DOI:
10.1039/D4MD00387J
(Research Article)
RSC Med. Chem., 2024,
15, 2718-2728
Chemical synthesis and immunological evaluation of cancer vaccines based on ganglioside antigens and α-galactosylceramide†
Received
24th May 2024
, Accepted 20th June 2024
First published on 21st June 2024
Abstract
iNKT cells – often referred as the “Swiss Army knife” of the immune system – have emerged as central players in cancer vaccine therapies. Glycolipids activating iNKT cells, such as α-galactosylceramide (αGalCer), can enhance the immune response against co-delivered cancer antigens and have been applied in the design of self-adjuvanting anti-tumor vaccines. In this context, this work focuses on the chemical synthesis of ganglioside tumor-associated carbohydrate antigens (TACAs), namely GM3 and (Neu5Gc)GM3 antigens, their conjugation to αGalCer, and their formulation into liposomes as an efficient platform for their in vivo delivery. Liposomes containing GM3–αGalCer, (Neu5Gc)GM3–αGalCer, and equimolar amounts of the two conjugates have been fully characterized and their ability to activate iNKT cell has been confirmed ex vivo in mouse and human cell assays. The candidates were tested in in vivo immunization studies, demonstrating an ability to induce both TH1 and TH2 cytokines leading to the production of all subclasses of IgG antibodies. Notably, the study also demonstrated that serum antibodies raised against the two TACAs, alone and in combination, were cross-reactive. This finding has consequences for future vaccine designs – even if a highly tumor-selective antigen is chosen, the resulting antibody response may be broader than anticipated.
Introduction
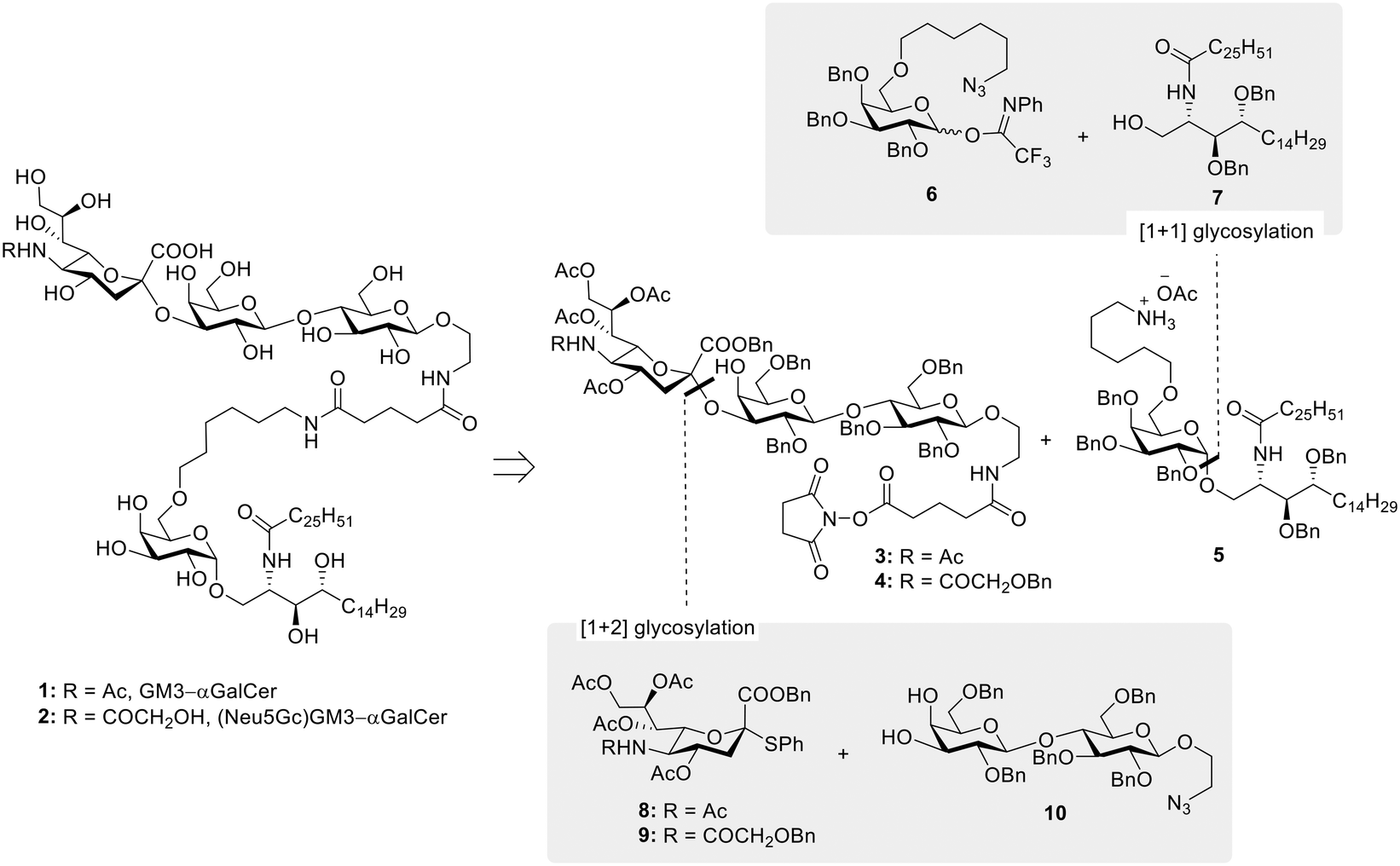
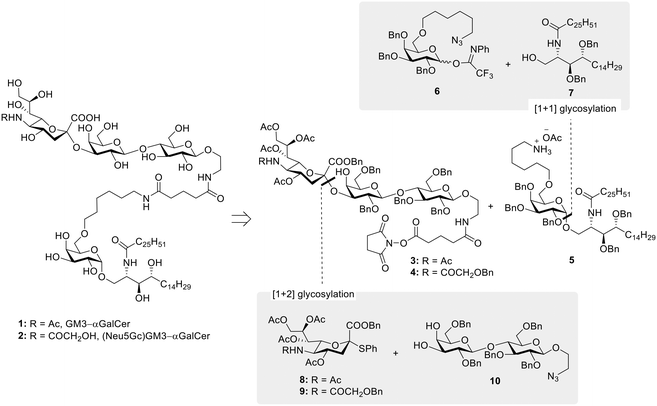
Since their discovery in the early 1990s and the observation of their anti-tumor activity,1,2 invariant natural killer T (iNKT) cells have been considered an attractive target in cancer immunotherapy.3–5 iNKT cells harbor characteristics of both natural killer (NK) and T cells and uniquely place themselves at the interface between innate and adaptive immunity, where they are able to orchestrate the generation of a range of immune responses. Unlike conventional T cells, iNKT recognize glycolipids presented by the non-polymorphic MHC class I-like CD1d protein,6 with α-galactosylceramide (αGalCer) identified as their prototypical agonist.7 To carry out its function, αGalCer first associates with CD1d in antigen-presenting cells (APCs). The αGalCer/CD1d cell surface complex is then engaged by the iNKT T cell receptor (TCR) to form the active TCR/αGalCer/CD1d ternary complex of which the X-ray crystal structures, both mouse and human, were solved in 2005.8–10 CD1d binds αGalCer by hosting its two lipid chains within two hydrophobic pockets (A′ and F′). In this way, the sugar moiety is carefully anchored by a series of intermolecular hydrogen bonds and extends above the surface of the lipid binding grooves for recognition by the TCR of iNKT cells. The binding is controlled by contacts between the α1 CD1d helix and the TCR complementarity determining regions (CDR), which interact with the 3-OH and 4-OH of galactose and establish hydrogen bond networks with the 2-OH of the sugar and the 3′-OH of the phytosphingosine chain.11,12 Stimulated iNKT cells rapidly produce TH1, TH2, and TH17-type cytokines, hence shaping the immune response of other effector cells, i.e. transactivation of NK cells, stimulation of DCs, activation of CD4+ and CD8+ T cells, and maturation of B cells.13 Although promising, the use of αGalCer alone for clinical therapy has not yet been successful due to cytokine antagonism effect and induction of iNKT cell anergy upon repeated αGalCer administration.14 Because of the limited effects of αGalCer in clinical studies, research has also focused on the development of αGalCer analogs with more distinct iNKT cell activating properties – i.e. TH1/TH2 skewing,11 on the optimization of delivery systems – e.g. liposomes,15,16 and on the use of αGalCer as a “universal helper” in vaccine development.17–19 In particular, the adjuvant properties of αGalCer have gained a substantial interest in glycoimmunology as a way to overcome the natural immunotolerance towards carbohydrate antigens. Indeed, in the past decades, the standard design of carbohydrate-based vaccines, especially for infectious diseases, has been based on the conjugation of carbohydrate antigens to immunogenic proteins and/or peptides. These systems are degraded to glycopeptides in APCs and presented via the classical MHC pathway leading to a T cell dependent immune response and the production of high affinity antibodies. While several studies have shown that also tumor-associated carbohydrate antigens (TACAs) can be exploited in the production of protein/peptide-based glycoconjugates as cancer vaccines, their lack of success in the clinic, associated with the known drawbacks related to the use of carrier proteins,20 have prompted the search for novel approaches in carbohydrate-based cancer vaccine design. In this, the conjugation of αGalCer to TACAs has been highlighted as a novel strategy towards fully synthetic cancer vaccines. Notable TACA–αGalCer designs include reports by Yin et al.21 and Broecker et al.22 exploring, respectively, the use of sialyl Tn (sTn) and Tn antigen covalently linked with αGalCer and their formulation into liposomes, to induce robust and specific anti-sTn and anti-Tn IgG antibody responses. More recently, the field of TACA–αGalCer conjugates was also extended to the GM3 ganglioside antigen.23 Specifically, the class of ganglioside TACAs, sialylated glycosphingolipids overexpressed in neuroectoderm-derived cancers (e.g. melanoma, neuroblastoma), often correlates with tumor aggressiveness,24 and has received attention as a valuable immunotherapeutic target for cancer treatment, both in passive and active approaches.25 The GM3 ganglioside has been identified in a number of metastatic cell lines, generally in a higher surface density in comparison to its distribution in normal tissues.26,27 More interestingly, the structurally similar (Neu5Gc)GM3 ganglioside is almost undetectable in normal human tissues, but it is highly expressed in several human cancers,28,29 making it an excellent target for antibody therapies and cancer vaccine development.30–32 This unique characteristic of (Neu5Gc)GM3 is the result of a deletion in the gene encoding the key enzyme for Neu5Gc synthesis (CMP-Neu5Ac hydroxylase),33,34 which has made humans unable to produce Neu5Gc glycoconjugates. The incorporation of Neu5Gc in human cancers is thus attributed to nonhuman Neu5Gc from meat and dairy products.35 With these premises, we have been interested in developing the synthesis of GM3 and (Neu5Gc)GM3 conjugates with αGalCer, with the objective of obtaining fully synthetic vaccine constructs which could be formulated in liposomes, an effective system to ensure co-delivery of antigen and adjuvant to the same APC. Herein we present the straightforward and efficient chemical synthesis of the desired conjugates 1 and 2 (Fig. 1), combining improved approaches in both ganglioside and αGalCer chemistry. Furthermore, we describe the liposomal formulation of our GM3–αGalCer and (Neu5Gc)GM3–αGalCer constructs, and their immunological evaluation in vitro and in vivo.
 |
| | Fig. 1 Global scheme for the synthesis of 1 and 2. | |
Results and discussion
Chemical synthesis of GM3–αGalCer 1 and (Neu5Gc)GM3–αGalCer 2
The synthesis of TACA–αGalCer constructs 1 and 2 required the preparation of the suitably amine-functionalized αGalCer 5 and the NHS-ester equipped GM3 and (Neu5Gc)GM3 gangliosides, compounds 3 and 4, respectively (Fig. 1). αGalCer 5 is functionalized at position C-6′′ with a 6-carbon amino linker, a modification that ensures retention of its adjuvant activity,36 while carrying a handle for derivatization. Similarly, both ganglioside TACAs are equipped with an ethanolamine linker at the anomeric position, further extended by an N-succinimidyl glutarate moiety. Different approaches have been developed previously for the synthesis of αGalCer and its derivatives, specifically addressing the low yield and poor selectivity during glycosylation to introduce the relatively unreactive ceramide. Notably, most successful approaches involve glycosylation with azide-protected phytosphingosine, followed by post-glycosylation acylation to introduce the fatty acid chain.11,37,38 Conversely, protocols which directly react the acylated phytosphingosine generally suffer from low reaction yields and poor α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β selectivities when employing (benzoyl) esters as temporary protecting groups for the phytosphingosine diol,39–41 thus generally requiring the introduction of silyl ether protecting groups.42,43 In our case, the synthesis of the glycolipid was planned to involve direct glycosylation of the acylated phytosphingosine moiety 7, equipped with benzyl ether groups to ensure higher reactivity, while allowing higher synthetic flexibility. Regarding the preparation of ganglioside TACAs, GM3 and (Neu5Gc)GM3, the stereoselective and high yielding synthesis of the trisaccharide scaffold was planned to be achieved by glycosylation of benzyl ether protected lactosyl diol 10 and thioglycoside sialyl donors 8 and 9, respectively (Fig. 1). Specifically, sialyl donors 8 and 9 were synthesized as the less common C-2 benzyl ester derivatives, instead of the C-2 methyl esters, in order to simplify the deprotection of the final conjugates.
:β selectivities when employing (benzoyl) esters as temporary protecting groups for the phytosphingosine diol,39–41 thus generally requiring the introduction of silyl ether protecting groups.42,43 In our case, the synthesis of the glycolipid was planned to involve direct glycosylation of the acylated phytosphingosine moiety 7, equipped with benzyl ether groups to ensure higher reactivity, while allowing higher synthetic flexibility. Regarding the preparation of ganglioside TACAs, GM3 and (Neu5Gc)GM3, the stereoselective and high yielding synthesis of the trisaccharide scaffold was planned to be achieved by glycosylation of benzyl ether protected lactosyl diol 10 and thioglycoside sialyl donors 8 and 9, respectively (Fig. 1). Specifically, sialyl donors 8 and 9 were synthesized as the less common C-2 benzyl ester derivatives, instead of the C-2 methyl esters, in order to simplify the deprotection of the final conjugates.
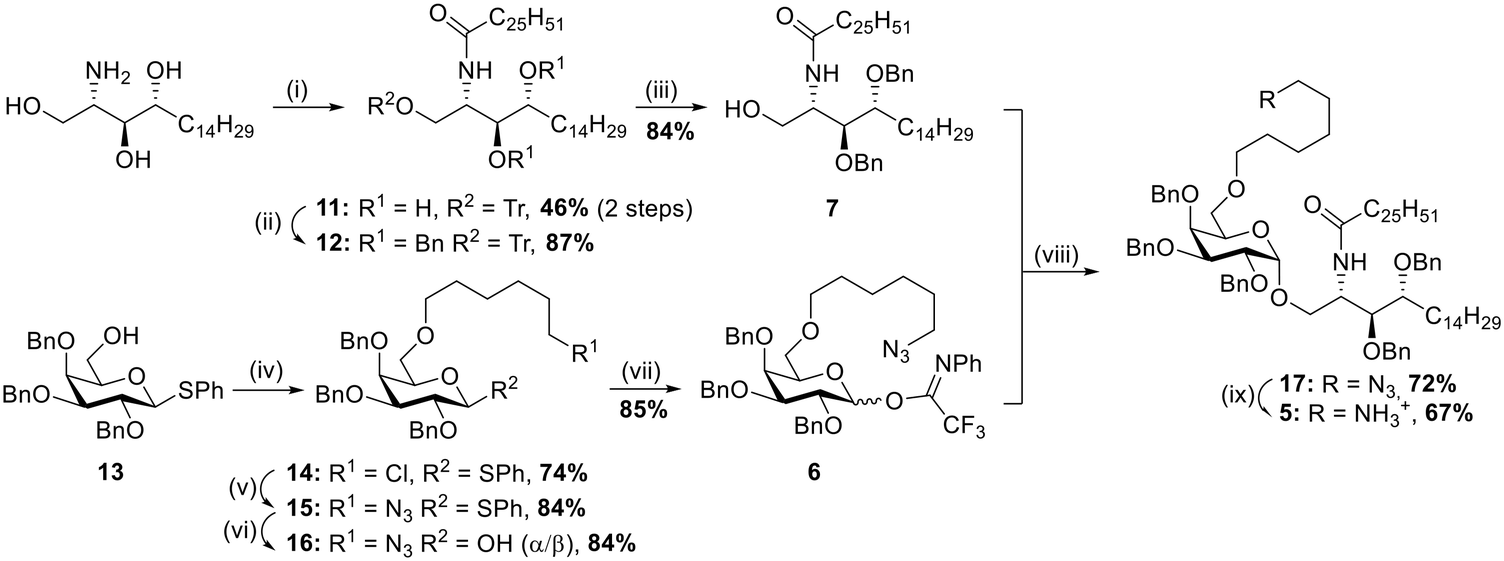
Synthesis of linker equipped αGalCer 5
The synthesis of αGalCer 5 (Scheme 1) started with the preparation of the acylated phytosphingosine moiety. Phytosphingosine was initially treated with N-(hexacosanoyloxy)succinimide44 and Et3N in THF under heating, then the primary OH group was selectively tritylated (trityl chloride, DMAP, pyridine, 80 °C) to afford compound 11 in 46% yield over two steps. To our delight, benzylation of the two free hydroxyl groups on the acylated phytosphingosine scaffold with benzyl bromide and NaH in DMF yielded the desired benzyl protected compound 12 in 87% yield, without formation of the N-benzylated byproduct. To the best of our knowledge, this is the first example of preparation of a benzyl ether protected and acylated phytosphingosine that does not require the use of azide-phytosphingosine or further manipulations of the ceramide scaffold. The synthetic advantage here is threefold: 1) the direct benzylation of the acylated scaffold circumvents unnecessary protection/deprotection steps of the amine functionality, 2) benzyl groups provide a favourable electron-donating character during the glycosylation step, 3) the protecting groups can be easily removed in one final global deprotection step. Removal of the trityl protecting group proceeded uneventfully by treatment with p-toluenesulfonic acid in CH2Cl2/MeOH (→7, 84%). With the desired acylated sphingosine 7 in hand, attention was devoted to the synthesis of the galactoside moiety with the C-6 alkylation of thioglycoside 13 (ref. 45) carried out under basic conditions in the presence of 6-chlorohexyl 4-methylbenzenesulfonate.46 The reaction afforded compound 14 in 74% yield. Nucleophilic substitution to introduce the azide was performed with NaN3 in DMF under heating (→15, 84%). A first glycosylation attempt with thioglycoside 15 and benzylated ceramide 7 was carried out in the presence of NIS/TfOH at −20 °C in THF/Et2O. The reaction yielded the desired product 17 in 77% yield, although as a 2:1 α:β mixture. While the procedure allowed access to the desired αGalCer scaffold, a more efficient glycosylation was investigated. Namely, 15 was converted to its corresponding N-phenyl 2,2,2-trifluoroacetimidate donor47 by hydrolysis of the thioglycoside moiety under standard conditions (NBS, acetone/H2O, →16, 84%) and then reaction with 2,2,2-trifluoro-N-phenylacetimidoyl chloride in the presence of Cs2CO3 (→6, 85%). Gratifyingly TMSOTf-promoted glycosylation with glycosyl donor 6 and acceptor 7 at −20 °C and in THF/Et2O afforded the desired product 17 in 72% yield and with complete α-stereoselectivity. The developed strategy thus allowed for the easy and straightforward formation of a fully protected αGalCer bearing an important functionalization handle (i.e. terminal azide) in only 9 reaction steps and in a global yield of 11%. Further derivatization of compound 17 was achieved by reduction of the azide by treatment with zinc under acidic conditions, affording αGalCer 5 in 67% yield.
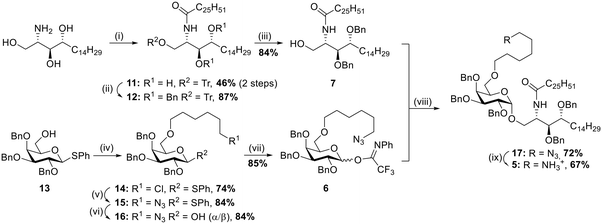 |
| | Scheme 1 (i) a. Hexacosanoic acid-NHS ester, Et3N, THF, 50 °C, o/n, b. TrCl, pyridine, DMAP, 80 °C, o/n; (ii) BnBr, NaH (60% dispersion in mineral oil), TBAI, anhydr. DMF, 0 °C to RT, 2 h; (iii) p-TsOH, MeOH, 3 h; (iv) 6-chlorohexyl 4-methylbenzenesulfonate, NaH (60% dispersion in mineral oil), anhydr. DMF, 0 °C to RT, 3.5 h; (v) NaN3, DMF, 80 °C, o/n; (vi) NBS, acetone/H2O, 0 °C, 1 h; (vii) 2,2,2-trifluoro-N-phenylacetimidoyl chloride, Cs2CO3, anhydr. CH2Cl2, RT; (viii) TMSOTf, anhydr. THF/Et2O, −20 °C, 1 h; (ix) Zn, AcOH, CH2Cl2, RT, 45 min. | |
Synthesis of TACAs derivatives 3–4 and conjugation to αGalCer
To access the desired TACA–αGalCer derivatives 3 and 4, linker equipped-lactose 10 (ref. 48) was straightforwardly prepared in 7 reaction steps and in 22% global yield following literature procedures. Sialyl donor 8 was also prepared in 4 steps (38% global yield, ESI†) exclusively as the α-anomer with the key reaction being the SN2-like substitution of the corresponding glycosyl chloride with thiophenol under basic conditions. Conversely, sialyl donor 9 was synthesized from 8. First, deacetylation and amide hydrolysis were achieved by treatment with methanesulfonic acid under reflux, then the crude mixture was treated with readily synthesized O-benzylglycolic acid succinimidyl ester under basic conditions. Finally, the obtained compound was acetylated under standard conditions (Ac2O, pyridine) to afford sialyl donor 9 in 56% over three steps (Scheme 2). Sialyl donors 8 and 9 were reacted with lactose acceptor 10 in the presence of IBr/AgOTf in CH3CN/CH2Cl2 at −78 °C and −40 °C,49 respectively. Both glycosylations afforded the desired products 18 and 19 in high yields, 85% and 71% respectively, with complete stereo- and regioselectivity. The developed protocols for accessing the GM3 and (Neu5Gc)GM3 scaffolds also reduced the number of glycosylation steps to only one by making use of a suitably protected lactose building block, as opposite to recent reports involving more lengthy approaches.23 Moreover, both glycosylation reactions proceeded in high yields even in the presence of the natural acetamide functionality on the sialic acid donor, thus greatly simplifying the number of manipulations required following glycosylation. Selective reduction of the azide moiety proceeded smoothly when both compounds 18 and 19 were treated with zinc under acidic conditions and subsequently reacted with readily prepared disuccinimidyl glutarate50 in the presence of Et3N to afford the N-hydroxylsuccinimide functionalized derivatives 3 and 4 in 79% and 67% yield, respectively. Conjugation with αGalCer 5 proceeded uneventfully and promoted by the addition of Et3N (→20, 74%; →21, 80%) to yield the desired GM3 and (Neu5Gc)GM3 scaffolds covalently linked to αGalCer. While initial deprotections were carried out by first removing the acetyl ester groups under Zemplén conditions (MeONa, MeOH) and subsequent removal of the benzyl ether and benzyl ester groups by hydrogenolysis (H2, 5% Pd/C), it was later found that inverting the order of the reactions afforded cleaner reactions and higher yield, allowing the isolation of compounds 1 and 2 in 88% and 85% yield, respectively.
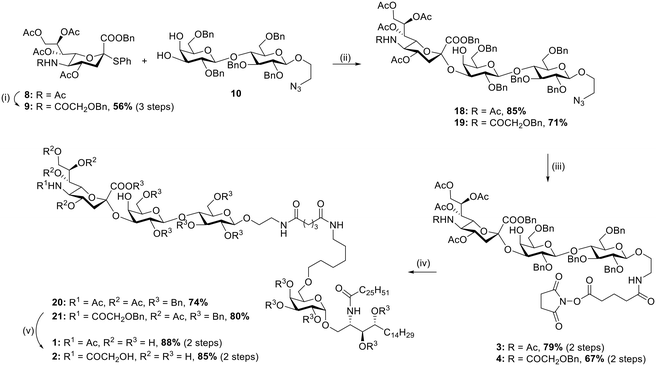 |
| | Scheme 2 (i) a. MsOH, anhydr. MeOH, 75 °C, o/n, b. O-benzylglycolic acid NH-ester, CH3CN/H2O, RT, 2 h, c. Ac2O, pyridine, RT, o/n; (ii) IBr/AgOTf, anhydr. CH3CN/CH2Cl2, 3 Å MS, −78 °C for 18, −40 °C for 19, 2 h; (iii) a. Zn, AcOH, anhydr. CH2Cl2, RT, 1 h, b. disuccinimidyl glutarate, anhydr. DMF, Et3N, RT, 2 h; (iv) 5, Et3N, anhydr. DMF, RT, 4 h; (v) a. H2, Pd/C (5% wt), AcOH, EtOH, RT, o/n, b. MeONa, MeOH, H2O, RT, o/n. | |
Formulation in liposomes
Conjugates 1 and 2 were formulated as liposomes. While presenting the GM3 and (Neu5Gc)GM3 TACAs in a multivalent manner, this type of formulation can itself contribute in shaping the immune response in vivo: when administered to mice (subcutaneous (sc) injection or oral uptake) smaller lipid vesicles (<150 nm) tend to promote the development of a TH2 response, whereas larger lipid vesicles (>200 nm) shift the response towards the production of IFN-γ, a typical TH1 response.51,52 The different activity is proposed to be related to differences in the trafficking of the vesicles by APCs, with small liposomes being transported to the late endosomes and larger ones to early endosomes. Size-defined liposomes containing either conjugate 1 (I), conjugate 2 (II), equimolar amounts of 1 and 2 (III), equimolar amounts of the GM3 ganglioside and αGalCer (IV), or exclusively αGalCer (V) were prepared by lipid extrusion and subsequently characterized (ESI†). The liposomes contained 1,2-diastearoyl-sn-glycero-3-phosphocholine (DSPC) and cholesterol (Chol) (Table 1) and were extruded through 200 nm polycarbonate filters. The physiochemical properties of each liposomal formulation were evaluated by dynamic light scattering (DLS) confirming a homogeneous particle population (approx. size of 200 nm) and small polydispersity index (PDI), and by zeta potential measurement, showing a negative surface charge in all groups I–IV. The contents of the liposomes were further analyzed by RP-HPLC-MS/MS to evaluate the recovery of each individual component. This ensures that the same amount of active ingredient is administered in each assay when comparing liposomes I–V.
Table 1 Composition of liposomes I–V
|
|
Compound(s) |
Formulation |
Mol ratio |
|
I
|
GM3–αGalCer (1) |
DSPC:Chol:1 |
58.2:38.9:2.9 |
|
II
|
(Neu5Gc)GM3–αGalCer (2) |
DSPC:Chol:2 |
58.2:38.9:2.9 |
|
III
|
Equimolar mix of (1) and (2) |
DSPC:Chol:1:2 |
56.2:38:2.9:2.9 |
|
IV
|
GM3 and αGalCer |
DSPC:Chol:GM3:αGalCer |
56.2:38:2.9:2.9 |
|
V
|
αGalCer |
DSPC:Chol:αGalCer |
58.2:39.0:2.9 |
Immunological evaluation
The liposomes activate murine and human DCs and consequently induce iNKT proliferation ex vivo.
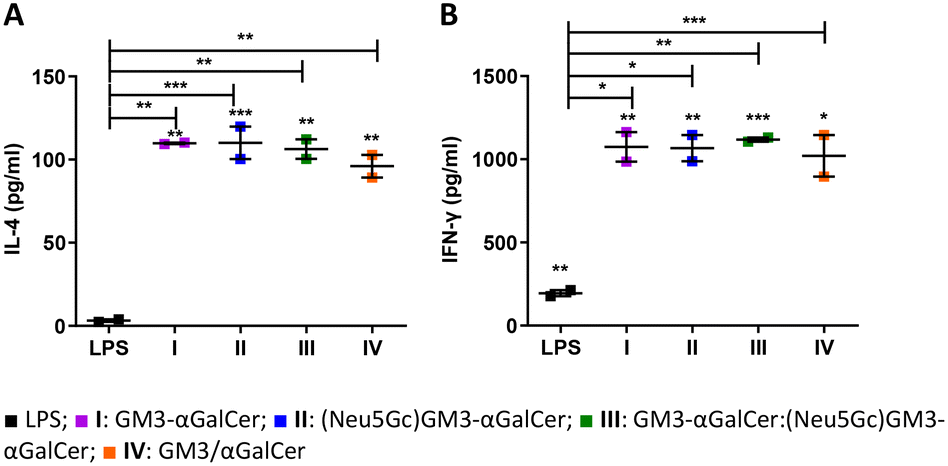
Copious amounts of cytokines are produced by iNKT cells within hours following activation. This includes an initial burst in IL-4 production followed by IFN-γ.53 Moreover, iNKT cells constitutively express receptors for several cytokines such as IL-12,54 making them ready to quickly respond to activated APCs. The potency of liposomes I–IV was initially investigated ex vivo. Murine bone marrow-derived dendritic cells (BMDCs) were first pulsed with either I–IV or LPS, co-cultured with iNKT cells and then incubated for 48 hours. A significant increase in IL-4 and IFN-γ was detected when DCs and iNKT cells were activated with the liposomal formulations (Fig. 2A and B).
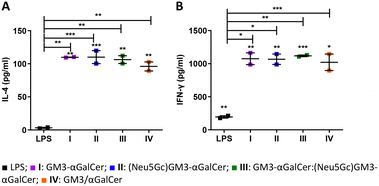 |
| | Fig. 2
Ex vivo production of IL-4 and IFN-γ induced by the vaccine candidates in co-culture supernatant (A and B). BMDCs were isolated from naïve mice and treated with either 100 ng mL−1 LPS or 10 ng mL−1I–IV. Thereafter, isolated iNKT cells were added to the culture. Secretion of cytokines in the supernatant was measured by cytometric bead array (CBA). | |
Furthermore, iNKT cells that were co-cultured with DCs pulsed with I–IV produced significantly higher levels of both IL-4 and IFN-γ compared to the co-culture with LPS-activated DCs. This further confirms the potential of I–IV in activating an immune response.
Afterwards, we sought to determine whether the liposome formulations I–V were also able to activate human iNKT cells ex vivo. MUTZ3-derived human immature dendritic cells55,56 were matured in the presence of I–V before co-culturing with human T cells (Fig. 3A–C and S7†). DCs matured in the presence of I–III showed increased expression of CD1d, while no change was observed in the presence of groups IV and V. Expression of the maturation markers CD83, HLA-DR, CD209, and CD1a was induced to similar levels on all matured DCs irrespective of the presence of liposomes (Fig. 3, panel A and S7†). Increased frequencies of iNKT cells were detected in samples co-cultured with DCs primed with III and, to a lesser extent, I compared to II, IV, and V (Fig. 3B and C). Thus human iNKT cells proliferate extensively in response to activation with the GM3–αGalCer conjugate (I) and in particular the combination of GM3–αGalCer and (Neu5Gc)GM3–αGalCer (III), while stimulation with the (Neu5Gc)GM3–αGalCer (II) conjugate alone or the individual unconjugated components (IV and V) appear less effective.
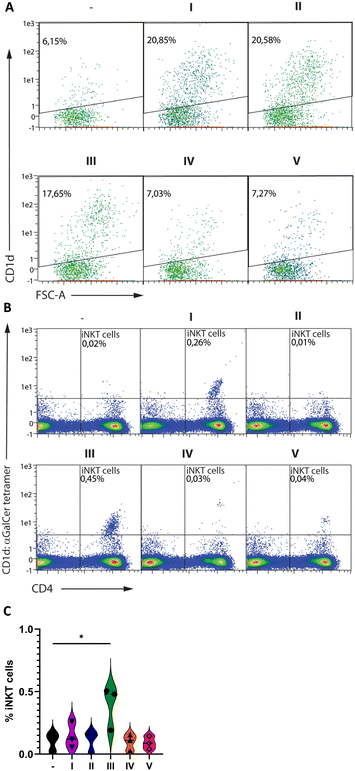 |
| | Fig. 3 Maturation of human DCs and activation of human iNKT cells (A–C). Human MUTZ-3-derived DCs were primed in the presence or absence of the liposomal formulations I–V before analysis by flow cytometry (A) or co-culture with human CD3+ lymphocytes and subsequent analysis for iNKT cells by flow cytometry (B and C). For all data, gating was performed on singlets and viable cells (based on staining with live/dead yellow – Fig. S6†). A and B show representable dot plots from three independent experiments, while C summarizes the percentage of positive cells from three independent experiments using cells from different donors. | |
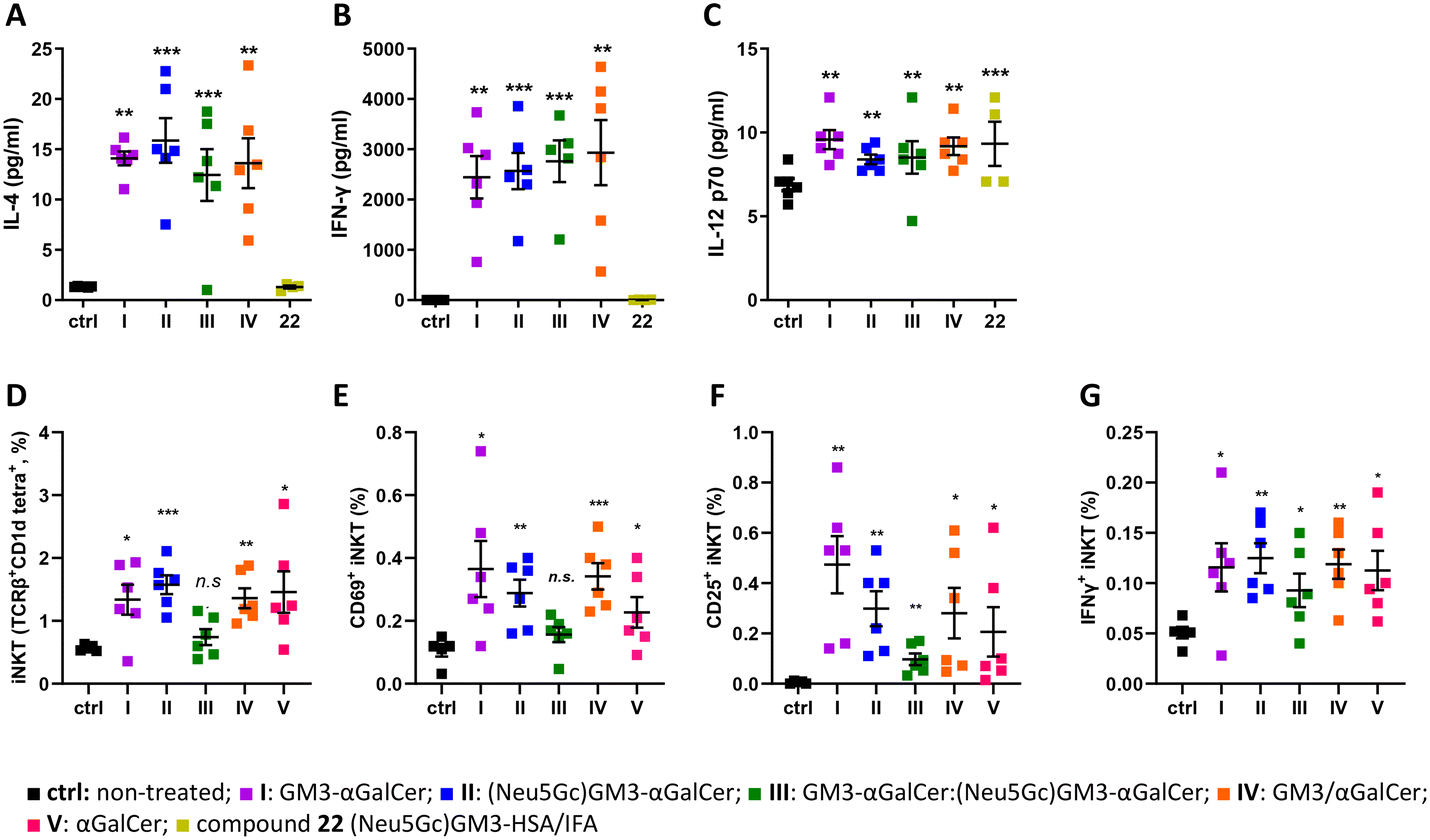
The liposomes induce cytokines, iNKT numbers, and iNKT activation in vivo.
Next, we set out to determine whether liposomes I–IV promote cytokine production in vivo. The levels of IL-4, IFN-γ, and IL-12p70 were evaluated following the administration of liposomes in mice. Groups of six female C57BL/6 mice were immunized sc with liposomes I–V and with 22, a glycoconjugate composed of the (Neu5Gc)GM3 antigen and the carrier protein HSA, namely (Neu5Gc)GM3–HSA (ESI†), emulsified in incomplete Freund's adjuvant (IFA). To evaluate the production of IL-4, IFN-γ, and IL-12p70 cytokines in response to administration of the liposomes, sera samples were collected 24 hours after injection for groups I–IV and 22. All groups of mice except for the one treated with (Neu5Gc)GM3–HSA 22 produced high levels of all three cytokines compared to the untreated control mice, indicating a rapid activation of iNKT cells (Fig. 4A–C). The levels of cytokines were not different between groups I–IV. All groups of mice induced the production of higher levels of IFN-y, IL-4 and IL-12p70 which indicates that the vaccine candidates can promote a cytokine milieu leading to the activation of both TH1 and TH2 responses. Low levels of IL-4 and IL-12p70 are presumably due to different in vivo kinetics of these two cytokines compared to IFN-γ following immunization. In addition to the production of cytokines, the in vivo response of iNKT cells is characterized by the induction of a variety of activation markers such as CD25 and CD69.57 Mice splenocytes were analyzed by flow cytometry 48 hours after administration of liposomes I–V to evaluate the effect of the liposome on iNKT cells in vivo. iNKT cells were identified by staining with anti-TCRβ antibody and mCD1d:αGalCer tetramer. The TCRβint mCD1d:αGalCer tetramer+ cells were co-stained with anti-CD69, CD25, and IFN-γ mAbs to identify subpopulations of activated iNKT cells (ESI,† Fig. S3). All groups of mice that were treated with liposomes I–V showed an increase in expression of CD25 and CD69 on iNKT cells and intracellular levels of IFN-γ, suggesting that a considerable proportion of iNKT cells had undergone the activation pathway (Fig. 4D–G). Although administration of III did not result in a significant increase in the percentage of splenic iNKT cells, the increase in CD69, CD25, and IFN-γ expression on cells suggests that III generates cytokine production but do not stimulate iNKT cells activation and proliferation at 48 hours post-administration to the extent observed for the other liposome formulations. Understanding the optimal interaction between iNKT cells and immune stimulating liposomes is therefore important to further elucidate. Comparing the iNKT cell expansion in the human ex vivo (Fig. 3) and mouse in vivo (Fig. 4) systems, an interesting difference is revealed; while I and III showed higher expansion of human iNKT cells than II, the opposite trend was observed in the mouse system. Furthermore, no overall difference in the efficiency of the conjugates (I–III) compared to the unconjugated components (IV) or αGalCer alone (V) was observed for the activation of mouse iNKT cells, indicating that while murine iNKT cell stimulation by these liposomal formulations is mainly driven by αGalCer, the sugar antigens affect stimulation of the human iNKT cells. Previous reports have also highlighted how the TCRs of mouse and human iNKT cells have different fine specificities that happen to translate to differences in iNKT cell activation and proliferation, encouraging further investigation of the difference between human and mouse iNKT cells.
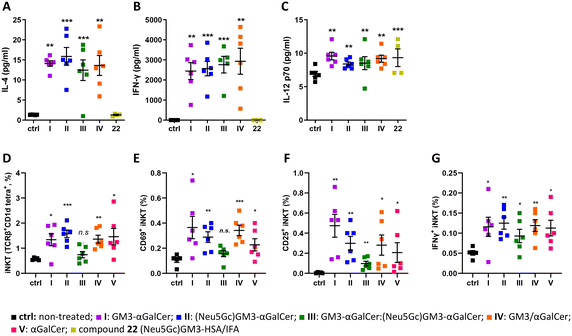 |
| | Fig. 4 (A–C) Serum concentrations of cytokines evaluated by CBA 24 hours after the first injection. Data are indicated as the average value ± SEM. Asterisks without brackets show significant difference to the untreated control group using one-way ANOVA followed by Dunnett's multiple comparison test. Asterisks with brackets indicate significant differences to the LPS group using an unpaired two-tailed Student's t-test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. (D–G) Liposomes I–V induced generation and activation of splenic iNKT cells evaluated at 48 hours after the first injection by flow cytometry. (D) Percentage of iNKT cells (TCRβint CD1d:αGalCer tetramer+) among total splenic cells and (E–G) absolute numbers of splenic iNKT cells with the activated phenotype (CD25+, CD69+, IFN-γ+). Data are indicated as the average value ± SEM of 6 mice per group. Asterisks show significant difference to placebo-treated mice using one-tailed unpaired Welch's t test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. | |
The liposomes induce antibody responses.
Groups of six female C57BL/6 mice were immunized sc with liposomes I–V at biweekly intervals (days 1, 15, 29) and sera were collected on days 0, 14, 28, and 42 (ESI,† Fig. S2). An additional group was instead immunized with glycoconjugate 22 emulsified in IFA as a vaccine control. The conjugation of carbohydrate antigens to protein carriers is a well-established strategy for overcoming their poor immunogenicity through the MHC presentation of glycopeptide epitopes to CD4+ T cells.58 Group 22 followed the same immunization schedule as the groups treated with liposomes I–IV. Moreover, one group of mice remained untreated throughout the experiment to serve as negative control. First, it was examined whether the immunization with I–IV was capable of stimulating B cells to produce anti-GM3 or anti-(Neu5Gc)GM3 antibodies. IgM and IgG antibody responses were measured by ELISA using (Neu5Gc)GM3–HSA 22 or GM3–HSA 23 as coating antigens (ESI†). Mice immunized with I–IV generated carbohydrate-specific antibodies of the IgM class (Fig. 5A and C). With respect to IgG, all vaccine candidates efficiently generated an anti-IgG response, which gradually increased following the course of the experiment (Fig. 5B and D). The IgM and IgG levels on specified weeks are shown in Fig. S4 (ESI†). The decrease in levels of IgM following the second and third immunization moves in parallel with the gradual increase in levels of IgG, indicating an induction of isotype switching. Moreover, the immunization protocol induced an IgG response which remained high two weeks after the last injection. Mice immunized with (Neu5Gc)GM3–HSA 22 produced the highest levels of IgM and IgG as a result of activation of the helper T cell pathway due to its glycoprotein nature and the recognized effect of IFA on shaping the immune response.59 The IgG response following the second and third immunizations with I–III was more pronounced compared to group IV, which might be the result of the covalent bond between TACAs and αGalCer in the former. Moreover, both the IgM and IgG antibodies generated following immunization with formulations I and IV, containing GM3 only, cross-reacted with the (Neu5Gc)GM3–HSA coated ELISA microplates (Fig. S4†). The same cross-reactivity was detected for the antibodies produced in mice immunized with formulations II and III, presenting the (Neu5Gc)GM3 antigen, which were found to bind to the GM3 antigen in ELISA (Fig. S4†). The isotype distribution of IgG antibodies was also investigated. In mice, TH2-type responses mainly induce the generation of IgG1, while TH1 ones favor isotype switching to IgG3.60 Mice immunized with liposomes I–III produced higher levels of anti-GM3 and anti-(Neu5Gc)GM3 IgG3 and IgG2b while displaying minimal levels of IgG1 and IgG2a (Fig. 5E–L). A comparable magnitude of switching was recently reported in a similar study23 and can partly be explained by the suppression of the TH2-like response of iNKT cells by the GM3 ganglioside.61 Although the highest levels of IgG1 and IgG2a were detected in mice immunized with the conventional glycoconjugate 22, there was a low isotype switching to IgG3 and IgG2b. This pattern of class switching following injection of a glycoprotein conjugate with IFA is likely due to the presence of IFA and the accompanying cytokine milieu.
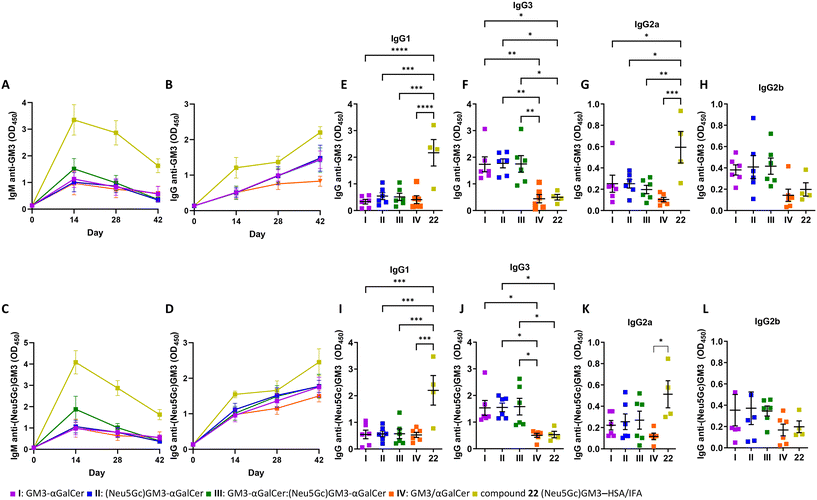 |
| | Fig. 5 Stimulation of antibody production and isotype by liposomal formulations I–IV and 22. The GM3 (A and B) and (Neu5Gc)GM3 (C and D) IgM and IgG responses on day 14, 28 and 42 after immunization. Data represents mean value ± SEM, n = 6 for I–IV and 4 for 22 emulsified in IFA. The GM3 (E–H) and (Neu5Gc)GM3 (I–L) specific IgG subtypes at day 42 as measured by ELISA. Mice were immunized sc at biweekly intervals and sacrificed on day 42. Data are represented as the average value ± SEM in each group. n = 6 for I–IV and 4 for 22 emulsified in IFA. | |
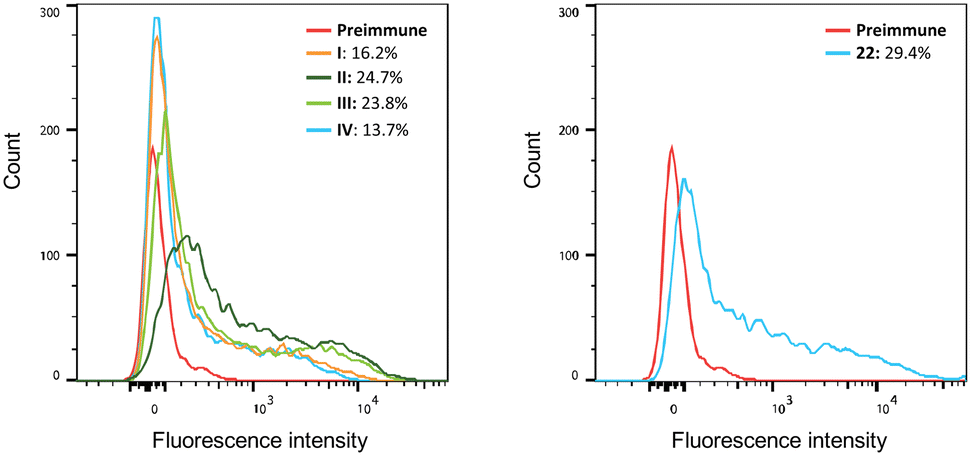
The ability of antisera prepared on day 42 to bind to the B16F10 cell line, known to express the GM3 antigen,62 was determined by flow cytometry. Serum obtained from mice vaccinated with all vaccine candidates displayed increased binding levels to B16F10 cells (Fig. 6). Immunization with liposomes I–III led to higher binding capacities (positive cells: 16.2%, 24.7%, and 23.8%, respectively) compared to group IV containing non-conjugated GM3 ganglioside and αGalCer (13.7% positive cells). A 29.4% binding was detected in sera obtained from mice vaccinated with the protein conjugate 22. Finally, the ability of antisera obtained by immunization to activate the complement system on the surface of B16F10 cells was evaluated. The acute monocytic leukemia THP-1 cell line that does not express GM3 and (Neu5Gc)GM3 antigens was used as the negative control. B16F10 cells were first incubated with the antisera prepared on day 42. Next, complement protein was added to the cells and the percentage of dead cells was determined. The complement cascade was efficiently activated on the surface of B16F10 cells resulting in their killing (ESI,† Fig. S5). This result demonstrates that the antibodies produced in the immunized mice are capable of specifically binding to the surface of B16F10 cells and not to a cell line such as THP-1 which lacks expression of the specific ganglioside TACAs GM3 and (Neu5Gc)GM3.
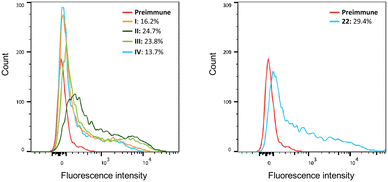 |
| | Fig. 6 Binding of serum obtained from immunized mice to B16F10 cells. Pooled sera obtained from all groups of mice were incubated with B16F10 cells, and PE-conjugated anti mouse IgG was used for the detection of the binding using flow cytometry. Serum of mice before the first immunization (pre-immune) was used as the negative control. | |
Conclusions
An efficient synthetic strategy for the preparation of ganglioside–αGalCer conjugates has been developed. In particular, a straightforward approach was applied to the synthesis of linker-functionalized αGalCer, including a key stereoselective glycosylation step with the notoriously unreactive ceramide moiety. In the same way, the desired ganglioside TACAs, namely GM3 and (Neu5Gc)GM3 were obtained in high yields and efficiently conjugated to αGalCer. Contrarily to recent approaches23 the developed methodology for accessing these compounds requires only one glycosylation step and, at the same time, ensures α-selectivity.
The liposomal formulations elicited strong and consistent production of IgM and IgG antibodies. The observed isotype switching to IgG in immunized mice is presumably due to the help provided by iNKT cells and not helper T cells because the formulations did not contain any helper peptide epitope and it is well established that iNKT cells can help B cells initiate antibody responses, affinity maturation, and isotype switching.15 Moreover, the antibodies exhibited binding to the B16F10 cell line and efficient activation of the complement system on its surface.
We showed that our liposomal formulations induced production of both TH1- and TH2-associated cytokines such as IFN-γ and IL-4, leading to the production of all subclasses of IgG antibodies in mice. Moreover, data using human DCs, indicate that the liposome formulations with ganglioside–αGalCer conjugates are resulting in enhanced maturation of human DC, whereby CD1d is strongly upregulated, while the non-conjugated GM3 with αGalCer does not lead to increased CD1d. Due to distinct fine specificities of the iNKT TCR–agonist–CD1d interaction in mice and humans, differences are often observed in the activation ad proliferation of mouse and human iNKT cells when stimulated with α-GalCer analogues.19 In light of this, the results with human DCs and iNKT cells are encouraging with regards to the possibility of translating the ganglioside–αGalCer liposomal platform from a mouse model to the human setting. In future studies, it could be of interest to investigate the apparent differences in iNKT cell activation by the different vaccine constructs in murine and human DC cells, including the the role played by the sugar antigen processing in the human immune response. The current data highlights the importance on human test systems for translation of iNKT targeting glycolipid cancer vaccines. These results further emphasize the potential of the synthesized constructs in shaping the immune response and opens new venues for designing a variety of vaccine candidates by introducing elements which can be used to redirect the therapy towards either activation or suppression of the immune system.
The serum antibodies raised against the two TACAs, GM3 and (Neu5Gc)GM3, were cross-reactive. While generally considered highly specific for their designated antigen, previous screenings of 27 anti-glycan antibodies and 80 different glycans (and glycoproteins) have highlighted the cross-reactivity of several antibodies, which were thought to be highly selective.63 The cross-reactivity of anti-GM3 and anti-(Neu5Gc)GM3 antibodies, raised by the presentation of the carbohydrate epitopes of the GM3 and (Neu5Gc)GM3 TACAs, is thus not completely surprising. This finding is an important element to consider in future carbohydrate-based cancer vaccine designs as the antibody responses might be broader than anticipated even when highly tumor-selective TACAs epitopes are employed.
Ethics
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of the Technical University of Denmark and approved by the Animal Ethics Committee of the the Danish Animal Experiments Inspectorate.
Author contributions
Conceptualization: C. R. and M. H. C.; experiment design: C. R., M. H. C., S. G., A. E. H., J. R. H., D. C., and T. L. A.; synthesis and characterization: C. R. and H. J.; liposomes fabrication and characterization: S. T., P. W., U. B. K., G. C.; immunological evaluation (mice) and data analysis: S. T. and P. W.; immunological evaluation (human iNKT cells) and data analysis: S. L. S., A. L. B., L. H., and S. G.; writing original draft: C. R.; manuscript review & editing: M. H. C., S. T., G. C., S. L. S., A. L. B., and L. H., S. G., D. C.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. R. acknowledges financial support from the European Commission's ARGONAUT project (grant agreement no. 841278) and Lundbeck Foundation Post-Doc Fellowship (grant no. R347-2020-2307). M. H. C. and C. R. acknowledge financial support from the Novo Nordisk Foundation (grant no. NNF18OC0053048 and NNF20OC0065094) and the Danish Council for Independent Research (grant no. 9041-00080B). Vaccine research in the M. H. C. laboratories is further supported by grants from DTU (PoC) and SPARK Denmark. S. G., S. L. S, L. H., and A. L. B. acknowledge financial support from the Novo Nordisk Foundation (grant no. NNF19SA0056783, NNF19SA0035474, NNF20SA0066621, and NNF21SA0072683). P. W. acknowledges financial support from the China Scholarship Council. The authors thank Chloé Stoll, Emilie Ljungberg, Helena Prpić, Juliane Sørensen, Kirstine Bækgaard Andersen, Ninon Cholvy, Olivia Mathilde Ottendal Bartram, and Sajedeh Alavi, for the large-scale synthesis of carbohydrate building blocks during their internships in the laboratory.
References
- T. Kawano, J. Cui, Y. Koezuka, I. Toura, Y. Kaneko, H. Sato, E. Kondo, M. Harada, H. Koseki, T. Nakayama, Y. Tanaka and M. Taniguchi, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5690–5693 CrossRef CAS PubMed.
- O. Lantz and A. Bendelac, J. Exp. Med., 1994, 180, 1077–1106 CrossRef PubMed.
- Y. Zhang, R. Springfield, S. Chen, X. Li, X. Feng, R. Moshirian, R. Yang and W. Yuan, Front. Immunol., 2019, 10, 11–15 CrossRef PubMed.
- L. A. King, R. Lameris, T. D. de Gruijl and H. J. van der Vliet, Front. Immunol., 2018, 9, 1–7 CrossRef PubMed.
- M. Bedard, M. Salio and V. Cerundolo, Front. Immunol., 2017, 8, 1–12 Search PubMed.
- M. Brigl and M. B. Brenner, Annu. Rev. Immunol., 2004, 22, 817–890 CrossRef CAS PubMed.
- E. Kobayashi, K. Motoki, T. Uchida, H. Fukushima and Y. Koezuka, Oncol. Res., 1995, 7, 529–534 CAS.
- D. M. Zajonc, C. Cantu, J. Mattner, D. Zhou, P. B. Savage, A. Bendelac, I. A. Wilson and L. Teyton, Nat. Immunol., 2005, 6, 810–818 CrossRef CAS PubMed.
- M. Koch, V. S. Stronge, D. Shepherd, S. D. Gadola, B. Mathew, G. Ritter, A. R. Fersht, G. S. Besra, R. R. Schmidt, E. Y. Jones and V. Cerundolo, Nat. Immunol., 2005, 6, 819–826 CrossRef CAS PubMed.
- N. A. Borg, K. S. Wun, L. Kjer-Nielsen, M. C. J. Wilce, D. G. Pellicci, R. Koh, G. S. Besra, M. Bharadwaj, D. I. Godfrey, J. McCluskey and J. Rossjohn, Nature, 2007, 448, 44–49 CrossRef CAS PubMed.
- A. Banchet-Cadeddu, E. Hénon, M. Dauchez, J.-H. Renault, F. Monneaux and A. Haudrechy, Org. Biomol. Chem., 2011, 9, 3080–3104 RSC.
- E. Girardi and D. M. Zajonc, Immunol. Rev., 2012, 250, 167–179 CrossRef PubMed.
- V. Cerundolo, J. D. Silk, S. H. Masri and M. Salio, Nat. Rev. Immunol., 2009, 9, 28–38 CrossRef CAS PubMed.
- V. V. Parekh, S. Joyce, L. Van Kaer, V. V. Parekh, M. T. Wilson, D. Olivares-villagómez, A. K. Singh and L. Wu, J. Clin. Invest., 2005, 115, 2572–2583 CrossRef CAS PubMed.
- S. Deng, L. Bai, R. Reboulet, R. Matthew, D. A. Engler, L. Teyton, A. Bendelac and P. B. Savage, Chem. Sci., 2014, 5, 1437–1441 RSC.
- R. Verbeke, I. Lentacker, K. Breckpot, J. Janssens, S. Van Calenbergh, S. C. De Smedt and H. Dewitte, ACS Nano, 2019, 13, 1655–1669 CAS.
- M. Speir, I. F. Hermans and R. Weinkove, Drugs, 2017, 77, 1–15 CrossRef CAS PubMed.
- Z. Liu and J. Guo, Carbohydr. Res., 2017, 452, 78–90 CrossRef CAS PubMed.
- C. Romanò and M. H. Clausen, Eur. J. Org. Chem., 2022, e202200246 CrossRef.
- R. M. Wilson and S. J. Danishefsky, J. Am. Chem. Soc., 2013, 135, 14462–14472 CrossRef CAS PubMed.
- X. G. Yin, X. Z. Chen, W. M. Sun, X. S. Geng, X. K. Zhang, J. Wang, P. P. Ji, Z. Y. Zhou, D. J. Baek, G. F. Yang, Z. Liu and J. Guo, Org. Lett., 2017, 19, 456–459 CrossRef CAS PubMed.
- F. Broecker, S. Götze, J. Hudon, D. C. K. Rathwell, C. L. Pereira, P. Stallforth, C. Anish and P. H. Seeberger, J. Med. Chem., 2018, 61, 4918–4927 CrossRef CAS PubMed.
- X.-G. Yin, J. Lu, J. Wang, R.-Y. Zhang, X.-F. Wang, C.-M. Liao, X.-P. Liu, Z. Liu and J. Guo, J. Med. Chem., 2021, 64, 1951–1965 CrossRef CAS PubMed.
- S. Groux-Degroote, Y. Guérardel and P. Delannoy, ChemBioChem, 2017, 18, 1146–1154 CrossRef CAS PubMed.
- J. L. Daniotti, A. A. Vilcaes, V. T. Demichelis, F. M. Ruggiero and M. Rodriguez-Walker, Front. Oncol., 2013, 3, 1–12 Search PubMed.
- S. I. Hakomori and K. Handa, Glycoconjugate J., 2015, 32, 1–8 CrossRef CAS PubMed.
- M. Nishikawa, M. Kurano, T. Nitta, H. Kanoh, J. I. Inokuchi and Y. Yatomi, Sci. Rep., 2019, 9, 1–9 CrossRef CAS PubMed.
- J. L. Daniotti, R. D. Lardone, A. A. Vilcaes and J. L. Daniotti, Front. Oncol., 2016, 5, 1–11 Search PubMed.
- Y. N. Malykh, R. Schauer and L. Shaw, Biochimie, 2001, 83, 623–634 CrossRef CAS PubMed.
- U. Krengel and P. A. Bousquet, Front. Immunol., 2014, 5, 1–11 CAS.
- J. Heimburg-Molinaro, M. Lum, G. Vijay, M. Jain, A. Almogren and K. Rittenhouse-Olson, Vaccine, 2011, 29, 8802–8826 CrossRef CAS PubMed.
- V. Padler-Karavani, N. Hurtado-Ziola, M. Pu, H. Yu, S. Huang, S. Muthana, H. A. Chokhawala, H. Cao, P. Secrest, D. Friedmann-Morvinski, O. Singer, D. Ghaderi, I. M. Verma, Y. T. Liu, K. Messer, X. Chen, A. Varki and R. Schwab, Cancer Res., 2011, 71, 3352–3363 CrossRef CAS PubMed.
- A. N. Samraj, H. Läubli, N. Varki and A. Varki, Front. Oncol., 2014, 4, 1–13 Search PubMed.
- H. H. Chou, H. Takematsu, S. Diaz, J. Iber, E. Nickerson, K. L. Wright, E. A. Muchmore, D. L. Nelson, S. T. Warren and A. Varki, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11751–11756 CrossRef CAS PubMed.
- S. Bashir, L. K. Fezeu, S. Leviatan Ben-Arye, S. Yehuda, E. M. Reuven, F. Szabo De Edelenyi, I. Fellah-Hebia, T. Le Tourneau, B. M. Imbert-Marcille, E. B. Drouet, M. Touvier, J. C. Roussel, H. Yu, X. Chen, S. Hercberg, E. Cozzi, J. P. Soulillou, P. Galan and V. Padler-Karavani, BMC Med., 2020, 18, 1–19 CrossRef PubMed.
- N. Kamada, H. Iijima, K. Kimura, M. Harada, E. Shimizu, S. I. Motohashi, T. Kawano, H. Shinkai, T. Nakayama, T. Sakai, L. Brossay, M. Kronenberg and M. Taniguchi, Int. Immunol., 2001, 13, 853–861 CrossRef CAS PubMed.
- S. Figueroa-Pérez and R. R. Schmidt, Carbohydr. Res., 2000, 328, 95–102 CrossRef PubMed.
- W. Du and J. Gervay-Hague, Org. Lett., 2005, 7, 2063–2065 CrossRef CAS PubMed.
- C. Xia, Q. Yao, J. Schümann, E. Rossy, W. Chen, L. Zhu, W. Zhang, G. De Libero and P. G. Wang, Bioorg. Med. Chem. Lett., 2006, 16, 2195–2199 CrossRef CAS PubMed.
- J. M. H. Cheng, S. H. Chee, Y. Dölen, M. Verdoes, M. S. M. Timmer and B. L. Stocker, Carbohydr. Res., 2019, 486, 107840 CrossRef CAS PubMed.
- W. Ma, J. Bi, C. Zhao, Z. Zhang, T. Liu and G. Zhang, Bioorg. Med. Chem., 2020, 28, 1–10 Search PubMed.
- M. Cavallari, P. Stallforth, A. Kalinichenko, D. C. K. Rathwell, T. M. A. Gronewold, A. Adibekian, L. Mori, R. Landmann, P. H. Seeberger and G. De Libero, Nat. Chem. Biol., 2014, 10, 950–956 CrossRef CAS PubMed.
- J.-J. Park, J. H. Lee, S. C. Ghosh, G. Bricard, M. M. Venkataswamy, S. A. Porcelli and S.-K. Chung, Bioorg. Med. Chem. Lett., 2008, 18, 3906–3909 CrossRef CAS PubMed.
- J. Janssens, T. Decruy, K. Venken, T. Seki, S. Krols, J. Van Der Eycken, M. Tsuji, D. Elewaut and S. Van Calenbergh, ACS Med. Chem. Lett., 2017, 8, 642–647 CrossRef CAS PubMed.
- D. Magaud, C. Grandjean, A. Doutheau, D. Anker, V. Shevchik, N. Cotte-Pattat and J. Robert-Baudouy, Carbohydr. Res., 1998, 314, 189–199 CrossRef CAS PubMed.
- K. Mori, Y. Shikichi, S. Shankar and J. Y. Yew, Tetrahedron, 2010, 66, 7161–7168 CrossRef CAS.
- B. Yu and H. Tao, Tetrahedron Lett., 2001, 42, 2405–2407 CrossRef CAS.
- A. Chernyak, S. Oscarson and D. Turek, Carbohydr. Res., 2000, 329, 309–316 CrossRef CAS PubMed.
- A. Meijer and U. Ellervik, J. Org. Chem., 2002, 67, 7407–7412 CrossRef CAS PubMed.
- D. M. Marsden, R. L. Nicholson, M. Ladlow and D. R. Spring, Chem. Commun., 2009, 7107–7109 RSC.
- J. M. Brewer, L. Tetley, J. Richmond, F. Y. Liew and J. Alexander, J. Immunol., 1998, 161, 4000–4007 CrossRef CAS.
- R. A. Schwendener, Ther. Adv. Vaccines, 2014, 2, 159–182 CrossRef CAS PubMed.
- D. B. Stetson, M. Mohrs, R. L. Reinhardt, J. L. Baron, Z.-E. Wang, L. Gapin, M. Kronenberg and R. M. Locksley, J. Exp. Med., 2003, 198, 1069–1076 CrossRef CAS PubMed.
- M. C. Leite-de-moraes, A. Hameg, A. Arnould, F. Machavoine, Y. Koezuka, E. Schneider, A. Herbelin and M. Dy, J. Immunol., 1999, 163, 5871–5876 CrossRef CAS.
- S. J. A. M. Santegoets, A. J. M. van den Eertwegh, A. A. van de Loosdrecht, R. J. Scheper and T. D. de Gruijl, J. Leukocyte Biol., 2008, 84, 1364–1373 CrossRef CAS PubMed.
- A. J. Masterson, C. C. Sombroek, T. D. De Gruijl, Y. M. F. Graus, H. J. J. Van Der Vliet, S. M. Lougheed, A. J. M. Van Den Eertwegh, H. M. Pinedo and R. J. Scheper, Blood, 2002, 100, 701–703 CrossRef CAS PubMed.
- L. Van Kaer, Nat. Rev. Immunol., 2005, 5, 31–42 CrossRef CAS PubMed.
- P. O. Livingston, Immunol. Rev., 1995, 145, 147–156 CrossRef CAS PubMed.
- J. C. C. Chang, J. P. Diveley, J. R. Savary and F. C. Jensen, Adv. Drug Delivery Rev., 1998, 32, 173–186 CrossRef CAS PubMed.
- R. L. Coffman, B. W. P. Seymour, D. A. Lebman, D. D. Hiraki, J. A. Christiansen, B. Shrader, H. M. Cherwinski, H. F. J. Savelkoul, F. D. Finkelman, M. W. Bond and T. R. Mosmann, Immunol. Rev., 1988, 102, 5–28 CrossRef CAS PubMed.
- J. E. Park, D. Y. Wu, M. Prendes, S. X. Lu, G. Ragupathi, N. Schrantz and P. B. Chapman, Immunology, 2008, 123, 145–155 CrossRef CAS PubMed.
- D. Dorvignit, K. F. Boligan, E. Relova-Hernández, M. Clavell, A. López, M. Labrada, H. U. Simon, A. López-Requena, C. Mesa and S. von Gunten, Sci. Rep., 2019, 9, 1–12 CrossRef CAS PubMed.
- J. C. Manimala, T. A. Roach, Z. Li and J. C. Gildersleeve, Glycobiology, 2007, 17, 17C–23C CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hao
Jiang
a,
Sahar
Tahvili
a,
Peng
Wei
*a,
Hao
Jiang
a,
Sahar
Tahvili
a,
Peng
Wei