
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Covalent porous catalysts for electrochemical reduction of CO2
Shuanglong
Lu
 a,
Hongyin
Hu†
a,
Huimin
Sun†
a,
Fulin
Yang†
a,
Han
Zhu
a,
Mingliang
Du
a,
Yinghua
Jin
b and
Wei
Zhang
*b
a,
Hongyin
Hu†
a,
Huimin
Sun†
a,
Fulin
Yang†
a,
Han
Zhu
a,
Mingliang
Du
a,
Yinghua
Jin
b and
Wei
Zhang
*b
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, Jiangnan University, Wuxi 214122, Jiangsu, China
bDepartment of Chemistry, University of Colorado Boulder, Boulder, CO 80309, USA. E-mail: wei.zhang@colorado.edu
First published on 9th April 2024
Abstract
The electrocatalytic CO2 reduction reaction (eCO2RR) stands out as a highly promising approach to simultaneously resolving the issue of the elevated atmospheric CO2 concentration and its utilization to produce value-added products. One of the present challenges is the rational design of efficient catalysts toward the eCO2RR and the understanding of their structure–activity relationship. Covalent porous catalysts (CPCs) with atomically dispersed active sites embedded in the porous organic backbone, including polymer-based catalysts, such as covalent organic frameworks (COFs) and conjugated microporous polymers (CMPs), and molecule-based catalysts, such as porous organic cages (POCs) have received increasing attention recently. They feature extremely high atom utilization, customizable porosity, a customizable backbone, and distinct electronic properties. In this review, the design principles of CPCs, both in their selection of metal sites (metallic centers, heterometallic centers and their axial coordination) and optimization of organic backbones (chelating sites, their electronic modulation and microstructures) will be covered, aiming to provide guidance to tune their eCO2RR performance. Then representative examples will be discussed to reveal their structure–activity relationships. The perspective and key challenges in CPC based eCO2RR electrocatalysts will be finally proposed.
1. Introduction
Due to the ongoing emissions from fossil fuels, carbon dioxide (CO2) continues to build up in the atmosphere. According to the forecast by Mauna Loa Observatory in Hawaii, the global annual mean CO2 concentration has exceeded 419 ppm in 2022, with an almost 100 ppm increase in the past 60 years.1–3 The rising trend of CO2 concentration will be kept in the next few decades. The elevated atmospheric CO2 concentration would give rise to a series of severe environmental crises, including ocean acidification, extreme weather events, the greenhouse effect, and species extinction.4,5 Meanwhile, CO2 could be used as a useful feedstock to synthesize various valuable chemicals. Consequently, the capture and conversion of CO2 into value-added products and fuels represents a key and meaningful route for the mitigation of these environmental problems and the utilization of carbon sources.6–11To date, some strategies have been proposed for the conversion of CO2 into value-added products, including chemical transformation,12–14 photocatalytic reduction,15–19 electrocatalytic reduction,20–23 and biological conversion.24–27 Among them, the electrocatalytic CO2 reduction reaction (eCO2RR) stands out as a highly promising pathway due to the following merits: (a) simple devices and mild reaction conditions; (b) high environmental compatibility; (c) possible combination of other renewable energy sources (e.g., solar, wind, water, and biomass energy); and (d) highly tunable pathways to different products.18,28–30 However, how to effectively activate and selectively convert CO2 is challenging, given its linear and chemically stable nature with poor electron affinity. The dissociation energy for breaking the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is higher than 750 kJ mol−1. Therefore, a relatively large overpotential is required to initiate CO2 activation, resulting in low energy efficiency. Furthermore, the eCO2RR is a complex process involving multi-step complicated proton/electron transfer, yielding diverse gaseous and liquid products such as formic acid (HCOOH), carbon monoxide (CO), methanol (CH3OH), methane (CH4), ethylene (C2H4), and ethanol (CH3CH2OH).31–39 The thermodynamic redox potentials of these products are closely aligned, resulting in a limited selectivity for a specific product. Moreover, the eCO2RR is invariably accompanied by the competing hydrogen evolution reaction (HER) due to their similar potential windows, which presents another significant obstacle restricting the catalytic efficiency and potential application.40,41 Thus, the rational design of electrocatalysts with high activity, selectivity, and stability becomes crucial in the practical eCO2RR. Until now, many types of electrocatalysts have been developed for eCO2RR, including noble metals, transition metals, metal oxides, metal alloys, and metal chalcogenides.42–51 Most of them could provide enhanced catalytic efficiency and considerable stability with increasingly clear structure–activity relationships.
O bond is higher than 750 kJ mol−1. Therefore, a relatively large overpotential is required to initiate CO2 activation, resulting in low energy efficiency. Furthermore, the eCO2RR is a complex process involving multi-step complicated proton/electron transfer, yielding diverse gaseous and liquid products such as formic acid (HCOOH), carbon monoxide (CO), methanol (CH3OH), methane (CH4), ethylene (C2H4), and ethanol (CH3CH2OH).31–39 The thermodynamic redox potentials of these products are closely aligned, resulting in a limited selectivity for a specific product. Moreover, the eCO2RR is invariably accompanied by the competing hydrogen evolution reaction (HER) due to their similar potential windows, which presents another significant obstacle restricting the catalytic efficiency and potential application.40,41 Thus, the rational design of electrocatalysts with high activity, selectivity, and stability becomes crucial in the practical eCO2RR. Until now, many types of electrocatalysts have been developed for eCO2RR, including noble metals, transition metals, metal oxides, metal alloys, and metal chalcogenides.42–51 Most of them could provide enhanced catalytic efficiency and considerable stability with increasingly clear structure–activity relationships.
To further enhance the utilization of active species and make it easier to illustrate the structure–activity relationships, atomically dispersed active sites are emerging as novel electrocatalysts toward the eCO2RR.52–55 In particular, covalent porous catalysts (CPCs) with atomically dispersed active sites embedded in the porous organic skeleton have received increasing attention recently.56–60 They feature properties such as extremely high atom utilization, customizable porosity, a tunable backbone, and distinct electronic properties. Compared to classic electrocatalysts, many research studies have proved that porous catalysts could enrich CO2 and enhance the atomic utilization of metals. Furthermore, the electronic structure of the active sites could be facilely tailored. Different from the analogous Metal–Organic Frameworks (MOFs) with similar properties, CPCs are all formed via covalent bonds. They could endow them with exceptional structural stabilities.61–64 The CPCs here are mainly classified into two categories, one is polymer-based catalysts, such as covalent organic frameworks (COFs) and conjugated microporous polymers (CMPs), and the other is molecule-based catalysts, such as porous organic cages (POCs). They all have the versatility of their organic building blocks, organic linkers and variable side groups in their skeleton.59,65–70 Taking advantage of suitable chelating groups, diverse metallic sites could be incorporated into their architectures to serve as active sites toward the eCO2RR.64,71–73 Moreover, their microstructures, such as their porosity and nanostructure, could also be effectively regulated in accordance with specific requirements.74–79 Apparently, these characteristics make CPCs an ideal platform for the development and fabrication of electrochemical CO2RR catalysts.
Some excellent review articles on the porous frameworks for eCO2RR have already been published.56,58,80–84 For example, Wang et al. recently published a comprehensive review on functional porous frameworks, including MOFs and COFs and their derived porous materials.58 They summarized typical rational design strategies based on several key factors and different categories of materials. Nevertheless, other covalent porous catalysts (CPCs), such as conjugated microporous polymers (CMPs) and porous organic cages (POCs), were not covered. Moreover, with the rapid development in this area, more research studies have been published recently, which also need to be summarized timely. In this review, we mainly focus on several typical covalent porous catalysts toward the eCO2RR, including COFs, CMPs and POCs. The design principles both for the selection of metal sites and optimization of organic backbones in CPCs will be covered, aiming to provide guidance to regulate their eCO2RR performance (Fig. 1). Then representative examples will be summarized to reveal their structure–activity relationships. Finally, the perspective and key challenges in CPC based eCO2RR electrocatalysts will be proposed.
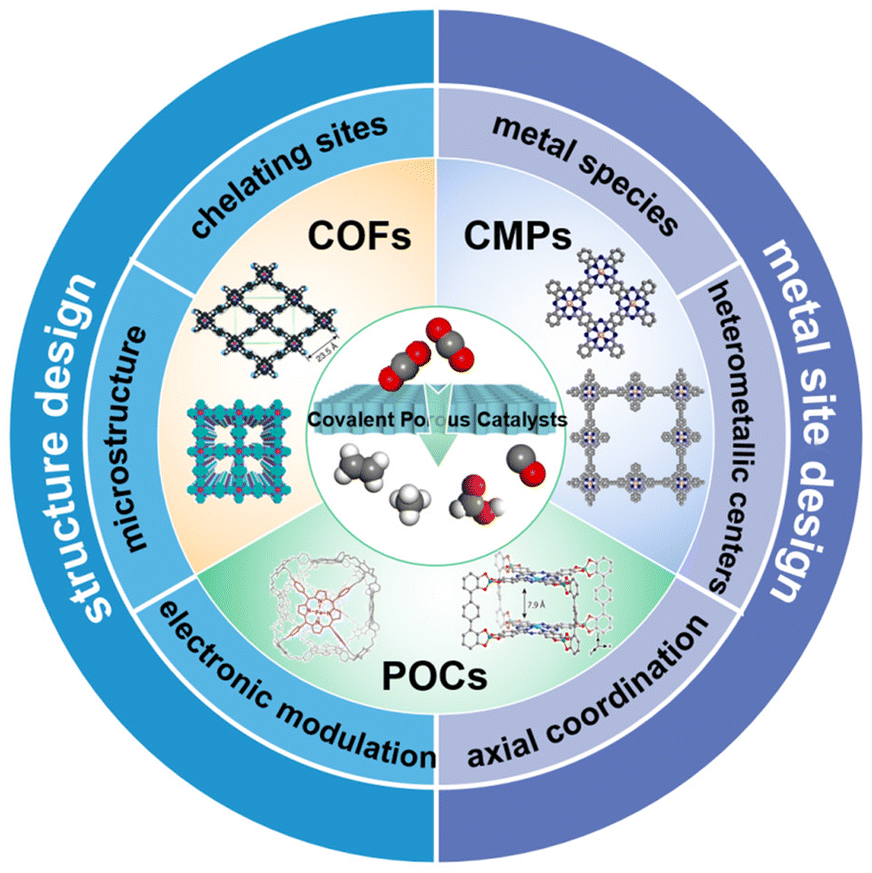 | ||
| Fig. 1 Classification and design principle of the CPCs toward the eCO2RR. | ||
2. Design principles for CPCs with high eCO2RR performance
CPCs were made by polymerization of organic monomers with typical chelating sites to anchor metal species as active sites. It is proposed that metal sites within CPCs are responsible for eCO2RR activity, and through the tuning of organic building blocks, metal species and microstructures, their eCO2RR activity could be efficiently regulated.79,85–87 In this section, we will first give a brief introduction on eCO2RR, including the general mechanism, characteristic tools and fundamental parameters. Then we will classify the design principles into two categories, the metal catalytic sites and organic backbones, revealing the underlying factors that govern eCO2RR performance in CPCs.2.1 General introduction of the eCO2RR
The eCO2RR involves multi-step complicated proton/electron transfer reactions, which result in multiple products.31,88–91 The catalysis starts from the adsorption of CO2 molecules, followed by the protonated intermediates, *OCHO or *COOH. HCOOH could be obtained through another protonation step and desorption, while CO could form through further reduction, where *CO is a key intermediate during the eCO2RR. Catalysts with a weak *CO binding energy usually produce CO as the main product. The strong binding energy of *CO may lead to the coverage of active sites (Fig. 2). Therefore, a suitable binding energy of *CO may induce subsequent protonation to give protonated *CHO and *COH, which will undergo further reduction to CH3OH and CH4. HCOH could be obtained through two step proton/electron transfer from the intermediate *OCHO or *COOH. It is worth noting that as only spatial separated metallic centers exist in CPCs, C2+ products, such as CH3COOH, CH3CHO, CH3CH2OH, etc., are rarely reported.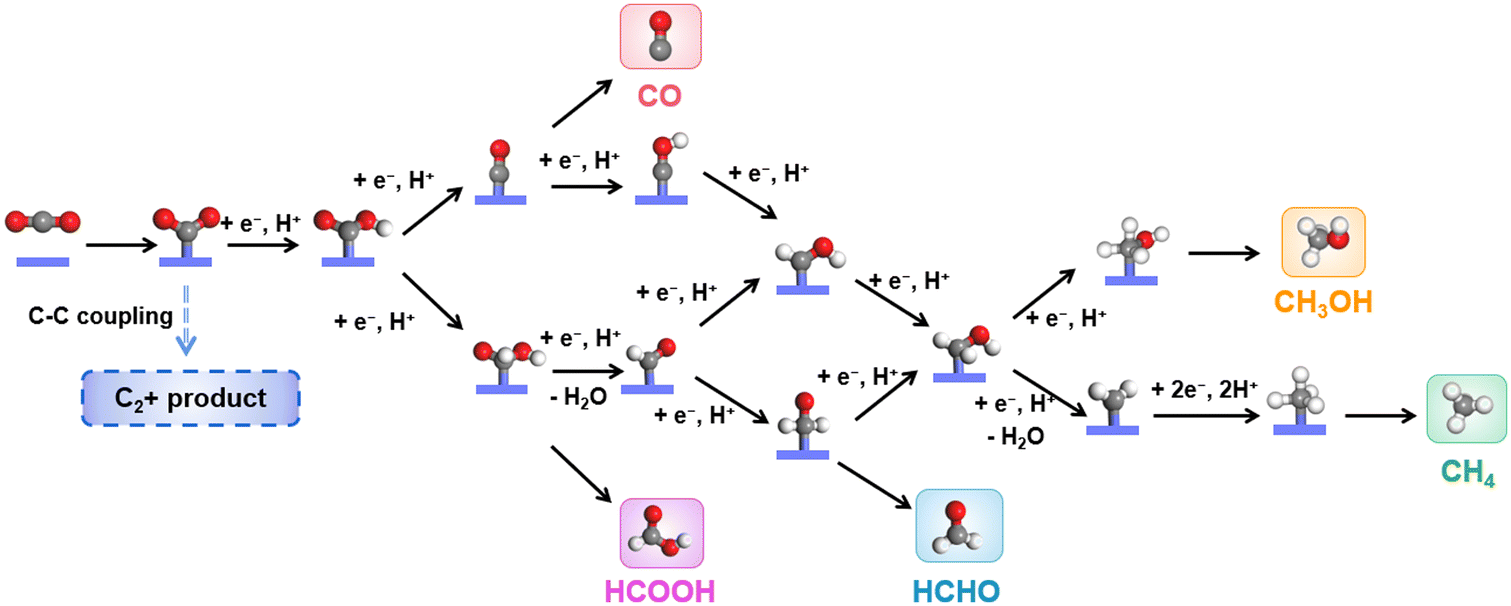 | ||
| Fig. 2 Possible pathways for CO2 reduction over CPCs. | ||
There are mainly three types of electrocatalytic reactors for the eCO2RR, i.e., an H-cell, flow cell and membrane electrode assembly (MEA) cell.92–94 The most commonly used reactor in fundamental studies is an H-cell. It is provided with low cost and easy operation. However, due to the limited solubility of CO2 molecules in the aqueous electrolyte, the current density is usually low in an H-cell. The flow cell could achieve a relatively high current density by using the gas diffusion electrode, which also has broad applications in many laboratories. The MEA cell is quite close to the real energy conversion devices, which holds great potential for scalability and practical application, though it has not been widely applied in fundamental research. Electrochemical measurement methods, such as cyclic voltammetry (CV), linear sweep voltammetry (LSV) and chronoamperometry, are typically used to evaluate the activity and stability of electrocatalysts toward the eCO2RR. Meanwhile, gaseous products, such as CO, H2, CH4, etc., could be quantified by online or offline gas chromatography (GC), while liquid products, such as HCOOH, CH3CH2OH, etc., could be quantified by nuclear magnetic resonance spectroscopy (NMR). Based on the analysis of the electrochemical curves and the quantitative results, several fundamental parameters are generally used for the comparison and evaluation of electrocatalysts’ selectivity, activity and stability toward the eCO2RR, including the onset potential, overpotential (η), Tafel slope, normalized current density, faradaic efficiency (FE), turnover number (TON), and turnover frequency (TOF).
2.2 Metal catalytic sites in CPCs
Isolated metal sites can be introduced into chelating sites before or after the synthesis of CPCs. Typically, the isolated metal species are considered as the active sites in CPCs for eCO2RR. Many previously reported works are devoted to understanding the origin of catalytic activity and designing excellent catalysts based on isolated metal centers. It should be pointed out that some metallic centers, such as Cu, could transform into metal clusters during electrocatalysis, which serves as actual active sites for the reaction. Even though it won't be discussed deeply here, more attention should be paid during the research to avoid misconceptions.(1) Metallic centers. The selection of the metallic centers could provide facile regulation of CPCs toward the eCO2RR.71,83 The metallic centers are believed to be one of the key factors that determine the catalytic activity and selectivity of CPCs. Typically, metallic centers are responsible for different affinities toward CO2 molecules and diverse binding energies of adsorption intermediates during proton–electron transfer processes. The previously reported metallic centers in CPCs are mainly from the first transition series metals, such as Mn, Fe, Co, Ni, and Cu (Fig. 3a).
 | ||
| Fig. 3 Design principles of CPCs. (a) Typical metal centers and (b) typical chelating groups of CPCs toward the eCO2RR; (c) illustration of the design principle of the CPCs toward the eCO2RR. | ||
(2) Heterometallic centers. Although the mono-metallic centers in CPCs could already show relatively high catalytic performance toward the eCO2RR, the kinetics of proton-coupled electron transfer and in-time release of the product still need to be enhanced.95 The incorporation of bimetallic sites is believed to be plausible to improve this problem through the synergistic effects between bimetallic sites. Besides the heterometallic centers, the deliberately designed metal vacancies in the coordination sites are also proposed as efficient modulation strategies to influence the electrocatalytic performance of CPCs. The uncoordinated ligands could serve as both electron and proton regulators during the eCO2RR.
(3) Axial coordination of metallic centers. The axial coordination of metallic centers represents one of the effective strategies to modulate the intrinsic adsorption behavior of intermediates during the eCO2RR.96,97 It usually involves the introduction of M–X or M–O bonds in the axial direction of metallic active sites. This unique spatial coordination configuration endows metallic centers with a denser distribution of charges, enabling oriented electron transfer. The electronic configuration of metal centers is believed to exert a significant influence on the catalytic activity, selectivity, and stability of electrocatalysts.
2.3 Organic backbones in CPCs
To incorporate metallic centers inside CPCs, organic chelating sites with the binding ability to metal ions are needed.(1) Chelating sites. Typical ligands such as porphyrin, phthalocyanine, salen, bipyridine, and 1,10-phenanthroline have been reported in previous research to coordinate with metal ions in CPCs (Fig. 3b). These chelating ligands are decisive to the local coordination structures of metallic centers, determining the preferred reaction pathways during the eCO2RR.98–100 They usually have coordination atoms with lone pair electrons (e.g., N, O). The M–N4, especially in porphyrin/phthalocyanine-based CPCs, is the most commonly used symmetric chelating type with metal ions. Besides, the asymmetric M–N2O2 chelating type is typical in salen-based CPCs, and bipyridine-based or 1,10-phenanthroline-based CPCs with M(OAc)x as the metal ion sources. When metal halides are used as metal precursors, the asymmetric M–N2X2 chelating type could also be constructed. The asymmetric coordination structures could break the uniform charge distribution on the metallic centers, inducing oriented electron transfer. These chelating sites with different metallic centers could provide an ideal platform for studying the structure–activity relationships during the eCO2RR.
(2) Electronic modulation. As the organic backbones of CPCs are constructed with different organic building blocks, the electronic character of active sites could be tuned by the modification of reticular structures.101–103 Efficient charge transport along the skeleton's covalent backbone promotes the electronic connectivity between remote functional groups and the active sites, which could be used to modulate the catalytic performance of the metallic centers. The tunable sites could be located at the selected building blocks, suitable covalent linkages and variable side groups. More importantly, strategies such as establishing a comprehensive π-conjugated main chain and constructing donor–acceptor heterojunctions are developed in previous research, as a result of which, the electron transfer capability could be effectively enhanced, together with the electronic modulation of the metallic centers.
(3) Microstructures. The microstructures of CPCs are typically determined by the inherent properties of the organic backbones, such as their porosity and nanostructure.104,105 Noteworthily, CPCs usually feature high porosity with adjustable pore sizes, pore distribution, and variable substituent groups in their cavities. These features have a great influence on the mass transfer and exposure of active sites during the electrocatalysis. According to the definition of the International Union of Pure and Applied Chemistry (IUPAC), the pore size could be classified into micropores (<2 nm), mesopores (2–50 nm) and macropores (>50 nm). It is proposed that a pore distribution between 1–10 nm is the highly suitable pore size range to facilitate the diffusion of CO2 molecules and the penetration of the electrolyte. It is worth noting that even though compared to conventional catalysts, CPCs possess fully exposed active sites, not all electroactive sites in the porous structures could fully participate in electrocatalysis. The pore structure of CPCs is relatively intricate, leading to challenges during the eCO2RR process, such as massive pH gradients, a variable local CO2 concentration, and insufficient electron supply, which may hinder the complete utilization of active sites to a certain extent, particularly those located deeper inside the pores. To further enhance the catalytic efficiency, some nanostructures of CPCs, such as nanosheets, nanofibers or hollow tubes, could be obtained through morphology regulation. The elaborate design of these low dimensional nanostructures could ensure sufficient exposure and utilization of active sites.
3. Catalytic eCO2RR by CPCs
3.1 COFs
COFs are one of the emerging and promising CPCs due to their high structure tunability, well-defined pore structures and high specific surface areas. COFs can be designed and customized by selecting suitable building blocks at the molecular level toward the eCO2RR. Introducing different chelating groups can provide various anchoring sites and coordination environments for different metal species.In 2015, Chang and co-workers first reported the utilization of COFs as catalysts for eCO2RR. A model framework (COF-366-Co) was constructed via an imine condensation between [5,10,15,20-tetrakis(4-aminophenyl)porphinato]cobalt [Co(TAP)] and 1,4-benzenedicarboxaldehyde (BDA) (Fig. 4).106 The performance of the obtained catalysts was investigated in a buffer solution (pH = 7.3). Particularly, at a potential of −0.67 V, the catalyst demonstrated a FECO of 90%. The researchers proposed that a larger pore size would allow for higher electrochemical and chemical accessibility of the catalytic cobalt porphyrin active sites. They synthesized an extended COF-367-Co analogue by replacing BDA with biphenyl-4,4′-dicarbaldehyde (BPDA). It is then proved by CV studies that the extended organic framework COF-367-Co had a higher surface concentration of electroactive cobalt porphyrin sites compared to COF-366-Co. Moreover, given the low solubility of CO2, it is speculated that not all cobalt porphyrin sites in the framework are fully engaged in electrocatalysis. Through the dilution of the active sites, the TOF values for CO generation could be significantly enhanced at the expense of decreased FECO.
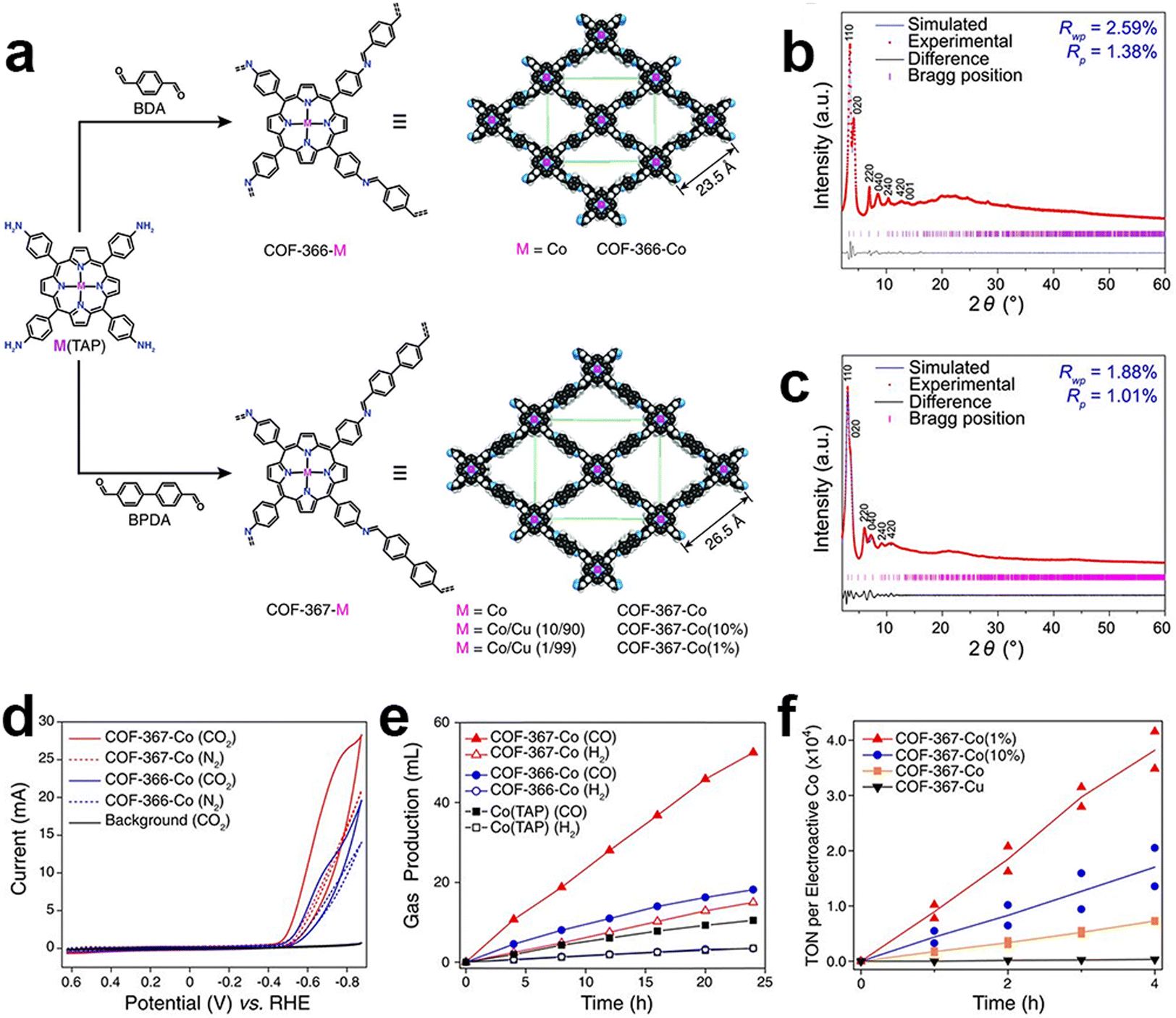 | ||
| Fig. 4 Schematic illustration, characterization and performance of COF-366-M and COF-367-M. (a) Schematic illustration of the preparation of COF-366-M and COF-367-M; PXRD patterns of (b) COF-366-M and (c) COF-367-M; (d) cyclic voltammograms of COF-366-Co and COF-367-Co; (e) the volume of CO and H2 produced by COF-367-Co, COF-366-Co and Co(TAP); (f) TON of CO production by COF-367-Co(1%), COF-367-Co(10%), COF-367-Co and COF-367-Cu.106 | ||
This work has inspired the research interest in the field of COF-based electrocatalysts toward the eCO2RR. Various research has been reported within the past few years. The metal species in COFs are the first critical factor, which deserve to be carefully considered. Their types and content could significantly determine the composition, faradaic efficiency and yield rate of products.
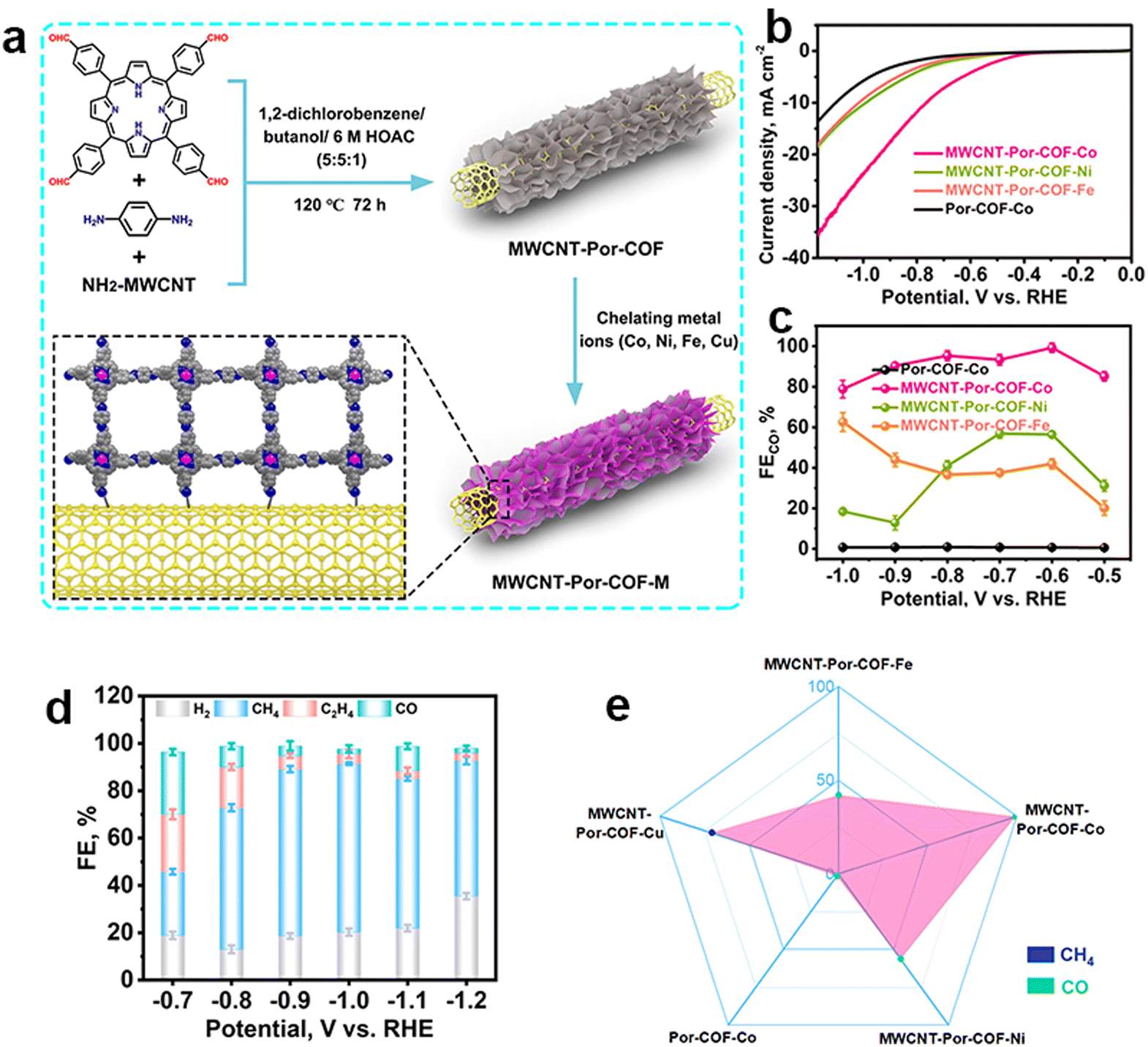 | ||
| Fig. 5 Schematic illustration of the metallic selection in COFs and their different performance. (a) Schematic illustration of the preparation of MWCNT-Por-COF and MWCNT-Por-COF-M; (b) LSV curves and (c) FECO of MWCNT-Por-COF-Fe, MWCNT-Por-COF-Co, MWCNT-Por-COF-Ni and Por-COF-Co; (d) FE of MWCNT-Por-COF-Cu; (e) the main product occupation of MWCNT-Por-COF-M.107 | ||
In addition to the above-mentioned metallic sites, Mn ions could serve as the chelated metal centers toward the eCO2RR. Julio Lloret-Fillol and colleagues incorporated a mononuclear Mn center into the pyridine-based COF (denoted as COFbpyMn).108 In this structure, the coordination geometry of Mn within the COF forms an octahedral structure, with three carbonyl groups facing the front and one bromine atom completing the first coordination shell. It is accepted that employing various coordination modes for metal centers can effectively modulate the electronic configuration of the metal centers. The embedded Mn species in the porous channels of COFs could change the redox pathway of the Mn molecular units, in terms of both steric hindrance, imposed by the rigidity of the COF structure, and the electronic effect of the organic framework. The redox process during the eCO2RR was thus modulated compared with the molecular counterpart. The COFbpyMn material exhibited a low eCO2RR onset potential (190 mV) and a high current density (>12 mA cm−2, at an overpotential of 550 mV) in aqueous solution. Additionally, precise control of the electronic configuration of metal sites within COFs can be achieved through the axial coordination of metal centers. P. Kubiak et al. developed a simple bottom-up solvent-free synthesis method to prepare iron porphyrin COF films.109 The iron centers in the porphyrin units were proposed to have an axially coordinated Cl. In this work, the molecular counterpart FeTAPPCl electrode demonstrated higher catalytic activity in the CV test, while the COF network could aid in the retainment of FeTAPPCl molecules, leading to the increased CO production during electrolysis in organic electrolytes. Nevertheless, the influence of axial coordination on the electrocatalytic performance still needs to be further explored.
 | ||
| Fig. 6 Schematic illustration of the donor–acceptor network design of COFs. (a and b) Schematic illustration of electron transfer between donor blocks and acceptor blocks; (c) schematic illustration of the preparation of the TTF-Por(Co)-COF; (d) LSV curves of the TTF-Por(Co)-COF and COF-366-Co in the N2-saturated and CO2-saturated electrolytes; (e) FECO of the TTF-Por(Co)-COF and COF-366-Co;110 (f) schematic illustration of the preparation of the TT-Por(Co)-COF; (g) FECO of TT-Por(Co)-COF and COF-366-Co.112 | ||
Other than the building blocks, the linkages in COFs could also exert an influence on the electronic states of metallic centers. The Zeng group employed a linking approach to synthesize two thiomorpholine-linked and dithiine-linked COFs with intriguing electronic and redox properties (Fig. 7a).115 Within these sulfur-containing heterocyclic materials, the low electronegativity of sulfur atoms and their vacant d orbitals offer opportunities for electronic conjugation modulation (Fig. 7b–g). The dithiine-linked COF exhibited the highest TOF value of 1548.2 h−1 at −1.0 V toward CO production via the eCO2RR. In comparison to the thiomorpholine-linked COF, the dithiine-linked COF not only displayed superior activity but also exhibited enhanced selectivity. It was proposed that a higher content of sulfur atoms contributed to higher conductivity and notable redox capability. Another work by the Zeng group reported a multi-tiered synthetic post-modification strategy to construct catalytic COFs for eCO2RR (Fig. 7h).116 The imine linkage was converted into amine and ionic bonds was introduced into COFs simultaneously (N+-NHCOF (N+: ionic modification; NH: reduction modification)). Through this dual modification process, the binding affinity of CO2 and its electronic state are finely tuned, thereby enabling control over the activity and selectivity of the CO2 reduction reaction. It is noteworthy that COFs possessing dual functional groups exhibit remarkable selectivity, achieving a maximum FECO of 97.32%, with a TOF value of 9922.68 h−1.
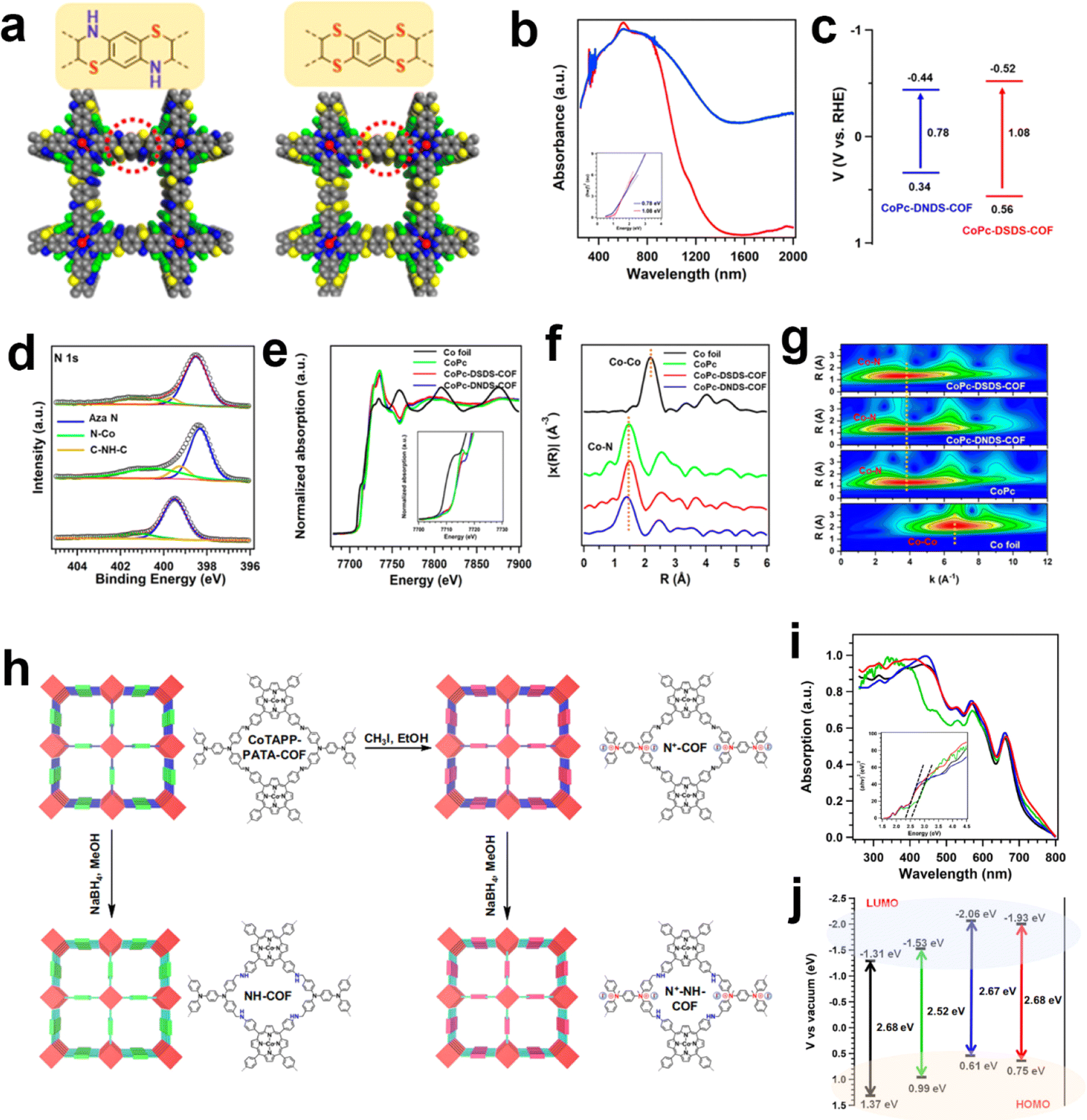 | ||
| Fig. 7 Schematic illustration of the linkage modulation of COFs. (a) The structure difference between thiomorpholine-linked and dithiine-linked COFs; (b) solid-state UV–vis absorption spectra and the (c) band structure diagram of thiomorpholine-linked and dithiine-linked COFs; (d) high-resolution XPS spectra of N 1s for the thiomorpholine-linked COF, dithiine-linked COF and CoPc. (e) XANES spectra of the Co K-edge, (f) the corresponding Co K-edge k3-weighted Fourier transform spectra and (g) WT contour spectra for the k3-weighted EXAFS data of the standard Co foil, CoPc, thiomorpholine-linked COF and dithiine-linked COF.115 (h) The synthesis of the N+-COF, NH-COF and N+-NH-COF from the base COF; (i) the UV-vis absorption (inset: Tauc plots) and (j) the energy gap (HOMO and LUMO) of the CoTAPP-PATA-COF (black), N+-COF (green), NH-COF (blue) and N+-NH-COF (red).116 | ||
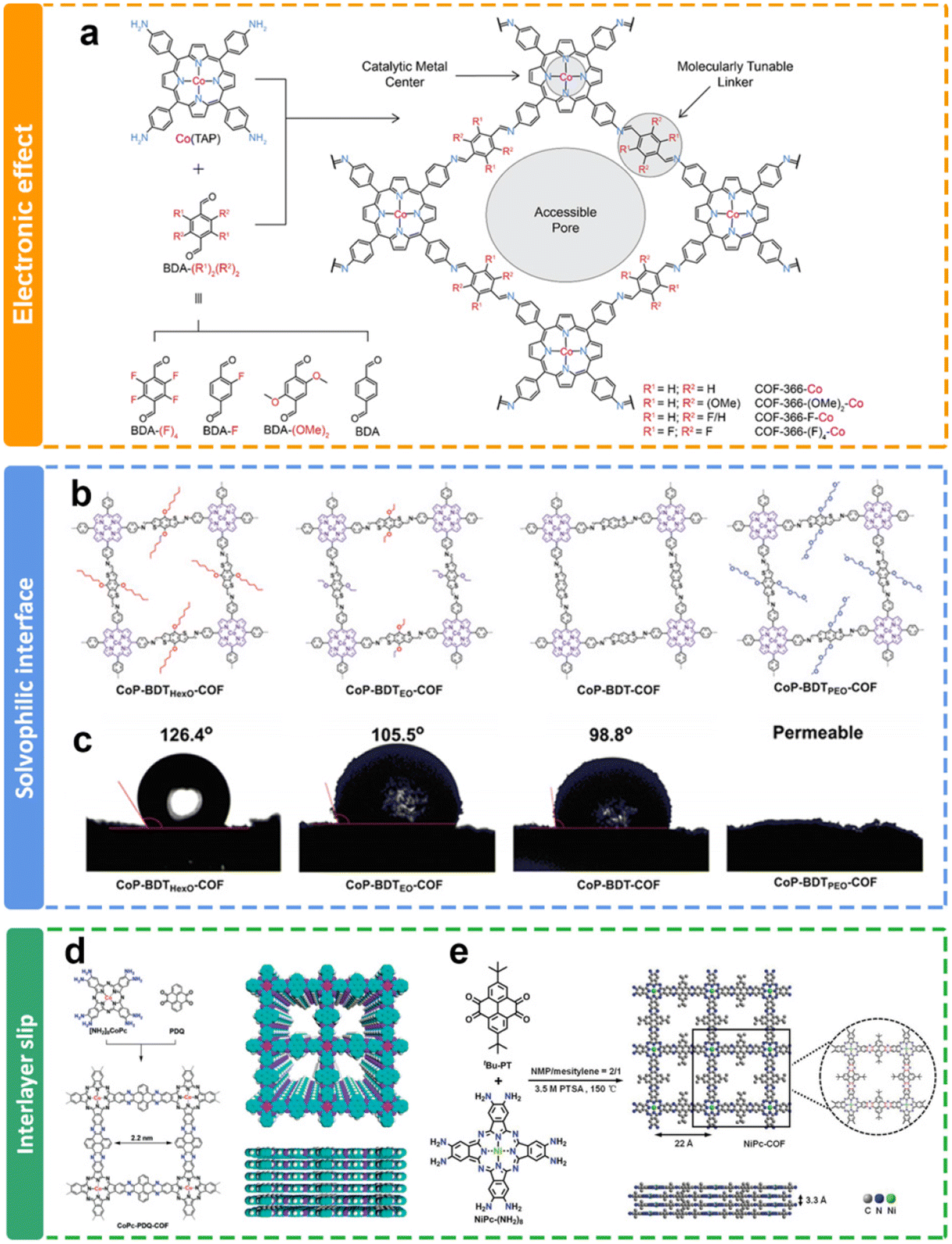 | ||
| Fig. 8 Schematic illustration of the side chain modulation of COFs. (a) Design of cobalt-porphyrin derived covalent organic frameworks;117 (b) schematics and (c) images of contact angles of crosslinked metalloporphyrin networks with different electron-flow and mass-transport interfaces;119 (d) synthesis of phenazine-linked CoPc-based COFs;120 (e) synthesis of phenazine-linked NiPc-based COFs with tert-butyl groups.121 | ||
The internal environment of the COF channel, such as hydrophilic and hydrophobic, could be further improved through the selection of different side groups in the channels of COFs. Jiang and co-workers have adopted a bottom-up approach to integrate electronic conduction and mass transfer interfaces within the interconnected networks (Fig. 8b).119 Through topology-guided polycondensations under solvothermal conditions, crystalline solvophobic π networks with varying solvophobicities have been synthesized (Fig. 8c). The eCO2RR involves the migration of CO2/solution through nanochannels to reach the localized reaction center within the local environment. It is noteworthy that both solvophobic and solvophilic interfaces were proved to enhance CO2 absorption, while the solvophilic interface further increased water adsorption, leading to an increased occurrence of H2 generation as a side reaction and subsequently reducing the faradaic efficiency of CO production.
Furthermore, the existence of side chain functional groups may cause large steric hindrance to induce interlayer slip, and prevent the overaccumulation of layers of COFs, which may benefit the exposure of active sites for eCO2RR. The Jiang group122 and Cao group121 reported similar phenazine-linked Pc-based COFs (Fig. 8d and e). They all delivered remarkable performance toward the eCO2RR due to their structural extended p-conjugation. Interestingly, the COFs in Cao's research has a tert-butyl side chain. The PXRD results demonstrated that the introduction of flexible side groups (tert-butyl groups) not only prevented the growth of large crystalline particles, but also gave rise to less-ordered edges. It is proposed that more active sites could be exposed due to the interlayer slip. It exhibits an impressively elevated CO selectivity exceeding 93% across a wide range of applied potentials spanning from −0.6 V to −1.1 V. Additionally, these COF nanosheets deliver a substantial jco of 35 mA cm−2 at −1.1 V in aqueous solution.
Experimentally, Jiang et al. synthesized two polyimide-linked phthalocyanine COFs with different pore sizes through the polymerization between 2,3,9,10,16,17,23,24-octacarboxyphthalocyanine cobalt(II) tetraanhydrides with 1,4-phenylenediamine (CoPc-PI-COF-1) or 4,4′-diaminodiphenyl (CoPc-PI-COF-2) (Fig. 9b).124 They have similar structures except for the pore size. Because of the identical Co(II) electroactive sites within the CoPc-PI-COFs, along with comparable permanent porosity and CO2 adsorption capabilities, both frameworks exhibited analogous FECO values of 87% to 97% in a 0.5 M KHCO3 solution when employed as cathodes at potentials ranging from −0.60 to −0.90 V. Nevertheless, CoPc-PI-COF-1 presented a jCO of −21.2 mA cm−2 at −0.90 V and an elevated electrical conductivity relative to CoPc-PI-COF-2. The exclusive structural disparity between CoPc-PI-COF-1 and CoPc-PI-COF-2 lied in the longer bridging unit within CoPc-PI-COF-2, a factor potentially contributing to diminished conductivity in the latter and improved CO2 concentration in the catalytic microenvironment. Similarly, another work by the Jiang group also demonstrated that the smaller pore size of COFs may be favorable for the enhancement of catalytic performance toward the eCO2RR.127 They successfully prepared two cobalt porphyrin-based COFs with pore sizes of 1.1 nm and 1.6 nm, respectively. They presented similar FEco values, while the COF with a smaller pore size exhibited more than 2-fold JCO values when compared with the COF with a relatively larger pore size. They proposed that the larger charge-transfer resistance due to the longer electron transfer path caused by the additional benzene ring in the skeleton is possibly responsible for the relatively worse catalytic performance in the COF.
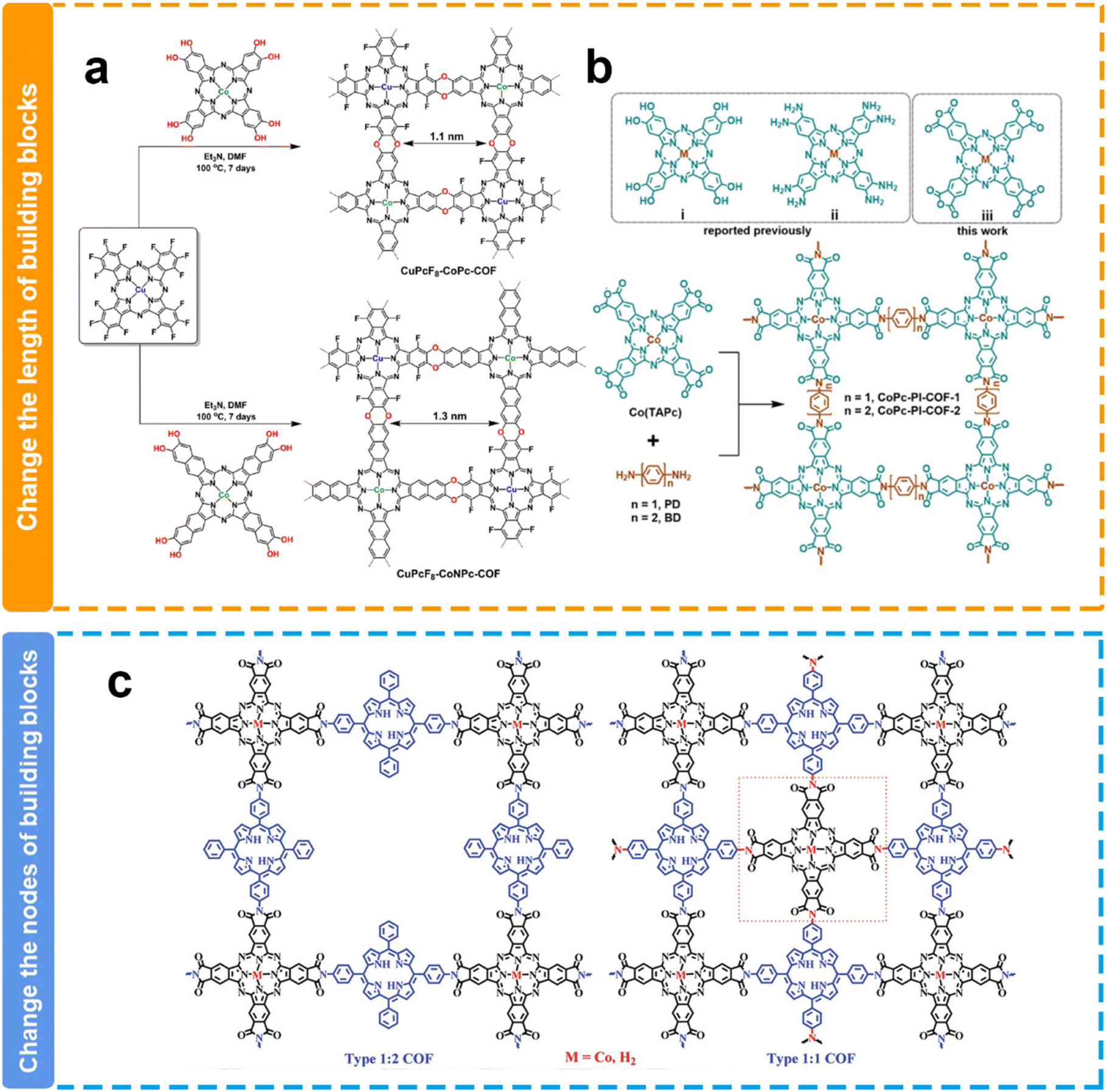 | ||
| Fig. 9 Schematic illustration of the pore size modulation of COFs. (a and b) Schematic illustration of the modulation of the COF pore size via length building blocks;124,125 (c) schematic illustration of the modulation of the COF pore size via the nodes of building blocks.126 | ||
However, the opposite result has been reported by Huang's group (Fig. 9a). The pore size of CuPcF8-CoPc-COF (1.1 nm) is smaller than that of CuPcF8-CoNPc-COF (1.3 nm), while CuPcF8-CoPc-COF shows lower activity and selectivity (TOF = 1.28 s−1, FECOmax = 91%) than CuPcF8-CoNPc-COF (TOF = 2.87 s−1, FECOmax = 97%).125 The authors attributed this to the larger pore size of CuPcF8-CoNPc-COF that affords higher CO2 capacity within COF skeletons to enhance the accessibility of cobalt centers and the larger conjugation π–π stacking structure facilitates electron transfer during the catalytic process. In line with the result, Peng et al. also reported in their research that a COF with a larger pore size could deliver higher eCO2RR performance (Fig. 9c).126 They constructed COFs with different pore sizes through changing the nodes of building blocks. 2,3,9,10,16,17,23,24-Octacarboxyl phthalocyanine cobalt(II) (CoTAPc) as the node is coupled with 5,15-di(4-aminophenyl)-10,20-diphenylporphyrin (DAPor) or 5,15,10,20-tetra(4-aminophenyl)porphyrin (TAPor) to form two COFs with different pore sizes. The COF with a larger pore size demonstrated higher TOF values. This difference might be attributed to their distinct pore sizes, resulting in varying CO2 mass transport capabilities.
The structural evolution in the dimension could significantly alter the microstructures of COFs. Compared to 2D COFs, 3D COFs possess a richer structural dimensionality, endowing them with greater porosity and surface area. This diversity in structure not only provides a broader space for the adsorption and diffusion of reactant molecules, but also creates numerous active sites. Recently, Cao et al. reported a 3D COF based on the condensation with tetra(4-formylphenyl)methane (TFPM), 5,10,15,20-tetrakis(4-aminophenyl)porphyrin cobalt (Co-TAPP), and 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (TAPP) (Fig. 10a).128 Within a voltage range of −0.6 to −1.1 V, CO and H2 are the predominant products (Fig. 10b and c) Notably, the 3D COF exhibited a remarkable FECO of 92.4% at −0.6 V, surpassing the FECO of the 2D COF (82.4%). Furthermore, across a broader voltage window, the FECO could maintain over 80%. Another 3D COF was reported by Jiang et al.130 They successfully synthesized a 3D polyimide-linked phthalocyanine COF composed of interpenetrating pts nets (Fig. 10d). It was prepared via the polymerization reaction between 2,3,9,10,16,17,23,24-octacarboxyphthalocyanine tetraanhydride M(TAPc) (M = Co, H2) and 1,3,5,7-tetra(4-aminophenyl)adamantane (TAPA). Notably, the distinctive 3D porous structure had an impressive electrochemical surface concentration of 183 nmol cm−2 on the electrode, accounting for approximately 32.7% of the total metal phthalocyanine subunits. Consequently, a jCO of 31.7 mA cm−2 was achieved at 0.90 V, surpassing the performance of nearly all COF catalysts reported under similar conditions. Furthermore, this material demonstrated a TOF value of 0.6 s−1 at 0.80 V during a continuous 20-hour test.
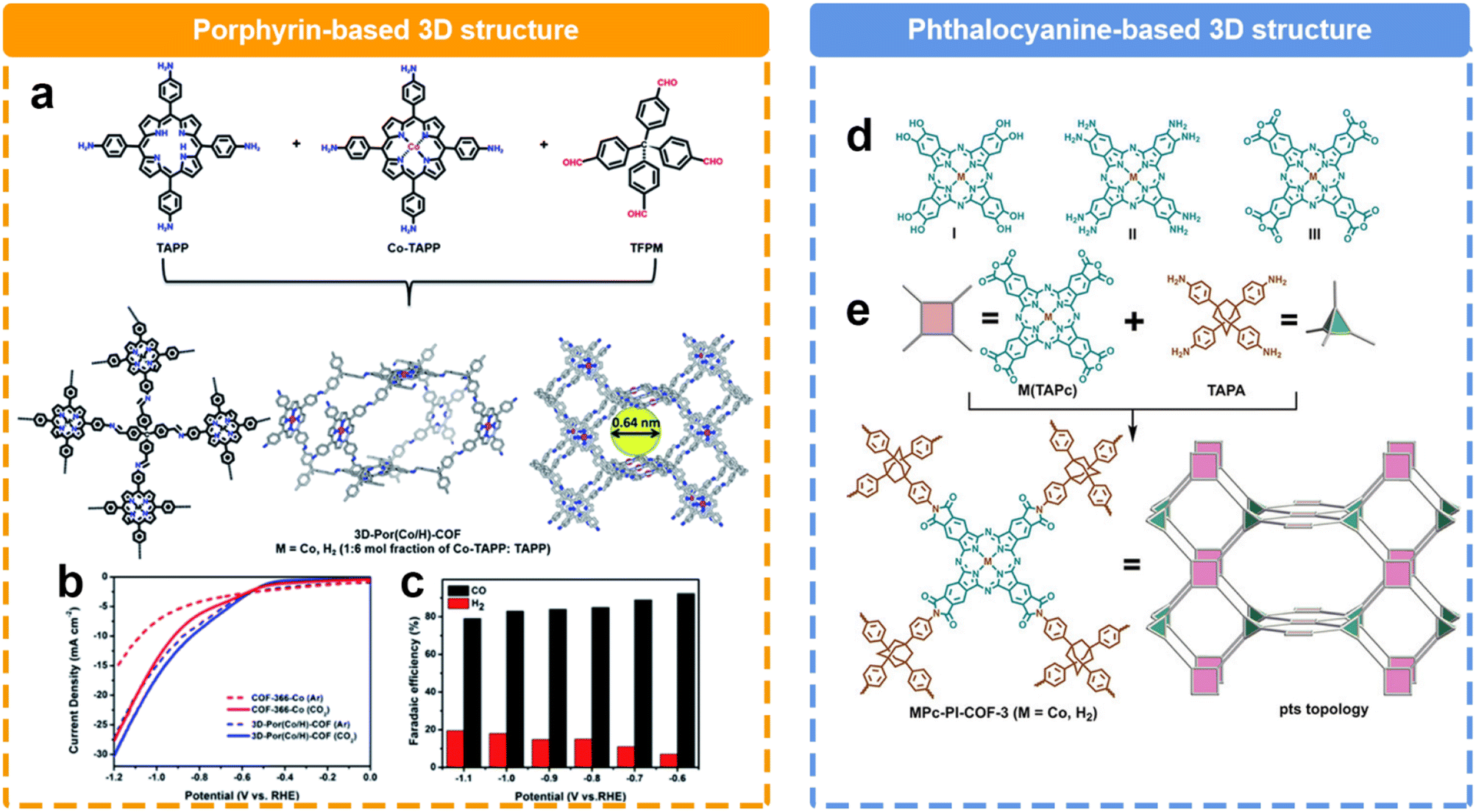 | ||
| Fig. 10 Schematic illustration of the synthesis of different 3D COFs and their performance. (a) Schematic illustration of prepare porphyrin-based 3D COF; (b) LSV curves and (c) FE of porphyrin-based 3D COF;128 (d) Different kinds of phthalocyanine building blocks, (e) Schematic illustration of the preparation of phthalocyanine-based 3D COF.129 | ||
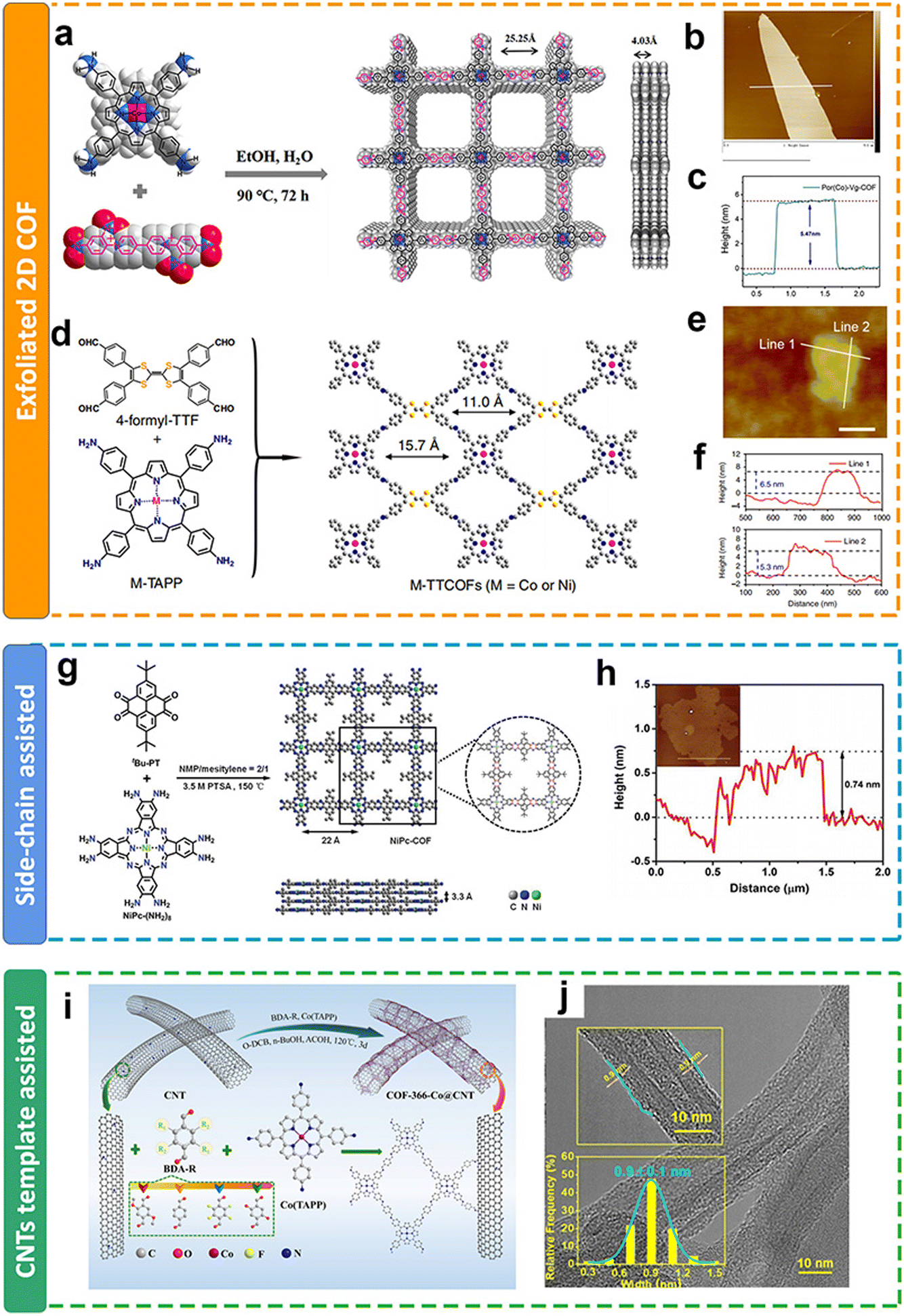 | ||
| Fig. 11 Schematic illustration of the thickness modulation for 2D COFs. (a and d) Schematic illustration of the preparation of metalloporphyrin-based COFs; (b and e) AFM images and (c and f) the thickness of metalloporphyrin-based COFs after exfoliation;111,113 (g) schematic illustration of the preparation of metalloporphyrin-based COFs with side chain groups; (h) AFM images and the thickness of COFs with side chain groups after exfoliation;121 (i) schematic illustration of the preparation of metalloporphyrin-based COFs on CNTs; (j) TEM images and the thickness of metalloporphyrin-based COFs on CNTs.118 | ||
For instance, HCOOH and CO are both products from CO2 reduction involving 2 electron transfer but through different pathways. Thus, designing COFs to effectively modulate the sequence of proton coupling and oxygen atom removal in the eCO2RR process is beneficial for obtaining more valuable reduction products. Qiao et al. reported a Co-coordinated COF with a high nitrogen content (N-COF), which enables efficient CO2 reduction to HCOOH.132 The COF was synthesized through direct condensation of hexaaminotriphenylene hexahydrochloride (HATP) and hexaketocyclohexane octahydrate (HKH). The COF furnishes a coordinating environment for cobalt single atoms anchored between two nitrogen atoms. The coordination of cobalt in Co-N-COF reduces the bandgap significantly from 1.8 eV in N-COF to 0.67 eV, resulting in exceptional catalytic activity. The Co-N-COF exhibits remarkable activity with a JHCOOH value of ∼446 mA cm−2, a high FEHCOOH value of 97.4%, and outstanding stability over 100 hours. Moreover, the purification of the electrocatalytically produced formic acid solutions reaches up to ∼100 wt%. The abundant N atoms around the Co species in the skeleton may be responsible for the production of HCOOH, a hydrogenated product also through two electron transfer.
It is interesting to find that CH4, as the complete reduction product involving proton coupling and oxygen atom removal, has been reported in several research studies. The metallic centers used in this research are all Cu species. It is proposed that the strong adsorption strength of typical intermediates on Cu species during the eCO2RR may be the main reason for the production of this deeply reduced product. The Lan group reported anthraquinone-based COFs for efficient eCO2RR. Tunable 1D superstructures (nanofibers and hollow tubes) of COFs could be achieved using different side groups in the building blocks, which were then post-modified with Cu ions within the N and O atoms (Fig. 12a).133 These porous nanostructures are proposed to have the features of exceptional chemical stability, substantial surface area, and capability for CO2 enrichment and activation. The Cu-incorporated COF nanofibers and hollow tubes exhibited superior faradaic efficiency for CH4 production in flow cells, with a FECH4 value of 77% (at −0.9 V, 128.1 mA cm−2) and 61% (at −1.0 V, 99.5 mA cm−2), respectively (Fig. 12b–e). Notably, the study also found that the nanofiber morphology was more favorable for CH4 production, while the hollow tube morphology had a particular control effect on the generation of ethylene, which provides a general methodology for exploring morphology-controlled COFs for electrocatalytic CO2 reduction. The morphology-dependent catalytic performance was also reported by Lan et al. in the tailoring of porphyrin-based COFs incorporated with Cu ions (COF-366-Cu) (Fig. 12f).134 They successfully prepared three distinct types of nanostructures, including hollow nanospheres (HS), nanosheets (NS) and nanofibers (NF). The nanostructured Cu-COF showed higher CH4 selectivity (Fig. 12g and h). Specifically, COF-366-Cu (HS), COF-366-Cu (NF) and COF-366-Cu (NS) exhibit a superior FECH4 of 68.2% (285.4 mA cm−2 at −0.9 V), 71.0% (350.8 mA cm−2 at −0.9 V) and 64.2% (−276.0 mA cm−2 at −0.9 V) in the flow-cell, respectively. Besides, the achieved FECH4+C2H4 (82.8% at −0.9 V) of COF-366-Cu (HS) is superior to most of the reported COFs and copper-based electrocatalysts. Due to the unique catalytic mechanism of Cu, Cu single sites will convert into nanoclusters during the catalytic process which have been previously reported. The stronger adsorption energy between Cu clusters and CO intermediates can further reduce CO intermediates to more deeper reduction products on the active sites. This is why C2H4 could be produced in their works. Cu clusters, rather than chelated Cu metallic sites, are the real active sites during the catalysis, so this content won't be discussed further in this review.
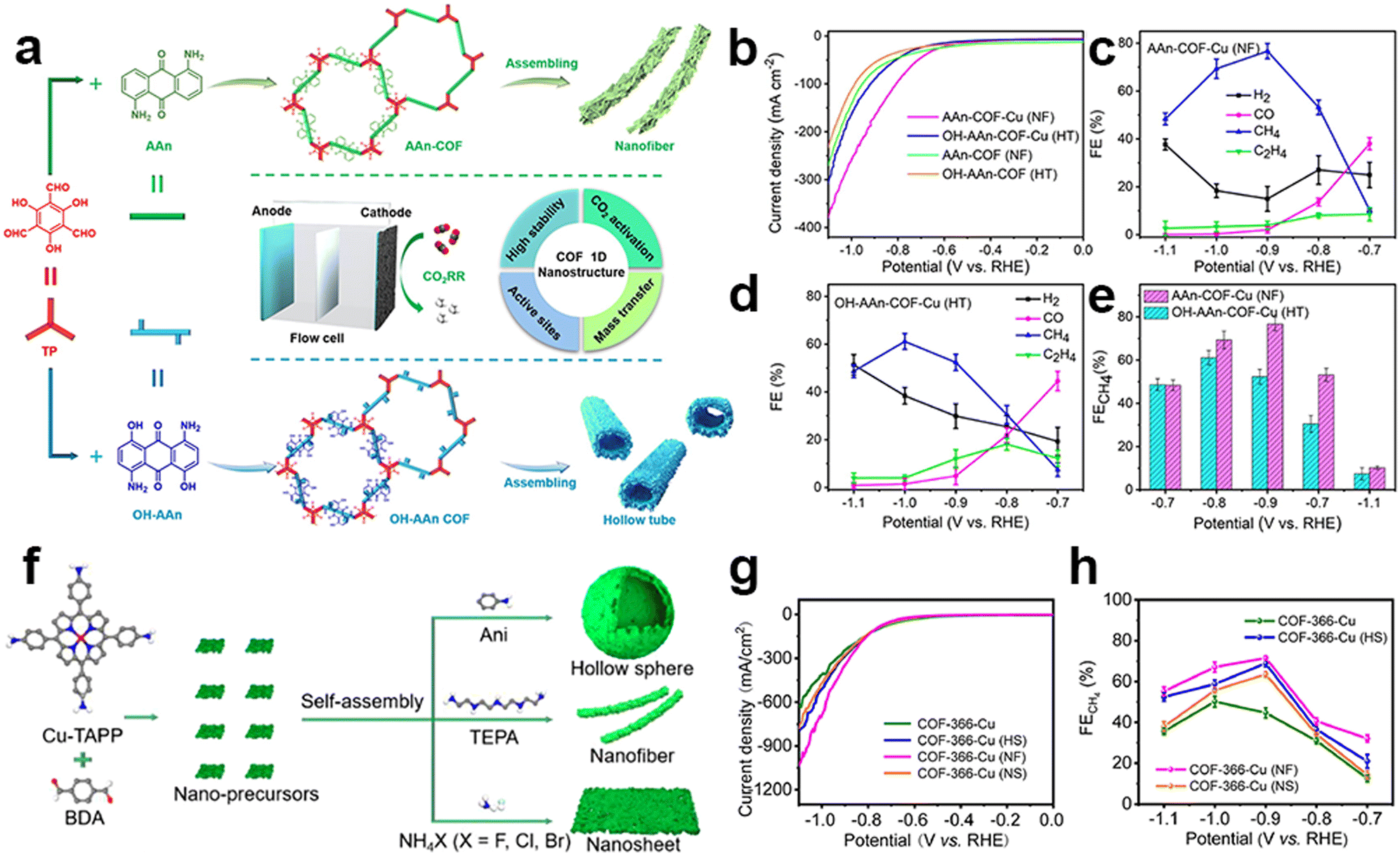 | ||
| Fig. 12 Schematic illustration of super-structured COFs and their performance. (a) The schematic representation of anthraquinone COF-based nanofibers and hollow tubes in the electrocatalytic eCO2RR; (b) LSV curves of AAn-COF-Cu (NF), OH-AAn-COF-Cu (HT), AAn-COF (NF) and OH-AAn-COF(HT); (c–e) FE of AAn-COF-Cu(NF) and OH-AAn-COF-Cu(HT).133 (f) The schematic representation of porphyrin COF-based nanostructures; (g) LSV curves and (h) FECO values of COF-366-Cu, COF-366-Cu(HS), COF-366-Cu(NF) and COF-366-Cu(NS).134 | ||
Another work by the Lan group using exfoliation to synthesize large-scale (∼1.0 mm) and ultrathin (∼3.8 nm) nanosheets of Cu-based COFs toward the efficient eCO2RR to CH4.131 The functionalized exfoliating agent, 2,4-diamino-6-chloro-1,3,5-triazine (Dct), could also serve as a co-catalyst during the eCO2RR. It may enhance the CO2 absorption/activation, stabilize active intermediates, and facilitate CO production, thereby increasing the concentration of CO around Cu active sites. The optimized COF structure comprises continuous units of Cu-porphyrin and Dct. At 0.90 V, the FECH4 reaches 80%, with a current density of ∼220.0 mA cm−2, which is nearly double that of the bare COF counterpart. The ultrathin layer of Cu-COFs was also achieved by the template-directed synthesis reported by the same group. They synthesized a series of honeycomb-like porous crystalline hetero-electrocatalysts by growing a copper-porphyrin-based COF on the surface of a porous MOF template. The specially designed heterostructures with an integrated porous MOF-template and ultrathin COF-coating enable efficient CO2 adsorption/activation and conversion into CH4. The optimized heterogeneous structure delivered a superior FECH4 of 76.7%, with an excellent current density of 398.1 mA cm−2.
In the synthesis of products with more than two C atoms, the C–C coupling step for ethylene and ethanol requires the coupling of *CO with *CO or *CHO, a process that necessitates adjacent active sites to facilitate its occurrence. Since most metal active sites in COFs are isolated, there have been few reports on the synthesis of products with more than two C atoms through the eCO2RR using COFs. Recently, Chen et al. successfully synthesized acetic acid, a C2 product, using a PcCu-TFPN COF.135 They proposed that during their synthesis, the C–C coupling step is achieved between *CH3 and CO2. They compared PcCu-TFPN with copper single-atom catalysts (CuSAC) and copper-porphyrin COFs. According to the DFT calculations, the Cu atoms in PcCu-TFPN possess a higher electron density compared to CuSAC and Cu porphyrin, primarily due to the presence of more nitrogen atoms and stronger conjugation effects in the phthalocyanine structure. During the CO2RR process, CuSAC exhibits unstable adsorption of *CO, with longer Cu–C bond lengths that promote the desorption of CO, making it difficult to proceed with further hydrogenation reactions. On the other hand, although the Cu porphyrin COF can also further hydrogenate *CO to *CH3, the higher electron density of Cu atoms in PcCu-TFPN, combined with the strong conjugation effect of phthalocyanine, leads to the transfer of d electrons from Cu to C, resulting in a lower oxidation state for C. Consequently, this strengthens the interaction between PcCu-TFPN and *CH3. Such single-site catalytic centers could also help avoid the generation of unwanted byproducts like ethylene and ethanol, providing a feasible strategy for future eCO2RR.
Typical COFs as electrocatalysts in the eCO2RR are summarized in Table 1.
| Main product | Material | Metal center | Chelating site | Design principle | Highest FE | TOF | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|---|---|
| CO | COF-366-Co | Co | Porphyrin | Microstructures and heterometallic centers | 90% | ∼2500 h−1 | H type cell and 0.5 M KHCO3 | 106 |
| COF-367-Co | Co and Cu | 70% | ∼4400 h−1 | |||||
| CO | FeDhaTph-COF | Fe | Porphyrin | Electronic modulation | 80% | >600 h−1 | Five-necked pear-shaped flask, 0.1 M TBAPF6 and 0.5 M TFE MeCN solution | 109 |
| CO | TTF-Por(Co)-COF | Co | Porphyrin | Electronic modulation | 95% | 676 h−1 | H type cell and 0.5 M KHCO3 | 110 |
| CO | CoPc-PDQ-COF | Co | Phthalocyanine | Electronic modulation | 96% | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 412 h−1 412 h−1 |
H type cell and 0.5 M KHCO3 | 122 |
| CO | COF-366-Co@CNT | Co | Porphyrin | Electronic modulation and microstructures | 92% | >10000 h−1 |
H type cell and 0.5 M KHCO3 | 118 |
| COF-366-(OMe)2-Co@CNT | 94% | 11877 h−1 |
||||||
| COF-366-(OH)2-Co@CNT | 90% | ∼10000 h−1 |
||||||
| COF-366-(F)4-Co@CNT | 90.50% | ∼8000 h−1 | ||||||
| CO | COF-366-Co | Co | Porphyrin | Electronic modulation and microstructures | 87% | N/A | H type cell and 0.5 M KHCO3 | 117 |
| COF-366-(OMe)2-Co | N/A | N/A | ||||||
| COF-366-F-Co | N/A | N/A | ||||||
| COF-366-(F)4-Co | N/A | N/A | ||||||
| CO | 2D NiPc-COF | Ni | Phthalocyanine | Electronic modulation | 99.10% | 3772 h−1 | H type cell and 0.5 M KHCO3 | 121 |
| CO | 2D TT-Por(Co)-COF | Co | Porphyrin | Electronic modulation | 91.40% | 481 h−1 | H type cell and 0.5 M KHCO3 | 112 |
| CO | COFbpyMn | Mn | 2,2-Dipyridine | Electronic modulation and microstructures | 62% | 627 h−1 | H type cell and 0.5 M NaHCO3 | 108 |
| CO | CoPc-PI-COF-3 | Co | Phthalocyanine | Electronic modulation and microstructures | 96% | 0.6 s−1 | H type cell and 0.5 M KHCO3 | 130 |
| CO | CuPcF8-CoPc-COF | Cu and Co | Phthalocyanine | Heterometallic centers and microstructures | 91% | 1.25 s−1 | H type cell and 0.5 M CsHCO3 | 125 |
| CuPcF8-CoNPc-COF | 97% | 2.87 s−1 | ||||||
| CO | 3D-Por(Co/H)-COF | Co | Porphyrin | Microstructures | 92.40% | 4610 h−1 | H type cell and 0.5 M KHCO3 | 128 |
| CO | CoPc-2H2Por COF | Co | Phthalocyanine | Heterometallic centers, microstructures, and electronic modulation | 95% | >800 h−1 | H type cell and 0.5 M KHCO3 | 126 |
| CoPc-H2Por COF | 94% | >400 h−1 | ||||||
| CO | CoP-BDTHexO-COF | Co | Porphyrin | Microstructures and electronic modulation | >90% | 8655 h−1 | H type cell and 0.5 M KHCO3 | 119 |
| CoP-BDTEO-COF | >90% | 5425 h−1 | ||||||
| CoP-BDT-COF | >90% | 3336 h−1 | ||||||
| CoP-BDTPEO-COF | ∼60% | 1351 h−1 | ||||||
| CO | TPPDA-CoPor-COF | Co | Porphyrin | Metal species and electronic modulation | 92% | 2.0 s−1 | H type cell and 0.5 M KHCO3 | 136 |
| TPPDA-NiPor-COF | Ni | 76% | ∼0.4 s−1 | |||||
| CO | Por(Co)-Vg-COF | Co | Porphyrin | Electronic modulation | 91% | N/A | H type cell and 0.5 M KHCO3 | 113 |
| CO | TPE-CoPor-COF | Co | Porphyrin | Electronic modulation and microstructures | 95% | 2.4 s−1 | H type cell and 0.5 M KHCO3 | 127 |
| TPTPE-CoPor-COF | 96% | 1.21 s−1 | ||||||
| CO | TAPP(Co)-B18C6-COF | Co | Porphyrin | Electronic modulation and microstructures | 93.20% | 1267 h−1 | H type cell and 0.5 M KHCO3 | 114 |
| CO | NiPc-TFPN COF | Ni | Phthalocyanine | Metal species and electronic modulation | 99.8% | 490 h−1 | H type cell and 0.5 M KHCO3 | 137 |
| CoPc-TFPN COF | Co | 96.1% | 369 h−1 | |||||
| ZnPc-TFPN COF | Zn | 22.9% | ∼50 h−1 | |||||
| CO | CoPc-DNDS-COF | Co | Phthalocyanine | Electronic modulation | 96.30% | 1076.4 h−1 | H type cell and 0.5 M KHCO3 | 115 |
| CoPc-DSDS-COF | 96.50% | 1548.2 h−1 | ||||||
| CO | CoPc-PI-COF-1 | Co | Phthalocyanine | Electronic modulation and microstructures | 97% | 2.2 s−1 | H type cell and 0.5 M KHCO3 | 124 |
| CoPc-PI-COF-2 | 96% | 1.9 s−1 | ||||||
| CO | CoTAPP-PATA-COF | Co | Porphyrin | Electronic modulation | 81.75% | 4166.19 h−1 | H type cell and 0.5 M KHCO3 | 116 |
| N+-COF | 92.07% | 7453.19 h−1 | ||||||
| NH-COF | 95.26% | 7604.63 h−1 | ||||||
| N+-NH-COF | 97.32% | 9922.68 h−1 | ||||||
| CO | Co-TTCOFs | Co | Porphyrin | Electronic modulation and metal species | 91.3% | 1.28 s−1 | H type cell and 0.5 M KHCO3 | 111 |
| Ni-TTCOFs | Ni | 20.9% | N/A | |||||
| CO | H2Pc-COFs | Metal free | Phthalocyanine | Electronic modulation and heterometallic centers | 34.21% | 1258.64 h−1 | H type cell and 0.5 M KHCO3 | 138 |
| 0.25 NiPc-COFs | Ni | 37.66% | 2790.41 h−1 | |||||
| 0.5 NiPc-COFs | 92.52% | 4713.53 h−1 | ||||||
| 0.75 NiPc-COFs | 95.37% | 3749.88 h−1 | ||||||
| NiPc-COFs | 75.68% | 3860.12 h−1 | ||||||
| CO | Co-pPPC@CNFs | Co | Phthalocyanine | Electronic modulation and microstructures | 74% | N/A | H type cell and 0.5 M KHCO3 | 139 |
| Co-anPPC@CNFs | 67% | N/A | ||||||
| CO | PcNi-im | Ni | Phthalocyanine | Electronic modulation | 100% | 3.06 s−1 | Flow cell 0.5 M KHCO3 | 140 |
| PcNi-pz | 98% | 2.37 s−1 | ||||||
| PcNi-tfpn | 99% | 2.45 s−1 | ||||||
| CO | Co-iBFBim-COF-I− | Co | Porphyrin | Electronic modulation | 99% | 3018 h−1 | MEA cell and 1 M KOH | 141 |
| CO | Co-Bpy-COF-Ru1/2 | Co | Porphyrin and 2,2-dipyridine | Electronic modulation | 84.6% | 681 h−1 | H type cell and 0.5 M KHCO3 | 142 |
| CO | pCoPc | Co | Phthalocyanine | Electronic modulation and metal species | 67% | N/A | Flow cell and 1 M KOH | 143 |
| pNiPc | Ni | 99% | N/A | |||||
| pCoNiPc | Co and Ni | >90% | N/A | |||||
| pCo1Ni6Pc | Co and Ni | >90% | N/A | |||||
| CO | MWCNT-Por-COF-Fe | Fe | Porphyrin | Metal species | ∼63% | >25 s−1 | H type cell and 0.5 M KHCO3 | 107 |
| MWCNT-Por-COF-Co | Co | 99.30% | 70.6 s−1 | |||||
| MWCNT-Por-COF-Ni | Ni | ∼60% | >20 s−1 | |||||
| CH4 | MWCNT-Por-COF-Cu | Cu | 71.20% | N/A | H type cell and 0.1 M KOH | |||
| CH4 | Cu-Tph-COF-Dct | Cu | Porphyrin | Microstructures and electronic modulation | ∼80% | N/A | Flow cell and 0.1 M KOH | 131 |
| Cu-Tph-COF-OH | 48.8% | |||||||
| CH4 | AAn-COF-Cu | Cu | Others | Microstructures, chelating sites and electronic modulation | 77% | N/A | Flow cell and 1 M KOH | 133 |
| OH-AAn-COF-Cu | 61% | |||||||
| CH4 | COF-366-Cu-HS | Cu | Porphyrin | Microstructures | 68.20% | N/A | Flow cell and 1 M KOH | 134 |
| COF-366-Cu-NF | 64.20% | |||||||
| COF-366-Cu-NS | 71% | |||||||
| HCOOH | Co-N-COF | Co | Others | Metal species and microstructures | 97% | N/A | H-type cell and 1.0 M KHCO3 | 132 |
| CH3COOH | PcCu-TFPN | Cu | Phthalocyanine | Electronic modulation | 90.3% | N/A | Flow cell and 0.1 M KHCO3 | 135 |
COF-based catalysts can obtain excellent eCO2RR performance by selecting suitable building blocks due to the well-defined and tunable structures. However, COF-based catalysts also face several challenges. The first is that the stacking between adjacent layers of 2D COFs may block the metal catalytic sites, especially in porphyrin- and phthalocyanine-based 2D COFs. Exfoliating 2D COFs into oligolayer sheets or building 3D COFs are assumed to be conductive to make metal centers readily accessible. Moreover, the poor intrinsic electric conductivity limits the eCO2RR activity of COFs. Developing highly conductive COFs or incorporating conductive carbon materials could help improve the intrinsic electric conductivity of COFs.
3.2 CMPs
CMPs represent an important class of amorphous organic porous materials. Since their discovery in 2007, CMPs have become an important subclass of porous materials, which combine the characteristics of extended π-conjugated polymers and permanent microporous scaffolds. They have the advantages of large specific surface area, high stability, and tunable nanostructure. Well-designed CMPs can selectively adsorb CO2, and much research has been devoted to exploring their applications in electrocatalytic CO2 reduction. Moreover, compared with COFs, their extended π-conjugated structures could deliver relatively better electrical conductivity, which is highly required in electrocatalysis.CMPs could provide rigid frameworks to accommodate electroactive metal species to effectively catalyze the eCO2RR. For example, Alexander J. Cowan et al. synthesized a bipyridine-based amorphous polymer CMP through the Sonogashira–Hagihara cross-coupling reaction of 1,3,5-triethynylbenzene with 5,5′-dibromo-2,2′-bipyridine and 1,4-dibromobenzene.144 Then the metal–organic CMP containing a manganese carbonyl electrocatalyst was obtained after the reflux with [Mn(CO)5Br]. [Mn(bpy)(CO)3Br] and its derivatives have previously been reported to be active toward the eCO2RR, while it will undergo dimerization prior to formation of the catalytically active species in solution. It was proved that after fixing it in the CMP framework, the dimerization could be prevented. According to CV and LSV curves, the Mn center of the complex remained electrochemically active within the CMP framework with CO as the main product.
In addition to the rigid framework of CMPs as a support, it is also widely believed that the highly conjugated structure in CMPs is conducive to electron transfer. Duan et al. prepared a two-dimensional conjugated nickel phthalocyanine polymer (NiPcP) by a simple one-step vacuum calcination method.145 In comparison with the NiPc monomer, NiPcP possesses a highly cross-linked conjugated network structure, a higher surface area, more accessible active sites and enhanced structural robustness. Consequently, NiPcP exhibits a remarkably high performance toward the eCO2RR to CO with a high FE > 98% in a wide potential range from −0.15 to −0.60 V. The maximum partial current density (jCO) reaches 236 mA cm−2 at −0.6 V, far superior to the NiPc monomer. Experimental and theoretical studies revealed that the cross-linked conjugated structure was responsible for its high performance in the CO2RR. Utilizing carbons such as graphene and carbon nanotubes (CNTs) to support CMPs is an effective way to improve electrical conductivity. However, the catalytic activity of MPc-containing CMPs often suffers from a limited molecular modulation strategy. As for NiPc, there is a strong correlation between the localized d electrons of the Ni atom and delocalized π electrons of the phthalocyanine ligand. The expansion of π-conjugation is expected to regulate the electron density of the Ni position. Liu et al. established nickel polyphthalocyanine (NiPPc) supported on carbon nanotubes (NiPPc/CNTs) for efficient eCO2RR (Fig. 13a).146 The XRD patterns and Raman spectra indicate the successful synthesis of coaxial NiPPc/CNT (Fig. 13b and d). It also displays a red shift in Q-band absorption in contrast to NiPc, indicating NiPPc with an extended π conjugation (Fig. 13c). Electrochemical results show that NiPPc/CNT exhibits a high current density of ∼300 mA cm−2 for CO2RR with a CO selectivity of 99.8%. A similar research study was conducted by Zang et al.,147 by introducing graphene oxide (GO) to modify the charge density distribution of CoPc (Fig. 13f). They designed and synthesized a conjugated phthalocyanine framework catalyst (CPF-Co) with abundant CoN4 centers. Meanwhile, they further optimized the coordination microenvironment of Co species through the axial coordination between oxygen in the partially reduced GO (LGO) and the catalytic sites (Fig. 13g–i). The optimized O–CoN4 structure could break the electron distribution symmetry of Co, thus reducing the energy barrier to the activation of CO2 to COOH*. By adjusting the content of oxygen, the appropriate support could also facilitate the charge transfer efficiency between the matrix layer and the catalytic sites. The optimized CPF-Co@LGO exhibited a high CO selectivity (97.6%), TOF value (2.81 s−1) and stability (24 h) at 21 mA cm−2 current density.
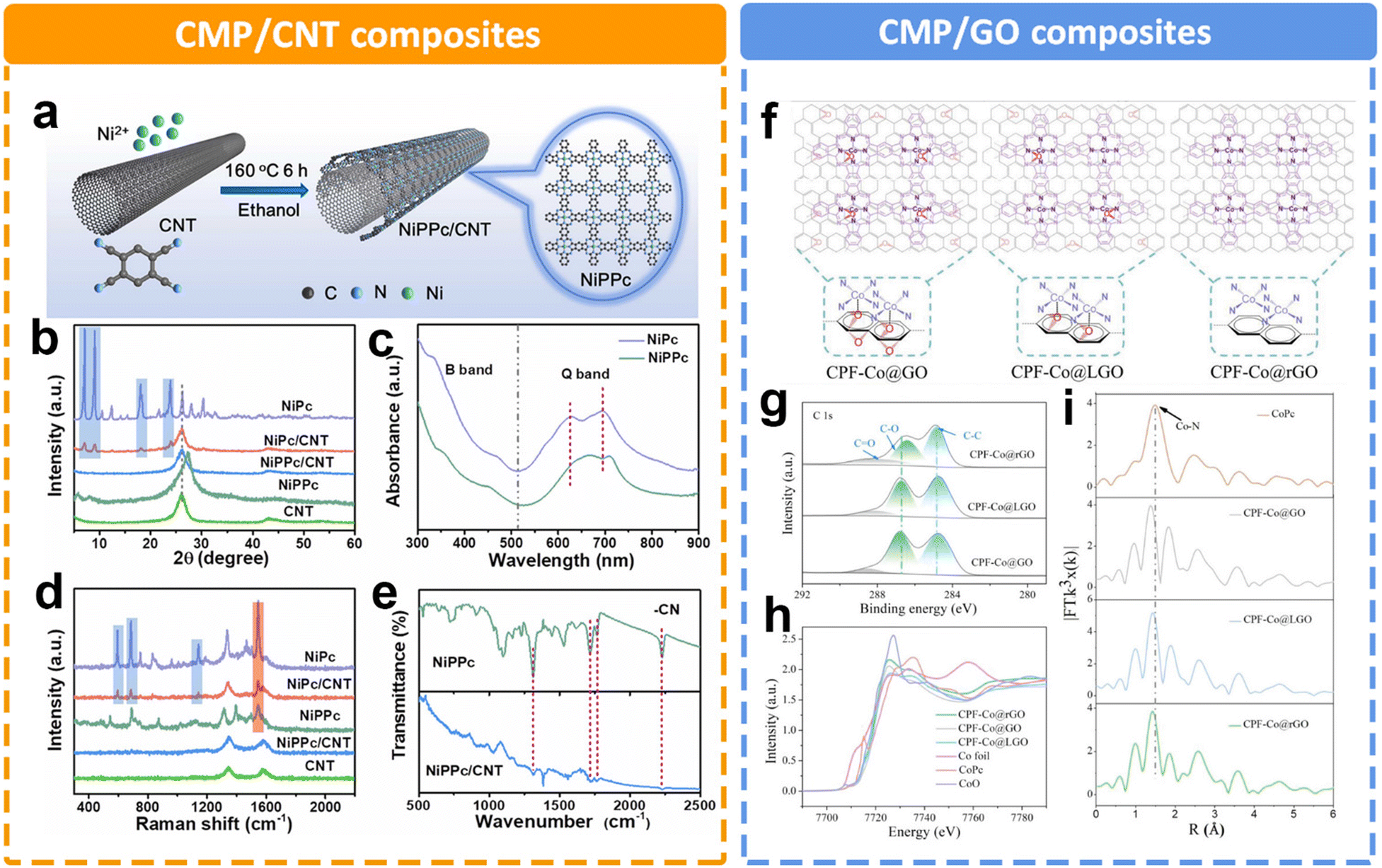 | ||
| Fig. 13 Schematic illustration of different CMP composites and their characterization. (a) Schematic illustration of synthesizing NiPPc/CNTs, (b) XRD patterns of samples, (c) UV-Vis spectra of NiPc and NiPPc in DMF, (d) Raman spectra of samples, and (e) FTIR spectra of NiPPc and NiPPc/CNT.146 (f) Schematic illustration of the CPF-Co@GO/LGO/rGO hybrid preparation and application to electrocatalytic CO2 to CO conversion. Structural characterization of CPF-Co@GO, CPF-Co@LGO, and CPF-Co@rGO, (g) the high-resolution C 1s XPS spectra, (h) Co K edge XANES spectra, and (i) Co K-edge FT of the EXAFS spectra.147 | ||
Other than the electronic modulation of MPc-containing CMPs from the support, another modulation strategy is the tuning of the metallic centers. The deliberately designed metal vacancies in Pc are proposed to influence the electrocatalytic performance of CMPs. Guo et al. adopted a solid-state ionothermal method to synthesize an ultrathin CMP sheath around CNTs by a Scholl coupling reaction of metal-free H2Pc with CoPc in a controllable manner to regulate atomic dispersion of metal centers (Fig. 14a).148 Copolymerization of H2Pc and CoPc via the dehydrogenation of periphery benzene rings produces a highly cross-linked two-dimensional network. The incorporated H2Pc unit serves not only as an electron donor improving the nucleophilicity of Co sites for CO2 binding, but also as a proton donor to transport hydrogen to the Co active sites (Fig. 14d). The optimized catalyst generates a jCO value of 15.2 mA cm−2 (−0.6 V) and a high FECO (max. 97% at −0.9 V) in a broad potential window (Fig. 14b and c).
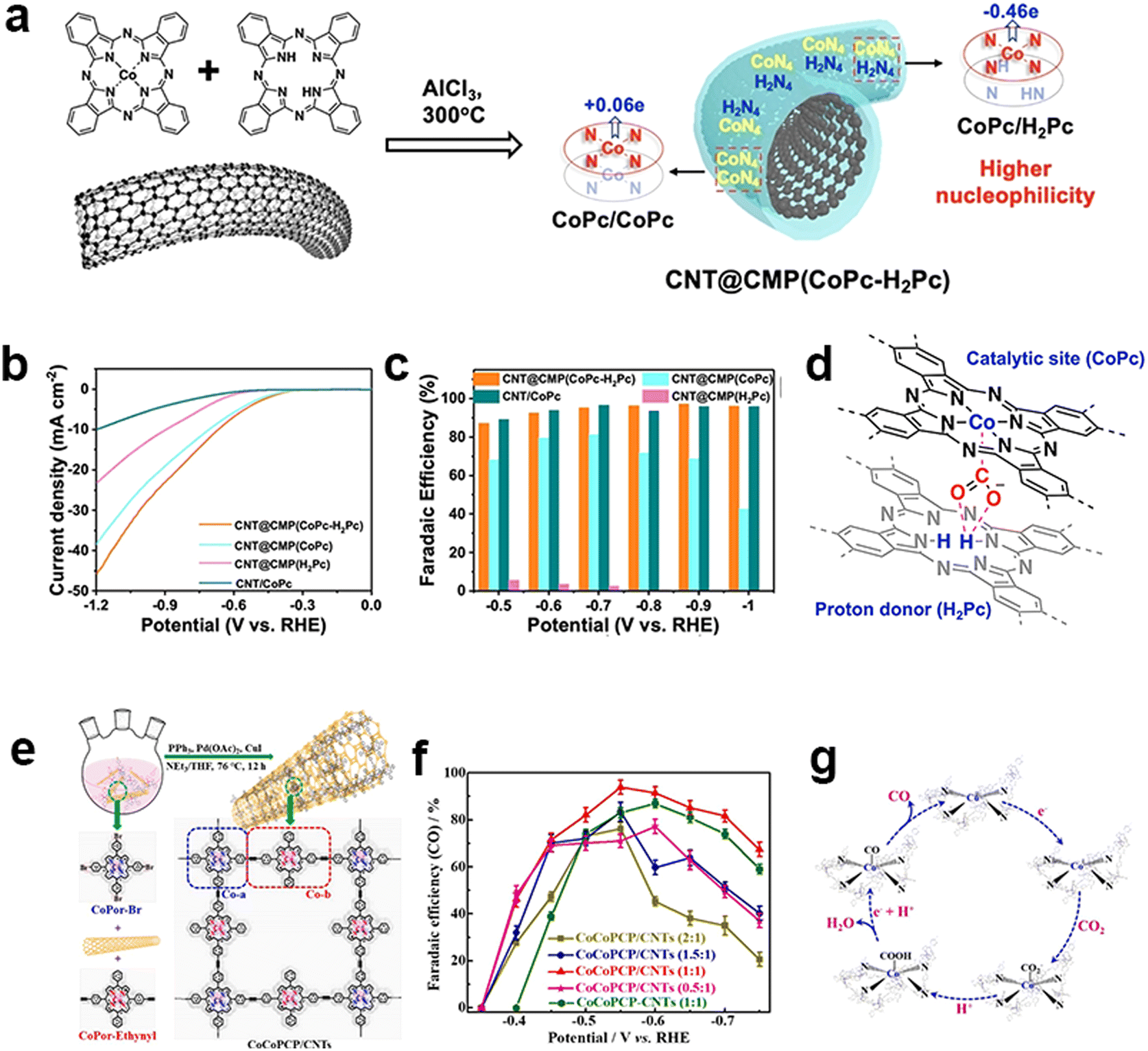 | ||
| Fig. 14 Heterometallic design of CMPs. (a) Ionothermal synthesis of CNT@CMP(CoPc-H2Pc) containing the stacked CoPc/CoPc and CoPc/H2Pc moieties with different Hirshfeld Co atomic charges, (b) polarization curves of CNT@CMP(CoPc-H2Pc), CNT@CMP(CoPc) and CNT@CMP(H2Pc) in CO2-saturated 0.5 M KHCO3 solutions and (c) their CO FEs at different applied potentials, (d) proposed proton-donor mechanism of H2Pc for the synergistic catalysis of CO2 reduction.148 (e) Schematic illustration of the in situ polymerization of CoCoPCP on CNTs by the Sonogashira coupling reaction, (f) CO faradaic efficiencies of the CoCoPCP/CNTs with different mass ratios and CoCoPCP-CNTs with a mass ratio of 1:1, and (g) the proposed mechanism (b) for the CO2 RR process of CoCoPCP/CNTs.149 | ||
The incorporation of bimetallic sites is another plausible strategy to achieve electronic modulation through the synergistic effects between bimetallic sites. Peng et al. constructed ethynyl-linked Co-porphyrin based CMPs on CNTs to fabricate a bi-metallic central electrocatalyst (CoCoPCP/CNTs) for eCO2RR (Fig. 14e).149 It bears four-linked Co-porphyrin units (Co-a) and two-linked Co-porphyrin bridging ones (Co-b), which could promote the charge transfer and electronic modulation of Co active sites. It synergistically improves the electrocatalytic activity and selectivity of the eCO2RR. The CoCoPCP/CNT composites have up to 94% FECO and excellent cycling stability (Fig. 14f). It could maintain a current density of −8.1 mA cm−2 within 24 hours and the FECO could be achieved at more than 90% for a long time.
As most reported CMPs have a randomly packed morphology with highly stacking and aggregated structures, their active centers embedded in the cavities could not be fully exposed during the eCO2RR. Reasonable nanostructural design is helpful to further improve their electrocatalytic efficiency and stability. Ye et al. reported an ultrathin CMP based on the squaraine-bridged cobalt tetraamino phthalocyanine structure (COP-SA) for high-performance eCO2RR towards CO (Fig. 15a),150 which has an extended π-conjugated skeleton with an ultrathin thickness of ∼1.7 nm and an abundant microporous structure (Fig. 15c). The ultrathin nanostructure avoids the agglomeration of molecular catalysts and is beneficial for the exposure of active sites. Theoretical calculation suggests that the squaraine unit will enhance *COOH adsorption and favor *CO desorption. These properties synergistically contribute to the high current density and FECO (Fig. 15f and g).
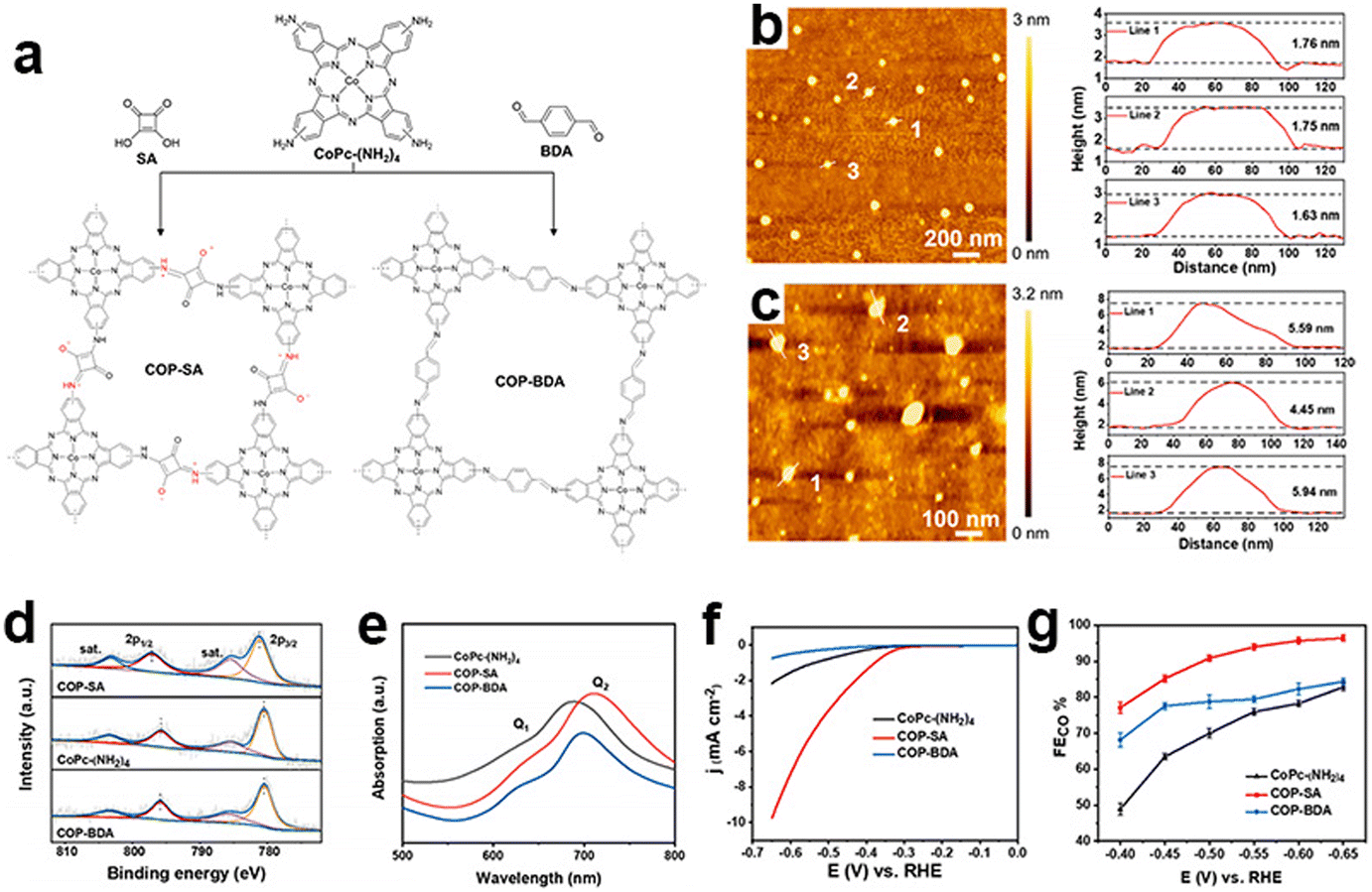 | ||
| Fig. 15 Ultrathin nanostructured CMPs. (a) Schematic illustration of the synthesis of COP-SA and COP-BDA, (b) the atomic force microscopy topographical image of COP-SA and the height profile of lines 1, 2 and 3, (c) the atomic force microscopy topographical image of COP-BDA and the height profile of lines 1, 2 and 3, (d) high resolution Co(2p) XPS spectra and (e) UV–vis spectra of CoPc-(NH2)4, COP-SA and COP-BDA, (f) LSV curves of CoPc-(NH2)4, COP-SA and COP-BDA, and (g) faradaic efficiencies from −0.4 to −0.65 V vs. RHE of CoPc-(NH2)4, COP-SA and COP-BDA.150 | ||
The main product of the eCO2RR catalyzed by CMPs is CO, and other hydrocarbon products are rarely observed in previous research. Their production generally requires sufficient and highly active redox centers to realize the challenging proton-coupled electron transfer steps. Lan and his coworkers reported a facile and scalable strategy to prepare a series of Cu-porphyrin based CMPs with different linkages by in situ copolymerization of dialdehyde-based ligands and pyrrole.151 These catalysts show efficient electrocatalytic performance toward the electrocatalytic conversion of CO2 to CH4. The obtained materials are composed of directly bonded auxiliary groups and Cu porphyrin centers. Specifically, the optimized catalyst with ferrocene as a linker exhibited excellent CO2-to-CH4 electroreduction performance (current density, −491.5 mA cm−2 and FECH4, 75.9%). DFT calculations revealed that the directly interacting ferrocene groups enhance the electron-cloud density of Cu in the porphyrin-center. The strong adsorption ability of ferrocene groups toward OH* promotes the proton-coupled electron transfer reaction process, which kinetically improves the preferential CO2-to-CH4 pathway.
CMPs may have better intrinsic electric conductivity compared with COFs due to their well-conjugated structures. However, due to their short-range ordered and layer stacked structures, the obtained CMPs may have variable properties which are not as expected. This may bring about the challenge in designing high-performance CMP-based catalysts toward the eCO2RR.
3.3 POCs
As discussed above, COFs and CMPs often have a large number of active sites blocked by interlayer stacking and steric hindrance, limiting their full utilization. In contrast, porous organic cages (POCs), which are discrete covalent-bonded molecular architectures, also known as covalent organic polyhedra, represent alternative promising candidates for CPCs. POCs possess the advantages of a large surface area, stable chemical properties and easy processing. Compared with COFs and CMPs, POCs have fully exposed structures that maximize the active sites and speed up the mass transfer process. However, due to the spatial separated structures of POCs, the intrinsic conductivity is limited. Moreover, the synthesis of POCs is challenging. Within the past few years, only a few examples of POCs used in the eCO2RR have been published, even though they have superiority in some aspects.The preparation of POCs based on redoxative porphyrins has been an attractive research subject due to the accessible active sites, favorable guest diffusion and confinement inside the cage, and reduced aggregation of catalytic centers. Chang et al. proposed a method to increase the electrochemically active surface area and facilitate mass transport, which was installing iron tetraphenylporphyrin (Fe-TPP) into a rhombicuboctahedral POC for electrochemical CO2-to-CO conversion (Fig. 16a).152 Fe-porphyrin based POCs could facilitate the diffusion of the substrate and electrolyte throughout an array of readily accessible catalytic metal centers (Fig. 16b). The porous architecture of POCs resulted in a larger amount of exposed electroactive iron centers compared with their monomeric counterpart Fe-TPP. The electrochemical test results showed enhanced stability and current density than Fe-TPP. Similarly, Jiang et al. conducted a comprehensive study of a functional cofacial porphyrin organic cage, CPOC-H2, through the linking of a 5,10,15,20-tetrakis(4-formylphenyl)porphyrin and chiral (2-aminocyclohexyl)-1,4,5,8-naphthalenetetraformyl diimide (Fig. 16c).153 The following post-synthetic metalation of the metal-free cage with different metal ions leads to the generation of four D–A-type cages. Among them, Co porphyrin based POCs have the most electron-rich environment with stronger physical adsorption capability for CO2 molecules (Fig. 16d). According to the DFT calculation, the Co–C Mayer bond order (0.93) of the *COOH intermediate is the highest, indicating the key influence of metallic centers on the electrocatalytic performance toward the eCO2RR. Co porphyrin based POCs supply a partial current density of 18.0 mA cm−2 at −0.9 V with the FECO of 90%.
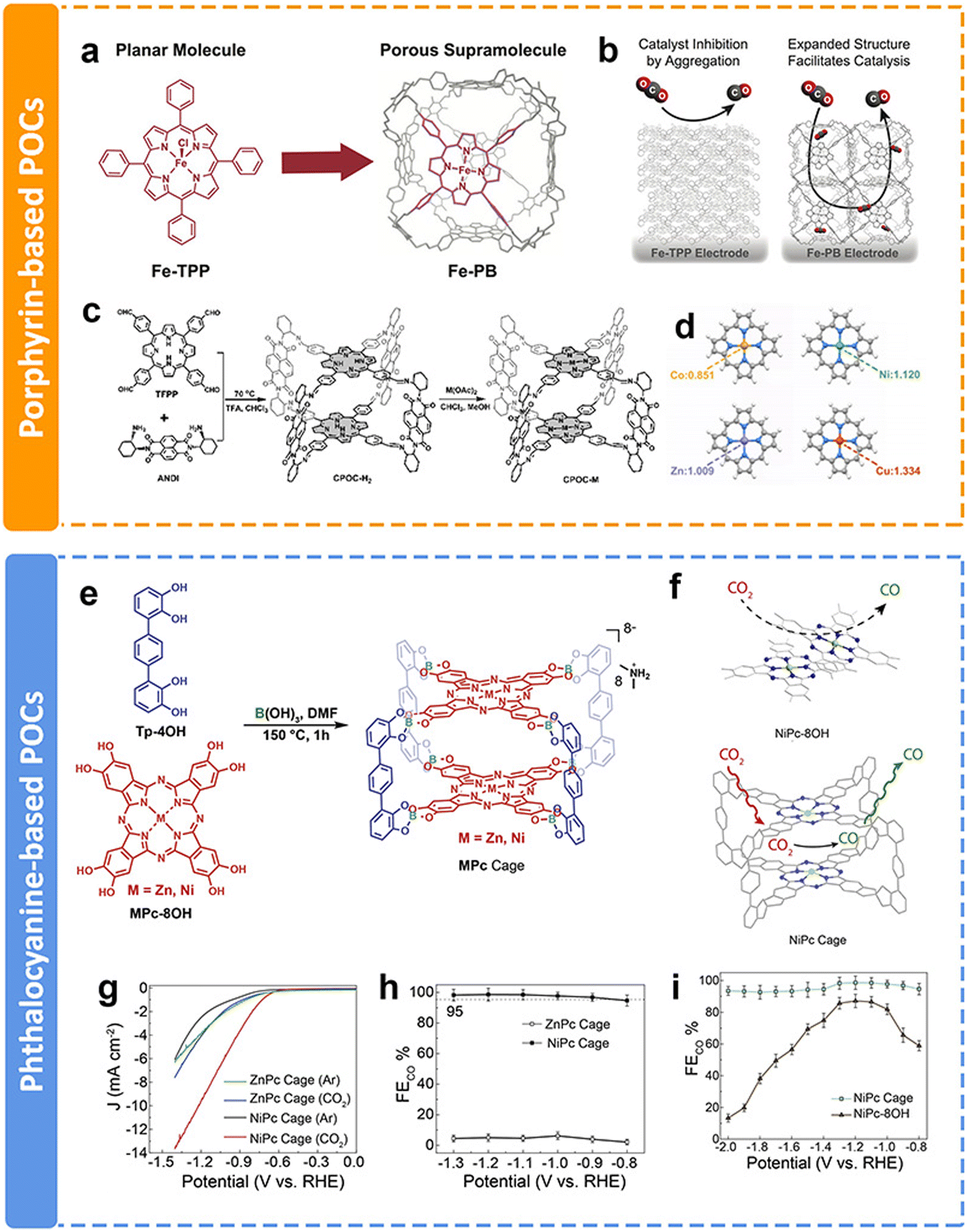 | ||
| Fig. 16 Schematic illustration of different POCs and their performance. (a) Supramolecular structure of Fe-PB composed of six Fe-TPP monomer building blocks, (b) proposed model by which the permanently porous supramolecular Fe-PB facilitates the diffusion of the substrate and the electrolyte around the catalytic iron centers;152 (c) schematic synthesis of CPOC-M (M = H2, Co(II), Ni(II), Cu(II), and Zn(II)), (d) NPA charge of Co, Ni, Cu and Zn atoms in the porphyrin without the introduction of CO2,153 (e) the synthesis of ZnPc and NiPc cages, (f) partially blocked and readily accessible CO2 reduction catalytic center in the aggregated NiPc-8OH monomer (left) and the NiPc cage (right), (g) LSV curve of ZnPc and NiPc cages in Ar- and CO2-saturated 0.1 M KHCO3 solutions, respectively, (h) CO faradaic efficiency of ZnPc and NiPc cages, and (i) CO faradaic efficiency of the NiPc cage and NiPc-8OH.154 | ||
In addition to porphyrin-based POCs, phthalocyanine-based POCs for eCO2RR have been developed recently in our group.154 Two kinds of well-defined phthalocyanine-based organic cages chelated with Ni and Zn ions were obtained via one-step dynamic spiroborate formation in high yields for the first time (Fig. 16e). The site-isolated redox-active metal centers, readily accessible intrinsic cavity, and the shape persistency of the cage structure precludes the aggregation of adjacent NiPc panels, which maintains the number of electrochemically accessible and active Pc centers (Fig. 16f). The Ni-metalated Pc (NiPc) cage exhibits high catalytic efficiency, selectivity, and stability toward the eCO2RR. LSV results show that the onset potential of the NiPc cage is −0.60 V, which is better than that of the ZnPc cage (−0.74 V) (Fig. 16g). It almost exclusively generates CO with a selectivity of 98.7% at −1.2 V (Fig. 16h). The selectivity is maintained at over 95% in long-term durability tests, which demonstrates the advantages of integrating electrocatalytically active NiPc moieties into a molecular cage structure (Fig. 16i). Additionally, due to the structural superiority of POCs, they could also be applied as additives to modify the electrocatalytic eCO2RR of other catalysts, allowing the facile contact of CO2 molecules with the active sites. This research will not be discussed in this review.
POCs exhibit the most well-defined catalyst structure but low intrinsic conductivity. It is assumed that well-defined POCs could have their active sites fully exposed during electrocatalysis. However, electrons are difficult to transfer directly between different POC molecules. Typically, incorporating POCs with conductive materials are needed. How to ensure their uniform and non-aggregated distribution on conductive materials is still challenging, which may be one of the key problems in developing efficient POC-based electrocatalysts for eCO2RR.
Typical CMPs and POCs as electrocatalysts in the eCO2RR are summarized in Table 2.
| Main product | Material | Metal center | Chelating sites | Design principle | Highest FE | TOF | Reaction conditions | Ref. |
|---|---|---|---|---|---|---|---|---|
| CO | COP-SA | Co | Phthalocyanine | Electronic modulation | 96.50% | 46.3 s−1 | Home-made three-electrode electrochemical cell and 0.5 M KHCO3 | 150 |
| COP-BDA | 84.4% | 1.5 s−1 | ||||||
| CO | NiPcP | Ni | Phthalocyanine | Electronic modulation | ∼100% | 23148 h−1 |
Three-compartment flow cell and 1 M KOH | 145 |
| CO | NiPPc/CNT | Ni | Phthalocyanine | Electronic modulation | 99.80% | N/A | Flow cell and 1 M KHCO3 | 146 |
| CO | CoPPc/CNT | Co | Phthalocyanine | Electronic modulation | 90% | 4900 h−1 | H type and 0.5 M NaHCO3 | 155 |
| CO | CoCoPCP/CNTs | Co | Porphyrin | Electronic modulation and heterometallic centers | 94% | 2.4 s−1 | H type cell and 0.5 M KHCO3 | 149 |
| CoH2PCP/CNTs | 85% | N/A | ||||||
| H2CoPCP/CNTs | 65% | |||||||
| CO | CNT@CMP(CoPc-H2Pc) | Co | Phthalocyanine | Electronic modulation and heterometallic centers | 97% | 97592 h−1 |
Flow cell and 0.5 M KHCO3 | 148 |
| CNT@CMP(CoPc) | 82% | N/A | ||||||
| CNT@CMP(H2Pc) | Metal free | 5% | ||||||
| CO | CPF-Co@GO | Co | Phthalocyanine | Electronic modulation | 79.1% | 1.07 s−1 | Type cell and 0.5 M KHCO3 | 147 |
| CPF-Co@LGO | 97.6% | 2.81 s−1 | ||||||
| CPF-Co@rGO | 70.4% | 2.27 s−1 | ||||||
| CH4 | Fc-CPP-Cu | Cu and Fe | Porphyrin and cyclopentadienyl | Electronic modulation and heterometallic centers | 75.90% | 0.013 s−1 | Flow cell and 1 M KOH | 151 |
| Bz-CPP-Cu | Cu | Porphyrin | 62.70% | N/A | ||||
| Bp-CPP-Cu | 45.80% | |||||||
| Tp-CPP-Cu | 44.30% | |||||||
| CO | Fe-PB | Fe | Porphyrin | Electronic modulation and microstructure | 100 ± 2% | 1.74 s−1 | Custom-made three-piece glass electrochemical cell and 0.5 M KHCO3 | 152 |
| Fe-TPP | 96 ± 3% | 0.94 s−1 | ||||||
| CO | CPOC-Co | Co | Porphyrin | Metal species and microstructure | 92% | 0.98 s−1 | H-type cell and 0.5 M KHCO3 | 153 |
| CO | NiPc cage | Ni | Phthalocyanine | Metal species and microstructure | 98.7% | 606 h−1 | Standard two-compartment electrochemical cell and 0.1 M KHCO3 | 154 |
| ZnPc cage | Zn | <10% | N/A |
4. Conclusion and perspectives
Covalent porous catalysts (CPCs) are considered promising catalysts for eCO2RR due to their well-defined structures, high specific surface areas and high atomic utilization. Moreover, their well-defined chemical structures at the molecular level provide excellent platforms for studying the catalytic mechanism for eCO2RR, which could guide the rational design of high-performance eCO2RR catalysts. In this review, we summarized the design principle and specific application in the eCO2RR of three typical covalent porous catalysts (COFs, CMPs and organic cages). We focus on the CPCs’ intrinsic activity enhancing methods including the design of metal centers, chelating groups, organic backbones, and side chain groups. It could be concluded that the key factors through these modulations lie in the electronic properties of the active metallic sites embedded in the porous structure and the porosity of the organic backbone. The electronic properties of the active metallic sites mainly determine the activity and selectivity, while the porosity of the organic backbone mainly determines the efficiency of CPCs. Moreover, compounded CPCs with carbon additives, such as CNTs and graphene, facilitate the electron transfer during the catalytic process. With the development of synthetic methods and suitable functional building blocks, a number of CPCs have shown remarkable eCO2RR performance in previous research. Some challenges still stand, which need further exploration.(1) Accurate relationship between CPC structures and eCO 2 RR performance. Due to the two-dimensional stacking structures of 2D COFs and CMPs, the active sites in the adjacent layers may be interfered by each other, which could alter or reduce their certain performance when compared with independent single metal sites. This problem is overlooked in current works when exploring the relationship between CPC structures and eCO2RR performance. It is expected that CPCs with clearer structures, combined with more advanced characterization methods and theoretical calculation, will help gain a better understanding. Moreover, multiscale theoretical simulations, such as finite element simulation, quantum mechanics, and machine learning, are needed to better understand the influences of the porosity of the catalyst and mass transfer on the reaction process.
(2) The large-scale practical application of CPCs for eCO 2 RR. The current density catalysed by CPCs during the eCO2RR still falls far short of meeting practical requirements due to the slow reaction kinetics and lower intrinsic electrical conductivity of CPCs. Due to the organic nature of CPC backbones, the conductivity of CPCs is relatively low, when compared with metal-based electrocatalysts. Low conductivity may cause high resistance and low efficiency during the eCO2RR. Moreover, due to the coordinated metal leaching and conjugated structure destruction, the stability of CPCs in the long-term operation also represents a challenge. Designing advanced reactors or combining CPCs with conductive substrates can significantly improve the catalytic efficiency of CPCs for eCO2RR. Enhancing the metal coordination strength and improving the robustness of the linkages between building blocks (such as sp2 carbon conjugated and cyclized linkages) are highly needed, which may improve the stability of CPCs for eCO2RR. Additionally, to realize large-scale synthesis, the high cost of monomers used for the construction of CPCs becomes another obstacle in their practical application. Therefore, an optimized and low-cost industrial supply of various monomers is also urgently needed in the future.
(3) The deep reduction pathway of the eCO 2 RR on CPCs with different metal centers. Although the deep reduction of CO2 to HCHO, CH3OH and CH4 has been reported on single atom catalysts besides Cu species, it is still difficult to achieve it on CPCs. Only several products, such as CO and HCOOH (2e− transfer products) and CH4 (8e− transfer products), have been reported on CPCs. The important semi-hydrogenated products, such as HCHO and CH3OH, have not been obtained by eCO2RR on CPCs at present, which is worth paying attention. The reduction degree is determined by the binding strength of the key intermediates on the active sites. We suspect that rational modulation of the charge density on metal centers may optimize the adsorption of key intermediates for deep reduction, even facilitating C–C coupling to obtain C2+ products.
(4) Real active sites on CPCs for eCO 2 RR. In the catalytic process of the eCO2RR, it has been observed that some metal species can transform into metal clusters, which serve as the actual active sites for the reaction. However, many studies have previously presumed that the coordinated metal centers are the active sites, leading to a divergence between the experimental results and analysis. To bridge this gap, it is essential to employ advanced analytical techniques that can monitor the evolution of metal species (including metal bonds, coordination bonds and valence states). By doing so, a more accurate understanding of the genuine active sites responsible for eCO2RR catalysis by CPCs can be achieved.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the National Natural Science Foundation of China (NSFC) (Grants No. 21905115, 52173201), the China Postdoctoral Science Foundation (Grants No. 2020M671334), the MOE & SAFEA, 111 Project (B13025), the National First-Class Discipline Program of Light Industry Technology and Engineering (LITE2018-19), the Fundamental Research Funds for the Central Universities (JUSRP11929), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX23_2556). W. Z. thanks the University of Colorado Boulder for financial support.References
- CenCO2PIP, Science, 2023, 382, eadi5177 CrossRef PubMed.
- J. M. Chen, Innovation, 2021, 2, 100127 CAS.
- L. Chen, G. Msigwa, M. Yang, A. I. Osman, S. Fawzy, D. W. Rooney and P. S. Yap, Environ. Chem. Lett., 2022, 20, 2277–2310 CrossRef CAS PubMed.
- S. A. Montzka, E. J. Dlugokencky and J. H. Butler, Nature, 2011, 476, 43–50 CrossRef CAS PubMed.
- M. R. Shaw, E. S. Zavaleta, N. R. Chiariello, E. E. Cleland, H. A. Mooney and C. B. Field, Science, 2002, 298, 1987–1990 CrossRef CAS PubMed.
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- L. Li, X. Li, Y. Sun and Y. Xie, Chem. Soc. Rev., 2022, 51, 1234–1252 RSC.
- K. S. Song, P. W. Fritz and A. Coskun, Chem. Soc. Rev., 2022, 51, 9831–9852 RSC.
- X. Jiang, X. Nie, X. Guo, C. Song and J. G. Chen, Chem. Rev., 2020, 120, 7984–8034 CrossRef CAS PubMed.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. M. Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- F. Julia-Hernandez, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef CAS PubMed.
- A. Banerjee, G. R. Dick, T. Yoshino and M. W. Kanan, Nature, 2016, 531, 215–219 CrossRef CAS PubMed.
- K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- T. Kong, Y. Jiang and Y. Xiong, Chem. Soc. Rev., 2020, 49, 6579–6591 RSC.
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC.
- S. Navarro-Jaen, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef CAS PubMed.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- B. A. Rosen, A. Salehi-Khojin, M. R. Thorson, W. Zhu, D. T. Whipple, P. J. A. Kenis and R. I. Masel, Science, 2011, 334, 643–644 CrossRef CAS PubMed.
- Y. Chen, X. Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B. H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2024, 19, 311–318 CrossRef CAS PubMed.
- P. P. Yang and M. R. Gao, Chem. Soc. Rev., 2023, 52, 4343–4380 RSC.
- S. Gleizer, R. Ben-Nissan, Y. M. Bar-On, N. Antonovsky, E. Noor, Y. Zohar, G. Jona, E. Krieger, M. Shamshoum, A. Bar-Even and R. Milo, Cell, 2019, 179, 1255–1263 CrossRef CAS PubMed.
- E. W. Yemm and A. J. Willis, Nature, 1954, 173, 726–726 CrossRef CAS PubMed.
- T. E. Miller, T. Beneyton, T. Schwander, C. Diehl, M. Girault, R. McLean, T. Chotel, P. Claus, N. S. Cortina, J.-C. Baret and T. J. Erb, Science, 2020, 368, 649–654 CrossRef CAS PubMed.
- A. Burlacot, O. Dao, P. Auroy, S. Cuine, Y. Li-Beisson and G. Peltier, Nature, 2022, 605, 366–371 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- L. Zhang, Z. J. Zhao, T. Wang and J. Gong, Chem. Soc. Rev., 2018, 47, 5423–5443 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- K. Zhao and X. Quan, ACS Catal., 2021, 11, 2076–2097 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- H.-Q. Liang, S. Zhao, X.-M. Hu, M. Ceccato, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2021, 11, 958–966 CrossRef CAS.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS PubMed.
- X. Zheng, P. De Luna, F. P. García de Arquer, B. Zhang, N. Becknell, M. B. Ross, Y. Li, M. N. Banis, Y. Li, M. Liu, O. Voznyy, C. T. Dinh, T. Zhuang, P. Stadler, Y. Cui, X. Du, P. Yang and E. H. Sargent, Joule, 2017, 1, 794–805 CrossRef CAS.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- C. Choi, X. Wang, S. Kwon, J. L. Hart, C. L. Rooney, N. J. Harmon, Q. P. Sam, J. J. Cha, W. A. Goddard III, M. Elimelech and H. Wang, Nat. Nanotechnol., 2023, 18, 160–167 CrossRef CAS PubMed.
- L. Xu, J. Feng, L. Wu, X. Song, X. Tan, L. Zhang, X. Ma, S. Jia, J. Du, A. Chen, X. Sun and B. Han, Green Chem., 2023, 25, 1326–1331 RSC.
- H. Chen, C. Ding, C. Kang, J. Zeng, Y. Li, Y. Li, Y. Li, C. Li and J. He, Green Chem., 2023, 25, 5320–5337 RSC.
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, J. Am. Chem. Soc., 2020, 142, 4154–4161 CrossRef CAS PubMed.
- P. Yang, Z. J. Zhao, X. Chang, R. Mu, S. Zha, G. Zhang and J. Gong, Angew. Chem., Int. Ed., 2018, 57, 7724–7728 CrossRef CAS PubMed.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Moller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef PubMed.
- J. Hao, Z. Zhuang, J. Hao, K. Cao, Y. Hu, W. Wu, S. Lu, C. Wang, N. Zhang, D. Wang, M. Du and H. Zhu, ACS Nano, 2022, 16, 3251–3263 CrossRef CAS PubMed.
- Y. Jiang, J. Shan, P. Wang, L. Huang, Y. Zheng and S.-Z. Qiao, ACS Catal., 2023, 13, 3101–3108 CrossRef CAS.
- J. Wang, S. Kattel, C. J. Hawxhurst, J. H. Lee, B. M. Tackett, K. Chang, N. Rui, C. J. Liu and J. G. Chen, Angew. Chem., Int. Ed., 2019, 58, 6271–6275 CrossRef CAS PubMed.
- X. Yang, J. H. Lee, S. Kattel, B. Xu and J. G. Chen, Nano Lett., 2022, 22, 4576–4582 CrossRef CAS PubMed.
- L. Liu, F. Wang, X. Chu, L. Zhang, S. Zhang, X. Wang, G. Che, S. Song and H. Zhang, Adv. Energy Mater., 2024, 14, 2301575 CrossRef CAS.
- R. Zhao, Y. Wang, G. Ji, J. Zhong, F. Zhang, M. Chen, S. Tong, P. Wang, Z. Wu, B. Han and Z. Liu, Adv. Mater., 2023, 35, 2205262 CrossRef CAS PubMed.
- O. Christensen, S. Zhao, Z. Sun, A. Bagger, J. V. Lauritsen, S. U. Pedersen, K. Daasbjerg and J. Rossmeisl, ACS Catal., 2022, 12, 15737–15749 CrossRef CAS.
- Y. Zhang, P. Li, C. Zhao, G. Zhou, F. Zhou, Q. Zhang, C. Su and Y. Wu, Sci. Bull., 2022, 67, 1679–1687 CrossRef CAS PubMed.
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y. H. Chou, X. Lu, P. V. Kumar, Y. H. Ng, D. Chu, C. Y. Toe, Z. Han and R. Amal, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef CAS PubMed.
- Y. Hou, Y.-B. Huang, Y.-L. Liang, G.-L. Chai, J.-D. Yi, T. Zhang, K.-T. Zang, J. Luo, R. Xu, H. Lin, S.-Y. Zhang, H.-M. Wang and R. Cao, CCS Chem., 2019, 1, 384–395 CrossRef CAS.
- L. Lin, Q. Zhang, Y. Ni, L. Shang, X. Zhang, Z. Yan, Q. Zhao and J. Chen, Chem, 2022, 8, 1822–1854 CAS.
- K. Wang, D. Qi, Y. Li, T. Wang, H. Liu and J. Jiang, Coord. Chem. Rev., 2019, 378, 188–206 CrossRef CAS.
- C. Wang, Z. Lv, W. Yang, X. Feng and B. Wang, Chem. Soc. Rev., 2023, 52, 1382–1427 RSC.
- J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS PubMed.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dinca, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lachelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- Y. Yusran, Q. Fang and V. Valtchev, Adv. Mater., 2020, 32, 2002038 CrossRef CAS PubMed.
- Q. Guan, L.-L. Zhou and Y.-B. Dong, Chem. Soc. Rev., 2022, 51, 6307–6416 RSC.
- X. Li, P. Yadav and K. P. Loh, Chem. Soc. Rev., 2020, 49, 4835–4866 RSC.
- Z. Li, T. He, Y. Gong and D. Jiang, Acc. Chem. Res., 2020, 53, 1672–1685 CrossRef CAS PubMed.
- H. Ma, Y. Chen, X. Li and B. Li, Adv. Funct. Mater., 2021, 31, 2101861 CrossRef CAS.
- X. Yang, Z. Ullah, J. F. Stoddart and C. T. Yavuz, Chem. Rev., 2023, 123, 4602–4634 CrossRef CAS PubMed.
- R. D. Mukhopadhyay, Y. Kim, J. Koo and K. Kim, Acc. Chem. Res., 2018, 51, 2730–2738 CrossRef CAS PubMed.
- H. Wang, Y. Jin, N. Sun, W. Zhang and J. Jiang, Chem. Soc. Rev., 2021, 50, 8874–8886 RSC.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Angew. Chem., Int. Ed., 2020, 59, 13722–13733 CrossRef CAS PubMed.
- Z. Li, X. Zhang, H. Cheng, J. Liu, M. Shao, M. Wei, D. G. Evans, H. Zhang and X. Duan, Adv. Energy Mater., 2019, 10, 1900486 CrossRef.
- Y. Cao, W. Peng, Y. Li, F. Zhang, Y. Zhu and X. Fan, Green Energy Environ., 2023, 8, 360–382 CrossRef CAS.
- Q. Pan, Z. Lei, Y. Zhao and W. Zhang, EnergyChem, 2023, 5, 100107 CrossRef CAS.
- L. Xiao, L. Qi, J. Sun, A. Husile, S. Zhang, Z. Wang and J. Guan, Nano Energy, 2024, 120, 109155 CrossRef CAS.
- S. Suleman, Y. Zhang, Y. Qian, J. Zhang, Z. Lin, O. Metin, Z. Meng and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, 202314988 CrossRef PubMed.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. H. Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, 202309480 CrossRef PubMed.
- S. Daliran, A. R. Oveisi, Y. Peng, A. López-Magano, M. Khajeh, R. Mas-Ballesté, J. Alemán, R. Luque and H. Garcia, Chem. Soc. Rev., 2022, 51, 7810–7882 RSC.
- P. De La Torre, L. An and C. J. Chang, Adv. Mater., 2023, 35, 2302122 CrossRef CAS PubMed.
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- J. Xu, Y. Xu and X. H. Bu, Small, 2021, 17, 2102331 CrossRef CAS PubMed.
- Z. Liang, H. Y. Wang, H. Zheng, W. Zhang and R. Cao, Chem. Soc. Rev., 2021, 50, 2540–2581 RSC.
- X. Cui, S. Lei, A. C. Wang, L. Gao, Q. Zhang, Y. Yang and Z. Lin, Nano Energy, 2020, 70, 104525 CrossRef CAS.
- D. H. Yang, Y. Tao, X. Ding and B. H. Han, Chem. Soc. Rev., 2022, 51, 761–791 RSC.
- P. Xiao and Y. Xu, J. Mater. Chem. A, 2018, 6, 21676–21695 RSC.
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Adv. Energy Mater., 2022, 12, 2003990 CrossRef CAS.
- X. Cui, M. Wu, X. Liu, B. He, Y. Zhu, Y. Jiang and Y. Yang, Chem. Soc. Rev., 2024, 53, 1447–1494 RSC.
- W. Ma, X. He, W. Wang, S. Xie, Q. Zhang and Y. Wang, Chem. Soc. Rev., 2021, 50, 12897–12914 RSC.
- F.-Y. Gao, R.-C. Bao, M.-R. Gao and S.-H. Yu, J. Mater. Chem. A, 2020, 8, 15458–15478 RSC.
- X. Tan, C. Yu, Y. Ren, S. Cui, W. Li and J. Qiu, Energy Environ. Sci., 2021, 14, 765–780 RSC.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, 202207252 CrossRef PubMed.
- Z. Wang, Y. Zhou, P. Qiu, C. Xia, W. Fang, J. Jin, L. Huang, P. Deng, Y. Su, R. Crespo-Otero, X. Tian, B. You, W. Guo, D. Di Tommaso, Y. Pang, S. Ding and B. Y. Xia, Adv. Mater., 2023, 35, 2303052 CrossRef CAS PubMed.
- R. He, N. Xu, I. M. u. Hasan, L. Peng, L. Li, H. Huang and J. Qiao, EcoMat, 2023, 5, 12346 CrossRef.
- Y. Kuang, H. Rabiee, L. Ge, T. E. Rufford, Z. Yuan, J. Bell and H. Wang, Energy Environ. Sci., 2023, 6, 12596 Search PubMed.
- Y. Li, H. Wang, X. Yang, T. O'Carroll and G. Wu, Angew. Chem., Int. Ed., 2024, e202317884 CAS.
- L. Zhang, N. Jin, Y. Yang, X. Y. Miao, H. Wang, J. Luo and L. Han, Nano-Micro Lett., 2023, 15, 228 CrossRef CAS PubMed.
- K. E. R. Cruz, Y. Liu, T. L. Soucy, P. M. Zimmerman and C. C. L. McCrory, ACS Catal., 2021, 11, 13203–13216 CrossRef.
- S. M. Lee, W. S. Cheon, M. G. Lee and H. W. Jang, Small Struct., 2022, 4, 2200236 CrossRef.
- J. Wang, Y. Song, C. Chen, X. Zhao and W. Fan, ACS Catal., 2023, 13, 15794–15810 CrossRef CAS.
- H. Wang, Y. Tong and P. Chen, Nano Energy, 2023, 118, 108967 CrossRef CAS.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Adv. Mater., 2023, 35, 2209298 CrossRef CAS PubMed.
- S. Wei, W. Liu, C. Yang, P. Bai, X. Kong, W. Sun and L. Xu, Mater. Chem. Front., 2023, 7, 4723–4743 RSC.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- H. Chen, X. Liang, Y. Liu, X. Ai, T. Asefa and X. Zou, Adv. Mater., 2020, 32, 2002435 CrossRef PubMed.
- H. Li, F. Pan, C. Qin, T. Wang and K. J. Chen, Adv. Energy Mater., 2023, 13, 2301378 CrossRef CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, et al. , Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- H. Dong, M. Lu, Y. Wang, H.-L. Tang, D. Wu, X. Sun and F.-M. Zhang, Appl. Catal., B, 2022, 303, 120897 CrossRef CAS.
- G. C. D. Bandomo, S. S. Mondal, F. Franco, A. Bucci, V. Martin-Diaconescu, M. A. Ortuño, P. H. van Langevelde, A. Shafir, N. López and J. Lloret-Fillol, ACS Catal., 2021, 11, 7210–7222 CrossRef.
- P. L. Cheung, S. K. Lee and C. P. Kubiak, Chem. Mater., 2019, 31, 1908–1919 CrossRef CAS.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- Q. Wu, M. J. Mao, Q. J. Wu, J. Liang, Y. B. Huang and R. Cao, Small, 2020, 17, 2004933 CrossRef PubMed.
- X. Zhang, Y.-Z. Yuan, H.-F. Li, Q.-J. Wu, H.-J. Zhu, Y.-L. Dong, Q. Wu, Y.-B. Huang and R. Cao, Mater. Chem. Front., 2023, 7, 2661–2670 RSC.
- S. An, C. Lu, Q. Xu, C. Lian, C. Peng, J. Hu, X. Zhuang and H. Liu, ACS Energy Lett., 2021, 6, 3496–3502 CrossRef CAS.
- X. Yang, X. Li, M. Liu, S. Yang, Q. Niu, L. Zhai, Z. Jiang, Q. Xu and G. Zeng, ACS Mater. Lett., 2023, 5, 1611–1618 CrossRef CAS.
- M. Liu, S. Yang, X. Yang, C.-X. Cui, G. Liu, X. Li, J. He, G. Z. Chen, Q. Xu and G. Zeng, Nat. Commun., 2023, 14, 3800 CrossRef CAS PubMed.
- C. S. Diercks, S. Lin, N. Kornienko, E. A. Kapustin, E. M. Nichols, C. Zhu, Y. Zhao, C. J. Chang and O. M. Yaghi, J. Am. Chem. Soc., 2018, 140, 1116–1122 CrossRef CAS PubMed.
- Y. Lu, J. Zhang, W. Wei, D.-D. Ma, X.-T. Wu and Q.-L. Zhu, ACS Appl. Mater. Interfaces, 2020, 12, 37986–37992 CrossRef CAS PubMed.
- T. He, C. Yang, Y. Chen, N. Huang, S. Duan, Z. Zhang, W. Hu and D. Jiang, Adv. Mater., 2022, 34, 2205186 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- M. D. Zhang, D. H. Si, J. D. Yi, S. S. Zhao, Y. B. Huang and R. Cao, Small, 2020, 16, 2005254 CrossRef CAS PubMed.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- C. L. Yao, J. C. Li, W. Gao and Q. Jiang, Chem. – Eur. J., 2018, 24, 11051–11058 CrossRef CAS PubMed.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- Y. Yue, P. Cai, K. Xu, H. Li, H. Chen, H.-C. Zhou and N. Huang, J. Am. Chem. Soc., 2021, 143, 18052–18060 CrossRef CAS PubMed.
- J. Yuan, S. Chen, Y. Zhang, R. Li, J. Zhang and T. Peng, Adv. Mater., 2022, 34, 2203139 CrossRef CAS PubMed.
- L. Gong, Y. Gao, Y. Wang, B. Chen, B. Yu, W. Liu, B. Han, C. Lin, Y. Bian, D. Qi and J. Jiang, Catal. Sci. Technol., 2022, 12, 6566–6571 RSC.
- S.-Y. Chi, Q. Chen, S.-S. Zhao, D.-H. Si, Q.-J. Wu, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2022, 10, 4653–4659 RSC.
- B. Han, Y. Jin, B. Chen, W. Zhou, B. Yu, C. Wei, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2022, 61, 202114244 CrossRef PubMed.
- B. Han, Y. Jin, B. Chen, W. Zhou, B. Yu, C. Wei, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, Angew. Chem., Int. Ed., 2021, 61, 202114244 CrossRef PubMed.
- Y. R. Wang, H. M. Ding, X. Y. Ma, M. Liu, Y. L. Yang, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, 202114648 CrossRef PubMed.
- S. Ali, R. Iqbal, F. Wahid, P. M. Ismail, A. Saleem, S. Ali, F. Raziq, S. Ullah, I. Ullah, Tahir, M. Zahoor, X. Wu, H. Xiao, X. Zu and L. Qiao, Fuel Process. Technol., 2022, 237, 107451 CrossRef CAS.
- M. Liu, Y. R. Wang, H. M. Ding, M. Lu, G. K. Gao, L. Z. Dong, Q. Li, Y. Chen, S. L. Li and Y. Q. Lan, Sci. Bull., 2021, 66, 1659–1668 CrossRef CAS PubMed.
- Y.-L. Yang, Y.-R. Wang, G.-K. Gao, M. Liu, C. Miao, L.-Y. Li, W. Cheng, Z.-Y. Zhao, Y. Chen, Z. Xin, S.-L. Li, D.-S. Li and Y.-Q. Lan, Chin. Chem. Lett., 2022, 33, 1439–1444 CrossRef CAS.
- X. F. Qiu, J. R. Huang, C. Yu, Z. H. Zhao, H. L. Zhu, Z. Ke, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2022, 61, 202206470 CrossRef PubMed.
- L. Gong, B. Chen, Y. Gao, B. Yu, Y. Wang, B. Han, C. Lin, Y. Bian, D. Qi and J. Jiang, Inorg. Chem. Front., 2022, 9, 3217–3223 RSC.
- M. Lu, M. Zhang, C. G. Liu, J. Liu, L. J. Shang, M. Wang, J. N. Chang, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2021, 60, 4864–4871 CrossRef CAS PubMed.
- M. Liu, X. Zhao, S. Yang, X. Yang, X. Li, J. He, G. Z. Chen, Q. Xu and G. Zeng, ACS Appl. Mater. Interfaces, 2023, 15, 44384–44393 CrossRef CAS PubMed.
- K. S. Song, P. W. Fritz, D. F. Abbott, L. N. Poon, C. M. Caridade, F. Gandara, V. Mougel and A. Coskun, Angew. Chem., Int. Ed., 2023, 62, 202309775 CrossRef PubMed.
- M. D. Zhang, J. R. Huang, W. Shi, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2023, 62, 202308195 CrossRef PubMed.
- Q. J. Wu, D. H. Si, Q. Wu, Y. L. Dong, R. Cao and Y. B. Huang, Angew. Chem., Int. Ed., 2023, 62, 202215687 CrossRef PubMed.
- Q. J. Wu, D. H. Si, S. Ye, Y. L. Dong, R. Cao and Y. B. Huang, J. Am. Chem. Soc., 2023, 145, 19856–19865 CrossRef CAS PubMed.
- Y. Zhang, X. Zhang, L. Jiao, Z. Meng and H. L. Jiang, J. Am. Chem. Soc., 2023, 145, 24230–24239 CrossRef CAS PubMed.
- C. L. Smith, R. Clowes, R. S. Sprick, A. I. Cooper and A. J. Cowan, Sustainable Energy Fuels, 2019, 3, 2990–2994 RSC.
- S. Wei, H. Zou, W. Rong, F. Zhang, Y. Ji and L. Duan, Appl. Catal., B, 2021, 284, 119739 CrossRef CAS.
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu, E. Cortés and M. Liu, Appl. Catal., B, 2022, 306, 121093 CrossRef CAS.
- Z. Wang, Y. Han, B. Li, P. Peng and S. Q. Zang, Small, 2023, 19, 2301797 CrossRef CAS PubMed.
- R. Wang, X. Wang, W. Weng, Y. Yao, P. Kidkhunthod, C. Wang, Y. Hou and J. Guo, Angew. Chem., Int. Ed., 2022, 61, 202115503 CrossRef PubMed.
- T. Wang, L. Xu, Z. Chen, L. Guo, Y. Zhang, R. Li and T. Peng, Appl. Catal., B, 2021, 291, 120128 CrossRef CAS.
- Y. Song, J.-J. Zhang, Z. Zhu, X. Chen, L. Huang, J. Su, Z. Xu, T. H. Ly, C.-S. Lee, B. I. Yakobson, B. Z. Tang and R. Ye, Appl. Catal., B, 2021, 284, 119750 CrossRef CAS.
- Qi. Li, Z.-M. Wang, Y. Chen, Y.-R. Wang, C. Guo, Q. Huang, L.-Z. Dong, S.-L. Li and Y.-Q. Lan, J. Mater. Chem. A, 2022, 10, 25356–25362 RSC.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- X. Liu, C. Liu, X. Song, X. Ding, H. Wang, B. Yu, H. Liu, B. Han, X. Li and J. Jiang, Chem. Sci., 2023, 14, 9086–9094 RSC.
- Y. Hu, S. Huang, L. J. Wayment, J. Wu, Q. Xu, T. Chang, Y.-P. Chen, X. Li, B. Andi, H. Chen, Y. Jin, H. Zhu, M. Du, S. Lu and W. Zhang, Cell Rep. Phys. Sci., 2023, 4, 101285 CrossRef CAS.
- N. Han, Y. Wang, L. Ma, J. Wen, J. Li, H. Zheng, K. Nie, X. Wang, F. Zhao, Y. Li, J. Fan, J. Zhong, T. Wu, D. J. Miller, J. Lu, S.-T. Lee and Y. Li, Chem, 2017, 3, 652–664 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |