
A review on sustainable iron oxide nanoparticles: syntheses and applications in organic catalysis and environmental remediation
Dinesh S.
Chaudhari†
 a,
Rohit P.
Upadhyay†
a,
Gajanan Y.
Shinde
a,
Manoj B.
Gawande
*ac,
Jan
Filip
b,
Rajender S.
Varma
*b and
Radek
Zbořil
*bc
a,
Rohit P.
Upadhyay†
a,
Gajanan Y.
Shinde
a,
Manoj B.
Gawande
*ac,
Jan
Filip
b,
Rajender S.
Varma
*b and
Radek
Zbořil
*bc
aDepartment of Industrial and Engineering Chemistry, Institute of Chemical Technology, Mumbai-Marathwada Campus, Jalna 431213, Maharashtra, India. E-mail: mb.gawande@marj.ictmumbai.edu.in
bRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute, (CATRIN), Palacký University Olomouc, Šlechtitelů 27, 779 00 Olomouc, Czech Republic. E-mail: rajvarma@hotmail.com
cCentre for Energy and Environmental Technologies (CEET), Nanotechnology Centre, VŠB–Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic. E-mail: radek.zboril@upol.cz
First published on 22nd May 2024
Abstract
Iron oxide nanoparticles have been intensively investigated owing to their huge potential as diagnostic, therapeutic, and drug-carrier agents in biomedicine, sorbents in environmental technologies, sensors of various inorganic and organic/biological substances, energy-generating and storing materials, and in assorted biotechnological and industrial processes involving microbiology, pigment industry, recording and magnetic media or (bio)catalysis. An eminent interest in exploring the realm of iron oxides is driven by their chemical and structural diversity, high abundance, low cost, non-toxicity, and broad portfolio of chemical procedures enabling their syntheses with desirable physicochemical features. The current review article centers its attention on the contemporary advancements in the field of catalysis and environmental technologies employing iron oxides in various chemical forms (e.g., hematite, magnetite, maghemite), sizes (∼10–100 nm), morphology characteristics (e.g., globular, needle-like), and nano architecture (e.g., nanoparticles, nanocomposites, core–shell structures). In particular, the catalytic applications of iron oxides and their hybrids are emphasized regarding their efficiency and selectivity in the coupling, oxidation, reduction, alkylation reactions, and Fischer–Tropsch synthesis. The deployment of iron oxides and their nanocomposites in environmental and water treatment technologies is also deliberated with their roles as nanosorbents for heavy metals and organic pollutants, photocatalysts, and heterogeneous catalysts (e.g., hydrogen peroxide decomposition) for oxidative treatment of various contaminants. The associated challenges and potential progress in iron-oxide-based catalytic and environmental technologies are highlighted as well. Young chemists, researchers, and scientists could find this review useful in enhancing the usefulness of nano iron oxides in their investigations and developing sustainable methodologies.
 Dinesh S. Chaudhari | Dinesh S. Chaudhari was born in 1995 in the state of Maharashtra, India. He obtained his Bachelor's degree (Chemistry) in 2016 and Master's degree (Medicinal Chemistry) in 2018 from Savitribai Phule Pune University, Pune. Afterward, he worked in the pharmaceutical industry as a Research Associate at Aurigene Discovery Technology Limited, Bangalore, and Aragen Life Sciences Pvt. Ltd, Hyderabad. He also worked as a Project Fellow on the DST Funded Project at CSIR-National Chemical Laboratory, Pune. He qualified MH-SET and IIT-GATE exams in chemical science subject. In 2022, he joined Prof. Manoj Gawande's group at the Institute of Chemical Technology, Mumbai-Marathwada Campus, Jalna, India. Presently, he is pursuing his Ph.D. in synthesizing carbon nitride-based single-atom photocatalysts and their application in various organic transformations. |
 Rohit P. Upadhyay | Rohit P. Upadhyay completed his M.Sc. in Organic Chemistry in 2018 from Ramnarain Ruia college affiliated to Mumbai university. He began his career as a research trainee in the R&D department of Colgate-Palmolive, where he worked for one year (2018–2019) in the formulation department in oral care. Subsequently, he joined Jyothy Labs, where he gained five months of FMCG industry experience in the R&D department. From 2021 to 2022, he worked at the Institute of Chemical Technology, Mumbai campus in the Chemical Engineering Department under Prof. Vilas G. Gaikar on a project funded by Bharat Petroleum Corporation Ltd. Currently, he is a Ph.D. candidate at ICT Mumbai, Marathwada Campus, Jalna under the supervision of Prof. Manoj B. Gawande. His research focuses on developing single-atom catalysts (SACs) supported on carbon-based material for organic transformations, with a particular interest in hydrogenation reactions and conversion of biomass precursors to value-added products. |
 Manoj B. Gawande | Manoj B. Gawande (FRSC-UK, FICS, FMASc) received his Ph.D. in 2008 from the Institute of Chemical Technology, Mumbai, India, and then undertook several research stints in Germany, South Korea, Portugal, Czech Republic, United States, and United Kingdom. He has worked as a Visiting Professor at CBC-SPMS, Nanyang Technological University, Singapore, in 2013. Presently, he is an Associate Professor at the Institute of Chemical Technology, Mumbai-Marathwada Campus, Jalna, India. He also holds a Visiting Professor position at Centre for Energy and Environmental Technologies (CEET), Nanotechnology Centre, VŠB–Technical University of Ostrava, Czech Republic. His research interests focus on single-atom catalysts, advanced nanomaterials, and sustainable technologies, as well as cutting-edge catalysis and energy applications. He is an Editor and an Editorial Board Member of several reputed journals including Guest Editor of Small, Advanced Materials Interfaces and ACS Sustainable Chemistry and Engineering. He has published over 170 research and review articles (citations ∼16 |
 Jan Filip | Dr Jan Filip received his PhD degrees in field of Mineralogy from Masaryk University in Brno, Czech Republic with dissertation focused on the crystal chemistry of borosilicate minerals and OH defects in nominally-anhydrous minerals. Currently, he leads the group “Environmental nanotechnologies” at the Regional Centre of Advanced Technologies and Materials (as a division of Czech Advanced Technology and Research Institute – CATRIN), Palacký University Olomouc, Czech Republic. His research interests cover development, optimization, and application of zero-valent iron nanoparticles; ferrates; metal oxide nanoparticles and biochar in technologies for water treatment, identification of nanoparticles in the environment, application of X-ray based techniques (X-ray diffraction, X-ray fluorescence, X-ray photoelectron spectroscopy, and related techniques), and Mössbauer spectroscopy in materials science. He actively participates in various national and international collaborative projects and he is the author and co-author of four book chapters, more than 150 papers in refereed journals that have received about 4500 citations, and several other applied results including verified technologies, utility models, and prototypes. He is a named inventor of five patents. His H-index has reached 36. |
 Rajender S. Varma | Prof. Rajender Varma (H-Index 137, Highly Cited Res. 2016, 18, 19, 20, 21, 22) born in India (Ph.D., Delhi University 1976) has been a senior scientist at U.S. EPA since 1999. He has over 50 years of multidisciplinary research experience ranging from eco-friendly synthetic methods using microwaves, ultrasound, etc. to greener assembly of nanomaterials and sustainable appliances of magnetically retrievable nanocatalysts in benign media. He is a member of the editorial advisory board of several international journals, published over 985 papers, and awarded 17 U.S. Patents, 11 books, 31 book chapters, and 3 encyclopedia contributions with 77 |
 Radek Zbořil | Radek Zboril received his PhD degree at Palacky’ University in Olomouc, Czech Republic. After his doctoral studies, he spent some time at universities around the world in locations such as Tokyo, Delaware, and Johannesburg. Currently, he is a Professor of Physical Chemistry and Scientific Director of the RCPTM Centre at Czech Advanced Technology and Research Institute of Palacky’ University in Olomouc, Czech Republic, and the head of Materials-Envi Lab at Centre for Energy and Environmental Technologies at VŠB-Technical University in Ostrava. His research is focused on nanotechnologies, materials science, and applications of nanomaterials in energy, catalysis, biomedicine, and environmental technologies. He is a Highly Cited Researcher (according to Clarivate Analytics), the Materials Science in Czech Republic Leader (Research.com) and a member of the ERC Advanced Grants Evaluation Panel. |
1. Introduction
For several decades, iron oxides have been intensively examined and manufactured for a broad scale of applications. An eminent interest in exploring the realm of iron oxides has been nourished by comprehension of their remarkable physical behaviors and their inherent physicochemical properties, most of which have been found appealing for various applications. Until now, iron oxides have been successfully applied in many branches of human activities comprising first media to store information, tools for diagnostic,1–3 therapeutic,4,5 and drug-delivery purposes in medicine,6–9 agents enhancing photocatalytic/catalytic performances in diverse chemical processes, materials improving efficiency of energy-generating cells (e.g., cells for the direct solar splitting of water and hydrogen generation),10 key components in spintronic/electronic devices and energy-storing units (e.g., Li-ion batteries),11,12 sorbents removing toxic inorganic and organic/biological pollutants from the environment,13,14 among many others.15–17 Such a broad portfolio of iron oxide applications is attributed to their high abundance, low cost, and diversity of chemical procedures to synthesize them with desirable physicochemical features. Moreover, once synthesized in the nano-dimension form, they are endowed with unique properties driven by finite-size (i.e., quantum, confinement-related) effects and surface effects. Iron oxides also exhibit favorable biochemical properties such as non-toxicity, biodegradability, and biocompatibility18 placing them among the most appropriate (nano)materials in various biomedical19,20 and biotechnological areas especially in view of their encounter with living organisms. The examples include contrast agents in NMR imaging, carriers for drugs and biomolecules, tools providing immobilization of proteins and cell separation, and therapeutic agents in magnetically-assisted hyperthermia, among others. Furthermore, in the context of sustainability and environmental consciousness, the utilization of abundant iron as a key element in various applications holds significant value. According to the ACS Green Chemistry Institute's Periodic Table of Endangered Transition Elements, iron stands out as the only ‘white space’ among the transition metals of consequence, thus aligning with the principles of green chemistry and sustainable practices (Fig. 1a). The unique position of iron in the periodic table not only reinforces its virtues but also underscores its importance in promoting environmentally friendly solutions. Fig. 1b indicates constantly high interest of scientific community in research of “Iron Oxide, nanoparticles” with nearly 3000 publications per year.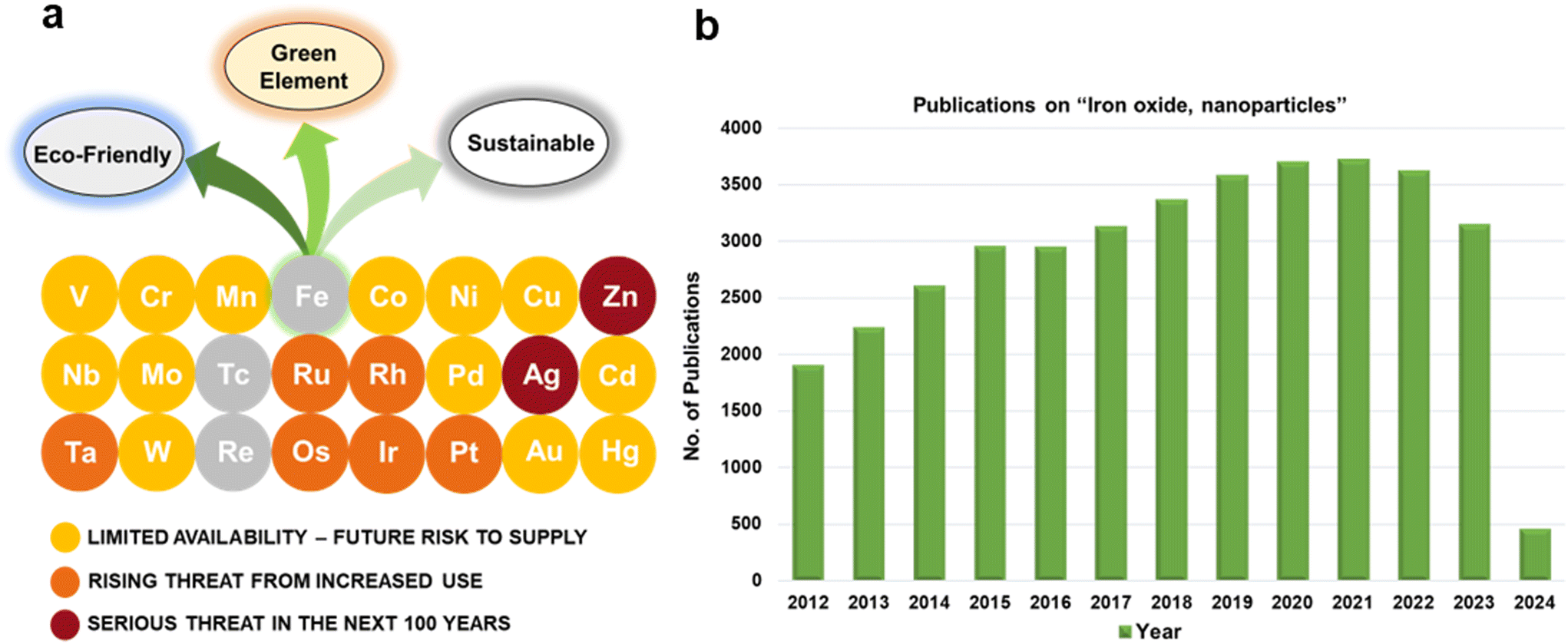 | ||
| Fig. 1 (a) Periodic table of endangered transition elements (source: ACS Green Chemistry Institute, Endangered Elements). (b) The number of publications for the keyword search “iron oxide, nanoparticles” as uncovered in Web of Science (dated: March 27, 2024). | ||
Till date, seventeen crystalline iron oxide phases have been found and described in detail. They are commonly categorized into two important classes, i.e., hydrated and non-hydrated iron oxide phases. Hydrated iron oxides of mineral nature contain water in their crystal structure, and comprise goethite (α-FeOOH), akaganeite (β-FeOOH), lepidocrocite (γ-FeOOH), feroxyhyte (δ-FeOOH), ferrihydrite (5Fe2O3·9H2O), high-pressure FeOOH, schwertmannite (Fe8O8(OH)6(SO4)n·nH2O), and green rust/green-rust-like compounds.21 Formally, they were frequently used as precursors for the synthesis of non-hydrated iron oxide forms as upon thermal treatment, they readily undergo phase transformations with molecular water leaving their crystal structure. In the realm of non-hydrated iron oxides, three classes have been acknowledged with respect to the degree of oxidation of Fe identified in their crystalline arrangement:22–24 (i) FeO (wüstite) with only Fe2+ ions; (ii) Fe2O3 (ferric oxide) with only Fe3+ ions, and (iii) Fe3O4 (magnetite) containing both Fe2+ and Fe3+ ions. While FeO and Fe3O4 each exhibit only one crystal pattern, iron(III) oxide demonstrates extensive polymorphism, i.e., indicating the presence of multiple crystal phases that possess identical chemical composition, however having distinct physical characteristics revealing their various crystal structures. Other than amorphous phase, five distinct polymorphs of ferric oxide, stable under ambient conditions, have been described so far: (i) α-Fe2O3 (mineralogically known as hematite); (ii) β-Fe2O3; (iii) γ-Fe2O3 (mineralogically known as maghemite); (iv) ε-Fe2O3, and (v) recently reported ζ-Fe2O3. The crystal structures of non-hydrated iron oxide phases are shown in Fig. 2.23 As the present review deals with the systematic description of the most important catalytical and environmental applications of nanocrystalline non-hydrated iron oxides, it is thus timely to briefly summarize their fundamental structural and physicochemical properties.
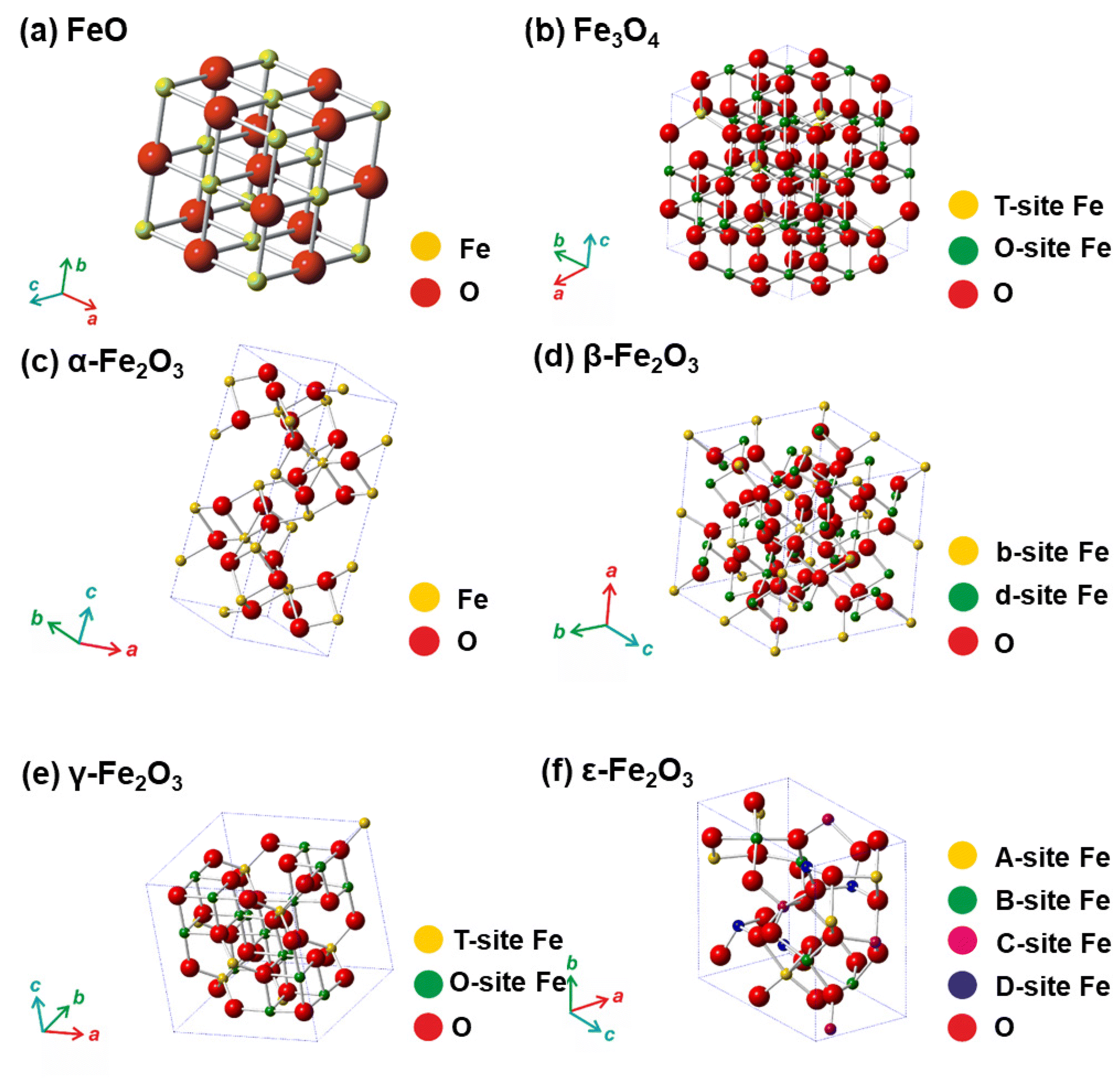 | ||
| Fig. 2 Crystal structures of non-hydrated iron oxides. In panel (a), Fe represents octahedral position in the framework of FeO. In panel (b), T-site Fe (tetrahedral) and O-site Fe (octahedral) in the Fe3O4 crystal structure. In panel (c), Fe represents octahedral position in the framework of α-Fe2O3. In panel (d), b-site Fe and d-site Fe represents octahedral b-sites and d-sites, respectively, with different polyhedra distortion in the crystalline arrangement of β-Fe2O3. In panel (e), T-site Fe and O-site Fe represents tetrahedral and octahedral position, respectively, in the γ-Fe2O3 crystalline arrangement. In panel (f), A-site Fe, B-site Fe, and C-site Fe stand for the octahedral sites with different polyhedra distortion and D-site Fe stands for the tetrahedral position in the crystalline framework of ε-Fe2O3. In all the panels, O stands for oxygen positions in the respective crystal structures. Panels (c), (d), (e), and (f) Reproduced with permission from ref. 23 Copyright 2011, American Chemical Society. | ||
α-Fe2O3, γ-Fe2O3, and Fe3O4 are naturally-occurring non-hydrated iron oxide phases, and a various synthetic pathways have been documented to produce both nanoscale and bulk forms with sophisticated control of particle size, particle size distribution (PSD), structure, surface modification, and hence, physicochemical features. Conversely, β-Fe2O3 and ε-Fe2O3 are viewed as rare non-hydrated Fe2O3 phases which barely exist naturally, and are challenging to prepare in the laboratory as single phases without contamination by other Fe2O3 admixtures. By virtue of being low surface energy and thermodynamical stability, they exist only as nanosized objects (e.g., as nanoparticles, nanotubes, thin films). To date, only a few reaction pathways are known for the production of β-Fe2O3 and ε-Fe2O3 phases and most of them deliver lower yields without proper control on attaining precise particle size and morphology.
Among all iron(III) oxide polymorphs, α-Fe2O3 phase possess the highest degree of thermodynamic stability.22,23 Notably, α-Fe2O3 shows a corundum-type crystal structure (similar to ilmenite (FeTiO3) and corundum) with hexagonally-closed packed oxygen (O) lattice wherein Fe3+ ions occupy two-thirds of the octahedral positions (see Fig. 2c). The crystalline arrangement of α-Fe2O3 is represented within the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c space group with lattice parameters a = 5.034 Å and c = 13.752 Å. α-Fe2O3 exhibits two magnetic transitions, (∼950 K Néel temperature) and (∼265 K, Morin transition temperature MTT). The strong magnetic dipolar anisotropy competes with local ion anisotropy which shows different thermal dependencies to drive the Morin transition. Upon cooling past the MTT, the spins undergo a 90° spin reorientation from the basal plane to the c-axis. Nonetheless, above MTT, due to effective spin–orbit coupling, the spins deviate slightly from the basal plane as a result slight canting is observed, inducing feeble magnetic moment pointing along the c-axis. The tilting of the spins arises due to Dzyaloshinsky–Moriya antisymmetric interaction prompted by the crystal fields arising from favorable positioning of O2− anions within the α-Fe2O3 crystal lattice. The temperature of Morin transition is influenced by many factors like lattice defects, exchange by foreign cations, divergences from stoichiometry, surface effects, structures, and particle size. In a weakly ferromagnetic state, α-Fe2O3 shows very small net magnetization, which limits its exploitation in magnetism-based applications. However, the n-type semiconductor (α-Fe2O3) exhibit the wide band gap (∼2.1 eV) among all the polymorphs of iron(III) oxide reported so far; α-Fe2O3 possesses very low hole mobility. Both empirical and theoretical investigations proven that the photoelectrochemical characteristics of α-Fe2O3 can be notably improved through modifying the morphology, particle size, doping, and substrate type during the growth of nanoscale α-Fe2O3 thin films, opening the entry to effectively apply iron(III) oxide polymorphs for the fabrication of electrodes in the photo electrolysis of H2O to produce H2.16 Besides, α-Fe2O3 has been found as a promising constituent in gas and humidity sensors and electrodes in lithium-ion batteries.25 In geophysics, hematite-ilmenite solid solutions have been used as a model system for description and explanation of the essence of lamellar magnetism, a phenomenon behind strong remanent magnetization imprinted by a planet's internal magnetic field and stable for hundreds of millions of years.26
c space group with lattice parameters a = 5.034 Å and c = 13.752 Å. α-Fe2O3 exhibits two magnetic transitions, (∼950 K Néel temperature) and (∼265 K, Morin transition temperature MTT). The strong magnetic dipolar anisotropy competes with local ion anisotropy which shows different thermal dependencies to drive the Morin transition. Upon cooling past the MTT, the spins undergo a 90° spin reorientation from the basal plane to the c-axis. Nonetheless, above MTT, due to effective spin–orbit coupling, the spins deviate slightly from the basal plane as a result slight canting is observed, inducing feeble magnetic moment pointing along the c-axis. The tilting of the spins arises due to Dzyaloshinsky–Moriya antisymmetric interaction prompted by the crystal fields arising from favorable positioning of O2− anions within the α-Fe2O3 crystal lattice. The temperature of Morin transition is influenced by many factors like lattice defects, exchange by foreign cations, divergences from stoichiometry, surface effects, structures, and particle size. In a weakly ferromagnetic state, α-Fe2O3 shows very small net magnetization, which limits its exploitation in magnetism-based applications. However, the n-type semiconductor (α-Fe2O3) exhibit the wide band gap (∼2.1 eV) among all the polymorphs of iron(III) oxide reported so far; α-Fe2O3 possesses very low hole mobility. Both empirical and theoretical investigations proven that the photoelectrochemical characteristics of α-Fe2O3 can be notably improved through modifying the morphology, particle size, doping, and substrate type during the growth of nanoscale α-Fe2O3 thin films, opening the entry to effectively apply iron(III) oxide polymorphs for the fabrication of electrodes in the photo electrolysis of H2O to produce H2.16 Besides, α-Fe2O3 has been found as a promising constituent in gas and humidity sensors and electrodes in lithium-ion batteries.25 In geophysics, hematite-ilmenite solid solutions have been used as a model system for description and explanation of the essence of lamellar magnetism, a phenomenon behind strong remanent magnetization imprinted by a planet's internal magnetic field and stable for hundreds of millions of years.26
β-Fe2O3, discovered in 1956, is metastable crystalline polymorph of Fe3+ oxide22,23 which adopts a cubic-body-centered (CBC) crystalline morphology of a bixbyite type with Ia space group (see Fig. 2d). Owing to the distinct crystallographic atmospheres, two distinct cation positions in the β-Fe2O3 crystal structure can be clearly recognized and are assigned as b-sites and d-sites. In the cubic unit cell with a = 9.393 Å, 24 Fe3+ ions fill the d-sites with the C2 symmetry and 8 Fe3+ ions occupy the b-sites with the C3i symmetry leaving no vacant cation position. Unlike other ferric oxide polymorphs, β-Fe2O3 is paramagnetic at room temperature (RT) while at 110 K, it is magnetically ordered, and below that it becomes antiferromagnetic, most probably of canted nature.27 Moreover, it undergoes a polymorphic transformation to α-Fe2O3 at ∼500 °C; however, if β-Fe2O3 nanoparticles are endowed with a hollow morphology, β-Fe2O3-to-γ-Fe2O3 transformation has been observed. Despite being a non-attractive magnetic material, the β-Fe2O3 phase offers itself a prospective contender within the realm of optoelectronics,28 serving as a viable option for chloroform sensing29 and enhancement of lithium-ion battery performance when employed as an anode.30 In view of the band gap width of ∼1.9 eV, β-Fe2O3 phase, prepared as a thin film has been successfully deployed for the photo-assisted H2 production as an alternative iron oxide photocatalysts.31
γ-Fe2O3, initially identified in 1925, has an inverse spinel-type cubic crystal arrangement with a lattice parameter of a = 8.351 Å and is formed by the topo tactic oxidation of Fe3O4.22,23 Two crystallographically distinctive cation sites (octahedral and tetrahedral) are established in its crystal structure (see Fig. 2e). To develop a neutral charge within the γ-Fe2O3 crystalline arrangement, certain octahedral sites are left unoccupied. Thus, γ-Fe2O3 is described with a stoichiometric formula specified as [(Fe3+)T(Fe5/33+o1/3)OO4]3/4, where o denotes vacancies. The vacancies within the octahedral sites can be divided randomly in accordance with cubic Fd3m, cubic P4132 or fully ordered tetragonal P43212 space groups. This modulation affects the γ-Fe2O3 crystal symmetry, transitioning from higher (cubic) to lower (tetragonal) symmetrical configurations. In addition, the ordering of vacancies over the octahedral sites is believed to minimize the electrostatic energy of the crystal, occurring dominantly under conditions of elevated temperatures and pressures and at reduced dimensions. Similarly, as in the case of α-Fe2O3 and β-Fe2O3, γ-Fe2O3 is a semiconductor with a bandgap of ∼2 eV. From the perspective of magnetism, γ-Fe2O3 is a strong ferrimagnet with two magnetic sublattices (i.e., tetrahedral and octahedral) and where a range of 780 to 980 K is estimated for the Curie temperature. The precise determination of Curie temperature is impossible due to the γ-Fe2O3 to α-Fe2O3 polymorphic conversion upon heating. The net magnetic moment of stoichiometric γ-Fe2O3 per formula unit is offered by the unequal occupancies of Fe3+ ions in the tetrahedral and octahedral magnetic sub lattices, oriented in the direction of the octahedral Fe3+ magnetic moments and amounting to 2.5μB. When the size of γ-Fe2O3 NPs falls down to a specific value (∼30 nm), it exhibits superparamagnetic through a robust magnetic reactivity under minor external magnetic fields and intense relaxation phenomenon. Moreover, in the nanoworld, ε-Fe2O3 is frequently identified as midway phase upon thermal processing on the γ-Fe2O3-to-α-Fe2O3 route.22,23 γ-Fe2O3 is considered as the first model and commonly explored nanomaterials deployed to construct the groundwork for superparamagnetic relaxation theory and other characteristics magnetic phenomena prevalent in the nanoscale domain such as emergence of collective magnetic excitations in the blocked state, theory of spin tilting and spin defeat, etc.22,23 Apart from theoretical studies, nanosized γ-Fe2O3 is the most significant form of ferric oxide, which has previously been used in assorted magnetism-based technologies (e.g., information storage pigments, magnetic fluids, gas sensors, magneto-optical devices, magneto-caloric refrigeration, etc.) and medical appliances (e.g., NMR imaging agent, medical diagnosis, cell labeling, and separation, targeted drug delivery, etc.).6–8,15
Similar to β-Fe2O3, ε-Fe2O3 is rare, metastable crystalline form of iron(III) oxide; initially observed in 1934 and primarily described in 1963.22,23,32 It occurs only in the nanoworld with a scarce natural abundance. The synthesis of ε-Fe2O3 as an individual nanosized object is challenging in view of the essential requirement of the maintenance of spatial restriction of NPs growth. Thus, synthetic protocols towards ε-Fe2O3 must involve some extent of agglomeration in either the precursor and/or the silica matrix used as support. ε-Fe2O3 crystallizes embracing an orthorhombic unit cell which falls within the Pna21 space group. Three octahedral and one tetrahedral sites are authorized in the ε-Fe2O3 crystalline arrangement differing significantly in the degree of polyhedral distortion (see Fig. 2f). ε-Fe2O3 is a 4-sublattice Néel P-type ferrimagnet. Generally, ε-Fe2O3 shows complex magnetic features which drastically changes at two distinct temperatures, i.e., at ∼490 K (Curie temperature) and ∼110 K. At RT, ε-Fe2O3 display the characteristics of a collinear ferrimagnet33 with an enormous coercivity of ∼2 T and a net magnetic moment of 1.3μB.34 At ∼110 K, another magnetic transition is noticed with drastic drop in the coercive field of ε-Fe2O3 and a notable decrease in the orbital component of the magnetic moment associated with Fe3+.32 Nonetheless, the magnetic state of ε-Fe2O3 at lower temperature remains unresolved and lacks a satisfactory explanation in current research. In addition, the magnetic transition at ∼110 K is attended by lattice distortions and a numerous morphological transformations.32 Upon thermal treatment, ε-Fe2O3 transforms to α-Fe2O3; the temperature of polymorphous transformation intensely depends on the restriction of size growth during structural change and varies from ∼600 to 1300 K.32 ε-Fe2O3 is considered as an extraordinary nanomaterial that exhibit a high coercive field at RT,34 millimeter-wave ferromagnetic resonance,35 and coupled magnetoelectric characteristics.36 It is important to note that these unique properties not observed in another iron oxide phase. A combination of these nanomaterial properties renders ε-Fe2O3 as an ideal matter for deployment in various applications that requires materials with high coercivity34 and/or necessitates the coupling of electrical and magnetic material features (e.g., memory elements with multiple state)36 and/or involving electromagnetic waves absorption.35
ζ-Fe2O3, the youngest iron(III) oxide polymorph unearthed in 2015 as a product of pressure transformation of β-Fe2O3,24 is the first example of a monoclinic phase in the iron(III) oxide family. Its crystal structure falls into the I2/a space group with a = 9.683 Å, b = 10.00 Å, and c = 8.949 Å; Fe3+ ions occupy six non-equivalent octahedral sites. At RT, ζ-Fe2O3 is paramagnetic with a transition to the antiferromagnetic state at the Néel temperature of ∼69 K. It has been suggested that ζ-Fe2O3 could have other interesting features such as electronic, optical, and transport that would enable its usage for empirical applications. However, more studies are needed to address its electronic structure and, therefore, physical properties.
FeO is often observed as an intermediate phase in the phase transformations of iron oxides and is frequently deficient with iron (i.e., FexO with x ranging from 0.83 to 0.96); if stoichiometric, it shows a cubic crystal morphology within the Fm3m space group and a = 4.296 Å (see Fig. 2a). For nonstoichiometric FexO, defects in its crystal structure cause an ordered distribution of vacancies in the cation sites. FexO can be readily oxidized to Fe3O4 and ultimately into γ-Fe2O3. FeO is an antiferromagnet with the Néel temperature of around 200 K; below the Néel temperature, the magnetocrystalline distortion establishes a rhombohedral crystal structure. FeO has been described in mineralogical and petrological studies as it a part of the solid solution with periclase (MgO) and its oxidation may form goethite-limonite systems.
Fe3O4, the oldest magnetic material ever studied, has an identical crystal structure to γ-Fe2O3, i.e., the cubic crystal structure of inverse spinel type with the two distinct cation sites, the T and O positions (a = 8.397 Å, Fd3m space group) (see Fig. 2b). Contrary to γ-Fe2O3, the O sites are occupied by Fe2+ ions and Fe3+ ions in equal ratio. Thus, Fe3O4 is described as (Fe3+)T(Fe2+Fe3+)OO4. Fe3O4 behaves as a ferrimagnetic material with a net magnetic moment of 4μB per unit formula and a Curie temperature of about 980 K; moreover, above 120 K (the Verwey transition temperature), Fe3O4 is a conductor. At RT, Fe3O4 is believed to show a half-metallic behavior, a feature required for spintronics materials. At very high temperatures (>1300 K), Fe3O4 transforms to α-Fe2O3 with an occasional occurrence of γ-Fe2O3 as an intermediate phase. When synthesized as nanoparticles or core/shell structures, non-stoichiometric phases of Fe3O4 are often observed, i.e., Fe3−δO4 with δ in the range from 0 (Fe3O4) to 1/3 (γ-Fe2O3). There have been several discussions in the literature reporting different threshold value of δ determining the transition between non-stochiometric Fe3O4 and non-stoichiometric γ-Fe2O3. Due to its robust magnetic reactivity in the presence of small external magnetic fields and superparamagnetic properties at nanoscale, Fe3O4 has been found to be effective in many biomedical applications such as contrast agents, carriers of drugs, and heating elements in magnetically-assisted hyperthermia for cancer treatment, and catalytical processes such as magnetically-separable catalysts.6,7,21,37,38
It is widely acknowledged that the physical and chemical features and applicability of nanocrystalline iron oxides can be altered by their size and morphology, surface functionalization, particle arrangement and texture, and cation substitution.6–8,10,13,15–17 In this review, the selected and vitally important applications of iron oxide nanomaterials are deliberated in the domain of catalysis and environmental remediation (see Fig. 3). Importantly, the emphasis has been mainly on challenging aspects in specific applications with the introductory aspects being briefly outlined under each section comprising recent review references.
 | ||
| Fig. 3 Applications of iron oxide nanoparticles in catalysis and environmental technologies. | ||
The review is structured into three main sections with section 2 being focused on applications of iron oxides in the field of catalysis with an emphasis on their roles in diverse reactions such as reduction-, oxidation-, coupling-, and alkylation reactions, Fischer–Tropsch synthesis, and multicomponent reactions (Fig. 3). Section 3 deliberates the use of iron oxides in the most recent environmental applications; accentuating the role of iron oxides as nanosorbents for toxic heavy metals and organic contaminants, their significance in photocatalysis processes deployed for environmental treatment while promoting H2O2 decomposition, and their immobilization capability (Fig. 3). The concluding section summarizes the current state of applicability of nanocrystalline iron oxides in the aforementioned fields and delineates challenges that may motivate future research in the chemistry of nanocrystalline iron oxides.
2. Catalytic applications of nanosized iron oxides
Heterogeneous catalysis mostly deals with various metal nanoparticles, supported catalysts, hybrid catalysts, and metal oxides,39–41 which includes Al2O3, MgO, CeO2, Fe3O4, α-Fe2O3, and γ-Fe2O3 among other iron oxides; often deployed themselves or as supports for important catalytic transformations.42–44 To date, various magnetic catalysts or magnetic supported catalysts, predominantly based on iron oxides have been employed in numerous organic transformations besides other applications.45–47In the case of iron oxides, up to now, four distinct crystalline polymorphic forms of ferric oxides namely α-, β-, γ-, and ε-Fe2O3 have been thoroughly characterized and their applications described.23 Because of their unique structural arrangements, each of the nanoscale polymorphic variants of Fe2O3 exhibits distinctive magnetic, biochemical, and catalytic features;48–52 α-Fe2O3 and related hybrid nanomaterials have been examined as heterogeneous catalysts.53–57 The catalytic applications of various forms of iron oxides and related hybrid catalysts are deliberated with emphasis on the non-magnetic supported or/iron oxides, especially hematite. The magnetically recyclable catalysts,58–65 electrocatalysts,66–70 and assorted Fenton-type reactions71–74 associated with iron oxides are not included here as they have been well discussed in the literature and being beyond the scope of this review, with the exception of environmental applications.75–82
Iron oxides are ideal oxide supports, as they are straightforward to synthesize, presence of highly reactive surface hydroxyl groups, facilitating effective adsorption and immobilization of metals and ligands. Additionally, they can be isolated through simple filtration post-reaction, rendering them relatively benign and greener heterogeneous catalysts.83,84 In this section, nanoiron oxides (e.g., hematite, goethite and their hybrid forms, like Au/α-Fe2O3, β-FeOOH@poly(dopamine)–Au–poly(dopamine), etc. and their catalytic applications are highlighted with few representative cases of their synthesis. A variety of catalytic transformations encompassing reduction-, oxidation-, coupling-, and alkylation reactions, Fischer Tropsch synthesis, multicomponent reactions, and other miscellaneous reactions are discussed (see Fig. 4).
 | ||
| Fig. 4 Selected catalytic applications of nanocrystalline iron oxides. | ||
2.1. Coupling reactions
Noble metal NPs, such as Pd, Pt, Au, and Ru, have garnered immense interest over the last decade because of their special features and promising applications in diverse fields like surface enhanced Raman scattering (SERS),85–87 surface plasmon resonance (SPR),88,89 and biomedicine90,91 and especially in catalysis82,92–95 encompassing C–C coupling reactions;96–98 Heck and Suzuki reactions being the most important examples.99–102 These reactions are generally catalyzed by palladium-based catalysts and are widely deployed in the synthesis of various pharmaceuticals.103–106 As it relates to catalysis, Pd is the widespread noble metal employed for its outstanding activity and selectivity in various coupling reactions under rather mild conditions.107–110 However, there are still lingering limitations associated with such processes often stemming from the recovery of the catalyst and challenges pertaining to cumbersome isolation of the products. To overcome these problems, dispersion of Pd nanocatalysts on solid supports have been explored.111–115The field of catalysis science has seen a notable development with the emergence of single-atom catalysts (SACs), where individual metal atoms are distributed on solid supports.122–124 This novel approach has garnered significant research interest.43,125 Recently, a highly efficient and scalable method for the synthesis of iron oxide-supported Pd SACs using a facile, eco-friendly ball milling process was reported for the Suzuki–Miyaura cross-coupling reaction.126 In a typical gram-scale synthesis of Pd1/FeOx-10 SACs, the metal oxide, and Pd nitrate precursors are directly milled together in a planetary ball mill for a given time and then calcinated at 400 °C under aerobic conditions for 2 h (see Fig. 5a). Using this approach, a kilogram scale synthesis (up to 1.002 kg) of Pd1/FeOx-1000 SACs in one batch is also achieved, which holds considerable potential for application in industrial scale-up. The transmission electron microscopy (TEM) images of Pd1/FeOx-10 catalyst revealed that the absence of visible Pd nanoparticles or clusters, which indicated the uniform dispersion of Pd isolated atoms on iron oxide support (Fig. 5b). This homogeneous distribution of Pd, Fe, and O atoms is further confirmed by the elemental mapping images, suggesting a well-dispersed atomic structure (Fig. 5c). Additionally, the aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) images (Fig. 5d) demonstrated that the atomic dispersion of Pd species on the iron oxide support, as highlighted by the yellow circles. This well characterized Pd1/FeOx-10 catalyst exhibited remarkable catalytic activity, achieving a high turnover frequency (TOF) of 7844 h−1 for the Suzuki–Miyaura cross-coupling reaction between bromobenzene and phenylboronic acid at 40 °C. Various aryl bromides containing electron-donating and withdrawing functional groups were efficiently converted into biphenyl products with high product yields (>95%) without the need for an inert atmosphere (Fig. 5e). This exceptional performance significantly surpassed other catalysts, including PdO, Pd/C, and Pd nanoparticles, highlighting the superior catalytic activity of the Pd1/FeOx-10 SACs. Furthermore, the catalyst displayed greater stability and recyclability, indicating its potential for practical applications in catalyzing cross-coupling reactions.
 | ||
| Fig. 5 (a) Schematic representation of the synthesis of Pd1/FeOx-10 catalysts; TEM (b), elemental mapping (c) and AC HAADF-STEM (d) images of Pd1/FeOx-10; (e) Pd1/FeOx-10 catalyzed Suzuki–Miyaura cross-coupling. Reproduced with permission from ref. 126 Copyright 2022, American Chemical Society. | ||
Iron oxide nanoparticles proved their high potential to act as a template for the production of advanced nanocatalyst.127–129 Fe2O3 containing zeolite-NaY (Fe2O3-Y) support have been developed for the fabrication of heterogeneous Pd/PdO–Fe2O3-Y nanocatalyst and deployed for the Suzuki–Miyaura cross-coupling reaction.130 The support was prepared by employing coprecipitation method (Fig. 6a). The SEM images of the supported Pd/PdO–Fe2O3-Y nanocatalyst on zeolite-NaY revealed that the presence of foreign particles (Fig. 6c), whereas untreated zeolite-NaY displayed clear crystal indicating absence of additional particles (Fig. 6b). TEM analysis further confirmed the deposition of ultrafine Pd/PdO NPs on Fe2O3-Y support (Fig. 6d and e). This Pd/PdO–Fe2O3-Y catalyst demonstrated remarkable efficiency in activating the robust C–Cl bond of aryl chlorides in the Suzuki–Miyaura cross-coupling reaction under mild conditions (Fig. 6f). Despite having an extremely low metal (Pd) loading of 0.0037 mol%, this catalyst exhibited higher reactivity, selectivity, and recyclability; ensued biaryl product yield of up to 92% with a good turnover number (249) was discerned. Particularly, biaryl product yield significantly improved when a small amount of water with methanol was used as a solvent system for the reaction. The presence of basic sites, together with zeolite-NaY and the transfer of charge from Fe to Pd, played a pivotal role in the activation of the C–Cl bond.
 | ||
| Fig. 6 (a) Schematic illustration of the preparation of Fe2O3-Y; (b, c) SEM images of untreated zeolite-NaY and Pd/PdO–Fe2O3-Y respectively; (d, e) TEM images of Pd/PdO–Fe2O3-Y; (f) Pd/PdO–Fe2O3-Y mediated Suzuki–Miyaura cross-coupling reaction. Reused with permission from ref. 130 Copyright 2023, American Chemical Society. | ||
Diao and co-workers131 synthesized hollow carbon nanonets (HCN) by combining hydrothermal carbonization of glucose with α-Fe2O3 NPs acting as a hard template; Pd NPs were immobilized onto the exterior of the hollow HCN via in situ precipitation-reduction method (Fig. 7). HCN have been attractive candidates for the catalyst support as they could be fabricated employing the pre-synthesized hematite NPs as the hard template. Carbon supported porous nanostructures have demonstrated significant efficacy as catalyst support, particularly for noble metal nanocatalysts.132–134 TEM images of α-Fe2O3, α-Fe2O3@C, HCN, and Pd/HCN are depicted in Fig. 7b–e. The α-Fe2O3 NPs with 50 nm diameter (Fig. 7a) have been prepared by hydrothermal precipitation, whereas the Pd nanoparticles with diameter ∼8 nm was formed when the reduction/precipitation method was employed to graft Pd NPs on HCN. Following their synthesis and comprehensive characterization, the Pd/HCN catalysts were then investigated for the Suzuki and Heck–Miyaura reaction under mild reaction parameters. Notably, outstanding yields of the respective products were achieved in an aqueous medium (irrespective of the halides counterparts) with good Turnover frequency (TOF) for Suzuki cross-coupling reaction. The reaction scope was further explored for Heck–Miyaura reaction of haloarenes with alkenes at 120 °C, in DMF (dimethyl formamide) using a base, K2CO3. Comparatively, iodobenzene gave excellent yields of products, while bromobenzene and chlorobenzene gave rather low yields of the coupling products. Furthermore, when compared with some reported Pd-supported catalysts, the catalytic performance of Pd/HCN in facilitating the Suzuki reaction, between phenyl boronic acid and iodobenzene, displayed notable efficiency in terms of both product yields and reaction time.131
 | ||
| Fig. 7 (a) Schematic illustration of synthesis of the Pd/HCN catalysts; (b–e) TEM images of α-Fe2O3 NPs, α-Fe2O3@C, HCN, and Pd/HCN catalysts respectively. Reproduced with permission from ref. 131 Copyright 2013, The Royal Society of Chemistry. | ||
A heterogeneous iron oxide catalyst on graphene oxide (GO) support (α-Fe2O3 nanocluster/GO) was synthesized for the Suzuki–Miyaura coupling reaction and hydrogenation of 4-hydroxynitrobenzene in an aqueous medium.135 For the assembly of the catalyst, the tris-trz-PEG ligand was employed to promote the fabrication and deposition of α-Fe2O3 on GO by supramolecular interactions (Fig. 8a). The TEM images revealed that the uniform deposition of α-Fe2O3, with a size of 1.8 nm, on the GO (Fig. 8b). The hexagonal morphology and a lattice fringe space (0.25 nm) correspond to the crystallographic plane (110) of the α-Fe2O3 crystal, which was affirmed by HR-TEM analysis (Fig. 8c). In context of the Suzuki–Miyaura coupling in aqueous medium, Handa et al. developed a novel Fe/ppm Pd nanoparticle catalyst.136 The highly active needle-shaped NPs of Fe/ppm Pd catalysts facilitated the Suzuki–Miyaura couplings across a broad range of electrophiles and nucleophiles. This innovative approach enabled the first ppm-level Pd catalysis under micellar conditions, offering significant value to the pharmaceutical industry by reducing the residual palladium in the final product.
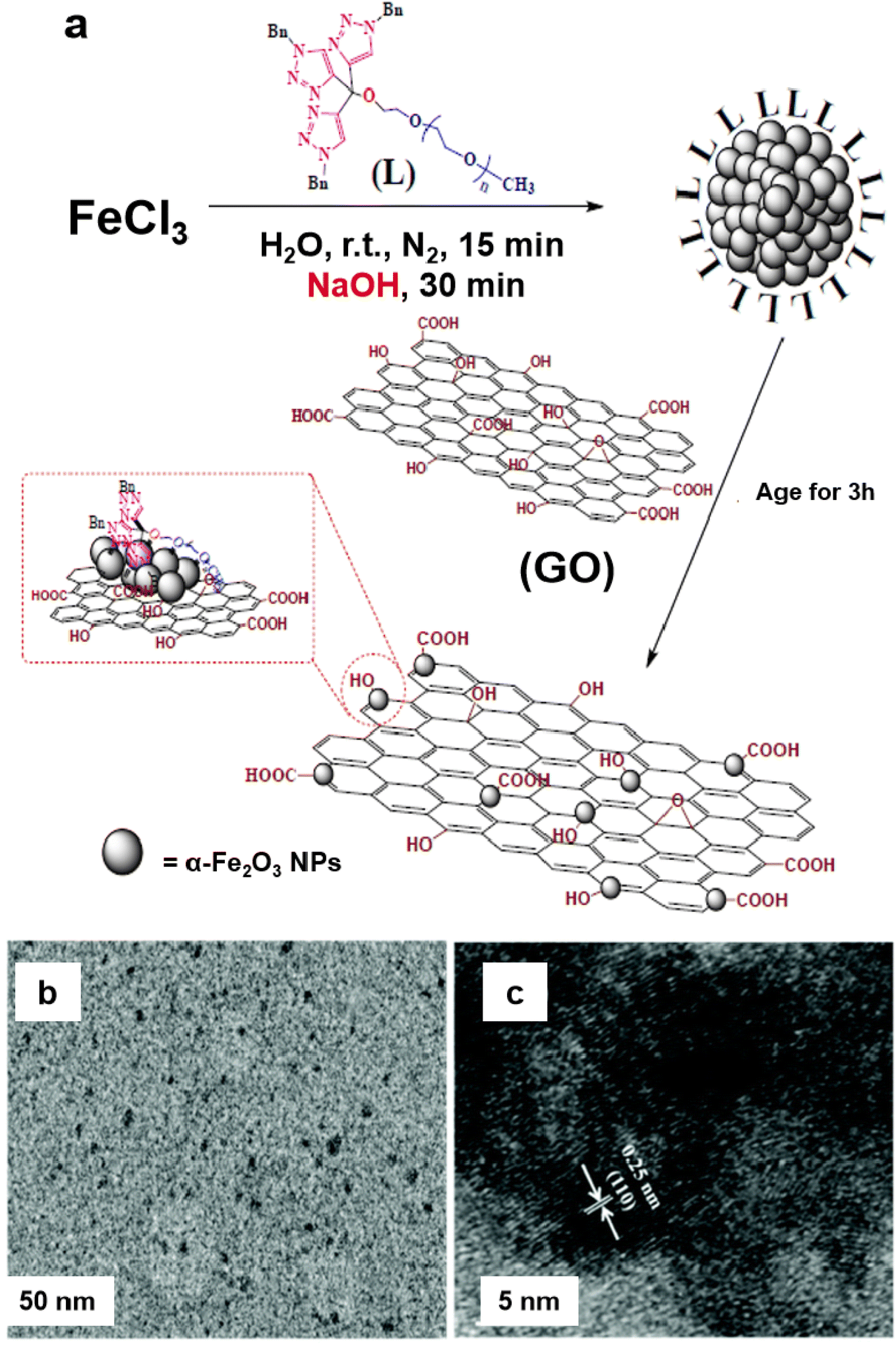 | ||
| Fig. 8 (a) Schematic representation for the synthesis of the α-Fe2O3/GO catalyst; (b, c) TEM and HR-TEM images of a-Fe2O3 nanocluster/GO catalyst. Reproduced with permission from ref. 135 Copyright 2017, The Royal Society of Chemistry. | ||
 | ||
| Fig. 9 (a) Schematic illustration of the synthesis of Fe2O3–Ovac; TEM and HR-TEM images of Fe2O3 (b, e), 0.5 Fe2O3–Ovac (c, f), 1.0 Fe2O3–Ovac (d, g); (h) O 1s XPS spectra; (i) X-ray absorption near-edge structure (XANES) spectra of Fe K-edge. Reused with permission from ref. 140 Copyright 2023, Springer Nature. | ||
The catalytic performance of the synthesized 1.0 Fe2O3–Ovac catalyst was examined under the optimized condition via aminocarbonylation and alkoxycarbonylation (Fig. 10a and b).140 The ideal 1.0 Fe2O3–Ovac catalyst demonstrated conversion of aryl halides and amines/alcohols into the corresponding amides/esters with excellent yields (75–99%). Additionally, the catalytically active 1.0 Fe2O3–Ovac catalyst was also explored for the synthesis of important drugs such as Moclobemide, CX-546, Nikethamide, and chiral compounds via carbonylation reactions (Fig. 10c), highlighting their potential for pharmaceutical applications. The catalytic activity of the 1.0 Fe2O3–Ovac catalysts was found to be critically dependent on the presence and density of oxygen vacancies, directly impacting their performance. Furthermore, the different Fe sites on the Fe2O3–Ovac surface emerged as a crucial factor in enhancing catalytic efficiency. The 1.0 Fe2O3–Ovac catalyst showed excellent recyclability, indicating its suitability for sustainable catalytic applications. The catalytic mechanism of the carbonylation reaction has been studied using DFT calculations. The Fe2O3–Ovac catalyst facilitated the elementary steps of PhI activation, CO insertion, and C–N/C–O coupling efficiently, leading to improved catalytic performance (Fig. 10d).
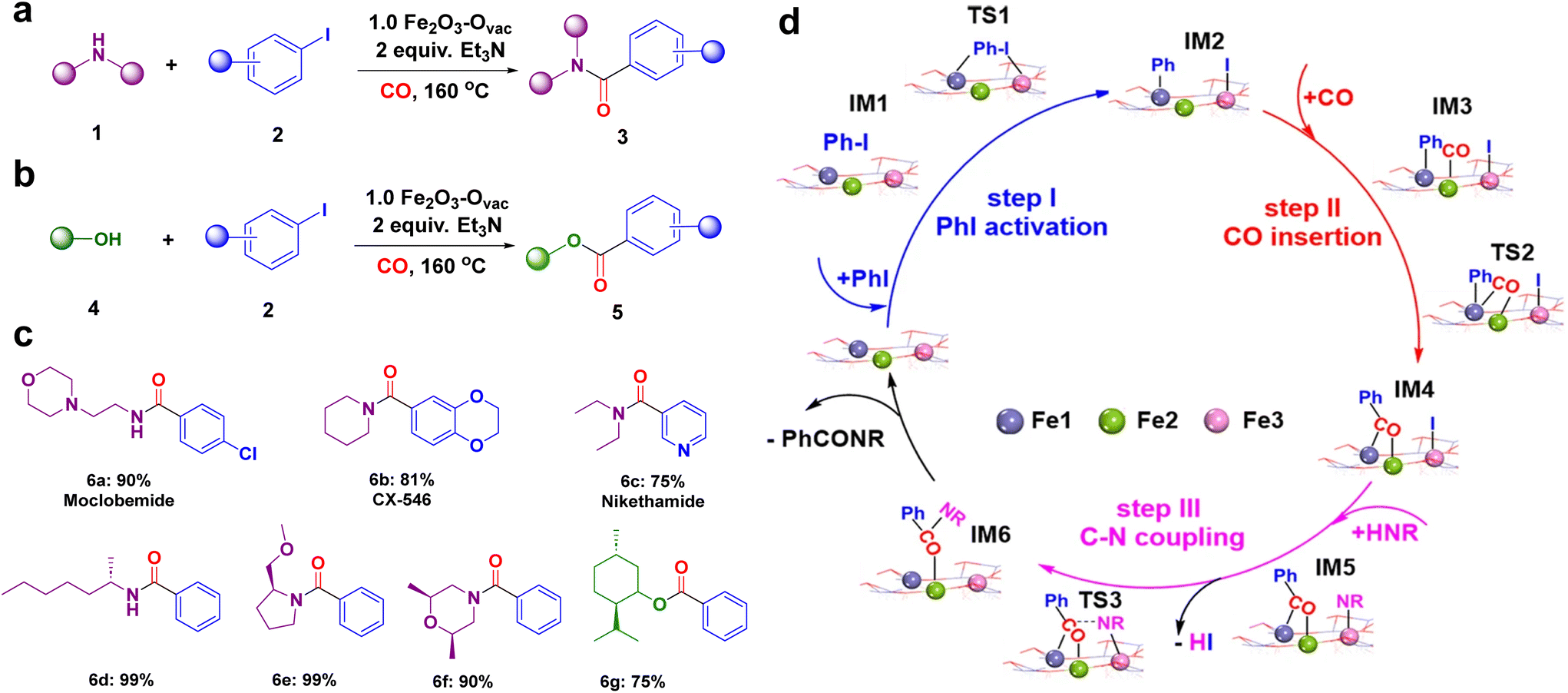 | ||
| Fig. 10 Catalytic applications of 1.0 Fe2O3–Ovac catalyst (a) the aminocarbonylation; (b) alkoxycarbonylation reaction; (c) synthesized important drugs and chiral compounds; (d) mechanism for the aminocarbonylation. Reused with permission from ref. 140 Copyright 2023, Springer Nature. | ||
The strategy of borrowing hydrogen is one of the benign approaches for the C–N coupling reaction as it circumvents the necessity for an external hydrogen source for the reduction reactions.141–144 The heterogeneous iron oxide nanocatalyst145 for the C–N/C–C bond formation reactions via borrowing hydrogen approach were reported to obtain quinoline and its derivatives (Fig. 11); hydrogen auto-transfer mechanism was confirmed by the deuterium labelling study. In the similar context, Sarno et al. prepared core–shell Fe3O4/Au nanocatalyst and deployed it for the N-alkylation reactions.146
 | ||
| Fig. 11 (a–c) The N-alkylation, C-alkylation and quinoline synthesis over nano-Fe2O3 catalyst. Reproduced with permission from ref. 145 Copyright 2019, The Royal Society of Chemistry. | ||
 | ||
| Fig. 12 (a) Schematic representation for synthesis of PdCu@GQD@Fe3O4 nanocatalyst; (b) TEM images PdCu@GQD@Fe3O4; (c) TEM images of GQD@Fe3O4 NPs and (d) Application of Sonogashira–Hagihara reaction over the catalyst PdCu@GQD@Fe3O4. Reproduced with permission from ref. 151 Copyright 2017, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
2.2. Reduction reactions
Reduction reactions are indispensable in various industries, for the syntheses of pharmaceuticals, dyes and rubber, and agricultural chemicals153–155 among others. Nanosized iron oxides have garnered attention among researchers for the catalytic hydrogenation of aromatic nitro groups, α,β-unsaturated aldehydes, nitric oxide (NO) in view of their inexpensive nature, ease of preparation and environmentally benign attributes.Goethite (FeOOH), maghemite (γ-Fe2O3) and hematite (α-Fe2O3) supported gold catalysts have been used for reduction of cinnamaldehyde and trans-benzalacetone.156 Among these studied catalysts, Au anchored on FeOOH (AF35G) presented excellent performance and selectivity for unsaturated alcohols to generate the corresponding unsaturated carbonyl compounds, while Au anchored on α-Fe2O3 (AF35H) and γ-Fe2O3 (AF35M) displayed inferior performances. It is crucial to emphasize that the catalytic performance for unsaturated alcohols in the reduction of cinnamaldehyde and benzalacetone are completely dependent on the characteristics of supporting material wherein both the performance and selectivity to unsaturated alcohols is enhanced due to the reducibility of the catalysts. Finally, it has been concluded that the active and selective sites are generated via transfer of electron from the reduced support to the metal nanoparticles.156
Iron oxide supported Au catalyst for the liquid phase hydrogenation of α,β-unsaturated aldehydes was reported.157 Various supports including hematite, goethite, and iron oxide modified alumina have been used for the immobilization of gold and the corresponding catalysts investigated for the reduction of crotonaldehyde and cinnamaldehyde at 100 °C and at 1 MPa of H2 in 2-propanol (Fig. 13). It is observed that iron oxides immobilized gold particles are more selective relative to γ-Al2O3 supported gold particles; hematite supported gold exhibits the high selectivity for unsaturated alcohols like cinnamyl alcohol (48%) and hydrocinnamyl alcohol (88%). Furthermore, no dependence of selectivity on substrates such as crotyl alcohol and cinnamyl alcohol has been discerned on gold particles.
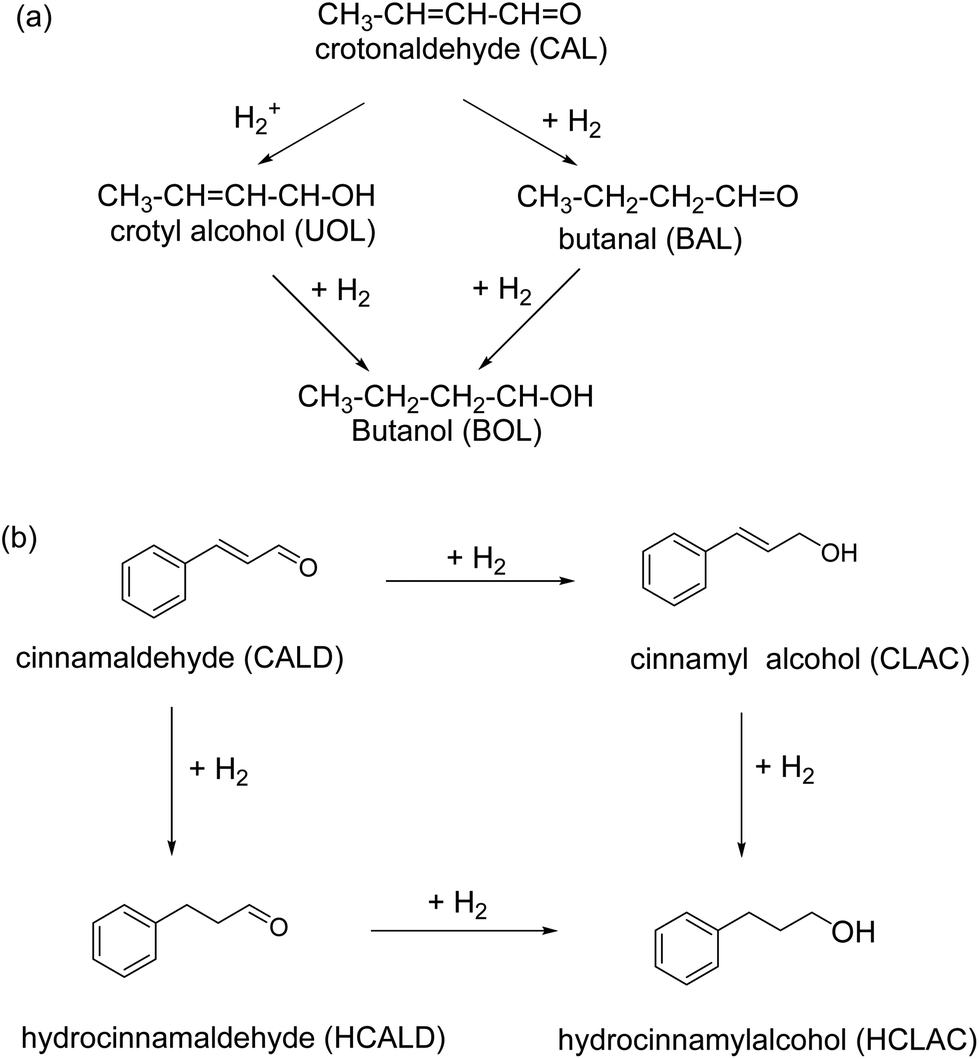 | ||
| Fig. 13 Hydrogenation of (a) crotonaldehyde, and (b) cinnamaldehyde using gold NPs supported on iron oxides. Reproduced with permission from ref. 157 Copyright 2009, Elsevier. | ||
The noble metal Pt was adorned on iron oxide via the atomic layer deposition and deployed for the selective hydrogenation of cinnamaldehyde to cinnamyl alcohol;158 Pt NPs with 2.7 nm size were evenly dispersed over the alumina support (Fig. 14a and b) wherein the Pt30Fe30 catalyst exhibited higher selectivity (84%) as compared to the pure Pt catalyst (45%) for cinnamyl alcohol (Fig. 14c).
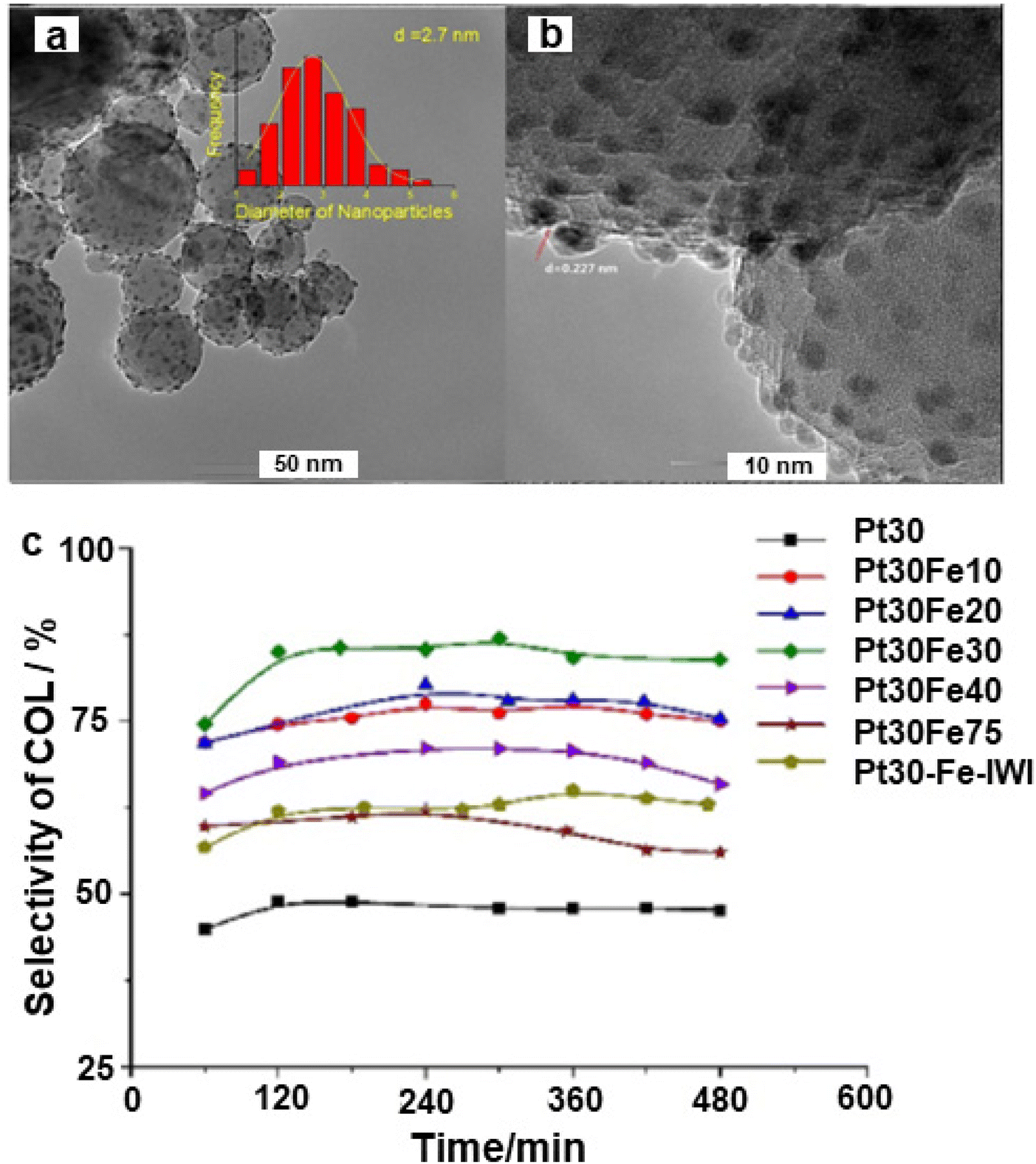 | ||
| Fig. 14 (a) TEM image of Pt30Fe30; (b) HR-TEM image of Pt30Fe30, and (c) Selectivity of cinnamyl alcohol. Reproduced with permission from ref. 158 Copyright 2017, Elsevier. | ||
The chemoselective hydrogenation of cinnamaldehyde to cinnamyl alcohol were also carried out by employing an iron oxide decorated catalyst (Pt–3FeOx/MoO3−y).159 The observed lattice fringes of 0.226 and 0.196 nm refers to Pt (111) and Pt (200), respectively. The size of Pt NPs was 3 nm which were well dispersed on FeOx (Fig. 15a and b). The Pt–3FeOx/MoO3−y catalyst showed a higher conversion and selectivity for cinnamyl alcohol due to the Pt–FeOx interfaces (Fig. 15c). The acidic Pt–FeOx interface had direct impact on the chemisorption and activation of the carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), leading to the selective cinnamaldehyde hydrogenation to cinnamyl alcohol (Fig. 15d).
O), leading to the selective cinnamaldehyde hydrogenation to cinnamyl alcohol (Fig. 15d).
 | ||
| Fig. 15 (a) TEM images of Pt–3FeOx/MoO3−y; (b) HAADF-STEM image of Pt–3FeOx/MoO3−y; (c) Performance of catalysts for conversion and selectivity, and (d) Mechanism for the hydrogenation of cinnamaldehyde over Pt–3FeOx/MoO3−y catalyst. Reproduced with permission from ref. 159 Copyright 2018, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
The hydrogenation of nitroarenes by heterogeneous catalysis has been an imperative chemical transformation. The selective hydrogenation of nitro compounds containing additional reducible groups such as alkene, carbonyl, and/or halide to produce functionalized amines poses a significant and challenging task in both contemporary chemical and pharmaceutical industry.160–163 In this context, Ma et al.164 fabricated TiO2 based iron oxide (Fe3O4@TiO2) heterogeneous photocatalyst for the selective hydrogenation of nitroarenes into amine derivatives. The Fe3O4@TiO2 photocatalyst was synthesized by the sol–gel approach followed by the pyrolysis process (Fig. 16a). The catalyst structure comprises Fe3O4 NPs supported on TiO2, forming a composite with a well-defined morphology evident by TEM and HRTEM analysis (see Fig. 16b and c). The lattice fringes observed in the HRTEM image (Fig. 16c) correspond to specific crystal planes within the iron oxide phase, with measurements of 2.5 Å and 2.9 Å corresponding to the (311) and (220) planes, respectively. Additionally, a lattice fringe of 3.5 Å was attributed to the TiO2 (100) plane. The prepared Fe3O4@TiO2 photocatalyst demonstrated remarkable efficiency in the selective hydrogenation of nitroaromatics to amines in the presence of other reducible groups under photothermal conditions (Fig. 16d). The exceptional performance of the Fe3O4@TiO2 catalyst was attributed to the robust interaction between Fe3O4 and the reducible TiO2 support, boosting the photothermal effect and enhancing the adsorption of reactants. Moreover, this catalyst was also utilized for gram-scale reactions, showcasing its viability for diverse applications in organic synthesis, offering a sustainable and efficient alternative for the conversion of nitro compounds to valuable amines.
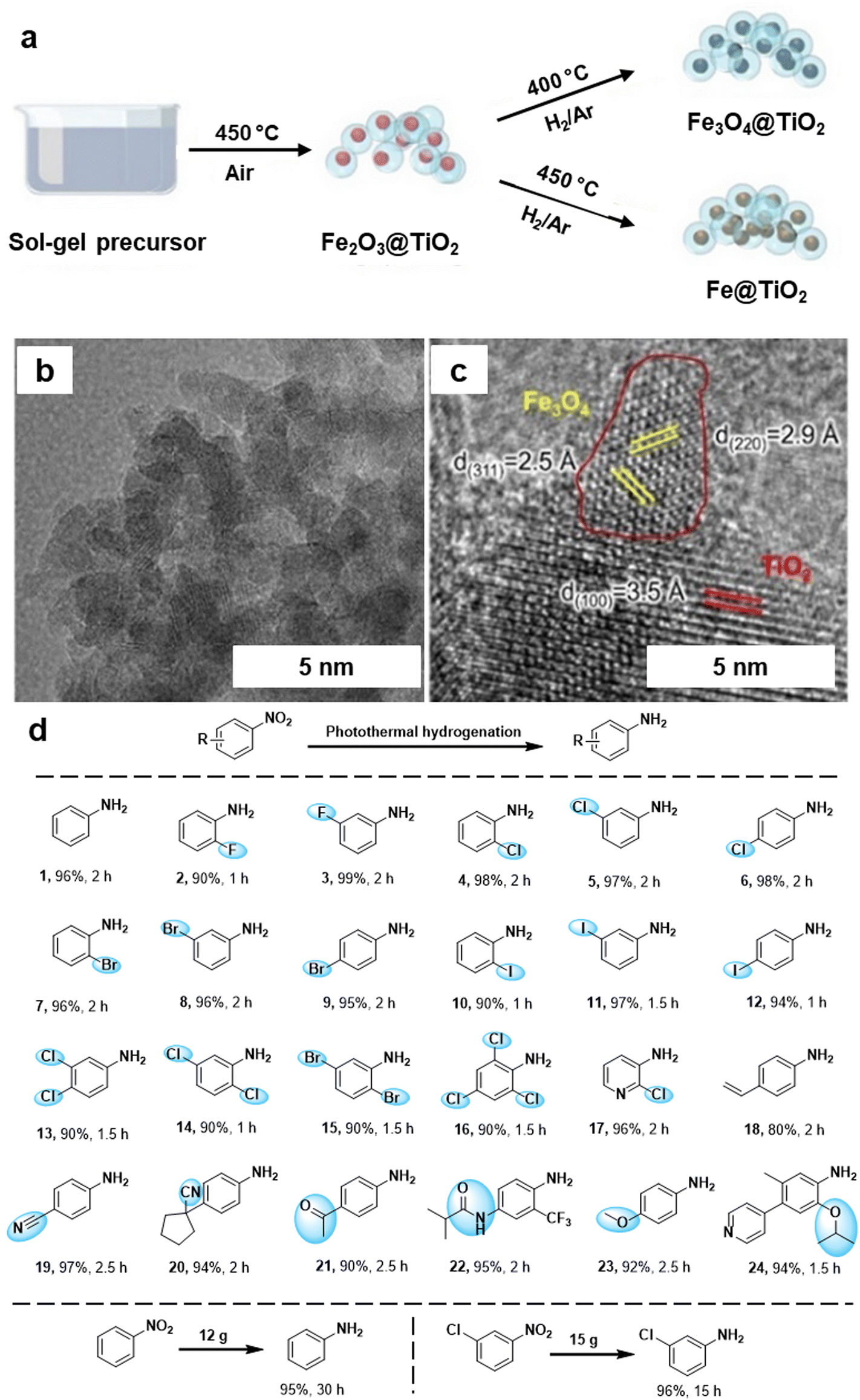 | ||
| Fig. 16 (a) Synthetic scheme for Fe3O4@TiO2 and Fe@TiO2; (b, c) TEM and HR-TEM images of the Fe3O4@TiO2 catalyst; (d) Application of the Fe3O4@TiO2 catalyst for photothermal hydrogenation of various nitroaromatics. Reused with permission from ref. 164 Copyright 2024, American Chemical Society. | ||
The oxygen defective nanocrystalline iron oxide catalysts (Fe3O4) were used for the reduction of several nitroarenes to respective anilines with excellent yield and selectivity. Among as-prepared catalysts, Fe3O4 showed high activity, excellent recycling stability, and 100% selectivity.165 Moreover, Pt SACs and pseudo SACs supported on iron oxide were developed by Zhang and coworkers and deployed for the selective hydrogenation of nitroarenes.166 The 0.08%Pt/FeOx-R250 catalysts exhibited broad compatibility with a diverse range of substituted nitroarenes containing various functional groups, showcasing their versatility and applicability. In addition, micro–mesoporous iron oxide also employed for the base free transfer hydrogenation of nitroarenes.167
The hydrogenation of N-heterocycles in aqueous media was studied using the Rh@Fe3O4 nanocatalyst,168 where the Rh particles were uniformly distributed on the Fe3O4 particles (7–9 nm) as scrutinized by TEM image analysis (Fig. 17a and b). The identification of Rh was confirmed by STEM (Fig. 17e) and <1 nm size of the Rh NPs (denoted by red circles) was affirmed by HR-TEM image of Rh@Fe3O4 (Fig. 17f). The heterogeneous Rh@Fe3O4 catalyst proved to be highly effective in the hydrogenation of N-heterocyclic compounds in water using tetrahydroxydiboron (THDB) as a reductant, demonstrating high selectivity for 1,2,3,4-tetrahydroquinoline (py-THQ) with an impressive turnover frequency of 1632 h−1 (Fig. 17h).
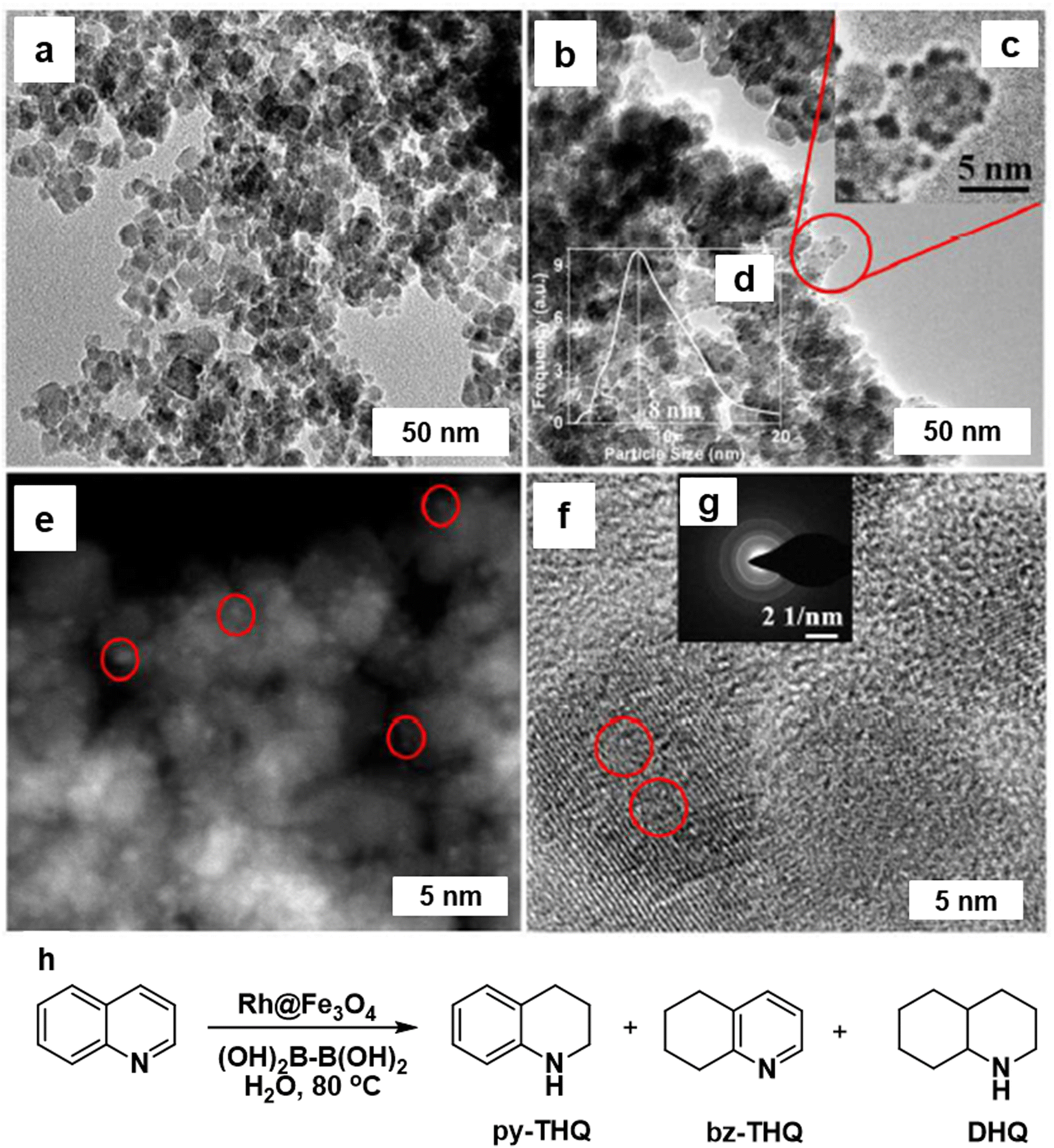 | ||
| Fig. 17 TEM images of (a) Fe3O4; and (b) Rh@Fe3O4; (e) STEM image of Rh@Fe3O4 particles; (f) HRTEM image of Rh@Fe3O4; (h) Rh@Fe3O4-mediated hydrogenation of quinoline using THDB. Reproduced with permission from ref. 168 Copyright 2018, The Royal Society of Chemistry. | ||
Afterwards, the Rh@Fe3O4 nanocatalyst was modified by incorporation of Co3O4 nanoparticles wrapped in N-doped g-C3N4 on Fe3O4 support.169 The synthesis of Co3O4/N-Gr/Fe3O4 nanocomposites is depicted in Fig. 18a wherein the Co3O4 NPs (15–20 nm) were wrapped in N-doped graphitic carbon on the surface of Fe3O4 (Fig. 18b and c). The lattice spacing d-value (2.43 Å) is associated with (111) plane of crystalline Co3O4 NPs as analyzed by HR-TEM (Fig. 18d). The core–shell morphology of this nanocomposite was confirmed by the HAADF-STEM imaging (Fig. 18e). This nanocomposite was deployed for the hydrogenation of nitroarenes, cinnamaldehyde, and quinoline (Fig. 18f). Notably, more than 99% hydrocinnamaldehyde (HCAL) chemoselectivity was obtained in toluene at 30 bar H2 (Fig. 18g).
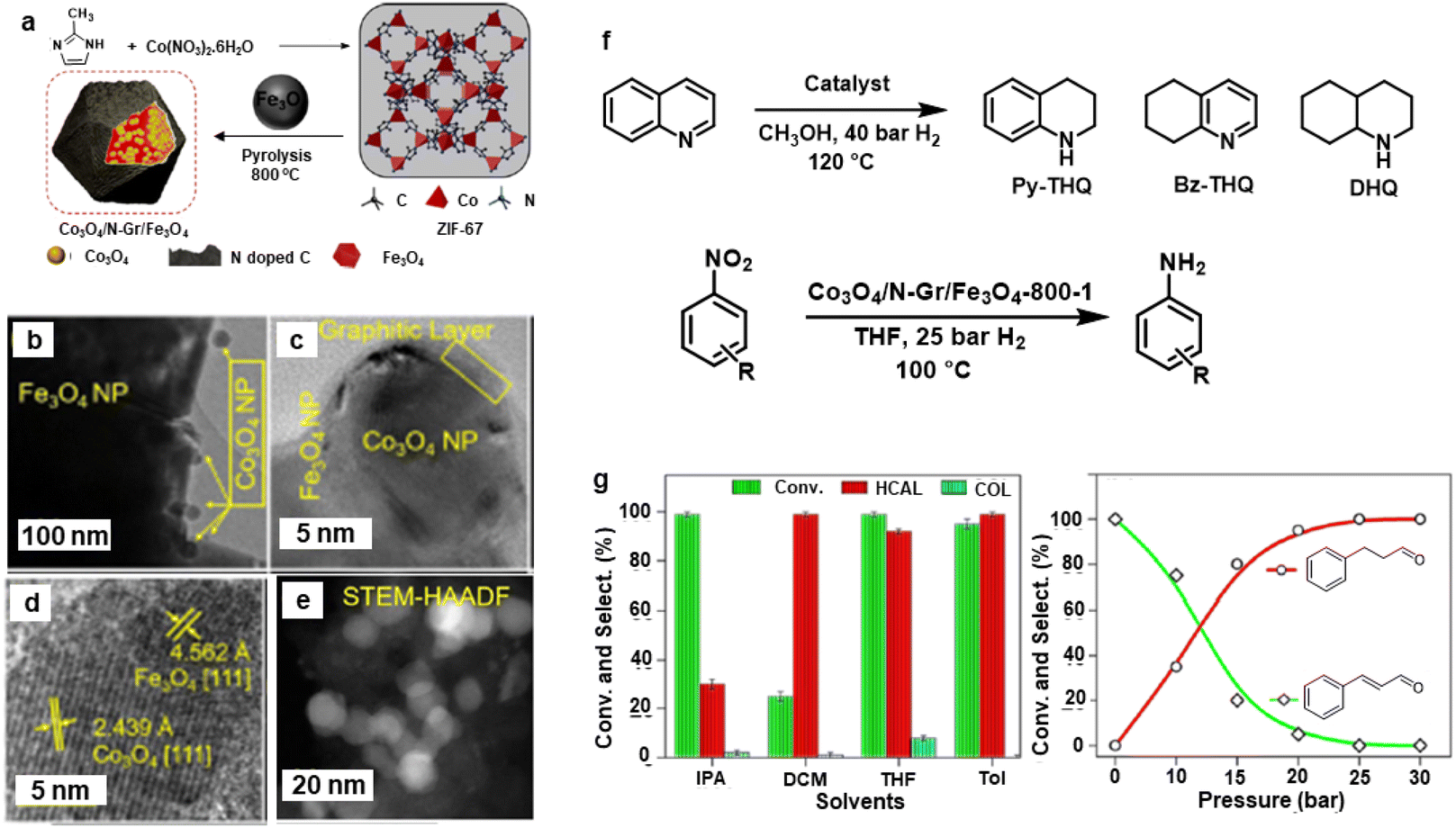 | ||
| Fig. 18 (a) Schematic for the synthesis of Co3O4/N-Gr/Fe3O4 nanocomposite; (b, c) TEM; (d) HR-TEM; (e) HAADF-STEM images of Co3O4/N-Gr/Fe3O4-800-1 nanocomposite; (f) Schemes for hydrogenation of quinoline and nitroarenes, and (g) Effect of solvents and pressure on hydrogenation of cinnamaldehyde. Reproduced with permission from ref. 169 Copyright 2021, American Chemical Society. | ||
Recently, silica-supported Fe/Fe–O NPs (Fe/Fe–O@SiO2) were utilized for the reduction of nitriles to amines.170 The catalyst was synthesized through the impregnation and thermal treatment of Fe salt over the silica (Fig. 19a). The globular and rod-shape structure (Fig. 19b and c) and Fe core NPs resulting from SiO2 matrix covered by ultrathin iron oxide (Fig. 19d and e) comprising the catalyst, Fe(OAc)2–SiO2-800, was investigated by the TEM, HR-TEM imaging, and elemental analysis, respectively. The as-prepared catalyst with the catalytic amount of aluminium additives was employed for the reduction of various nitriles (aromatic, aliphatic, and heterocyclic) to important primary amines (Fig. 19f). Moreover, this catalyst was investigated for its utility in the reduction of fatty nitriles to respective fatty amines which are valuable oleochemicals (Fig. 19g). Apart from the cinnamaldehyde, nitroarenes and quinoline reduction, iron oxide was also instrumental in the reduction of 5-hydroxymethylfurfural (HMF) to 2,bis(hydroxymethyl)furan (BHMF).171
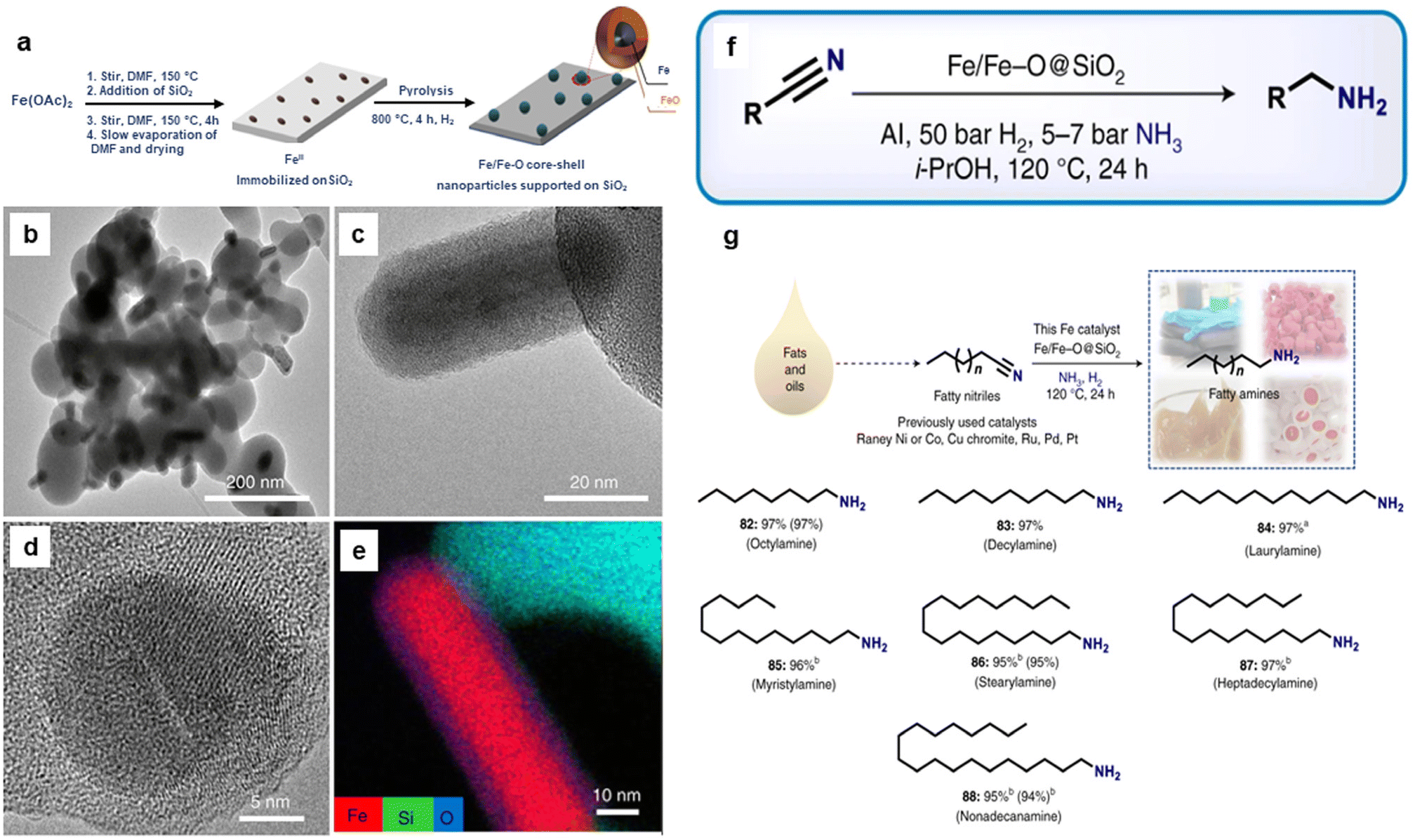 | ||
| Fig. 19 (a) Schematic representation for the preparation of Fe/Fe–O core–shell NPs; (b, c) TEM; (d) HR-TEM images of Fe(OAc)2–SiO2-800; (e) Elemental mapping image of Fe, Si and O; (f) Application of the catalyst for the various nitrile hydrogenation; and (g) Reduction of fatty nitriles. Reproduced with permission from ref. 170 Copyright 2021, Springer Nature. | ||
Selective catalytic reduction (SCR) of NO with ammonia at lower temperature has been reported via the deployment of Fe2O3 catalysts172 and various reaction parameters were inspected including temperature, oxygen concentration, and [NH3]/[NO] molar ratio for SCR activity. Notably, Fe2O3 catalysts displayed excellent catalytic performance for the SCR of NO with ammonia in a wide range of temperature from 150–270 °C; more than 95% NO conversion was achieved at 180 °C (molar ratio [NH3]/[NO] = 1). When the addition of SO2 on SCR catalytic performance were examined at 180 °C, the NO conversion was lowered from 99% to 58% in 240 min. Thermogravimetric and Fourier transform infrared spectroscopy analysis revealed172 that SO2 was responsible for the deactivation of Fe2O3 catalysts because the development of metal and ammonium sulfates at the surface of the catalyst during the de-NO reaction, leading to pore clogging and catalytic activity inhibition.
Similarly, FeW(x) (hematite supported tungsten) catalysts with diverse Fe/W molar ratios obtainable through co-precipitation procedure were introduced for the SCR of NOx with NH3.173 Among the catalysts subjected to testing, namely (FeW(1), FeW(2), FeW(3), FeW(4), FeW(5)), the catalyst FeW(5) demonstrated an excellent activity, achieving nearly quantitative conversion of NO, N2 selectivity from 250 to 450 °C, and robust H2O/SO2 strength at temperature more than 300 °C. It was observed that the insertion of WOx moiety enabled the lattice structure to alter to the hematite form and high distribution of WOx on the catalysts surface were noticed for the FeW(x). Though redundant WOx addition was contrary to the reaction performance and presumably shields the acid sites due to big quantity of tungsten oxide moiety, the best performance of FeW(5) catalysts were ascribed to its largest BET surface area, amount of surface adsorbed oxygen molecules, Brønsted acidity and quantity of weak acid sites thus enabling the SCR reaction. Overall, the study offers promising alternatives to substitute the conventional vanadium related catalysts by FeW(x) type catalysts for the control of NOx emission.173
Iron oxide/silver (FexOy/Ag) nanocomposite was synthesized by a single step process using goethite (α-FeOOH) rods as support.174 First, FeOOH was treated with sodium citrate followed by attachment of Ag via electrostatic interaction. Afterwards, Ag ions were reduced by NaBH4 to generate Ag NPs on the goethite rods surface (Fig. 20a).174 The structural configuration of the composite material was characterized by SEM and TEM images as depicted in Fig. 20; Ag NPs with size of 10–50 nm appears to be well-coated over surface of FeOOH nanorod indicating the high quality FexOy/Ag nanocomposite (Fig. 20b and c). The SAED pattern confirms the presence of spots because of Ag, Fe21.333O32 and FeOOH (see inset in Fig. 20d). The FeOOH layers Fe21.333O32 and crystalline lattices can be seen in HRTEM attributed to (111) phase (d = 0.235 nm) of Ag (Fig. 20e).174
 | ||
| Fig. 20 (a) Schematic diagram of the preparation of FexOy/Ag nanocomposite; FexOy/Ag nanocomposite: SEM image (b), TEM images (c, d) (inset: SAED), (e) HRTEM image. Reproduced with permission from ref. 174 Copyright 2016, Springer Nature. | ||
The as-synthesized and well characterized FexOy/Ag nanocomposite bearing varying contents of Ag were then studied for hydrogenation of nitroaromatic molecules and decomposition of organic dyes utilizing NaBH4 as a reductant; two nitroaromatic molecules, namely p-nitro aniline and p-nitrophenol were reduced completely in few minutes as illustrated by instant decolorization of yellowish green solution. The good catalytic activities of FexOy/Ag nanocomposite were attributed to the incorporated FexOy components wherein rapid electron transfer between BH4− ions and p-nitrophenol compound occurred not only on the surface of AgNPs but also on the interface between AgNPs and FexOy. Accordingly, FexOy can play important role in catalysis through a synergistic effect, resulting in better catalytic performance.174
2.3. Oxidation reactions
In the context of sustainable chemistry, catalytic oxidation of alcohols, carbonyls, CO, and other oxidizable entities including carbohydrates with molecular oxygen as an environmentally friendly oxidizing agent has garnered immense focus.175–179 Generally, these protocols are mainly performed on supported noble metal catalysts like Au, Pd, Rh and Ru.180–186 Since these metal-supported catalysts are expensive, the relatively non-toxic and abundant iron has been considered as a viable alternative as it is inexpensive and the second most easily available metal in the earth's crust.For the fabrication of ultrafine iron oxides (FeOx/MC), mesoporous carbon (MC) support was developed via a hard-template route with abundant surface functional groups.187 First, MC was synthesized through the modulated hard-template technique that encompasses carbonizing an organic–inorganic composite including sugar, citric acid, Al and P ions, and then the removal of on-site formed aluminophosphate template (Fig. 21a). Three different types of MC (pore sizes 6.5, 3.8, and 2.9 nm) were attained by adjusting the amount of citric acid and sugar in composite (abbreviated as MC-1, MC-2 and MC-3) and were deployed as support for the immobilization of iron oxides (Fe2O3). The as-synthesized catalysts were investigated for the selective oxidation of benzyl alcohols to their respective aldehydes (Fig. 21b). Notably, the individual components, Fe2O3 and MC supports, under the same preparative protocols were not active for the oxidation reaction. Under similar reaction parameters, all 5 wt% FeOx/MC catalysts with porous structure accomplished the conversions of more than 92% with a wide substrate scope as exemplified by the oxidation of an array of benzyl alcohols to the respective aldehydes with satisfactory yields.
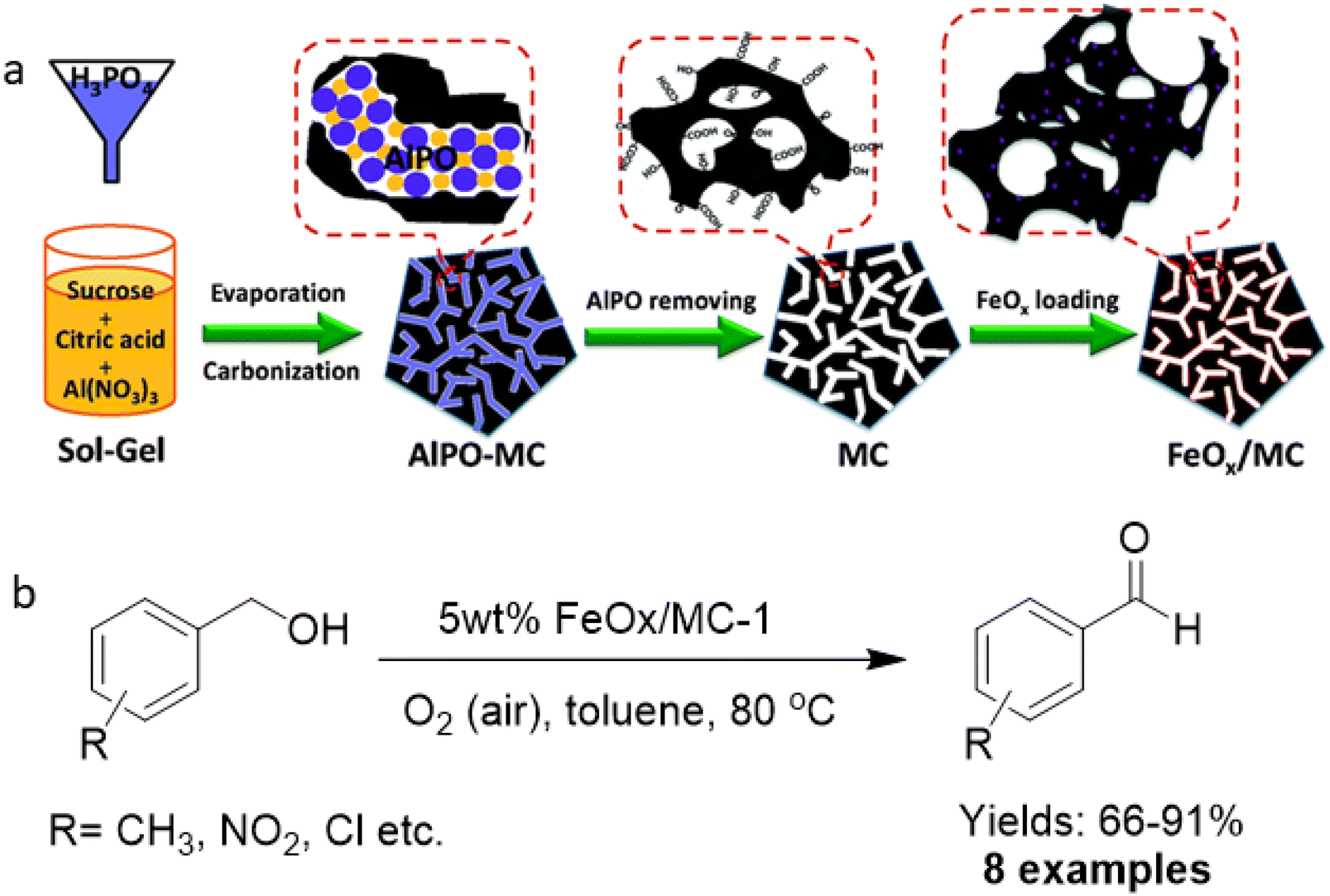 | ||
| Fig. 21 (a) Schematic diagram for the preparation of MC and FeOx/MC catalysts; (b) Oxidation of alcohols with 5 wt% FeOx/MC-1. Reproduced with permission from ref. 187 Copyright 2015, The Royal Society of Chemistry. | ||
Iron oxide nanoparticles showed their high potential also as the catalyst support for CO oxidation.188–190 The hydrothermal approach to well-ordered α-Fe2O3 nanoparticles using amino acids as additives were reported,191 wherein the nature of amino acids regulates the size and shape of the α-Fe2O3 NPs. The spindle shape α-Fe2O3 NPs were obtained, when acidic amino acid was used, whereas basic amino acid afforded rhombohedrally shaped α-Fe2O3 nanoparticles (see Fig. 22). Additionally, gold NPs were immobilized on the surfaces of α-Fe2O3 NPs to assess their catalytic activity for CO oxidation; spindle shaped Au/α-Fe2O3 displayed superior performance relative to the rhombohedral Fe2O3. Along with the morphological impact, particle size of α-Fe2O3 play an integral role in CO oxidation; larger spindle α-Fe2O3 support caused excellent performance compared to smaller counterparts whereas in case of rhombohedral Au/α-Fe2O3, medium-sized α-Fe2O3 nanocrystals displayed the maximum catalytic performance for CO oxidation.
 | ||
| Fig. 22 TEM images of α-Fe2O3 NPs. Samples (a–c) prepared with [Fe3+]/[L-lysine] molar ratios; samples (d–f), prepared with [Fe3+]/[D-asparagine] molar ratios. Reproduced with permission from ref. 191 Copyright 2009, Elsevier. | ||
The catalytic performance of hematite concave nanocubes for low temperature CO oxidation was also explored.192 These nanocubes with high index and facets were fabricated by hydrothermal process employing kinetically controlled growth approach. To investigate the details of morphological development of α-Fe2O3 concave nanocubes, time-dependent experiments were performed (see Fig. 23). Among the different catalysts, highly stable α-Fe2O3 nanocubes unveiled superior catalytic performance for CO oxidation.
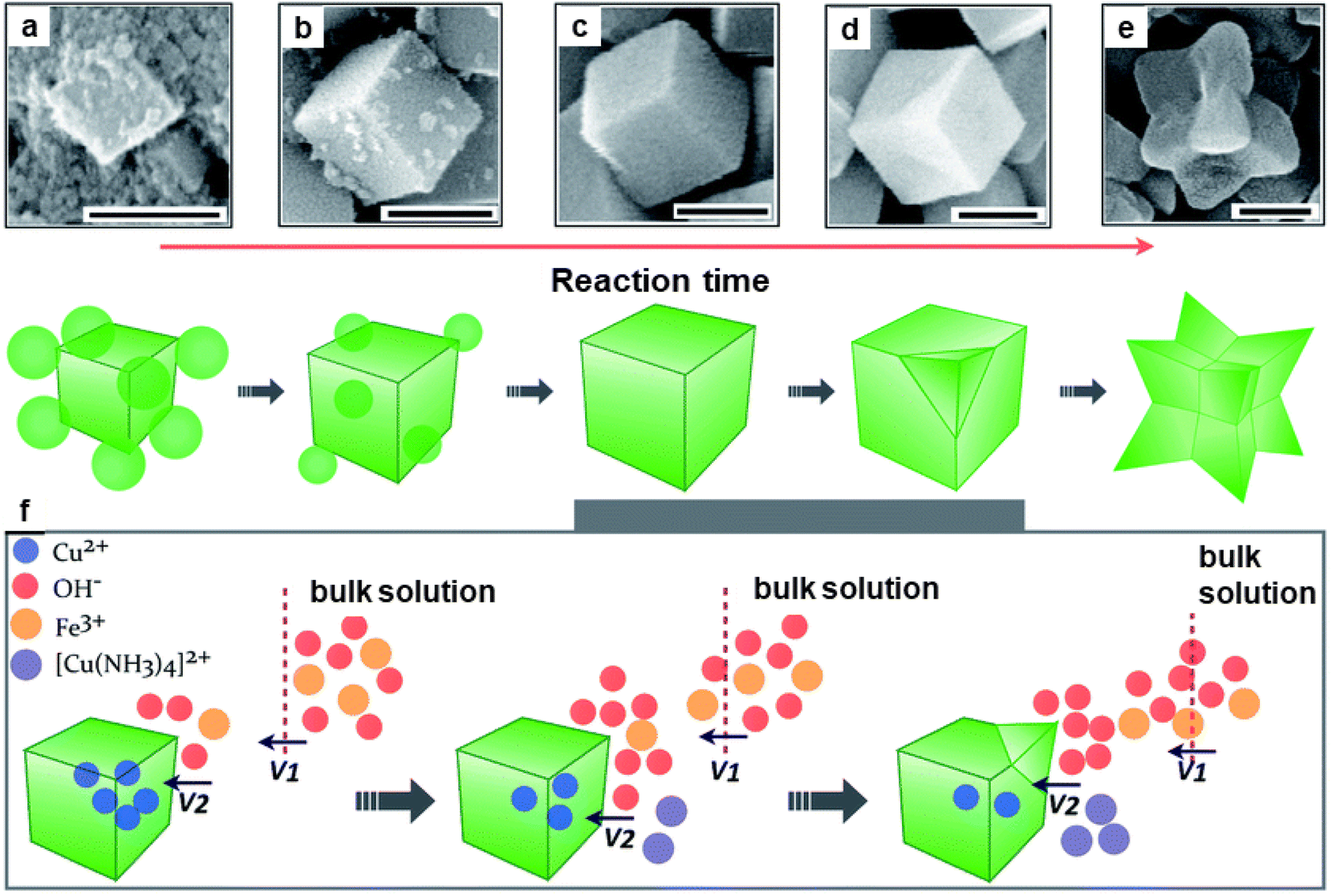 | ||
| Fig. 23 SEM images (a–e) of the nanocrystals collected at different reaction times at 200 nm; (f) Schematic illustration of the kinetically controlled overgrowth of concave nanocubes developed from pseudonanocubes. Reproduced with permission from ref. 192 Copyright 2014, The Royal Society of Chemistry. | ||
The synthesis of hematite (α-Fe2O3) and Pd-doped α-Fe2O3 NPs via a surfactant-free hydrolysis approach has been reported.193 Rod-like iron oxide NPs were yielded through hydrolysis of iron(III) chloride with diluted hydrochloric acid at 100 °C; further addition of citric acid in the mixture containing iron oxide NPs and (Pd (CH3CN)2Cl2), afforded Pd/iron oxide nanocomposites (see Fig. 24). This attractive protocol avoided the use of surfactant, template and higher temperatures culminating in good shape/size selectivity as well. A rod-like iron oxide/Pd nanocomposite displayed high catalytic performance at low-temperature for CO oxidation as compared to usage of sole α-Fe2O3.193 Pd NPs displayed an important role in low-temperature catalytic CO oxidation and catalytic performance was associated with the facile absorption of (i) oxygen on the reduced sites of the support (Fe2+) and (ii) CO adsorbed on Pd@Fe2O3 interface.
 | ||
| Fig. 24 (a) TEM images and the corresponding SAED patterns of Pd doped α-Fe2O3 rod-like particles; (b) HRTEM image of Pd@Fe2O3, where the lattice spacing ∼0.385 nm, which corresponds to Pd planes. Reproduced with permission from ref. 193 Copyright 2009, Elsevier. | ||
Oxidation of alcohol using heterogeneous catalyst is a crucial process in the production of high-value fine chemicals.194–196 Iron oxide NPs were successfully used also for catalytic oxidation of alcohol.197,198 Platinum nanowires (NWs) decorated on iron oxide (Pt@Fe2O3) were synthesized via the oxidation of FePt NWs in oleylamine and the resulting catalyst were examined for the oxidation of alcohols and alkenes.199 For the styrene derivatives, molecular oxygen was employed as an oxidizing agent to obtain benzaldehyde in an absolute yield of 31%; however, when tBuOOH was used as the sole oxidant for alcohol oxidation, higher yields (more than 80%) of carbonyl compounds could be obtained (Fig. 25).
 | ||
| Fig. 25 (a) Oxidation of styrene and (b) alcohols catalyzed by Pt@Fe2O3. Reproduced with permission from ref. 199 Copyright 2011, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
The lengths of PtFex NWs were several micrometers with diameters of ∼1.9 nm (see Fig. 26a and b). After the catalytic oxidation reaction, the diameters got enlarged to ∼2.8 nm (see Fig. 26c and d) and the Fe in the catalyst was oxidized to Fe2O3via the Kirkendall effect. This unsupported PtFex NWs catalyst was identified as more efficient catalyst relative to other reported Fe2O3 NPs and was reusable many times with negligible loss of catalytic performance.
 | ||
| Fig. 26 TEM and HR-TEM images of (A, B) PtFe NWs and after catalysis (C, D) Pt@Fe2O3 NWs. Reproduced with permission from ref. 199 Copyright 2011, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
The application of iron oxide NPs has been also investigated for the oxidative dehydrogenation reaction (ODR).200,201 ODR is an important transformation in the chemicals reactions to produce olefins which are vital raw materials for variety of industrially important compounds, including polymers, fibers and their analogues.202,203 In this context, Ondruschka and co-workers204 performed oxidative dehydrogenation of ethylbenzene on iron oxide containing multi-walled carbon nanotubes (MWCNTs) in an integral fixed-bed container using conventional heating and microwave (MW)-assisted methods. The SAED pattern confirmed the presence of hematite nanoparticles, which were arbitrarily found on the external surface of the MWCNTs (see Fig. 27). It has been observed that 3 wt% of iron oxide-MWCNTs was found to be the ideal catalyst for ethylbenzene conversion and selectivity of styrene, while all others were found to be more selective at 380–450 °C under operation in a MW field. The sample displayed excellent styrene selectivity under MW irradiation than the ones achieved by traditional heating under the identical reaction parameters.
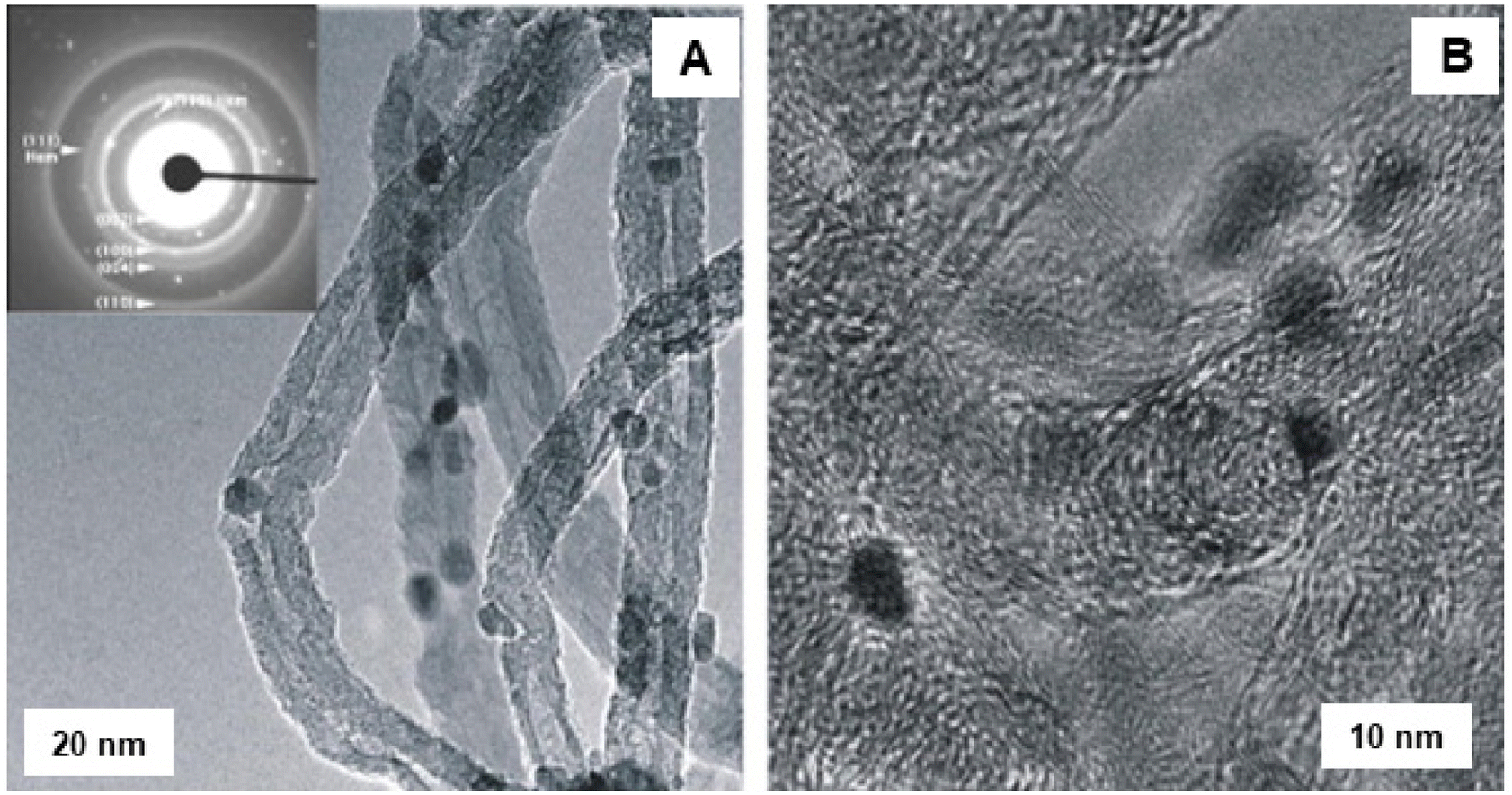 | ||
| Fig. 27 TEM images of fresh Fe2O3 modified MWCNTs: (A) LR image with SAED pattern; (B) HR-TEM image. Reproduced with permission from ref. 204 Copyright 2008, Elsevier. | ||
However, the influence of lanthanum (La), neodymium (Nd), zirconium (Zr), and aluminium (Al) on the structural properties of hematite and on its catalytic performance for the dehydrogenation of ethylbenzene has been studied;205 all dopants helped to enhance the specific surface area of the fresh and recycled catalysts thus rendering them more resistant against reduction. As the specific surface area rises, the area-dependent catalytic activity of hematite also increases except in the case of aluminum which essentially performed only as a textural agent. Notably, zirconium and lanthanum were the main dopants which decreased the selectivity of the catalytic processes, whereas other dopants did not affect the activity.
The iron-oxide NPs were synthesized within the macroporous resins through hydrolysis of Fe3+ ions, which were chemically adsorbed at the pore walls;206 the phase of the ensuing spherical iron oxide particles (2–5 nm) inserted in resin was identified as β-FeOOH. They were employed for the hydroxylation of phenol with H2O2 measured at 50% of conversion, when excellent catalytic activity for phenol hydroxylation was observed because of nanosized β-FeOOH particles within resins and exhibiting minimal aggregation.
The gold catalyst anchored on iron oxide nanoparticles were reported for the oxidation of formaldehyde and CO at low temperature.207 The catalyst being prepared by the dispersion–precipitation method,208,209 termed as FeA. For comparison, additional iron oxide support (denoted by FeB) was synthesized through precipitation technique. Both the samples were further decorated with gold nanoparticles and evaluated for the oxidation of formaldehyde and CO.207 Oxidation of CO was accomplished in a U-tube the reactor cooled in an ice bath containing NH4Cl and NaNO3 to obtain primary temperature of −25 °C. Afterwards the temperature was increased up to 25–80 °C and the initial concentration of CO was maintained by introducing air to keep the concentration 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm and the flow rate at 50 mL min−1 with gas hourly velocity (GHSV) of ca. 30000 h−1. The HCHO oxidation was achieved by reactant containing 450 ppm HCHO in a 20.8 vol% (O)/79.2 vol% (N) mixture with a total flow rate was set at 50 mL min−1 and product was analysed by GC. The catalytic activity of Au/FeA (2.5) and Au/FeB (2.5) in CO and HCHO oxidation were assessed (Fig. 28c and d). As depicted in Fig. 28c, the conversion of CO reached 89% at a temperature of −21 °C, attaining 100% conversion at −3 °C in case of Au/FeA (2.5), whereas value of only 22% was achieved at −21 °C and the total transformation only accomplished at a temperature of 23 °C on Au/FeB (2.5). In the case of oxidation of HCHO, Au/FeA (2.5) also displayed greater performance as compared to Au/FeB (2.5) at ambient temperature where 33% and 23% conversions were achieved, respectively (Fig. 28d). However, the catalyst with 2.5–3.5% loading of Au exhibits greater activity for CO conversion compare to others (see Fig. 28e and f). The superior performance of Au/FeA (2.5) was rationalized by considering higher BET specific surface area of Au/FeA (2.5) (99 m2 g−1) than that of Au/FeB (2.5) (67 m2 g−1). Further, TEM images of Au/FeA (2.5) showed the existence of smaller particles assumed to be useful for its higher catalytic applicability (Fig. 28a and b).207
000 ppm and the flow rate at 50 mL min−1 with gas hourly velocity (GHSV) of ca. 30000 h−1. The HCHO oxidation was achieved by reactant containing 450 ppm HCHO in a 20.8 vol% (O)/79.2 vol% (N) mixture with a total flow rate was set at 50 mL min−1 and product was analysed by GC. The catalytic activity of Au/FeA (2.5) and Au/FeB (2.5) in CO and HCHO oxidation were assessed (Fig. 28c and d). As depicted in Fig. 28c, the conversion of CO reached 89% at a temperature of −21 °C, attaining 100% conversion at −3 °C in case of Au/FeA (2.5), whereas value of only 22% was achieved at −21 °C and the total transformation only accomplished at a temperature of 23 °C on Au/FeB (2.5). In the case of oxidation of HCHO, Au/FeA (2.5) also displayed greater performance as compared to Au/FeB (2.5) at ambient temperature where 33% and 23% conversions were achieved, respectively (Fig. 28d). However, the catalyst with 2.5–3.5% loading of Au exhibits greater activity for CO conversion compare to others (see Fig. 28e and f). The superior performance of Au/FeA (2.5) was rationalized by considering higher BET specific surface area of Au/FeA (2.5) (99 m2 g−1) than that of Au/FeB (2.5) (67 m2 g−1). Further, TEM images of Au/FeA (2.5) showed the existence of smaller particles assumed to be useful for its higher catalytic applicability (Fig. 28a and b).207
 | ||
| Fig. 28 (a, b) TEM images of Au/FeA; (c, d) CO and HCHO conversion curves over Au/FeA and Au/GeB; (e, f) Effect of Au loading on the conversion of CO. Reproduced with permission from ref. 207 Copyright 2015, Elsevier. | ||
The iron oxide catalyst has been synthesized by the surface high-temperature synthesis (STS) approach210 and the ensuing materials were deployed for oxidation of H2S; iron oxide was also synthesized by the traditional method. The STS approach was well established for the generation of well-dispersed particles of active components which in turn showed high catalytic performance.211,212 The TEM images of FeOx/GFC glass-fiber catalysts (GFC) and traditionally synthesized FeOx/Al2O3 catalyst are shown in Fig. 29; FeOx/GFC shows the localized silica through large porous particles, while iron oxide exists mainly as flattened Fe2O3 islet with 1–2 nm long and a thickness of 0.5–1.0 nm. The conventional catalyst based on iron oxide in alumina is found with particle size (15–30 nm) over the surface of α-Al2O3 support. It has been affirmed that FeOx-based GFC, synthesized from Fe(III) nitrate and glycine by the STS method is more active for selective H2S oxidation with high conversion (55–60% at 200 °C) than conventional Fe2O3/Al2O3 granular catalyst.
 | ||
| Fig. 29 TEM images of the FeOx/GFC (left) and conventional FeOx/Al2O3 catalyst (right). Reproduced with permission from ref. 210 Copyright 2016, Elsevier. | ||
The porous silicate supported iron oxide NPs were synthesized by the continuous flow methodology and applied it for the oxidation of benzyl alcohol;213 synthesis of porous silicates supported iron oxide NPs being illustrated in Fig. 30a. The black stripes on gray silica grains of iron nanoparticles for FeAlSi and FeZrSi samples with 6–12 and 5–27 nm, respectively, were investigated by TEM analysis (Fig. 30b–e). The catalytic activity decreased with increasing iron content as depicted in Fig. 30f. The 51–66% conversion of benzyl alcohol and 69–85% selectivity to benzaldehyde for Fe/Al-SBA-15 catalyst was observed (Fig. 30g). Later, an ultra-small (<10 nm) iron oxide hydroxide nanocatalyst have been synthesized and deployed it for various oxidation reactions like alcohol oxidation, epoxidation of alkenes and organic sulfide oxidation.214
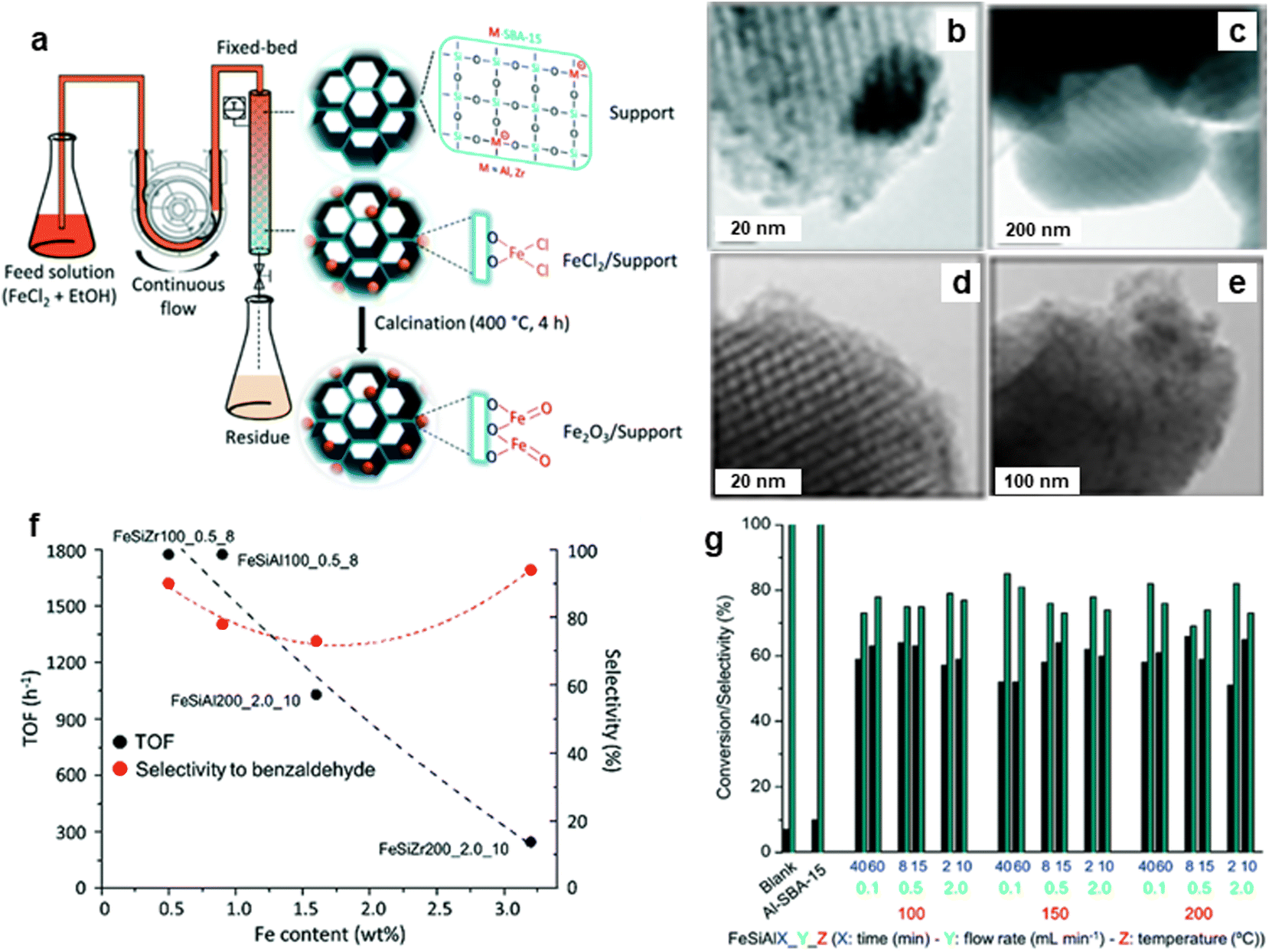 | ||
| Fig. 30 (a) Schematic for continuous flow synthesis of Fe2O3 NPs; (b–e) TEM images of FeAlSi150_0.1_40, FeZrSi150_0.1_40, FeSiAl200_0.1_40 and FeZrSi200_0.1_40 catalysts respectively, (f) Effect of Fe loading on TOF and selectivity, and (g) Performance of catalysts on conversion and selectivity. Reproduced with permission from ref. 213 Copyright 2018, The Royal Society of Chemistry. | ||
Adipic acid plays a fundamental role in the synthesis of polymers, fertilizers, lubricants, and insecticides.215,216 Recently Raja and coworkers217 have fabricated the iron and tungsten oxide-incorporated MC sphere (Fe–W-MC) with diverse carbon origins via in situ hydrothermal method. The SEM images of 10Fe 90 W-MC FF (for fructose) and 10Fe 90 W-MC GF (for glucose) revealed sphere like morphology (Fig. 31a and b). Notably, 10Fe 90 W-MC FF demonstrated that the carbon spheres produced with fructose was consistently spherical and evenly distributed, exhibiting a homogeneous surface (Fig. 31a). The HR-TEM images of 10Fe 90 W-MC FF (Fig. 31c and d) revealed that the distinctive d-spacing values of 0.29 and 0.23 nm, signifying the presence of Fe3O4 and Fe2O3 respectively. Additionally, lattice d-spacings of 0.31 nm were identified, corresponding to orthorhombic WO3. This well characterized 10Fe 90 W-MC FF catalyst was employed for the synthesis of adipic acid from cyclohexanone using H2O2 as an environmentally friendly oxidant under solvent-free conditions with the highest conversion of 93% in 12 h which is more than the traditional process of production of adipic acid from KA (ketone-alcohol) oil.218
 | ||
| Fig. 31 (a, b) SEM Images of 10Fe 90 W-FF and 10Fe 90 W-GF respectively; (c, d) HR-TEM images of 10Fe 90 W-FF. Reused with permission from ref. 217 Copyright 2022, Elsevier. | ||
Heterogeneous mesoporous Fenton nanocatalyst was synthesized using aluminum oxide anchored on iron-oxide as a support (Al2O3@Fe2O3)219via fabrication by self-assembly pathway (see Fig. 32a). TEM images of Al2O3@Fe2O3-2 core–shell nanoparticles displayed that the self-aggregation of tiny spherical nanocrystals with size of 15–20 nm (see Fig. 32b). The exterior of the α-Fe2O3 layer can be distinctly visible with (012) lattice fringes (see Fig. 32c). Furthermore, the Fast Fourier Transformation (FFT) pattern of Fig. 32c was displayed in Fig. 32d, which demonstrated the existence of γ-Al2O3 and α-Fe2O3. The diffraction spots observed in the FFT pattern accurately matched and indexed with the crystal structures of γ-Al2O3 (A) and α-Fe2O3 (F) (see Fig. 32e). The as prepared Al2O3@Fe2O3-2 catalyst has a very high BET surface area of 385 m2 g−1 with an average pore size of 3.2 nm. This mesoporous Al2O3@Fe2O3-2 nanocatalyst demonstrated significant catalytic activity in the one-pot transformation of cyclohexanone into adipic acid in a water-based system in presence of H2O2 as a green oxidant. Recently, TiO2@Fe2O3 catalyst prepared via sol–gel method was employed for the cyclohexene oxidation using molecular oxygen achieved an impressive 97% selectivity for adipic acid.220
 | ||
| Fig. 32 (a) Schematic synthesis of Al2O3@Fe2O3 mesoporous core–shell catalyst; (b–d) TEM images of Al2O3@Fe2O3-2; (e) Crystal structures of γ-Al2O3 (A) and α-Fe2O3(F). Reproduced with permission from ref. 219 Copyright 2013, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
Although oxygen gas is the most cost-effective and easily accessible oxidant, its utilization is limited due to the safety concerns associated in terms of its handling. Consequently, it is one of the least commonly employed reagents in chemical processes.221,222 By considering up-to-date research findings on this issue, pure oxygen can be safely utilized on a large scale in continuous-flow reactors for liquid-phase aerobic oxidations. Moreover, this approach offers significant advantages in terms of synthetic outcomes and manufacturing efficiencies. The pharmaceutical industry can draw valuable insights from the established protocols in the bulk chemicals sector for handling aerobic oxidation processes. For successful implementation of large-scale molecular oxygen processes, it is imperative to foster collaboration among process chemists, chemical engineers, and process safety experts.223
2.4. Alkylation reactions
Friedel–Crafts alkylation reactions are universally known for the synthesis of the industrially significant organic compounds. e.g., alkylation of benzene with benzyl chloride (BC) enables routes to benzylbenzene,224–226 whose derivatives serve as essential scaffolds for pharmaceuticals and fine chemicals.227,228 In this chemical reaction, iron oxide NPs exhibits an influential role.In this regard, a Fe/SBA-15/carbon materials (Fe2O3–SBA-15 and Fe–carbon) prepared by chemical vapor infiltration (CVI) process utilizing ferrocene as source of Fe and carbon raw materials.229 These Fe2O3 NPs, anchored on SBA-15 silica or metallic Fe consistently introduced in MC, were prepared by selective oxidation of the carbon constituent in air or by eliminating the silica template, respectively. Two sets of catalysts were prepared namely Fe2O3/SBA-15-t and Fe/carbon-t (where t represents minute), via two different processes. Fe2O3/SBA-15-t catalyst were obtained via calcination of the Fe/SBA-15/carbon composites at 500 °C in an aerobic atmosphere for 3 h to eliminate the carbon precursor with concurrent formation of Fe2O3 from Fe species. On the other hand, the Fe/carbon-t were prepared by calcination of the Fe/SBA-15/carbon materials at 800 °C in Ar for 2 h.229Fig. 33 depicts the TEM of different Fe2O3/SBA-15-t composites, where the catalyst synthesized at t = 20 min showed a fairly uniform distribution of Fe2O3 NPs with ∼2 nm (loading 21–22 wt%) in the mesoporous channels of SBA-15. Fe2O3-15–40 form small aggregates and Fe2O3 nanoparticles in Fe2O3/SBA-15-60 show lattice fringes of 0.275 nm (see Fig. 33b and c, inset), confirming the hexagonal hematite phase. Fe2O3-15–40 displayed excellent catalytic activity and selectivity for the benzene alkylation with BC. Importantly, Fe2O3/SBA-15 offered high selectivity and its catalytic performance was extremely sensitive to the reaction temperature. Both Fe2O3/SBA-15-20 and Fe2O3/SBA-15-40 catalysts exhibited good BC conversion (94%) within 5 and 10 min, respectively. The catalyst Fe2O3/SBA-15-40 synthesized by impregnation technique with the same Fe2O3 loading (denoted a Fe2O3/SBA-15-40-im) showed a much less conversion compared to the Fe2O3/SBA-15-40 catalyst.
 | ||
| Fig. 33 (a–d) TEM images of Fe2O3/SBA-15-20; Fe2O3/SBA-15-40; Fe2O3/SBA-15-60; and Fe2O3/SBA-15-90 catalysts respectively; inset in (c): high-resolution TEM image of (104) reflection of α-Fe2O3. Reproduced from ref. 229 Copyright 2009, Elsevier. | ||
For the purposes of Friedel–Crafts acylation, the α-Fe2O3 NPs (30 nm) were synthesized employing superabsorbent nanopolymer (SANP) via thermal treatment.230 The salient feature of this approach has been to use salep-g-poly acrylic acid (salep-g-PAA) as a less expensive and easily accessible coating material which possesses –OH and –COOH functional groups to increase the binding affinity to metal ions as well as to manage the size and structure of α-Fe2O3 excluding formation of clusters.231,232 A variety of aromatic ketones were formed in moderate to good yields (71–94%) with low mol% of the catalyst (1 mol%) in 1 h (see Fig. 34). Further, catalytic performance of α-Fe2O3 NPs was explored for Friedel–Crafts acylation reaction with p-methyltoluene and benzyl bromide afforded product with good yield (87%).
 | ||
| Fig. 34 Friedel–Crafts acylation using the hematite nanoparticles.230 | ||
Preparation of iron oxide NPs using a hydrothermal protocol has been reported for alkylation of benzene with BC to generate diphenylmethanes.233 The benzylation of benzene with BC was performed using α-Fe2O3 nanoparticles, and the corresponding product was obtained in excellent activity and selectivity; benzylation reaction over α-Fe2O3 NPs were also explored for substituted aromatic compounds, e.g., toluene and p-xylene (see Table 1). It has been observed that 100% conversion of BC was obtained within 3 min.
| Substrate | Time (min) | Temp. (K) | BC conversion (%) | Reaction products (selectivity (%)) |
|---|---|---|---|---|
| Benzene | 3 | 358 | 100 | Diphenylmethane (97.76%) |
| Toluene | 3 | 368 | 100 | o-Benzyltoluene (52.36%) |
| p-Benzyltoluene (47.64%) | ||||
| p-Xylene | 3 | 378 | 100 | 2-Benzyl-1,3-dimethylbenzene (99.93%) |
Activated hematite catalyzed Friedel–Crafts acylation of benzene (i.e., chlorobenzene and nitrobenzene) has been reported under benign conditions.234 The activation of hematite was achieved via sonication under air atmosphere, followed by heating at 200 °C, which certainly helped improve the activity of the catalyst. A range of benzene derivatives were smoothly transformed to the desired acylated compounds with moderate to good yields (Fig. 35) and the activated α-Fe2O3 can be recovered with minor loss in catalytic performance. The benefits of employing this solvent-free methodology encompass easy reaction set-up, mild reaction parameters, greater efficacy, and selectivity, which bodes well for the future use of the catalyst for various other greener organic transformations.
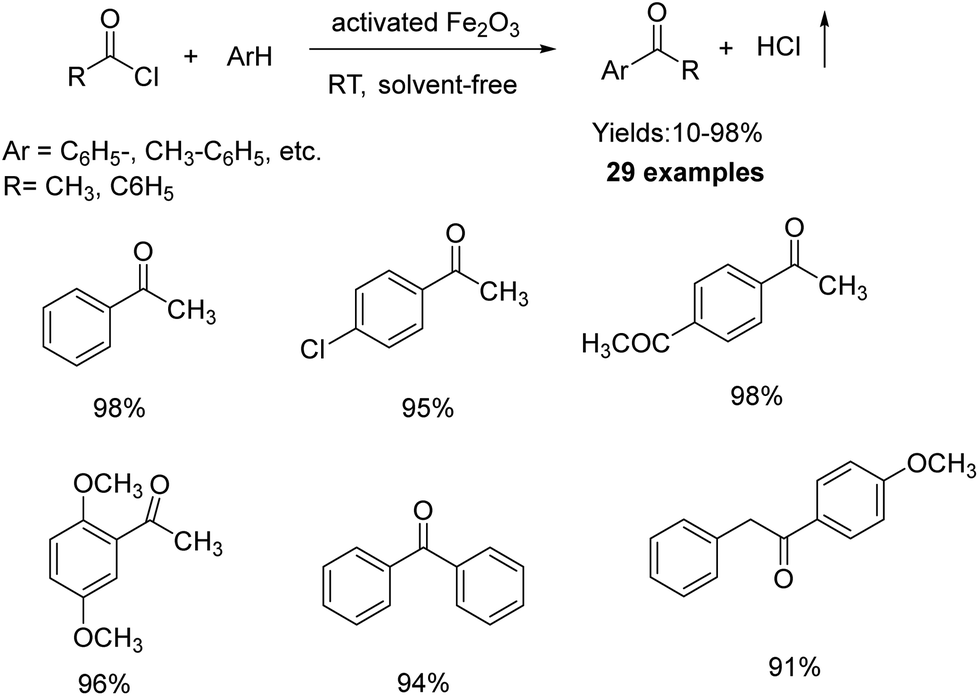 | ||
| Fig. 35 Friedel–Crafts acylation and benzoylation of aromatic compounds. Reproduced with permission from ref. 234 Copyright 2010, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
Recently, Guo and colleagues235 reported a blend of mechanically combined lipophilic α-Fe2O3 and CaCO3 NPs as a highly effective catalyst in the Friedel–Crafts acylation reaction. In this study, α-Fe2O3 nanoparticles were synthesized using a continuous precipitation method in a micro-mixer, while CaCO3 NPs were prepared in a flow reactor (see Fig. 36). The modified NPs were then deployed as catalysts in the acylation process, leading to a high yield of 97.8% under optimized conditions. Interestingly, the addition of CaCO3 NPs enhanced the catalytic efficiency compared to using only modified α-Fe2O3 NPs. This novel acylation method demonstrated effectiveness across various substrates.
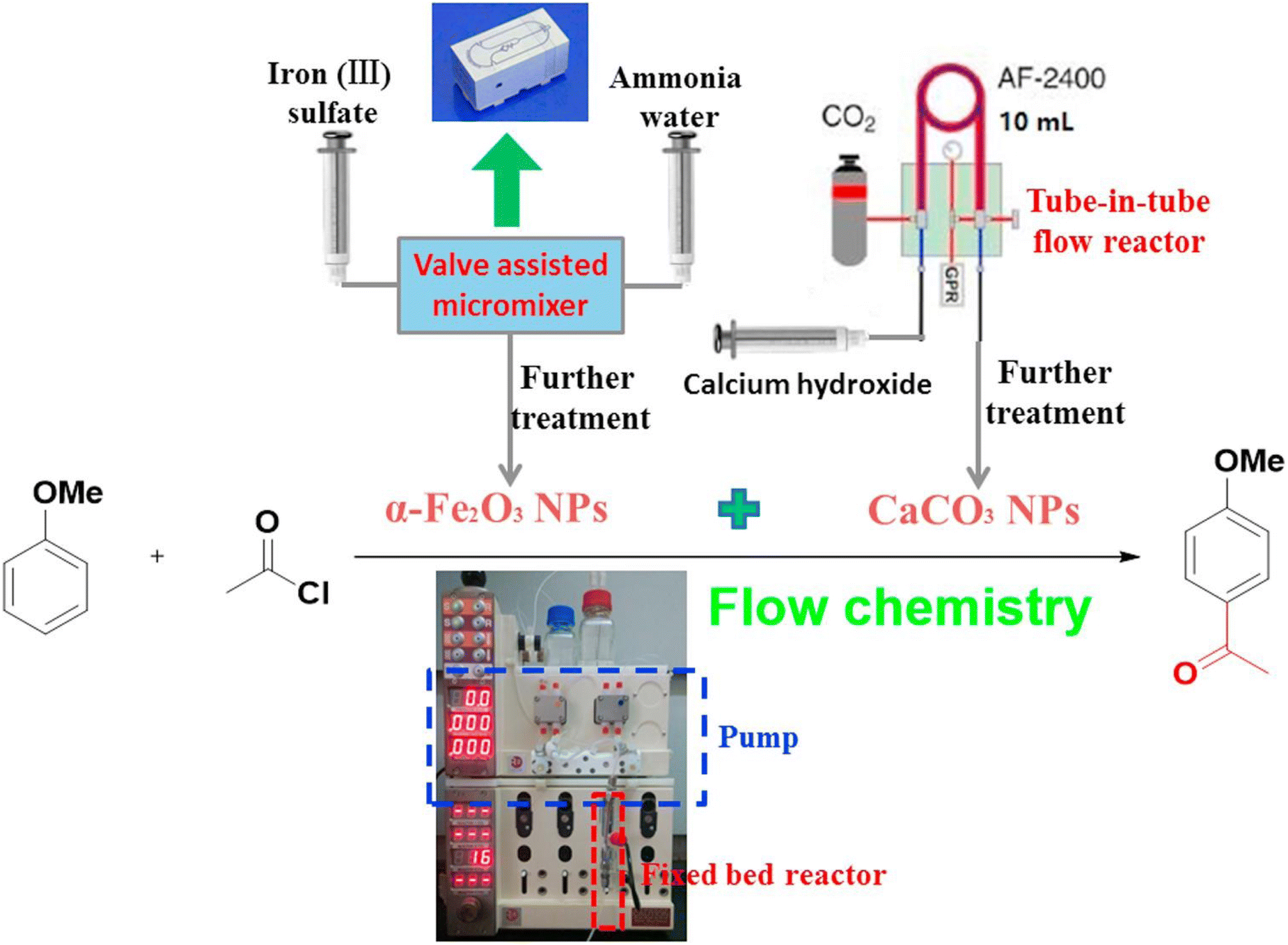 | ||
| Fig. 36 Schematic representation of continuous flow setup for Friedel–Crafts acylation reaction. Reproduced with permission from ref. 235 Copyright 2018, Elsevier. | ||
2.5. Fischer–Tropsch synthesis
The transformation of syngas into value-added linear hydrocarbons, popularly referred to as Fisher–Tropsch synthesis (FTS), is considered an important chemical transformation in heterogeneous catalysis.236–238 However, creating FTS catalyst for the production of an array of compounds with greater performance and selectivity still remains a formidable task for chemists, the use of iron-based catalyst can potentially offer a viable solution because such inexpensive, earth-abundant materials have displayed good performance and have capacity to produce various hydrocarbon compounds such as diesel, wax, gasoline to C2–C4 olefins or paraffins.239–243The graphene oxide incorporated monodispersed iron oxide NPs were employed as a catalysts for FTS.244 In this process, it was discovered that the thermal behavior of supporting materials was important for good catalytic performance and selectivity especially for C5+ hydrocarbons. The size and the structure of monodispersed iron oxide NPs and the final catalyst, Fe/PGO (pyrolytic graphene oxide), are depicted in Fig. 37. It was noted that the iron oxide nanoparticles were keeping the cubic morphology with a size of about 8 nm (see Fig. 37a) and can be accumulated into a “supper lattice” assembly because of their homogeneous distribution;245 iron oxide NPs were very well-dispersed throughout the carbon support (see Fig. 37b). Because of the heat processing, the amount of “O” and “S” species decreased, and the degree of graphitization increased. It is noteworthy to highlight that oxygen groups onto the surface could change the surface features of the active iron form, which provides diverse catalytic performance.
 | ||
| Fig. 37 (A and B) TEM images of iron oxide NPs and Fe/PGO respectively (scale bars: 50 nm). Reproduced with permission from ref. 244 Copyright 2013, Elsevier. | ||
The prepared catalyst has been employed for the CO conversions as time-on-stream function of FTS at 270 °C and their product selectivity's are depicted in Table 2. At the fixed iron loading for all catalysts (10%), when graphitization temperature was increased to 380 °C ((Fe/PGO-380), and 600 °C (Fe/PGO-600)), clearly an improved CO conversion was observed. As increasing treatment temperature up to 800 °C, only marginal increase in the activity was noticed when compared to Fe/PGO-600, signifying the similar physical properties of PGO-600 and PGO-800; comparable selectivity patterns were discerned for activated carbon containing catalysts, Fe/AC-HNO3 and Fe/AC-HNO3-800, based on PGO resources.
| Catalysts | Conversion% | CO2 yield (%) | Selectivity in hydrocarbon wt.% | Olefin/paraffin in C2–C4 | |||
|---|---|---|---|---|---|---|---|
| CH4 | Methanol | C2–C4 | C5+ | ||||
| a Reaction parameters: 80 mg catalyst having 10% iron, 3 MPa syngas,1 270 °C. The C2–C4 oxygenates were included in the C2–C4 products. | |||||||
| Fe/PGO | 10.2 | 0.6 | 21.4 | 9.7 | 52.0 | 16.9 | 0.24 |
| Fe/PGO-380 | 30.4 | 5.1 | 21.2 | 8.6 | 46.6 | 23.6 | 0.77 |
| Fe/PGO-600 | 35.0 | 10.8 | 12.7 | 3.2 | 39.1 | 45.0 | 1.57 |
| Fe/PGO-800 | 36.7 | 10.1 | 12.6 | 3.2 | 36.8 | 47.4 | 1.44 |
| Fe/AC-HNO3 | 7.8 | 0.7 | 22.5 | 6.7 | 47.1 | 23.7 | 0.80 |
| Fe/AC-HNO3-800 | 13.6 | 1.7 | 16.0 | 4.1 | 40.8 | 39.1 | 1.28 |
The dynamic behavior of the model iron-based Fisher–Tropsch catalysts has been demonstrated wherein Ag and Cu delafossite model systems were synthesized via co-precipitation method under basic conditions using metal nitrates as precursors, followed by calcination at elevated temperatures.246 The catalysts were characterized using AAS, XRD, and Mössbauer spectroscopy and the catalytic activity was assessed in a slurry reactor. Upon activation of the catalysts under different conditions, via H2 and CO-activations (commonly employed for FT catalysts), the evolution of the different phases was monitored using in situ XRD to understand the dynamic performance of such catalytic systems. The presence of either Ag or Cu assisted in the generation of magnetite during the H2-activation step, which ultimately resulted into conversion of magnetite into metallic iron, possibly via H2-spill-over. However, for CO-activation, the influence of as synthesized catalysts (Cu, and Ag catalyst) seems to be less prominent. Silver nanoparticles displayed higher catalytic activity generating not only the olefinic products but also relatively higher amount of undesired CO2 byproduct. The generation of CO2 has been attributed to the existence of superparamagnetic iron species formed during the activation step and/or reaction conditions. Alternatively, hydrogen transfer from the group 11 metal surface could partly reduce the magnetite surface, aiding in the carburization process; Fig. 38 illustrates the changes in iron-based FT catalyst with group 11 metals presents.
 | ||
| Fig. 38 Schematic diagram for the transformations over the iron-based FT catalyst. Reproduced with permission from ref. 246 Copyright 2013, Elsevier. | ||
The contribution of hematite in the precipitated iron-based reported for FT catalysts, by Duvenhage and co-workers;247 where the co-precipitation method was used originally reported by the same group. By simply varying the hold time between the precipitation and the filtration steps, the content, and the crystallite size of hematite in the catalysts could be tuned. In their catalytic studies, the low amount of hematite content (1.5 to 25 wt%) did not improve the performance of the catalysts presumably due to lower surface area and pore volume. However, catalysts with hematite content between 25 to 45 wt% not only displayed better catalytic performance but higher reducibility and improved attrition resistance.
The influence of support for the iron-based FT catalysts were analyzed by preparing four supports namely SBA-1 (silica), activated carbon (carbon), CMK-3 (carbon) and CNTs (carbon) and incorporated Fe-species via incipient wetness impregnation method.248 After a calcination step under nitrogen, these materials were treated with CO as a part of the activation step; finally, they were characterized and studied for the FT synthesis. During the calcination step, hematite was formed when silica was used as a support, while the decomposition of iron nitrate in carbon materials mostly formed Fe3O4, which cannot exclude the presence of maghemite phase. The CO activation step allowed the facile carbidisation process to yield Fe-carbide. Among these hybrid systems, silica-supported catalysts exhibited the lowest performance compared to carbon-supported catalysts presumably because of the variation in phase compositions. The greater catalytic performance was obtained with carbon-nanotube supported catalysts (Fig. 39b). The variation in performance was ascribed to the existence of the Fe-oxides as well as the formation of Fe-carbide during the CO-activation step (Fig. 39a). In case of CNTs, the highest activity was explained by the formation of Fe-carbide-magnetite core–shell nanoparticles, which act as “nodules” in a confined area exhibiting higher performance. It is crucial to emphasize that the catalysts without CO-activation did not catalyze the reaction, confirming that the iron-carbide-magnetite core–shell serve as efficient catalytic centers.
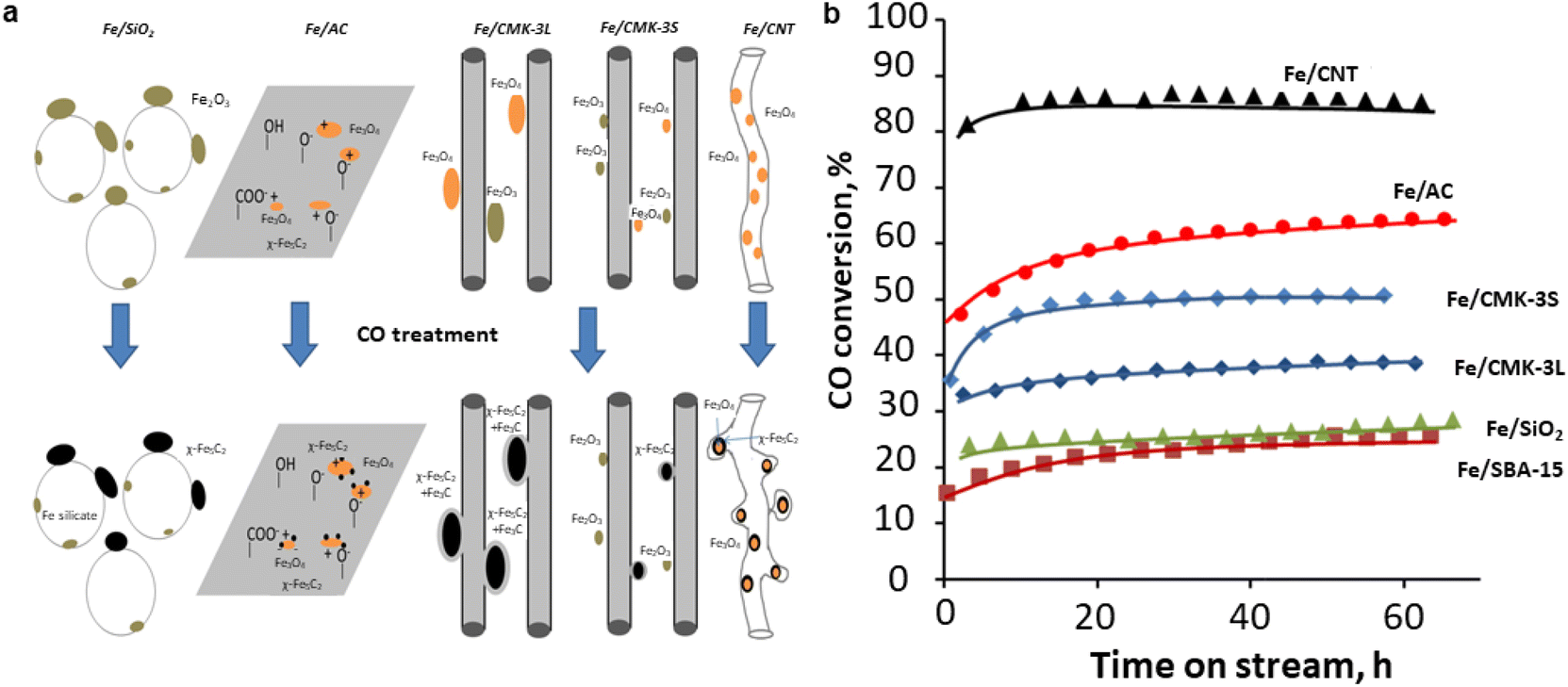 | ||
| Fig. 39 (a) Evolution of iron species over silica and carbon support for CO activation; (b) Catalytic performance of iron catalysts. Reproduced from ref. 248 Copyright 2014, Elsevier. | ||
A relatively new and sophisticated inelastic neutron scattering (INS) technique was deployed to probe the mechanistic details of the hydrogen pre-treatment process for iron-based FTS catalysts used in CO hydrogenation.249 The study was performed using hematite (prepared by co-precipitation method) as pre-catalysts, which were first activated using H2, at two different temperatures (623 and 723 K), and then subsequently investigated for CO hydrogenation using INS technique, coupled with TEM, XRD and other material characterization techniques. The INS spectra indicated the presence of carbonaceous layers with primarily saturated hydrocarbons for the samples heated at 623 K. However, this activation step for the high temperature sample exhibited larger quantity of amorphous carbons on the surface, thereby reducing the catalytic performance. The research showed that the activation step might be disadvantageous for such catalytic systems although a detailed mechanistic investigation is warranted.
The surface characteristics of 1D nanostructured iron oxides exhibit significant distinctions when compared to their counterparts with spherical shapes.250,251 Among them, nanotubes are of particular importance due to its rolling, which imposes stress and allows the electron wave functions to be different within nanotubes interior and exterior walls. In FTS, the selectivity of the product is the main issue because of Anderson–Schulz–Flory distribution of hydrocarbons.252 Well-defined pore size, structure and surface properties have significant effect on the selectivity of FTS products.253,254 Thus, nanotubes with unique properties such as mesoporous structure, uniform PSD, and surface properties are attractive for FTS. Recently, Sun and co-workers has synthesized Fe2O3 nanorods and nanotubes by the hydrothermal process of ferric chloride in the existence of KH2PO4 at 220 °C at certain reaction time.255 TEM images of nanorods prepared after 6 h, show a diameter of around 80 nm and a length of 270 nm. The nanotubes synthesized after treatment for 48 h, show outer diameters, inner diameters, and lengths between 60–80, 40–60 and 260 nm respectively (Fig. 40).
 | ||
| Fig. 40 (a, b) SEM and TEM analysis of Fe2O3 nanorods; (c, d) SEM and TEM images of Fe2O3 nanotubes. Reproduced with permission from ref. 255 Copyright 2016, The Royal Society of Chemistry. | ||
The catalytic FTS reaction under simulated industrial conditions at 280 °C, 2.0 MPa, and a H2/CO ratio of 2 revealed that the activity of Fe2O3 nanorods increases primarily for 16 h and reaches a maximum value of 39.5% and then falls slowly up to 12.9% at 120 h. On the other hand, for Fe2O3 nanotubes the CO conversion values reached a maximum within 16 h, and remained constant during the 120 h period. The ratio of olefins to paraffin (O/P) for Fe2O3 nanorods and nanotubes was found to be 1.85 and 3.0, respectively along with the selectivity for C5+ products (63.4% and 72.1%, respectively). These findings clearly indicate superior selectivity's of nanotubes over nanorods. Reduction and carburization of Fe-based catalysts allegedly activate catalyst for FT synthesis due to carbide formation.256
Co- and Fe-based catalysts257 were investigated for FT process wherein Fe-based catalyst were synthesized by reportedly similar method in which a Fe(100)/Zn(10) precursor (atomic ratios) mixture was obtained by semi-batch-co-precipitation of Fe and Zn nitrates at constant pH to produce porous Fe–Zn oxy/hydroxycarbonate powders. The obtained powders were heated at 350 °C for 1 h and further facilitated by potassium carbonate and copper nitrate by incipient wetness impregnation method, followed by calcinations at 400 °C for 4 h. Samples with a Cu/Fe a (0.01) and K/Fe with atomic ratios of 0.02, 0.04 and 0.1 were also synthesized; denoted as “Fe2K”, “Fe4K” and “Fe10K” respectively.
All prepared catalysts were deployed in FT process under various reaction conditions. During the hydrogenation of CO2, conversion of H2 and CO2 were found of 20% and 9%, respectively. The degree of the conversion remains constant over the entire period of the reaction (60–80 h). In fact, in addition to water, gaseous and liquid hydrocarbons, a significant amount of CO was observed in the product pool. Subsequent to subjecting the catalyst to testing in the presence of CO2, the activity of the catalysts with H2/CO mixtures were evaluated for comparative study analysis when CO and H2 conversions were identified as 12% and 22%, respectively, which are greater than with the similar H/C ratio of H2/CO2 mixture. These findings suggest that CO2 has lesser performance than CO under similar conditions for an iron-based catalyst. Remarkably, it was found that in contrast to the COx conversion rates, the hydrocarbon distributions achieved from the hydrogenation of CO and CO2 were completely different. For CO, a direct Anderson–Schulz–Flory (ASF) distribution of hydrocarbons was accomplished, with probability of chain growth for the C3–C10 species of 0.71 and a small negative deviation from the linear trend of species C1 and C2. These results are completely consistent with the existing literature,258–260 which suggests that the alkali-doped Fe-based catalysts show less selectivity for CH4 during the hydrogenation of CO.261 In contrast, for CO2, the yield of hydrocarbons (C1–C3) is substantially better and the chain growth probability of species C3–C10 decreased to a degree of 0.65. The other fractions of olefins, specifically C2–C10 products, are also found as a function of the carbon number. In this case, the olefin fractions decrease monotonically with C number, tending to an asymptote value close to 70% for C4–C10 species. It indicates that the distribution of product and the yield of olefin in the hydrocarbons are constant over the course of continuous CO hydrogenations and, after catalytic tests with CO2, the mixture H2/CO quickly reaches a steady state after switching back.
An et al.262 synthesized Fe3O4@iron carbide NPs by pyrolysis of Fe-containing MOFs for FTS. The synthesis of the Fischer–Tropsch catalyst Fe-MIL-88B/C and Fe-MIL-88B-NH2/C is illustrated in Fig. 41a. The formation of surface carbonates comes from the CO in the feed gas, as shown in Fig. 41b; core–shell Fe NPs were uniformly distributed on iron carbide. The lattice planes (311) and (111) correspond to the inner core Fe3O4 and the outer shell χ-Fe5C2, respectively (Fig. 41c). In the first 30 hours, the catalyst activity increased and then remains constant with no evidence of quenching even after 200 hours. The selectivity to olefins and long-chain hydrocarbons remains unchanged during test (Fig. 41d and e).
 | ||
| Fig. 41 (a) Schematic representation for the synthesis of Fe3O4@Fe5C2 nanocatalyst; (b) Schematic diagram for production of Fe3O4@Fe5C2 catalysts and Fischer Tropsch surface chemistry; (c) HR-TEM images of Fe-MIL-88B-NH2/C; (d, e) The catalytic activity of Fe-MIL-88B-NH2/C nanocatalyst. Reproduced with permission from ref. 262 Copyright 2016, American Chemical Society. | ||
Recently, Wu et al.263 reported boron carbon nitride supported iron oxide nanocatalyst (Fe@BCNNSs) through thermal decomposition for the FTS. The TEM images affirmed that the black iron oxide nanoparticles were evenly dispersed across boron carbon nitride sheet (BCNNs) with particle size of 2–18 nm (Fig. 42a and b). The lattice space of dark particle and the encapsulated layer was 0.21 nm and 0.34 nm, respectively (Fig. 42c and d). The conversion of CO enhanced remarkably with increasing reaction temperature at 320 °C and reached 95% at 340 °C over Fe@BCNNSs catalyst (Fig. 42f). The selectivity for different hydrocarbons is also illustrated (Fig. 42e). The increased stability of the catalyst is due to the special encapsulated structure even after 1000 hours of operation. The BCNNSs shell prevents catalyst deactivation under high temperature conditions.
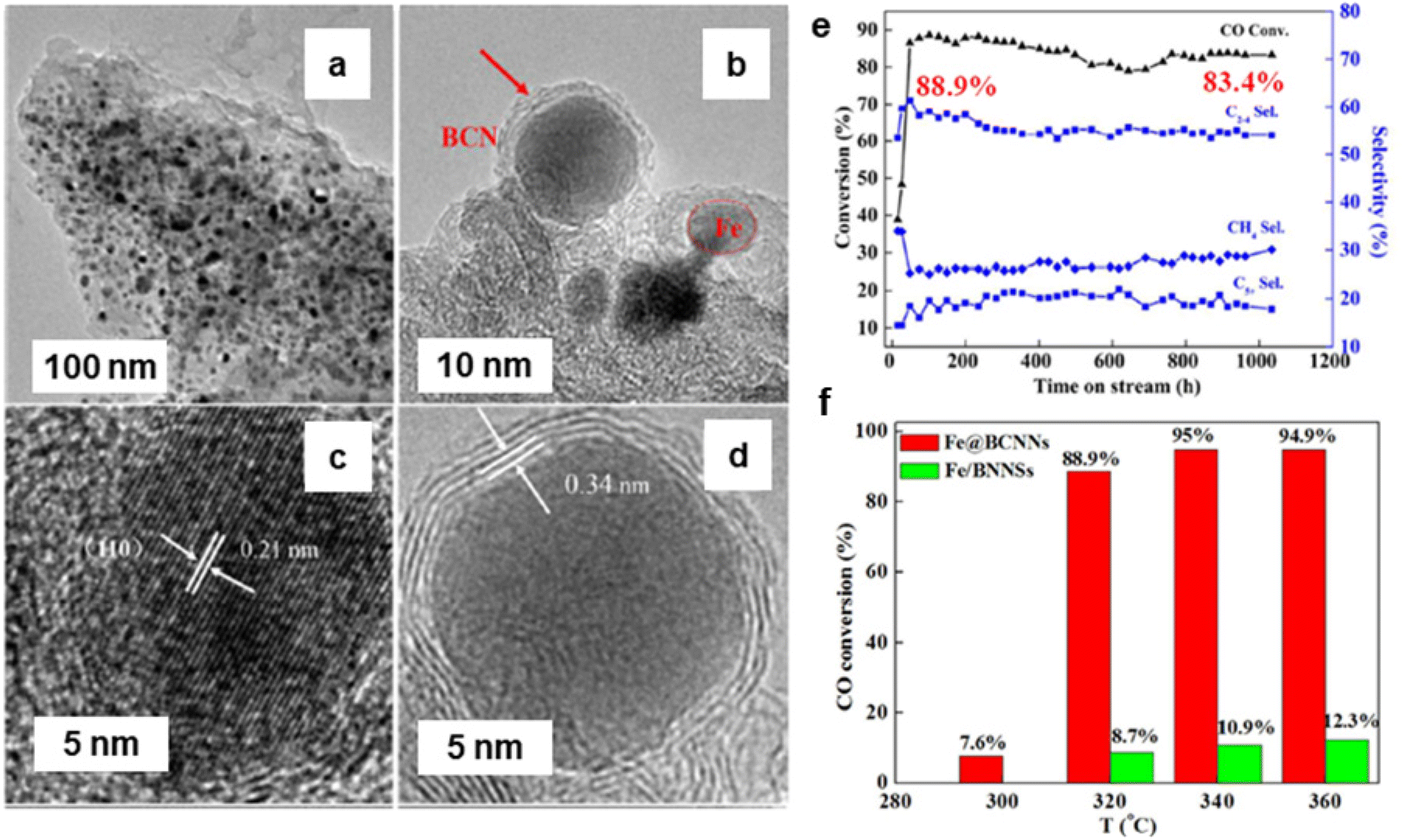 | ||
| Fig. 42 (a, b) TEM images of Fe@BCNNSs nanocatalyst; (c, d) HR-TEM images Fe@BCNNSs of nanocatalyst; (e, f) Activity of the Fe@BCNNSs of nanocatalyst. Reproduced with permission from ref. 263 Copyright 2017, American Chemical Society. | ||
2.6. Multicomponent reactions
Multicomponent reactions (MCRs) involve the simultaneous reaction of three or more reactants within a single vessel to produce a final product that incorporates features from all the reactive components.264 3,4-Dihydropyrano[c]chromenes and its analogues are important building blocks in numerous areas including chemistry, biology and pharmacology;265–269 representative compounds from this class display spasmolytic, diuretic, anti-cancer, anticoagulant, and anti-anaphylactic activities.270,271 Additionally, they can be employed as a cognitive activator for the therapeutic interventions of neurodegenerative diseases, such as Alzheimer's disease, Huntington's disease, amyotrophic lateral sclerosis, etc.272,273In this context, the combustion-based preparation of α–Fe2O3 nanopowder were developed274 and their catalytic performance was studied for single step preparation of 3,4-dihydropyrano[c]chromenes under mild and benign reaction conditions. The α–Fe2O3 nanopowder was found to be very effective for the three-component condensation of aldehyde, malononitrile and 4-hydroxycoumarin at RT (Fig. 43). This appears to be an ideal alternative for the preparation of 3,4-dihydropyrano[c]chromenes as the reactions are rapid, clean, and the target compounds are obtained with satisfactory yields. The recyclability of α-Fe2O3 catalyst was also inspected wherein the catalyst can be recovered readily and they are reusable till five tested cycles.
 | ||
| Fig. 43 One-pot preparation of 3,4-dihydropyrano[c]chromenes catalyzed by α-Fe2O3. Reproduced with permission from ref. 274 Copyright 2010, Elsevier. | ||
Afterwards, iron oxide nanocatalyst was used for benzimidazoles synthesis.275 The acidic nature of the prepared iron oxide catalyst activates aldehyde which participates in the nucleophilic addition of amine. In the next steps, Michael addition and air oxidation lead to the benzimidazole product. Recently, a benign nanocatalyst (NiFe2O4/geopolymer) were prepared for the synthesis of imidazole derivatives via ultrasonication.276 The nanocomposite was prepared by two steps including (i) synthesis of NiFe2O4 NPs by coprecipitation method, and (ii) synthesis of bentonite-based geopolymer and the addition of NiFe2O4 NPs to geopolymer (Fig. 44a). The cubic shape and particle size (30 nm) of NiFe2O4 NPs were studied by FE-SEM analysis (Fig. 44b and c). Fig. 44d and e shows the presence and dispersion of NiFe2O4 NPs on geopolymer. The stepwise mechanism is depicted in Fig. 44f. The Lewis acid sites of the nanocomposite activates the aldehyde while ammonium chloride was used as a nitrogen source for the synthesis of imidazole derivatives.
 | ||
| Fig. 44 (a, b) Field emission SEM images; (c, d) TEM images; (e) Application; (f) The mechanism for the preparation of imidazole derivatives. Reproduced with permission from ref. 276 Copyright 2020, Springer Nature. | ||
The Biginelli reaction is notably acknowledged for its role in the syntheses of dihydropyrimidones/thiones and essential compounds with biological significance.264 Bhaumik and coworkers277 developed chemically conjugated catalyst that has been engineered through the thio–ene click reaction, incorporating Fe3O4 NPs into mesoporous SBA-15 (Fe3O4@SBA-15). The HR-TEM image confirms honey-comb like hexagonal pattern (see Fig. 45a) with a pore size ranging from 6–7 nm and selected area electron diffraction (SAED) pattern affirms the existence of crystalline Fe3O4 (see Fig. 45b). This as prepared Fe3O4@SBA-15 heterogeneous catalyst was utilized in the one-pot Biginelli condensation process (Fig. 45c) enabling the synthesis of diverse and valuable 3,4-dihydropyrimidin-2(1H)-ones.
 | ||
| Fig. 45 (a) TEM images of Fe3O4@SBA-15 (FFT pattern is shown in the inset); (b) SAED Pattern of Fe3O4@SBA-15. (c) Biginelli condensation reaction over Fe3O4@SBA-15. Reproduced with permission from ref. 277 Copyright 2012, The Royal Society of Chemistry. | ||
Additionally, highly innovative and efficient green catalyst i.e. Fe3O4@[Ni(bpy)2(py-tmos)] was developed for the Biginelli condensation reaction278 and this well characterized nanocatalyst was employed for the synthesis of a broad spectrum of 3,4-dihydropyrimidin-2(1H)-ones from the mixture of urea, aromatic-aldehyde and ethyl-acetoacetate. The synthesis of α-aminonitriles, critical intermediates for α-amino acid derivatives, amides, diamines, peptides, proteins, and heterocycles, was accomplished through the Strecker reaction, using methylarene oxidation. This reaction, considered one of the oldest and most cost-effective methods, involves the nucleophilic addition of cyanide ions to imines. Similarly, wet impregnation method was deployed for the fabrication of nanocrystalline spinel ferrites i.e. ZnFe2O4.279 It is represented by MFe2O4 (M = Ni, Zn, Mn, Co, Mg, etc.), and these heterogeneous Lewis acid catalysts are attracting significant attention for their unique structures, superior selectivity, and catalytic activities compared to monometallic counterparts.280 The most favorable outcome has been achieved when using ZnFe2O4 as a dual catalyst, among these metal oxides (ZnFe2O4 > NiFe2O4 > Fe3O4). Due to high Lewis acidity of as prepared ZnFe2O4 catalyst further utilized for the Strecker reaction in presence of KI. Additionally, KI could serve as a scavenger for any elemental iodine that might be produced during the reaction, preventing side reactions and improving the yield of the desired product. This as prepared KI/ZnFe2O4 catalytic system utilized for oxidation of methyl arenes using aqueous tert-butyl hydroperoxide (TBHP) as the oxidizing agent and trimethylsilyl cyanide (TMSCN) as a nitrile source, achieving a yield of up to 88% within 2 h.
2.7. Miscellaneous reactions
In this section, a variety of miscellaneous catalytic reactions, exploring iron oxide NPs are described. Nanomaterials with a core/shell assembly can be developed with various elements/components to prepare multifunctional materials to fine tune their catalytic performances.92,281,282 Besides composition, the size,283 shape,284 and assemblies also affect the catalytic activity. The anisotropic nanocomposites display versatile fundamental properties and considerable efforts are being made for the synthesis of novel functional 1D core/shell nanomaterials.285,286A reusable rod-like β-FeOOH@poly(dopamine)–Au–poly(dopamine) nanomaterials were prepared for reduction of Rhodamine B (RhB).287 Initially, β-FeOOH was coated with poly(dopamine)(PDA) to generate a core/shell nanomaterial, followed by the decoration of Au nanoparticles on β-FeOOH@PDA nanorods. Another layer of PDA then coated onto the ensuing particles to avoid the leaching of Au nanoparticles (see Fig. 47A). TEM images of the nanomaterials during various steps are depicted in Fig. 47B, which shows an average size of the β-FeOOH nanorods amounting to 260 nm. The obtained β-FeOOH@PDA core/shell nanoparticles were properly distributed on to Cu grid leaving out agglomeration. Next, the catalytic applicability of β-FeOOH@PDA–Au was examined for the reduction of Rhodamine B (RhB) where the amount of catalyst used seemingly played a pivotal role as the catalytic activity strongly improved with the increasing quantity of the catalyst. Due to the thin shell layer of PDA, the reusability of β-FeOOH@PDA–Au–PDA radically increased; excellent yield of 98% was obtained even after five cycles when using β-FeOOH@PDA–Au–PDA as catalysts, whereas the β-FeOOH@PDA–Au demonstrated that it was maintained at only 73%.
 | ||
| Fig. 46 (a, b) SEM images of Fe3O4-bpy-Ni(II). Reproduced with permission from ref. 278 Copyright 2018, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
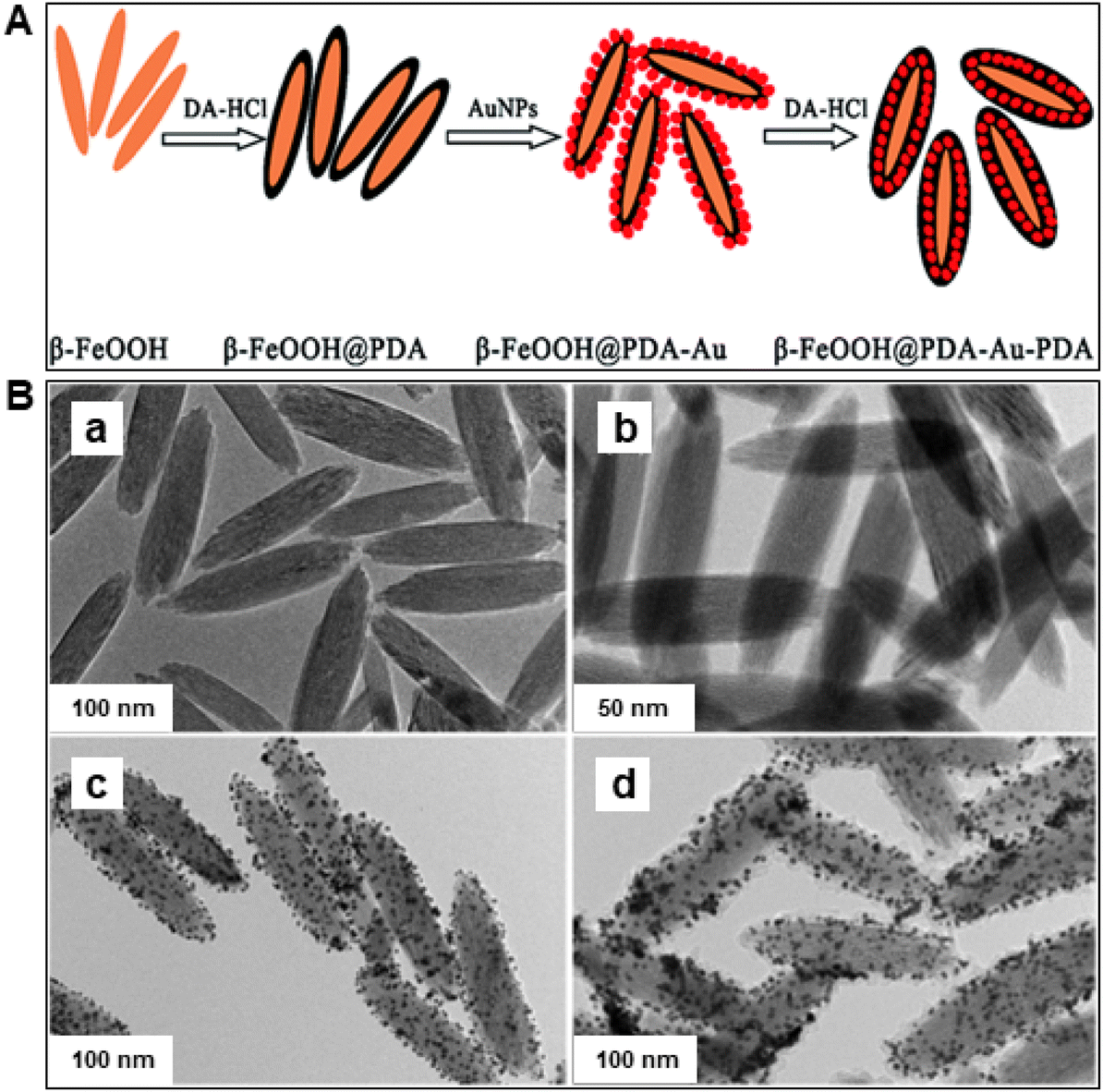 | ||
| Fig. 47 (A) Synthesis route of β-FeOOH@PDA–Au–PDA nanostructure; (B) TEM images of prepared catalysts: (a) β-FeOOH, (b) β-FeOOH@PDA, (c) β-FeOOH@PDA–Au, and; (d) β-FeOOH@PDA–Au–PDA. Reproduced with permission from ref. 287 Copyright 2015, The Royal Society of Chemistry. | ||
Earth surface contains its fair share of toxic conditions, which renders iron prevailing in the form of Fe(III) (oxyhydr) oxide minerals. Nonetheless, because of the microbial respiration, this subsurface environment become anoxic which could turn to reduction of iron species, including aq. Fe(II) and aq. Fe(III). The co-occurrence of aq. Fe(II) and Fe(III) (oxyhydr) oxides is normal at redox interfaces within the enduring of Fe(II)-containing primary rocks, the upsurge of anoxic spring water, and microbial iron species reduction. The chemical reaction of aqueous Fe(II) with iron oxides occurs macroscopically via cation sorption at principally passive oxide surfaces.288–291 Scherer and co-workers292 reported Fe atom switch within hematite and aq. Fe(II) under mild reaction conditions, where aqueous Fe(II) has been exchanged with Fe(III) of goethite devoid of any substantial phase alteration although it not clear that the aqueous Fe(II) endures related interchange reactions with hematite Fe(III). In this study, an enriched 57Fe tracer was used to confirm that aqueous Fe(II) exchange with structural Fe(III) hematite at RT, and it was found that the quantity of interchange was affected by size of the particle, pH, and the concentration of Fe(II). In their experimental studies, hematite (80 nm, 27 m2 g−1) in an aqueous medium of Fe(II) at pH = 7.0, was kept for thirty days, which resulted in ∼5% of its structural Fe(III) atoms interchanging within Fe(II) in solution phase; being comparable to nearly one surface iron coating. Notably, hematite nanoparticles (50 nm, 54 m2 g−1) displayed roughly 25% exchange (∼3× surface Fe) in aqueous Fe(II), indicating that structural hematite Fe(III) was convenient for liquid at existence of Fe(II). Importantly, the magnitude of exchange in hematite rises up with pH up to 7.5 and then starts to drop, presumably due to saturation of surface site sorbed by Fe(II). However, exchange levels decreased when aq. Fe(II) was enhanced above surface saturation limit. Overall, it can be concluded that even though hematite being the supreme thermodynamically stable iron oxide, Fe(II) can catalyze Fe atom interchange among aq. Fe(II) and bulk hematite. Remarkably, HR-TEM images of the hematite NPs before and after the reaction remained unchanged (see Fig. 48) and the absence of any new phases were affirmed by XRD analyses.292
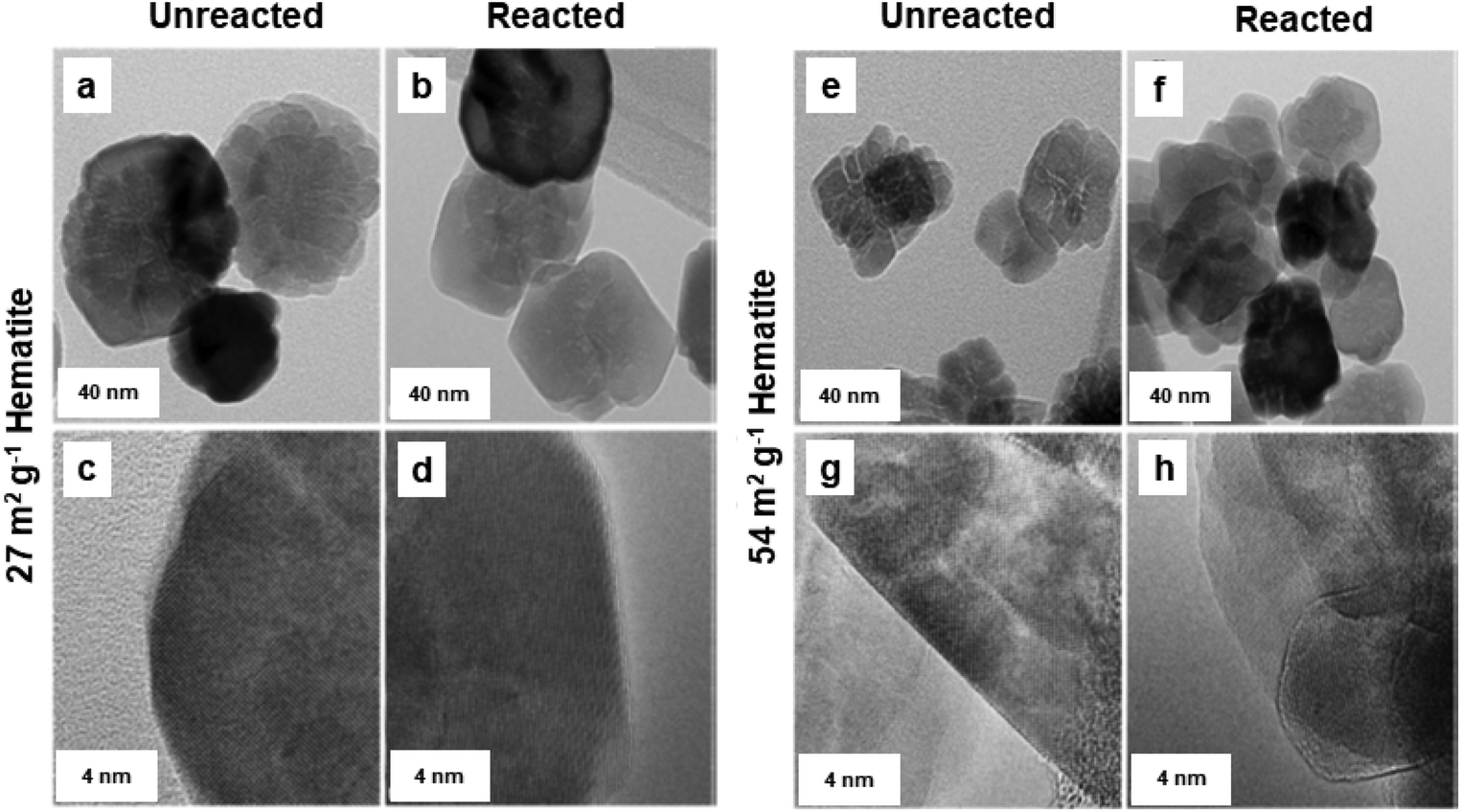 | ||
| Fig. 48 HRTEM images at two length scales for 27 m2 g−1 (a–d) and 54 m2 g−1 (e–h) hematite. Reproduced with permission from ref. 292 Copyright 2015, American Chemical Society. | ||
The strategy exploiting the graphene-based materials with designer features entails better knowledge of the interfacial interaction amidst iron oxide particles and graphene sheets.293,294 Therefore, it is essential to know how the oxidation–reduction reactions of Fe NPs are influenced by the graphene support. This acquaintance is necessary in consideration of the activity of the nanocomposites depending crucially on a specific phase of the iron oxides (i.e., oxidation state of the iron and the crystal structure of the oxides).
The spectroscopic investigation295 was carried out as to how the specific surface sites influence the reducibility of α-Fe2O3/graphene nanomaterials with H2; the steadiness of the Fe2O3 nanoparticles concerning reduction was enriched by the interaction of graphene nanosheets than the bulk Fe2O3. Post annealing TEM images, disclose the presence of both core/shell and homogeneous Fe2O3 nanoparticles with the final development of unusual channels into the graphene sheets (see Fig. 49). Because of heat treatment under Fe2O3 and H2, it was partly reduced to FeO and to some extent the Fe nanoparticles drill the few layers thick graphite (FLGs) and were grafted on the graphene surface. Overall, in this work, empirical evidence for the function of FeOx during the construction of trenches in the FLGs was established comparatively at lower temperature.
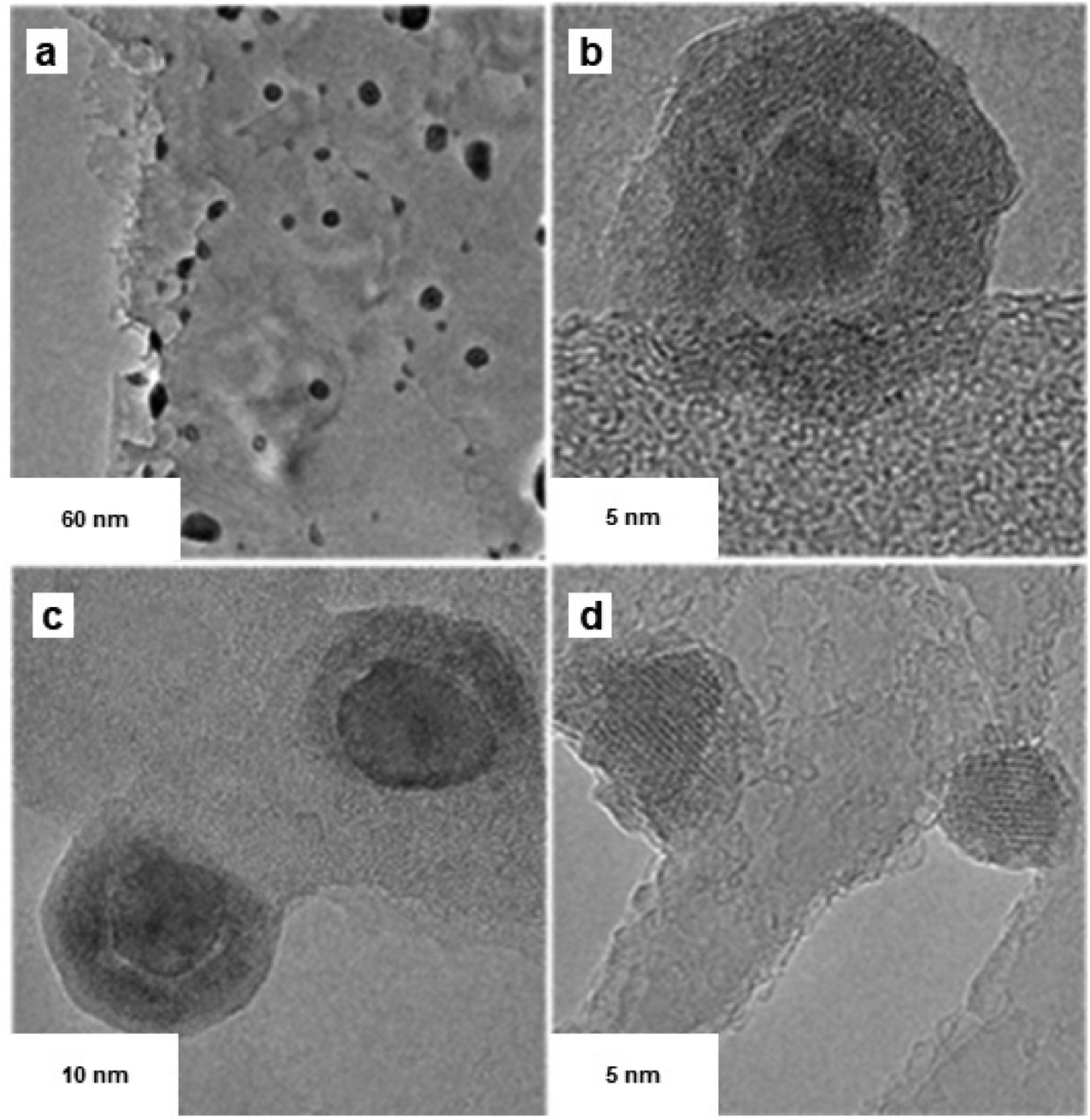 | ||
| Fig. 49 Post-annealing TEM image of Fe/FLGs (a) large-scale image; (b) core–shell iron NPs; (c) early stage of trenching and; (d) nanotrench creation in FLGs (few-layer thick graphite). Reproduced with permission from ref. 295 Copyright 2013, American Chemical Society. | ||
In petroleum refineries, valuable light fuels are obtained from bulk volume of atmospheric residue and vacuum-distilled residual oil.293 In general, to produce the lighter fuels from the residual oil, various conventional processes are used including thermal cracking,296,297 catalytic cracking298,299 and hydrocracking.300,301
Iron oxide nanocatalysts (as free α-Fe2O3 NPs) as well as SiO2-supported α-Fe2O3 crystals were reported302 for the catalytic cracking of bottom product from the vacuum distillation unit, i.e., vacuum residue (VR) in supercritical water (SCW). Various reaction parameters such as reaction period, temperature, catalyst to oil weight ratio, and water to oil weight ratio were investigated on the yield of coke and maltene obtained from VR cracking including asphaltene conversion. It was found that the α-Fe2O3@SiO2 catalyst was more durable and efficient when compared to free NPs in case of VR cracking. In a plausible reaction mechanism, a cycle starting with oxidative cracking of hydrocarbons with lattice oxygen (obtained from reduction of α-Fe2O3 to Fe3O4), followed by liberation of the active H2via SCW gas shift reaction into VR hydrocracking and manufacture of aliphatic compounds, was reported; the reduced species of the Fe3O4 catalyst were converted back into α-Fe2O3 by the liberated “O” atom from SCW (see Fig. 50).
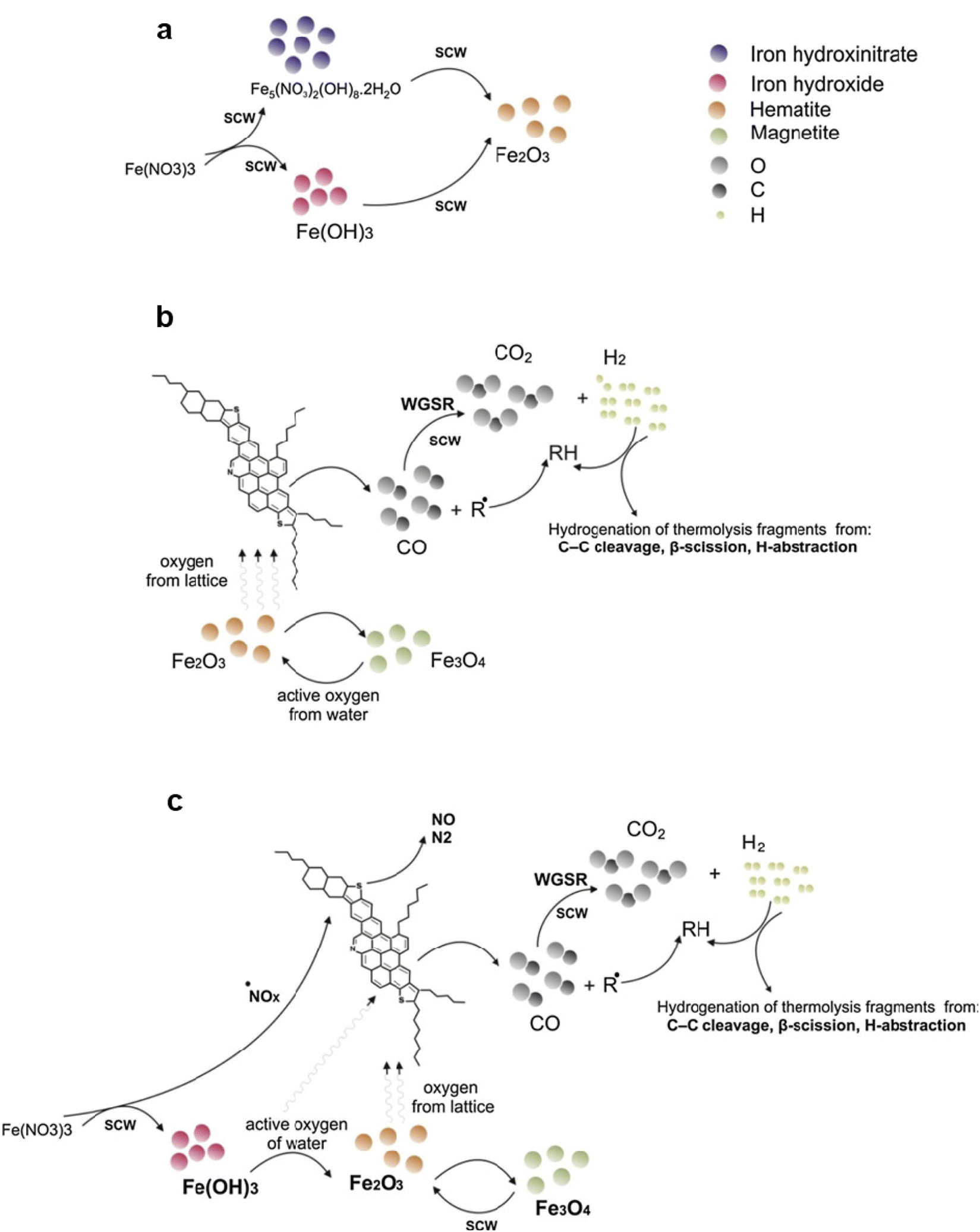 | ||
| Fig. 50 Schematic representation of catalytic cracking in the SCW: (a) generation of hematite from iron nitrate in SCW; (b) cracking of VR in the presence of hematite and; (c) cracking of VR in the presence of iron nitrate in SCW. Reproduced with permission from ref. 302 Copyright 2015, Elsevier. | ||
Recently, iron containing nitrogen-functionalized graphene nanoflakes (N-GNFs) were prepared303 following a procedure by Pristavita et al.304 for producing an active oxygen reduction reaction catalyst. By taking benefit of the exceptional performance of the plasma atmosphere in the GNFs preparation step, they produced iron-based catalyst in a one pot method. The methane was employed as a carbon source which upon decomposition with argon (Ar) thermal plasma produced GNFs via homogeneous nucleation. An inconsequential quantity of N is added at the time of the growth resulting in N content up to 2%. The Fe is added in two distinct phases, one in oxide and other in iron acetate. It is assumed that the velocity variation and the residence time of the excited species in the plasma recombination zone would affect the Fe functionalization of the GNFs. Further, iron functionalization is predicted to be governed by the duration of exposure of the GNFs to iron vapour; shorter exposure time leads to the generation of an inadequate number of catalytic sites, whereas longer duration of exposure results into the Fe overload of the samples (considering the restricted number of the N-based host sites).
Aromatic and aliphatic halogenated chemicals are essential to the pharmaceutical industry and fine chemical synthesis, but because of their potential harm to the environment and human health, they must be used responsibly and disposed of appropriately. Regulatory frameworks and industry practices are adapting for more sustainable chemical and pharmaceutical production. Metal NPs play a key role in removing halogenated disinfection byproducts, aiding environmental remediation.305 In this context, a precisely structured mesoporous metal oxide material named MFT-1 consisting of TiO2–Fe2O3 was prepared using sodium dodecyl sulfate as a structured-directing agent by sol–gel method.306 The HRTEM and FFT image of mesoporous TiO2–Fe2O3 (MFT-1) catalyst revealed a highly ordered two-dimensional (2D) hexagonal mesophase with a 6-fold symmetry (Fig. 51a). The FESEM image showed that the sample consists of small spherical NPs around 40 nm in diameter (see Fig. 51b). This mesoporous MFT-1 catalyst exhibited good catalytic performance in dehalogenation of aryl iodides, bromides, and chlorides, even in the presence of functional groups like –F, –CN, –CH3, –OCH3, and –NO2 (see Fig. 51c). This material offers a promising avenue for the development of sustainable technologies in the treatment and remediation of halogenated organic compounds.
 | ||
| Fig. 51 (a) HRTEM image of MFT-1, FFT image is shown in the inset; (b) FESEM image of MFT-1 (c) Dehalogenation reaction using MFT-1 nanocatalyst. Reproduced with permission from ref. 306 Copyright 2012, American Chemical Society. | ||
In a similar context, a new approach was deployed for the hydro-dehalogenation of haloaromatic compounds, utilizing Pd/Fe2O3 nanohybrids anchored on Amberlite resin (Pd/Fe2O3@ARF-110).129 The HRTEM image and energy-dispersive X-ray spectroscopy (EDS) analysis confirmed that the crystalline Pd and Fe2O3 NPs, with an average dimension of about 4–5 nm, coexisting within the resin matrix with a molar ratio approximately equal to 1:1 (see Fig. 52a–d). The arrangement of Pd and Fe2O3 NPs on the polymer surface was optimized to boost the catalytic activity, showing potential for addressing persistent organic pollutants (POPs). Effective electron exchange between the iron oxide and palladium NPs aids in breaking B–H bonds of NaBH4, resulting in the generation of active hydrogen and impressive catalytic efficiency. Particularly, the Pd/Fe2O3@ARF-110 nanocomposites showed outstanding performance in dehalogenating various haloaromatic compounds (see Fig. 52e). Moreover, the heterogeneous Pd/Fe2O3@ARF-110 catalyst attests to be conveniently separable and recyclable, exhibiting nearly identical efficiency over five consecutive runs (see Fig. 52f).
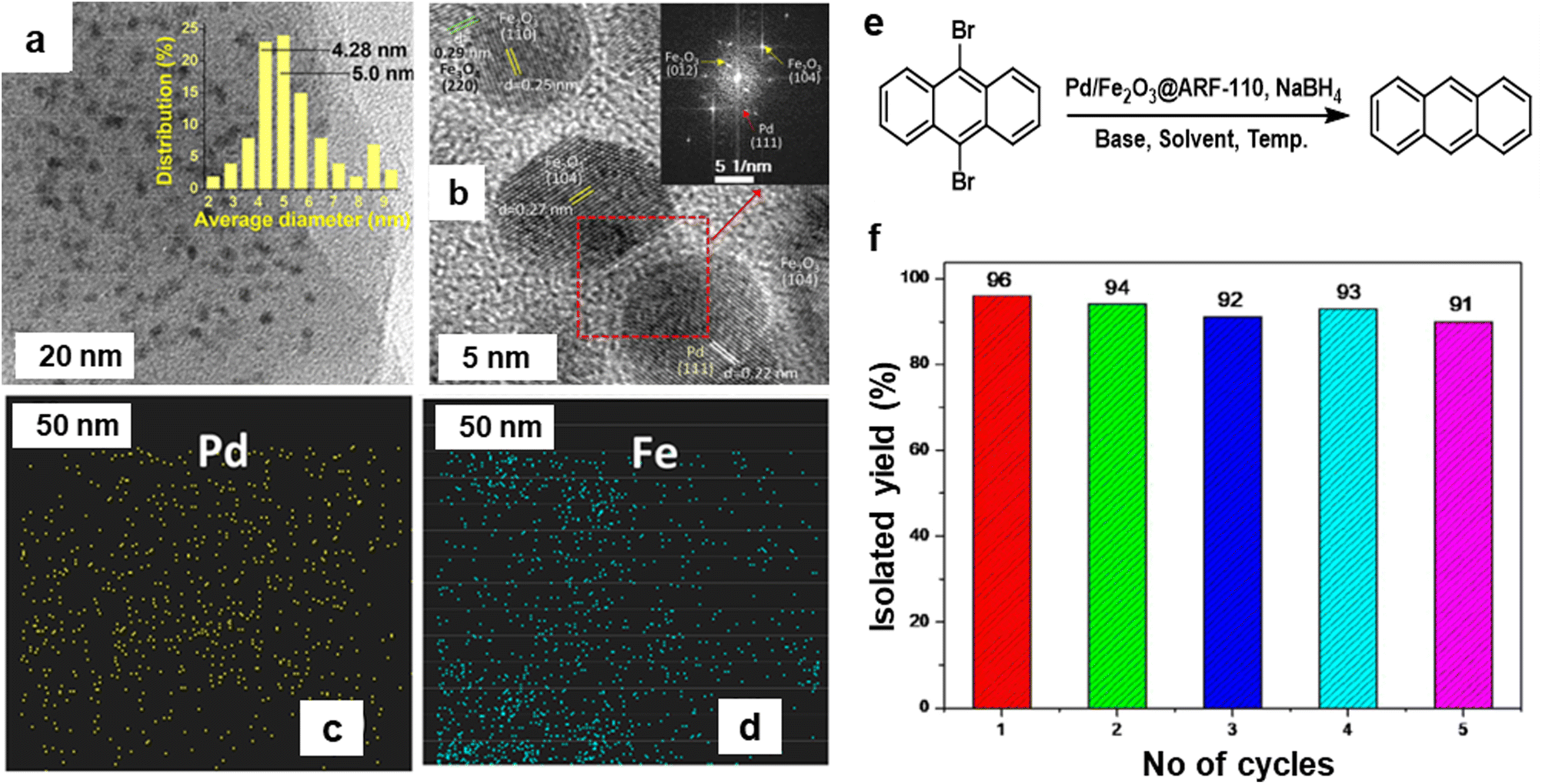 | ||
| Fig. 52 (a, b) HRTEM images of Pd and Fe-oxide NPs; (c, d) EDS elemental mapping of Pd and Fe respectively; (e) Hydrodebromination of 9,10-dibromoanthracene employing Pd/Fe2O3@ARF-110 nanohybrids (f) Recycle study of Pd/Fe2O3@ARF-110 catalyst. Reproduced with permission from ref. 129 Copyright 2019, American Chemical Society. | ||
3. Nanosized iron oxides in environmental applications
Starting with the industrial revolution in the late 18th century and beginning of 19th century, a boom in the anthropogenic activities including mining, heavy industry, metallurgy, textile manufacture, production of chemicals, agriculture, steam power, etc. had a severe impact on environment. Air, water, and soil were contaminated with toxic inorganic and organic compounds with a little effort to decrease pollution and/or treat the affected environment. During 19th century, several laws have been introduced in big cities such as London and Paris to inflict companies polluting air and water; however, these laws and regulations were not strictly obeyed. In the 20th century, the human race witnessed increase in automobile industry, armament industry, industrial chemistry, pharmaceutical industry, plastic industry, electronics, energy production, transportation, and nuclear science, representing additional sources of pollution. The issue of atmospheric pollution was firstly brought to public attention after the World War II with worries caused by information about radioactive fallout from atomic testing and conflict. In the 1970s, several ecological movements were established to force governments of developed countries to limit pollution of the environment. Currently, the problem of pollution is addressed worldwide by international and local advisory and executive boards introducing regulations and laws in the area of pollution limits and protection of the environment with law suits and penalties. The protection of the environment constitutes a topic of utmost importance and is heavily discussed within the scientific community over the broad spectrum of fields.Over the past five decades, substantial emphasis has been placed on the issue of water pollution which is frequently identified as prominent worldwide contributor to fatalities and diseases. The water contaminants include a broad portfolio of inorganic, organic, and biological compounds/entities such as heavy metals, nitrates, phosphates, arsenates, ammonia, pharmaceutical drugs, surfactants, endocrine disrupting chemicals, antibiotics, hormones, perchlorate, chlorinated solvents, volatile organic substances, detergents, and pathogens, to name a few. Diverse methodologies have been introduced for water treatment including physical, chemical, and biological processes and their combinations. Currently, nanotechnology strategies are regarded as promising alternatives to complement or substitute traditional water treatment technologies as they often offer enhancement in the effectiveness of the treatment process, are environmentally friendly and, to some extent, low-cost.
Many efforts have focused on treatment of outer and underground water which are the source of potable water through which toxic pollutants enter the human body. Various techniques have been suggested for treatment of surface and underground water such as adsorption, exchange of ion, reverse osmosis, membrane filtration, photo-oxidation and Fe/Mn removal processes and their combinations. Among a long list of traditionally used materials and specifically designed nanostructures, iron oxides, especially in the form of NPs, hold a paramount position due to relatively high abundance, availability in nanosized forms via a diversity of sophisticatedly controlled chemical pathways, high surface area, robustness, eco-friendly attributes, and cost-effectiveness. Herein, the focus has been on environmental uses of iron oxide nanomaterials in sewage treatment, magnetic separation, photocatalytic applications, and activation of hydrogen peroxide and their use as supports via immobilization of biomaterials and enzymes (see Fig. 53).13,71,307–313
 | ||
| Fig. 53 Examples of environmental applications of iron oxide nanomaterials. | ||
3.1. Iron oxide nanomaterials for wastewater treatment
Due to worrying prognosis of the chronic shortage of drinking water around the globe in near future, an eminent scientific interest is devoted to the development and advancement of wastewater treatment technologies, especially using iron oxide nanoparticles.71,311,314,315 Their distinct features such as greater surface area, excellent magnetic behavior and response to external magnetic fields, non-toxicity, biocompatibility, and biodegradability, rank them among most promising materials employed for waste water treatment. Numerous environmental and sustainable purification methods have been developed for sewage processing, which commonly exploit iron oxide nanoparticles as nanosorbents. The most recent applications in sewage processing and the knowledge gaps that restricts their extensive applications are delineated here.The steadiness of iron oxide nanomaterials can be significantly enriched through modifying the surface with appropriate functional units, such as amine, cysteine, carboxylic and phosphonic acid (see Fig. 54).316,317 Several options exist for adornment of various functional groups onto iron oxide NPs surface, still search for a robust protocol is continually being explored. It is important to highlight that the usage of iron oxide nanoparticles is strongly linked with their natural properties, which are dictated by immobilized entities on their surface.
 | ||
| Fig. 54 Immobilization of polymers and functional groups on the iron oxide NPs surface. Reproduced with permission from ref. 317 Copyright 2010, Elsevier. | ||
The numerous techniques have been adopted for the elimination of heavy metal ions from industrial wastewater includes adsorption, nanofiltration, exchange of ion, electrodialysis, and chemical precipitation.331 Among these approaches, adsorption is the elegant because of its greatest effectiveness, ease of use and minimal costs.332 Here, iron oxide NPs are broadly used owing to their great performance and sustainable properties.333 A variety of iron oxide NPs varying in particle size, PSD, morphology, structure, and aggregation degree have been successfully designed via diverse protocols to improve their adsorption performances.334–338 In general, heavy metal ions withdrawal by sorption relies on the degree of oxidation of the heavy metal, adsorption capability of the sorbent against these species, pH of the solution, sorbent-to-heavy metal ratio, and salts and molecules present in the aqueous phase that can compete for adsorption sites. Therefore, an effective approach to improve the removal efficacy is to adjust the size, structure, and physiochemical properties of the sorbent or the degree of oxidation of heavy metal species at the time of their withdrawal.
For heavy metal ions, these unique characteristics of iron oxide nanoparticles with diverse morphologies and three-dimensional (3D) structures show greater adsorption ability as compared to the bulk counterparts (see Fig. 55).339,340 Although several types of 3D iron oxides nanoparticles comprising Fe3O4, α-Fe2O3, α-FeOOH, and γ-Fe2O3 have been successfully synthesized by various methods,339–343 the assembly of 3D iron oxides with higher adsorption potential for heavy metal ions remains a substantial challenge. Cui et al.344 have documented the fabrication of 3D Mn doped α-Fe2O3 porous nano-system engineered by calcination of carbon spheres including Fe(II) and Mn(II) ions. This synthesis was achieved by hydrothermally treating the mixture of glucose, Fe(II), and Mn(II) ions. The prepared and well-characterized iron oxide nanostructures show greater capabilities for elimination of Pb(II), As(III), and Cr(VI), ions from wastewater. Nassar and co-workers345 have observed maximum adsorption performance (36.0 mg g−1) for Pb(II) ions with Fe3O4 nanoparticles. It was proposed that the smaller sized Fe3O4 nanosorbents were promising for distribution of metal ions onto the adsorbent's surface, thus rendering them useful for extraction and retrieval of metal ions from wastewater. Despite that the extremely significant surface-forced phenomena in water medium, aggregation, induced by high surface area to volume ratios of nanoparticles, may impede various imperative environmental processes, along with ion uptake.346 Besides aggregation, numerous other interactions with ions could be invoked in wastewater, which could influence the adsorption of metals. In particular, the high concentration of phosphates in the wastewater can make it good adsorbent and even surpass metals for adsorption sites.347 These causes along with the nature of the pollutants can confine the efficiency of nanosorbents, and consequently the investigation of considerably selective transformation protocols for nanomaterials is becoming a hot bed of research activity for enhancing the efficacy of nanosorbents.
 | ||
| Fig. 55 (a) SEM image of the iron oxide NPs. Inset: EDX analysis; (b, c) SEM and TEM images of flowerlike iron oxide NPs respectively. Reproduced with permission from ref. 339 Copyright 2006, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
Several interesting findings have been reported on heavy metal decontamination by iron oxide NPs and 3D type iron oxide nanostructures.339,340 Arsenic (As) is classified as a carcinogen for humans and is viewed as one of the most toxic elements. The rising levels of arsenic in nature is an escalating problem that several nations currently face. Because of extreme health impacts on the population (irritation of the digestive tract, nausea, diarrhea, skin cancer, increased risk of cancers of the lung, kidneys, and liver), the influence on the health of humans significantly relies on the oxidized state of arsenic. Beside processes of natural origin such as weathering of rocks, artificial interferences like mining, utilization of arsenic containing toxicants, factory wastes, and inappropriate discarding chemical effluents heavily contribute to the increased levels of arsenic in nature.348 Thus, to increase the performance of As extraction from the environment, engineered iron-oxide-based environmentally friendly materials349–351 have been developed and examined. These materials comprise monodispersed Fe3O4 nanocrystals,352 hybrids of Fe3O4 and rGO sheets,353 thiol-functionalized iron oxide NPs,354 (GO)/Fe(OH)3 composites,355 high-surface area superparamagnetic Fe3O4 NPs,356 and superparamagnetic γ-Fe2O3 nanoparticles with mesoporous nature of particle arrangement promoted by strong magnetic interparticle interactions.357 Recently, a hypothesis has emerged suggesting that if the surface of Fe(III) oxide nanoparticles is functionalized with NaOH in liquid phase, new hydroxide reactive functional groups are generated which promotes increase in the ion exchange process, resulting in the enhancement of removal capacity towards arsenic species.358
A magnetic hybrid material fabricated by incorporating iron oxide NPs into a chitosan matrix with the assistance of eucalyptus extract as a reductant and utilized it for arsenic removal.359 Bangari et al. synthesized magnetite-coated boron nitride nanocomposite for As(V) ion extraction from aqueous medium.360 The diameter of the BNNS–Fe3O4 nanocomposite (10–22 nm) was analyzed by HR-TEM analysis (Fig. 56a). The HAADF image display the uniform distribution of boron nitride on Fe3O4 (Fig. 56b). The adsorption ability of BNNS–Fe3O4 nanocomposite were higher relative to those of BNNSs nanocomposites for As(V) ion removal (Fig. 56c). The several active sites in the BNNS–Fe3O4 nanocomposite enhanced the percent removal than BNNSs nanocomposites; highest adsorption ability of both adsorbents being 0.2 g L−1 (Fig. 56d). Recently, magnetite iron oxide NPs nanodevice coated with meglumine ligand by co-precipitation method were developed for the adsorption of heavy metals (As, Cr and B).361
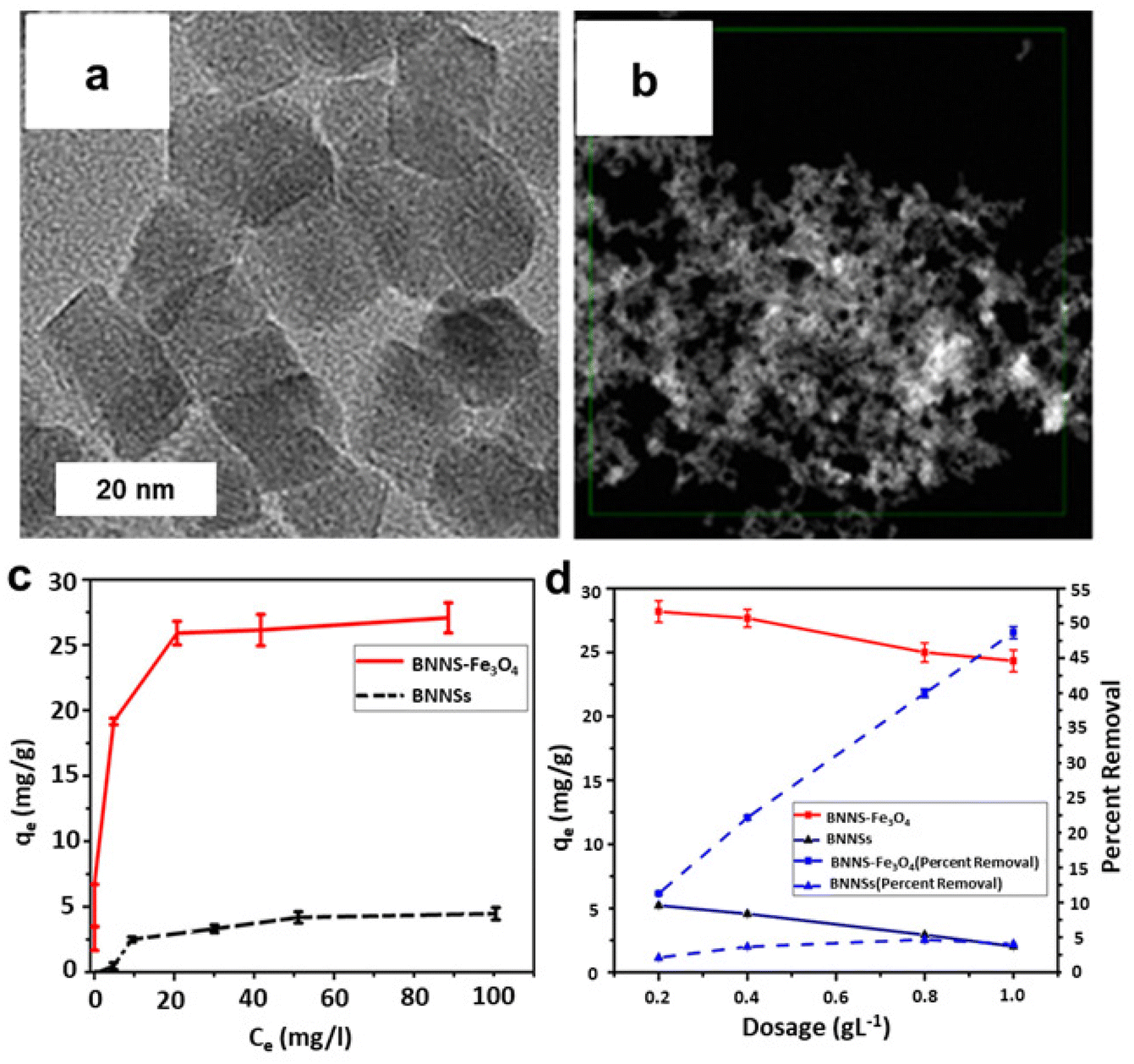 | ||
| Fig. 56 (a, b) HR-TEM and HAADF image of BNNS-Fe3O4 nanocomposite; (c) Adsorption isotherm of BNNSs and BNNS–Fe3O4 nanocomposite; (d) Effect of adsorbent dose on As(V) ions removal. Reproduced with permission from ref. 360 Copyright 2019, American Chemical society. | ||
If γ-Fe2O3 nanoparticles are distributed on the CMK-3 surface, such a hybrid shows eminent capability to extract Cr(VI) from the aquatic medium362 (Fig. 57). The synthesis method involves two steps: AcOH fumes react with an immobilized Fe cation in the carbon matrix to generate an iron acetate intermediate, following by its pyrolysis to produce iron(III) oxide nanoparticles. Iron-oxide/carbon hybrids, combining organized forms of interlinked carbon rods, were reportedly displayed high specific surface areas ∼1043 m2 g−1, evenly immobilized γ-Fe2O3 NPs with equal sizes (∼20 nm) and superparamagnetic characteristics at RT. Because of the explicit surface characteristics of magnetic carbon hybrids and synergistic impact of both equivalents, these hybrids exhibited greater Cr(VI) extraction ability, which was significantly better compared to pure CMK-3 carbon.
 | ||
| Fig. 57 γ-Fe2O3/CMK-3 hybrid for Cr(VI) extractions from aquatic solutions. Reproduced with permission from ref. 362 Copyright 2012, American Chemical Society. | ||
Tuning the morphology and degree of porosity is frequently adopted as another approach to enhance the sorption capabilities of iron oxides as exemplified by the synthesis of α-Fe2O3 in the form of very thin and porous nanofibers and its assessment for extraction of Cr(VI) from the aquatic environment.363 Such α-Fe2O3 nanofibers displayed greater adsorption efficiency (16.17 mg g−1), extraction rate, and excellent recovery without loss of performance; they can be recycled after centrifugation and treatment with diluted NaOH. The Cr(VI) species could be removed by a monolayer adsorption on the surface of porous α-Fe2O3 nanofibers.
A single-step preparation of magnetic–silica nanoparticles by using alkaline sodium silicate solution has been reported by Gole and co-workers.364 This methodology not only created iron oxide but simultaneously decorated Fe3O4 nanoparticles with silica coating. The biocompatible and stable silica-shell magnetic nanoparticles serve as anchors for various functional groups; iron oxide-silica surface modified with 3-amino propyl triethoxy silane (APTES) are employed for tagging of Rhodamine B (Rh B) dye molecules (flurophore tagging). The bare magnetic and silica-coated nanoparticles have been used for the zinc loading and the utility of such nanocomposites have been explored for water purification by arsenic removal in the form of arsenate.364
Iron ferrite NPs365 were synthesized via a biogenic strategy and utilized for the Pb(II) and Cr(III) heavy metal removal. The particle size (20–45 nm) of the magnetic inverse spinel iron oxide nanoparticles (MISFNPs) was confirmed by TEM analysis (Fig. 58a) and the adsorption and desorption study suggested that the MISFNPs was stable and reusable up to four cycles (Fig. 58b). The adsorption percentage of the MISFNPs was higher relative to bulk Fe3O4 because of the nano size and high surface area of the MISFNPs (Fig. 46d). The highest adsorption of Pb(II) and Cr(II) reached up to 0.5 g L−1 with increased NPs dosage and then decreased due to lower active sites (Fig. 58c). Pan et al.366 reported iron oxide nanoparticles coated with organics and utilized them for the elimination of Cr(VI) from water. The DNA modified Fe3O4@Au magnetic NPs has been employed for the electrochemical detection of Ag+ and Hg2+.367
 | ||
| Fig. 58 (a) HR-TEM images of MISFNPs; (b) Study of the stability and reusability of the MISFNPs; (c) Adsorption activity; (d) Effect of MISFNPs dosage on Pb(II) and Cr(III) adsorption. Reproduced with permission from ref. 365 Copyright 2016, Elsevier. | ||
A nanocomposite (mPCS@Fe2O3) was prepared by combining mesoporous carbon sphere with Fe2O3 and employed the catalyst to remove antimony (Sb) from wastewater.368 The SEM images of mPCS and mPCS@Fe2O3 revealed spherical morphology (Fig. 59a and b) and they were well dispersed on mPCS with particle size <6 nm, as confirmed by HR-TEM studies (Fig. 59c and d). The adsorption kinetics suggested that the adsorption of Sb increased during its initial 40 min and afterwards, the adsorption attains equilibrium within 120 min (Fig. 59e). The co-existing anions contained in the Sb solution did not affect the removal of Sb metal. The PO43− ion slightly effected the Sb removal because of its similar structure to Sb(III) (Fig. 59f). Xiang et al.369 reported carbon-supported zero valent Fe NPs for the photocatalytic extraction of U(VI) from sewage.
 | ||
| Fig. 59 (a, b) SEM images of mPCS and mPCS@ Fe2O3; (c, d) HR-TEM images of mPCS@ Fe2O3 and mPCS@ Fe2O3/Sb(III) catalysts respectively; (e) Kinetic curves for the extraction of Sb; (f) Impact of present anions on adsorption of Sb. Reproduced with permission from ref. 368 Copyright 2018, American Chemical Society. | ||
A magnetic Fe2O3/AlO(OH) nanocomposite was synthesized using a template-free route by utilizing ferrous sulphate and magnesium acetate. Initially, urchin-like lepidocrocite γ-FeOOH was prepared in single step, then α,γ-Fe2O3 was obtained via high temperature calcination and finally, the target compound was formed by immobilizing α-,γ-Fe2O3 and Al(NO3)3·9H2O utilizing ultrasound-assisted chemical precipitation method370 (Fig. 60a). The composite material comprising Fe2O3 and AlO(OH) displayed admirable efficiency to extract heavy metal ions from drinking or wastewater.371,372 The obtained hierarchical material exhibited superfast adsorption rates and greater adsorption performance for the extraction of As(V), Cr(VI) and Pb(II) ions from aquatic medium in 30 min; highest adsorption extents being 37.45 mg g−1, 21.6 mg g−1, and 29.55 mg g−1, respectively. Notably, Fe2O3/AlO(OH) materials displayed excellent recyclability for As(V) removal, even after three cycles 75% adsorption rate was retained. The high activity and recyclability could be because of the extensive surface area of the materials and special morphology of Fe2O3@AlO(OH)370 (Fig. 60b). It is speculated that this inexpensive and benign synthetic approach for Fe2O3@AlO(OH) nanomaterial might be useful for other types of environmental applications.
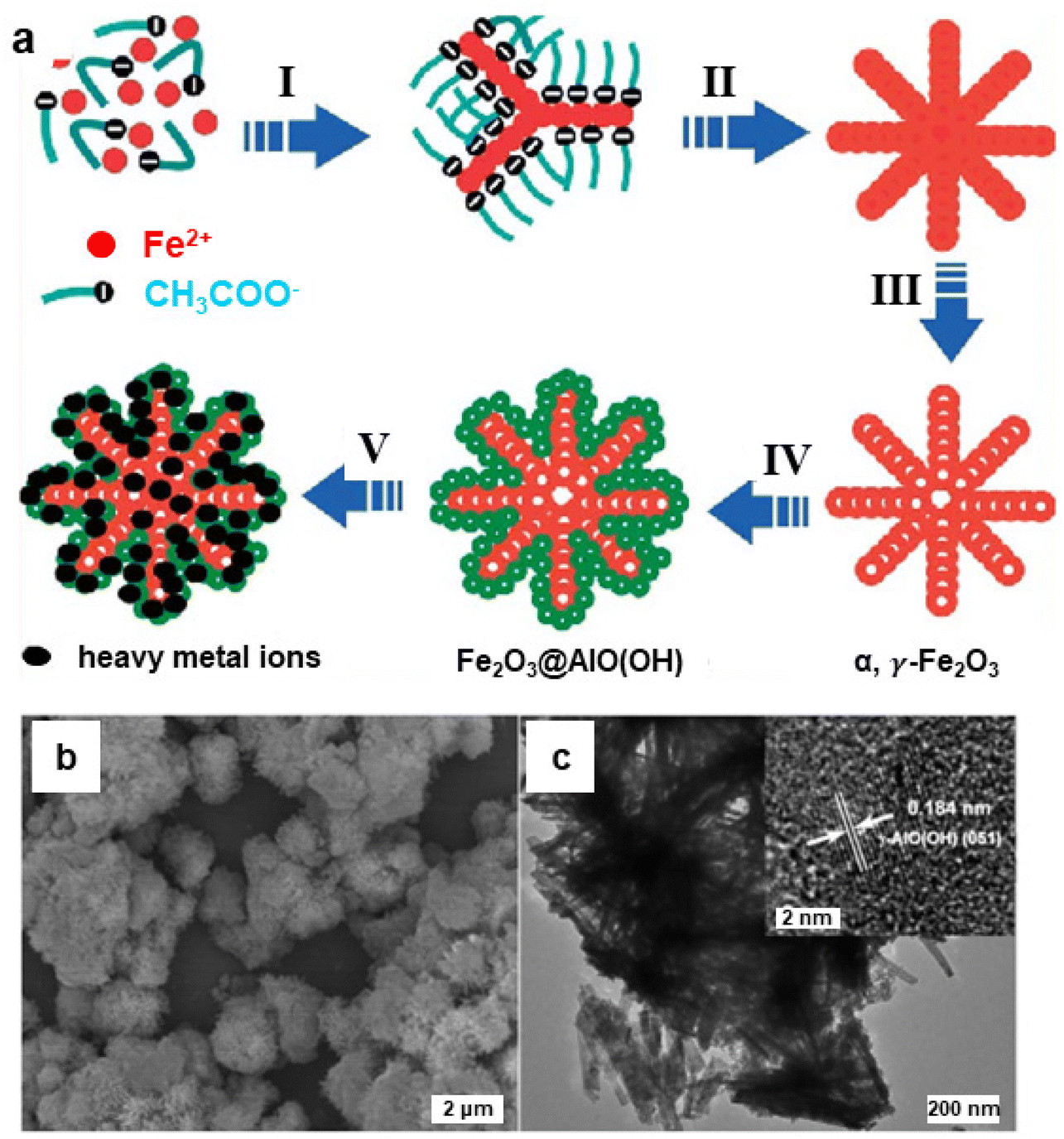 | ||
| Fig. 60 (a) Schematic illustration of the preparation of Fe2O3@AlO(OH) hierarchical porous structure; (I) generation of nucleation centers; (II) nanocrystal development and directed linking to 3D urchin-like γ-FeOOH superstructure; (III) calcination and generation of porous magnetic α, γ-Fe2O3; (IV) formation of mesoporous γ-AlO(OH) from α,γ-Fe2O3; (V) adsorption of heavy metal ions from aquatic media; (b, c) SEM images of Fe2O3@AlO(OH). Reproduced with permission from ref. 370 Copyright 2013, The Royal Society of Chemistry. | ||
The growing universal need for pure and clean water coerce the research experts to discover functional processes for drinking water production.373,374 Among these, membrane filtration is one of the most efficient and simple pathways to deliver high-quality of safe drinking water. Nevertheless, membrane fouling is greatest drawbacks that inhibits the widespread utilization of membrane process. A new type of membrane, created from interwoven Fe2O3 and TiO2 nanowires (FeTi–NWM) has been reported375 with anti-fouling ability for humic acid (HA) removal from water. TiO2 and iron oxide nanowires were employed to make TiO2 nanowires by a filtration and hot-pressing technique (Fig. 61). HA is a primary contaminant responsible for the fouling of membranes in water treatment processes.376 A stainless-steel reactor housing nanowire-membrane based circulating water filtration technique was introduced.376 The ensuing results revealed that under solar illumination the FeTi–NWM accomplished nearly complete HA extraction within 2 h short-term trial at a starting HA concentration of 200 mg L−1, which is comparable with HA extraction by Ti–NWM (89%). After long-term trial of 12 h, the FeTi–NWM displayed 98% HA abstraction, though the Ti–NWM could show only 55% extraction at the end. The enhanced HA removal provided by FeTi–NWM relative to Ti–NWM and its exceptional anti-fouling potential under solar light illumination can be ascribed to: (i) an improved absorption of HA by iron oxide nanowires and (ii) the created Fe2O3/TiO2 heterojunctions that enhances the photo-induced charge transfer and enhance visible light performance. Outstandingly, in comparison to Ti–NWM without Fe2O3 nanowires, FeTi–NWM displayed greater HA extraction.
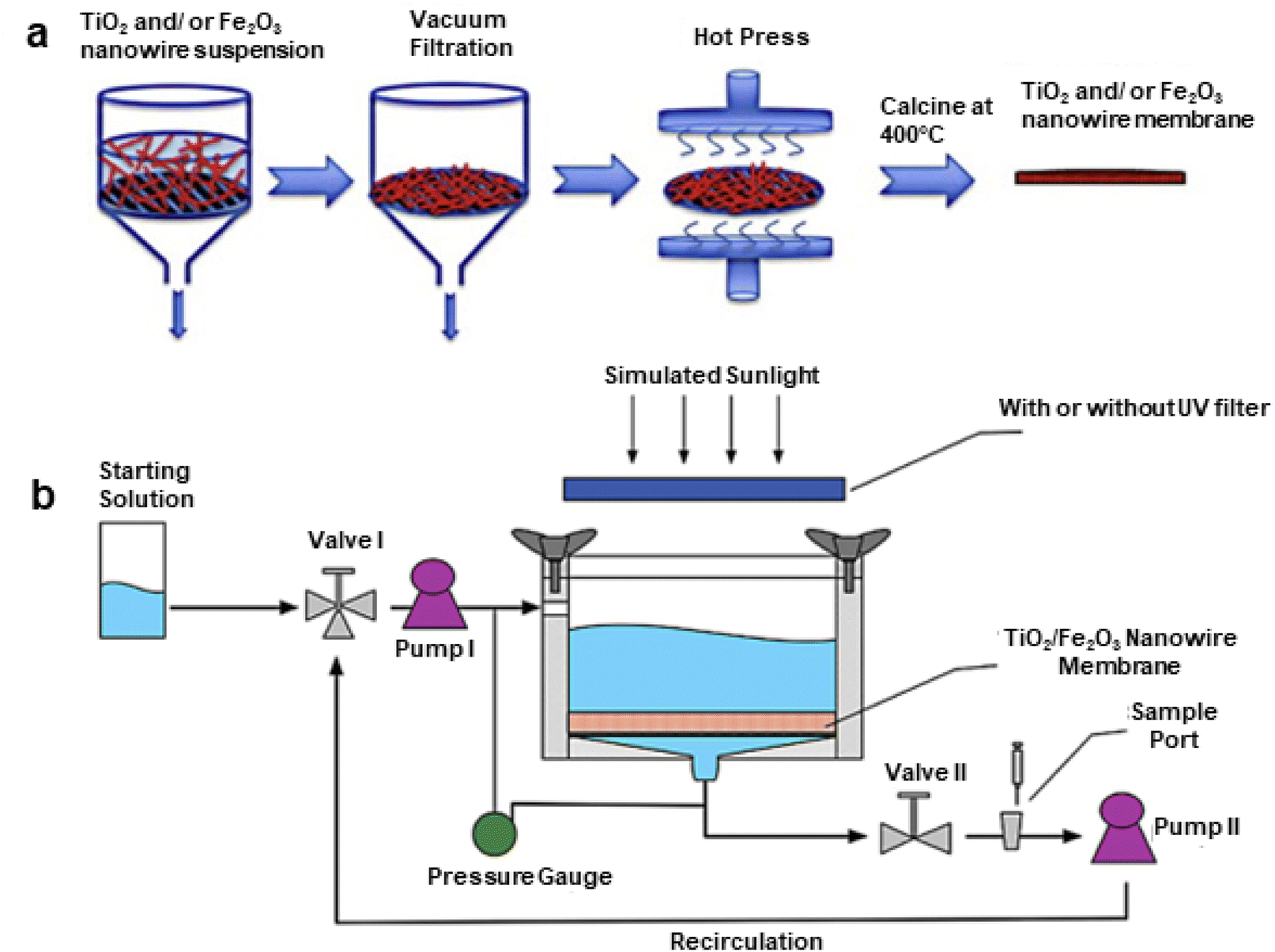 | ||
| Fig. 61 (a) Schematic representation of process of synthesis of Ti–NWM and FeTi–NWM; (b) Schematic representation for water filtration process for HA extraction through nanowire membranes under sunlight irradiation. Reproduced with permission from ref. 375 Copyright 2015, Elsevier. | ||
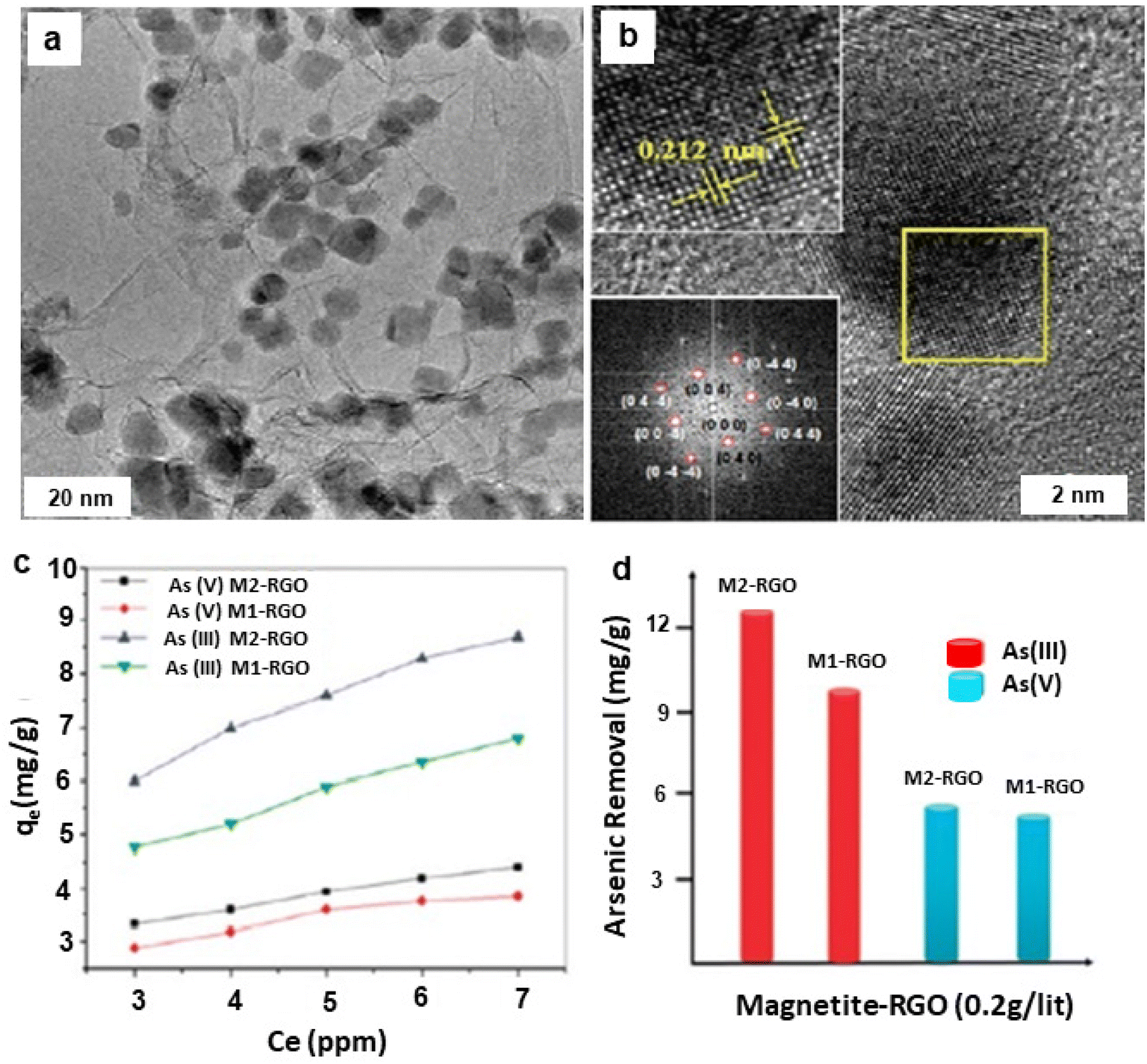 | ||
| Fig. 62 (a, b) TEM and HR-TEM images of M-RGO (inset: SAED pattern); (c) adsorption isotherms over the Fe3O4–RGO composite; (d) maximal adsorption ability of M-RGO composites for the extraction of As from water. Reproduced with permission from ref. 353 Copyright 2010, American Chemical Society. | ||
The surface area, temperature, and pH of the contaminated solution determine the ultimate adsorption capacity.378 Certain supplementary additives boost the adsorption as shown by Luo et al.379 for As(III) and As(V) ions, where the inclusion of MnO2 with the magnetite/graphene material helps for the oxidation of As(III) to As(V) without any additives. As(V) was firmly absorbed at pH ranging from 2 to 10 which was not the case for the basic magnetite/graphene which afforded stable adsorption at lower pH range. Similarly, the addition of ZnO in maghemite/graphene composite, enhanced the decomposition of methylene blue (MB) with a high efficacy (95%) and was consistently effective after 5 cycles as shown by Kumar and co-workers.380 The endocrine disruptors like dibutyl phthalate, bisphenol A, atrazine, and 1-naphthol, could be actively eliminated (85–100%) from contaminated aqueous streams by using graphene/Fe2O3 based nanocomposites prepared by simple method.381 These materials are well suited for the adsorption of endocrine disruptors from water due to the large surface area and an exposed graphene surface. Although synthetic steps for the hybrids containing graphene sheets and core/double shell NPs of Fe/Fe2O3/Si–S–O type were involved but they were found to be active in the extraction of Cr(VI).382 The Fe/Fe2O3/Si–S–O material was prepared by thermo-decomposition procedure having a core@double-shell arrangement of the NPs with Fe as the core, Fe2O3 as the first shell and amorphous Si–S–O material as the second shell (Fig. 63). This iron oxide based graphene nanomaterial displayed extremely quick extraction of Cr(VI) from the sewage. Importantly, the material was retrievable for several runs and it can be employed to treat solutions with lower pH levels which make it ideal material to eliminate the heavy metals from wastewater.
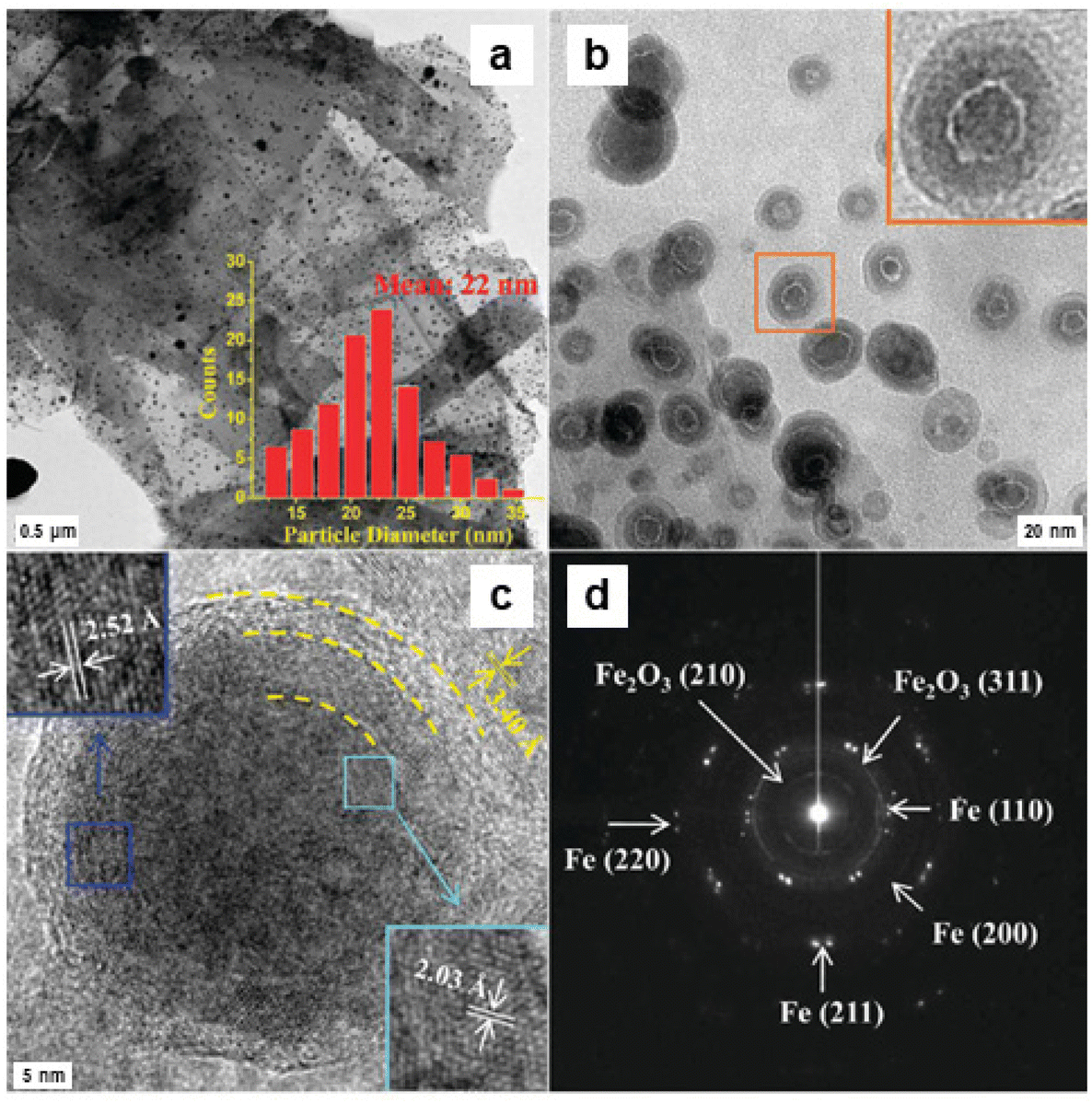 | ||
| Fig. 63 TEM images of the MGNCs (a) The nanoparticles are equally spread on the graphene sheet; (b) magnified image of (a) shows core@shell structure of the NPs; (c) HR-TEM of a single particle displaying an Fe core covered by a double shell structure; (d) SAED pattern of the core@shell nanoparticles. Reproduced with permission from ref. 337 Copyright 2011, American Chemical Society. | ||
The size and morphology of modified NPs can contribute to the design of the magnetic graphene nanocomposites with unique characteristics, such as electroconductivity, flexibility, higher magnetic resistance, negative permittivity, and superparamagnetism.383–385 The propensity of these materials to organic compounds and ions has also been used in sensing applications. More importantly, it offers suitable conditions for the immobilization of biomolecules like hemoglobin,386 horseradish peroxidase387 and such materials could be used for the detection of H2O2 at low sensing limits of ∼0.5 μM. These type of materials have also been employed for the extractions of numerous heavy metal ions such as Cr(VI),388,389 U(VI)390 and Co(II)391 and organic constituents including tetracycline,392 1-naphthol, 1-aminonaphthalene, methylene blue,393 polychlorinated biphenyls, polyaromatic hydrocarbons, phthalates,394 ciprofloxacin, and norfloxacin.395,396
Mixed composite of nanocrystalline γ-Fe2O3 and cerium oxide (CeO2/γ-Fe2O3) was developed for the decomposition of organophosphorus pesticides, parathion methyl, and the chemical fighting agent, soman, and nerve agents, VX.404 The composite was prepared by simple reverse co-precipitation process in which Fe(III)/Fe(II) salts with aq. NH3 was employed for the preparation of Fe3O4 core, with further precipitation of Ce2(CO3)3 on the surfaces of Fe3O4 (again in the reversed mode); calcination finally delivered cerium oxide while Fe3O4 got converted to γ-Fe2O3 (Fig. 64).404
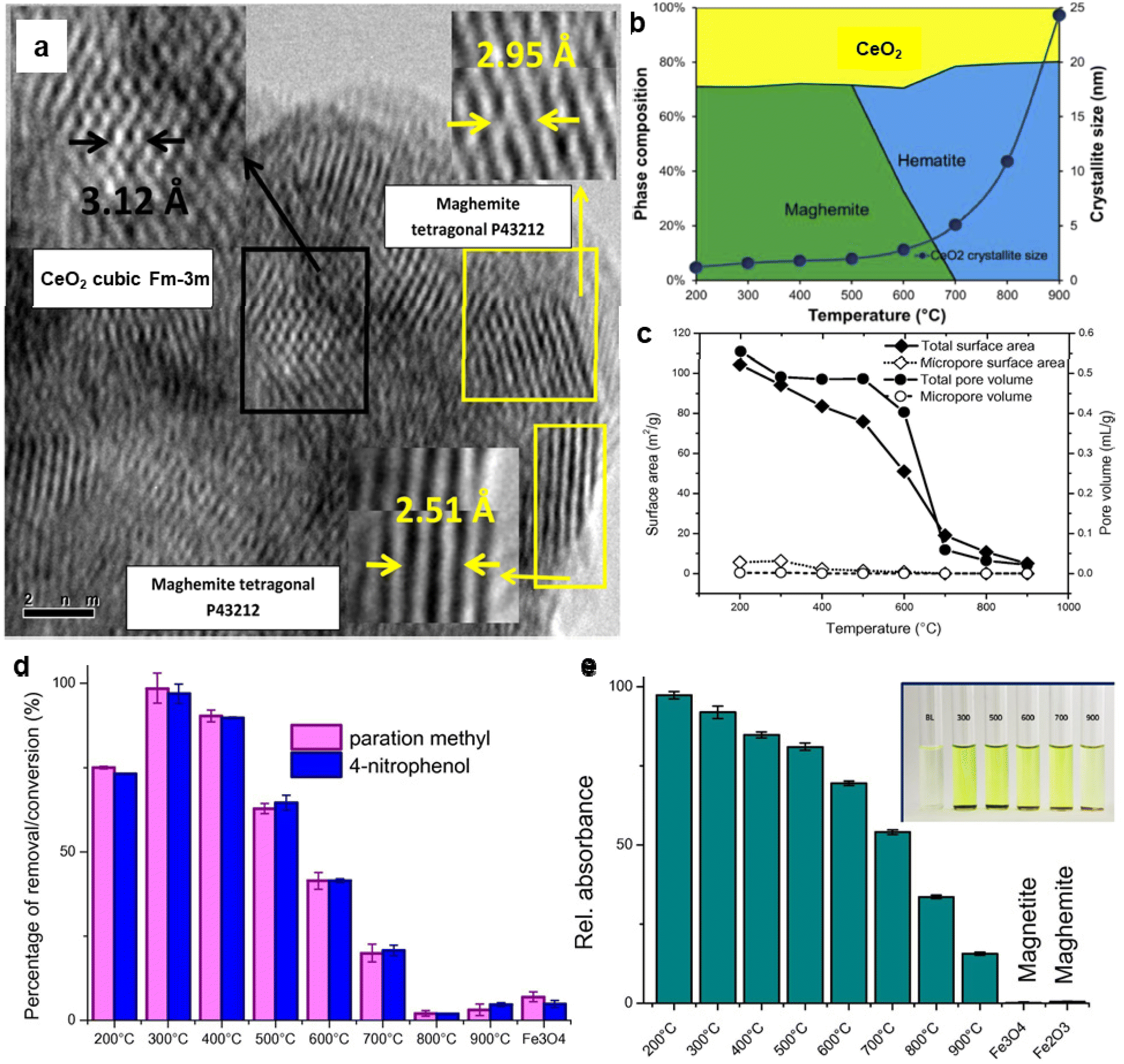 | ||
| Fig. 64 (a) HRTEM image of CeO2/γ-Fe2O3 nanocatalyst; (b) phase combinations of annealed composite sorbents and; (c) N2 isotherm and BJH plot; (d) Impact of calcination temperature on the degradation performance for parathion methyl in 2 h; (e) The phosphatase-like performance of the sorbents inset: images of chosen samples. Reproduced with permission from ref. 404 Copyright 2014, Elsevier. | ||
HR-TEM confirmed the existence of CeO2 nanoparticles decorating the γ-Fe2O3 surface (Fig. 64a). The phase combination of the reactive sorbents produced at distinct temperatures is depicted in Fig. 64b. Increase in calcination temperature decreased the SSA and pore volume, and significant modifications occurred at 500 and 700 °C temperature range (see Fig. 64c). Degradation efficiencies were assessed by a test in 2 h using all types of sorbents; correlation of the degradation efficacy on the calcination temperature was discerned at 300 °C (see Fig. 64d). The highest efficacy was obtained using sorbents annealed at temperatures <500 °C, and the phosphatase-like performance lowered with rise in annealing temperature (see Fig. 64e). In general, CeO2/γ-Fe2O3 nanomaterials synthesized at temperatures of 300–400 °C displayed the higher decomposition performance without any change in its magnetic properties.404
Xiong et al.405 described the preparation of nanocomposites comprising cellulose and iron oxide NPs by co-precipitation employing ionic liquid (IL) as co-solvent; as-synthesized γ-Fe2O3 NPs exhibited uniform particle size distribution. The individual adsorption capacities of cellulose@γ-Fe2O3, pure γ-Fe2O3 and IL processed cellulose were investigated, and cellulose@γ-Fe2O3 was found to be the best candidate.405 The adsorption ability of nanospheres for the withdrawal of Pb(II) and Methylene Blue (MB) were 21.5 and 40.5 mg g−1, respectively which is comparable to the other reported magnetic materials. Additionally, utilizing an external magnet, magnetic nanoadsorbent could be easily recycled. The excellent removal capacities render the cellulose@γ-Fe2O3 as extremely capable nanoadsorbent for water treatment. In a similar context, magnetic iron oxide nanosorbent, and polymer functionalized nanoparticles were utilized for MB adsorption.406–408
Water bodies confront pollution from iodinated contrast media (ICM), primarily released by pharmaceutical companies.409 The microbial processes involved in organohalide respiration are essential for developing effective bioremediation strategies and promoting the sustainable cleanup of sites contaminated with chloroethene.410 To tackle this problem, the reductive dehalogenation of trichloroethene (TCE) has been scrutinized using aquifer material rich in iron oxides in a continuous flow column.411 The study observed sustained dehalogenation effectiveness over a period of 1000 days of column operation. In a similar context, recent research employed nanoscale zero-valent iron (nZVI) for the reduction-based removal of halogens, specifically focusing on the iodinated contrast medium diatrizoate (DTA).412 The study reported a 100% removal efficiency of diatrizoate (DTA) within a 24-hour timeframe.
The iron oxide NPs (17 nm) prepared by Chang et al.413 was employed for the extraction of Reactive Black 5 (RB5, Azo dye) from polluted water in the textile industry. The adsorption of RB5 to iron oxide NPs was ascribed to electrostatic attraction and the highest withdrawal capacity was 18 mg g−1 at 45 °C; withdrawal capability of NPs being 90% after 10 cycles. Mazarío and co-workers314 reported superparamagnetic nanosorbents for the adsorptive elimination of methyl orange and lead (Pb) from water. The physio-chemical interactions of the pollutant with the nanosorbent were demonstrated by a pseudo-second order kinetic model. The highest adsorption ability for methyl orange was found to be 240 mg gNS−1. In a similar line, iron oxide was also utilized for the degradation of organic matters.414 Recently, core–shell magnetic NPs were synthesized and deployed them for binding of the organic pollutant such as polychlorinated biphenyls.415
 | ||
| Fig. 65 TEM images of (a) Fe3O4; (b) Fe3O4@PDA; (c) UiO-67-bpydc; and (d) Fe3O4@UiO-67-bpydc. Reproduced with permission from ref. 418 Copyright 2021, American Chemical Society. | ||
In recent years, the utilization of hydrogen as a renewable substitute for fossil-based energy has been gaining momentum, and photoelectrochemical (PEC) water splitting stands out as an appealing and promising method for converting sunlight into hydrogen without the direct emission of greenhouse gases. However, achieving economic viability in PEC necessitates the enhancement of electrode efficiency and durability as the primary challenge.419 Mondal and colleagues420 have developed a cost-effective bifunctional Fe2O3@C and FeP@C electrocatalyst for solar-driven water splitting (see Fig. 66a). As Fe2O3@C was synthesized through a straightforward one-step pyrolysis process at different temperatures 600, 700, and 800 °C under inert conditions. The resulting brown colored powders synthesized at different temperature were identified by various shades and labeled as Fe-A, Fe-B, and Fe-C. TEM images of the Fe2O3@C nanohybrid revealed that the densely packed arrangement of Fe2O3 in Fe2O3@C (see Fig. 66b–d). The HRTEM images confirmed that the distinct lattice fringes, with a measured d-spacing of ∼0.252 nm (see Fig. 66e–g). Additionally, selected area electron diffraction (SAED) patterns, as shown in the insets of Fig. 66(e–g), indicated that the polycrystalline nature of Fe2O3 in all the nanostructures. The Fe2O3@C/Si composite has demonstrated significant activity in catalyzing the oxygen evolution reaction (OER), achieving a photoanodic current density of 2.5 mA cm−2 at 1.65 V vs. RHE (versus the reversible hydrogen electrode).
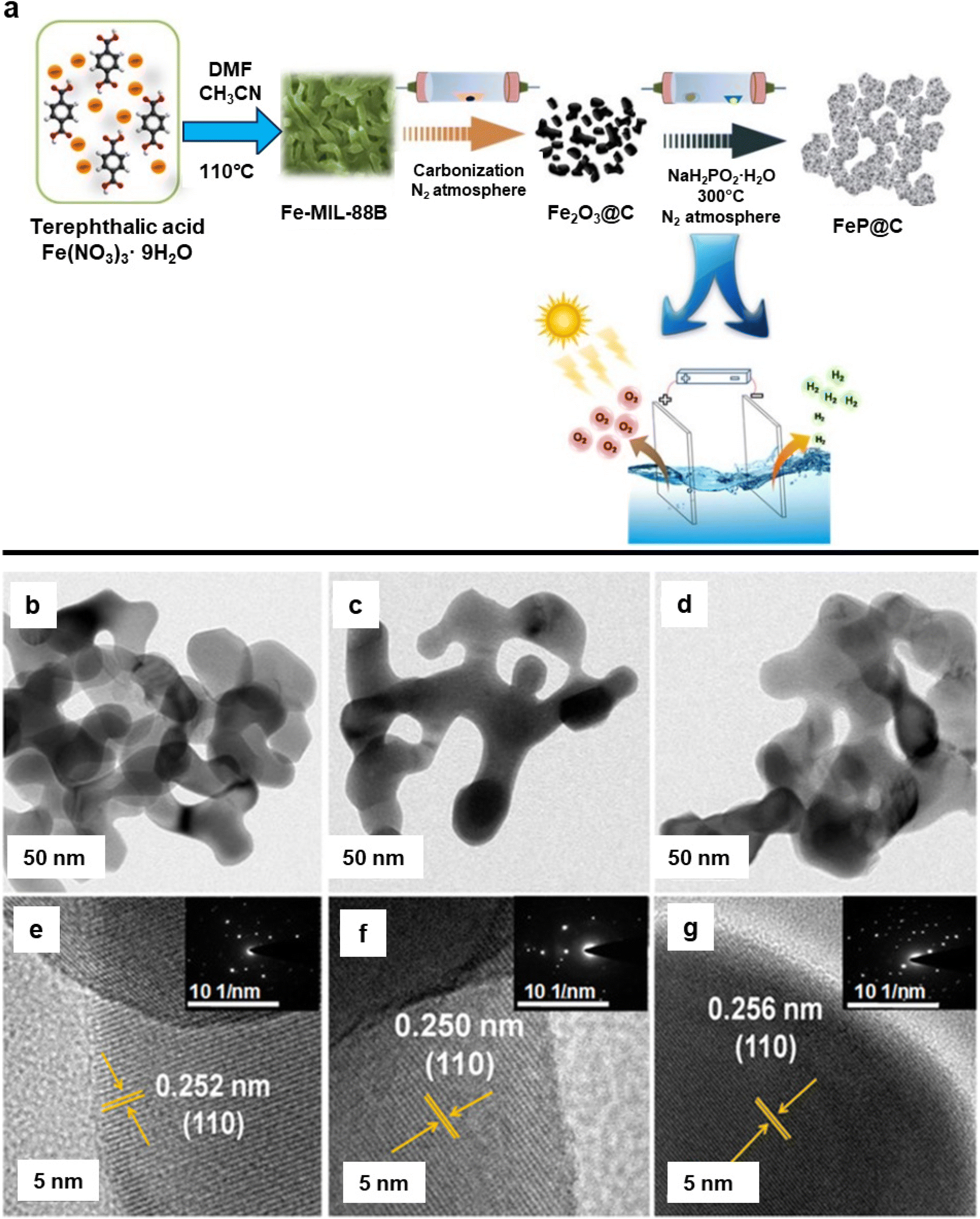 | ||
| Fig. 66 (a) Synthesis of bifunctional photoelectrodes, namely Fe2O3@C and FeP@C, intended for neutral water splitting; (b–d) TEM images of the Fe-A, Fe-B, and Fe-C respectively; (e–g) HRTEM images of the Fe-A, Fe-B, and Fe-C respectively, inset SAED patterns of the Fe-A, Fe-B, and Fe-C respectively; reproduced with permission from ref. 420 Copyright 2018, Wiley-VCH GmbH & Co. KGaA, Weinheim. | ||
Oxytetracycline (OTC) is a frequently utilized antibiotic for addressing infectious diseases in animals in intensive farming systems and aquaculture due to its broad-spectrum antibiotic properties but it became a major problem when OTC release was encountered in soil and groundwater.421,422 Traditional methods for detecting oxytetracycline (OTC) primarily rely on liquid chromatography, capillary electrophoresis, enzyme-linked immunosorbent assay, and high-performance liquid chromatography (HPLC). While these conventional techniques are known for their reliability, sensitivity, and stability, their widespread application is hindered by certain drawbacks, including the need for expensive equipment, high costs, and lengthy sample preparation times. Limited research has been conducted on MOF-derived materials in the context of electrochemical sensing and aptasensors applications.423 A chain of Fe3O4@mC nanocomposites derived through the pyrolysis of Fe-MOF (525-MOF) was successfully explored for detection of OTC.424 The as-synthesized catalyst was analyzed by TEM to examine the morphological transformations of the Fe3O4@mC nanocomposites under different calcination temperatures. In the initial temperature range (350 °C) (see Fig. 67a and b), the majority of the Fe2O3 exhibits a uniform distribution, resembling sphere-like structures with an average size of 5–8 nm. Moving to the second stage (550 °C) (see Fig. 67c and d), larger spherical particles are observed, forming, and embedding into porous carbon nanosheets, with most particles ranging in size between 10 and 12 nm. Further elevation of the calcination temperature to 700 and 900 °C results in the enlargement of iron oxide particle sizes due to crystal aggregation (see Fig. 67e–h), as evidenced by TEM images. Red arrows indicated that the Fe2O3 or Fe3O4 NPs; yellow arrows indicated the mesopores within the amorphous carbon sheet (see Fig. 67a, c, e and g). As reported Fe3O4@mC900 stands out as the optimal candidate for achieving highly sensitive and selective detection of OTC.
 | ||
| Fig. 67 TEM images of (a, b) Fe3O4@mC350, (c, d) Fe3O4@mC550, (e, f) Fe3O4@mC700, and (g, h) Fe3O4@mC900 nanocomposites (insets: corresponding SAED patterns). Reproduced with permission from ref. 424 Copyright 2013, The Royal Society of Chemistry. | ||
Hydrogen sulfide (H2S) is a malodorous gas emitted from various natural and human activities. Gupta et al.425 have developed an innovative method to convert H2S waste into value-added products by utilizing FeBDC and FeBTC derived iron oxide MOF's, labelled as IO-D and IO-T. They have detailed a process for synthesizing iron-based MOFs by sonication of organic linkers with freshly prepared iron hydroxide. The removal of H2S using iron oxide adsorbents primarily involves oxidizing H2S into sulfur and sulfates. Notably, the adsorption capacity of iron oxide adsorbents surpassed that of the corresponding MOF. Among them, FeBDC (IO-D) exhibited the highest adsorption capacity, reaching 36.2 mg g−1 under moist conditions.
3.2. Photocatalytic applications of iron oxides in environmental technologies
Photocatalysis, a cutting-edge technology employed in photocatalytic decomposition of organic pollutants, has garnered attention in view of its sustainability attributes.426–429 By definition, photocatalysis entails speeding up the reaction by the light in the existence of catalyst. In catalyzed photolysis, a substrate absorbs light, whereas in the photogenerated catalysis, the catalyst performance depends on the capacity to form electron–hole pairs, which undergo secondary reactions.However, some difficulties restrict the extensive implementation of iron oxide NPs for the photodegradation of hazardous chemicals due to difficulty in partition of compounds after completion of the action. The operational procedure is usually costly due to labor, time and raw materials intended for precipitation, further decantation or centrifugation at the termination of process, and the low quantum-yield of treatment that limits the rate and efficacy of the process.430 Great strides, however, have been made to increase the photocatalytic performance by reducing the size of the photocatalyst, thereby increasing the surface area, thus merging photocatalyst with some new and efficient metal nanoparticles;431–433 a variety of iron oxide nanoparticles have been employed for photocatalytic reactions with varied applications.434 As an example, Zhou et al.435 have reported 1D nanorods of goethite (α-FeOOH) and hematite (α-Fe2O3) for photocatalytic decomposition of Rhodamine B (RhB) by visible light under the existence of H2O2. Nanometer-scale α-FeOOH and α-Fe2O3 particles were found to be more effective compare to microscale counterparts in respect of surface area normalized reaction rate. Furthermore, α-Fe2O3 nanorods displayed the highest catalytic performance compared to other samples. The perceived photocatalytic efficiency of iron oxide NPs was ascribed to the combined impact of particle composition, porosity, size, and deviations in morphology.
The Fe2O3/CQDs (Carbon Quantum Dots) composites for photodegradation of toxic gas under visible light have been reported by Liu and Kang.436Fig. 68a shows three photographs of distinct colors of (i) CQDs aq. solution, (ii) reaction mixture including CQDs, FeCl3 and (NH2)2CO and (iii) product solution. The complete disappearance of the dust color of CQDs is indicative of the formation of Fe2O3/CQDs. The deployment of Fe2O3/CQDs composites for photocatalytic toxic gas decomposition has been described in this report for the first time. The synthesized Fe2O3/CQDs nanomaterials displayed improved photocatalytic performance for decomposition of toxic gas under visible light when compared to Fe2O3; photoexcited electrons from iron oxide particles could be transferred without restraint in the conducting network of CQDs (Fig. 68b).436 Moreover, it was proposed that an improvement in the photoactivity of iron oxide is encouraged by the presence of CQDs as they support for active segregation of photogenerated electrons and holes. The electron–hole pairs can then react with adsorbed oxidants/reductants, generating plentiful active oxygen radicals with strong oxidizing ability for the degradation of toxic gases. In addition, CQDs display up conversion photoluminescence by lower wavelength of light than absorbed light, thereby exciting Fe2O3 to form electron–hole pairs. There have been other reports on grafting iron oxide NPs in the mesoporous carbon matrix for such potential applications.362,437–440
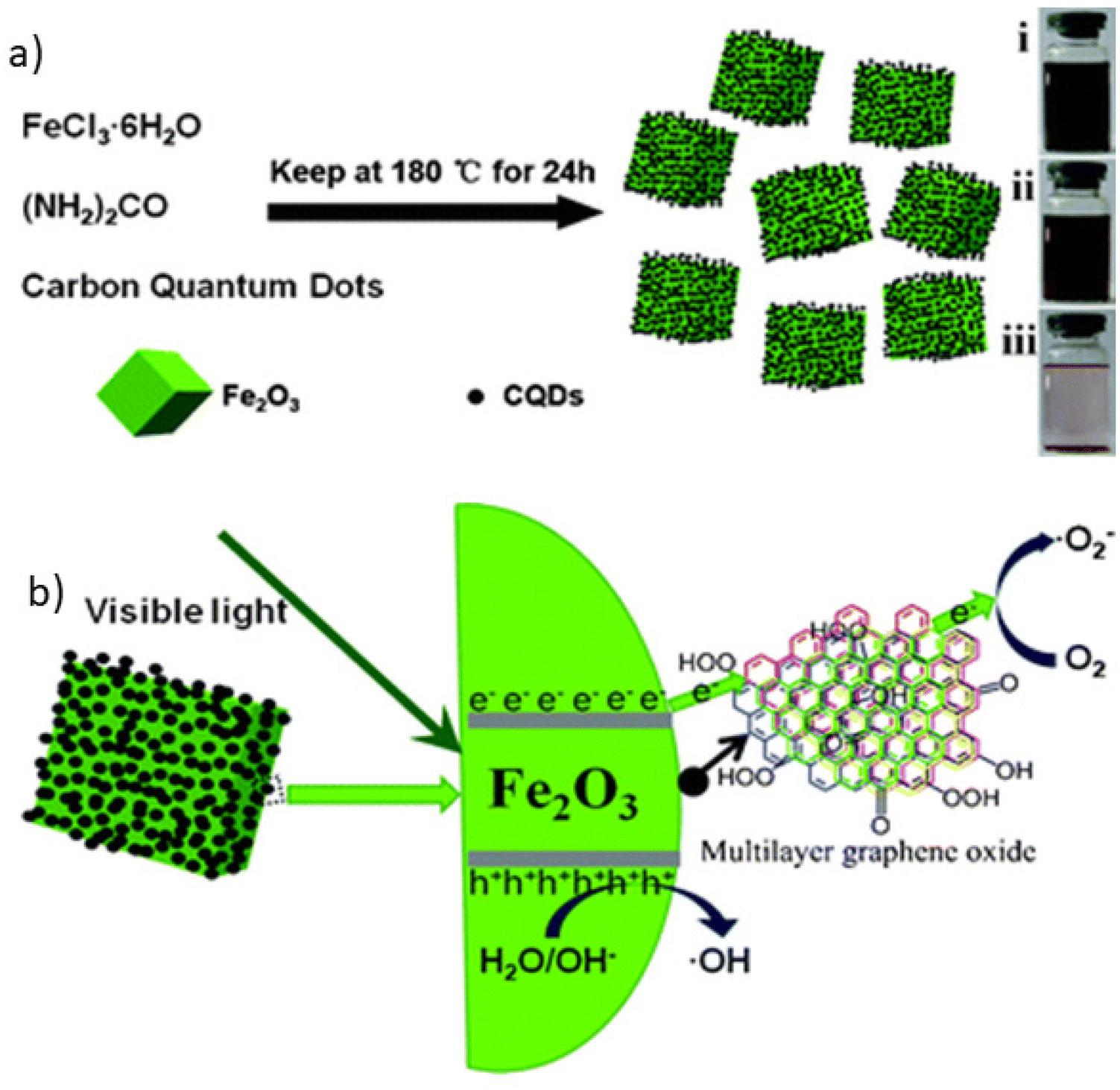 | ||
| Fig. 68 (a) Preparation of Fe2O3/CQDs composites by the hydrothermal treatment; (b) Schematic of the photocatalytic process of Fe/CQDs composites in the presence of visible light. Reproduced with permission from ref. 436 Copyright 2011, The Royal Society of Chemistry. | ||
Among iron oxides, hematite (α-Fe2O3, with a bandgap of ∼2.1 eV) is one of most widely explored oxides for photocatalysis;441–443 various protocols have been applied for the synthesis of controlled films of hematite with considerable photocatalytic activity although some of them have drawbacks including formation of less amount of the product. Diab and Mokari434 reported the scaled-up general synthesis of mesoporous α-Fe2O3 to exploit their photoelectrochemical (PEC) properties. The presented methodology has been established on direct thermal degradation of iron acetate on a substrate to form a mesoporous film of α-Fe2O3 with excellent pore size regulation. An improvement of the PEC features of iron oxide was attained by using CoO areas, which were developed by thermolysis of the Co salt on the hematite surface to get α-Fe2O3/CoO nanomaterials. The cobalt material was deposited by two methods: (a) by thermal decomposition and (b) by adsorption of Co salt. The photoconductivity of pristine hematite was 0.25 mA cm−2 at 1.23 V versus reversible hydrogen electrode (RHE), whereas alteration of hematite surface employing thermolysis protocol displayed 180% enhancement by the adsorption process. Furthermore, the beginning potential was changed by 130 and 70 mV when hematite surface was amended by the thermolysis and adsorption process, respectively.
Fenton and photo-Fenton processes have been proposed as potent methods for treating organic pollutants in wastewater. Hydroxyl (HO˙) and superoxide (O–O˙) radical are formed in the Fenton reactions (eqn (1)–(3)), which then significantly decompose and mineralize the organic contaminants to H2O, CO2, and other inorganic compounds. In particular,
| Fe2+ + H2O → Fe3+ + HO˙, | (1) |
| Fe3+ + H2O2 ↔ FeOOH2+ + H+, | (2) |
| FeOOH2+ → Fe2+ + HO2, | (3) |
Despite exhibiting powerful oxidation capability, Fenton reagents face several drawbacks like very low pH range for efficient reactions (2.5–4.0), formation of ferric hydroxide sludge at values of pH higher than 4.0, difficulty in recovery of the catalyst, and higher costs for iron ions removal from the process after the termination of the reaction.444 Moreover, an impulsive oxidation of Fe2+ to Fe3+ ions frequently occur by dissolved oxygen in water via diverse partially oxidized meta-stable Fe2+/Fe3+ intermediates which eventually transforms into stable iron oxides like hematite, maghemite, and magnetite. Thus, an idea has been proposed to replace the dissolved iron with solid catalysts.
Photo-Fenton reaction is a fusion of the Fenton reagents and ultraviolet and/or visible light that promotes formation of an extra ˙OH radicals through photo-reduction of Fe3+ to Fe2+ ions and H2O2 photolysis. Iron oxides have been identified as promising candidates for photocatalysts as they, among others, show semiconductor properties. However, hole–electron recombination process in iron oxides can be very fast, impairing the photocatalyst performance. In iron oxides, semiconducting mechanisms involves two steps. Firstly, a photon with an energy equal of higher than the energy band gap between the valence and conduction band is adsorbed by iron oxides, generating an electron–hole pair, i.e., Fe2O3 + hν → Fe2O3 (e− + h+). Even though the excited electron–hole pair can recombine with a heat dissipated, few of the excited electron and holes can promote the redox reactions occurring on the surface of iron oxide semiconducting particle. Several redox processes may then occur in an aqueous environment, containing iron oxide particles with generated electron–hole pairs and organic substance (OS) such as (eqn (4)–(7)).445
| ecb− + O2 → O2˙−, | (4) |
| hvb+ + O2˙− → 1O2, | (5) |
| ecb− + >Fe3+ → >Fe2+, | (6) |
| hvb+ + OSad → OSad˙− | (7) |
It is well known that irradiation may also trigger the heterogeneous Fenton process on Fe-containing nanoparticles, encouraging occurrence of the photo-reduction of >Fe3+OH to >Fe2+; >Fe2+ can then react with H2O2 to produce ˙OH radicals at the surface of the nanoparticle, i.e.446
| >Fe3+OH + hν → >Fe2+ + ˙OH, | (8) |
| >Fe2+ + H2O2 → >Fe3+ + OH− + ˙OH. | (9) |
However, α-Fe2O3 shows a poor Fenton reaction performance. Thus, there have been several attempts to increase the reaction activity of α-Fe2O3. One of the approaches is to dope α-Fe2O3 structure with sulfur (in the form of FeS or FeS2) as has been described by Guo et al.;447 sulfur in the α-Fe2O3 structure promotes photo-Fenton reaction. Firstly, S-doping alters the interfacial reaction process of Fe ions with H2O2, i.e.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Fe3+ + H2O2 → [Fe3+O2H]2+ + H+, Fe3+ + H2O2 → [Fe3+O2H]2+ + H+, | (10) |
| [Fe3+O2H]2+ → Fe2+ + ˙O2H, | (11) |
| Fe2+ + H2O → Fe2+(H2O2), | (12) |
| Fe2+(H2O2) → Fe4+O + H2O. | (13) |
It confirms that electron transfer among the peroxide species (˙O–O) and Fe ions is increased at the interface which results in a higher Fenton reactivity. Secondly, doping sulfur of α-Fe2O3 promotes formation of trap states within the band gap of α-Fe2O3. When illuminated under UV or visible light, α-Fe2O3 photocatalytic processes occur.447
| α-Fe2O3 + hν → ecb− + hvb+, | (14) |
| dye + hν → dye*, | (15) |
| dye* + α-Fe2O3 → dye+˙ + ecb−, | (16) |
| ecb− + hvb+ → recombination, | (17) |
| ecb− + Fe3+ → Fe2+, | (18) |
| Fe3+–OH + hvb+ → Fe4+O + H+, | (19) |
| Fe3+–OH + hvb+ → Fe3+ + ˙OH, | (20) |
| Fe2+ + H2O2 → Fe2+(H2O2), | (21) |
| Fe2+(H2O2) → Fe4+O + H2O, | (22) |
| ˙OH˙(Fe4+O) + organics → degradation intermediates. | (23) |
The formed trap states act as defect state with a tendency to catch the charge carriers immediately after they are formed, thus retarding their recombination process. The photo-Fenton reactivity of S-doped α-Fe2O3 has been then assessed for the decomposition of phenol and acid orange, showing much higher performance compared to undoped α-Fe2O3.447
A nanocomposite comprising TiO2 nanowires, Fe2O3 NPs, and GO sheets has been synthesized to address the problem of membrane fouling in membrane water treatment technologies, produced by humic acid.448 The removal of humic acid involves two steps, i.e., adsorption and photo-degradation (see Fig. 69). Humic acid is first adsorbed on the Fe2O3 nanoparticle sites. Then, under irradiation, electron–hole pairs are formed on the surface layers of the TiO2 nanowires. The holes oxidize water and generate ˙OH radicals which oxidize nearby adsorbed HA. The electrons are preferentially carried to graphene oxide sheets (as a result of strong electric conductivity of graphene oxide) and upon reaction with oxygen, they promote formation of superoxide anion radicals (O2˙−). Thus, membrane photocatalytic activity is enhanced due to splitting of electrons and holes. The 12 h test confirmed a higher removal of humic acid (92%) and a decrease in pressure across membrane were observed under irradiation compared to dark conditions, implying an anti-fouling ability of the novel membrane. Very recently, Qiu and co-workers449 employed iron for the photocatalytic remediation of heavy metals. The combination of iron oxide and graphene also increased the adsorption for the decomposition of ammonium perchlorate.450
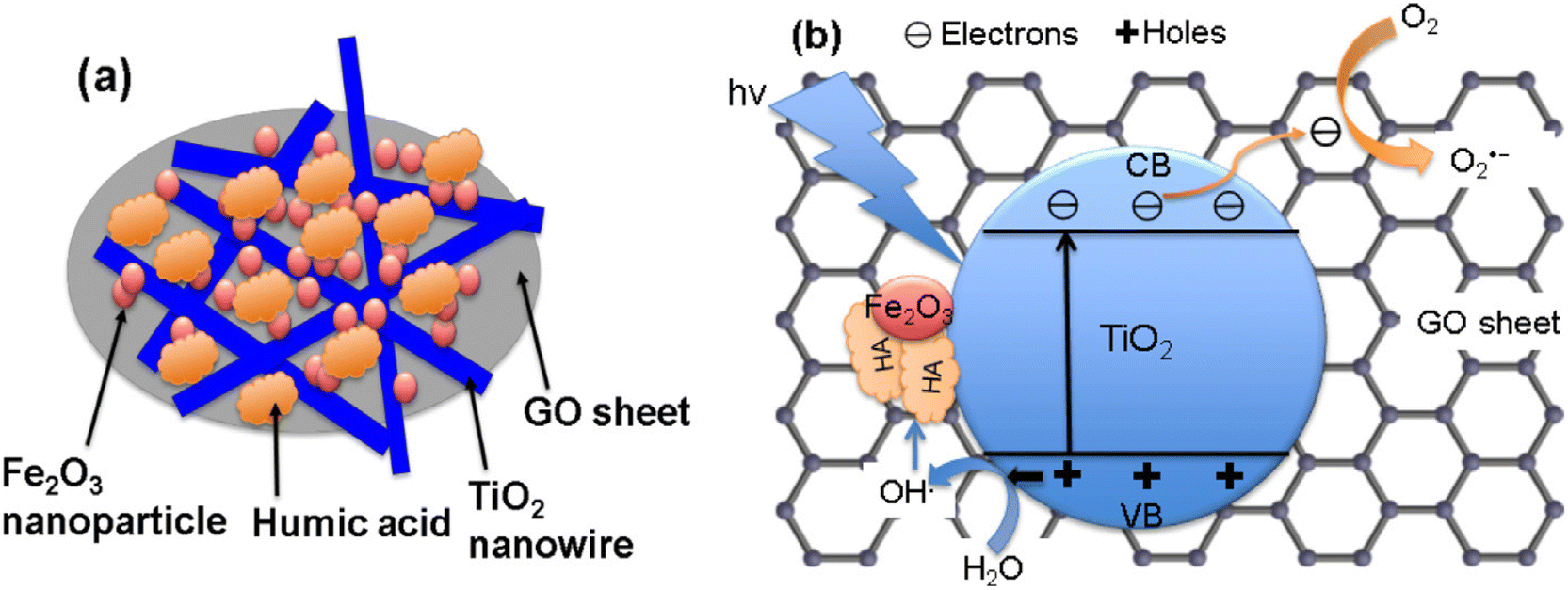 | ||
| Fig. 69 (a, b) Schematic representations of membrane surface with humic acid adsorption and plausible mechanism for humic acid photo-degradation over the membrane surface. Reproduced with permission from ref. 448 Copyright 2016, Elsevier. | ||
3.3. H2O2 decomposition promoted by iron oxides for environmental applications
In general, the Fenton process was initiated with the invention of short-lived, enormously reactive hydroxyl radical (˙OH) with a potential of 2.8 V (ref. 451) from the interaction between Fe2+ and H2O2; hydroxyl radical then helps for the material decomposition as discussed above.452–454 Over the last few years, tremendous emphasis has been directed towards the progress of Fenton reactions specially focused on role of pH increase, in situ delivery of H2O2 though oxygen reduction, in situ offering of Fe2+ though the electrochemical oxidation of Fe, prevention loss of Fe and regeneration of Fe(II) by enlightening the sludge of iron origins (see Table 3).455| Sr. no. | Fenton's reaction types | Reagents | Iron loss | pH | Light |
|---|---|---|---|---|---|
| 1 | Photo Fentons | Complex free iron ions | Yes | Acidic to neutral | Yes |
| 2 | Classic Fentons | Fe2+, H2O2 | Yes | 2–4 | No |
| 3 | Fenton like | Fe3+, H2O2 | Yes | 2–4 | No |
| 4 | Electro-Fentons | Electrogenerated Fe2+ | Yes | 2–4 | |
| 5 | Heterogeneous Fentons | H2O2, solid iron oxide, free iron | No | Broad range | No |
| 6 | Heterogeneous Photo-Fenton | H2O2, solid iron oxide | No | Broad Range | Yes |
| 7 | Heterogeneous Photoelectron-Fenton | H2O2, solid iron oxide | No | Broad Range | Yes |
The catalytic decomposition of H2O2 has been widely examined because of its major application in water treatment technologies. Hydrogen peroxide along with iron oxides has the capability to decompose organic pollutants in wastewater and contaminated soils; thus, it is viewed as a challenge for chemists to design unique and inexpensive catalyst materials/nanomaterials.75,456 The rate of the H2O2 decomposition primarily depends on several experimental factors like concentration of H2O2, pH, and the nature of catalyst.457,458 The reactions of Fe(II) with O2 and H2O2 are essential as they directly produce the highly reactive oxidants i.e. hydroxyl radical (˙OH) which is the key reactive species in various advanced oxidation processes.458–460
There are numerous scientific literatures detailing the exploitation of various iron oxide nanoparticles for the heterogeneous Fenton reaction including mainly hematite, magnetite, goethite, and maghemite NPs.77,461–463 Tang and co-workers462 reported BiFeO3 NPs for the oxidation of bisphenol A (BPA) with benign oxidant. To initiate the catalytic efficacy of BiFeO3 nanoparticles, in situ surface tempering of ligands was employed and BPA removal was enhanced through incorporating chelates including formic acid, tartaric acid, glycine, nitrilotriacetic acid (NTA) and EDTA. The catalytic efficiency order was found to be, blank < tartaric acid < formic acid < glycine < NTA < EDTA. Among these ligands, EDTA serve as the highly active complexing agent, and 0.25 mmol L−1 of EDTA removes 91% BPA. The degradation of BPA was reliant on the quantity of additional EDTA, the starting conc. of H2O, pH, and temperature. Furthermore, EDTA strongly induced the performance of BPA decomposition, resulting from the three different reaction sites on the BiFeO2 surface, such as bare BiFeO3 sites, EDTA-adsorbed sites, and intermediate blocked sites (see Fig. 70).462
 | ||
| Fig. 70 Degradation of BPA showing distinct reaction sites. Reproduced with permission from ref. 462 Copyright 2011, American Chemical Society. | ||
The mechanism of hydrogen peroxide decomposition utilizing iron oxide as a catalyst has been clearly delineated459,464 where degree of H2O2 decomposition is in direct proportion to the iron oxide concentration.465 However, salient feature responsible for the enhanced catalytic efficacy in heterogeneous catalysis involves greater surface area, obtained with the help of NPs. Hydrogen peroxide with iron oxide nanoparticles have been especially found attractive for wastewater treatment; H2O2 decomposes catalytically by means of Fe2+ at acidic pH to generate hydroxyl radicals.466
Fe/H2O2 oxidations have been prominent because of the fact that Fe is an earth-abundant and relatively nontoxic metal, combined with the ease of handling of H2O2, which additionally degrades into environmentally benign byproducts when present in excess safe products. It has been referred to as the most economical alternative when compared to other oxidation methods; the process uses simple equipment and mild operational conditions (atmospheric pressure and room temperature).467 H2O2 decomposition on iron oxide nanoparticles is a well-established platform for environmental applications in wastewater treatment, organic compounds, and dyes degradation.468–471
Mesoporous iron oxides are important and versatile materials as they possess high surface area, equal pore size distributions, nanosized walls, substantial pore volume, and surface functionality.472 An organized mesoporous Mg-modified hematite with an ultra-high surface area was synthesized by hard templating techniques in mesoporous silica KIT-6 and was employed for the methylene blue degradation.473 The concentration of methylene blue in the supernatant was obtained at an absorbance peak of 664 nm and the concentration of ˙OH was determined by the terephthalic acid by a fluorescence method.474 Further, these results confirmed that approximately 2/3 of the Mg cations were detached at the time of leaching, and as a result produces more structural defects (Fig. 71). The activated mesoporous Fe2O3 displays outstanding catalytic efficiency for MB degradation, achieving over 95% extraction of 60 mg L−1 MB after 3 h. The rate constant of employed meso-Mg/Fe2O3 catalyst is 1.972 h−1 which is 1.22, 3.02 and 4.53 times higher compared to meso-Fe2O3, con-Mg/Fe2O3,473 and γ-Fe2O3 respectively. The leaching concentration of Mg and the catalytic activity increased with the reuse of meso-Mg/Fe2O3 catalyst, which is fully disparate with the multi-component catalytic mechanism in typical heterogeneous Fenton-like processes, like Fe–Cu and Fe–Zn composites.473
 | ||
| Fig. 71 Schematic representation of mesoporous Mg–Fe2O3 by leaching of Mg ions; arrows indicate the Mg leaching sites. Reproduced with permission from ref. 473 Copyright 2015, The Royal Society of Chemistry. | ||
The heterogeneous catalyst (Fe3O4/usGO) for Fenton-like degradation of MB as dye pollutant was reported by Song et al.475 Various iron oxide NPs have been employed for the removal of methylene blue,476 antibiotics degradation477 and also for safeguarding of human health by Fenton reaction.478
To increase the reactivity of the system, a strategy was suggested to enhance the contact of Fe(0) with Fe3O4 nanoparticles by grinding;479 it was found that assembling such architectures promote electron transfer from the metal to the reaction center. In particular, at the Fe(0)/Fe3O4 interface, the metal transfers electrons to iron oxide which serves as an intermediary with the reaction center. Iron oxide is a semiconductor showing a high electric conductivity, thus promoting the movement of electrons; moreover, its octahedral sites can readily adapt Fe2+ and Fe3+ species in the redox equilibrium. The development of the reactive species is schematically depicted in Fig. 72. Reactive networks are also synthesized by mechanical milling and thermal treatment leading to Fe(0)/α-Fe2O3 and Fe(0)/γ-Fe2O3 interface mixtures, but with an inferior efficiency for H2O2 in comparison with Fe(0)/Fe3O4. This implies that Fe2+ species are accountable for the activity and thus, are more effective in the oxidation of organic pollutants in wastewaters. Recently, Qin et al.480 studied the degradation of pollutant such as p-nitrophenol by regulating the concentration of Fe2+ and ligand as a tripolyphosphate.
 | ||
| Fig. 72 The formation of the reactive species at the Fe(0)/Fe3O4 interface. Reproduced with permission from ref. 479 Copyright 2006, Elsevier. | ||
3.4. Biomolecule immobilization on iron oxide nanoparticles towards advanced environmental applications
Iron oxide nanoparticles have displayed a substantial potential for immobilizing the biomaterials, enzymes, etc.481,482 The biosorption capability of diverse macro- and microbial enzymes or biomolecules has extensively employed to eliminate many contaminants; assorted types of iron oxide nanomaterials offer higher surface areas and number of active sites for synergy.483–485The research on biofuels is advancing at a fast pace as an emerging field of study to offer substitute sources of renewable energy. Nanotechnology advancements have aimed to increase the efficacy of biofuel production, thereby increasing energy security.486 Recently, magnetic single walled carbon nanotubes (mSWCNTs) have developed487 by introducing an iron oxide NPs into SWCNTs. In this protocol, the enzyme amyloglucosidase (AMG) was immobilized onto mSWCNTs utilizing to combine the two technologies i.e., nanotechnology and biofuel generation. Up on immobilization on mSWCNTs, enzyme preserves its catalytic performance in the ideal enzymatic hydrolysis of starch over repeated applications, subsequently, the material can be removed from the reacting environment by using an external magnet. These ensuing results, open the path for higher efficacy and lower principal expense of carbon nanotube enzyme bioreactors in industrial enzyme protocols, together with the special features of carbon nanotubes. There are several other innovative examples of post-synthetic modifications of iron oxides nanoparticles. Carbon-coated cobalt NPs that are chemically functionalized were employed for the immobilization of enzyme.488 Li and co-workers489 have investigated Ni-NTA (2,2′,2′′-nitrilotriacetic acid) functionalized iron oxide NPs that were deployed for the selective immobilization of histidine-tagged enzyme; other promising examples of immobilization of enzymes and their applications have been documented.490–492
Another crucial arena is the immobilization of biomaterials onto a sturdier and porous support that has been significantly investigated for varied applications; examples can be found for fungi and microalgae and exploitation for hydrogen production.493–497 Indeed, immobilized cells have garnered a huge attention since the 1970s, because of their clear merits over dispersed cells.498 First, the immobilization of native biomaterial improves the physical properties and proposes advanced degree of action.499 In addition, opposition to environmental influences like pH, temperature, and toxicity can be improved.500 Moreover, immobilization promotes the use cyclic biomaterials, offering simplified liquid–solid separation, and reducing the blocking in continuous-flow systems. These benefits could meet the technical requirements for the utilization of such supports, where it is crucial to use economical, simple, easy, and suitable methods for immobilization.
Peng and co-workers501 have reported that Saccharomyces cerevisiae supported on chitosan-coated magnetic NPs (SICCM) can be deployed as an adsorbent for Cu(II) ions from aqueous media. The removal efficacy of over 90% is achievable within 20 min, with the highest adsorption capability being 134 mg g−1. It can therefore be assumed that the dispersion of biomaterials on appropriate support is important for the application of bio adsorbents in bulk-scale technologies. Additional research, however, is needed to improve the adsorbent, primarily by choosing suitable strains that have great adsorption capability for specific heavy metals and organic compounds.
In a similar context, recently the modified iron oxide nanoparticles have been used for the environmental application such as the immobilization of cadmium from contaminated soil,502 arsenic removal from wastewater,503 nano-remediation of contaminated aquifers504,505 and also, for the immobilization of phosphorous from soil.506,507
Although an array of immobilization media and protocols have been examined, relatively scant information is available on combination of nanotechnology-based iron oxides with other biological matrices for environmental use. An ideal fusion could result in the development of innovative applications by using the inherent advantages of both the nanomaterial and biomaterials. Consequently, it is crucial to investigate not only the mass scale applications of adsorption technique but also ideal immobilization methods with a greater potential and constancy. Further research ought to exploit the potential of magnetic nanomaterials in combination with biological components for novel biotechnology applications like biosensors, immobilization of proteins for magnetic separation and environmental enhancement.
4. Conclusions and future directions
In this review, we have strived to comprehensively describe the usage of non-hydrated iron oxides in catalytic and environmental applications. It is amply demonstrated that the position of non-hydrated iron oxide phases in these areas is paramount and superior compared to other materials. The review deals with the identification of various parameters that can be synthetically controlled and, hence, they can be finely tuned in a way to be targeted for specific catalytic and environmental technologies.Iron oxide nanoparticles have been extensively deployed as highly efficient catalysts and/or catalytic supports for diverse organic transformations like reduction, oxidation, coupling, and alkylation reactions, Fischer Tropsch synthesis, or multicomponent reactions. A remarkable impact has also been made by iron oxide nanoparticles to promote the production of crucial organic compounds. Nonetheless, there are still several challenges associated with iron oxide NPs and their applications in catalysis that need to be circumvented in the future. Despite of having diverse physical, chemical, and biological procedures to prepare various iron oxide NPs, there is still an obstacle to regulate their surface properties and immobilization of surface fractions in the framework of organic/inorganic composites, thus tuning their stability and the composite's strength. Therefore, future research ought to be undertaken on computational design including artificial intelligence approaches and optimizing routes for the controllable and scalable synthesis of iron oxide nanoparticles. There is also a significant challenge in exploring the reaction mechanisms and identifying individual reaction steps. Here, the computational approaches and advanced in situ techniques should be largely applied. Finally, the possible use of magnetic iron oxides as advanced supports for single atom embedding need to be more intensively studied thus opening the doors towards scalable and readily separable iron oxide based single atom catalysts.
One of the foremost societal and scientific challenge is the generation of large amounts of wastewater, which needs routine wastewater treatment operations to prevent harm to people, animals, soils, and plants as well as the spread of numerous illnesses. Thus, the substantial portion of this review article is dedicated to the use of iron oxide NPs in advanced environmental and water treatment technologies including e.g., sorption technologies, photocatalytic applications, hydrogen peroxide assisted degradation of pollutants and other techniques. When compared to traditional adsorbents, iron-oxide nanoparticles operate more effectively and efficiently in wastewater treatment in terms of their abundance, low toxicity, and low costs. However, the future research should examine and assess the possibility of sorbent regeneration, and reusability potential to exploit the use of iron oxide nanoparticles for wastewater treatment. Similarly, the tailored surface modification of iron oxide NPs towards selective removal of particular contaminants represents another important challenge. There is a paucity of information on the interaction of iron oxides with biological matrices, which would affect their efficiency in environmental applications. Behind the successful progress of the heterogeneous nanocrystalline iron oxides for the use in photo-Fenton process, skillful coupling of this technology with traditional techniques including filtration, coagulation, and membrane technology is needed. In addition, further pilot-scale studies are required to investigate the full-scale application of iron oxide NPs in remediation and photocatalytic technologies.
Author contributions
MBG, RV, and RZ conceptualized the idea, supervised, and edited the complete manuscript. DSC and RPU wrote the draft, edited the manuscript, and compiled the review. GYS and JF co-wrote the manuscript part.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
DSC and RPU express special thanks to the Institute of Chemical Technology, Mumbai-Marathwada Campus, Jalna, for providing facilities for pursuing a doctoral degree. M. B. G. gratefully acknowledge the funding support from Department of Science and Technology, SERB, Core Research Grant (CRG/2021/001738). This article has been produced with the financial support of the European Union under the REFRESH – Research Excellence for Region Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition. Furthermore, the work was supported by ERDF/ESF project TECHSCALE (No. CZ.02.01.01/00/22_008/0004587).References
- D. Liu, W. Wu, J. Ling, S. Wen, N. Gu and X. Zhang, Adv. Funct. Mater., 2011, 21, 1498–1504 CrossRef CAS.
- W. Mekseriwattana, W. Phanchai, T. Thiangtrongjit, O. Reamtong, T. Puangmali, P. Wongtrakoongate, A. Roig and K. P. Katewongsa, ACS Appl. Nano Mater., 2023, 6, 13952–13964 CrossRef CAS.
- M. Ognjanović, M. Radović, M. Mirković, Ž. Prijović, M. del Puerto Morales, M. Čeh, S. Vranješ-Đurić and B. Antić, ACS Appl. Mater. Interfaces, 2019, 11, 41109–41117 CrossRef PubMed.
- K. X. Vazquez-Prada, S. S. Moonshi, Y. Wu, F. Akther, B. W. C. Tse, K. A. Sokolowski, K. Peter, X. Wang, G. Xu and H. T. Ta, Small, 2023, 19, 2205744 CrossRef CAS PubMed.
- T. Vangijzegem, V. Lecomte, I. Ternad, L. Van Leuven, R. N. Muller, D. Stanicki and S. Laurent, Pharmaceutics, 2023, 15, 236 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167 CrossRef CAS.
- T. Vangijzegem, D. Stanicki and S. Laurent, Expert Opin. Drug Delivery, 2019, 16, 69–78 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- Z. Zhang, L. Weng, Q. Rao, S. Yang, J. Hu, J. Cai and Y. Min, J. Power Sources, 2019, 439, 227107 CrossRef CAS.
- M. Chen, F.-M. Liu, H. Zhao, S.-S. Chen, X. Qian, Z.-Y. Yuan and R. Wan, Phys. Chem. Chem. Phys., 2022, 24, 27114–27120 RSC.
- D. Mohan and C. U. Pittman, J. Hazard. Mater., 2007, 142, 1–53 CrossRef CAS PubMed.
- A. M. H. Shadi, M. A. Kamaruddin, N. M. Niza, F. M. Omar and M. S. Hossain, J. Environ. Chem. Eng., 2022, 10, 107753 CrossRef CAS.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef CAS PubMed.
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- M. I. Idris, J. Zaloga, R. Detsch, J. A. Roether, H. Unterweger, C. Alexiou and A. R. Boccaccini, Sci. Rep., 2018, 8, 7286 CrossRef PubMed.
- H. Cai, X. An, J. Cui, J. Li, S. Wen, K. Li, M. Shen, L. Zheng, G. Zhang and X. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 1722–1731 CrossRef CAS PubMed.
- H. Guo and A. S. Barnard, J. Mater. Chem. A, 2013, 1, 27–42 RSC.
- R. Zboril, M. Mashlan and D. Petridis, Chem. Mater., 2002, 14, 969–982 CrossRef CAS.
- L. Machala, J. Tuček and R. Zbořil, Chem. Mater., 2011, 23, 3255–3272 CrossRef CAS.
- J. Tuček, L. Machala, S. Ono, A. Namai, M. Yoshikiyo, K. Imoto, H. Tokoro, S.-i. Ohkoshi and R. Zbořil, Sci. Rep., 2015, 5, 15091 CrossRef PubMed.
- C. Wu, P. Yin, X. Zhu, C. OuYang and Y. Xie, J. Phys. Chem. B, 2006, 110, 17806–17812 CrossRef CAS PubMed.
- P. Robinson, R. J. Harrison, S. A. McEnroe and R. B. Hargraves, Nature, 2002, 418, 517–520 CrossRef CAS PubMed.
- O. Malina, J. Tuček, P. Jakubec, J. Kašlík, I. Medřík, H. Tokoro, M. Yoshikiyo, A. Namai, S.-i. Ohkoshi and R. Zbořil, RSC Adv., 2015, 5, 49719–49727 RSC.
- C.-W. Lee, K.-W. Lee and J.-S. Lee, Mater. Lett., 2008, 62, 2664–2666 CrossRef CAS.
- M. M. Rahman, A. Jamal, S. B. Khan and M. Faisal, Superlattices Microstruct., 2011, 50, 369–376 CrossRef CAS.
- G. Carraro, D. Barreca, M. Cruz-Yusta, A. Gasparotto, C. Maccato, J. Morales, C. Sada and L. Sánchez, ChemPhysChem, 2012, 13, 3798–3801 CrossRef CAS PubMed.
- G. Carraro, C. Maccato, A. Gasparotto, T. Montini, S. Turner, O. I. Lebedev, V. Gombac, G. Adami, G. Van Tendeloo, D. Barreca and P. Fornasiero, Adv. Funct. Mater., 2014, 24, 372–378 CrossRef CAS.
- J. Tuček, R. Zbořil, A. Namai and S.-i. Ohkoshi, Chem. Mater., 2010, 22, 6483–6505 CrossRef.
- J. Tucek, S.-i. Ohkoshi and R. Zboril, Appl. Phys. Lett., 2011, 99, 253108 CrossRef.
- J. Jin, S. Ohkoshi and K. Hashimoto, Adv. Mater., 2004, 16, 48–51 CrossRef CAS.
- A. Namai, S. Sakurai, M. Nakajima, T. Suemoto, K. Matsumoto, M. Goto, S. Sasaki and S.-i. Ohkoshi, J. Am. Chem. Soc., 2009, 131, 1170–1173 CrossRef CAS PubMed.
- M. Gich, C. Frontera, A. Roig, J. Fontcuberta, E. Molins, N. Bellido, C. Simon and C. Fleta, Nanotechnology, 2006, 17, 687–691 CrossRef CAS.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
- O. K. Arriortua, M. Insausti, L. Lezama, I. Gil de Muro, E. Garaio, J. M. de la Fuente, R. M. Fratila, M. P. Morales, R. Costa, M. Eceiza, M. Sagartzazu-Aizpurua and J. M. Aizpurua, Colloids Surf., B, 2018, 165, 315–324 CrossRef CAS PubMed.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125 RSC.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- Y. Liu, W. Wang, X. Xu, J.-P. Marcel Veder and Z. Shao, J. Mater. Chem. A, 2019, 7, 7280–7300 RSC.
- S. Jung, S. Bae and W. Lee, Environ. Sci. Technol., 2014, 48, 9651–9658 CrossRef CAS PubMed.
- R. Lang, X. Du, Y. Huang, X. Jiang, Q. Zhang, Y. Guo, K. Liu, B. Qiao, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 11986–12043 CrossRef CAS PubMed.
- A. Aitbekova, E. D. Goodman, L. Wu, A. Boubnov, A. S. Hoffman, A. Genc, H. Cheng, L. Casalena, S. R. Bare and M. Cargnello, Angew. Chem., Int. Ed., 2019, 58, 17451–17457 CrossRef CAS PubMed.
- D. Toulemon, B. P. Pichon, C. Leuvrey, S. Zafeiratos, V. Papaefthimiou, X. Cattoën and S. Bégin-Colin, Chem. Mater., 2013, 25, 2849–2854 CrossRef CAS.
- Q. Wang, Z. Shang, H. Wang and A. Wei, Nanoscale, 2023, 15, 5074–5082 RSC.
- F. Zareh, M. Gholinejad, H. Sheibani and J. M. Sansano, Environ. Sci. Pollut. Res., 2023, 30, 69362–69378 CrossRef CAS PubMed.
- A. Navrotsky, L. Mazeina and J. Majzlan, Science, 2008, 319, 1635–1638 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- M. Afrouz, F. A. Nouraldinvand, S. G. Elias, M. T. Alebrahim, T. M. Tseng and H. Zahedian, Sci. Rep., 2023, 13, 775 CrossRef CAS PubMed.
- Z. Sun, J. Ma, Y. Liu, H. Wang, W. Cao, N. Zhu and Z. Lou, Sep. Purif. Technol., 2021, 277, 119506 CrossRef CAS.
- M. Tang, L. Wang, H. Li, X. Huang, C. Du and S. Lu, J. Environ. Sci., 2024, 137, 478–487 CrossRef PubMed.
- S. Dong, X. Yan, W. Li, Y. Liu, X. Han, X. Liu, J. Feng, C. Yu, C. Zhang and J. Sun, J. Ind. Eng. Chem., 2022, 108, 254–262 CrossRef CAS.
- J. Li, J. You, Z. Wang, Y. Zhao, J. Xu, X. Li and H. Zhang, J. Environ. Chem. Eng., 2022, 10, 108329 CrossRef CAS.
- A. A. Marciniak, E. C. S. Santos, R. J. Caraballo-Vivas, O. C. Alves, M. E. H. Maia da Costa, F. Garcia and C. J. A. Mota, Energy Fuels, 2024, 38, 628–636 CrossRef CAS.
- L. Chen, M. Arshad, Y. Chuang, T.-B. Nguyen, C.-H. Wu, C.-W. Chen and C.-D. Dong, J. Alloys Compd., 2023, 947, 169577 CrossRef CAS.
- R. K. Sahoo, A. K. Manna, A. Das, A. Mitra, M. Mohapatra, S. Nath Sarangi, P. Garg, U. Deshpande and S. Varma, Appl. Surf. Sci., 2022, 577, 151954 CrossRef CAS.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- M. B. Gawande, Y. Monga, R. Zboril and R. K. Sharma, Coord. Chem. Rev., 2015, 288, 118–143 CrossRef CAS.
- M. B. Gawande, R. Luque and R. Zboril, ChemCatChem, 2014, 6, 3312–3313 CrossRef CAS.
- R. Hudson, Y. Feng, R. S. Varma and A. Moores, Green Chem., 2014, 16, 4493–4505 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Commun., 2013, 49, 752–770 RSC.
- J. Rahimi, M. Naderi, M. T. Ijdani, M. Heidari, M. Azizi and A. Maleki, Mater. Today Chem., 2023, 28, 101375 CrossRef CAS.
- X. Xiao, S. Lee, H. Ma, J. Yang, W.-S. Han and T. Yu, Dalton Trans., 2022, 51, 11485–11490 RSC.
- A. Marandi and N. Koukabi, Colloids Surf., A, 2021, 621, 126597 CrossRef CAS.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- J. Qi, W. Zhang, R. Xiang, K. Liu, H.-Y. Wang, M. Chen, Y. Han and R. Cao, Adv. Sci., 2015, 2, 1500199 CrossRef PubMed.
- J. Gautam, K. Kannan, M. M. Meshesha, B. Dahal, S. Subedi, L. Ni, Y. Wei and B. L. Yang, J. Colloid Interface Sci., 2022, 618, 419–430 CrossRef CAS PubMed.
- N.-U.-A. Babar, Y. F. Joya, H. Khalil, F. Hussain and K. S. Joya, Int. J. Hydrogen Energy, 2021, 46, 7885–7902 CrossRef CAS.
- S. Yuan, Y. Xue, R. Ma, Q. Ma, Y. Chen and J. Fan, Sci. Total Environ., 2023, 866, 161444 CrossRef CAS PubMed.
- S. Rahim Pouran, A. A. Abdul Raman and W. M. A. Wan Daud, J. Cleaner Prod., 2014, 64, 24–35 CrossRef CAS.
- Y. Cheng, S. Zhang, Z. Wang, B. Wang, J. You, R. Guo and H. Zhang, Sep. Purif. Technol., 2023, 318, 123971 CrossRef CAS.
- S. Xin, S. Huo, Y. Xin, M. Gao, Y. Wang, W. Liu, C. Zhang and X. Ma, Appl. Catal., 2022, 312, 121442 CrossRef CAS.
- C. Lai, F. Huang, G. Zeng, D. Huang, L. Qin, M. Cheng, C. Zhang, B. Li, H. Yi, S. Liu, L. Li and L. Chen, Chemosphere, 2019, 224, 910–921 CrossRef CAS PubMed.
- M. C. Pereira, L. C. A. Oliveira and E. Murad, Clay Miner., 2012, 47, 285–302 CrossRef CAS.
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC.
- K. Rusevova, F.-D. Kopinke and A. Georgi, J. Hazard. Mater., 2012, 241–242, 433–440 CrossRef CAS PubMed.
- S. Lian, H. Li, X. He, Z. Kang, Y. Liu and S. Tong Lee, J. Solid State Chem., 2012, 185, 117–123 CrossRef CAS.
- D. Saberi, M. Sheykhan, K. Niknam and A. Heydari, Catal. Sci. Technol., 2013, 3, 2025–2031 RSC.
- M. B. Gawande, V. D. B. Bonifácio, R. S. Varma, I. D. Nogueira, N. Bundaleski, C. A. A. Ghumman, O. M. N. D. Teodoro and P. S. Branco, Green Chem., 2013, 15, 1226–1231 RSC.
- M. B. Gawande, A. K. Rathi, I. D. Nogueira, R. S. Varma and P. S. Branco, Green Chem., 2013, 15, 1895–1899 RSC.
- M. B. Gawande, A. K. Rathi, J. Tucek, K. Safarova, N. Bundaleski, O. M. N. D. Teodoro, L. Kvitek, R. S. Varma and R. Zboril, Green Chem., 2014, 16, 4137–4143 RSC.
- Y. Gao, D. Ma, G. Hu, P. Zhai, X. Bao, B. Zhu, B. Zhang and D. S. Su, Angew. Chem., Int. Ed., 2011, 50, 10236–10240 CrossRef CAS PubMed.
- K. J. Datta, M. B. Gawande, K. K. R. Datta, V. Ranc, J. Pechousek, M. Krizek, J. Tucek, R. Kale, P. Pospisil, R. S. Varma, T. Asefa, G. Zoppellaro and R. Zboril, J. Mater. Chem. A, 2016, 4, 596–604 RSC.
- X. Qian, X.-H. Peng, D. O. Ansari, Q. Yin-Goen, G. Z. Chen, D. M. Shin, L. Yang, A. N. Young, M. D. Wang and S. Nie, Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- H. Yilmaz, D. Yilmaz, I. C. Taskin and M. Culha, Adv. Drug Delivery Rev., 2022, 184, 114184 CrossRef CAS PubMed.
- C.-Y. Zhang, B.-C. Zhao, R. Hao, Z. Wang, Y.-W. Hao, B. Zhao and Y.-Q. Liu, J. Hazard. Mater., 2020, 385, 121510 CrossRef CAS PubMed.
- S. Liu, G. Chen, P. N. Prasad and M. T. Swihart, Chem. Mater., 2011, 23, 4098–4101 CrossRef CAS.
- J.-H. Choi, J.-H. Lee, J. Son and J.-W. Choi, Sensors, 2020, 20, 1003 CrossRef CAS PubMed.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- J. Zhu and R. B. Lennox, ACS Appl. Nano Mater., 2021, 4, 3790–3798 CrossRef CAS.
- K. J. Datta, K. K. R. Datta, M. B. Gawande, V. Ranc, K. Čépe, V. Malgras, Y. Yamauchi, R. S. Varma and R. Zboril, Chem. – Eur. J., 2016, 22, 1577–1581 CrossRef CAS PubMed.
- M. Sankar, Q. He, R. V. Engel, M. A. Sainna, A. J. Logsdail, A. Roldan, D. J. Willock, N. Agarwal, C. J. Kiely and G. J. Hutchings, Chem. Rev., 2020, 120, 3890–3938 CrossRef CAS PubMed.
- K. Sakamoto, S. Masuda, S. Takano and T. Tsukuda, ACS Catal., 2023, 13, 3263–3271 CrossRef CAS.
- T.-H. Yang, J. Ahn, S. Shi, P. Wang, R. Gao and D. Qin, Chem. Rev., 2021, 121, 796–833 CrossRef CAS PubMed.
- A. K. Rathi, M. B. Gawande, J. Pechousek, J. Tucek, C. Aparicio, M. Petr, O. Tomanec, R. Krikavova, Z. Travnicek, R. S. Varma and R. Zboril, Green Chem., 2016, 18, 2363–2373 RSC.
- S. Lapointe, A. Sarbajna and V. H. Gessner, Acc. Chem. Res., 2022, 55, 770–782 CrossRef CAS PubMed.
- K. Hasan, I. A. Shehadi, K. Ahmed Bagudu, N. Osama Mohamed Elmabrouk, A. Elgamouz, S. P. Patole and R. A. Al-Qawasmeh, Appl. Surf. Sci., 2022, 571, 151369 CrossRef CAS.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181–5203 RSC.
- G. S. Lee, D. Kim and S. H. Hong, Nat. Commun., 2021, 12, 991 CrossRef CAS PubMed.
- K. Hong, M. Sajjadi, J. M. Suh, K. Zhang, M. Nasrollahzadeh, H. W. Jang, R. S. Varma and M. Shokouhimehr, ACS Appl. Nano Mater., 2020, 3, 2070–2103 CrossRef CAS.
- K. Sakoda, J. Mihara and J. Ichikawa, Chem. Commun., 2005, 4684–4686 RSC.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- W. Yao, G. Zhao, Y. Wu, L. Zhou, U. Mukherjee, P. Liu and M.-Y. Ngai, J. Am. Chem. Soc., 2022, 144, 3353–3359 CrossRef CAS PubMed.
- M. J. Buskes and M.-J. Blanco, Molecules, 2020, 25, 3493 CrossRef CAS PubMed.
- S. Sá, M. B. Gawande, A. Velhinho, J. P. Veiga, N. Bundaleski, J. Trigueiro, A. Tolstogouzov, O. M. N. D. Teodoro, R. Zboril, R. S. Varma and P. S. Branco, Green Chem., 2014, 16, 3494–3500 RSC.
- A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed.
- J. Colavida, J. A. Lleberia, A. Salom-Català, A. Gual, A. Collado, E. Zangrando, J. M. Ricart, C. Godard, C. Claver, J. J. Carbó and S. Castillon, ACS Catal., 2020, 10, 11458–11465 CrossRef CAS.
- D. Wang, C. A. Salazar and S. S. Stahl, Organometallics, 2021, 40, 2198–2203 CrossRef CAS PubMed.
- A. K. Rathi, M. B. Gawande, V. Ranc, J. Pechousek, M. Petr, K. Cepe, R. S. Varma and R. Zboril, Catal. Sci. Technol., 2016, 6, 152–160 RSC.
- L. Sun, Q. Li, M. Zheng, S. Lin, C. Guo, L. Luo, S. Guo, Y. Li, C. Wang and B. Jiang, J. Colloid Interface Sci., 2022, 608, 2463–2471 CrossRef CAS PubMed.
- Á. Mastalir and Á. Molnár, ChemCatChem, 2023, e202300643 CrossRef.
- N. Y. Baran, T. Baran and M. Nasrollahzadeh, Sci. Rep., 2023, 13, 12008 CrossRef CAS PubMed.
- X. Pei, Y. Li, L. Lu, H. Jiao, W. Gong and L. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 44418–44426 CrossRef CAS PubMed.
- J. Boström, D. G. Brown, R. J. Young and G. M. Keserü, Nat. Rev. Drug Discovery, 2018, 17, 709–727 CrossRef PubMed.
- A. Taheri Kal Koshvandi, M. M. Heravi and T. Momeni, Appl. Organomet. Chem., 2018, 32, e4210 CrossRef.
- I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS.
- V. S. Shende, V. B. Saptal and B. M. Bhanage, Chem. Rec., 2019, 19, 2022–2043 CrossRef CAS PubMed.
- G. Wang, P. Hao, Y. Chang, Q. Zhang, W. Liu, B. Duan, H. Zhan and S. Bi, Nanoscale, 2022, 14, 2256–2265 RSC.
- X. Chen, D. Qian, G. Xu, H. Xu, J. Dai and Y. Du, Colloids Surf., A, 2019, 573, 67–72 CrossRef CAS.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zbořil, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS.
- B. Singh, V. Sharma, R. P. Gaikwad, P. Fornasiero, R. Zbořil and M. B. Gawande, Small, 2021, 17, 2006473 CrossRef CAS PubMed.
- H. B. Kale, A. D. Kute, R. P. Gaikwad, P. Fornasiero, R. Zbořil and M. B. Gawande, Coord. Chem. Rev., 2024, 502, 215602 CrossRef CAS.
- H. Zhang, X. Zhang, S. Shi, Q. He, X. He, T. Gan and H. Ji, ACS Appl. Mater. Interfaces, 2022, 14, 53755–53760 CrossRef CAS PubMed.
- X. Wang, J. Li, Q. Xue, X. Han, C. Xing, Z. Liang, P. Guardia, Y. Zuo, R. Du, L. Balcells, J. Arbiol, J. Llorca, X. Qi and A. Cabot, ACS Nano, 2023, 17, 825–836 CrossRef CAS PubMed.
- M. J. Baruah, A. Dutta, S. Biswas, G. Gogoi, N. Hoque, P. K. Bhattacharyya and K. K. Bania, ACS Appl. Nano Mater., 2022, 5, 1446–1459 CrossRef CAS.
- K. Biswas, S. Chattopadhyay, Y. Jing, R. Che, G. De, B. Basu and D. Zhao, Ind. Eng. Chem. Res., 2019, 58, 2159–2169 CrossRef CAS.
- T. J. Bora, N. Hazarika, N. K. Gour, S. Lee, Y.-B. Park, S. Biswas, A. Devi and K. K. Bania, ACS Appl. Nano Mater., 2023, 6, 17972–17985 CrossRef CAS.
- M. Zhu, Y. Wang, C. Wang, W. Li and G. Diao, Catal. Sci. Technol., 2013, 3, 952–961 RSC.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073–2094 CrossRef CAS.
- B. Hu, L. Warczinski, X. Li, M. Lu, J. Bitzer, M. Heidelmann, T. Eckhard, Q. Fu, J. Schulwitz, M. Merko, M. Li, W. Kleist, C. Hättig, M. Muhler and B. Peng, Angew. Chem., Int. Ed., 2021, 60, 6807–6815 CrossRef CAS PubMed.
- D. Hu, H. Xu, Z. Yi, Z. Chen, C. Ye, Z. Wu, H. F. Garces and K. Yan, ACS Sustainable Chem. Eng., 2019, 7, 15339–15345 CrossRef CAS.
- C. Wang, L. Salmon, R. Ciganda, L. Yate, S. Moya, J. Ruiz and D. Astruc, Chem. Commun., 2017, 53, 644–646 RSC.
- S. Handa, Y. Wang, F. Gallou and B. H. Lipshutz, Science, 2015, 349, 1087–1091 CrossRef CAS PubMed.
- P. Arora, P. Kumar, V. Tomar, M. Sillanpää, R. Kumar Joshi and M. Nemiwal, Inorg. Chem. Commun., 2022, 145, 109982 CrossRef CAS.
- M. Shahabi Nejad, N. Seyedi, H. Sheibani and S. Behzadi, Mol. Divers., 2019, 23, 527–539 CrossRef CAS PubMed.
- G. Li, L. Yang, J.-J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS PubMed.
- S. Liu, T. Li, F. Shi, H. Ma, B. Wang, X. Dai and X. Cui, Nat. Commun., 2023, 14, 4973 CrossRef CAS PubMed.
- C. Gonzalez-Arellano, K. Yoshida, R. Luque and P. L. Gai, Green Chem., 2010, 12, 1281–1287 RSC.
- E. Podyacheva, O. I. Afanasyev, D. V. Vasilyev and D. Chusov, ACS Catal., 2022, 12, 7142–7198 CrossRef CAS.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- X. Q. Ng, Y. Zhao and V. Isoni, ACS Sustainable Chem. Eng., 2023, 11, 12389–12396 CrossRef CAS.
- M. Nallagangula, C. Sujatha, V. T. Bhat and K. Namitharan, Chem. Commun., 2019, 55, 8490–8493 RSC.
- M. Sarno, C. Cirillo and M. Iuliano, ChemistryOpen, 2019, 8, 520–531 CrossRef PubMed.
- W. Pickhardt, E. Siegfried, S. Fabig, M. F. Rappen, M. Etter, M. Wohlgemuth, S. Grätz and L. Borchardt, Angew. Chem., Int. Ed., 2023, 62, e202301490 CrossRef CAS PubMed.
- P. Singh and A. C. Shaikh, Chem. Commun., 2023, 59, 11615–11630 RSC.
- X. Zeng, C. Wang, W. Yan, J. Rong, Y. Song, Z. Xiao, A. Cai, S. H. Liang and W. Liu, ACS Catal., 2023, 13, 2761–2770 CrossRef CAS PubMed.
- M. Tarahomi, H. Alinezhad and B. Maleki, Appl. Organomet. Chem., 2019, 33, e5203 CrossRef CAS.
- M. Gholinejad, J. Ahmadi, C. Nájera, M. Seyedhamzeh, F. Zareh and M. Kompany-Zareh, ChemCatChem, 2017, 9, 1442–1449 CrossRef CAS.
- S. Handa, B. Jin, P. P. Bora, Y. Wang, X. Zhang, F. Gallou, J. Reilly and B. H. Lipshutz, ACS Catal., 2019, 9, 2423–2431 CrossRef CAS.
- M. B. Gawande, A. K. Rathi, P. S. Branco, I. D. Nogueira, A. Velhinho, J. J. Shrikhande, U. U. Indulkar, R. V. Jayaram, C. A. A. Ghumman, N. Bundaleski and O. M. N. D. Teodoro, Chem. – Eur. J., 2012, 18, 12628–12632 CrossRef CAS PubMed.
- M. B. Gawande, A. K. Rathi, P. S. Branco, T. M. Potewar, A. Velhinho, I. D. Nogueira, A. Tolstogouzov, C. A. A. Ghumman and O. M. N. D. Teodoro, RSC Adv., 2013, 3, 3611–3617 RSC.
- Y. Li, N. Li, W. Jiang, G. Ma and M. M. Zangeneh, Int. J. Biol. Macromol., 2020, 163, 2162–2171 CrossRef CAS PubMed.
- C. Milone, C. Crisafulli, R. Ingoglia, L. Schipilliti and S. Galvagno, Catal. Today, 2007, 122, 341–351 CrossRef CAS.
- J. Lenz, B. C. Campo, M. Alvarez and M. A. Volpe, J. Catal., 2009, 267, 50–56 CrossRef CAS.
- Q. Hu, S. Wang, Z. Gao, Y. Li, Q. Zhang, Q. Xiang and Y. Qin, Appl. Catal., B, 2017, 218, 591–599 CrossRef CAS.
- Y. Shu, T. Chen, H. C. Chan, L. Xie and Q. Gao, Chem. – Asian J., 2018, 13, 3737–3744 CrossRef CAS PubMed.
- Y. Wang, R. Qin, Y. Wang, J. Ren, W. Zhou, L. Li, J. Ming, W. Zhang, G. Fu and N. Zheng, Angew. Chem., Int. Ed., 2020, 59, 12736–12740 CrossRef CAS PubMed.
- Y. Zhong, P. Liao, J. Kang, Q. Liu, S. Wang, S. Li, X. Liu and G. Li, J. Am. Chem. Soc., 2023, 145, 4659–4666 CrossRef CAS PubMed.
- J. Feng, S. Handa, F. Gallou and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 8979–8983 CrossRef CAS PubMed.
- W. Liu, H. Feng, Y. Yang, Y. Niu, L. Wang, P. Yin, S. Hong, B. Zhang, X. Zhang and M. Wei, Nat. Commun., 2022, 13, 3188 CrossRef CAS PubMed.
- J. Ma, X. Mao, C. Hu, X. Wang, W. Gong, D. Liu, R. Long, A. Du, H. Zhao and Y. Xiong, J. Am. Chem. Soc., 2024, 146, 970–978 CrossRef CAS PubMed.
- H. Niu, J. Lu, J. Song, L. Pan, X. Zhang, L. Wang and J.-J. Zou, Ind. Eng. Chem. Res., 2016, 55, 8527–8533 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- K. J. Datta, A. K. Rathi, M. B. Gawande, V. Ranc, G. Zoppellaro, R. S. Varma and R. Zboril, ChemCatChem, 2016, 8, 2351–2355 CrossRef CAS.
- M. N. Shaikh, M. A. Aziz, A. N. Kalanthoden, A. Helal, A. S. Hakeem and M. Bououdina, Catal. Sci. Technol., 2018, 8, 4709–4717 RSC.
- M. N. Shaikh, M. M. Abdelnaby, A. S. Hakeem, G. A. Nasser and Z. H. Yamani, ACS Appl. Nano Mater., 2021, 4, 3508–3518 CrossRef CAS.
- V. G. Chandrashekhar, T. Senthamarai, R. G. Kadam, O. Malina, J. Kašlík, R. Zbořil, M. B. Gawande, R. V. Jagadeesh and M. Beller, Nat. Catal., 2022, 5, 20–29 CrossRef CAS.
- I. Elsayed, M. A. Jackson and E. B. Hassan, ACS Sustainable Chem. Eng., 2020, 8, 1774–1785 CrossRef CAS.
- X. Wang and K. Gui, J. Environ. Sci., 2013, 25, 2469–2475 CrossRef CAS PubMed.
- X. Li, J. Li, Y. Peng, T. Zhang, S. Liu and J. Hao, Catal. Sci. Technol., 2015, 5, 4556–4564 RSC.
- L. Zhang, J. Li, X. Chu and G.-c. Zhao, J. Cluster Sci., 2016, 27, 227–239 CrossRef CAS.
- A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851–863 CrossRef CAS PubMed.
- K. Schröder, B. Join, A. J. Amali, K. Junge, X. Ribas, M. Costas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 1425–1429 CrossRef PubMed.
- Y. Hou, M. Niu and W. Wu, Ind. Eng. Chem. Res., 2020, 59, 16899–16910 CrossRef CAS.
- Z. He, Y. Hou, H. Li, J. Wei, S. Ren and W. Wu, Chem. Eng. Sci., 2023, 280, 119055 CrossRef CAS.
- Z. He, Y. Hou, H. Li, Y. Wang, S. Ren and W. Wu, Renewable Energy, 2023, 211, 403–411 CrossRef CAS.
- R. Weingarten, A. Rodriguez-Beuerman, F. Cao, J. S. Luterbacher, D. M. Alonso, J. A. Dumesic and G. W. Huber, ChemCatChem, 2014, 6, 2229–2234 CrossRef CAS.
- A. Villa, G. M. Veith, D. Ferri, A. Weidenkaff, K. A. Perry, S. Campisi and L. Prati, Catal. Sci. Technol., 2013, 3, 394–399 RSC.
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed.
- Y. Liu, H.-Y. Ma, D. Lei, L.-L. Lou, S. Liu, W. Zhou, G.-C. Wang and K. Yu, ACS Catal., 2019, 9, 8306–8315 CrossRef CAS.
- Z. Wang and H. Wang, ACS Appl. Mater. Interfaces, 2023, 15, 36676–36687 CrossRef CAS PubMed.
- G. Qi, T. E. Davies, A. Nasrallah, M. A. Sainna, A. G. R. Howe, R. J. Lewis, M. Quesne, C. R. A. Catlow, D. J. Willock, Q. He, D. Bethell, M. J. Howard, B. A. Murrer, B. Harrison, C. J. Kiely, X. Zhao, F. Deng, J. Xu and G. J. Hutchings, Nat. Catal., 2022, 5, 45–54 CrossRef CAS.
- F. Gu, X. Qin, M. Li, Y. Xu, S. Hong, M. Ouyang, G. Giannakakis, S. Cao, M. Peng, J. Xie, M. Wang, D. Han, D. Xiao, X. Wang, Z. Wang and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202201540 CrossRef CAS PubMed.
- L. Geng, M. Zhang, W. Zhang, M. Jia, W. Yan and G. Liu, Catal. Sci. Technol., 2015, 5, 3097–3102 RSC.
- S. Dey, S. Sun and N. S. Mehta, Carbon Capture Sci. Technol., 2021, 1, 100013 CrossRef CAS.
- B. Zheng, T. Gan, S. Shi, J. Wang, W. Zhang, X. Zhou, Y. Zou, W. Yan and G. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 27029–27040 CrossRef CAS PubMed.
- S. Neumann, H. H. Doebler, S. Keil, A. J. Erdt, C. Gutsche, H. Borchert, J. Kolny-Olesiak, J. Parisi, M. Bäumer and S. Kunz, ACS Catal., 2020, 10, 4136–4150 CrossRef CAS.
- G.-H. Wang, W.-C. Li, K.-M. Jia, B. Spliethoff, F. Schüth and A.-H. Lu, Appl. Catal., A, 2009, 364, 42–47 CrossRef CAS.
- H. Liang, X. Jiang, Z. Qi, W. Chen, Z. Wu, B. Xu, Z. Wang, J. Mi and Q. Li, Nanoscale, 2014, 6, 7199–7203 RSC.
- X. C. Jiang and A. B. Yu, J. Mater. Process. Technol., 2009, 209, 4558–4562 CrossRef CAS.
- S. Najafishirtari, K. Friedel Ortega, M. Douthwaite, S. Pattisson, G. J. Hutchings, C. J. Bondue, K. Tschulik, D. Waffel, B. Peng, M. Deitermann, G. W. Busser, M. Muhler and M. Behrens, Chem. – Eur. J., 2021, 27, 16809–16833 CrossRef CAS PubMed.
- H. Jiang, X. Chen, D. Cui, K. Lu, X. Kong and X. Zhang, Materials, 2023, 16, 3139 CrossRef CAS PubMed.
- D. Xu, J. Li, B. Li, H. Zhao, H. Zhu, J. Kou, F. Zhang, Z. Dong and J. Ma, Chem. Eng. J., 2022, 434, 134545 CrossRef CAS.
- C. Zhou, W. Shi, X. Wan, Y. Meng, Y. Yao, Z. Guo, Y. Dai, C. Wang and Y. Yang, Catal. Today, 2019, 330, 92–100 CrossRef CAS.
- K. Yang, Y. Liu, J. Deng, X. Zhao, J. Yang, Z. Han, Z. Hou and H. Dai, Appl. Catal., B, 2019, 244, 650–659 CrossRef CAS.
- H. Hong, L. Hu, M. Li, J. Zheng, X. Sun, X. Lu, X. Cao, J. Lu and H. Gu, Chem. – Eur. J., 2011, 17, 8726–8730 CrossRef CAS PubMed.
- J. Sheng, W.-C. Li, W.-D. Lu, B. Yan, B. Qiu, X.-Q. Gao, R.-P. Zhang, S.-Z. Zhou and A.-H. Lu, Appl. Catal., B, 2022, 305, 121070 CrossRef CAS.
- T. Ye, J. H. Carter, B. Chen, X. Li, Y. Ye, S. H. Taylor and G. J. Hutchings, Catal. Commun., 2022, 162, 106383 CrossRef CAS.
- S. Kumar, A. Lyalin, Z. Huang and T. Taketsugu, ChemistrySelect, 2022, 7, e202103795 CrossRef CAS.
- J. H. Carter, T. Bere, J. R. Pitchers, D. G. Hewes, B. D. Vandegehuchte, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Green Chem., 2021, 23, 9747–9799 RSC.
- B. Nigrovski, U. Zavyalova, P. Scholz, K. Pollok, M. Müller and B. Ondruschka, Carbon, 2008, 46, 1678–1686 CrossRef CAS.
- M. de S. Ramos, M. de S. Santos, L. P. Gomes, A. Albornoz and M. do C. Rangel, Appl. Catal., A, 2008, 341, 12–17 CrossRef CAS.
- D. Wang, Z. Liu, F. Liu, X. Ai, X. Zhang, Y. Cao, J. Yu, T. Wu, Y. Bai, T. Li and X. Tang, Appl. Catal., A, 1998, 174, 25–32 CrossRef CAS.
- Z. Tang, W. Zhang, Y. Li, Z. Huang, H. Guo, F. Wu and J. Li, Appl. Surf. Sci., 2016, 364, 75–80 CrossRef CAS.
- W. Cheng, J. Xu, Y. Wang, F. Wu, X. Xu and J. Li, J. Colloid Interface Sci., 2015, 445, 93–101 CrossRef CAS PubMed.
- W. Cheng, J. Xu, W. Ding, Y. Wang, W. Zheng, F. Wu and J. Li, Mater. Chem. Phys., 2015, 153, 187–194 CrossRef CAS.
- P. Mikenin, S. Zazhigalov, A. Elyshev, S. Lopatin, T. Larina, S. Cherepanova, D. Pisarev, D. Baranov and A. Zagoruiko, Catal. Commun., 2016, 87, 36–40 CrossRef CAS.
- P. E. Mikenin, P. G. Tsyrul'nikov, Y. S. Kotolevich and A. N. Zagoruiko, Catal. Ind., 2015, 7, 155–160 CrossRef.
- I. V. Desyatykh, A. A. Vedyagin, Y. S. Kotolevich and P. G. Tsyrul'nikov, Combust., Explos. Shock Waves, 2011, 47, 677–682 CrossRef.
- A. Yepez, P. Prinsen, A. Pineda, A. M. Balu, A. Garcia, F. L. Y. Lam and R. Luque, React. Chem. Eng., 2018, 3, 757–768 RSC.
- M. Amini, Y. Mousazade, Z. Zand, M. Bagherzadeh and M. M. Najafpour, Sci. Rep., 2021, 11, 6642 CrossRef CAS PubMed.
- M. Lang and H. Li, ChemSusChem, 2022, 15, e202101531 CrossRef CAS PubMed.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- S. P. Mekala, M. Prabu, S. D. Gawali, K. Gopakumar, P. Gogoi, A. R. Bhatkar, G. Mohapatra, E. Unnikrishanan and T. Raja, Catal. Commun., 2022, 168, 106466 CrossRef CAS.
- A. Rahman, M. Mupa and C. Mahamadi, Catal. Lett., 2016, 146, 788–799 CrossRef CAS.
- A. K. Patra, A. Dutta and A. Bhaumik, Chem. – Eur. J., 2013, 19, 12388–12395 CrossRef CAS PubMed.
- N. Ameur, R. Bachir, S. Bedrane and A. Choukchou-Braham, J. Chin. Chem. Soc., 2017, 64, 1096–1103 CrossRef CAS.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC.
- C. A. Hone, D. M. Roberge and C. O. Kappe, ChemSusChem, 2017, 10, 32–41 CrossRef CAS PubMed.
- A. Vinu, D. P. Sawant, K. Ariga, K. Z. Hossain, S. B. Halligudi, M. Hartmann and M. Nomura, Chem. Mater., 2005, 17, 5339–5345 CrossRef CAS.
- A. Pineda, N. Lázaro, A. M. Balu, A. Garcia, A. A. Romero and R. Luque, Mol. Catal., 2020, 493, 111092 CrossRef CAS.
- B. Török, C. Schäfer and A. Kokel, Heterogeneous Catalysis in Sustainable Synthesis, 2022, 317–378 Search PubMed.
- C. Anand, P. Srinivasu, S. Alam, V. V. Balasubramanian, D. P. Sawant, M. Palanichamy, V. Murugesan and A. Vinu, Microporous Mesoporous Mater., 2008, 111, 72–79 CrossRef CAS.
- V. R. Choudhary, S. K. Jana and V. S. Narkhede, Appl. Catal., A, 2002, 235, 207–215 CrossRef CAS.
- Z. Lei, S. Bai, L. Dang, H. Xia, Q. Xu, Y. Cao, L. An, M. Zhao, A.-Y. Lo and S.-B. Liu, Microporous Mesoporous Mater., 2009, 123, 306–313 CrossRef CAS.
- H. Saeidian, F. M. Moghaddam, A. Pourjavadi, S. Barzegar, R. Soleyman and A. Sohrabi, J. Braz. Chem. Soc., 2009, 20, 466–471 CrossRef CAS.
- A. Pourjavadi, R. Soleyman and G. R. Barajee, Starch/Stärke, 2008, 60, 467–475 CrossRef CAS.
- G. R. Bardajee, A. Pourjavadi, R. Soleyman and N. Sheikh, Nucl. Instrum. Methods Phys. Res., Sect. B, 2008, 266, 3932–3938 CrossRef CAS.
- N. D. Cuong, N. D. Hoa, T. T. Hoa, D. Q. Khieu, D. T. Quang, V. V. Quang and N. V. Hieu, J. Alloys Compd., 2014, 582, 83–87 CrossRef.
- H. Sharghi, M. Jokar, M. M. Doroodmand and R. Khalifeh, Adv. Synth. Catal., 2010, 352, 3031–3044 CrossRef CAS.
- Z. Fang, W. He, T. Tu, N. Lv, C. Qiu, X. Li, N. Zhu, L. Wan and K. Guo, J. Chem. Eng., 2018, 331, 443–449 CrossRef CAS.
- Q. Zhang, J. Kang and Y. Wang, ChemCatChem, 2010, 2, 1030–1058 CrossRef CAS.
- Y. Tang, Y. Liu, J. Chen, C. He, X. Lu, R. Zhang and X. Liu, React. Chem. Eng., 2023, 8, 1229–1249 RSC.
- K. Wei, S. Yue, X. Hou, Y. Jiang, C. Jiang, Y. Yao, J. Yin and J. Ma, Int. J. Energy Res., 2022, 46, 10864–10874 CrossRef CAS.
- E. de Smit and B. M. Weckhuysen, Chem. Soc. Rev., 2008, 37, 2758–2781 RSC.
- Y. Gao, L. Shao, S. Yang, J. Hu, S. Zhao, J. Dang, W. Wang, X. Yan and P. Yang, Catal. Commun., 2023, 181, 106720 CrossRef CAS.
- X. Liu, T. Lin, P. Liu and L. Zhong, Appl. Catal., B, 2023, 331, 122697 CrossRef CAS.
- Y. Xu, X. Li, J. Gao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li and M. Ding, Science, 2021, 371, 610–613 CrossRef CAS PubMed.
- J. Weiß, Q. Yang, U. Bentrup, E. V. Kondratenko, A. Brückner and C. Kubis, ChemCatChem, 2022, 14, e202200577 CrossRef.
- H. Zhao, Q. Zhu, Y. Gao, P. Zhai and D. Ma, Appl. Catal., A, 2013, 456, 233–239 CrossRef CAS.
- J. Park, K. An, Y. Hwang, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, N.-M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- Z. H. Chonco, A. Ferreira, L. Lodya, M. Claeys and E. van Steen, J. Catal., 2013, 307, 283–294 CrossRef CAS.
- D. J. Duvenhage, C. Schmidt and H. W. Wright, J. Mater. Sci., 2014, 49, 2810–2823 CrossRef CAS.
- K. Cheng, V. V. Ordomsky, M. Virginie, B. Legras, P. A. Chernavskii, V. O. Kazak, C. Cordier, S. Paul, Y. Wang and A. Y. Khodakov, Appl. Catal., A, 2014, 488, 66–77 CrossRef CAS.
- R. Warringham, N. G. Hamilton, I. P. Silverwood, C. How, P. B. Webb, R. P. Tooze, W. Zhou, C. D. Frost, S. F. Parker and D. Lennon, Appl. Catal., A, 2015, 489, 209–217 CrossRef CAS.
- G. R. Patzke, F. Krumeich and R. Nesper, Angew. Chem., Int. Ed., 2002, 41, 2446–2461 CrossRef CAS PubMed.
- S. K. Mohapatra, S. E. John, S. Banerjee and M. Misra, Chem. Mater., 2009, 21, 3048–3055 CrossRef CAS.
- R. A. Friedel and R. B. Anderson, J. Am. Chem. Soc., 1950, 72, 1212–1215 CrossRef CAS.
- H. Becker, R. Güttel and T. Turek, Catal. Sci. Technol., 2016, 6, 275–287 RSC.
- H. Preising and D. Enke, Colloids Surf., A, 2007, 300, 21–29 CrossRef CAS.
- J. Sun, Y. Chen and J. Chen, Catal. Sci. Technol., 2016, 6, 7505–7511 RSC.
- S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 5743–5750 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, L. Fratalocchi and L. Lietti, Catal. Today, 2016, 277, 161–170 CrossRef CAS.
- H. Ando, Y. Matsumura and Y. Souma, J. Mol. Catal. A: Chem., 2000, 154, 23–29 CrossRef CAS.
- M. K. Gnanamani, W. D. Shafer, D. E. Sparks and B. H. Davis, Catal. Commun., 2011, 12, 936–939 CrossRef CAS.
- Y. Yao, X. Liu, D. Hildebrandt and D. Glasser, Ind. Eng. Chem. Res., 2011, 50, 11002–11012 CrossRef CAS.
- S. Li, A. Li, S. Krishnamoorthy and E. Iglesia, Catal. Lett., 2001, 77, 197–205 CrossRef CAS.
- B. An, K. Cheng, C. Wang, Y. Wang and W. Lin, ACS Catal., 2016, 6, 3610–3618 CrossRef CAS.
- J. Wu, L. Wang, B. Lv and J. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 14319–14327 CrossRef CAS PubMed.
- R. Shakya, B. D. Kurmi and P. Patel, Appl. Organomet. Chem., 2023, 37, e7075 CrossRef CAS.
- R. Pratap and V. J. Ram, Chem. Rev., 2014, 114, 10476–10526 CrossRef CAS PubMed.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonia, Molecules, 2020, 25, 505 CrossRef PubMed.
- S. Agarwal, A. Sethiya, J. Soni, N. Sahiba and P. Teli, Appl. Organomet. Chem., 2022, 36, e6604 CrossRef CAS.
- E. Sheikhhosseini and M. Yahyazadehfar, Front. Chem., 2023, 10, 984502 CrossRef PubMed.
- V. Raj and J. Lee, Front. Chem., 2020, 8, 623 CrossRef CAS PubMed.
- L. Bonsignore, G. Loy, D. Secci and A. Calignano, Eur. J. Med. Chem., 1993, 28, 517–520 CrossRef CAS.
- M. Costa, T. A. Dias, A. Brito and F. Proença, Eur. J. Med. Chem., 2016, 123, 487–507 CrossRef CAS PubMed.
- C. Konkoy, D. Fick, S. Cai, N. Lan and J. Keana, PCT Int. ApplWO0075123, 2000 Search PubMed; Chem. Abstr. 2001 134 29313a Search PubMed.
- S. Luo, C. Ma, M.-Q. Zhu, W.-N. Ju, Y. Yang and X. Wang, Front. Cell. Neurosci., 2020, 14, 21 CrossRef CAS PubMed.
- H. Nagabhushana, S. S. Saundalkar, L. Muralidhar, B. M. Nagabhushana, C. R. Girija, D. Nagaraja, M. A. Pasha and V. P. Jayashankara, Chin. Chem. Lett., 2011, 22, 143–146 CrossRef CAS.
- S. A. Mirfarjood, M. Mamaghani and M. Sheykhan, J. Nanostruct. Chem., 2017, 7, 359–366 CrossRef CAS.
- Z. Hajizadeh, F. Radinekiyan, R. Eivazzadeh-keihan and A. Maleki, Sci. Rep., 2020, 10, 11671 CrossRef CAS PubMed.
- J. Mondal, T. Sen and A. Bhaumik, Dalton Trans., 2012, 41, 6173–6181 RSC.
- D. Girija, H. S. Bhojya Naik, B. Vinay Kumar, C. N. Sudhamani and K. N. Harish, Arabian J. Chem., 2019, 12, 420–428 CrossRef CAS.
- L. Ghandi, M. Kazemi Miraki, M. Karimi, I. Radfar and A. Heydari, Appl. Organomet. Chem., 2019, 33, e4616 CrossRef.
- E. Eidi, M. Z. Kassaee and Z. Nasresfahani, Appl. Organomet. Chem., 2016, 30, 561–565 CrossRef CAS.
- K. S. Soppimath, D. C. W. Tan and Y. Y. Yang, Adv. Mater., 2005, 17, 318–323 CrossRef CAS.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D.-L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- S. Bashir, A. K. Wahab and H. Idriss, Catal. Today, 2015, 240, 242–247 CrossRef CAS.
- Y. Wang, D. Wang, Q. Fu, D. Liu, Y. Ma, K. Racette, Z. He and F. Liu, Mol. Pharm., 2014, 11, 3766–3771 CrossRef CAS PubMed.
- S. Liu, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Appl. Mater. Interfaces, 2012, 4, 6378–6385 CrossRef CAS PubMed.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- Y. Mao, W. Jiang, S. Xuan, Q. Fang, K. C.-F. Leung, B. S. Ong, S. Wang and X. Gong, Dalton Trans., 2015, 44, 9538–9544 RSC.
- S. Dixit and J. G. Hering, Chem. Geol., 2006, 228, 6–15 CrossRef CAS.
- B.-H. Jeon, B. A. Dempsey and W. D. Burgos, Environ. Sci. Technol., 2003, 37, 3309–3315 CrossRef CAS PubMed.
- B.-H. Jeon, B. A. Dempsey, W. D. Burgos and R. A. Royer, Colloids Surf., A, 2001, 191, 41–55 CrossRef CAS.
- R. Sitko, E. Turek, B. Zawisza, E. Malicka, E. Talik, J. Heimann, A. Gagor, B. Feist and R. Wrzalik, Dalton Trans., 2013, 42, 5682–5689 RSC.
- A. J. Frierdich, M. Helgeson, C. Liu, C. Wang, K. M. Rosso and M. M. Scherer, Environ. Sci. Technol., 2015, 49, 8479–8486 CrossRef CAS PubMed.
- T. Yan, J. Xu, L. Wang, Y. Liu, C. Yang and T. Fang, RSC Adv., 2015, 5, 75129–75140 RSC.
- M. Ding, Y. Tang and A. Star, J. Phys. Chem. Lett., 2013, 4, 147–160 CrossRef CAS PubMed.
- V. Papaefthimiou, I. Florea, W. Baaziz, I. Janowska, W. H. Doh, D. Begin, R. Blume, A. Knop-Gericke, O. Ersen, C. Pham-Huu and S. Zafeiratos, J. Phys. Chem. C, 2013, 117, 20313–20319 CrossRef CAS.
- J. B. Joshi, A. B. Pandit, K. L. Kataria, R. P. Kulkarni, A. N. Sawarkar, D. Tandon, Y. Ram and M. M. Kumar, Ind. Eng. Chem. Res., 2008, 47, 8960–8988 CrossRef CAS.
- L. Carbognani, M. F. González, F. Lopez-Linares, C. S. Stull and P. Pereira-Almao, Energy Fuels, 2008, 22, 1739–1746 CrossRef CAS.
- C. Xu, H. E and K. H. Chung, Energy Fuels, 2003, 17, 631–636 CrossRef CAS.
- H. Gao, G. Wang, H. Wang, J. Chen, C. Xu and J. Gao, Energy Fuels, 2012, 26, 1870–1879 CrossRef CAS.
- J. G. Speight, Catal. Today, 2004, 98, 55–60 CrossRef CAS.
- N. Hallale and F. Liu, Adv. Environ. Res., 2001, 6, 81–98 CrossRef CAS.
- M. Hosseinpour, S. Fatemi and S. J. Ahmadi, Fuel, 2015, 159, 538–549 CrossRef CAS.
- U. Legrand, J. L. Meunier and D. Berk, Appl. Catal., A, 2016, 528, 36–43 CrossRef CAS.
- R. Pristavita, J.-L. Meunier and D. Berk, Plasma Chem. Plasma Process., 2011, 31, 393–403 CrossRef CAS.
- A. K. Ilunga, B. B. Mamba and T. T. I. Nkambule, J. Water Process. Eng., 2021, 44, 102402 CrossRef.
- A. K. Patra, A. Dutta and A. Bhaumik, ACS Appl. Mater. Interfaces, 2012, 4, 5022–5028 CrossRef CAS PubMed.
- M. Hua, S. Zhang, B. Pan, W. Zhang, L. Lv and Q. Zhang, J. Hazard. Mater., 2012, 211–212, 317–331 CrossRef CAS PubMed.
- S. Yang, Y. Guo, N. Yan, D. Wu, H. He, Z. Qu, C. Yang, Q. Zhou and J. Jia, ACS Appl. Mater. Interfaces, 2011, 3, 209–217 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- W. Li, D. Liu, J. Wu, C. Kim and J. D. Fortner, Environ. Sci. Technol., 2014, 48, 11892–11900 CrossRef CAS PubMed.
- O. V. Kharissova, H. V. R. Dias and B. I. Kharisov, RSC Adv., 2015, 5, 6695–6719 RSC.
- D. S. Monje, O. S. Ruiz, G. C. Valencia and D. F. Mercado, Environ. Sci. Pollut. Res., 2022, 29, 57127–57146 CrossRef CAS PubMed.
- L. Jia, Y. Yu, Z.-p. Li, S.-n. Qin, J.-r. Guo, Y.-q. Zhang, J.-c. Wang, J.-c. Zhang, B.-g. Fan and Y. Jin, Bioresour. Technol., 2021, 332, 125086 CrossRef CAS PubMed.
- A. Gallo-Cordova, J. Lemus, F. J. Palomares, M. P. Morales and E. Mazarío, Sci. Total Environ., 2020, 711, 134644 CrossRef CAS PubMed.
- K. Q. Jabbar, A. A. Barzinjy and S. M. Hamad, Environ. Nanotechnol., Monit. Manage., 2022, 17, 100661 CAS.
- C. Fang and M. Zhang, J. Mater. Chem., 2009, 19, 6258–6266 RSC.
- A. M. G. C. Dias, A. Hussain, A. S. Marcos and A. C. A. Roque, Biotechnol. Adv., 2011, 29, 142–155 CrossRef CAS PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, ChemSusChem, 2011, 4, 1712–1730 CrossRef CAS PubMed.
- Y. Fan, J. Su, L. Xu, S. Liu, C. Hou, Y. Liu and S. Cao, Environ. Res., 2023, 231, 116307 CrossRef CAS PubMed.
- S. M. Korotta-Gamage and A. Sathasivan, Chemosphere, 2017, 167, 120–138 CrossRef CAS PubMed.
- Y. Masue, R. H. Loeppert and T. A. Kramer, Environ. Sci. Technol., 2007, 41, 837–842 CrossRef CAS PubMed.
- S. Daer, J. Kharraz, A. Giwa and S. W. Hasan, Desalination, 2015, 367, 37–48 CrossRef CAS.
- M. Iram, C. Guo, Y. Guan, A. Ishfaq and H. Liu, J. Hazard. Mater., 2010, 181, 1039–1050 CrossRef CAS PubMed.
- R. D. Ambashta and M. Sillanpää, J. Hazard. Mater., 2010, 180, 38–49 CrossRef CAS PubMed.
- M. E. Mahmoud, M. S. Abdelwahab and E. M. Fathallah, Chem. Eng. J., 2013, 223, 318–327 CrossRef CAS.
- Y. H. Huang, P. K. Peddi, C. Tang, H. Zeng and X. Teng, Sep. Purif. Technol., 2013, 118, 690–698 CrossRef CAS.
- L.-n. Shi, X. Zhang and Z.-l. Chen, Water Res., 2011, 45, 886–892 CrossRef CAS PubMed.
- H. Liu, Q. Wang, C. Wang and X.-z. Li, Chem. Eng. J., 2013, 215–216, 90–95 CrossRef CAS.
- H. Hu, Z. Wang and L. Pan, J. Alloys Compd., 2010, 492, 656–661 CrossRef CAS.
- F. Almomani, R. Bhosale, M. Khraisheh, A. Kumar and T. Almomani, Appl. Surf. Sci., 2020, 506, 144924 CrossRef CAS.
- F. Fu and Q. Wang, J. Environ. Manage., 2011, 92, 407–418 CrossRef CAS PubMed.
- I. Ali, Chem. Rev., 2012, 112, 5073–5091 CrossRef CAS PubMed.
- P. Xu, G. M. Zeng, D. L. Huang, C. L. Feng, S. Hu, M. H. Zhao, C. Lai, Z. Wei, C. Huang, G. X. Xie and Z. F. Liu, Sci. Total Environ., 2012, 424, 1–10 CrossRef CAS PubMed.
- Z. Wei, R. Xing, X. Zhang, S. Liu, H. Yu and P. Li, ACS Appl. Mater. Interfaces, 2013, 5, 598–604 CrossRef CAS PubMed.
- B. Wang, H. Wu, L. Yu, R. Xu, T.-T. Lim and X. W. Lou, Adv. Mater., 2012, 24, 1111–1116 CrossRef CAS PubMed.
- H. Li, W. Li, Y. Zhang, T. Wang, B. Wang, W. Xu, L. Jiang, W. Song, C. Shu and C. Wang, J. Mater. Chem., 2011, 21, 7878–7881 RSC.
- C.-Y. Cao, J. Qu, W.-S. Yan, J.-F. Zhu, Z.-Y. Wu and W.-G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- M. E. Mahmoud, A. A. Yakout, K. H. Hamza and M. M. Osman, J. Ind. Eng. Chem., 2015, 25, 207–215 CrossRef CAS.
- L.-S. Zhong, J.-S. Hu, H.-P. Liang, A.-M. Cao, W.-G. Song and L.-J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- J.-S. Hu, L.-S. Zhong, W.-G. Song and L.-J. Wan, Adv. Mater., 2008, 20, 2977–2982 CrossRef CAS.
- M. B. Gawande, S. N. Shelke, R. Zboril and R. S. Varma, Acc. Chem. Res., 2014, 47, 1338–1348 CrossRef CAS PubMed.
- B. Kong, J. Tang, Z. Wu, J. Wei, H. Wu, Y. Wang, G. Zheng and D. Zhao, Angew. Chem., Int. Ed., 2014, 53, 2888–2892 CrossRef CAS PubMed.
- G. Ali, Y. J. Park, A. Hussain and S. O. Cho, Nanotechnology, 2019, 30, 095601 CrossRef CAS PubMed.
- H.-J. Cui, J.-K. Cai, J.-W. Shi, B. Yuan, C.-L. Ai and M.-L. Fu, RSC Adv., 2014, 4, 10176–10179 RSC.
- N. N. Nassar, J. Hazard. Mater., 2010, 184, 538–546 CrossRef CAS PubMed.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- Y. Feng, J.-L. Gong, G.-M. Zeng, Q.-Y. Niu, H.-Y. Zhang, C.-G. Niu, J.-H. Deng and M. Yan, Chem. Eng. J., 2010, 162, 487–494 CrossRef CAS.
- S. R. Kanel, T. K. Das, R. S. Varma, S. Kurwadkar, S. Chakraborty, T. P. Joshi, A. N. Bezbaruah and M. N. Nadagouda, ACS Environ. Au, 2023, 3, 135–152 CrossRef CAS PubMed.
- T. A. Kurniawan, M. E. T. Sillanpää and M. Sillanpää, Crit. Rev. Environ. Sci. Technol., 2012, 42, 1233–1295 CrossRef CAS.
- J. Tuček, K. C. Kemp, K. S. Kim and R. Zbořil, ACS Nano, 2014, 8, 7571–7612 CrossRef PubMed.
- T. Mishra and D. K. Mahato, J. Environ. Chem. Eng., 2016, 4, 1224–1230 CrossRef CAS.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–967 CrossRef PubMed.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I.-C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- W. Yantasee, C. L. Warner, T. Sangvanich, R. S. Addleman, T. G. Carter, R. J. Wiacek, G. E. Fryxell, C. Timchalk and M. G. Warner, Environ. Sci. Technol., 2007, 41, 5114–5119 CrossRef CAS PubMed.
- K. Zhang, V. Dwivedi, C. Chi and J. Wu, J. Hazard. Mater., 2010, 182, 162–168 CrossRef CAS PubMed.
- L. Feng, M. Cao, X. Ma, Y. Zhu and C. Hu, J. Hazard. Mater., 2012, 217–218, 439–446 CrossRef CAS PubMed.
- M. Kilianová, R. Prucek, J. Filip, J. Kolařík, L. Kvítek, A. Panáček, J. Tuček and R. Zbořil, Chemosphere, 2013, 93, 2690–2697 CrossRef PubMed.
- E. Iakovleva, P. Maydannik, T. V. Ivanova, M. Sillanpää, W. Z. Tang, E. Mäkilä, J. Salonen, A. Gubal, A. A. Ganeev, K. Kamwilaisak and S. Wang, J. Cleaner Prod., 2016, 133, 1095–1104 CrossRef CAS.
- M. Martínez-Cabanas, M. López-García, J. L. Barriada, R. Herrero and M. E. Sastre de Vicente, Chem. Eng. J., 2016, 301, 83–91 CrossRef.
- R. S. Bangari, A. K. Singh, S. Namsani, J. K. Singh and N. Sinha, ACS Appl. Mater. Interfaces, 2019, 11, 19017–19028 CrossRef CAS PubMed.
- S. Scurti, S. Dattilo, D. Gintsburg, L. Vigliotti, A. Winkler, S. C. Carroccio and D. Caretti, ACS Omega, 2022, 7, 10775–10788 CrossRef CAS PubMed.
- M. Baikousi, A. B. Bourlinos, A. Douvalis, T. Bakas, D. F. Anagnostopoulos, J. Tuček, K. Šafářová, R. Zboril and M. A. Karakassides, Langmuir, 2012, 28, 3918–3930 CrossRef CAS PubMed.
- T. Ren, P. He, W. Niu, Y. Wu, L. Ai and X. Gou, Environ. Sci. Pollut. Res., 2013, 20, 155–162 CrossRef CAS PubMed.
- M. Kokate, K. Garadkar and A. Gole, J. Mater. Chem. A, 2013, 1, 2022–2029 RSC.
- L. P. Lingamdinne, Y.-Y. Chang, J.-K. Yang, J. Singh, E.-H. Choi, M. Shiratani, J. R. Koduru and P. Attri, Chem. Eng. J., 2017, 307, 74–84 CrossRef CAS.
- Z. Pan, X. Zhu, A. Satpathy, W. Li, J. D. Fortner and D. E. Giammar, Environ. Sci. Technol., 2019, 53, 11913–11921 CrossRef CAS PubMed.
- P. Miao, Y. Tang and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 3940–3947 CrossRef CAS PubMed.
- J. Wang, Y. Chen, Z. Zhang, Y. Ai, L. Liu, L. Qi, J. Zhou, Z. Hu, R. Jiang, H. Bao, S. Ren, J. Liang, H. Sun, D. Niu and Q. Liang, ACS Sustainable Chem. Eng., 2018, 6, 12925–12934 CrossRef CAS.
- S. Xiang, W. Cheng, F. Chi, X. Nie, T. Hayat and N. S. Alharbi, ACS Appl. Nano Mater., 2020, 3, 1131–1138 CrossRef CAS.
- X. Yang, X. Wang, Y. Feng, G. Zhang, T. Wang, W. Song, C. Shu, L. Jiang and C. Wang, J. Mater. Chem. A, 2013, 1, 473–477 RSC.
- Y. Zhu, H. Hou, G. Tang and Q. Hu, Eur. J. Inorg. Chem., 2010, 2010, 872–878 CrossRef.
- Y. Lin, W. Cai, H. He, X. Wang and G. Wang, RSC Adv., 2012, 2, 1769–1773 RSC.
- T. Josephine, Drinking Water Treatment and Challenges in Developing Countries, in The Relevance of Hygiene to Health in Developing Countries, ed. P. Natasha and H. Afsatou Ndama Traore, IntechOpen, Rijeka, 2019, ch. 5 Search PubMed.
- S. H. Teo, C. H. Ng, A. Islam, G. Abdulkareem-Alsultan, C. G. Joseph, J. Janaun, Y. H. Taufiq-Yap, S. Khandaker, G. J. Islam, H. Znad and M. R. Awual, J. Cleaner Prod., 2022, 332, 130039 CrossRef CAS.
- Q. Zhang, G. Rao, J. Rogers, C. Zhao, L. Liu and Y. Li, Chem. Eng. J., 2015, 271, 180–187 CrossRef CAS.
- W. Yuan and A. L. Zydney, J. Membr. Sci., 1999, 157, 1–12 CrossRef CAS.
- T. Peik-See, A. Pandikumar, L. H. Ngee, H. N. Ming and C. C. Hua, Catal. Sci. Technol., 2014, 4, 4396–4405 RSC.
- G. Sheng, Y. Li, X. Yang, X. Ren, S. Yang, J. Hu and X. Wang, RSC Adv., 2012, 2, 12400–12407 RSC.
- X. Luo, C. Wang, S. Luo, R. Dong, X. Tu and G. Zeng, Chem. Eng. J., 2012, 187, 45–52 CrossRef CAS.
- S. V. Kumar, N. M. Huang, N. Yusoff and H. N. Lim, Mater. Lett., 2013, 93, 411–414 CrossRef.
- A. Sinha and N. R. Jana, Chem. – Asian J., 2013, 8, 786–791 CrossRef CAS PubMed.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef CAS PubMed.
- H. Y. Koo, H.-J. Lee, H.-A. Go, Y. B. Lee, T. S. Bae, J. K. Kim and W. S. Choi, Chem. – Eur. J., 2011, 17, 1214–1219 CrossRef CAS PubMed.
- J. Zhu, Z. Luo, S. Wu, N. Haldolaarachchige, D. P. Young, S. Wei and Z. Guo, J. Mater. Chem., 2012, 22, 835–844 RSC.
- Y. He, Q. Sheng, J. Zheng, M. Wang and B. Liu, Electrochim. Acta, 2011, 56, 2471–2476 CrossRef CAS.
- K. Zhou, Y. Zhu, X. Yang and C. Li, Electroanalysis, 2011, 23, 862–869 CrossRef CAS.
- K. Yang, L. Feng, X. Shi and Z. Liu, Chem. Soc. Rev., 2013, 42, 530–547 RSC.
- H. Jabeen, K. C. Kemp and V. Chandra, J. Environ. Manage., 2013, 130, 429–435 CrossRef CAS PubMed.
- P. Zong, S. Wang, Y. Zhao, H. Wang, H. Pan and C. He, Chem. Eng. J., 2013, 220, 45–52 CrossRef CAS.
- M. Liu, C. Chen, J. Hu, X. Wu and X. Wang, J. Phys. Chem. C, 2011, 115, 25234–25240 CrossRef CAS.
- Y. Zhang, B. Chen, L. Zhang, J. Huang, F. Chen, Z. Yang, J. Yao and Z. Zhang, Nanoscale, 2011, 3, 1446–1450 RSC.
- J. Guo, R. Wang, W. W. Tjiu, J. Pan and T. Liu, J. Hazard. Mater., 2012, 225–226, 63–73 CrossRef CAS PubMed.
- A. A. Karamani, A. P. Douvalis and C. D. Stalikas, J. Chromatogr. A, 2013, 1271, 1–9 CrossRef CAS PubMed.
- Y. Tang, H. Guo, L. Xiao, S. Yu, N. Gao and Y. Wang, Colloids Surf., A, 2013, 424, 74–80 CrossRef CAS.
- K. C. Kemp, H. Seema, M. Saleh, N. H. Le, K. Mahesh, V. Chandra and K. S. Kim, Nanoscale, 2013, 5, 3149–3171 RSC.
- H. Li, Y. Jia, A. Wang, W. Cui, H. Ma, X. Feng and J. Li, Chem. – Eur. J., 2014, 20, 499–504 CrossRef CAS PubMed.
- J. K. Biswas, A. Mitra, M. P. Shah and S. Rodriguez-Couto, in Development in Wastewater Treatment Research and Processes, ed. M. Shah, S. Rodriguez-Couto and J. Biswas, Elsevier, 2022, pp. 1–22 Search PubMed.
- A. M. Gutierrez, T. D. Dziubla and J. Z. Hilt, Rev. Environ. Health, 2017, 32, 111–117 CAS.
- S. Nizamuddin, M. T. H. Siddiqui, N. M. Mubarak, H. A. Baloch, E. C. Abdullah, S. A. Mazari, G. J. Griffin, M. P. Srinivasan and A. Tanksale, in Nanoscale Materials in Water Purification, 2019, pp. 447–472 Search PubMed.
- N. Nassar, Iron Oxide Nanoadsorbents for Removal of Various Pollutants from Wastewater: An Overview, 2012, pp. 81–118 Search PubMed.
- M. Li, C. Liu, Z. Zhang, S. Cao, H. Liu, S. Shen and W. Wang, Sep. Purif. Technol., 2022, 281, 119978 CrossRef CAS.
- Z.-Y. Ma, Y.-P. Guan, X.-Q. Liu and H.-Z. Liu, J. Appl. Polym. Sci., 2005, 96, 2174–2180 CrossRef CAS.
- P. Janoš, P. Kuráň, V. Pilařová, J. Trögl, M. Šťastný, O. Pelant, J. Henych, S. Bakardjieva, O. Životský, M. Kormunda, K. Mazanec and M. Skoumal, Chem. Eng. J., 2015, 262, 747–755 CrossRef.
- R. Xiong, Y. Wang, X. Zhang and C. Lu, RSC Adv., 2014, 4, 22632–22641 RSC.
- C. Păcurariu, O. Paşka, R. Ianoş and S. G. Muntean, Clean Technol. Environ. Policy, 2016, 18, 705–715 CrossRef.
- X. Zheng, H. Zheng, R. Zhao, Y. Sun, Q. Sun, S. Zhang and Y. Liu, Materials, 2018, 11, 1312 CrossRef PubMed.
- S. Sharma, A. Hasan, N. Kumar and L. M. Pandey, Environ. Sci. Pollut. Res., 2018, 25, 21605–21615 CrossRef CAS PubMed.
- H. Yan, T. Zhang, Y. Yang, J. Li, Y. Liu, D. Qu, L. Feng and L. Zhang, J. Environ. Manage. Today, 2024, 351, 119931 CrossRef CAS PubMed.
- I. Dolinová, M. Štrojsová, M. Černík, J. Němeček, J. Macháčková and A. Ševců, Environ. Sci. Pollut. Res. Int., 2017, 24, 13262–13283 CrossRef PubMed.
- M. F. Azizian, I. P. G. Marshall, S. Behrens, A. M. Spormann and L. Semprini, J. Contam. Hydrol., 2010, 113, 77–92 CrossRef CAS PubMed.
- P. P. He, C. S. He, Q. Liu and Y. Mu, RSC Adv., 2017, 7, 27214–27223 RSC.
- M. Chang and Y. H. Shih, J. Environ. Manage., 2018, 224, 235–242 CrossRef CAS PubMed.
- L. Zhang, Y. Zhu, J. Zhang, G. Zeng, H. Dong, W. Cao, W. Fang, Y. Cheng, Y. Wang and Q. Ning, Waste Manage., 2019, 95, 289–297 CrossRef CAS PubMed.
- A. M. Gutierrez, F. C. Leniz, X. Wang, T. D. Dziubla and J. Z. Hilt, Mat. Sci. Eng., B, 2022, 277, 115577 CrossRef CAS PubMed.
- P. Falcaro, R. Ricco, A. Yazdi, I. Imaz, S. Furukawa, D. Maspoch, R. Ameloot, J. D. Evans and C. J. Doonan, Coord. Chem. Rev., 2016, 307, 237–254 CrossRef CAS.
- X. Li, K. Chen, R. Guo and Z. Wei, Chem. Rev., 2023, 123, 10432–10467 CrossRef CAS PubMed.
- D. Lu, M. Qin, C. Liu, J. Deng, G. Shi and T. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 5357–5367 CrossRef CAS PubMed.
- J. R. McKone, N. S. Lewis and H. B. Gray, Chem. Mater., 2014, 26, 407–414 CrossRef CAS.
- S. C. Shit, I. Mondal, S. Pendem, L. Bai, J. Y. Park and J. Mondal, ChemElectroChem, 2018, 5, 2842–2849 CrossRef CAS.
- T. Ma, L. Zhou, L. k. Chen, Z. Li, L. Wu, P. Christie and Y. Luo, J. Agric. Food Chem., 2016, 64, 8045–8053 CrossRef CAS PubMed.
- S. Santaeufemia, E. Torres, R. Mera and J. Abalde, J. Hazard. Mater., 2016, 320, 315–325 CrossRef CAS PubMed.
- A. Vasilescu and J.-L. Marty, TrAC, Trends Anal. Chem., 2016, 79, 60–70 CrossRef CAS.
- Y. Song, F. Duan, S. Zhang, J.-Y. Tian, Z. Zhang, Z.-W. Wang, C.-S. Liu, W.-M. Xu and M. Du, J. Mater. Chem. A, 2017, 5, 19378–19389 RSC.
- N. K. Gupta, J. Bae and K. S. Kim, J. Environ. Chem. Eng., 2021, 9, 106195 CrossRef CAS.
- X. Wang, C. Liu, X. Li, F. Li and S. Zhou, J. Hazard. Mater., 2008, 153, 426–433 CrossRef CAS PubMed.
- F. Li, X. Wang, Y. Li, C. Liu, F. Zeng, L. Zhang, M. Hao and H. Ruan, J. Colloid Interface Sci., 2008, 321, 332–341 CrossRef CAS PubMed.
- F. B. Li, X. Z. Li, C. S. Liu and T. X. Liu, J. Hazard. Mater., 2007, 149, 199–207 CrossRef CAS PubMed.
- W. Feng and D. Nansheng, Chemosphere, 2000, 41, 1137–1147 CrossRef CAS PubMed.
- J. Bandara, U. Klehm and J. Kiwi, Appl. Catal., B, 2007, 76, 73–81 CrossRef CAS.
- S. Linley, Y. Liu, C. J. Ptacek, D. W. Blowes and F. X. Gu, ACS Appl. Mater. Interfaces, 2014, 6, 4658–4668 CrossRef CAS PubMed.
- S. Vasantharaj, S. Sathiyavimal, P. Senthilkumar, F. LewisOscar and A. Pugazhendhi, J. Photochem. Photobiol., B, 2019, 192, 74–82 CrossRef CAS PubMed.
- S. Qasim, A. Zafar, M. S. Saif, Z. Ali, M. Nazar, M. Waqas, A. U. Haq, T. Tariq, S. G. Hassan, F. Iqbal, X.-G. Shu and M. Hasan, J. Photochem. Photobiol., B, 2020, 204, 111784 CrossRef CAS PubMed.
- M. Diab and T. Mokari, Inorg. Chem., 2014, 53, 2304–2309 CrossRef CAS PubMed.
- X. Zhou, H. Yang, C. Wang, X. Mao, Y. Wang, Y. Yang and G. Liu, J. Phys. Chem. C, 2010, 114, 17051–17061 CrossRef CAS.
- H. Zhang, H. Ming, S. Lian, H. Huang, H. Li, L. Zhang, Y. Liu, Z. Kang and S.-T. Lee, Dalton Trans., 2011, 40, 10822–10825 RSC.
- Y. Zhai, Y. Dou, X. Liu, B. Tu and D. Zhao, J. Mater. Chem., 2009, 19, 3292–3300 RSC.
- X. Dong, H. Chen, W. Zhao, X. Li and J. Shi, Chem. Mater., 2007, 19, 3484–3490 CrossRef CAS.
- Z. Wu, W. Li, P. A. Webley and D. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- A.-H. Lu, J.-J. Nitz, M. Comotti, C. Weidenthaler, K. Schlichte, C. W. Lehmann, O. Terasaki and F. Schüth, J. Am. Chem. Soc., 2010, 132, 14152–14162 CrossRef CAS PubMed.
- H. G. Cha, H. S. Noh, M. J. Kang and Y. S. Kang, New J. Chem., 2013, 37, 4004–4009 RSC.
- C. Baumanis, J. Z. Bloh, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2011, 115, 25442–25450 CrossRef CAS.
- W. Zhou, L. Lin, W. Wang, L. Zhang, Q. Wu, J. Li and L. Guo, J. Phys. Chem. C, 2011, 115, 7126–7133 CrossRef CAS.
- M. I. Pariente, F. Martínez, J. A. Melero, J. Á. Botas, T. Velegraki, N. P. Xekoukoulotakis and D. Mantzavinos, Appl. Catal., B, 2008, 85, 24–32 CrossRef CAS.
- J. R. Chueca, R. M. Abad, M. Melero, N. Miguel and J. Narvión, Water Technol., 2012, 337, 72–77 Search PubMed https://dialnet.unirioja.es/servlet/articulo?codigo=3896468 .
- C. Ruales-Lonfat, J. F. Barona, A. Sienkiewicz, M. Bensimon, J. Vélez-Colmenares, N. Benítez and C. Pulgarín, Appl. Catal., B, 2015, 166–167, 497–508 CrossRef CAS.
- L. Guo, F. Chen, X. Fan, W. Cai and J. Zhang, Appl. Catal., B, 2010, 96, 162–168 CrossRef CAS.
- G. Rao, Q. Zhang, H. Zhao, J. Chen and Y. Li, Chem. Eng. J., 2016, 302, 633–640 CrossRef CAS.
- L. Liu, A. Li, M. Cao, J. Ma, W. Tan, S. L. Suib and G. Qiu, ACS EST Eng., 2022, 2, 1376–1385 CrossRef CAS.
- F. Yang, J. Pei and H. Zhao, Langmuir, 2022, 38, 3844–3851 CrossRef CAS PubMed.
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS PubMed.
- E. Brillas, I. Sirés and M. A. Oturan, Chem. Rev., 2009, 109, 6570–6631 CrossRef CAS PubMed.
- H. Vafaei Molamahmood, W. Geng, Y. Wei, J. Miao, S. Yu, A. Shahi, C. Chen and M. Long, Chemosphere, 2022, 291, 133037 CrossRef CAS PubMed.
- T. Tatarchuk, N. Danyliuk, I. Lapchuk, W. Macyk, A. Shyichuk, R. Kutsyk, V. Kotsyubynsky and V. Boichuk, J. Mol. Liq., 2022, 366, 120267 CrossRef CAS.
- A. Bozzi, T. Yuranova, J. Mielczarski and J. Kiwi, New J. Chem., 2004, 28, 519–526 RSC.
- S.-S. Lin and M. D. Gurol, Environ. Sci. Technol., 1998, 32, 1417–1423 CrossRef CAS.
- C.-P. Huang and Y.-H. Huang, Appl. Catal., A, 2008, 346, 140–148 CrossRef CAS.
- W. P. Kwan and B. M. Voelker, Environ. Sci. Technol., 2002, 36, 1467–1476 CrossRef CAS PubMed.
- A. L.-T. Pham, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2012, 46, 1055–1062 CrossRef CAS PubMed.
- M. Hartmann, S. Kullmann and H. Keller, J. Mater. Chem., 2010, 20, 9002–9017 RSC.
- S.-P. Sun and A. T. Lemley, J. Mol. Catal. A: Chem., 2011, 349, 71–79 CrossRef CAS.
- N. Wang, L. Zhu, M. Lei, Y. She, M. Cao and H. Tang, ACS Catal., 2011, 1, 1193–1202 CrossRef CAS.
- T. Jiang, A. S. Poyraz, A. Iyer, Y. Zhang, Z. Luo, W. Zhong, R. Miao, A. M. El-Sawy, C. J. Guild, Y. Sun, D. A. Kriz and S. L. Suib, J. Phys. Chem. C, 2015, 119, 10454–10468 CrossRef CAS.
- M. Yang and M. Jonsson, J. Phys. Chem. C, 2014, 118, 7971–7979 CrossRef CAS.
- M. D. Gurol and S.-S. Lin, J. Adv. Oxid. Technol., 2002, 5, 147–154 CAS.
- C. Walling and A. Goosen, J. Am. Chem. Soc., 1973, 95, 2987–2991 CrossRef CAS.
- C. Gregor, M. Hermanek, D. Jancik, J. Pechousek, J. Filip, J. Hrbac and R. Zboril, Eur. J. Inorg. Chem., 2010, 16, 2343–2351 CrossRef.
- K. C. Park, F. Wang, S. Morimoto, M. Fujishige, A. Morisako, X. Liu, Y. J. Kim, Y. C. Jung, I. Y. Jang and M. Endo, Mater. Res. Bull., 2009, 44, 1443–1450 CrossRef CAS.
- H. Lim, J. Lee, S. Jin, J. Kim, J. Yoon and T. Hyeon, Chem. Commun., 2006, 463–465 RSC.
- M. Gui, V. Smuleac, L. E. Ormsbee, D. L. Sedlak and D. Bhattacharyya, J. Nanopart. Res., 2012, 14, 861 CrossRef PubMed.
- S. Hsieh and P.-Y. Lin, J. Nanopart. Res., 2012, 14, 956 CrossRef.
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS.
- C. Zheng, X. Cheng, P. Chen, C. Yang, S. Bao, J. Xia, M. Guo and X. Sun, RSC Adv., 2015, 5, 40872–40883 RSC.
- K.-i. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS.
- S. Song, Y. Wang, H. Shen, J. Zhang, H. Mo, J. Xie, N. Zhou and J. Shen, ACS Appl. Nano Mater., 2019, 2, 7074–7084 CrossRef CAS.
- N. Singh, S. Riyajuddin, K. Ghosh, S. K. Mehta and A. Dan, ACS Appl. Nano Mater., 2019, 2, 7379–7392 CrossRef CAS.
- S. Liu, L. Mei, X. Liang, L. Liao, G. Lv, S. Ma, S. Lu, A. Abdelkader and K. Xi, ACS Appl. Mater. Interfaces, 2018, 10, 29467–29475 CrossRef CAS PubMed.
- P. A. O'Day, A. Pattammattel, P. Aronstein, V. J. Leppert and H. J. Forman, Environ. Sci. Technol., 2022, 56, 7006–7016 CrossRef PubMed.
- F. C. C. Moura, G. C. Oliveira, M. H. Araujo, J. D. Ardisson, W. A. A. Macedo and R. M. Lago, Appl. Catal., A, 2006, 307, 195–204 CrossRef CAS.
- C. Zhang, C. Kong, P. G. Tratnyek and C. Qin, Environ. Sci. Technol., 2022, 56, 4367–4376 CrossRef PubMed.
- N. Shakeel, R. Perveen, M. Imran Ahamed, A. Ahmad and Inamuddin, Fuel, 2023, 341, 127579 CrossRef CAS.
- S. Rawat, N. Misra, S. S. Meena, S. S. Shelkar, N. Kumar N, N. K. Goel and V. Kumar, Environ. Sci. Pollut. Res., 2022, 29, 82524–82540 CrossRef CAS PubMed.
- M. Paljevac, M. Primožič, M. Habulin, Z. Novak and Ž. Knez, J. Supercrit. Fluids, 2007, 43, 74–80 CrossRef CAS.
- L. Cai, L. Chen, L. Zhang, G. Lu, X. He, S. Zhang, H. Sun, Z. Liu and B. Zhao, ChemPlusChem, 2013, 78, 1437–1439 CrossRef CAS PubMed.
- J. Virkutyte and R. S. Varma, Chem. Sci., 2011, 2, 837–846 RSC.
- S. K. Lee, H. Chou, T. S. Ham, T. S. Lee and J. D. Keasling, Curr. Opin. Biotechnol., 2008, 19, 556–563 CrossRef CAS PubMed.
- W. J. Goh, V. S. Makam, J. Hu, L. Kang, M. Zheng, S. L. Yoong, C. N. B. Udalagama and G. Pastorin, Langmuir, 2012, 28, 16864–16873 CrossRef CAS PubMed.
- V. Zlateski, R. Fuhrer, F. M. Koehler, S. Wharry, M. Zeltner, W. J. Stark, T. S. Moody and R. N. Grass, Bioconjugate Chem., 2014, 25, 677–684 CrossRef CAS PubMed.
- W. Wang, D. I. C. Wang and Z. Li, Chem. Commun., 2011, 47, 8115–8117 RSC.
- Y. Ren, J. G. Rivera, L. He, H. Kulkarni, D.-K. Lee and P. B. Messersmith, BMC Biotechnol., 2011, 11, 63 CrossRef CAS PubMed.
- R. F. H. Dekker, Appl. Biochem. Biotechnol., 1990, 23, 25–39 CrossRef CAS.
- A. R. Herdt, B.-S. Kim and T. A. Taton, Bioconjugate Chem., 2007, 18, 183–189 CrossRef CAS PubMed.
- X.-m. Li, Q. Yang, Y. Zhang, W. Zheng, X. Yue, D.-b. Wang and G.-m. Zeng, Water Sci. Technol., 2010, 62, 947–955 CrossRef CAS PubMed.
- B. S. Fernandes, N. K. Saavedra, S. I. Maintinguer, L. D. Sette, V. M. Oliveira, M. B. A. Varesche and M. Zaiat, Appl. Biochem. Biotechnol., 2013, 170, 1348–1366 CrossRef CAS PubMed.
- L. Lima de Oliveira, I. C. Silveira Duarte, I. K. Sakamoto and M. B. Amâncio Varesche, J. Environ. Manage., 2009, 90, 1261–1268 CrossRef CAS PubMed.
- C. Jolivalt, S. Brenon, E. Caminade, C. Mougin and M. Pontié, J. Membr. Sci., 2000, 180, 103–113 CrossRef CAS.
- X.-y. Jiang, G.-m. Zeng, D.-l. Huang, Y. Chen, F. Liu, G.-h. Huang, J.-b. Li, B.-d. Xi and H.-l. Liu, World J. Microbiol. Biotechnol., 2006, 22, 909–913 CrossRef CAS.
- A. P. McHale and S. McHale, Biotechnol. Adv., 1994, 12, 647–652 CrossRef CAS PubMed.
- S. R. Couto, Biotechnol. Adv., 2009, 27, 227–235 CrossRef PubMed.
- M. Shin, T. Nguyen and J. Ramsay, Appl. Microbiol. Biotechnol., 2002, 60, 218–223 CrossRef CAS PubMed.
- L. Peng, T. Xie, Y. Lu, H. Fan and D. Wang, Phys. Chem. Chem. Phys., 2010, 12, 8033–8041 RSC.
- J. Lin, M. Sun, B. Su, G. Owens and Z. Chen, Sci. Total Environ., 2019, 659, 491–498 CrossRef CAS PubMed.
- A. Sigdel, J. Lim, J. Park, H. Kwak, S. Min, K. Kim, H. Lee, C. H. Nahm and P.-K. Park, Environ. Sci.: Water Res. Technol., 2018, 4, 1114–1123 RSC.
- C. Bianco, J. E. Patino Higuita, T. Tosco, A. Tiraferri and R. Sethi, Sci. Rep., 2017, 7, 12992 CrossRef PubMed.
- M. Sarcletti, H. Park, J. Wirth, S. Englisch, A. Eigen, D. Drobek, D. Vivod, B. Friedrich, R. Tietze, C. Alexiou, D. Zahn, B. A. Zubiri, E. Spiecker and M. Halik, Mater. Today, 2021, 48, 38–46 CrossRef CAS.
- G. F. Koopmans, T. Hiemstra, C. Vaseur, W. J. Chardon, A. Voegelin and J. E. Groenenberg, Sci. Total Environ., 2020, 711, 135220 CrossRef CAS PubMed.
- H. Cui, B. Bao, Y. Cao, S. Zhang, J. Shi, J. Zhou and J. Zhou, Environ. Pollut., 2022, 292, 118323 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |