
Advancing lithium–sulfur battery efficiency: utilizing a 2D/2D g-C3N4@MXene heterostructure to enhance sulfur evolution reactions and regulate polysulfides under lean electrolyte conditions†
Vijay K.
Tomer
 *a,
Otavio Augusto Titton
Dias
a,
Abdelaziz M.
Gouda
b,
Ritu
Malik
*a and
Mohini
Sain
*a
*a,
Otavio Augusto Titton
Dias
a,
Abdelaziz M.
Gouda
b,
Ritu
Malik
*a and
Mohini
Sain
*a
aDepartment of Mechanical & Industrial Engineering, University of Toronto, Toronto, Canada. E-mail: vj.kumar@utoronto.ca; ritu.kumar@utoronto.ca; m.sain@utoronto.ca
bSolar Fuels Group, Department of Chemistry, University of Toronto, Toronto, Canada
First published on 16th April 2024
Abstract
Lithium–sulfur batteries (LSBs) show promise for achieving a high energy density of 500 W h kg−1, despite challenges such as poor cycle life and low energy efficiency due to sluggish redox kinetics of lithium polysulfides (LiPSs) and sulfur's electronic insulating nature. We present a novel 2D Ti3C2 Mxene on a 2D graphitic carbon nitride (g-C3N4) heterostructure designed to enhance LiPS conversion kinetics and adsorption capacity. In a pouch cell configuration with lean electrolyte conditions (∼5 μL mg−1), the g-C3N4-Mx/S cathode exhibited excellent rate performance, delivering ∼1061 mA h g−1 at C/8 and retaining ∼773 mA h g−1 after 190 cycles with a Coulombic efficiency (CE) of 92.7%. The battery maintained a discharge capacity of 680 mA h g−1 even at 1.25 C. It operated reliably at an elevated sulfur loading of 5.9 mg cm−2, with an initial discharge capacity of ∼900 mA h g−1 and a sustained CE of over 83% throughout 190 cycles. Postmortem XPS and EIS analyses elucidated charge–discharge cycle-induced changes, highlighting the potential of this heterostructured cathode for commercial garnet LSB development.
New conceptsWe designed a novel 2D/2D heterostructure composed of MXene nanosheets stacked with graphitic carbon nitride (g-C3N4) to enhance the conversion reaction kinetics and adsorption capacity of lithium polysulfides (LiPSs). The g-C3N4 in the nanocomposite is derived from a cheap precursor (melamine) and exhibits a high pyridine nitrogen content, which inhibits polysulfide shuttling by demonstrating strong interactions with LiPSs while the presence of Ti2+ ions (from Ti3C2 MXene) functions as catalytic sites (Sx–Ti⋯Li) for effectively grafting the LiPSs and accelerating conversion kinetics. Under lean electrolyte (∼5 μL mg−1) conditions, the g-C3N4-Mx/S cathode exhibited excellent rate performance, delivering ∼1061 mA h g−1 at C/8 and retaining ∼773 mA h g−1 after 190 cycles with a Coulombic efficiency (CE) of 92.7%. The battery maintained a discharge capacity of 680 mA h g−1 even at 1.25 C. It operated reliably at an elevated sulfur loading of 5.9 mg cm−2, with an initial discharge capacity of ∼900 mA h g−1 and a sustained CE of >83% throughout 190 cycles. By employing post-mortem analyses, we have correlated the excellent electrochemical results with the chemical and structural changes in the battery composition. Overall, we have devised a straightforward route for the synthesis of the sulfur cathode that not only differs from the existing host matrices in terms of the synthesis method and composition but also shows superior electrochemical performances in the extremely flexible pouch cell format. |
Introduction
To align with the imperative of low-carbon and environmentally sustainable development, there is a predominant focus on advancing a new generation of efficient and eco-friendly energy storage systems.1,2 The pursuit of cost-effective batteries with higher current density than conventional lithium-ion batteries is a significant area of interest.3,4 Among various contenders, lithium-sulfur batteries (LSBs) have garnered considerable attention due to their outstanding energy density (2600 W h kg−1), elevated theoretical discharge capacity (1675 mA h g−1), cost-effectiveness, abundance of terrestrial resources, and environmentally friendly characteristics.5,6 The Li–S redox process involves intricate multi-step chemical and phase transformations between solid sulfur, liquid polysulfides, and solid lithium sulfide (Li2S), introducing unique challenges for LSBs. A pivotal concern is the occurrence of “shuttle effects”, involving the formation of soluble intermediate polysulfides during battery operation.7,8 Notably, the diffusion of lower-order LiPSs (Li2S2 and Li2S) from the cathode to the anode contributes to capacity fade, and passivation on both electrodes and reduces Coulombic efficiency (CE) thereby impeding the cycle performance of the battery.9 The intrinsic insulation properties of sulfur (5 × 10−30 S cm−1) and Li2S (10−13 S cm−1) further impede efficient electron transfer, causing sluggish electrochemical reactions.10 Addressing these challenges necessitates the rational design of S-hosting materials with enhanced cathode conductivity and effective LiPS adsorption capabilities as the cathode hosts exhibit strong physical and chemical interactions with LiPSs and demonstrate improved capacity retention by mitigating the shuttle phenomenon and enhancing redox kinetics.11Among diverse host materials for LSBs, encompassing various multidimensional carbon (1D CNTs, 2D graphene, and 3D porous carbon)12 and its derivatives (acetylene black, active carbon, and biocarbon),13 polar materials (metal oxides/sulfides/phosphides/selenides/nitrides/bromides),14–16 and polymeric materials (organic polymers and organic frameworks),17,18 graphite-phase carbon nitride (g-C3N4) stands out as a promising candidate.19 It is a two-dimensional, metal-free conjugated polymer semiconductor with a distinctive energy band structure and offers promise due to its straightforward one-step polymerization process.19–21 Notably, g-C3N4 exhibits a high nitrogen content of approximately 61 wt%, predominantly in the form of pyridine nitrogen which serves as an active site for interacting with lithium polysulfide (LiPS), resulting in significant advancements in reducing the shuttle effect and improving the anchoring mechanism.22,23 Leveraging these advantages, the use of g-C3N4 as the sulfur host material in LSBs holds great promise, offering a potential and effective strategy for enhancing LSBs’ overall performance. Liu et al.24 synthesized a Nb2O5/g-C3N4 composite through electrospinning and double crucible gas–solid reactions, finding that ultralong 1D Nb2O5 nanofibers prevent g-C3N4 aggregation, forming tight heterojunctions. The nanocomposite efficiently adsorbs polysulfide, catalyzing its conversion, yielding an initial discharge capacity of 900 mA h g−1 at 0.5 C, with <0.1% decay after 500 cycles. Chen et al.25 created a g-C3N4 network in N, S co-doped carbonized wood fibers using an etching induction strategy. Benefiting from a large specific surface area and interfacial C–N–C bonds, the composite improved polysulfide redox kinetics, achieving capacities of 1590.8 mA h g−1 at 0.5 C and 976.9 mA h g−1 at 2.0 C. Zou et al.26 prepared N-doped porous carbon-coated graphitic carbon nitride heterojunction composites. The g-C3N4/g-C3N4/C heterojunction and conductive porous carbon served as an S host, ensuring an initial capacity of 752 mA h g−1 at 1 C, a reversible capacity of 496 mA h g−1 after 400 cycles, and a higher rate capacity of 468 mA h g−1 at 2 C for Li–S batteries. Additionally, Liu et al.27 assembled graphitized g-C3N4 with highly dispersed nickel as a catalyst to accelerate lithium polysulfide reaction kinetics. The Li–S battery exhibited a reversible capacity of 1271.6 mA h g−1 at 0.1 C, retaining 53% even after 500 cycles at 1.0 C. While these examples underscore the efficacy of g-C3N4 as a sulfur host, the high pyridine nitrogen content of g-C3N4 alone introduces irreversibility due to its strong affinity for Li atoms. Consequently, there is an urgent need for a suitable counterpart material to create a heterostructured hybrid with superior sulfur hosting performance in LSBs.
Within the intriguing 2D materials that can effectively be embedded in the layered sheet structure of g-C3N4, the use of layered 2D transition metal carbide (MXene) emerges as a promising strategy to counter the polysulfide shuttle phenomenon in lithium–sulfur batteries.28,29 MXene's attributes, including high electronic conductivity, abundant surface functional groups, and active surfaces bonding with polysulfides, distinguish it for this purpose.28 However, the persistent issue of anisotropy and restacking in Ti3C2 MXene nanosheets, attributed to van der Waals interactions, poses a major constraint observed in many 2D materials.28 This limitation restricts ion accessibility to interlayer sites and hampers the full utilization of surface areas for Li+ intercalation/de-intercalation.30 A layered heterostructured composite of g-C3N4 and MXene is envisioned, expected to mitigate restacking on MXene nanosheets and facilitate channels for Li+ ion intercalation during charge–discharge cycles.31 Additionally, hydroxyl terminal groups on MXenes are anticipated to adsorb LiPSs, initiating redox reactions like the Wackenroder reaction.32 These reactions connect with long-chain soluble LiPSs, forming poly-thionates and reducing them to insoluble Li2S concurrently. These chemisorption and redox processes effectively suppress the shuttle effect of LiPSs, minimizing sulfur loss, and consequently, enhancing the cycling performance of Li–S batteries.33
Another prominent but less studied factor that guides the practicality of LSBs is the performance evaluation under lean electrolytic conditions.34 Although great progress has been made in designing LSBs with long cycle life (>1000 cycles) and superior rate performances (>10 C), these improvements have been achieved with an excess electrolyte ratio (>10 μL mg−1) which inevitably reduces the actual energy density of LSBs.35 Therefore, a lean electrolyte is an essential condition for practical LSBs. To promote the development of practical LSBs, it is necessary to meet an E/S ratio below 5.0 μL mg−1.36 Recently, an electrolessly developed tin-plated sulfur nanocomposite shows a high gravimetric capacity of 520–663 mA h g−1 at a low electrolyte-to-capacity ratio of 3.75 μL mA h−1.37 Similarly, the core–shell polysulfide/carbon cathode with a high sulfur loading (12 mg cm−2) exhibits a high peak charge-storage capacity (832 mA h g−1), and long-term cyclability with high capacity retention approaching (200 cycles) at a low E/S ratio (4 μL mg−1).38 Reducing the amount of electrolyte in a battery, although inversely proportional to its energy density, can increase battery impedance, slow redox kinetics, and decrease sulfur utilization, eventually causing premature battery failure. This effect is observed when the electrolyte reaches saturation with LiPSs, leading to sulfur conversion following a quasi-solid mechanism that limits its maximum theoretical capacity. To solve this issue while leveraging the advantageous characteristics of both g-C3N4 and MXene, we designed a 2D/2D g-C3N4/MXene heterostructure to serve as a sulfur host, featuring prominently exposed active sites conducive to LiPS adsorption and catalytic conversion. The resultant g-C3N4-Mx/S composite electrodes, integrated into a pouch cell assembly, exhibited robust cycling performance. They showcased a capacity of ∼1061 mA h g−1 at C/8 and sustained ∼773 mA h g−1 after 190 cycles, maintaining a CE of 92.7%. Notably, under lean electrolyte conditions (∼5 μL mg−1), the discharge capacity remained at 680 mA h g−1, even at a high rate of 1.25 C. Furthermore, the pouch cell demonstrated exceptional flexibility, retaining an uncompromised open circuit voltage (OCV) even at a folding angle of 360°, underscoring its potential applications in wearable devices.
Experimental section
Materials
Melamine (99.99%), N-methyl-2-pyrrolidone (NMP), lithium bis(trifluoromethane) sulfonimide (LiTFSI), 1,3-dioxolane (DOL), 1,2-dimethoxyethane (DME), and lithium nitrate (LiNO3) were sourced from Sigma. Additional materials, including conducting carbon (CC, Super C45), polyvinylidene fluoride (PVDF) as the binder, Al foil (a cathode current collector), Cu foil (an anode current collector), a tri-layered (PP/PE/PP) 25 μm thick separator (Celgard 2325), and tabs (Al and Ni), were obtained from MTI (USA). Pure lithium foil (100 μm) as an anode, lithium sulfide (Li2S 99.99%), and MXene (Ti3C2) were purchased from Nanochemazone (Canada). All materials were utilized as received without additional treatment.Synthesis of cathode active materials
g-C3N4 powder was synthesized through the thermal polymerization of melamine. In a standard procedure, 5 g of melamine underwent heating to 600 °C at a ramp rate of 5 °C min−1 for 4 h in a covered ceramic crucible. Post-cooling to room temperature, the resulting light-yellow solid underwent grinding, yielding bulk g-C3N4 powder. This powder was further dispersed in a solution of deionized water and HCl and sonicated for 4 h to acquire protonated delaminated g-C3N4 nanosheets (NSs). Simultaneously, 0.1 g of MXene was added to a water/ethanol mixture, subjected to sonication for 30 min, and subsequently dried at 60 °C to eliminate residual ethanol. To form the composite via the electrostatic self-assembly method, 2 g of powdered g-C3N4 NSs were dispersed in absolute methanol (100 mL), followed by the addition of 20 mg of MXene. The resulting mixture underwent sonication for 1 h and drying at 60 °C overnight, resulting in the g-C3N4-Mx composite. For sulfur-based composites, the powdered g-C3N4 and g-C3N4-Mx materials were separately ground with sulfur (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 weight ratio) for 1 h and subjected to heating at 160 °C for 15 h, yielding g-C3N4/S and g-C3N4-Mx/S composites, respectively (Scheme 1).
:7 weight ratio) for 1 h and subjected to heating at 160 °C for 15 h, yielding g-C3N4/S and g-C3N4-Mx/S composites, respectively (Scheme 1).
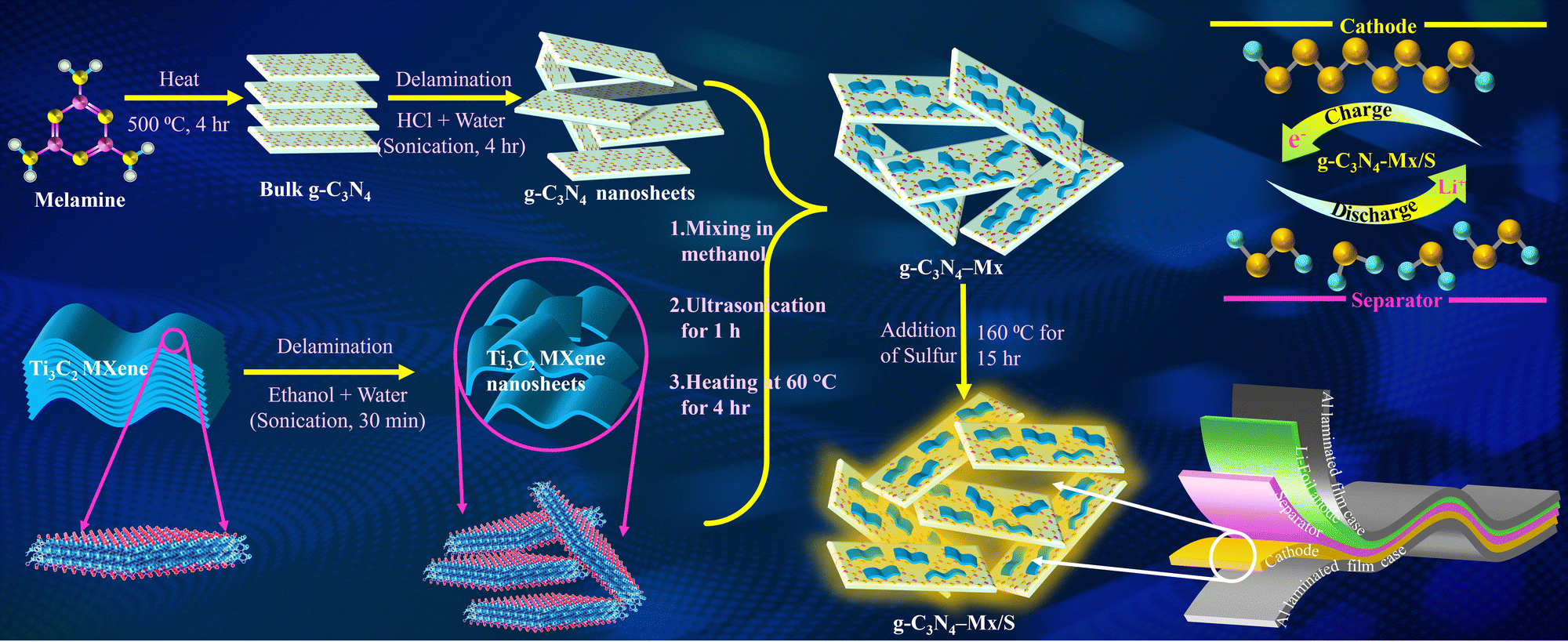 | ||
| Scheme 1 Schematic illustration for the synthesis of g-C3N4-Mx/S nanocomposites and the structure of pouch cell demonstrating arrangements of electrodes and a separator. | ||
Material characterization
Crystalline structures were analyzed employing a Philips P.W. 1830 powder X-ray diffractometer with a Cu Kα-ray source, scanning within the range of 10° to 70°. The microstructure and morphology were scrutinized using a JEOL 2010 transmission electron microscope and a QUANTA FEG 250 scanning electron microscope equipped with energy-dispersive X-ray spectroscopy (EDS). Fourier transform infrared (FTIR) spectroscopy data were collected across the 500–4000 cm−1 range using a Tensor 27 instrument. Raman spectroscopy (Bruker Senterra Infinity 1) assessed carbon defects. X-ray photoelectron spectroscopy (XPS) using a PerkinElmer Phi 5500 ESCA spectrophotometer probed the chemical and electronic states of the surface. The Brunauer–Emmett–Teller (BET) surface area and pore size were determined by N2 adsorption/desorption measurements (Quantachrome Nova 1200 Analyzer) following sample degassing under vacuum at 150 °C for 2 h. The sulfur content in composite materials was gauged through thermogravimetric differential scanning calorimetry (TGA, Q50) at a 5 °C min−1 heating rate from 25 to 600 °C. Conductivity measurements utilized sheets (10 × 20 mm) with the Ossila four-point probe technique. The zeta potential of a dilute aqueous solution of delaminated g-C3N4 and MXene was measured with a zeta potential analyzer (ZetaPlus, Brookhaven Instruments Corporation).Lithium–sulfur battery assembly
:1 volume ratio of 1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME), maintained a volume at 5 μL mg−1 sulfur. Sealed pouch cells underwent pressing for 2 h under an Ar atmosphere and were subsequently transferred to a vacuum oven for 12 h at 50 °C for aging.
:5 molar ratio) were combined in a DOL and DME solution (1:1, v/v) with vigorous stirring at 60 °C for 24 h in an Ar-filled glovebox. The resulting Li2S6 solution had a concentration of 5 mmol L−1. Before the polysulfide adsorption test, pure g-C3N4 and g-C3N4-Mx hybrids were dried at 60 °C under vacuum for 12 h. Subsequently, g-C3N4 and g-C3N4-Mx hybrids, each weighing 20 mg, were immersed in a 5 mL of Li2S6 solution for static adsorption over 12 h. The color variation of the supernatant over time was observed, and the adsorption capability of g-C3N4 and g-C3N4-Mx for Li2S6 was tested via UV-vis spectroscopy. A blank Li2S6 solution served as the reference.
Results and discussion
Active material characterization
Scheme 1 illustrates the schematic diagram of the g-C3N4/MXene composite. Melamine, upon high-temperature calcination, undergoes thermal polymerization and cross-linking of its monomers, resulting in the formation of bulk-layered g-C3N4.39 Comprising continuous tri-s-triazine structural units, g-C3N4 offers numerous LiPS adsorption sites and robust anchoring for Li2S. Ultrasonication of bulk g-C3N4 for several hours enhances interlayer spacing, enabling uniform exfoliation and dispersion of agglomerated g-C3N4 in the polar solvent (H2O), yielding g-C3N4 nanosheets.40 Post-ultrasonic treatment reduces the interlayer van der Waals force of g-C3N4, facilitating compounding with MXene.41 The positively charged zeta potential (ζ) of exfoliated g-C3N4 (+19.12 mV) tends to adsorb Ti-ions of MXene (ζ = −23.28 mV) through electrostatic interactions (Fig. S1, ESI†).33 During the mixing process, the difference in the surface charge between MXene and g-C3N4 easily leads to self-assembly by electrostatic interactions. With a high N-content (∼57%) consisting of periodic heptazine subunits that are bridged by planar tertiary amino groups to form large in-plane pores,42 the 2D layered structure of g-C3N4 deemed highly promising for efficient Li+ intercalation (theoretical specific capacity: ∼520 mA h g−1, and the corresponding electrochemical reaction can be presented as g-C3N4 + 2Li+ + 2e− = Li2-g-C3N4. The resulting g-C3N4-Mx composite is then combined with sulfur, producing the sulfur-based cathode composite termed as the active material.33 Nitrogen atoms of g-C3N4, acting as a potent polar adsorbent, effectively anchor polysulfides, mitigating the shuttle effect in Li–S batteries, thus reducing sulfur loss and enhancing the sulfur utilization ratio in the charge–discharge cycling process.19In Fig. 1a, pure g-C3N4 exhibits two distinct peaks at 13.2° and 27.5°, attributed to the (100) and (002) faces of g-C3N4, corresponding to the stacking motifs of the in-plane tri-s-triazine structure and the planar stacking of the conjugated aromatic system, respectively.40 The well-aligned diffraction peaks of the orthorhombic sulfur phase (JCPDS card no. 08-0247) in both g-C3N4/S and g-C3N4-Mx/S confirm the crystalline nature of sulfur within the respective host matrix.12 The absence of significant peaks for MXene is observed, attributed to its lower doping and crystallinity. Chemical structure analysis using FT-IR (Fig. 1b) reveals that the characteristic peak at 735 cm−1 originates from the stretching vibration of the heptazine ring. At the same time, bands at 1227–1521 cm−1 correspond to the aromatic C–N stretching and CN heterocycle stretching modes of g-C3N4.43 The broad peaks in the region of 2800–3500 cm−1 are associated with the N–H characteristic vibration of –NHx and the O–H characteristic vibration of the remaining hydroxyl group or absorbed H2O molecules. Additionally, the band at 2319 cm−1 corresponds to the signals from S–H stretching modes.44
 | ||
| Fig. 1 (a) XRD patterns, (b) FTIR spectra, and (c) Raman spectra of g-C3N4, g-C3N4/S, and g-C3N4-Mx/S. TEM images of (d) g-C3N4 and (e) g-C3N4-Mx/S materials. (f) SEM images of g-C3N4-Mx/S. (g) Color mapping of g-C3N4-Mx/S demonstrating the uniform distribution of C, N, S, and Ti elements. | ||
Raman spectroscopy, a non-destructive technique for chemical analysis, provides detailed insights into the chemical structure and molecular interactions of materials. The Raman spectra in Fig. 1c reveal peaks at 706, 768, and 1230 cm−1, corresponding to the breathing modes of the s-triazine ring in g-C3N4.45 Notably, all samples display peaks at 1412 and 1570 cm−1, attributed to the d-band and G-band, respectively. The d-band signifies disordered carbon or defects, while the G-band reflects the stretching motion of all carbon atoms in the aromatic ring, indicating the symmetry and graphitization degree (sp2) of the material.46 The calculated relative intensity ratios of the d-band and G-band (ID/IG) for pure g-C3N4, g-C3N4/S, and g-C3N4-Mx/S composites are approximately 1.02, 1.008, and 1.0, respectively. The decreasing ID/IG ratios with sulfur and MXene addition indicate a higher degree of crystalline graphitic structure and improved electronic conduction,47 aligning with the XRD pattern in Fig. 1a. For a comprehensive analysis of material-specific surface area changes, nitrogen adsorption–desorption experiments were conducted, and the results are depicted in Fig. S2 (ESI†). All three materials display a small adsorption volume at low relative pressures, a prominent H3 hysteric loop at medium relative pressures (0.30–0.80), and a distinct upward trend (type IV) at high relative pressures (0.90–1.0).48 These features indicate the simultaneous presence of micropores, predominantly mesopores, and some macropores in the sample. The specific surface area (SSA) of pure g-C3N4 was measured to be 65 m2 g−1 which decreases with the addition of sulfur and an SSA of 54 m2 g−1 was observed for the g-C3N4-Mx/S composite thus demonstrating the reduction in the pore volume on account of further loading of other components. The cathode host's conductivity is crucial, considering sulfur and its discharge products (Li2S) are insulators. In this study, the four-point probe method revealed the conductivity of the g-C3N4-Mx/S composite ranging between 10−2 and 10−3 S cm−1, significantly higher than that of pure g-C3N4/S materials, which falls within the range of 10−12 to 10−13 S cm−1.
The morphologies of the prepared active materials were scrutinized through TEM and SEM, complemented by elemental mapping. The TEM image in Fig. 1d highlights the sheet-like structure of g-C3N4, signifying thermal exfoliation at 600 °C effectively split bulk g-C3N4 into small nanosheets. The TEM image in Fig. 1e reveals that the morphology of g-C3N4-Mx/S with the 1% content of Ti3C2 MXene was not distinguished from that of the g-C3N4 nanosheets. This could be attributed to the thin-flake morphology of both the g-C3N4-Mx/S composites and the g-C3N4 NSs, and it is hard to distinguish from the TEM images. The SEM images in Fig. 1f illustrate the composites’ well-defined 2D layered morphology, indicating superior sulfur dispersion in g-C3N4-Mx/S. Elemental mapping in Fig. 1g vividly demonstrates uniform C, N, and S dispersion throughout g-C3N4-Mx/S, with mappings precisely conforming to sample shapes, confirming the uniform sulfur deposition on g-C3N4-Mx surfaces. Additionally, Mx prevents agglomeration by encapsulating g-C3N4, tightly coupling them, enhancing material dispersion, increasing the average surface roughness, and creating a larger reaction surface area with more channels, serving as a loading platform with N-containing functional groups to trap sulfur particles. The highly integrated g-C3N4-Mx/S with N-containing functional groups effectively confines LiPS, ensuring exceptional cycling performance. To determine the weight ratio of sulfur in the g-C3N4-Mx/S composite, TGA analysis was performed (Fig. S3, ESI†). The weight loss in the temperature range of 180–350 °C, attributed to the sulfur component's vapor, suggests a sulfur content of approximately 69.5%. This aligns with the experimentally executed mass ratio of g-C3N4-Mx to sulfur (3:7). The high sulfur content indicates that sulfur-rich g-C3N4-Mx/S possesses a high energy density, particularly crucial for the cathode material of Li–S batteries.
The elemental composition and valence information of the g-C3N4-Mx/S surface were acquired through XPS. The XPS survey spectra in Fig. S4 (ESI†) depict peaks at approximately 162.5, 226.1, 282.8, 398.1, 455.3, and 532.2 eV, corresponding to S 2p, S 2s, C 1s, N 1s, Ti 2p, and O 1s binding energies, respectively. The high-resolution fitted spectra in Fig. 2 for C 1s reveal five peaks: –Ti–C– (281.7 eV) and –C–C– (282.65 eV) from external and graphitic carbon, conjugated –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–/–CC– (284.9 eV) at the heptazine unit structure's edge, CO (285.9 eV) after thermal annealing of g-C3N4, and –S–CN (286.9 eV) likely originating from sulfur and nitrogen-containing heptazine rings' interaction (sp2 bonds).49 The high-resolution XPS spectrum of N 1s exhibits four peaks: pyridinic-N (CN–C, 397.4 eV), pyrrolic-N (–S–NC–, 398.6 eV), graphitic-like N/amino N (C3–N and N–H, 401.1 eV), and oxidized-N (N–O, 404.4 eV).50 The abundant electron-rich pyridinic N content in g-C3N4-Mx/S enhances LiPS adsorption by providing binding sites for positively charged Li+ of LiPSs, forming Li–N interactions, hindering LiPS migration, and further inhibiting the shuttle effect.51 The S 2p spectrum of g-C3N4-Mx/S shows peaks at 162.1 (S 2p3/2) and 163.2 eV (S 2p1/2), attributed to –S–S– bonds (S8 molecules).52,53 For Ti 2p, the spectrum was deconvoluted into four components: peaks at 455.2 eV (Ti–C, 2p3/2), 456.8 eV (Ti–O, 2p3/2), 459.3 eV (Ti–C, 2p1/2), and 462.1 eV (Ti–O, 2p1/2). Besides N, C, S, and Ti, oxygen (O) at 531.2 eV was detected, originating from g-C3N4 oxidation during heat treatment in air.54 These oxygenated groups contribute to chemically adsorbing sulfur, effectively preventing polysulfides from dissolving into electrolytes.
N–/–CC– (284.9 eV) at the heptazine unit structure's edge, CO (285.9 eV) after thermal annealing of g-C3N4, and –S–CN (286.9 eV) likely originating from sulfur and nitrogen-containing heptazine rings' interaction (sp2 bonds).49 The high-resolution XPS spectrum of N 1s exhibits four peaks: pyridinic-N (CN–C, 397.4 eV), pyrrolic-N (–S–NC–, 398.6 eV), graphitic-like N/amino N (C3–N and N–H, 401.1 eV), and oxidized-N (N–O, 404.4 eV).50 The abundant electron-rich pyridinic N content in g-C3N4-Mx/S enhances LiPS adsorption by providing binding sites for positively charged Li+ of LiPSs, forming Li–N interactions, hindering LiPS migration, and further inhibiting the shuttle effect.51 The S 2p spectrum of g-C3N4-Mx/S shows peaks at 162.1 (S 2p3/2) and 163.2 eV (S 2p1/2), attributed to –S–S– bonds (S8 molecules).52,53 For Ti 2p, the spectrum was deconvoluted into four components: peaks at 455.2 eV (Ti–C, 2p3/2), 456.8 eV (Ti–O, 2p3/2), 459.3 eV (Ti–C, 2p1/2), and 462.1 eV (Ti–O, 2p1/2). Besides N, C, S, and Ti, oxygen (O) at 531.2 eV was detected, originating from g-C3N4 oxidation during heat treatment in air.54 These oxygenated groups contribute to chemically adsorbing sulfur, effectively preventing polysulfides from dissolving into electrolytes.
 | ||
| Fig. 2 (a–e) XPS spectrum showing the complete scan and the corresponding spectra for the elements present in the g-C3N4-Mx/S composite. | ||
To evaluate adsorption ability, g-C3N4, and g-C3N4-Mx materials were immersed individually in a Li2S6 solution under magnetic stirring (30 min) and allowed to attain equilibrium for 24 h in an Ar-filled glove box. The adsorbents were then retrieved, and optical photographs of the solutions were captured (the inset of Fig. 3). Remarkably, the original bright-yellow color of the Li2S6 solution becomes nearly colorless after immersion in g-C3N4-Mx, indicating effective absorption of most polysulfides. In contrast, pristine g-C3N4 has a minimal effect on the color change of the Li2S6 solution. UV-vis measurements for Li2S6 reveal a strong absorption band in the 225–325 nm region, assigned to S62− polysulfide species (Fig. 3). According to the visible color tests, g-C3N4 exhibits lower absorbance than neat Li2S6, while g-C3N4-Mx demonstrates the lowest absorbance, likely due to the catalytic effect from adding MXene. This validates the robust affinity between polysulfides and Ti, particularly effective in suppressing polysulfide dissolution in the electrolyte (Sx–Ti⋯Li interactions).55 This robust affinity significantly contributes to enhancing the cycling stability of the sulfur electrode.
 | ||
| Fig. 3 UV-Vis spectra for blank Li2S6 and Li2S6 added with g-C3N4 and g-C3N4-Mx (after 24 h). The inset shows the digital picture of the effect of active materials on the adsorption of LiPSs in Li2S6 solution. | ||
Electrochemical characterization
The electrochemical performance of g-C3N4/S and g-C3N4-Mx/S was evaluated in a pouch cell configuration. In Fig. 4a, CV curves of g-C3N4/S and g-C3N4-Mx/S composites are presented within a voltage range of 1.6 V to 2.8 V at a sweep rate of 0.05 mV s−1. During the cathodic scan (discharge process), two primary reduction peaks manifest for both g-C3N4/S and g-C3N4-Mx/S at approximately 2.3 V and 2.0 V, corresponding to the cathodic reduction processes: the reduction of elemental sulfur to high-order polysulfides (S8 → Li2Sx (4 ≤ x ≤ 8)) and the formation of Li2S2/Li2S from low-order polysulfides (Li2Sx → Li2S2/Li2S (4 ≤ x ≤ 8)), respectively.56,57 In the subsequent anodic scan (charge process), two oxidation peaks at around 2.38 and 2.45 V correspond to the inverse processes and are associated with the oxidation of Li2S2/Li2S to long-chain polysulfides (Li2Sn (n > 2)) and further to sulfur. Compared to g-C3N4/S, the primary cathodic peaks of g-C3N4-Mx/S exhibit a slight shift to a higher reduction potential, while the anodic peaks shift to a lower oxidation potential. This suggests a reduction in polarization in g-C3N4-Mx/S, facilitating polysulfide conversion and improving the reversibility of the electrode during cycling.58 Furthermore, the smaller potential difference (ΔE) between cathodic and anodic peak current densities in g-C3N4-Mx/S (0.38 V) that in g-C3N4/S (0.46 V) reflects its improved tendency to effectively suppress the diffusion of liquid intermediate products during CV cycles. Additionally, during the scanning process for g-C3N4-Mx/S (Fig. 4b), the cathodic peak position, peak current, and peak area exhibit minimal changes over the 3 cycles, indicating superior capacity retention and good reversibility in the cell. CV curves at various current rates are depicted in Fig. 4c and d for g-C3N4-Mx/S and g-C3N4/S cathodes, respectively. The cathodic processes involve peak C1 and peak C2, attributed to S8 transformation to long-chain LiPSs (Li2Sn, 4 ≤ n ≤ 8) and the subsequent reduction of long-chain LiPSs to Li2S2/Li2S, respectively.33,59 The anodic process is represented by peak A1 and peak A2, corresponding to the reverse transformation. The redox peaks at higher current rates are very slightly broadened for both materials, indicating slow kinetics of LiPS conversion. Overall, g-C3N4-Mx/S exhibits higher peak current densities than g-C3N4/S, indicating enhanced Li+ transportability and superior rate capability. Both cathodic and anodic peak currents for both materials display a linear relationship with the square root of the scan rate (ν0.5), indicative of diffusion-limited discharge/charge reactions (Fig. 4e–h). The Li+ diffusion coefficient can be delineated by the Randles Sevcik equation:60Ip = (2.69 × 105)·n1.5·A·D0.5·CLi+0.5·v0.5, where n is the number of charge transfers, A is the active electrode area, D is the Li+ diffusion coefficient, and CLi+ is the concentration of Li+ in the bulk. Since n, A, and c0 are constants, , a larger
, a larger  implies a higher D. This equation elucidates a linear relationship between the peak current and the square root of the scanning rate, with the slope indicative of lithium-ion diffusion.61 In g-C3N4-Mx/S, the slopes for both reduction peaks (peaks C1 and C2) and oxidation peaks (peaks A1 and A2) surpass those for g-C3N4/S at the same electrode area for each sulfur reduction and oxidation reaction. This larger slope value correlates with accelerated Li ion diffusion and improved LiPS redox kinetics during discharge/charge processes due to the presence of MXene. MXene not only fosters LiPS transformation but also expedites rapid Li+ migration.62 EIS on the g-C3N4-Mx/S and g-C3N4/S cathodes further elucidated the kinetics of the electrode processes at the electrode/electrolyte interface. As depicted in Fig. 4i, the Nyquist plots outline three frequency regions: a high-frequency segment indicating electrolyte resistance (Re), a middle-frequency semicircle corresponding to charge transfer impedance (Rct), and a low-frequency spike representing Warburg impedance (W) associated with Li+ diffusion in the cathode.63,64 The semicircle diameter and Rct value of the g-C3N4-Mx/S cathode cell (34.9 Ω) markedly reduced compared to those of the g-C3N4/S cathode cell (58.3 Ω), indicating diminished charge transfer resistance due to enhanced electrical conductivity and improved electrical contact upon Ti3C2 incorporation into g-C3N4. Additionally, the Re of g-C3N4-Mx/S is 4 Ω, nearly half compared to that of g-C3N4/S (8.1 Ω), affirming exceptional cell conductivity. This outcome underscores lower electrochemical impedance in the g-C3N4-Mx/S host, attributed to the presence of Ti3C2 as chemically active sites that avidly adsorb LiPSs, facilitate charge and ion transfer, and diminish electrode–electrolyte interface resistance and charge transfer resistance. Parameters from the Nyquist plot are detailed in Table S1 (ESI†).
implies a higher D. This equation elucidates a linear relationship between the peak current and the square root of the scanning rate, with the slope indicative of lithium-ion diffusion.61 In g-C3N4-Mx/S, the slopes for both reduction peaks (peaks C1 and C2) and oxidation peaks (peaks A1 and A2) surpass those for g-C3N4/S at the same electrode area for each sulfur reduction and oxidation reaction. This larger slope value correlates with accelerated Li ion diffusion and improved LiPS redox kinetics during discharge/charge processes due to the presence of MXene. MXene not only fosters LiPS transformation but also expedites rapid Li+ migration.62 EIS on the g-C3N4-Mx/S and g-C3N4/S cathodes further elucidated the kinetics of the electrode processes at the electrode/electrolyte interface. As depicted in Fig. 4i, the Nyquist plots outline three frequency regions: a high-frequency segment indicating electrolyte resistance (Re), a middle-frequency semicircle corresponding to charge transfer impedance (Rct), and a low-frequency spike representing Warburg impedance (W) associated with Li+ diffusion in the cathode.63,64 The semicircle diameter and Rct value of the g-C3N4-Mx/S cathode cell (34.9 Ω) markedly reduced compared to those of the g-C3N4/S cathode cell (58.3 Ω), indicating diminished charge transfer resistance due to enhanced electrical conductivity and improved electrical contact upon Ti3C2 incorporation into g-C3N4. Additionally, the Re of g-C3N4-Mx/S is 4 Ω, nearly half compared to that of g-C3N4/S (8.1 Ω), affirming exceptional cell conductivity. This outcome underscores lower electrochemical impedance in the g-C3N4-Mx/S host, attributed to the presence of Ti3C2 as chemically active sites that avidly adsorb LiPSs, facilitate charge and ion transfer, and diminish electrode–electrolyte interface resistance and charge transfer resistance. Parameters from the Nyquist plot are detailed in Table S1 (ESI†).
 | ||
| Fig. 4 (a) CV profiles of g-C3N4/S and g-C3N4-Mx/S cathodes. (b) CV curves of the g-C3N4-Mx/S cathode for 3 consecutive cycles. CV curves at different scan rates of (c) g-C3N4-Mx/S and (d) g-C3N4/S. Plots of the peak currents versus the square root of scanning rates from CV curves of g-C3N4-Mx/S and g-C3N4/S cathodes for (e) peak C1, (f) peak C2, (g) peak A1, and (h) peak A2. (i) EIS spectra of the freshly prepared uncycled cell. | ||
The electrochemical performance of the as-prepared g-C3N4-Mx/S and g-C3N4/S cathodes was evaluated in pouch cell batteries. The sulfur content in the cathodes is ∼69%, and the capacity is calculated according to the sulfur weight. The charge–discharge voltage profiles for cells prepared by using g-C3N4-Mx/S and g-C3N4/S cathodes at a rate of C/8 are shown in Fig. 5a. As can be seen, two discharging voltage plateaus and one major charging voltage plateau were observed which are consistent with the multistep reduction of sulfur indicated by CV curves in Fig. 4a. The polarization potential (ΔE), calculated as the gap between the anode and cathode peaks, is significantly lower for g-C3N4-Mx/S (213 mV) than that for g-C3N4/S (272 mV), reflecting reduced polarization in the g-C3N4-Mx/S electrode facilitated by Ti-N active site catalysis. Additionally, the capacity of the lower discharge plateau (QL) and higher discharge plateau (QH) is calculated from discharge curves. The higher QL/QH signifies superior electrocatalytic activity for polysulfides, where QH corresponds to sulfur conversion into high-ordered soluble polysulfides, and QL represents efficient reduction of polysulfides to Li2S.65 In Fig. 5b, the capacity ratio (QL/QH) of g-C3N4-Mx/S (2.12) surpasses that of g-C3N4/S (1.86), indicating enhanced sulfur utilization owing to the presence of a higher amount of lower ordered polysulfides and the superior catalytic activity of Sx–Ti⋯Li in the g-C3N4-Mx/S electrode. The charge–discharge profiles of g-C3N4-Mx/S at C/8 over 190 cycles (Fig. 5c) reveal the persistent maintenance of the discharge plateaus and charging curves, demonstrating rapid mass transport and reaction kinetics. A comparison of long-term cycle performances between g-C3N4/S and g-C3N4-Mx/S (Fig. 5d) illustrates initial capacity decay attributed to the activation process. During the process of charging–discharging, the non-activated sulfur may aggregate on the g-C3N4-Mx surface leading to the formation of a non-stable solid electrolyte interface (SEI) layer.66 As can be seen, the g-C3N4-Mx/S witnessed enhanced sulfur utilization and uniform depreciation in capacity with an initial discharge capacity of 1061 mA h g−1 and a CE of 92.7%. The capacity drops to 773 mA h g−1 after 190 cycles with a capacity retention ratio of 73%. In contrast, g-C3N4-Mx/S achieves an initial discharge capacity of 749 mA h g−1 for 105 cycles only. The rate performance of the pouch cells prepared using g-C3N4-Mx/S and g-C3N4/S cathodes in Fig. 5e indicates higher discharge capacity for g-C3N4-Mx/S than that for g-C3N4/S for all current rates, with g-C3N4-Mx/S recovering well at C/8 which is ascribed to the porous structures of g-C3N4 ensuring fast Li+ transportation and chemical confinement for LiPSs.67,68 In conjugation with results presented in Fig. 5e, the galvanostatic charge–discharge profiles at different rates (C/8 to 1.25 C) for g-C3N4-Mx/S in Fig. 5f demonstrate excellent preservation of discharge plateaus even at a higher rate of 1.25 C, affirming swift mass transport and reaction kinetics. The cathode exhibits high reversibility, maintaining capacities of 1013, 830, and 682 mA h g−1 at C/4, 0.75 C, and 1.25 C, respectively, corresponding to 95.5%, 79%, and 65% of the original capacity.
 | ||
| Fig. 5 (a) Initial charge–discharge curves of the g-C3N4/S and g-C3N4-Mx/S cathodes at C/8. (b) The capacity contributions of QH and QL, and the QL/QH ratio of the g-C3N4-Mx/S and g-C3N4/S at C/8. (c) Galvanostatic charge–discharge profiles of the g-C3N4-Mx/S cathode for 190 cycles. (d) Cycle performance and coulombic efficiency of g-C3N4/S and g-C3N4-Mx/S cathodes for 105 and 190 cycles, respectively, (e) Rate capabilities of g-C3N4/S and g-C3N4-Mx/S cathodes at various current rates. (f) Galvanostatic charge–discharge profiles of the g-C3N4-Mx/S cathode at different current rates. | ||
To better understand the performance of g-C3N4-Mx/S-based LSBs, we have compared its capacity and CE with that of commercial porous carbon-based sulfur (C/S) composites. As can be seen in Fig. S5 (ESI†), the C/S cathode (S loading = 4.7 mg cm−2) based LSB shows an initial higher capacity (1273 mA h g−1) at C/8 than that of the g-C3N4-Mx/S cathode (1061 mA h g−1); however, with further cycling, the capacity decreases rapidly to 703 mA h g−1 demonstrating a capacity retention of 55.2% after the end of 125 cycles. This concludes the fact that g-C3N4 exhibited better performance as a host material than the porous carbon because the g-C3N4 possesses an ample electron-rich pyridinic N content (Fig. 2b). In LSBs, N-rich active sites in g-C3N4 are particularly beneficial. These sites enhance the chemical interaction between sulfur chains and oxygen functional groups on the g-C3N4 matrix during sulfur loading, forming more thiosulfate and sulfate species (Fig. 7c, discussed later). This promotes a uniform distribution of sulfur in the carbon host during charge/discharge processes. Additionally, the highly positively charged Li cations in Li2Sn can directly bind to the electron-rich pyrrolic and pyridinic nitrogen atoms with lone pair electrons in the N-rich g-C3N4, leading to a Li–N interaction (Fig. 7d, discusses later) and enhanced LiPS adsorption. Moreover, N-active sites adjacent to oxygen functional groups improve the interaction between the oxygen functional groups and LiPS, effectively reducing the polysulfide shuttle.
 | ||
| Fig. 6 Upper row: digital pictures of the OCV for the pouch cell consisting of the g-C3N4-Mx/S cathode at different folding angles. Bottom row: pictures of LEDs lighten up by a pouch cell at different folding angles demonstrating the excellent flexible feature of the developed pouch cell. | ||
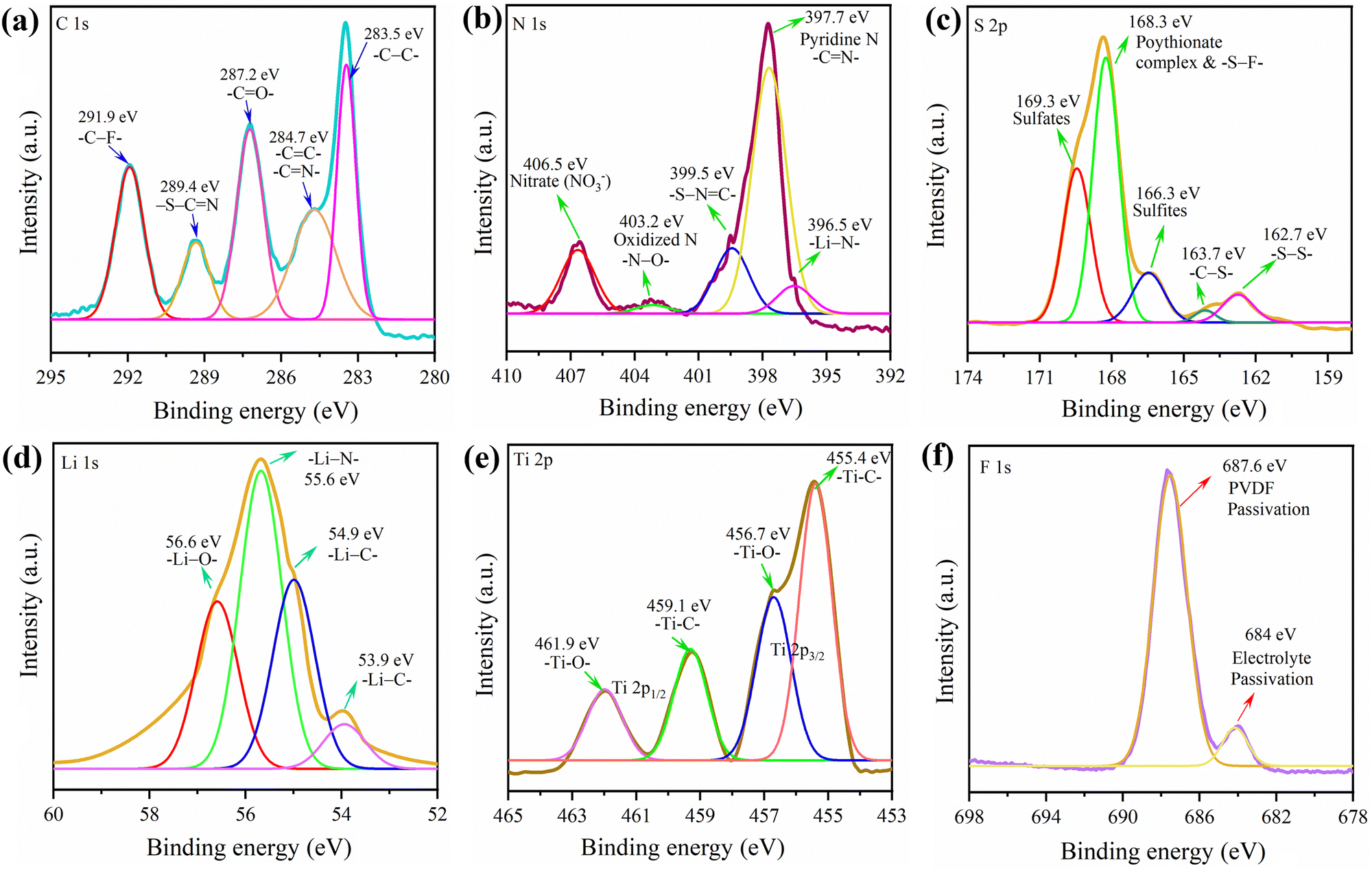 | ||
| Fig. 7 (a–f) XPS spectra of the g-C3N4-Mx/S cathode obtained from the pouch cell cycled for 190 charge–discharge cycles. | ||
For practical success in Li–S batteries, emphasizing high areal sulfur loading is crucial. Hence, pouch cells with sulfur loadings of 3.8, 4.6, and 5.9 mg cm−2 underwent testing (Fig. S5, ESI†) at C/8 for the g-C3N4-Mx/S cathode. The 5.9 mg cm−2 cell exhibited the lowest initial discharge capacity of 795 mA h g−1 (CE of 83.2%), reducing to 573 mA h g−1 after 190 cycles with a 72% retention rate. In the case of mild sulfur doping (3.8 and 4.6 mg cm−2), the cycled Li is a negligible fraction of the entire Li anode and the stripping/plating behavior is relatively facile at an electrolyte dosage of 5 μL mg−1, due to which the cell performance, including capacity, CE, and cycle life, is less affected by the Li anode. However, at a high sulfur loading (5.9 mg cm−2), the Li plating process is not as mild owing to the depleted electrolyte,69 which causes a decrease in the battery capacity due to the inevitable “shuttle effect”. The results highlight the g-C3N4-Mx/S's outstanding electrochemical performance under high-loading (4.6 mg cm−2) and lean electrolyte (5 μL mg−1) conditions, showcasing robust LiPS inhibition and accelerated kinetic conversion for enhanced sulfur utilization.
Comparisons with other pouch cells (Table S2, ESI†) demonstrated that the g-C3N4-Mx/S cathode outperforms most previously reported cathodes. The exceptional electrochemical performance of the integrated 2D/2D structure, consisting of g-C3N4/MXene nanosheets, arises from several key factors. Primarily, this structure facilitates faster electron transport during charge/discharge cycles and ensures a homogeneous distribution of sulfur between the g-C3N4 and MXene nanosheets, thereby enabling high sulfur utilization. Furthermore, the 2D structure of the g-C3N4 nanosheets prevents the stacking of MXene layers, creating additional active sites for polysulfide adsorption during the charge–discharge process. MXene also enhances the charge transfer process within g-C3N4, catalyzing the conversion of polysulfides. Additionally, the hydrophilic and polar nature of MXene prevents the stacking of g-C3N4, promoting an even distribution of components and effectively trapping polysulfides, suppressing the “shuttle effect”, respectively. To showcase the superior mechanical flexibility of the designed g-C3N4-Mx/S electrodes, various folding angles for pouch cells were tested. Fig. 6 reveals impressive open circuit voltage (OCV) stability at 2.3 V, successfully powering the red LED bulb formation of “LIS” at varying bending angles, with no discernible change in LED brightness or OCV readings at horizontal (90°), vertical (180°), or crumpled (360°) bending angles, demonstrating favorable electrical performance for practical utility in wearable and portable electronic device applications.
Postmortem analysis
To understand the electrochemical kinetics of the cathodes after cycling, the EIS spectra of the cycled cell after the 190th cycle (g-C3N4-Mx/S) and 105th cycle (g-C3N4/S) at C/8 were recorded (Fig. S6, ESI†). Post-cycling, the Re of the g-C3N4-Mx/S cathode cell increased from 4 to 4.9 Ω, markedly lower than that of the g-C3N4/S cathode cell (rising from 8.1 to 8.6 Ω), indicating restrained polysulfide diffusion from g-C3N4-Mx/S. Additionally, the reduced Rct for both cells (Table S1, ESI†) post-cycling implies enhanced electrolyte penetration, shortening charge, and ion transport paths.70,71 The impedance spectra affirm that the 2D/2D g-C3N4-Mx/S enhances sulfur cathode conductivity and mitigates the shuttle effect by retaining more polysulfides within the cathode. The lithiation mechanism of the g-C3N4-Mx/S cathode was further explored through XPS analysis post the 190th discharge cycle. The pouch cell was torn down in an argon-filled glovebox, and a 10 mm × 10 mm segment of the g-C3N4-Mx/S cathode was cut, washed with DME, and kept for overnight drying before XPS measurements. The XPS complete survey scan in Fig. S7 (ESI†) exhibits well-defined peaks and some new bond formations for C, N, S, Ti, and O, consistent with the pure active material, while additional elements (Li and F) were also observed. In Fig. 7, for the C 1s spectra, peaks at 283.5 eV, 284.7, 287.2, and 289.4 eV corresponded to –C–C, –CN–/–CC–, CO, and –S–CN, mirroring the active material C 1s spectra in Fig. 2. However, a new peak at 291.5 eV, representing –C–F– due to electrolyte passivation, emerged. Deconvoluting the N 1s spectra revealed peaks at 397.7 eV for pyridinic-like N (–CN–) in the g-C3N4 structure, 399.5 eV for –S–NC–, and 403.2 for oxidized N (N–O).72 The 396.5 eV peak confirmed Li–N bond formation during cathode lithiation, contributing to lithium storage.73 Additionally, the 406.5 eV peak indicated the nitrate (NO3−) presence on the cathode, formed by LiNO3 reduction during cycling.70 For S 2p, various new reduction products and intermediates were identified after cycling including sulfites at 166.3 eV, and S–F bonds at 168.3 eV.74 Compared to the S-spectra in Fig. 2, more sulfates were observed in the cycled cathode which could be a result of oxidized S-species (O–S) from the reaction between LiPSs and oxygen species. The O–S bond surfaced due to sulfate species interacting with residual oxygen-containing species, and the S–F bond indicated a covalent interaction between F-containing electrolytes and polysulfides.75 Besides, the doublet peaks at around 163 eV in Fig. 2 for elemental sulfur were shortened in the cycled cell which could be due to the presence of a partial amount of ‘dead’ sulfur on the cathode after 190 cycles.76 The Ti 2p spectrum was deconvoluted into four components and the peaks at 455.4 eV (Ti–C, 2p3/2), 456.7 eV (Ti–O, 2p3/2), 459.1 eV (Ti–C, 2p1/2), and 461.9 eV (Ti–O, 2p1/2) are aligned well to the spectrum in Fig. 2. In Li 1s spectra, peaks at 53.9, 54.9, 55.6, and 56.6 denoted Li–C, Li–S, Li–N, and Li–O bond formations, signifying the CN bond involvement in lithium storage in addition to Li–S bonds during discharge.44,77,78 F 1s peaks at 684.4 and 687.6 eV reflected the electrolyte and PVDF passivation, suggesting an interaction between the electrolyte and g-C3N4-Mx/S during cycling. Overall, the XPS analysis of the cycled cell affirmed polysulfide adsorption on the g-C3N4-Mx/S surface.
Conclusions
In summary, we synthesized a novel heterostructure comprising 2D g-C3N4 nanosheets and 2D Ti3C2 MXene to serve as a host matrix for a sulfur cathode in Li–S pouch cells. Analytical findings reveal the g-C3N4-Mx/S composite's elevated pyridine N content, fostering robust interactions with LiPSs. The presence of Ti2+ ions functions as catalytic sites (Sx–Ti⋯Li) for effectively grafting the LiPSs, accelerating conversion kinetics and suppressing the shuttle effect, thereby enhancing electrochemical performance in Li–S batteries. The g-C3N4-Mx/S electrode exhibits outstanding performance, achieving a discharge capacity of 1061 mA h g−1 at C/8 with excellent high-rate and long-term cycling stability (73% capacity retention after 190 cycles). It attains a high initial discharge capacity of 795 mA h g−1 at a high sulfur loading of 5.9 mg cm−2 after 190 cycles (a CE of 83.2%), with a retention rate of 72%. Postmortem XPS and EIS analyses further support the cell's excellent performance by revealing new bond formations and changes in cell resistance. Postmortem XPS investigations indicate a significant reduction in the binding energy peaks of elemental S8, suggesting the accumulation of dead sulfur on the cathode side during cycling. These outcomes illustrate that g-C3N4-Mx/S heterostructures serve as multifunctional polysulfide mediators, chemically adsorbing LiPS, accelerating Li-ion diffusion, catalyzing LiPS conversion, and lowering the energy barrier for Li2S precipitation/decomposition. This realization of the “adsorption–diffusion–conversion” of polysulfides ensures prolonged cycling life and high-rate capability.Author contributions
VKT: methodology, investigation, data curation, validation, and writing – original draft. OTD and AG: formal analysis and conceptualization. RM: methodology, data curation, and visualization. MS: conceptualization, review and editing, supervision, and project administration.Conflicts of interest
All the authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors would like to thank MITACS (Canada), Ms Denise Byrne at Powertrain Engineering Research & Development Centre (PERDC), and Ford Motor Company of Canada for providing financial and in-kind support for this project.References
- J. Xiao, J. Han, C. Zhang, G. Ling, F. Kang and Q. H. Yang, Adv. Energy Mater., 2022, 12, 2100775 CrossRef CAS.
- M. Jafari, A. Botterud and A. Sakti, Renewable Sustainable Energy Rev., 2022, 158, 112077 CrossRef CAS.
- J. Xu, X. Cai, S. Cai, Y. Shao, C. Hu, S. Lu and S. Ding, Energy Environ. Mater., 2023, 6, e12450 CrossRef CAS.
- F. M. N. U. Khan, M. G. Rasul, A. S. M. Sayem and N. K. Mandal, J. Energy Storage, 2023, 71, 108033 CrossRef.
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed.
- G. Zhou, H. Chen and Y. Cui, Nat. Energy, 2022, 7, 312–319 CrossRef CAS.
- W. Xue, Q. B. Yan, G. Xu, L. Suo, Y. Chen, C. Wang, C. A. Wang and J. Li, Nano Energy, 2017, 38, 12–18 CrossRef CAS.
- M. Wang, Z. Bai, T. Yang, C. Nie, X. Xu, Y. Wang, J. Yang, S. Dou and N. Wang, Adv. Energy Mater., 2022, 12, 2201585 CrossRef CAS.
- T. Liu, H. Hu, X. Ding, H. Yuan, C. Jin, J. Nai, Y. Liu, Y. Wang, Y. Wan and X. Tao, Energy Storage Mater., 2020, 30, 346–366 CrossRef.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- H. Liang, Z. Zeng, Z. Qiao, Y. Li and C. Wang, Phys. Chem. Chem. Phys., 2024, 26, 5858–5867 RSC.
- V. K. Tomer, R. Malik, J. Tjong and M. Sain, Coord. Chem. Rev., 2023, 481, 215055 CrossRef CAS.
- A. Benítez, J. Amaro-Gahete, Y. C. Chien, Á. Caballero, J. Morales and D. Brandell, Renewable Sustainable Energy Rev., 2022, 154, 111783 CrossRef.
- J. Du, Q. Li, J. Chai, L. Jiang, Q. Zhang, N. Han, W. Zhang and B. Tang, Dalton Trans., 2022, 51, 9584–9590 RSC.
- H. Wang, N. Deng, S. Wang, X. Wang, Y. Li, Q. Zeng, S. Luo, X. Cui, B. Cheng and W. Kang, J. Mater. Chem. A, 2022, 10, 23433–23466 RSC.
- S. Huang, Z. Wang, Y. Von Lim, Y. Wang, Y. Li, D. Zhang and H. Y. Yang, Adv. Energy Mater., 2021, 11, 2003689 CrossRef CAS.
- R. Yan, T. Ma, M. Cheng, X. Tao, Z. Yang, F. Ran, S. Li, B. Yin, C. Cheng and W. Yang, Adv. Mater., 2021, 33, 2008784 CrossRef CAS PubMed.
- Z. J. Zheng, H. Ye and Z. P. Guo, Energy Environ. Sci., 2021, 14, 1835–1853 RSC.
- W. Sun, Z. Song, Z. Feng, Y. Huang, Z. J. Xu, Y. C. Lu and Q. Zou, Nano-Micro Lett., 2022, 14, 1–23 CrossRef PubMed.
- D. Nepak, V. K. Tomer and K. Kailasam, in Metal-free Functionalized Carbons in Catalysis, ed. A. Villa and N. Dimitratos, Royal Society of Chemistry, UK, 2018, pp. 67–102 Search PubMed.
- R. Malik, V. K. Tomer and N. Joshi, ACS Appl. Mater. Interfaces, 2018, 10, 34087–34097 CrossRef CAS PubMed.
- S. A. Thomas, M. R. Pallavolu, M. E. Khan and J. Cherusseri, J. Energy Storage, 2023, 68, 107673 CrossRef.
- D. Zhao, Z. Xu, X. Yu, M. Chen, O. Wu, K. Zhou, W. Zhou, L. Ma and N. Wang, Chem. Eng. J., 2023, 474, 145983 CrossRef CAS.
- H. Liu, X. Li, L. Ma, F. Sun, B. Yue, Q. Ma, J. Wang, G. Liu, H. Yu, W. Yu, X. Dong and Z. Su, Colloids Surf., A, 2023, 670, 131572 CrossRef CAS.
- Y. Chen, Y. Wu, L. Li, Y. Liao, S. Luo, Y. Wu and Y. Qing, Chem. Eng. J., 2023, 461, 141988 CrossRef CAS.
- Y. Zou, H. Zou, Z. Ao, Y. Lv, N. Chen and Y. Huang, J. Alloys Compd., 2023, 940, 168772 CrossRef CAS.
- W. W. Liu, S. T. Niu, Z. Q. Xu, R. Zou, C. Y. Cui, Y. X. Lei, X. B. Zhang and F. Ran, Appl. Surf. Sci., 2023, 609, 155327 CrossRef CAS.
- L. Liao, S. Wang, H. Duan and Y. Deng, J. Energy Storage, 2024, 75, 109555 CrossRef.
- Y. Wang, T. Guo, E. Alhajji, Z. Tian, Z. Shi, Y. Z. Zhang and H. N. Alshareef, Adv. Energy Mater., 2023, 13, 2202860 CrossRef CAS.
- Y. P. Zhu, Y. Lei, F. Ming, E. Abou-Hamad, A. H. Emwas, M. N. Hedhili and H. N. Alshareef, Nano Energy, 2019, 65, 104030 CrossRef CAS.
- E. Abd-Alkuder Salman, K. Abaid Samawi, M. Fawzi Nassar, G. Abdulkareem-Alsultan and E. Abdulmalek, J. Electroanal. Chem., 2023, 945, 117629 CrossRef CAS.
- Y. Zhang, Q. Lu, L. Zhang, L. Zhang, G. Shao and P. Zhang, Small Struct., 2024, 5, 2300255 CrossRef CAS.
- Y. Zhang, C. Ma, W. He, C. Zhang, L. Zhou, G. Wang and W. Wei, Prog. Nat. Sci.: Mater. Int., 2021, 31, 501–513 CrossRef CAS.
- H. Kwon, J. Baek and H. T. Kim, Energy Storage Mater., 2023, 55, 708–726 CrossRef.
- H. Shi, W. Sun, J. Cao, S. Han, G. Lu, Z. A. Ghazi, X. Zhu, H. Lan and W. Lv, Adv. Funct. Mater., 2023, 33, 2306933 CrossRef CAS.
- M. Zhao, B. Q. Li, H. J. Peng, H. Yuan, J. Y. Wei and J. Q. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636–12652 CrossRef CAS PubMed.
- C. Y. Kung and S. H. Chung, Mater. Horiz., 2023, 10, 4857–4867 RSC.
- Y. C. Wu and S. H. Chung, J. Mater. Chem. A, 2023, 11, 9455–9463 RSC.
- R. Malik, N. Joshi and V. K. Tomer, Coord. Chem. Rev., 2022, 466, 214611 CrossRef CAS.
- J. Liu, H. Wang and M. Antonietti, Chem. Soc. Rev., 2016, 45, 2308–2326 RSC.
- T. Su, Z. D. Hood, M. Naguib, L. Bai, S. Luo, C. M. Rouleau, I. N. Ivanov, H. Ji, Z. Qin and Z. Wu, Nanoscale, 2019, 11, 8138–8149 RSC.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1–17 Search PubMed.
- V. K. Tomer, N. Thangaraj, S. Gahlot and K. Kailasam, Nanoscale, 2016, 8, 19794–19803 RSC.
- Y. Hu, W. Chen, T. Lei, Y. Jiao, J. Huang, A. Hu, C. Gong, C. Yan, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 2000082 CrossRef CAS.
- V. K. Tomer, K. Singh, H. Kaur, M. Shorie and P. Sabherwal, Sens. Actuators, B, 2017, 253, 703–713 CrossRef CAS.
- J. H. Kim, H. J. Eun, J. Jang, S. Eom, J. H. Ahn, M. Wu, J. Suk and S. Moon, ChemElectroChem, 2023, 10, e202300019 CrossRef CAS.
- X. Zhang, T. Yang, Y. Zhang, X. Wang, J. Wang, Y. Li, A. Yu, X. Wang and Z. Chen, Adv. Mater., 2023, 35, 2208470 CrossRef CAS PubMed.
- S. Duhan and V. K. Tomer, in Advanced Sensor and Detection Materials, ed. A. Tiwari and M. M. Demir, John Wiley and Sons Ltd, 2014, pp. 149–192 Search PubMed.
- H. Zou, Y. Zou, Y. Lv, Z. Ao, N. Chen and Y. Huang, ACS Appl. Energy Mater., 2022, 5, 10067–10075 CrossRef CAS.
- Y. Du, R. Huang, X. Lin, S. Khan, B. Zheng and R. Fu, Langmuir, 2020, 36, 14507–14513 CrossRef CAS PubMed.
- J. H. Zeng, Y. F. Wang, S. Q. Gou, L. P. Zhang, Y. Chen, J. X. Jiang and F. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 34783–34792 CrossRef CAS PubMed.
- J. Jiang, H. Wang, J. Zhao, J. Li, G. Liu and Y. Zhang, J. Alloys Compd., 2022, 899, 163240 CrossRef CAS.
- X. Wen, K. Xiang, Y. Zhu, L. Xiao, H. Liao, W. Chen, X. Chen and H. Chen, J. Alloys Compd., 2020, 815, 152350 CrossRef CAS.
- J. W. Park, S. C. Jo, M. J. Kim, I. H. Choi, B. G. Kim, Y. J. Lee, H. Y. Choi, S. Kang, T. Y. Kim and K. J. Baeg, NPG Asia Mater., 2021, 13, 1–12 CrossRef.
- J. He and A. Manthiram, Energy Storage Mater., 2019, 20, 55–70 CrossRef.
- W. Deng, J. Phung, G. Li and X. Wang, Nano Energy, 2021, 82, 105761 CrossRef CAS.
- O. Leonet, Á. Doñoro, A. Fernández-Barquín, A. Kvasha, I. Urdampilleta and J. A. Blázquez, Front. Chem., 2022, 0, 420 Search PubMed.
- D. Li, J. Liu, W. Wang, S. Li, G. Yang, P. Wang, K. Zhu and Z. Li, Appl. Surf. Sci., 2021, 569, 151058 CrossRef CAS.
- Q. Zhao, Q. Zhu, Y. Liu and B. Xu, Adv. Funct. Mater., 2021, 31, 2100457 CrossRef CAS.
- S. J. Kim, Y. Jeoun, J. Park, S. H. Yu and Y. E. Sung, Nanoscale, 2020, 12, 15466–15472 RSC.
- J. Shen, X. Xu, J. Liu, Z. Liu, F. Li, R. Hu, J. Liu, X. Hou, Y. Feng, Y. Yu and M. Zhu, ACS Nano, 2019, 13, 8986–8996 CrossRef CAS PubMed.
- L. Ding, Q. Lu, A. D. C. Permana, S. Ostwald, M. Hantusch, K. Nielsch and D. Mikhailova, Energy Technol., 2021, 9, 2001057 CrossRef CAS.
- D. Qu, G. Wang, J. Kafle, J. Harris, L. Crain, Z. Jin, D. Zheng, D. Qu, G. Wang, J. Kafle, J. Harris, L. Crain, D. Zheng and Z. Jin, Small Methods, 2018, 2, 1700342 CrossRef.
- J. T. Kim, A. Rao, H. Y. Nie, Y. Hu, W. Li, F. Zhao, S. Deng, X. Hao, J. Fu, J. Luo, H. Duan, C. Wang, C. V. Singh and X. Sun, Nat. Commun., 2023, 14, 1–10 Search PubMed.
- Y. Xie, J. Ao, L. Zhang, Y. Shao, H. Zhang, S. Cheng and X. Wang, Chem. Eng. J., 2023, 451, 139017 CrossRef CAS.
- D. Wang, K. Ma, J. Hao, W. Zhang, C. Wang, C. Xu, H. Shi, Z. Ji, X. Yan and Y. Gu, Nano Energy, 2021, 89, 106426 CrossRef CAS.
- H. Zhang, Q. Liu, S. Ruan, C. Ma, X. Jia, W. Qiao, L. Ling and J. Wang, Appl. Surf. Sci., 2022, 578, 152022 CrossRef CAS.
- J. Zhang, J. Y. Li, W. P. Wang, X. H. Zhang, X. H. Tan, W. G. Chu and Y. G. Guo, Adv. Energy Mater., 2018, 8, 1702839 CrossRef.
- S. Fu, H. Wang, Y. Zhong, S. Schaefer, M. Li, M. Wu and H. Wang, Adv. Mater., 2023, 35, 2302771 CrossRef CAS PubMed.
- S. S. Zhang, J. Electrochem. Soc., 2012, 159, A920–A923 CrossRef CAS.
- H. Pan, K. S. Han, M. H. Engelhard, R. Cao, J. Chen, J.-G. Zhang, K. T. Mueller, Y. Shao, J. Liu, H. Pan, K. S. Han, R. Cao, J. Chen, J.-G. Zhang, K. T. Mueller, Y. Shao, J. Liu and M. H. Engelhard, Adv. Funct. Mater., 2018, 28, 1707234 CrossRef.
- T. Pakki, E. H. Mohan, N. Y. Hebalkar, J. Adduru, S. V. Bulusu, A. Srinivasan, K. M. Mantravadi and N. R. Tata, J. Mater. Sci., 2019, 54, 9075–9087 CrossRef CAS.
- R. Li, Y. Zeng, L. Song, J. Lv, C. Wang, C. Zhou, S. Cai, T. Chen, S. Yue, K. Ma and H. Yue, Small, 2023, 2305283 Search PubMed.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Electrochem. Soc., 2012, 159, A1816–A1821 CrossRef CAS.
- S. Ponnada, M. S. Kiai, D. B. Gorle and A. Nowduri, Nanoscale Adv., 2021, 3, 4492–4501 RSC.
- X. Gao, C. Zheng, Y. Shao, V. R. Shah, S. Jin, J. Suntivich and Y. L. Joo, ACS Appl. Mater. Interfaces, 2023, 15, 19011–19020 CrossRef CAS PubMed.
- F. Zhao, J. Xue, W. Shao, H. Yu, W. Huang and J. Xiao, J. Energy Chem., 2023, 80, 625–657 CrossRef CAS.
- M. Zhang, W. Chen, L. Xue, Y. Jiao, T. Lei, J. Chu, J. Huang, C. Gong, C. Yan, Y. Yan, Y. Hu, X. Wang and J. Xiong, Adv. Energy Mater., 2020, 10, 1903008 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison with previously published works and results obtained from Nyquist plots; BET sorption isotherms; TGA; CV cycling performance; complete XPS scan and EIS spectra for the cycled cell. See DOI: https://doi.org/10.1039/d4mh00200h |
| This journal is © The Royal Society of Chemistry 2024 |