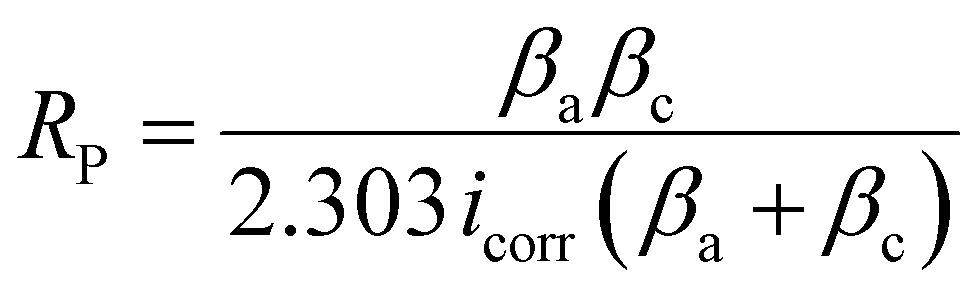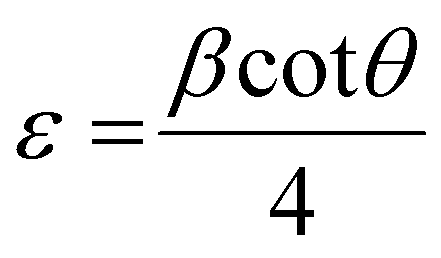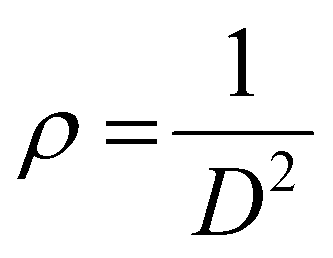DOI:
10.1039/D3MA00941F
(Paper)
Mater. Adv., 2024,
5, 5156-5166
High-entropy layered hydroxide for efficient and sustainable seawater oxidation†
Received
31st October 2023
, Accepted 29th April 2024
First published on 30th April 2024
Abstract
Developing efficient and stable electrocatalysts for high-current-density seawater oxidation remains challenging due to competitive chlorine evolution reaction at the anode. High-entropy layered hydroxides with different compositions, unique layered structures, and high mixing entropy have the potential to act as efficient as well as sustainable electrocatalysts for direct seawater splitting. Here, we report the development of a novel high-entropy NiCrCoFeMo layered hydroxide (HE-LH) material using a single-step hydrothermal method, which was found to be superior to its quaternary and ternary counterparts due to the availability of a larger number of electroactive sites and better chloride-restricting ability of interlayer anions. The HE-LH achieved a 100 mA cm−2 current density at overpotentials of only 242, 258 and 281 mV in 1 M KOH, 1 M KOH + seawater and 6 M KOH + seawater, respectively. The robustness due to the anti-corrosion ability of HE-LH has also been explored in highly alkaline (6 M KOH) real seawater, which showed stability over 140 h at an operating current density of 500 mA cm−2. The generation of β-NiOOH, also as active sites, during a prolonged electrooxidation process in seawater-based electrolyte was found to be beneficial to maintain such high activity. The combination of HE-LH with Pt/C in a two-electrode system also offered a high current density of 500 mA cm−2 at a cell voltage of only 1.92 V in highly alkaline (6 M KOH) real seawater electrolyte and exhibited stability over 50 h. With these excellent performances, the developed novel HE-LH is believed to have the potential for practical exploration in seawater electrolysers.
1. Introduction
The growing population worldwide, along with the ever-growing energy demand, has become a cause for worry due to rapid fossil fuel depletion causing severe environmental pollution.1 Having high energy density and zero-emission features, green hydrogen is considered to be leading clean energy candidate to fulfil growing global energy demands.2,3 Among various innovative technologies, water electrolysis has emerged as a promising one to produce carbon-free high-purity hydrogen gas.4–7 Electrochemically driven water splitting involves two half-cell reactions; the oxygen evolution reaction (OER) at the anode and the hydrogen evolution reaction (HER) at the cathode, among which the OER mechanism is of most concern due to its comparatively sluggish kinetics. Moreover, the consumption of large volumes of fresh water during electrolysis cannot be overlooked when considering hydrogen energy dependence in the future.8 In this regard, the splitting of seawater has recently drawn great attention due to its unlimited availability. However, seawater splitting is much more complex to handle, especially due to the presence of chlorides causing severe corrosion aided by the chlorine evolution reaction (CER) at the anode.9,10 The maximum potential difference between the competitive OER and CER is known to be 480 mV under alkaline conditions, and thus, the overpotential must be minimized to avoid the CER.11,12 Thereby, the rational design of electrocatalysts with small overpotential values along with an anti-corrosive nature is highly desired to enable use with seawater-based electrolytes.13–17
Recently, high-entropy materials consisting of five or more components with equiatomic or non-equiatomic tuneable compositions with a maximized entropy effect and also a synergistic effect from multiple elements, have become a new interest for researchers as efficient electrocatalysts.18 The reason behind their activity lies in the high entropy of the system, lattice-distortion-created defects accompanied by modulated electronic structure, and enhanced active sites, resulting in optimized adsorption free energy for boosted electrochemical performances.19 Owing to these unique features, various high entropy (HE) materials, such as high-entropy alloys,19,20 high-entropy phosphides,21,22 high-entropy oxides,23 and high-entropy oxyhydroxide24 have been reported as effective electrocatalysts, especially for the OER. Transition-metal-based layered double hydroxides (LDH) are best known for their two-dimensional layered structure and the accessibility of a large number of active sites for electrolytes during the electrochemical process.25 The easy modulation of cationic layers as well as intercalated anions between the two adjacent layers leads to desirable structural engineering in terms of electrocatalytic activity. The binary NiFe-LDH has been the primary choice, as explored extensively in various reports.11,26 The insertion of a third transition metal to form layered triple hydroxides (LTH) has also been studied and showed interesting effects in boosting electrocatalytic activities.27,28 Such LTHs have also been found to have the ability to restrict chlorides to a better extent compared to their binary counterparts during exploration of seawater-based electrolytes.29 The studies indicated that the insertion of additional transition metals with high valence states in the cationic layers maintaining the layered hydroxide structure also modulates anions in between layers to compensate extra cationic charges. While the addition of high-valence metal cations in the layers provides additional active sites along with desired electronic modulations, extra anionic species in between layers may retard the chlorides, allowing longer sustainability during seawater oxidation. In the current year, few reports are available on high-entropy LDHs for efficient OER activity.30–33 Wang and his co-workers30 developed high-entropy MnFeCoNiCu-LDH decorated with Au single atoms along with oxygen vacancies, which exhibited a low overpotential of 213 mV at 10 mA cm−2 current density, and very importantly, delivered good stability over 700 h at an operating current density of 100 mA cm−2 in alkaline fresh water. Li and his co-workers31 reported the development of HE-LDH composed of transition metals Ni, Fe, Mn, Zn, and Co in the form of metal hydroxides. This composition exhibited a small η10 value of 183 mV, although it increased to 327 mV at 100 mA cm−2 towards the OER mechanism and showed moderate stability over 28 h at a current density of 250 mA cm−2 in alkaline fresh water. Ting and his group32 developed high-entropy FeNiCoMnCr LDH, which showed excellent activity in alkaline freshwater, exhibiting a low overpotential of 218 mV at a current density of 50 mA cm−2, and the formation of highly active NiOOH was considered to be reason behind this excellent activity. The electrocatalyst delivered great stability over 600 h at a current density of 400 mA cm−2 in alkaline medium. Ding and his group33 revealed the monolayer HE-LH fabrication derived from the M-ZIF structure with well-defined geometric morphology and composition. Such HE-LH exhibited an overpotential value of 259 mV at 100 mA cm−2 and showed good stability over 1000 h at an operating current density of 20 mA cm−2 in alkaline fresh water.
It is important to mention here that though above discussed reports establish the fact that HE-LDHs are great candidates for the OER activity that show good stability under alkaline conditions, explorations in alkaline seawater investigating their activity, stability and anti-corrosion ability have still not been conducted. Knowing these facts, we herein developed novel high-entropy NiCrCoFeMo layered hydroxides (HE-LH) via a simple hydrothermal method and investigated their superiority towards longer seawater oxidation compared to their quaternary NiCrCoFe-LH (Q-LH) and ternary NiCrCo-LH (T-LH) counterparts. The developed HE-LH electrocatalyst exhibits excellent OER activity with an ultralow overpotential of 242 mV in alkaline fresh water and 258 mV in alkaline seawater at 100 mA cm−2 current density, keeping the overpotential well within the limit of 480 mV for avoiding the CER even at high current densities. The investigation also reveals the increased anti-corrosion properties of HE-LH compared to its counterparts and showed robust stability over 180 h at 500 mA cm−2 current density in alkaline (1 M KOH) seawater electrolyte. The practicability was further checked in highly alkaline (6 M KOH) seawater electrolyte, which also revealed outstanding stability over 140 h at an industrial-level current density of 500 mA cm−2.
2. Experimental section
2.1. Chemicals
Nickel(II) chloride hexahydrate (NiCl2·6H2O), chromium(III) chloride hexahydrate (CrCl3·6H2O), cobalt(II) chloride hexahydrate (CoCl2·6H2O), iron(III) chloride (FeCl3, anhydrous), sodium molybdate dihydrate (Na2MoO4·2H2O), ethanol, HCl, KOH, NaCl, commercial RuO2 and Pt/C were purchased from Sigma-Aldrich. All reagents and chemicals were of A.R. grade and were used without further purification.
2.2. Synthesis of materials
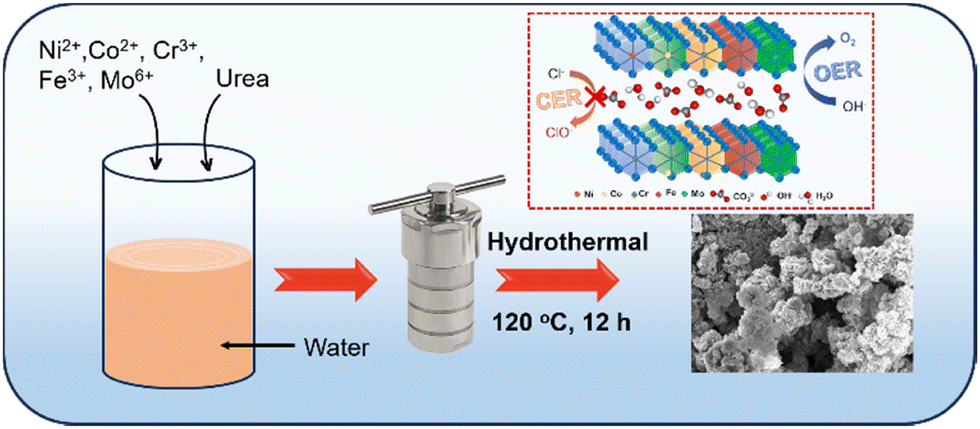
To synthesize high-entropy NiCrCoFeMo layered hydroxides, NiCl2·6H2O (0.0297 g, 5 mmol), CrCl3·6H2O (0.0333 g, 5 mmol), CoCl2·6H2O (0.0178 g, 3 mmol), FeCl3 (0.02 g, 5 mmol) and Na2MoO4·2H2O (0.0423 g, 7 mmol) were dissolved in 25 mL ultra-pure water, and the solution was stirred for 5 minutes. After that, urea (0.375 g, 0.25 mol) was added to the same solution under vigorous stirring for 15 minutes. The prepared homogeneous solution was transferred into a 50-mL stainless-steel Teflon-lined autoclave, and the reaction was carried out at 120 °C for 12 hours in a hot-air oven. After the completion of the reaction, the autoclave was allowed to cool naturally to room temperature, and the product was collected via centrifugation, washed several times with distilled water and ethanol, and dried under vacuum at 60 °C overnight. The as-obtained product was labelled as HE-LH.
For comparison, other layered hydroxides were prepared in the same way as HE-LH. For the quaternary NiCrCoFe layered hydroxides, all salts were used except Na2MoO4·2H2O, and the ternary NiCrCo layered hydroxides were synthesized without using FeCl3 and Na2MoO4·2H2O. These products are abbreviated as Q-LH and T-LH, as tabulated in Table S1 (ESI†).
2.3. Electrochemical measurements
All electrochemical experiments were performed with a Metrohm Autolab204 at room temperature using 1 M KOH (pH = 13.9) as the electrolyte in standard three-electrode configuration. The as-prepared catalyst, saturated Hg/HgO and a Pt wire were used as the working electrode, reference electrode and counter electrode, respectively, in three-electrode configurations. The working electrode was prepared by making a homogeneous ink by mixing the powder sample (2 mg) in 900 μL of a homogeneous solution of isopropanol and water followed by the addition of 100 μL of PVDF solution as a binder under ultrasonication and drop-casted on bare Ni foam. Prior to drop-casting, bare Ni foams were etched using 3 M HCl solution to remove the oxide layer, followed by washing with DI water and ethanol. The mass loading of the electrocatalysts on the treated Ni foam was maintained in the range of 0.8–1 mg cm−2. Commercial 20 wt% RuO2 and 10 wt% Pt/C were used with a mass loading of 1 mg cm−2 for comparative electrochemical study.
Linear sweep voltammetry (LSV) polarization curves were recorded at a scan rate of 2 mV s−1 from 0 to 1 V with respect to the standard Hg/HgO reference electrode in a three-electrode system. All LSV curves are shown with 90% iR compensation to avoid the uncompensated series resistance of the electrochemical circuit (Rs) All measured potentials were converted to the reversible hydrogen electrode (RHE) scale and calibrated with 90% iR compensation using eqn (1) and (2):
| | | ERHE = Eapply + EoHg/HgO + (0.059 × pH) | (1) |
The overpotential (
η) and Tafel slope (
b) of water oxidation were calculated using following
eqn (3) and (4):
| | η = a + b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logj logj | (4) |
Electrochemical impedance spectroscopy (EIS) of the as-prepared sample was performed at 250 mV overpotential in the frequency range of 10 kHz to 50 mHz. Cyclic voltammetry (CV) was carried out in the non-faradaic region at different sweep rates from 40 mV s−1 to 200 mV s−1 to calculate the double-layer capacitance (Cdl). The electrochemical active surface area (ECSA) was derived from Cdl values related by eqn (5):
where
Cs is the specific charge resistance of the working electrode, which has a value of 40 μF cm
−2 in alkaline solutions. Mass activity (
jmass) and specific activity (
jECSA) were calculated using following
eqn (6) and (7):
| | | Mass activity (MA) = j/m | (6) |
| | | Specific activity (SA) = j/ECSA | (7) |
Turn-over frequency (TOF) was determined based on the number of total active sites for all electrodes using the following
eqn (8):
| | | TOF (S−1) = (J × A)/(α × F × n) | (8) |
where
j (A cm
−2) is the current density at a particular overpotential,
A (cm
2) is the geometric surface area of the working electrode,
F is the Faraday constant (96
485 C mol
−1),
α is the electron transfer number and
n =
Qs/
F is the number of active sites during the electrochemical reaction process. Here, the surface charge (
Qs) is calculated from the integrated area of the redox peak of the CV plot. Chronopotentiometry (CP) and chronoamperometry (CA) tests were conducted to check the long-term stability and durability of the catalyst during water electrolysis.
Corrosion studies of HE-LH, Q-LH and T-LH were evaluated in 1 M KOH + 1 M NaCl medium using a three-electrode setup. The sample electrodes were immersed into an electrolyte solution for several hours to reach a stable open circuit potential (OCP). Electrochemical impedance spectroscopy (EIS) measurements were carried out at the OCP over the frequency range of 100 kHz to 0.01 Hz with an amplitude of 5 mV. The corrosion characteristics were further analyzed using Nyquist and Bode plots. The polarization test was carried out in the potential range from −0.4 to −0.3 V vs. the Hg/HgO reference electrode (the potential range was calculated based on the OCP). From the Tafel analysis of polarization curve, the corrosion potential (Ecorr), anodic slope (βa), cathodic slope (βc), and corrosion rate were calculated.
| | 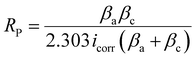 | (9) |
2.4. Iodometry test
To check if any hypochlorite formation occurred during the electrolysis process, iodometry tests were conducted using 10 mL electrolyte solution after the long-term stability test in 1 M KOH + seawater electrolyte. First, the electrolyte solution was made acidic (pH = 2) by adding the required amount of 0.5 M H2SO4 solution. Then, to that solution, 5 mL of freshly prepared KI solution was added. No yellow colour was observed, which confirmed that no chlorine evolution occurred during the long-term stability test.
3. Results and discussion
3.1. Characterization of electrocatalysts
High-entropy NiCrCoFeMo-LH and its quaternary NiCrCoFe-LH and ternary NiCrCo-LH counterparts were synthesized by adding metal precursors in optimum proportions via a simple hydrothermal method, as illustrated in Scheme 1 and described in the ESI.†
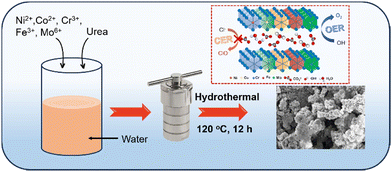 |
| | Scheme 1 Schematic illustration of the synthesis of high-entropy NiCrCoFeMo layered hydroxide (HE-LH) and its ability to promote sustainable seawater oxidation. | |
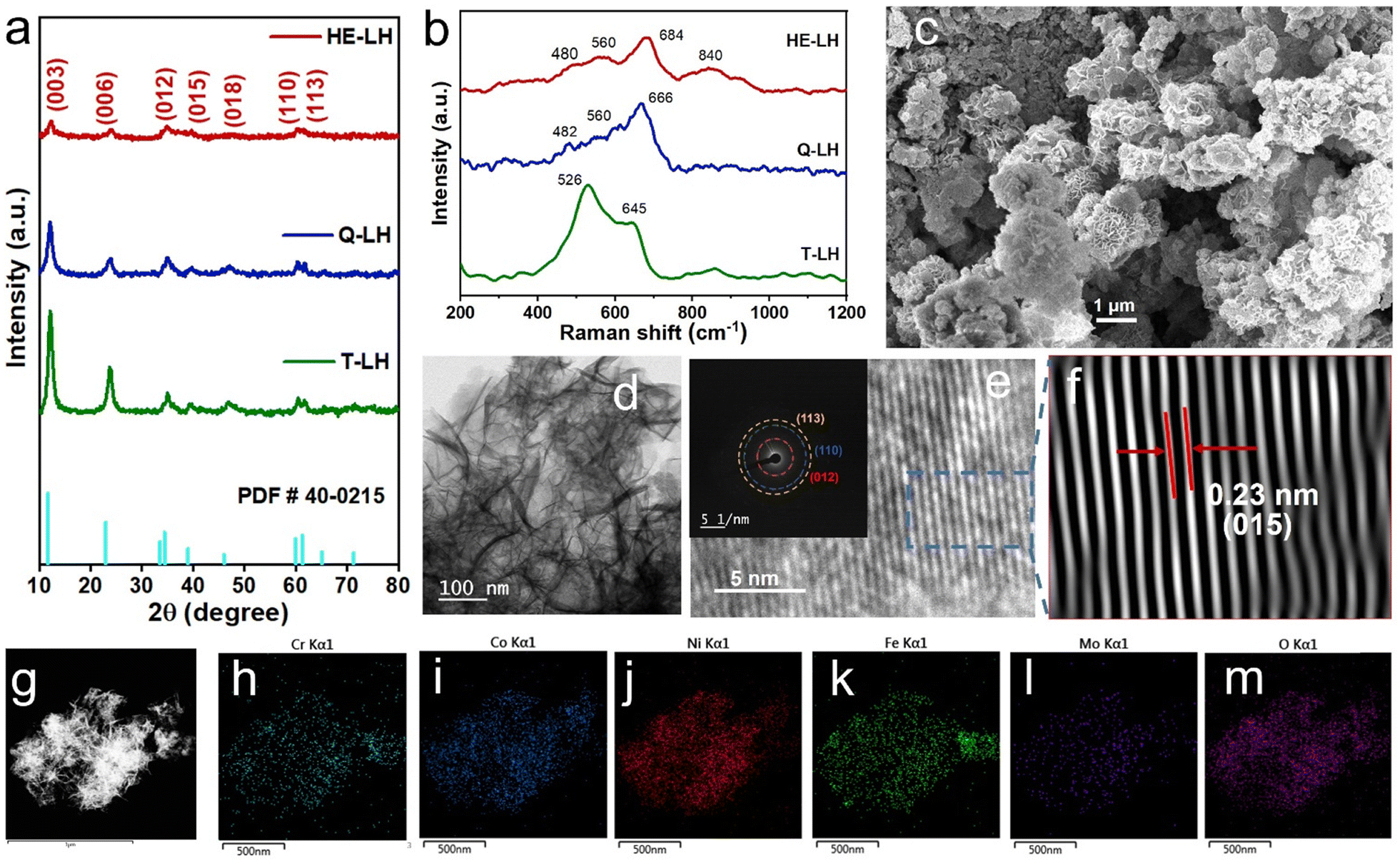
The X-ray powder diffraction patterns of all samples revealed the formation of phase pure layered hydroxides consisting of (003), (006), (009), (012), (015), (018), (110) and (113) planes, which nicely matched JCPDS file no. 40-0215, corresponding to characteristic layered double hydroxide materials (Fig. 1a).34 Interestingly, with the addition of 3, 4 or 5 different metal cations to layered hydroxide structures starting from T-LH to Q-LH to finally HE-LH, respectively, considerable peak broadening, along with a decrease in peak intensity can be observed, as shown in the magnification of the (003) plane in Fig. S1 (ESI†). This phenomenon definitely indicates lattice alteration generating stress and strain due to the insertion of multiple metal cations with different valence states. The lattice distortion can be evaluated in terms of microstrain (ε) and dislocation density (ρ) values as per Williamson and Smallman theory
| | 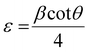 | (10) |
| | 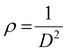 | (11) |
where,
β is full width at half maximum of characteristic diffraction peak and
D is the crystallite size obtained using
35 the Scherrer equation. The calculated values, as tabulated in Table S2 (ESI
†), show increasing microstrain values and dislocation densities upon the insertion of the Fe cation as the fourth metal in Q-LH and the Mo cation as the fifth metal into T-LH, confirming the introduction of defects in the lattice.
36,37 The Fourier transform infrared spectroscopy (FTIR) of all three samples reflects the evidence of water and carbonate molecules between layers, as shown in Fig. S2 (ESI
†). The broad spectrum at 3400 cm
−1 and a relatively weak band near 1650 cm
−1 can be assigned to the stretching vibrations of the hydroxyl group and bending vibration modes of water molecules, respectively. Two strong adsorption bands at 2360 cm
−1 and 669 cm
−1 correspond to the asymmetric and bending vibrations of adsorbed carbon dioxide molecules present in LH materials. Another broad band near 780 cm
−1 can be assigned to the stretching and bending modes of multiple MO bonds of metals with different oxidation states. The presence of carbonate anions in the interlayer positions of the LHs was confirmed by a sharp absorbance peak at 1360 cm
−1, which was probably generated by the slow hydrolysis of urea.
38,39 In any case, the presence of stronger bands can be noted in the HE-LH sample for obvious reasons. Raman spectroscopy provided additional information about the surface of the layered hydroxides. In the spectrum of T-LH, broad vibrational bands with peaks located at 526 and 645 cm
−1 are believed to be mainly due to the presence of M–OH and M–O bonds, which remain but are somewhat blue-shifted in Q-LH and HE-LH, as shown in
Fig. 1b.
40 This blueshift may be due to the defects or dislocations in Q-LH and especially in HE-LH due to the insertion of multiple different metal cations with different valence states.
41 The appearance of a band at 840 cm
−1 in HE-LH can be attributed to Mo–O vibrations due to the presence of Mo as the fifth metal.
42
 |
| | Fig. 1 (a) XRD patterns, (b) Raman spectra, (c) SEM images and (d) TEM images of HE-LH. (e) HRTEM images showing lattice fringes and SAED pattern (inset). (f) IFFT images showing the d spacing of adjacent planes. (g) HAADF-STEM images and corresponding EDAX mapping (h)–(m) of elements present in HE-LH. | |
The morphology of the LHs was further investigated with the help of field emission scanning electron microscopy (FESEM). An interesting evolution in the morphology of the LHs can be observed proceeding from T-LH to Q-LH and finally HE-LH. T-LH with Ni, Co and Cr species in LH leads to a nanowire-like morphology as shown in Fig. S3 (ESI†), and the corresponding energy-dispersive X-ray (EDX) mapping indicates the presence of all three metal species homogenously distributed. The addition of Fe as the fourth metal in Q-LH results in an evenly distributed nanosheet-like morphology with homogeneous distribution of all metal species throughout, as shown in Fig. S4 (ESI†). The addition of Mo as the fifth metal to HE-LH leads to the formation of nanosheet-composed spheres, as shown in Fig. 1c. To get more details about the morphology of HE-LH, transmission electron microscopy (TEM) was conducted, which shows the presence of fine flakes (Fig. 1d). The high-resolution transmission electron microscopy (HRTEM) and inverse fast Fourier transform (IFFT) images show well-defined lattice fringes in which the lattice spacing of 0.23 nm corresponds to the (015) plane, as shown in Fig. 1e and f. The presence of defects can be noted very well in the IFFT images, as expected for HE-LH. The corresponding SAED pattern shows diffused rings for (012), (110) and (113) planes with low crystallinity, in line with XRD patterns. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images (Fig. 1g), along with energy-dispersive X-ray spectroscopy (EDS) mapping, confirms the homogeneous distribution of elements Ni, Cr, Co, Fe, Mo and O (Fig. 1h–m). The atomic percentages of the metals present in the LHs are represented in a pie-chart in Fig. S5 (ESI†), and especially for the HE-LH samples, the presence of Ni, Cr, Co, Fe and Mo in 34.38%, 21.46%, 12.37%, 17.92% and 13.87%, respectively, confirms the formation of the high-entropy layered hydroxide material.
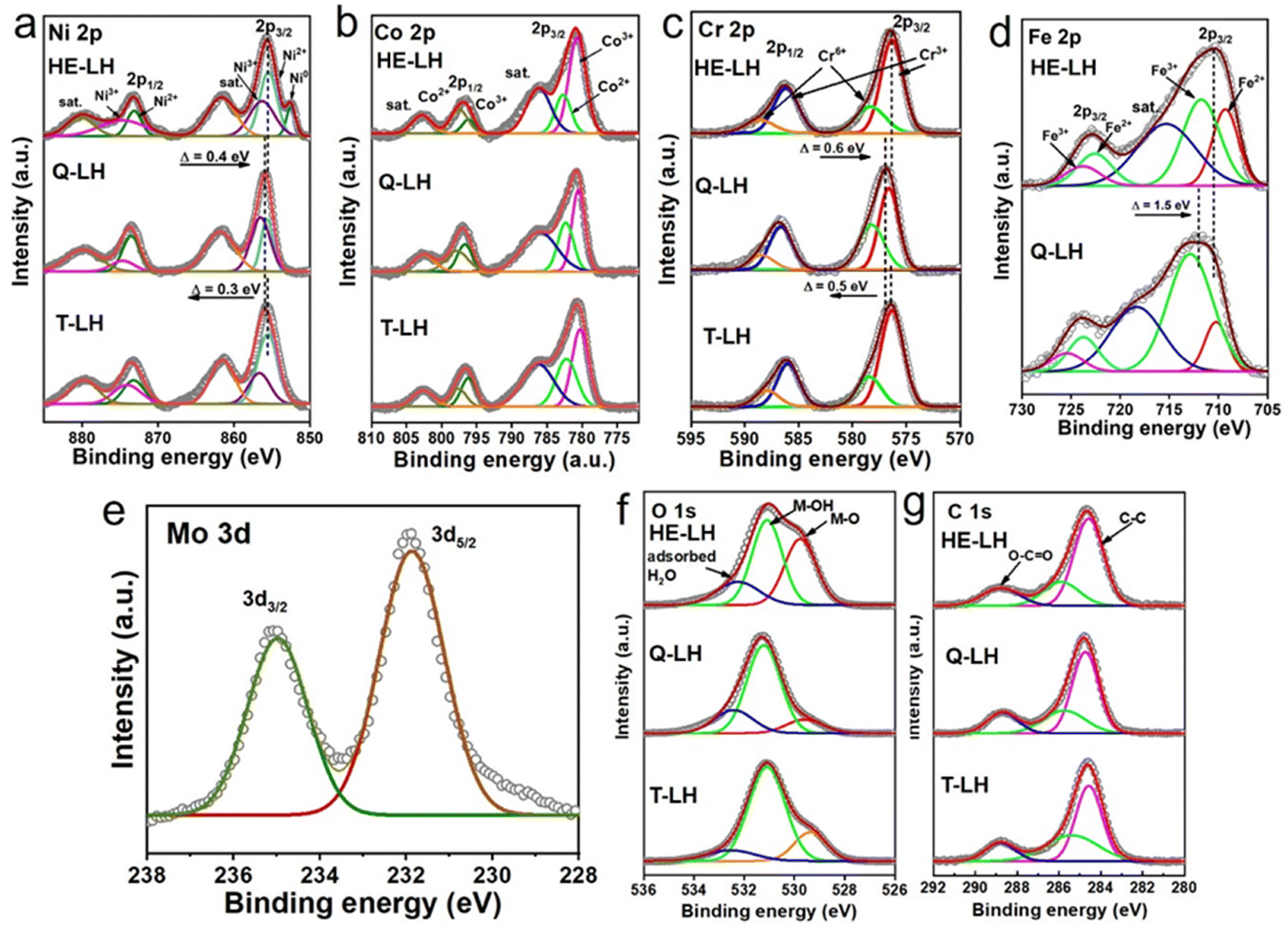
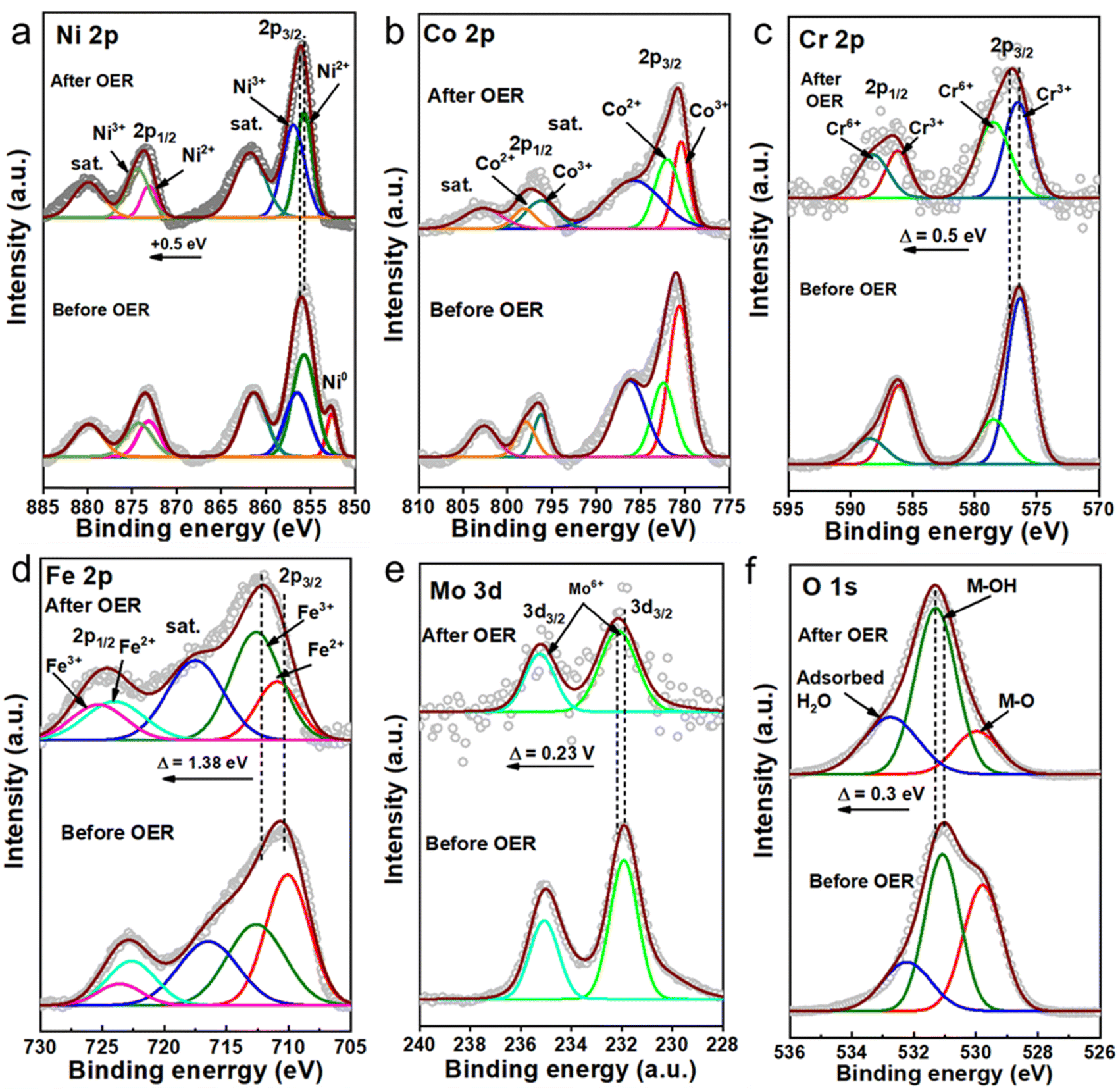
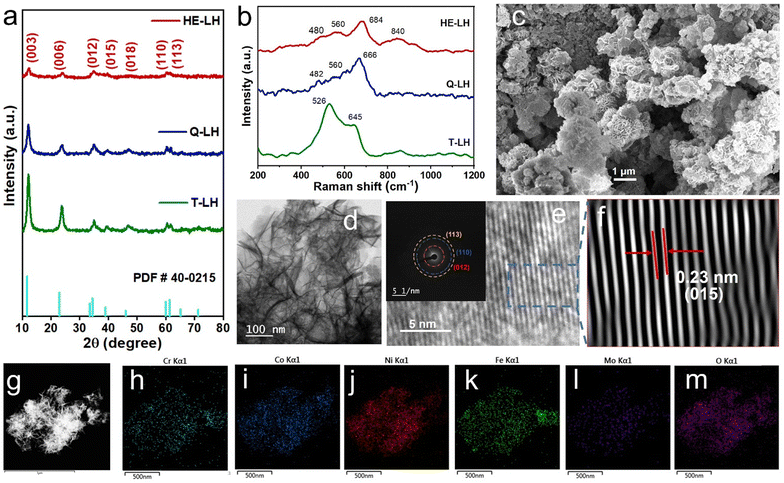
The elemental states and their local environment were thoroughly analysed with the help of X-ray photoelectron spectroscopy (XPS) and are presented in Fig. 2. The full XPS survey of the LHs indicated the presence of the respective metal species as expected (Fig. S6, ESI†). The high resolution XPS spectrum of Ni 2p for T-LH shows two major peaks at 855.5 eV and 873.3 eV for Ni 2p3/2 and Ni 2p1/2, respectively, along with two satellite peaks located at 861.5 eV and 879.8 eV. Careful deconvolution of the peaks indicates the coexistence of Ni2+ and Ni3+.43 The high-resolution Co 2p spectrum for T-LH also confirms the existence of Co2+ and Co3+ with two major peaks at 780.6 eV (2p3/2) and 796.7 eV (2p1/2), along with the presence of two satellite peaks at 786.1 eV and 802.6 eV, respectively.44 In the case of the Cr 2p spectrum for T-LH, careful deconvolution indicates peaks located at 576.3 eV (2p3/2) and 586.0 eV (2p1/2) corresponding to Cr3+ along with peaks at 578.4 eV (2p3/2) and 587.9 eV (2p1/2) for Cr6+.24 The addition of Fe in Q-LH leads to electronic transition among the metal species; a small shift in Ni 2p by 0.3 eV towards higher binding energy along with an increase in the Ni3+/Ni2+ ratio from 0.613 to 1.04 can be observed (Fig S7a, ESI†). A similar trend can be found for the Cr 2p spectrum as well, with an increase in the Cr6+/Cr3+ ratio from 0.35 to 0.57. On the contrary, a decrease in the ratio of Co3+/Co2+ from 1.32 to 1.17 indicates possible electronic transfer from Fe to Co, while the Fe 2p spectrum for Q-LH after deconvolution reveals the presence of Fe3+ (82.3%) as the major species.45 The addition of Mo as the fifth metal in HE-LH further leads to considerable alterations among the metal species, and it likely to be present in the form of Mo6+ based on the deconvoluted peaks located at 231.9 and 235 eV corresponding to Mo 3d5/2 and Mo 3d3/2, respectively. Interestingly, a reverse effect can be observed in case of Ni 2p for HE-LH with decrease in the Ni3+/Ni2+ ratio from 1.04 to 0.88 accompanied by a peak shifting towards lower binding energy by 0.4 eV. Additionally, the appearance of Ni0 can also be observed. Similarly, a tendency of slight reduction of Cr6+ to Cr3+ can also be noted. Unlike Ni and Cr, a slight increase in the Co3+/Co2+ ratio from 1.17 to 1.77 and a significant decrease in the Fe3+/Fe2+ ratio from 4.65 to 1.61 can clearly be seen. These striking alterations among the metal species, especially in HE-LH upon addition of Mo6+ in the system, indicates a possible strong electronic transition from Mo to Fe and Mo to Ni. It is anticipated that this electronic transition was possible due to the low electronegativity of Mo6+ (1.86) compared to Ni3+ (2.06) and Fe3+ (1.96).46 Indeed, it is expected that such electronic modulation will favour high activity of HE-LH compared to other layered hydroxides. Further, the high-resolution XPS spectra of O 1s can be deconvoluted into three major peaks located at 529.8 eV, 531.1 eV and 532.3 eV, which are ascribed to lattice M–O, M–OH and adsorbed water, respectively, as represented in Fig. 2f.47 An increase in the percentage of lattice oxygen was noticed in the case of HE-LH. The deconvolution of C 1s spectra shown in Fig. 2g, gives three characteristic peaks at 284.5 eV, 285.8 eV and 288.8 eV corresponding to C–C, O–C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, respectively, confirming the presence of carbonate anions in between the layers.
O bonds, respectively, confirming the presence of carbonate anions in between the layers.
 |
| | Fig. 2 High-resolution XPS spectra of (a) Ni 2p, (b) Co 2p, (c) Cr 2p, (d) Fe 2p, (e) Mo 3d, (f) O 1s and (g) C 1s for HE-LH, Q-LH and T-LH. | |
3.2. Electrocatalytic OER performances in 1 M KOH
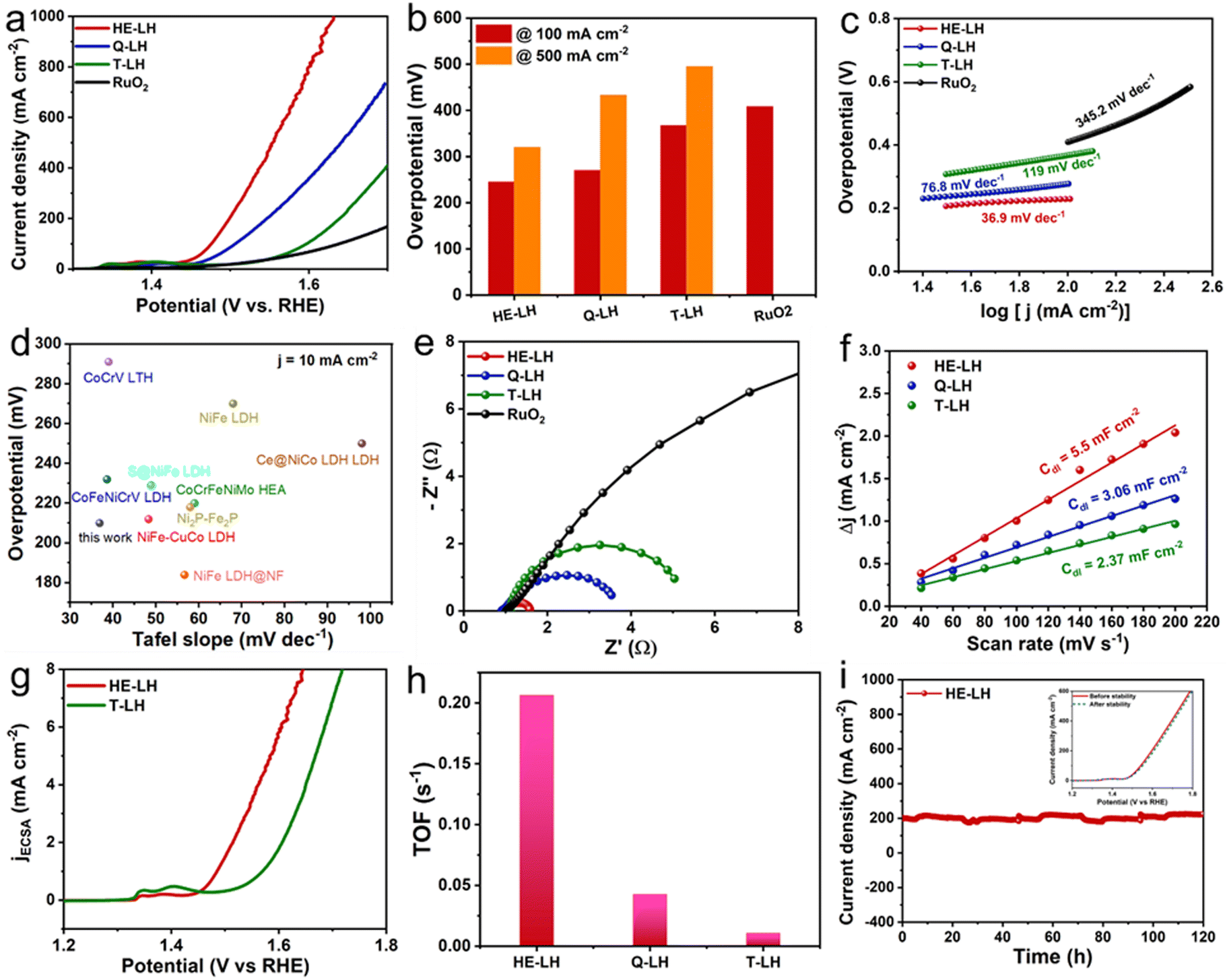
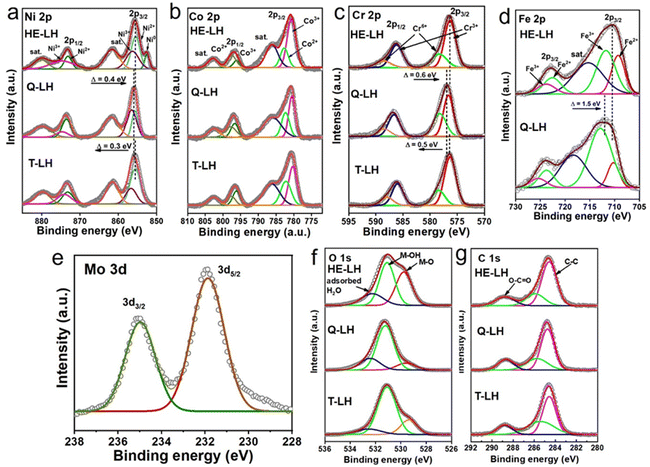
The performance of all the developed LHs and commercial RuO2 as a conventional electrocatalyst were analysed using electrodes prepared via drop-casting on Ni foam and using in a three-electrode system at room temperature in 1 M KOH (pH = 13.9) containing fresh water as electrolyte. The linear sweep voltammetry (LSV) of the electrocatalysts revealed that HE-LH was clearly the best performer for the OER among all the electrocatalysts. Prior to the LSV polarization curves, all electrodes were activated using scanning cyclic voltammetry (CV) at a 40 mV s−1 scan rate. The iR-compensated LSV polarization curve, shown in Fig. 3a, demonstrates the achievement of an ultralow potential of 242 mV at 100 mA cm−2 current density for HE-LH, which is far better than the results for Q-LH (270 mV), T-LH (367 mV) and RuO2 (408 mV). Impressively, HE-LH requires only 320 mV and 400 mV to reach 500 and 1000 mA cm−2 current densities, as shown in Fig. 3b, which are much smaller than values for the other LHs, RuO2 and bare NF (Fig. S8, ESI†). Indeed, the achievement of the high-entropy system in HE-LH involving multiple metal sites with different valence states is believed to be responsible for this outstanding electrocatalytic activity. The mass activity of HE-LH was also found to be the highest among them at 116.8 A g−1 at an overpotential of 250 mV, indicating the superior inherent catalytic activity of HE-LH, as shown in Fig. S9 (ESI†). Tafel slopes (Fig. 3c) obtained from respective LSV plots exhibit a considerably smaller value of only 36.9 mV dec−1 for HE-LH compared to the Q-LH (76.8 mV dec−1), T-LH (119 mV dec−1) and RuO2 (345.2 mV dec−1) electrocatalysts, demonstrating superior and faster kinetics for the OER mechanism. It is important to mention here that the developed HE-LH outperformed most of the recently reported transition-metal-based electrocatalysts for the OER in an alkaline medium, as shown in the comparison in Fig. 3d and tabulated in Table S8 (ESI†). The presence of multiple metal species may have a great influence on the electronic charge transfer ability of the materials, which can be evaluated with the help of electrochemical impedance spectroscopy (EIS). The charge transfer resistance (Rct), as estimated from the Nyquist plot in Fig. 3e and tabulated in Table S3 (ESI†), was found to be the smallest for HE-LH (0.58 Ω only), as expected, compared to those of Q-LH (2.61 Ω), T-LH (4.02 Ω) and RuO2 (16.03 Ω), indicating faster charge transfer ability owing to the strong electronic interaction between the five different metal species. The electrochemical active surface area (ECSA) was further calculated from the double-layer capacitance (Cdl), which was determined from CV curves taken at various scan rates in the non-faradaic region to evaluate the exposure of real active sites (Fig. S10, ESI†). As anticipated, the considerably higher Cdl value of HE-LH (5.5 mF cm−2) compared to those of Q-LH (3.06 mF cm−2) and T-LH (2.37 mF cm−2) indicates that a large number of exposed active sites are available to the electrolyte in HE-LH. The calculated ECSA was accordingly found to be sufficiently high for HE-LH (137.5 cm2), resulting in high activity. To understand the inherent abilities of the electrocatalysts, the specific activities of the electrocatalysts were evaluated using ECSA-normalised polarization curves, as shown in Fig. 3g, which also reflects the superior activity of HE-LH. The turnover frequency or TOF is another intrinsic parameter for defining excellence in electrocatalytic activity, as determined from the redox active area calculation (Fig. S11, ESI†). The calculated TOF (s−1) at 1.5 V vs. RHE, as shown in Fig. 3h, reveals that HE-LH possesses a TOF value five times higher than that of Q-LH and almost 20 times higher than that of T-LH. In addition to the determination of the inherent activity parameters of the electrocatalysts, the robustness of the electrocatalyst is another important factor that cannot be omitted. The durability of HE-LH was examined by performing a chronoamperometry (CA) study at an applied potential of 1.50 V vs. RHE in Fig. 3i, which shows its outstanding stability, maintaining a current density of 200 mA cm−2 over 120 h without no sign of degradation, as was also observed from the LSV plots before and after the stability test, which exhibited no noticeable variations. This excellent stability indicates the great phase stability of HE-LH, making it a potential electrocatalyst for further exploration in harsher conditions.
 |
| | Fig. 3 OER activity in 1 M KOH electrolyte: (a) LSV polarization curves taken at 2 mV s−1 scan rate. (b) Comparison of overpotential values at 100 and 500 mA cm−2 current densities for different electrocatalysts. (c) Tafel plots derived from LSV polarization curves of various electrocatalysts. (d) Comparison of overpotentials at 10 mA cm−2 current density and Tafel plot between HE-LH and other recently reported transition-metal-based OER electrocatalysts. (e) Nyquist plots of different electrocatalysts. (f) Double-layer capacitance (Cdl) plot of different catalysts. (g) ECSA-normalised specific activity plot for HE-LH and T-LH. (h) Bar diagram showing TOF (s−1) values for all the different catalysts at a potential of 1.5 V vs. RHE. (i) Chronoamperometry tests at a constant potential of 1.50 V vs. RHE (inset: LSV curves before and after stability test). | |
3.3. OER performance in alkaline simulated and real seawater electrolyte media
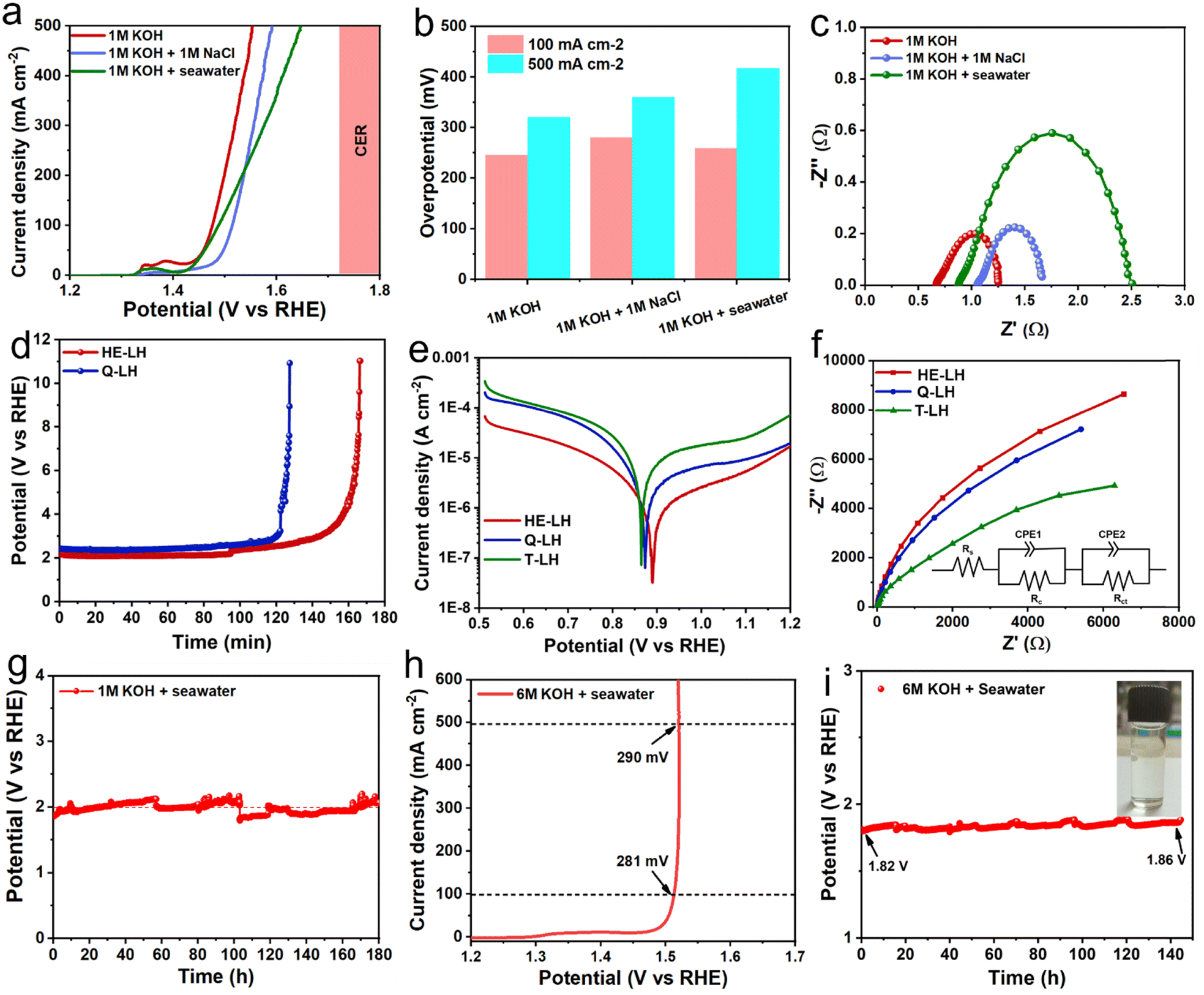
After showcasing the excellent OER activity using freshwater containing 1 M KOH as the electrolyte, the OER activity of HE-LH was further explored in an alkaline simulated seawater electrolyte (1 M KOH + 1 M NaCl) and alkaline real seawater electrolyte. The LSV curves, as shown in Fig. 4a, show the remarkable catalytic activity of HE-LH in chloride-containing simulated- and real-seawater-based electrolytes with η100 values of 280 and 258 mV, respectively, which are not far from overpotentials achieved in freshwater. Importantly, HE-LH reached an industrial-level current density of 500 mA cm−2 at 360 and 417 mV in the 1 M KOH + 1 M NaCl and 1 M KOH + seawater electrolytes, which is well below the value of 480 mV as the limiting overpotential for the CER, as shown in Fig. 4b. However, while the Nyquist plot shown in Fig. 4c indicates comparable Rct values in 1 M KOH + 1 M NaCl (0.68 Ω) and alkaline freshwater (0.58 Ω); in alkaline real seawater, the value is found to be higher (1.67 Ω) due to the presence of multiple interfering elements in seawater. Considering the presence of corrosive chlorides, the durability of the electrocatalyst is a major concern. As mentioned earlier, the introduction of metal cations in bimetallic LDH increases their stability against the chlorides due to electrostatic retardation by interlayer anionic species.29 Based on this context, the ability of HE-LH to retard the chlorides in alkaline saline water was compared with that of Q-LH by performing a chronopotentiometry study at an applied current density of 1000 mA cm−2. To achieve such high current density, in both cases, the potential was maintained at >2 V, which is far beyond the limiting potential of 1.72 V for the CER, as shown in Fig. 4d. Interestingly, in this CER-dominated region, HE-LH was stable for longer than Q-LH and T-LH. Specifically, at an applied current density of 500 mA cm−2, while T-LH survived for 2.5 h and Q-LH for only 5 h, HE-LH exhibited great stability and continued to operate for more than 60 h in the 1 M KOH + 1 M NaCl electrolyte (Fig. S12, ESI†). In comparison, bare Ni foam could not survive even 30 min at a comparatively low current density of 200 mA cm−2, demonstrating the great capability of the developed HE-LH (Fig. S13, ESI†). The superiority of HE-LH compared to Q-LH or T-LH in high-chloride-content electrolyte may originate from the electrostatic repulsion effect on the chlorides due to the presence of large numbers of anionic species, especially carbonates, in interlayers.27
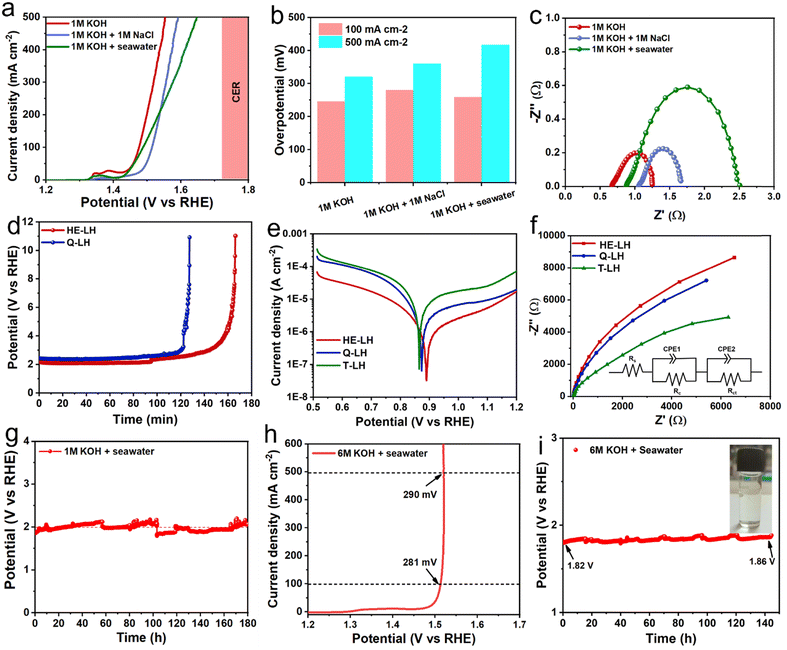 |
| | Fig. 4 OER activity in alkaline seawater electrolyte. (a) Comparison of the LSV plots for HE-LH in different electrolyte media and (b) overpotentials obtained at current densities of 100 and 500 mA cm−2. (c) Nyquist plots obtained at 1.51 V vs. RHE for HE-LH in different electrolytes. (d) Chronopotentiometry study at an applied current density of 1000 mA cm−2 in the 1 M KOH + 1 M NaCl electrolyte. (e) Potentiodynamic polarization plots of HE-LH, Q-LH and T-LH in the 1 M KOH + 1 M NaCl electrolyte for corrosion study. (f) Nyquist plots taken at OCP for corrosion studies using the different electrocatalysts (inset image shows fitted equivalent circuit). (g) Chronopotentiometry study at a constant current density of 500 mA cm−2 for HE-LH in the 1 M KOH + seawater electrolyte. (h) LSV curve for HE-LH in the 6 M KOH + seawater electrolyte and (i) CP test at an operating current density of 500 mA cm−2 (inset: iodometry test). | |
To investigate the anti-corrosion properties of the electrocatalysts, potentiodynamic polarization plots in the 1 M KOH + 1 M NaCl electrolyte were analysed to determine the corrosion potential (Ecorr) and corrosion current (Icorr) values, as presented in Fig. 4e. A higher Ecorr and more negative Icorr indicate better anti-corrosion ability of the electrocatalysts.48,49 In this regard, HE-LH showed a higher Ecorr of 0.89 V and lower Icorr of 2.7 × 10−8 A cm−2 than Q-LH (Ecorr = 0.873 V and Icorr = 6.3 × 10−8 A cm−2) and T-LH (Ecorr = 0.862 V and Icorr = 7.39 × 10−8 A cm−2), as also tabulated in Table S4 (ESI†), indicating better corrosion inhibition properties. The anti-corrosive nature of HE-LH is also reflected by its calculated highest polarization resistance (Rp) value along with lowest corrosion rate compared to the other LHs in high-chloride-content electrolyte. Furthermore, Nyquist plots from EIS spectroscopy taken under OCP conditions (Fig. 5f) were evaluated after fitting with an appropriate electronic circuit to understand the corrosion resistance ability of the electrocatalysts. The important parameters, such as solution resistance (Rs), corrosion resistance (Rc), charge transfer resistance (Rct) and capacitance values of the constant phase elements (Y1, Y2) of the electrocatalysts were determined and are compared in Table S5 (ESI†). The higher Rc and Rct values along with the lower capacitance (Y0) for HE-LH compared to the other LHs further confirms its better corrosion resistance ability.50,51 Owing to these good corrosion resistance properties, the durability of HE-LH in alkaline (1 M KOH) real seawater was extensively examined. The chronopotentiometry (CP) test at an operating current density of 500 mA cm−2 shows stability over 180 h maintaining the oxidation potential around 2 V without much degradation (Fig. 4g). The activity and stability of HE-LH was further examined in highly alkaline (6 M KOH) real seawater conditions, in which the electrocatalyst also delivered great performance, achieving a high current density of 500 mA cm−2 at only 290 mV, which is much smaller than the limiting overpotential value of 480 mV, as shown in Fig. 4h. Consequently, the CP test upon applying such a high current density led to sustainable performance over 140 h during a continuous run (Fig. 4i). An iodometry test was used to test for the possible presence of hypochlorite formed in the electrolyte during the process, and no characteristic yellow coloration was observed, indicating restricted CER.
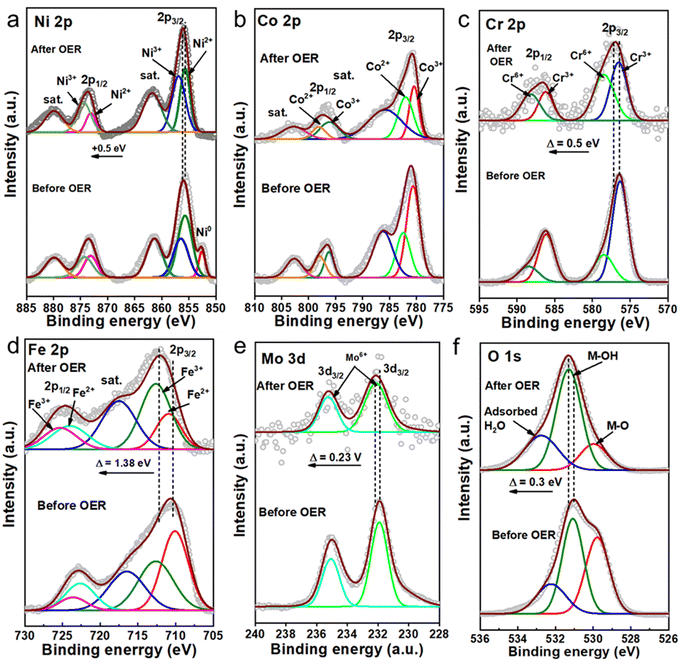 |
| | Fig. 5 High-resolution XPS spectra of Ni 2p (a), Co 2p (b), Cr 2p (c), Fe 2p, (d) Mo 3d, (e) and O1s (f) of low-crystallinity HE-LH before and after the electrochemical activity test. | |
Owing to its great electrocatalytic ability for seawater oxidation, HE-LH was further combined with Pt/C as a cathode in a two-electrode system as HE-LH‖Pt/C, and the performance was compared with that of conventional RuO2‖Pt/C, as shown in Fig. S14 (ESI†). While the conventional combination of RuO2‖Pt/C requires a cell voltage of 1.97 V to reach 100 mA cm−2 current density, HE-LH‖Pt/C needs a cell voltage of only 1.78 V to reach the same in freshwater containing 1 M KOH. The HE-LH‖Pt/C combination also requires considerably lower voltages of 1.80 V and 1.76 V to reach 100 mA cm−2 current density in alkaline (1 M KOH) and highly alkaline (6 M KOH) seawater-based electrolyte. Impressively, the cell combination achieved a 500 mA cm−2 current density at a cell voltage of 1.92 V (iR uncompensated voltage = 2.2 V) and exhibited excellent stability over 50 h without much degradation in performance, as represented in Fig. S14b (ESI†).
To examine and understand possible surface alterations during seawater oxidation, extensive post-characterization of HE-LH was carried out. The FE-SEM image of used HE-LH clearly shows the good retention of the nanosheet morphology after the durability test under harsh conditions, and importantly, the corresponding EDX elemental mapping indicates the presence and homogeneous distribution of all elements (Fig. S15, ESI†). In line with the EDX analysis, the XPS survey for HE-LH after the stability test also revealed the presence of all the elements, as presented in Fig. 5. The high-resolution XPS spectrum for Ni 2p after the stability test reveals the disappearance of Ni0, while the careful deconvolution of the peaks indicates that the Ni3+/Ni2+ ratio also increases from 0.88 to 1.41, as tabulated in Table S6 (ESI†), along with a peak-shift towards higher binding energy by 0.5 eV. The observations indicate the possible formation of NiOOH during the prolonged oxidation mechanism, which is considered to be the active site for electrooxidation, as reported elsewhere.24,33 Like Ni, the core XPS spectrum for Cr 2p also indicates partial transformation from Cr3+ to Cr6+ with an increase in the Cr6+/Cr3+ ratio from 0.39 to 1.04 accompanied by peak shifting towards higher binding energy by 0.5 eV. A similar trend can also be observed for Fe 2p with a significant peak shift towards higher binding energy by 1.38 eV, indicating depletion of electron density from Fe-sites. A slight shift in the peaks corresponding to Mo 3d by 0.23 V towards higher binding energy can also be seen due to long exposure to electrooxidation conditions. The deconvoluted O 1s spectra confirms the presence of M–O, M–OH and adsorbed water and after the prolonged stability test, and the considerable increase in the M–OH/M–O ratio from 1.13 to 3.56 supports the formation of MOOH during the OER.52 The stability of HE-LH in such harsh alkaline seawater was also examined by performing ICP-AES analysis of the electrolyte before and after the stability test (Table S7, ESI†). A trace amount of leaching of Cr and Mo can be identified; the possible formation of CrO42− or MoO42− is considered to be beneficial to achieve sustainable seawater oxidation due to their chloride retardation ability, as also reported elsewhere.53–55 Furthermore, the surface reconstruction of HE-LH was carefully evaluated with the help of surface-sensitive Raman spectroscopy, as represented in Fig. S16 (ESI†). The clear development of strong bands at 480 and 560 cm−1 can be attributed to Eg bending vibration and A1g stretching vibration modes, respectively, for Ni–O in NiOOH.56 The IE1g/IA1g ratio of 1.53 indicates the possible generation of the β-NiOOH phase, which acts as an active phase for the OER mechanism.57,58 Moreover, the broad band in the range of 900–1100 cm−1 was assigned to the NiOO− superoxide species under the positive polarization conditions.59
4. Conclusion
In conclusion, we have described the controlled fabrication of a phase pure high-entropy NiCrCoFeMo layered hydroxide as an efficient and anti-corrosive electrocatalyst that is much superior to its quaternary and ternary layered hydroxide counterparts. The presence of five metals with different valence states offers an increased number of active sites with desirable electronic modulation between them, resulting in better electrocatalytic activity; additionally, the presence of a large number of anionic species in the interlayers enhances the electrostatic repulsion against chlorides, making the material more sustainable than its counterparts during direct seawater splitting. Having these features, the HE-LH electrocatalyst achieved a high current density of 100 mA cm−2 at an overpotential of only 258 mV and exhibited outstanding stability for more than 100 h in alkaline (1 M KOH) and highly alkaline (6 M KOH) real seawater electrolytes. The post-characterization revealed the considerable stability of the electrocatalyst, along with the generation of β-NiOOH as an active phase during the prolonged electrooxidation process in seawater-based electrolyte. Moreover, the in situ formation of CrO42− or MoO42 due to the leaching of trace amounts of Mo and Cr during prolonged stability testing may also provide the much-needed stability in the course of seawater oxidation. Impressively, the combination of HE-LH‖Pt/C in a two-electrode system achieved a 500 mA cm−2 current density at a cell voltage of only 1.92 V in highly alkaline (6 M KOH) seawater electrolyte and exhibited 50 h stability. Based on these capabilities, it is believed that HE-LH has great potential to consider for practical implementation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
P. R., S. P. and S. K. gratefully acknowledge financial support from the Council of Scientific and Industrial Research (HCP44/01) and Department of Science and Technology, India (DST/TMD/HFC/2k18/60) for this work. S. K. thanks University Grant Commission (UGC, India) for providing a fellowship to carry out her PhD program.
References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed
 .
.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed .
- A. Sartbaeva, V. L. Kuznetsov, S. A. Wells and P. P. Edwards, Energy Environ. Sci., 2008, 1, 79 RSC .
- H. Song, M. Wu, Z. Tang, J. S. Tse, B. Yang and S. Lu, Angew. Chem., Int. Ed., 2021, 60, 7234–7244 CrossRef CAS PubMed .
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC .
- N. Sinha and P. Roy, Inorg. Chem., 2023, 62, 3349–3357 CrossRef CAS PubMed .
- S. Pal, T. Ahmed, S. Khatun and P. Roy, ACS Appl. Energy Mater., 2023, 6, 7737–7784 CrossRef CAS .
- C. J. Vörösmarty, P. B. McIntyre, M. O. Gessner, D. Dudgeon, A. Prusevich, P. Green, S. Glidden, S. E. Bunn, C. A. Sullivan, C. R. Liermann and P. M. Davies, Nature, 2010, 467, 555–561 CrossRef PubMed .
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS .
- S. Khatun, H. Hirani and P. Roy, J. Mater. Chem. A, 2021, 9, 74–86 RSC .
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed .
- W. Tong, M. Förster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS .
- C. Das and P. Roy, Chem. Commun., 2022, 58, 6761–6764 RSC .
- H.-J. Liu, R.-N. Luan, L.-Y. Li, R.-Q. Lv, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 461, 141714 CrossRef CAS .
- R.-Y. Fan, J.-Y. Xie, H.-J. Liu, H.-Y. Wang, M.-X. Li, N. Yu, R.-N. Luan, Y.-M. Chai and B. Dong, Chem. Eng. J., 2022, 431, 134040 CrossRef CAS .
- N. Yu, Y. Ma, J.-K. Ren, Z.-J. Zhang, H.-J. Liu, J. Nan, Y.-C. Li, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 478, 147415 CrossRef CAS .
- W. K. Gao, M. Yang, J. Q. Chi, X. Y. Zhang, J. Y. Xie, B. Y. Guo, L. Wang, Y. M. Chai and B. Dong, Sci. China Mater., 2019, 62, 1285–1296 CrossRef CAS .
- Y. Ma, Y. Ma, Q. Wang, S. Schweidler, M. Botros, T. Fu, H. Hahn, T. Brezesinski and B. Breitung, Energy Environ. Sci., 2021, 14, 2883–2905 RSC .
- R. Yao, Y. Zhou, H. Shi, W. Wan, Q. Zhang, L. Gu, Y. Zhu, Z. Wen, X. Lang and Q. Jiang, Adv. Funct. Mater., 2020, 31, 2009613 CrossRef .
- P. Li, Y. Yao, W. Ouyang, Z. Liu, H. Yin and D. Wang, J. Mater. Sci. Technol., 2023, 138, 29–35 CrossRef CAS .
- D. Lai, Q. Kang, F. Gao and Q. Lu, J. Mater. Chem. A, 2021, 9, 17913–17922 RSC .
- X. Zhao, Z. Xue, W. Chen, Y. Wang and T. Mu, ChemSusChem, 2020, 13, 2038–2042 CrossRef CAS PubMed .
- M. Einert, M. Mellin, N. Bahadorani, C. Dietz, S. Lauterbach and J. P. Hofmann, ACS Appl. Energy Mater., 2022, 5, 717–730 CrossRef CAS .
- M. Shi, T. Tang, L. Xiao, J. Han, X. Bai, Y. Sun, S. Chen, J. Sun, Y. Ma and J. Guan, Chem. Commun., 2023, 59, 11971–11974 RSC .
- X. Zhang, Y. Zhao, Y. Zhao, R. Shi, G. I. N. Waterhouse and T. Zhang, Adv. Energy Mater., 2019, 9, 1900881 CrossRef .
- S. Dresp, T. Ngo Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC .
- S. Pal, K. Shimizu, S. Khatun, S. Singha, S. Watanabe and P. Roy, J. Mater. Chem. A, 2023, 11, 12151–12163 RSC .
- S. Khatun and P. Roy, Chem. Commun., 2022, 58, 1104–1107 RSC .
- S. Khatun, S. Pal, N. Sinha, C. Das, T. Ahmed and P. Roy, Chem. Commun., 2023, 59, 4578–4599 RSC .
- F. Wang, P. Zou, Y. Zhang, W. Pan, Y. Li, L. Liang, C. Chen, H. Liu and S. Zheng, Nat. Commun., 2023, 14, 6019 CrossRef CAS PubMed .
- S. Li, L. Tong, Z. Peng, B. Zhang and X. Fu, J. Mater. Chem. A, 2023, 11, 13697–13707 RSC .
- T. X. Nguyen, C.-C. Tsai, V. T. Nguyen, Y.-J. Huang, Y.-H. Su, S.-Y. Li, R.-K. Xie, Y.-J. Lin, J.-F. Lee and J.-M. Ting, Chem. Eng. J., 2023, 466, 143352 CrossRef CAS .
- Y. Ding, Z. Wang, Z. Liang, X. Sun, Z. Sun, Y. Zhao, J. Liu, C. Wang, Z. Zeng, L. Fu, M. Zeng and L. Tang, Adv. Mater., 2023, 2302860 CrossRef PubMed .
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed .
- G. K. Williamson and R. E. Smallman, Philos. Mag., 1956, 1, 34–46 CrossRef CAS .
- S. Raj, S. Anantharaj, S. Kundu and P. Roy, ACS Sustainable Chem. Eng., 2019, 7, 9690–9698 CrossRef CAS .
- S. Khatun, K. Shimizu, S. Singha, R. Saha, S. Watanabe and P. Roy, Chem. Eng. J., 2021, 426, 131884 CrossRef CAS .
- A. Karmakar, K. Karthick, S. S. Sankar, S. Kumaravel, R. Madhu and S. Kundu, J. Mater. Chem. A, 2021, 9, 1314–1352 RSC .
- T. Bhowmik, M. K. Kundu and S. Barman, ACS Appl. Energy Mater., 2018, 1, 1200–1209 CrossRef CAS .
- Y. Li, X. Zhang and Z. Zheng, Small, 2022, 18, 2107594 CrossRef CAS PubMed .
- D. Wang, Q. Li, C. Han, Q. Lu, Z. Xing and X. Yang, Nat. Commun., 2019, 10, 3899 CrossRef PubMed .
- J. Liu, P. Ding, Z. Zhu, W. Du, X. Xu, J. Hu, Y. Zhou and H. Zeng, Small, 2021, 17, 2101671 CrossRef CAS PubMed .
- C.-F. Li, L.-J. Xie, J.-W. Zhao, L.-F. Gu, J.-Q. Wu and G.-R. Li, Appl. Catal., B, 2022, 306, 121097 CrossRef CAS .
- J. Chen, H. Li, S. Chen, J. Fei, C. Liu, Z. Yu, K. Shin, Z. Liu, L. Song, G. Henkelman, L. Wei and Y. Chen, Adv. Energy Mater., 2021, 11, 2003412 CrossRef CAS .
- X. Meng, J. Han, L. Lu, G. Qiu, Z. L. Wang and C. Sun, Small, 2019, 15, 1902551 CrossRef CAS PubMed .
- A. Campero and J. A. D. Ponce, ACS Omega, 2020, 5, 25520–25542 CrossRef CAS PubMed .
- K.-H. Kim, J. W. Choi, H. Lee, B. C. Moon, D. G. Park, W. H. Choi and J. K. Kang, J. Mater. Chem. A, 2018, 6, 23283–23288 RSC .
- T. Chen, H. John, J. Xu, Q. Lu, J. Hawk and X. Liu, Corros. Sci., 2013, 77, 230–245 CrossRef CAS .
- C. Linder, S. G. Rao, R. D. Boyd, A. le Febvrier, P. Eklund, S. Munktell and E. M. Björk, ACS Appl. Energy Mater., 2022, 5, 10838–10848 CrossRef CAS .
- C. Q. Li, D. K. Xu, X.-B. Chen, B. J. Wang, R. Z. Wu, E. H. Han and N. Birbilis, Electrochim. Acta, 2018, 260, 55–64 CrossRef CAS .
- B. G. Prakashaiah, D. V. Kumara, A. A. Pandith, A. N. Shetty and B. E. A. Rani, Corros. Sci., 2018, 136, 326–338 CrossRef CAS .
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed .
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu, Z. Liu, Q. Wang, W. Ren, C. Sun, H.-M. Cheng and B. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed .
- J. Zhou, L. Liu, H. Ren, L. Li, W. Song, N. Li, X. Shi, C. Kou, Y. Sun, M. Han, H. Wang, J. Han, K. Liu, C. D. Momo, Y. Liu, D. Feng, W. Zhu, S. Chen, H. Jiang, Y. Liu and H. Liang, Inorg. Chem. Front., 2024, 11, 498–507 RSC .
- H. Liao, X. Zhang, S. Niu, P. Tan, K. Chen, Y. Liu, G. Wang, M. Liu and J. Pan, Appl. Catal., B, 2022, 307, 121150 CrossRef CAS .
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, Chem. Sci., 2016, 7, 2639–2645 RSC .
- Z. Lin, P. Bu, Y. Xiao, Q. Gao and P. Diao, J. Mater. Chem. A, 2022, 10, 20847–20855 RSC .
- L. Dai, C. Fang, F. Yao, X. Zhang, X. Xu, S. Han, J. Deng, J. Zhu and J. Sun, Appl. Surf. Sci., 2023, 623, 156991 CrossRef CAS .
- Y. Li, Y. Wu, M. Yuan, H. Hao, Z. Lv, L. Xu and B. Wei, Appl. Catal., B, 2022, 318, 121825 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, electrochemical details, plots, comparative tables. See DOI: https://doi.org/10.1039/d3ma00941f |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab