a better functional-group tolerance (Scheme 1c). In some cases, the radical-photogeneration process involved the photoinduced ligand-to-metal charge transfer of CeCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37 or FeCl3,38 or involved the hydrogen-atom transfer to an excited aryl ketone,39 decatungstate40 or riboflavin.41 In the case reported by Li and co-workers, light and electricity induced the same reaction sequence simultaneously and independently.43 It was also reported that visible light could be introduced to electrolysis to cleave a C/N/Cl–halogen bond44 or to illuminate a photoanode.45 Despite these exciting advances, to date photoelectrosynthesis is still rather underdeveloped, and its reaction types are very limited. Considering its characteristics of sustainability, mildness and robustness, such a hybrid strategy is highly promising, and the development of new reaction modes is of great significance. As a part of our ongoing interest in the green synthesis of heterocycles,46 we report herein a photoelectrocatalytic construction of polycyclic pyrimidin-4-ones through the dehydrogenative carbocyclization of unactivated alkenes with simple malonates under mild conditions, wherein the diffusion issue, which is related to electrochemistry, of a key transient intermediate might be addressed through fast fixation by an oxidative quenching photocatalytic cycle (Scheme 1d). Similar synergistic effects were observed in two photoreductant-assisted electrochemical alkylations of quinolines, wherein the photocatalysts (PCs) were not regenerated at an electrode.47,48
37 or FeCl3,38 or involved the hydrogen-atom transfer to an excited aryl ketone,39 decatungstate40 or riboflavin.41 In the case reported by Li and co-workers, light and electricity induced the same reaction sequence simultaneously and independently.43 It was also reported that visible light could be introduced to electrolysis to cleave a C/N/Cl–halogen bond44 or to illuminate a photoanode.45 Despite these exciting advances, to date photoelectrosynthesis is still rather underdeveloped, and its reaction types are very limited. Considering its characteristics of sustainability, mildness and robustness, such a hybrid strategy is highly promising, and the development of new reaction modes is of great significance. As a part of our ongoing interest in the green synthesis of heterocycles,46 we report herein a photoelectrocatalytic construction of polycyclic pyrimidin-4-ones through the dehydrogenative carbocyclization of unactivated alkenes with simple malonates under mild conditions, wherein the diffusion issue, which is related to electrochemistry, of a key transient intermediate might be addressed through fast fixation by an oxidative quenching photocatalytic cycle (Scheme 1d). Similar synergistic effects were observed in two photoreductant-assisted electrochemical alkylations of quinolines, wherein the photocatalysts (PCs) were not regenerated at an electrode.47,48
Results and discussion
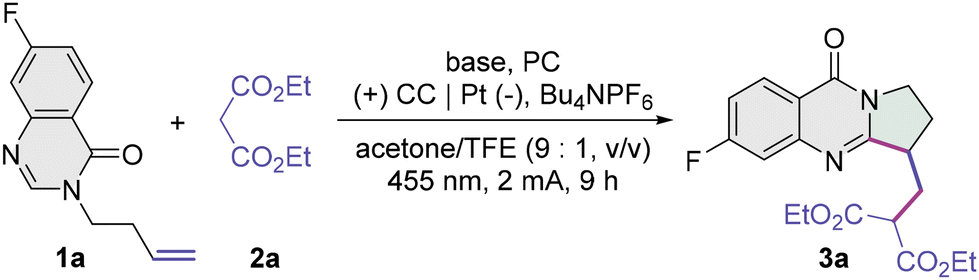
Exploration of the photoelectrocatalytic carbocyclization was begun using 7-fluoro-3-homoallylquinazolin-4-one 1a and diethyl malonate 2a as model substrates (Table 1; see the ESI† for full details). The desired product alkylated dihydropyrroloquinazolinone 3a was obtained in 54% NMR yield when the reaction was conducted in an undivided cell having a C cloth (CC) anode and a Pt plate cathode with K2CO3 (40 mol%) as the base, 4CzIPN (5 mol%) as the PC, Bu4NPF6 (1 equiv.) as the supporting electrolyte in acetone/2,2,2-trifluoroethanol (TFE, 9:1, v/v) under blue-light irradiation and 2.0 mA constant current conditions at room temperature for 9 h (Q = 2.24 F mol−1, entry 1). While tBuOK worked as well as K2CO3 (entry 2), the use of a weaker base such as KHCO3 led to a depreciation in the yield (entry 3). A base catalyst is essential for this transformation, and we failed to access the desired product 3a under base-free conditions (entry 4). A survey of solvents was then conducted. The use of acetone alone had a slightly deleterious effect on the reaction efficiency (entry 5), and the reaction in CH3CN gave a poor yield of 3a (entry 6). 1,2-Dichloroethane (DCE) proved ineffective for this transformation, probably due to the poor solubility of K2CO3 (entry 7). A comparable yield of 55% was achieved upon varying the ratio of acetone to TFE to 11:1 (entry 8). Interestingly, a slightly better yield was obtained when the photoreductant of Ir(ppy)3 was used as the PC (entry 9), whereas the use of Mes-Acr+ClO4−, a potent photooxidant, furnished 3a in a very low yield (entry 10). The yield was improved to 69% on reducing the current to 1.5 mA and prolonging the irradiation time to 12 h, suggesting the importance of the synchronization of electrocatalytic and photocatalytic steps (entry 11). Further increases in the yield were achieved by reducing the loadings of K2CO3 and Ir(ppy)3 and increasing the dosage of the electrolyte (entry 12). Whereas the absence of electricity led to no reactivity (entry 13), and pyrroloquinazolinone 3a was afforded in poor yields upon the removal of light (entry 14) or PC (entry 15) under otherwise optimized conditions.
|
|
||||
|---|---|---|---|---|
| Entry | Base | PC | Solvent | Yieldb (%) |
| 1 | K2CO3 | 4CzIPN | Acetone/TFE | 54 |
| 2 | tBuOK | 4CzIPN | Acetone/TFE | 50 |
| 3 | KHCO3 | 4CzIPN | Acetone/TFE | 20 |
| 4 | 4CzIPN | Acetone/TFE | 0 | |
| 5 | K2CO3 | 4CzIPN | Acetone | 47 |
| 6 | K2CO3 | 4CzIPN | CH3CN | 28 |
| 7 | K2CO3 | 4CzIPN | DCE | 0 |
| 8 | K2CO3 | 4CzIPN | Acetone/TFEc | 55 |
| 9 | K2CO3 | Ir(ppy)3 | Acetone/TFEc | 59 |
| 10 | K2CO3 | Mes-Acr+ClO4− | Acetone/TFEc | 16 |
| 11d | K2CO3 | Ir(ppy)3 | Acetone/TFEc | 69 |
| 12e | K2CO3 | Ir(ppy)3 | Acetone/TFEc | 75 (68)f |
| Entry | Deviation from entry 12 | Yieldb (%) |
|---|---|---|
|
a Reaction conditions: 1a (0.3 mmol), 2a (0.9 mmol), base (40 mol%, 0.12 mmol), PC (5 mol%, 0.015 mmol), Bu4NPF6 (0.3 mmol), acetone/TFE (9:1, v/v, 6.0 mL), 6 W blue LEDs (λmax = 455 nm), CC anode (15 × 15 mm), platinum plate cathode (15 × 15 mm), undivided cell, 2.0 mA, Ar, room temperature, 9 h.
b Yields were determined by 19F NMR analysis using trifluorotoluene as an internal standard.
c acetone/TFE (11:1, v/v).
d 1.5 mA, 12 h.
e K2CO3 (10 mol%, 0.03 mmol), Ir(ppy)3 (3 mol%, 0.009 mmol), Bu4NPF6 (1.5 equiv., 0.45 mmol), 1.5 mA, 12 h.
f Isolated yield.
|
||
| 13 | No electric current | 0 |
| 14 | No light | 40 |
| 15 | No PC | 38 |
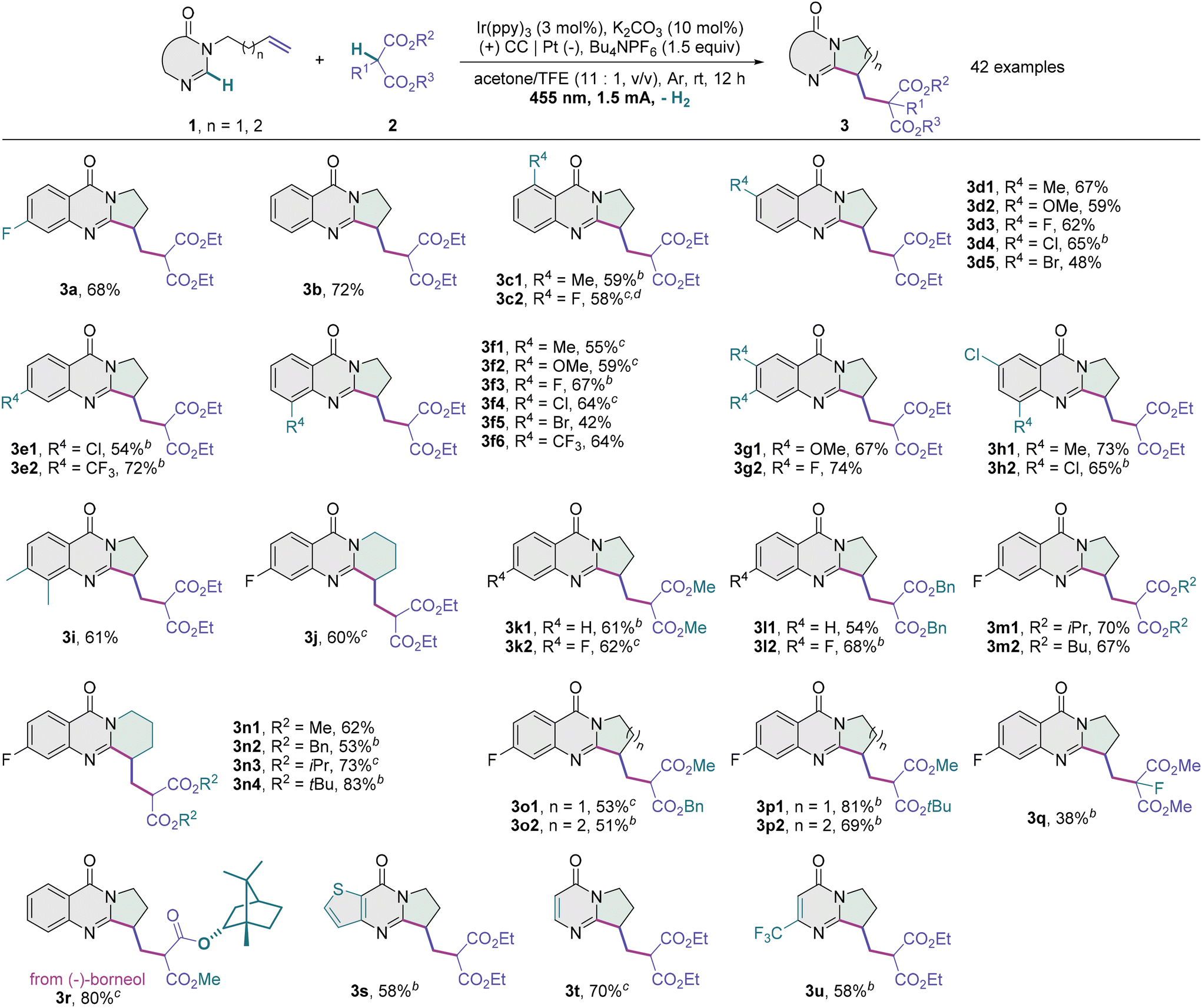
With the optimal conditions in hand, we evaluated the scope of this transformation (Table 2). While unsubstituted 3-homoallyl quinazolin-4-one reacted with diethyl malonate 2a to afford the desired product 3b in 72% yield, a variety of alkylated dihydropyrroloquinazolinones bearing an electron-releasing or electron-withdrawing unit at the 5- (3c1 and 3c2), 6- (3d1–3d5), 7- (3e1 and 3e2) or 8-position (3f1–3f6) could be prepared from the corresponding 3-homoallyl quinazolin-4-ones in moderate to high yields. The tested functionalities include methyl, methoxy, fluoro, chloro, bromo and trifluoromethyl groups, and their substituent effects are subtle and poorly characterized. Brominated quinazolin-4-ones (3d5 and 3f5) might be challenging substrates and prone to reductive dehalogenation (see the ESI†). These halide products possess complementary functional handles for subsequent cross-coupling. Such a photoelectrocatalytic protocol also tolerates disubstituted substrates such as 6,7-dimethoxy (3g1), 6,7-difluoro (3g2), 6-chloro-8-methyl (3h1), 6,8-dichloro (3h2) and 7,8-dimethyl quinazolin-4-ones (3i). Tetrahydropyridoquinazolinone 3j with a newly formed six-membered ring was furnished in a good yield from 3-(4-pentenyl) quinazolin-4-one with a five-carbon alkenyl chain. Using a longer alkenyl chain, we failed to obtain the desired seven- or eight-membered ring product (see the ESI†). As for C–H radical precursors, dimethyl, dibenzyl, diisopropyl, dibutyl and di-tert-butyl malonates (3k–3n), as well as asymmetric precursors like benzyl methyl malonate (3o1 and 3o2) and tert-butyl methyl malonate (3p1 and 3p2), were all competent to react with a quinazolin-4-one unit having an N-tethered alkene moiety, with the corresponding dihydropyrrolo- or tetrahydropyridoquinazolinones provided. A poor yield of the cyclized product 3q was obtained when dimethyl 2-fluoromalonate was used, probably owing to the steric effect. The potential for the late-stage editing of complex molecules was demonstrated by the carbocyclization of a malonate derived from (−)-borneol (3r). It is noteworthy that the photoelectrocatalytic conditions were mild enough to tolerate a thienoquinazolinone substrate (3s), and the alkenes linked to a monocyclic pyrimidin-4-one (3t) or 6-(trifluoromethyl)pyrimidin-4-one motif (3u) were productively engaged in this cyclization as well. Unfortunately, the phenylethynyl, homoallyl, (tert-butoxycarbonyl)amino and hydroxy substituents on the aryl moiety were not tolerated (see the ESI†) and represent some limitations of the present methodology.
|
a Reaction conditions: 1 (0.3 mmol), 2 (0.9 mmol), K2CO3 (0.03 mmol), Ir(ppy)3 (0.009 mmol), Bu4NPF6 (0.45 mmol), acetone/TFE (11:1, v/v, 6.0 mL), 6 W blue LEDs (λmax = 455 nm), CC anode (15 × 15 mm), platinum plate cathode (15 × 15 mm), undivided cell, 1.5 mA, Ar, room temperature, 12 h.
b 1.5 mmol of 2 was used.
c 0.15 mmol of K2CO3 was used.
d 24 h.
|
|---|
|
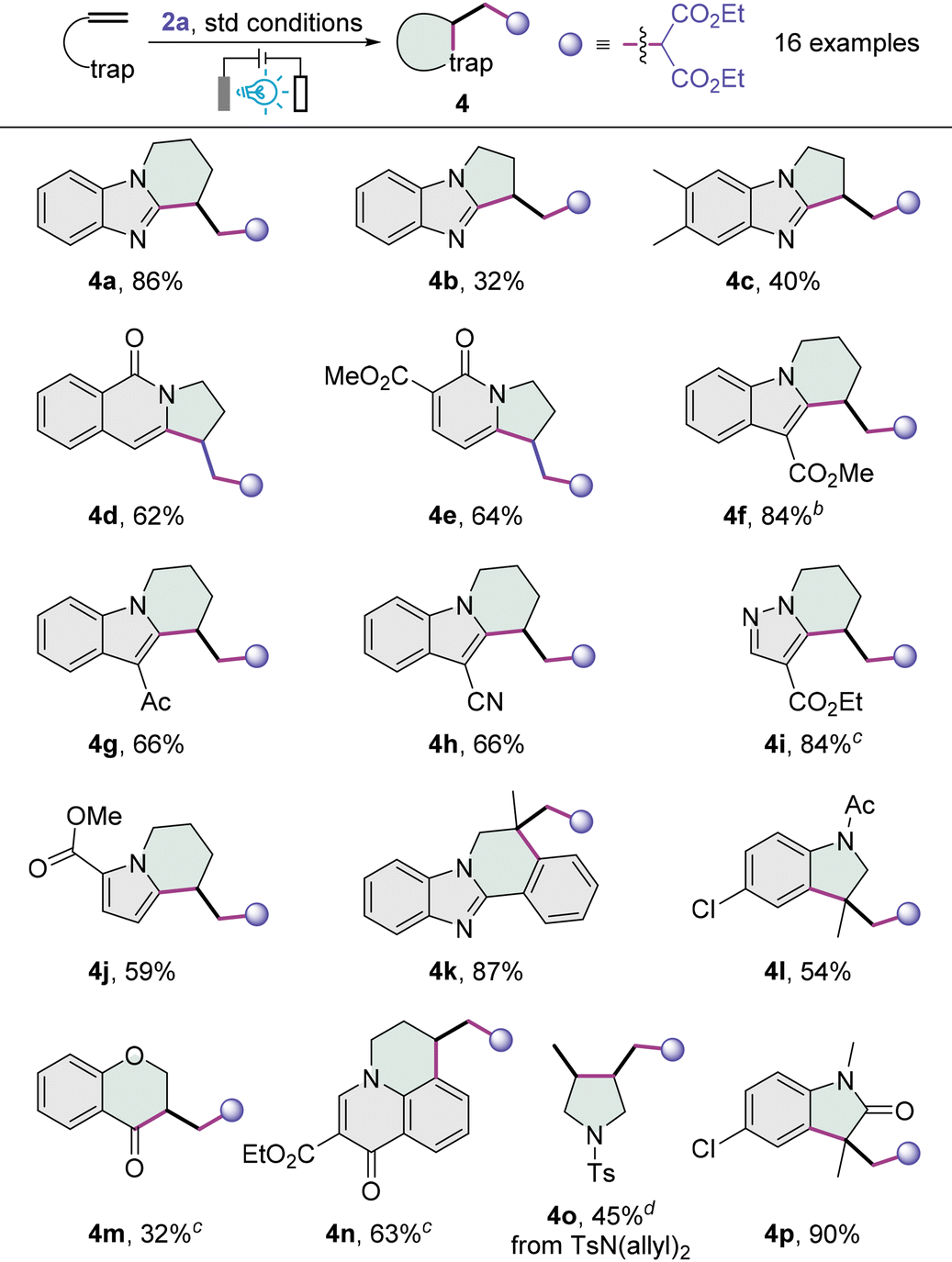
This new electrophotocatalytic strategy can be applied to a variety of unactivated alkenes (Table 3). Benzoimidazolic alkenes were first assessed, and with diethyl malonate 2a as the carbon radical precursor under optimal conditions, polycyclic product benzopiperidinoimidazole 4a was furnished in 86% yield from the N-(4-pentenyl) benzoimidazole. N-Homoallyl benzoimidazoles participated in this cyclizative cascade to afford modest yields of the corresponding benzopyrroloimidazoles 4b and 4c, which might be somewhat strained due to the fusion of two five-membered rings. The attempt to extend the reaction to N-homoallyl isoquinolin-1-one gave rise to the formation of pyrroloisoquinolinone 4d in a moderate yield, and pyrrolopyridinone 4e was also prepared from the corresponding 1-homoallyl pyridin-2-one. We then moved on to investigate the carbocyclization of indolic alkenes. Indolic substrates bearing an electron-withdrawing motif, such as carbethoxy, acetyl or cyano group, at the C3-position all worked well, providing the desired tetrahydropyridoindoles 4f–4h in good to high yields. The substrates featuring a pyrazole-4-carboxylate or pyrrole-2-carboxylate are both compatible with this transformation, and the desired pyridopyrazole 4i and 5,6,7,8-tetrahydroindolizine 4j were provided in 84% and 59% yields, respectively. While this carbocyclization is applicable to N-(2-methylallyl)-2-phenylbenzoimidazole, giving 5,6-dihydrobenzoimidazo[2,1-a]isoquinoline 4k in a very high yield, the use of N-allyl acetanilide afforded indoline 4l in 54% yield. Interestingly, when O-allyl salicylaldehyde was used as the substrate, the carbonyl group served as the inbuilt radical trap to furnish the chroman-4-one 4m albeit in a modest yield. The 6,7-dihydropyrido[3,2,1-ij]quinolin-1-one 4n rather than a 2,3-dihydropyrrolo[1,2-a]quinolin-5-one product was delivered when an N-homoallyl quinolin-4-one was employed. With the tosyl-protected diallylamine, the redox-neutral product 3,4-dialkylpyrrolidine 4o was afforded. Although we failed to extend this electrophotocatalytic protocol to several activated alkenes (see the ESI†), indolin-2-one 4p was obtained in an excellent yield from the parent N-arylacrylamide.
|
a Reaction conditions: alkene substrate (0.3 mmol), 2a (0.9 mmol), K2CO3 (0.03 mmol), Ir(ppy)3 (0.009 mmol), Bu4NPF6 (0.45 mmol), acetone/TFE (11:1, v/v, 6.0 mL), 6 W blue LEDs (λmax = 455 nm), CC anode (15 × 15 mm), platinum plate cathode (15 × 15 mm), undivided cell, 1.5 mA, Ar, room temperature, 12 h.
b 24 h.
c 0.15 mmol of K2CO3 were used.
d 0.45 mmol of K2CO3 were used.
|
|---|
|
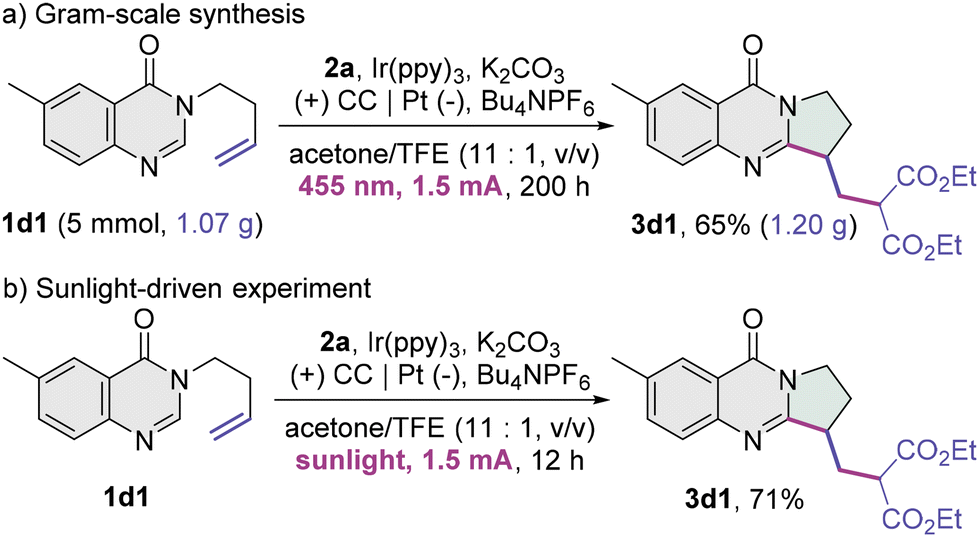
A gram-scale reaction of 6-methyl-3-homoallyl quinazolin-4-one 1d1 and diethyl malonate 2a was performed (Scheme 2a), and the fused quinazolin-4-one product 3d1 was delivered in a comparable yield. The yield of 3d1 was not compromised using sunlight as the light source (Scheme 2b). These results highlight the feasibility of the present protocol in practical utilization.
 | ||
| Scheme 2 Synthetic utility. | ||
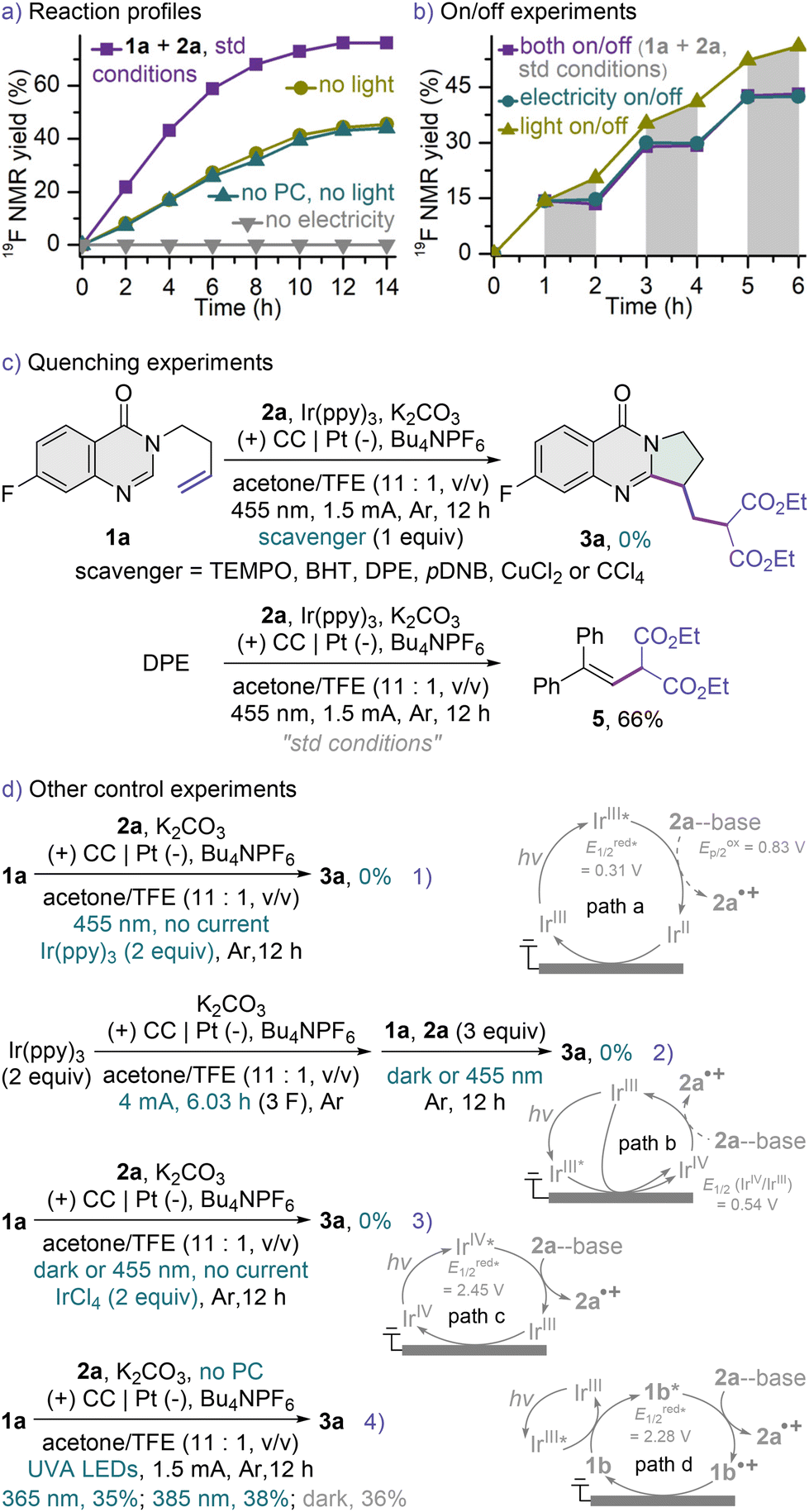
Attention was then turned towards gaining insight into the mechanism of this transformation (Scheme 3). Kinetic profiles were recorded using 19F NMR spectroscopy of the reactions run under standard, electrochemical or photoredox conditions (Scheme 3a and Fig. S4†). While the carbocyclization did not proceed in the absence of electric current (gray line), much lower reaction rates were observed in the electrolyses under dark conditions (dark yellow and dark cyan lines), confirming the unique catalytic power of this hybrid system and highlighting the importance of simultaneously harnessing the powers of both light and electricity (violet line). A mechanism of indirect electrolysis using ground-state Ir(ppy)3 as an electron shuttle could be ruled out, as comparable results were obtained in the electrochemical reactions with or without the PC (dark yellow and dark cyan lines). Similar trends were observed in the on/off experiments (Scheme 3b and Fig. S5†), in which the carbocyclization became slower under dark conditions and stopped in electricity-off cycles, suggesting a short chain mechanism. The radical and single-electron transfer (SET) nature of this protocol was evidenced by quenching experiments (Scheme 3c), in which the model reaction under otherwise optimal conditions ceased upon the addition of a quencher, and the tested quenchers include radical scavengers such as 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), butylated hydroxytoluene (BHT) and 1,1-diphenylethylene (DPE), a SET inhibitor of p-dinitrobenzene (pDNB), and electron scavengers like CuCl2 and CCl4. Subjecting DPE to our standard conditions afforded the malonate-DPE adduct 5 in 66% yield, indicating that the malonate radical might be involved in this transformation. Cyclic voltammetry (CV) tests revealed that the unactivated alkene 1a (Ep/2 = 1.92 V vs. SCE, Fig. S12†)22 has a higher oxidation potential than the base-activated diethyl malonate 2a (Ep/2 = 0.83 V vs. SCE, Fig. S10 and S11†),32,49 thus it is unlikely that 1a undergoes a preferential oxidation followed by the nucleophilic addition of 2a to the resulting alkene radical cation.50
 | ||
| Scheme 3 Mechanistic investigations. | ||
The excited-state reduction potential of Ir(ppy)3 is only 0.31 V vs. SCE (see the ESI†),51 thus precluding a proton-coupled electron transfer (PCET) from diethyl malonate 2a to excited PC in the presence of a base. Such results are in accordance with the facts that in Stern–Volmer quenching experiments 2a hardly quenched excited Ir(ppy)3 in acetone in the presence or absence of K2CO3 (Fig. S19†),32 and that 2 equiv. of Ir(ppy)3 as the photooxidant failed to promote the desired carbocyclization without electricity (Scheme 3d1). These combine to rule out the merging of electrolysis with the reductive quenching photocatalytic cycle of Ir(ppy)3 (path a).52 Although a transient photoexcited catalyst has not been reported to be heterogeneously oxidized at the anode,36 we still assessed the possibility of the oxidative quenching photocatalytic cycle53 serving as an electron shuttle between the anode and the reactants (path b). No reaction occurred using an excess of either electrochemically generated IrIV species (Scheme 3d2)54 or IrCl4 (Scheme 3d3) as the oxidant in the absence of the electric current, suggesting that an IrIV complex might not be a key oxidizing intermediate in this transformation. This was further evidenced by the CV tests of Ir(ppy)3, wherein the reduction wave for IrIV/IrIII (Fig. S13 and S14†) and the oxidation peak of IrIV/IrV (Fig. S13†) did not disappear or significantly decrease upon the addition of 2a and K2CO3.28b,55 Considering that the SET from base-activated 2a to the related IrIV complex (E1/2 (IrIV/IrIII) = 0.54 V vs. SCE, Fig. S7 and S8†)51 is thermodynamically unfavorable, a highly oxidized excited-state IrIV complex might be involved (path c).56 Although the excited-state reduction potential of the in situ electrochemically generated IrIV complex was estimated to be 2.45 V vs. SCE (see the ESI†), visible light failed to induce the excess IrIV-promoted carbocyclization (Schemes 3d2 and d3), suggesting that such an electron-primed photocatalytic mechanism (path c)34,37,42 is less likely yet could not be completely ruled out. Quinazolin-4-ones can absorb to some extent ultraviolet-A (UVA) light and serve as a photosensitizer,20 and the excited-state reduction potential of quinazolin-4-one 1b was estimated to be 2.28 V vs. SCE (see the ESI†). Nonetheless, an energy-transfer process leading to the triplet sensitization of 1b by excited Ir(ppy)3 might not be a productive pathway as well (path d), because no beneficial effect was observed when the electrochemical reaction without a PC was irradiated with UVA LEDs (Scheme 3d4). These negative results concerning the two highly oxidizing photooxidants (paths c and d) coupled to the PC screening data (Table S4†) revealed that the title reaction might not be excited-state reduction potential-dependent. The malonate radical might be generated at the anode.57
A mechanistic proposal consistent with these studies is presented in Scheme 4. In the beginning, diethyl malonate 2a undergoes a PCET to the anode to afford the malonate radical A, and such a polarity-reversed 1,3-dicarbonyl radical adds to the tethered alkene moiety, which is nucleophilic, of 3-homoallyl quinazolin-4-one 1a to give the alkyl radical intermediate B. B cyclizes to the ring closure N-radical C through the intramolecular radical trapping by the amidine moiety of the heterocyclic ring. Two factors weighed heavily against the oxidation of C by the reductive quenching photocatalytic cycle of Ir(ppy)3 followed by a deprotonation to furnish the pyrroloquinazolinone product 3a (path e). One is the extremely low excited-state reduction potential of the PC (E1/2 = 0.31 V vs. SCE),51 and the other is its much lower reduction potential (Ep/2 = −1.85 V vs. SCE, Fig. S9†)51 than that of product 3a (Ep/2 = −0.72 V vs. SCE, Fig. S15 and S16†), making the SET from intermediate C to excited Ir(ppy)3 thermodynamically unfavorable.47,48 On the other hand, the anodic oxidation of C and subsequent deprotonation could occur to deliver pyrroloquinazolinone 3a (path f).58 Nonetheless, because of the heterogeneous nature of electrochemistry, radical C might not be long-lived enough and might experience difficulty in wandering to the anode surface. In such a case, C is reduced by the oxidative quenching photocatalytic cycle (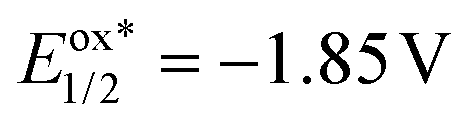 vs. SCE, see the ESI†)51 to give the nitranion intermediate D,47,48,59 and the IrIII catalyst is regenerated at the cathode (path g). The protonation of D gives tetrahydropyrroloquinazolinone E, which was detected by high-resolution mass spectrometry (HRMS, Fig. S22 and S23†). E is readily oxidized to pyrroloquinazolinone 3a after migrating to the anode.60 Therefore, we consider that the synergistic effect of this photoelectrocatalytic system originates from the fast anchoring of an active electrochemical intermediate by a photocatalytic cycle with a homogeneous, transient, fixed and highly biased excited-state potential. Significant hydrogen evolution was confirmed by the hydrogen detection experiments (Fig. S24†), which is associated with the cathodic reduction of protons to hydrogen to maintain electron conservation.
vs. SCE, see the ESI†)51 to give the nitranion intermediate D,47,48,59 and the IrIII catalyst is regenerated at the cathode (path g). The protonation of D gives tetrahydropyrroloquinazolinone E, which was detected by high-resolution mass spectrometry (HRMS, Fig. S22 and S23†). E is readily oxidized to pyrroloquinazolinone 3a after migrating to the anode.60 Therefore, we consider that the synergistic effect of this photoelectrocatalytic system originates from the fast anchoring of an active electrochemical intermediate by a photocatalytic cycle with a homogeneous, transient, fixed and highly biased excited-state potential. Significant hydrogen evolution was confirmed by the hydrogen detection experiments (Fig. S24†), which is associated with the cathodic reduction of protons to hydrogen to maintain electron conservation.
 | ||
| Scheme 4 The proposed mechanism. | ||
Conclusions
The work herein describes in detail the development of polycyclic pyrimidin-4-one synthesis using an electrolysis–photocatalysis–Brønsted base hybrid system through the dehydrogenative carbocyclization of unactivated alkenes with simple malonates. The reaction is mild, external-oxidant-free and atom-economical with H2 serving as the sole by-product, and exhibits a good functional-group tolerance. This new photoelectrocatalytic mode, which benefits from the fast anchoring of an active electrochemical intermediate by the oxidative quenching photocatalytic cycle, might provide clues for further development of novel photoelectrocatalytic reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (22061024), the Yunnan Ten Thousand Talent Program for Young Top-Notch Talents (YNWR-QNBJ-2018-133), the Open Project of Yunnan Precious Metals Laboratory (YPML-2023050262), the Scientific Research Funds of Yunnan Education Department (2023J0838), the Frontier Research Team of Kunming University 2023, and the Scientific Research Funds of Kunming University (XPZJ2204 and YJL23020). In addition, Minglin Tao would like to thank her family, especially her grandma Ms Wu, because she has always been deeply loved.References
- J. D. Salvo, H. Nagabukuro, L. A. Wickham, C. Abbadie, J. A. DeMartino, A. Fitzmaurice, L. Gichuru, A. Kulick, M. J. Donnelly, N. Jochnowitz, A. L. Hurley, A. Pereira, A. Sanfiz, G. Veronin, K. Villa, J. Woods, B. Zamlynny, E. Zycband, G. M. Salituro, T. Frenkl, A. E. Weber, S. D. Edmondson and M. Struthers, J. Pharmacol. Exp. Ther., 2017, 360, 346–355 CrossRef PubMed.
- J. Bolós, S. Gubert, L. Anglada, J. M. Planas, C. Burgarolas, J. M. Castelló, A. Sacristán and J. A. Ortiz, J. Med. Chem., 1996, 39, 2962–2970 CrossRef PubMed.
- J. Hiort, Chaidir, F. I. Bohnenstengel, B. W. Nugroho, C. Schneider, V. Wray, L. Witte, P. D. Hung, L. C. Kiet and P. Proksch, J. Nat. Prod., 1999, 62, 1632–1635 CrossRef CAS.
- A. H. Amin and D. R. Mehta, Nature, 1959, 184, 1317 CrossRef CAS PubMed.
- S. Johne, D. Gröger and M. Hesse, Helv. Chim. Acta, 1971, 54, 826–834 CrossRef CAS.
- S. E. Yeulet, P. G. Mantle, J. N. Bilton, H. S. Rzepa and R. N. Sheppard, J. Chem. Soc., Perkin Trans. 1, 1986, 1891–1894 RSC.
- (a) S. Devkota, M. S. Akhtar and Y. R. Lee, Adv. Synth. Catal., 2023, 365, 1465–1470 CrossRef CAS; (b) X. Liu, J. Chen, C. Yang, Z. Wu, Z. Li, Y. Shi, T. Huang, Z. Yang and Y. Wu, Org. Lett., 2021, 23, 6831–6835 CrossRef CAS PubMed; (c) Y. Shi, T. Huang, T. Wang, J. Chen, X. Liu, Z. Wu, X. Huang, Y. Zheng, Z. Yang and Y. Wu, Chem. – Eur. J., 2021, 27, 13346–13351 CrossRef CAS PubMed; (d) F. Dong, J.-Q. Liu and X.-S. Wang, Org. Lett., 2020, 22, 2887–2891 CrossRef CAS PubMed.
- (a) T. Huang, T. Wang, Y. Shi, J. Chen, X. Guo, R. Lai, X. Liu, Z. Wu, D. Peng, L. Wang, H. Li, L. Hai and Y. Wu, Org. Lett., 2021, 23, 1548–1553 CrossRef CAS PubMed; (b) Z.-H. Wang, H. Wang, H. Wang, L. Li and M.-D. Zhou, Org. Lett., 2021, 23, 995–999 CrossRef CAS PubMed; (c) Y. Feng, N. Tian, Y. Li, C. Jia, X. Li, L. Wang and X. Cui, Org. Lett., 2017, 19, 1658–1661 CrossRef CAS PubMed; (d) J. B. Lee, M. E. Kang, J. Kim, C. Y. Lee, J.-M. Kee, K. Myung, J.-U. Park and S. Y. Hong, Chem. Commun., 2017, 53, 10394–10397 RSC.
- (a) B. Rai, R. D. Shukla and A. Kumar, Green Chem., 2018, 20, 822–826 RSC; (b) C. Wang, L. Zhang, A. Ren, P. Lu and Y. Wang, Org. Lett., 2013, 15, 2982–2985 CrossRef CAS PubMed.
- (a) L. Q. Huang, C. L. Dong, Y. H. He and Z. Guan, Adv. Synth. Catal., 2022, 364, 3225–3237 CrossRef CAS; (b) F.-C. Jia, Z.-W. Zhou, C. Xu, Y.-D. Wu and A.-X. Wu, Org. Lett., 2016, 18, 2942–2945 CrossRef CAS PubMed.
- (a) L. Liu, Z. Xu, J. Lin, Z. Zhang, Y. Wu, P. Yang, Y. Hang, D. Song, W. Zhong and F. Ling, Adv. Synth. Catal., 2023, 365, 2248–2254 CrossRef CAS; (b) D. Wang, F. Xiao, F. Zhang, H. Huang and G. J. Deng, Chin. J. Chem., 2021, 39, 87–92 CrossRef CAS; (c) X. Chen, F. Xia, Y. Zhao, J. Ma, Y. Ma, D. Zhang, L. Yang and P. Sun, Chin. J. Chem., 2020, 38, 1239–1244 CrossRef CAS.
- M. Liu, M. Shu, C. Yao, G. Yin, D. Wang and J. Huang, Org. Lett., 2016, 18, 824–827 CrossRef CAS PubMed.
- (a) C. Lu, Z. Su, D. Jing, S. Jin, L. Xie, L. Li and K. Zheng, Org. Lett., 2019, 21, 1438–1443 CrossRef CAS PubMed; (b) F. Xie, Q.-H. Chen, R. Xie, H.-F. Jiang and M. Zhang, ACS Catal., 2018, 8, 5869–5874 CrossRef CAS; (c) Y. Liu, G. Q. Chen, C. W. Tse, X. Guan, Z. J. Xu, J. S. Huang and C. M. Che, Chem. - Asian J., 2015, 10, 100–105 CrossRef CAS PubMed.
- X. Zhou, Y. Ding and H. Huang, Chem. - Asian J., 2020, 15, 1678–1682 CrossRef CAS PubMed.
- (a) D. S. Reddy and A. G. Kutateladze, Org. Lett., 2019, 21, 2855–2858 CrossRef CAS PubMed; (b) X. Li, H. Huang, C. Yu, Y. Zhang, H. Li and W. Wang, Org. Lett., 2016, 18, 5744–5747 CrossRef CAS PubMed; (c) T. Abe and K. Yamada, Org. Lett., 2016, 18, 6504–6507 CrossRef CAS PubMed.
- J. Li, Z.-B. Wang, Y. Xu, X.-C. Lu, S.-R. Zhu and L. Liu, Chem. Commun., 2019, 55, 12072–12075 RSC.
- (a) K. Liang, D. Zhang, Y. Su, L. Lu, J. Hu, Y. H. Chen, X. Zhang, A. Lei and H. Yi, Chem. Sci., 2023, 14, 4152–4157 RSC; (b) X. Li, X. Li, W. Cui, Q. Wu, L. Wang, J. Lv and D. Yang, Org. Lett., 2023, 25, 3260–3265 CrossRef CAS PubMed; (c) W. Cui, G. Guo, Y. Wang, X. Song, J. Lv and D. Yang, Chem. Commun., 2023, 59, 6367–6370 RSC; (d) H. Y. Zhou, L. Q. Fei, J. L. Zhang, Y. M. Pan and H. T. Tang, Adv. Synth. Catal., 2023, 365, 1591–1595 CrossRef CAS.
- (a) P. Huang, Z. Yan, J. Ling, P. Li, J. Wang, J. Li, B. Sun and C. Jin, Green Chem., 2023, 25, 3989–3994 RSC; (b) W.-J. Wei, L. Zhan, C.-N. Jiang, H.-T. Tang, Y.-M. Pan, X.-L. Ma and Z.-Y. Mo, Green Chem., 2023, 25, 928–933 RSC; (c) K. Sun, D. Zhao, Q. Li, S. Ni, G. Zheng and Q. Zhang, Sci. China: Chem., 2023, 66, 2309–2316 CrossRef CAS; (d) Z. Zhang, P. Tan, S. Wang, H. Wang, L. Xie, Y. Chen, L. Han, S. Yang and K. Sun, Org. Lett., 2023, 25, 4208–4213 CrossRef CAS PubMed; (e) A. X. Huang, H. L. Zhu, F. L. Zeng, X. L. Chen, X. Q. Huang, L. B. Qu and B. Yu, Org. Lett., 2022, 24, 3014–3018 CrossRef CAS PubMed.
- (a) Y.-Z. Ren, C.-Z. Fang, B.-B. Zhang, L. He, Y.-L. Tu and X.-Y. Chen, Org. Lett., 2023, 25, 3585–3589 CrossRef CAS PubMed; (b) Z. Yang, X. Wu, J. Zhang, J.-T. Yu and C. Pan, Org. Lett., 2023, 25, 1683–1688 CrossRef CAS PubMed; (c) X. Wan, D. Wang, H. Huang, G.-J. Mao and G.-J. Deng, Chem. Commun., 2023, 59, 2767–2770 RSC; (d) Q.-W. Gui, F. Teng, P. Yu, Y.-F. Wu, Z.-B. Nong, L.-X. Yang, X. Chen, T.-B. Yang and W.-M. He, Chin. J. Catal., 2023, 44, 111–116 CrossRef CAS; (e) X. Chen, B. Liu, C. Pei, J. Li, D. Zou, Y. Wu and Y. Wu, Org. Lett., 2021, 23, 7787–7791 CrossRef CAS PubMed.
- (a) B. Sun, H. Ding, H. X. Tian, P. Y. Huang, C. Jin, C. L. Wu and R. P. Shen, Adv. Synth. Catal., 2022, 364, 766–772 CrossRef CAS; (b) J. Yang, B. Sun, H. Ding, P.-Y. Huang, X.-L. Tang, R.-C. Shi, Z.-Y. Yan, C.-M. Yu and C. Jin, Green Chem., 2021, 23, 575–581 RSC; (c) B. Sun, P. Huang, Z. Yan, X. Shi, X. Tang, J. Yang and C. Jin, Org. Lett., 2021, 23, 1026–1031 CrossRef CAS PubMed; (d) B. Sun, R. Shi, K. Zhang, X. Tang, X. Shi, J. Xu, J. Yang and C. Jin, Chem. Commun., 2021, 57, 6050–6053 RSC.
- (a) H. L. Wu, W. K. Zhang, C. C. Zhang, L. T. Wang, W. H. Yang, W. C. Tian, G. P. Ge, L. Y. Xie, R. Yi and W. T. Wei, Chem. – Eur. J., 2023, 29, e202301390 CrossRef CAS PubMed; (b) H. Liu, Z. Yang, J. T. Yu and C. Pan, Adv. Synth. Catal., 2022, 364, 1085–1090 CrossRef CAS.
- L. Liu, W. Zhang, C. Xu, J. He, Z. Xu, Z. Yang, F. Ling and W. Zhong, Adv. Synth. Catal., 2022, 364, 1319–1325 CrossRef CAS.
- (a) Z.-J. Liu, J.-F. Li, F.-P. Zhang, X.-T. Xu and M. Ye, Org. Lett., 2023, 25, 353–357 CrossRef CAS PubMed; (b) Y. Zhang, Q. Ni, B. Pan, L. Jiang and L. Qiu, Chin. Chem. Lett., 2023, 34, 108017 CrossRef CAS; (c) Q. Dherbassy, S. Manna, C. Shi, W. Prasitwatcharakorn, G. E. M. Crisenza, G. J. P. Perry and D. J. Procter, Angew. Chem., Int. Ed., 2021, 60, 14355–14359 CrossRef CAS PubMed.
- (a) Z. Li, L. Jiao, Y. Sun, Z. He, Z. Wei and W. W. Liao, Angew. Chem., Int. Ed., 2020, 59, 7266–7270 CrossRef CAS PubMed; (b) O. J. Turner, D. J. Hirst and J. A. Murphy, Chem. – Eur. J., 2020, 26, 3026–3029 CrossRef CAS PubMed; (c) P. Qian, Y. Deng, H. Mei, J. Han, J. Zhou and Y. Pan, Org. Lett., 2017, 19, 4798–4801 CrossRef CAS PubMed; (d) J. Zheng, Y. Zhang, D. Wang and S. Cui, Org. Lett., 2016, 18, 1768–1771 CrossRef CAS PubMed; (e) J. Zheng, Z. Deng, Y. Zhang and S. Cui, Adv. Synth. Catal., 2016, 358, 746–751 CrossRef CAS.
- T. Tian, Z. Li and C.-J. Li, Green Chem., 2021, 23, 6789–6862 RSC.
- (a) Q. Wang, X.-J. Meng, H.-T. Tang, Y.-M. Pan, W.-G. Duan and M.-X. He, Green Chem., 2023, 25, 2572–2576 RSC; (b) J. Wu, C. M. Young, A. A. Watts, A. M. Z. Slawin, G. R. Boyce, M. Bühl and A. D. Smith, Org. Lett., 2022, 24, 4040–4045 CrossRef CAS PubMed; (c) Y. Gao, W. Qin, M. Q. Tian, X. Zhao and X. H. Hu, Adv. Synth. Catal., 2022, 364, 2241–2247 CrossRef CAS; (d) K. Tamaribuchi, J. Tian, K. Akagawa and K. Kudo, Adv. Synth. Catal., 2022, 364, 82–86 CrossRef CAS.
- (a) X. Wang, H. Z. Miao, G. Q. Lin and Z. T. He, Angew. Chem., Int. Ed., 2023, 62, e202301556 CrossRef CAS PubMed; (b) C. Li, Z. Zhou and S. Ma, Chem. Sci., 2023, 14, 7709–7715 RSC; (c) F. A. Moghadam, E. F. Hicks, Z. P. Sercel, A. Q. Cusumano, M. D. Bartberger and B. M. Stoltz, J. Am. Chem. Soc., 2022, 144, 7983–7987 CrossRef CAS PubMed; (d) F. Cheng, T. Chen, Y.-Q. Huang, J.-W. Li, C. Zhou, X. Xiao and F.-E. Chen, Org. Lett., 2022, 24, 115–120 CrossRef CAS PubMed.
- (a) D. Wang, B. Yuan, J. Xu and L. Ackermann, Chem. – Eur. J., 2023, 29, e202300600 CrossRef CAS PubMed; (b) M. Chen, Z. J. Wu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2022, 61, e202115954 CrossRef CAS PubMed; (c) Z.-W. Hou, L. Li and L. Wang, Org. Chem. Front., 2022, 9, 2815–2820 RSC; (d) Z. W. Hou, T. Jiang, T. X. Wu and L. Wang, Org. Lett., 2021, 23, 8585–8589 CrossRef CAS PubMed.
- (a) Y. Tao, R. Hu, X. Jie and W. Su, ACS Catal., 2022, 12, 12670–12677 CrossRef CAS; (b) P. Hernández-Lladó, K. Garrec, D. C. Schmitt and J. W. Burton, Adv. Synth. Catal., 2022, 364, 1724–1731 CrossRef; (c) Z. Guan, S. Zhu, Y. Ye, X. Li, Y. Liu, P. Wang, H. Zhang, Z. Huang and A. Lei, Angew. Chem., Int. Ed., 2022, 61, e202207059 CrossRef CAS PubMed; (d) C. Wang, R. H. Liu, M. Q. Tian, X. H. Hu and T. P. Loh, Org. Lett., 2018, 20, 4032–4035 CrossRef CAS PubMed.
- (a) J. Zhang, C. Liu, Y. Qiao, M. Wei, W. Guan, Z. Mao, H. Qin, Z. Fang and K. Guo, Chem. Sci., 2023, 14, 2461–2466 RSC; (b) N. Ma, J. Yin, W. Huang, L. Fan and F. Gao, Chem. - Asian J., 2022, 17, e202200963 CrossRef CAS PubMed; (c) N. Katta, Q. Q. Zhao, T. Mandal and O. Reiser, ACS Catal., 2022, 12, 14398–14407 CrossRef CAS PubMed; (d) Z. J. Wu, S. R. Li and H. C. Xu, Angew. Chem., Int. Ed., 2018, 57, 14070–14074 CrossRef CAS PubMed.
- (a) H. Hou, Y. Pan, Y. Sun, Y. Han, C. Yan, Y. Shi and S. Zhu, Chem. – Eur. J., 2023, 29, e202301633 CrossRef CAS PubMed; (b) Q.-Q. Zhao, J. Rehbein and O. Reiser, Green Chem., 2022, 24, 2772–2776 RSC; (c) G. Lei, M. Xu, R. Chang, I. Funes-Ardoiz and J. Ye, J. Am. Chem. Soc., 2021, 143, 11251–11261 CrossRef CAS PubMed; (d) S. Baś, Y. Yamashita and S. Kobayashi, ACS Catal., 2020, 10, 10546–10550 CrossRef.
- Y. Hong, M. Y. Dong, D. S. Li and H. P. Deng, Org. Lett., 2022, 24, 7677–7684 CrossRef CAS PubMed.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2022, 122, 2487–2649 CrossRef CAS PubMed.
- (a) T. Shen, Y. L. Li, K. Y. Ye and T. H. Lambert, Nature, 2023, 614, 275–280 CrossRef CAS PubMed; (b) H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 18803–18809 CrossRef CAS PubMed; (c) T. Shen and T. H. Lambert, Science, 2021, 371, 620–626 CrossRef CAS PubMed; (d) H. Huang and T. H. Lambert, J. Am. Chem. Soc., 2021, 143, 7247–7252 CrossRef CAS PubMed.
- (a) X.-L. Lai and H.-C. Xu, J. Am. Chem. Soc., 2023, 145, 18753–18759 CrossRef CAS PubMed; (b) C.-Y. Cai, X.-L. Lai, Y. Wang, H.-H. Hu, J. Song, Y. Yang, C. Wang and H.-C. Xu, Nat. Catal., 2022, 5, 943–951 CrossRef CAS; (c) X.-L. Lai, M. Chen, Y. Wang, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 20201–20206 CrossRef CAS PubMed; (d) H. Yan, J. Song, S. Zhu and H.-C. Xu, CCS Chem., 2021, 3, 317–325 CrossRef CAS.
- (a) L. Qian and M. Shi, Chem. Commun., 2023, 59, 3487–3506 RSC; (b) H. Huang, K. A. Steiniger and T. H. Lambert, J. Am. Chem. Soc., 2022, 144, 12567–12583 CrossRef CAS PubMed; (c) S. Wu, J. Kaur, T. A. Karl, X. Tian and J. P. Barham, Angew. Chem., Int. Ed., 2022, 61, e202107811 CrossRef CAS PubMed.
- (a) Y. Yuan, J. Yang and J. Zhang, Chem. Sci., 2023, 14, 705–710 RSC; (b) Z. Tan, Y. Jiang, K. Xu and C. Zeng, J. Catal., 2023, 417, 473–480 CrossRef CAS; (c) Z. Yang, D. Yang, J. Zhang, C. Tan, J. Li, S. Wang, H. Zhang, Z. Huang and A. Lei, J. Am. Chem. Soc., 2022, 144, 13895–13902 CrossRef CAS PubMed; (d) Y. Wang, L. Li and N. Fu, ACS Catal., 2022, 12, 10661–10667 CrossRef CAS.
- (a) L. Zou, X. Wang, S. Xiang, W. Zheng and Q. Lu, Angew. Chem., Int. Ed., 2023, 62, e202301026 CrossRef CAS PubMed; (b) W. Wei, S. L. Homölle, T. von Münchow, Y. Li, I. Maksso and L. Ackermann, Cell Rep. Phys. Sci., 2023, 4, 101550 CrossRef CAS.
- (a) Q. Wan, Z.-W. Hou, X.-R. Zhao, X. Xie and L. Wang, Org. Lett., 2023, 25, 1008–1013 CrossRef CAS PubMed; (b) Q. Wan, C.-Y. Huang, Z.-W. Hou, H. Jiang and L. Wang, Org. Chem. Front., 2023, 10, 3585–3590 RSC; (c) W. Fan, X. Zhao, Y. Deng, P. Chen, F. Wang and G. Liu, J. Am. Chem. Soc., 2022, 144, 21674–21682 CrossRef CAS PubMed; (d) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed.
- (a) H. Bi, Y. Zhou, W. Jiang and J. Liu, Adv. Synth. Catal., 2022, 364, 1732–1737 CrossRef CAS; (b) L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
- W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed.
- (a) Y.-J. Chen, W.-H. Deng, J.-D. Guo, R.-N. Ci, C. Zhou, B. Chen, X.-B. Li, X.-N. Guo, R.-Z. Liao, C.-H. Tung and L.-Z. Wu, J. Am. Chem. Soc., 2022, 144, 17261–17268 CrossRef CAS PubMed; (b) C. P. Chernowsky, A. F. Chmiel and Z. K. Wickens, Angew. Chem., Int. Ed., 2021, 60, 21418–21425 CrossRef CAS PubMed; (c) N. G. W. Cowper, C. P. Chernowsky, O. P. Williams and Z. K. Wickens, J. Am. Chem. Soc., 2020, 142, 2093–2099 CrossRef CAS PubMed; (d) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed.
- L. Zeng, J. H. Qin, G. F. Lv, M. Hu, Q. Sun, X. H. Ouyang, D. L. He and J. H. Li, Chin. J. Chem., 2023, 41, 1921–1930 CrossRef CAS.
- (a) Y. Zhang, X. Sun, J.-H. Su, T. Li, C. Du, K. Li, Q. Sun, Z. Zha and Z. Wang, Org. Lett., 2023, 25, 5067–5072 CrossRef CAS PubMed; (b) P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed; (c) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed.
- (a) L. Luo, Y. Liu, W. Chen, X. Xue, S.-M. Xu, M. Li, H. Zhou, L. Ma, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, Chem Catal., 2023, 3, 100472 CrossRef CAS; (b) D. S. Choi, J. Kim, F. Hollmann and C. B. Park, Angew. Chem., Int. Ed., 2020, 59, 15886–15890 CrossRef CAS PubMed; (c) L. Zhang, L. Liardet, J. Luo, D. Ren, M. Gratzel and X. Hu, Nat. Catal., 2019, 2, 266–373 Search PubMed; (d) H. Tateno, Y. Miseki and K. Sayama, Chem. Commun., 2019, 55, 9339–9342 RSC.
- (a) D. Chen, X. Yang, D. Wang, Y. Li, L. Shi and D. Liang, Org. Chem. Front., 2023, 10, 2482–2490 RSC; (b) R. Yang, D. Chen, S. Lin, X. Yang, X. Hu, S. Gao, L. Shi and D. Liang, Mol. Catal., 2023, 547, 113360 CrossRef CAS; (c) X. Hu, M. Tao, Z. Ma, Y. Zhang, Y. Li and D. Liang, Adv. Synth. Catal., 2022, 364, 2163–2168 CrossRef CAS; (d) J.-Q. He, Z.-X. Yang, X.-L. Zhou, Y. Li, S. Gao, L. Shi and D. Liang, Org. Chem. Front., 2022, 9, 4575–4582 RSC.
- X. L. Lai, X. M. Shu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed.
- K. Wang, X. Liu, S. Yang, Y. Tian, M. Zhou, J. Zhou, X. Jia, B. Li, S. Liu and J. Chen, Org. Lett., 2022, 24, 3471–3476 CrossRef CAS PubMed.
- M.-Y. Dong, C.-Y. Han, D.-S. Li, Y. Hong, F. Liu and H.-P. Deng, ACS Catal., 2022, 12, 9533–9539 CrossRef CAS.
- (a) Y.-D. Du, S. Wang, H.-W. Du, X.-Y. Chang, X.-Y. Chen, Y.-L. Li and W. Shu, Nat. Commun., 2023, 14, 5339 CrossRef CAS PubMed; (b) G. A. Lutovsky, E. F. Plachinski, N. L. Reed and T. P. Yoon, Org. Lett., 2023, 25, 4750–4754 CrossRef CAS PubMed; (c) C. M. You, C. Huang, S. Tang, P. Xiao, S. Wang, Z. Wei, A. Lei and H. Cai, Org. Lett., 2023, 25, 1722–1726 CrossRef CAS PubMed; (d) Y. Yu, Y. Yuan and K. Y. Ye, Chem. Commun., 2023, 59, 422–425 RSC.
- (a) T.-Y. Shang, L.-H. Lu, Z. Cao, Y. Liu, W.-M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) X.-Y. Lu, M.-T. Gao, L.-J. Yu, H.-Y. Pan, X. Zhang, R. Huang, K. Yang, F.-Y. Shui, Y.-W. Song and G.-X. Yang, Org. Chem. Front., 2023, 10, 1788–1795 RSC; (b) Y. Saga, Y. Nakayama, T. Watanabe, M. Kondo and S. Masaoka, Org. Lett., 2023, 25, 1136–1141 CrossRef CAS PubMed; (c) P. Xu, X. Y. Wang, Z. Wang, J. Zhao, X. D. Cao, X. C. Xiong, Y. C. Yuan, S. Zhu, D. Guo and X. Zhu, Org. Lett., 2022, 24, 4075–4080 CrossRef CAS PubMed; (d) Y. Yuan, M. Zhang, X. Tang, J. L. Piper, Z.-H. Peng, J.-A. Ma, J. Wu and F.-G. Zhang, Org. Lett., 2023, 25, 883–888 CrossRef CAS PubMed.
- (a) D. Forster, W. Guo, Q. Wang and J. Zhu, ACS Catal., 2023, 13, 7523–7528 CrossRef CAS; (b) W. Zuo, L. Zuo, X. Geng, Z. Li and L. Wang, Org. Lett., 2023, 25, 6062–6066 CrossRef CAS PubMed; (c) Y. Song, B. Zheng, S. Yang, Y. Li, Q. Liu and L. Pan, Org. Lett., 2023, 25, 2372–2376 CrossRef CAS PubMed; (d) J. Q. Chen, X. Luo, M. Chen, Y. Chen and J. Wu, Org. Lett., 2023, 25, 1978–1983 CrossRef CAS PubMed.
- (a) C. L. Lee, L. Wu, J.-S. Huang and C.-M. Che, Chem. Commun., 2019, 55, 3606–3609 RSC; (b) D. Y. Shopov, B. Rudshteyn, J. Campos, D. J. Vinyard, V. S. Batista, G. W. Brudvig and R. H. Crabtree, Chem. Sci., 2017, 8, 1642–1652 RSC; (c) D. Y. Shopov, B. Rudshteyn, J. Campos, V. S. Batista, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2015, 137, 7243–7250 CrossRef CAS PubMed; (d) J.-U. Rohde and W.-T. Lee, J. Am. Chem. Soc., 2009, 131, 9162–9163 CrossRef CAS PubMed.
- X.-Y. Qian, S.-Q. Li, J. Song and H.-C. Xu, ACS Catal., 2017, 7, 2730–2734 CrossRef CAS.
- (a) L. Moggi, G. Varani, M. F. Manfrin and V. Balzani, Inorg. Chim. Acta, 1970, 4, 335–341 CrossRef CAS; (b) E. M. Glebov, V. F. Plyusnin, N. I. Sorokin, V. P. Grivin, A. B. Venediktov and H. Lemmetyinen, J. Photochem. Photobiol., A, 1995, 90, 31–37 CrossRef CAS; (c) M. Kinauer, M. Diefenbach, H. Bamberger, S. Demeshko, E. J. Reijerse, C. Volkmann, C. Würtele, J. van Slageren, B. de Bruin, M. C. Holthausen and S. Schneider, Chem. Sci., 2018, 9, 4325–4332 RSC.
- Q. Wan, Z. Zhang, Z.-W. Hou and L. Wang, Org. Chem. Front., 2023, 10, 2830–2848 RSC.
- Q. Tian, J. Zhang, L. Xu and Y. Wei, Mol. Catal., 2021, 500, 111345 CrossRef CAS.
- D. A. Schiedler, Y. Lu and C. M. Beaudry, Org. Lett., 2014, 16, 1160–1163 CrossRef CAS PubMed.
- (a) L. Jin, X. Chen, S. Huang, A. Wang, Z. Chen, Z. Le and Z. Xie, Mol. Catal., 2023, 547, 113305 CrossRef CAS; (b) Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS; (c) L. Cao, H. Huo, H. Zeng, Y. Yu, D. Lu and Y. Gong, Adv. Synth. Catal., 2018, 360, 4764–4773 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, and copies of NMR spectra. See DOI: https://doi.org/10.1039/d4gc00352g |
| This journal is © The Royal Society of Chemistry 2024 |