
Phase stabilization via A-site ion anchoring for ultra-stable perovskite light emitting diodes†
Shuo
Ding
 abc,
Zhuoyuan
Kong
abd,
Yipeng
Shen
abd,
Piaoyang
Shen
ab,
Chunyan
Wu
ab,
Lei
Qian
ab,
Xinyu
Zhang
c,
Long
Hu
e,
Hao
Chen
*c and
Chaoyu
Xiang
*ab
abc,
Zhuoyuan
Kong
abd,
Yipeng
Shen
abd,
Piaoyang
Shen
ab,
Chunyan
Wu
ab,
Lei
Qian
ab,
Xinyu
Zhang
c,
Long
Hu
e,
Hao
Chen
*c and
Chaoyu
Xiang
*ab
aLaboratory of Optoelectronic Information Materials and Devices, Ningbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, Zhejiang 315201, China. E-mail: xiangchaoyu@nimte.ac.cn
bHangzhou Bay Laboratory of Advanced Nano-Optoelectronic Materials and Devices, Qianwan Institute of CNITECH, Ningbo, Zhejiang 315300, China
cDepartment of Mechanical, Materials and Manufacturing Engineering, The University of Nottingham Ningbo China, Ningbo, Zhejiang 315100, China. E-mail: hao.chen@nottingham.edu.cn
dNano Science and Technology Institute, University of Science and Technology of China, Suzhou 215123, China
eSchool of Materials Science and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 6th August 2024
Abstract
A novel ion anchoring strategy stabilizes the perovskite phase, yielding ambient stable perovskite films and ultra-stable perovskite light-emitting diodes (PeLEDs) with an unprecedented operational half-lifetime over 37.2 years at 100 cd m−2 and exceeding 27% efficiency, marking a new stability benchmark for next-generation display and lighting applications.
New conceptsPerovskite light emitting diodes (PeLEDs) hold immense promise for next generation display technology, yet the limited operational stability still hinders their applications. The degradation of perovskites is prominently mediated through phase transition from the luminescent perovskite phase into non-luminescent phases via ion migration and decomposition. Therefore, we demonstrated a novel method for phase stabilization via ion anchoring. With a highly fluorinated molecule, 1H,1H-perfluorohexylamine (PFHA), strong coordination with all perovskite ions spontaneously is established, thereby achieving significant ion immobilization and phase stabilization. Thereby, a highly stable perovskite film was fabricated maintaining a photoluminescent quantum yield (PLQY) exceeding 95% over a period of 28 days under ambient conditions. Moreover, ultra-stable and efficient green PeLEDs with a champion external quantum efficiency (EQE) of 27.1% and an equivalent T50 lifetime over 326![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 252 hours (37.2 years) at 100 cd m−2 were fabricated, thereby setting a new benchmark across all classes of PeLEDs. This work confirms the stability potential of PeLEDs and charts a way for PeLED applications in display and lighting industries. 252 hours (37.2 years) at 100 cd m−2 were fabricated, thereby setting a new benchmark across all classes of PeLEDs. This work confirms the stability potential of PeLEDs and charts a way for PeLED applications in display and lighting industries.
|
Introduction
Metal halide perovskites (MHPs) have emerged as highly promising electroluminescent materials attributed to their tuneable bandgap characteristics, exceptional photoluminescent properties, and facile, cost-effective solution processability.1 The external quantum efficiency (EQE) of perovskite light-emitting diodes (PeLEDs) has experienced remarkable advancements since the pioneering report of room-temperature application in 2014,2 approaching 30% without the implementation of optical outcoupling,3,4 thereby underscoring a compelling potential for optoelectronic applications. Despite these breakthroughs, considerable challenges persist regarding the operational stability of PeLEDs, as evidenced by the operational lifetimes of typical high-efficiency devices confined to the order of hours or even minutes.5,6 Therefore, to align with the requisites of display and lighting applications, it is imperative to achieve critical enhancements in their operational lifetime.In summary, MHPs constitute a category of ionic materials characterized by their ABX3 crystal structure, wherein A represents monovalent cations such as methylammonium (MA+), formamidinium (FA+), or Cs+, B denotes divalent metal cations predominantly Pb2+ or Sn2+, and X symbolizes halide anions. The degradation of organometal halide perovskites is typically mediated through the phase transition, characterized by the conversion of luminescent perovskite phases into non-luminescent PbX2 phases, as shown in Fig. 1(A). This transition is fundamentally rooted in the ionic constitutions with a relatively flexible lattice framework and consequent high ion mobility of MHPs.7 Early studies have quantified the migration abilities of different ions with theoretical computations or in situ experimental measurements, whereas the ion migration activation energies of A-site, B-site, and X-site ions were found to be 0.4–0.8 eV, >2 eV and 0.1–0.6 eV, respectively,8–11 underscoring the importance of suppressing ion migration of both A-site and X-site ions. However, extensive research endeavours have been dedicated to X-site halide migration suppression to date,12–15 yet the study of A-site ion migration suppression remains relatively nascent.16 Moreover, A-site ions, particularly MA+ and FA+, tend to decompose upon exposure to external stimuli such as moisture and oxygen, which inevitably triggers the degradation of the MHP film.17 Consequently, to decisively confront the phase stability of MHPs and the stability challenges of PeLEDs, there arises a pressing requirement to develop a straightforward methodology to stabilize all perovskite ions spontaneously, with special focus on the stabilization of A-site ions.
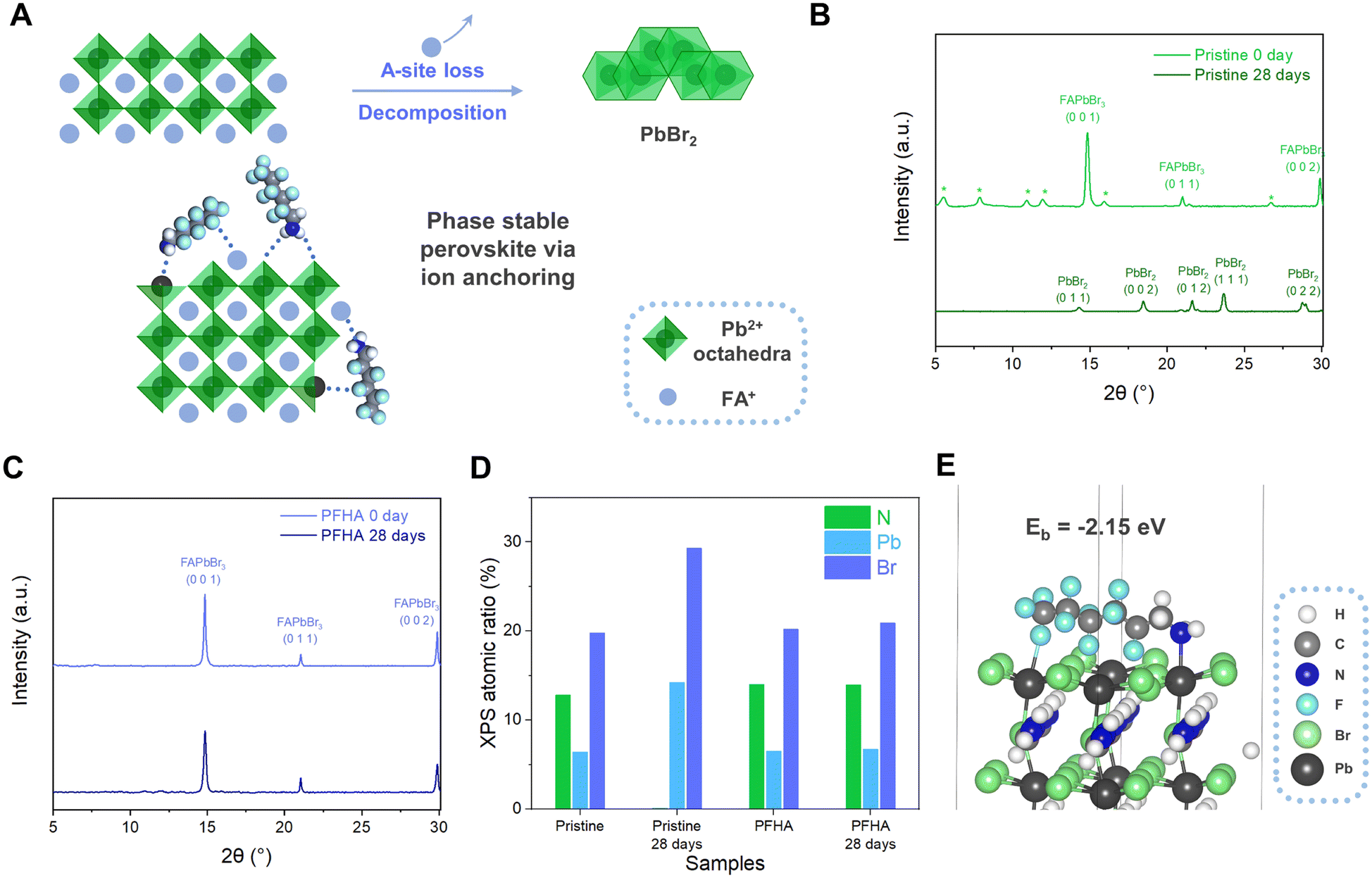 | ||
| Fig. 1 Phase stabilization via an ion anchoring effect. (A) Typical degradation process of the organometal halide perovskite and scheme of the ion anchoring effect. (B) XRD patterns of the pristine perovskite film before and after ambient storage for 28 days (*: low dimensional phases). (C) XRD patterns of the PFHA anchored perovskite film before after storage in ambient conditions for 28 days. (D) XPS atomic ratio changes for the pristine and PFHA perovskite film before after ambient storage for 28 days. (E) Representative configuration of PFHA coordination on the FAPbBr3 perovskite (0 0 1) surface. | ||
In this work, we demonstrated a simple and efficacious phase stabilization method via an ion anchoring strategy, thereby hindering ion migration, and substantially improving the operational lifetime of PeLEDs. To this end, we selected a highly fluorinated molecule, 1H,1H-perfluorohexylamine (PFHA), to establish strong coordination with all perovskite ions spontaneously, particularly with A-site ions, and effectively stabilize the perovskite phase and inhibit the ion migration process (Fig. 1(A)). Therefore, by PFHA anchoring, we successfully fabricated exceptionally stable MHP films, maintaining a photoluminescent quantum yield (PLQY) exceeding 95% over a period of 28 days under ambient conditions. Moreover, ultra-stable and efficient green PeLEDs were fabricated with a champion EQE of 27.1% and a T50 lifetime of 103.17 hours at 10000 cd m−2 (equivalent to over 326252 hours or 37.2 years at 100 cd m−2), thereby setting a new benchmark across all classes of PeLEDs. This work not only substantiates the stability potential of PeLEDs, but also charts a pathway for their eventual industrialization for display and lighting applications.
Experimental
Materials
Molybdenum oxide (MoOx, 99.9995%) was purchased from Alfa Aesar. Lead bromide (PbBr2, 99.999%) and polyvinyl pyrrolidone (PVP, average molecular weight 40000) were purchased from Sigma. Ethanolamine (99%) was purchased from Energy Chemical. Formamidine hydrobromide (FABr, 99%), 4-fluoro-phenethylammonium bromide (FPEABr, 99%), methylammonium bromide (MABr, 99%) and lithium fluoride (LiF, 99.9%) were purchased from Xi’an Polymer Light. 1H,1H-Perfluorohexylamine (PFHA, 97%), 1H,1H-perfluorohexylaminium bromide (PFHABr), chlorobenzene (CB, 99.8%), carbon tetrachloride (CT, 99%) and potassium trifluoromethanesulfonate (CF3SO3K, 98%) were purchased from Macklin. Nickel acetate tetrahydrate (99.9%) was purchased from Acros. Hexylamine (HA, 99%) was purchased from TCI. N-Methyl pyrrolidone (NMP, 99.9%) was purchased from Aladdin. Ethanol (99.5%) was purchased from Meryer. 2,4,6-Tris(3′-(pyridin-3-yl)biphenyl-3-yl)-1,3,5-triazine (TmPPPyTz) and 2-(4-(9,10-di(naphthalen-2-yl)anthracen-2-yl)phenyl)-1-phenyl-1H-benzo[d]imidazole (ADN-PBIP) were purchased from Lumtec. All chemicals were used as received without purification.
Preparation of the perovskite precursor
The perovskite precursor solution was prepared by dissolving FPEABr, FABr, and PbBr2 (0.5 mmol mL−1) and MABr, with a molar ratio of 2:3:4:0.05 in NMP. 2 mg ml−1 CF3SO3K was added as a solid in the solution. Then, the solution was stirred overnight and filtered with a 0.22 μm PTFE filter before use.
Device fabrication
For PeLEDs, pre-patterned indium tin oxide (ITO) glass substrates (HT15, 15 Ω sq−1, Liaoning Advanced Election Technology Co., Ltd) were cleaned by ultrasonication in deionized water, acetone, and isopropanol for 15 min, respectively. The substrates were dried with nitrogen flow, followed by ultraviolet ozone treatment for 5 min. Nickel oxide (NiOx) was prepared by spin-coating the precursor (prepared by dissolving nickel acetate tetrahydrate (24.886 mg mL−1) and ethanolamine (6.108 μL mL−1) in ethanol and stirring at 65 °C for 4 hours) onto the substrates at 6000 rpm and annealed at 250 °C for 1 hour. PVP (2 mg mL−1 in DMF) was then spin-coated at 8000 rpm and annealed at 120 °C for 10 minutes. The substrates were then transferred to a nitrogen-filled glove box and the perovskite film was spin-coated from the precursor at 4000 rpm for 3 min at a controlled temperature of 16 ± 1 °C and annealed at 90 °C for 10 min. During the spin-coating of the perovskite film, 200 μL of CB was gently dropped as an anti-solvent 45 s after the start of spinning. Then, for ion anchoring devices, 100 μL PFHA dissolved in CB (2% by volume) was dropped onto the perovskite film for 2 s, spin-coated at 10000 rpm for 1 min, and annealed at 90 °C for 10 min to remove residual solvent. Finally, the substrates were transferred to a vacuum chamber and TmPPPyTz (0.1 Å s−1, 5 nm), ADN-PBIP (0.2 Å s−1, 40 nm), LiF (0.05 Å s−1, 1 nm), Al (1 Å s−1, 80 nm), and Ag (0.2 Å s−1, 20 nm) were sequentially evaporated through a shadow mask. The effective area was 0.04 cm2, defined by the overlapping area of the ITO and metal electrodes.
Characterization
Fourier transform infrared spectroscopy (FTIR) was performed with a Thermal Fisher IS50 Fourier transform infrared spectrometer. The FTIR samples were prepared by dissolving either PFHA 19.1 μL mL−1 (0.1 mmol mL−1), FABr 12.5 mg mL−1 (0.1 mmol mL−1), PbBr2 36.7 mg mL−1 (0.1 mmol mL−1) or a binary combination of them in CT, and were vortex stirred and filtered with 0.45 μm PVDF filters before measurements. XPS spectra were acquired with a Kratos AXIS ULTRA X-ray photoelectron spectrometer, wherein the control sample of F 1s XPS spectra is made by spin coating PFHABr (20 mg mL−1 in DMSO) onto an ITO substrate at 1000 rpm for 60 s, followed by 100 °C annealing for 10 min to remove all the residual solvents. SEM images of the perovskite films and EDX spectroscopy of the perovskite/PCBM surface were obtained using a HITACHI Regulus 8230 instrument. Capacitance–frequency (C–F) was measured with a Biologic-SP 50e electrochemical workstation. PLQY was measured by a Hamamatsu C9920-2 absolute PL quantum yield spectrometer. All the samples for PLQY tests were fabricated with the structure of ITO/NiOx/PVP/perovskite or ITO/NiOx/PVP/perovskite/TmPPPyTz/ADN-PBIP (with an electron transporting layer (ETL)), unless otherwise specified. X-Ray diffraction (XRD) was measured with a BRUKER D8 AdVance.The electroluminescence performance of the devices was measured with the same setup reported before.18 The electroluminescence spectra were acquired by an Ocean Optics USB 2000+ spectrometer at a constant current mode using a Keithley 2400 source meter. The J–L–V curves were measured under ambient conditions, where a Keithley 6485 picoammeter and a calibrated silicon detector (Edmund) measured the light intensities, and a Keithley 2400 source meter measured the sweep voltages and currents. Luminance was calibrated with a photometer (Spectra Scan PR655) assuming a Lambertian emission pattern. The operational lifetime test was conducted under ambient conditions at room temperature (20 ± 2 °C) using a commercialized lifetime test system (Guangzhou Jinghe Equipment Co., Ltd). The devices were encapsulated with capping glasses and Nagase UV epoxy resin XNR5516Z(C)-SA1.
Computational methods
Ab initio simulation was performed with the Cambridge Sequential Total Energy Package (CASTEP, ver. 20.1.1).19 All geometry optimizations were performed with the generalized gradient approximation (GGA) Perdew Burke Ernzerhof (PBE) exchange–correlation functional under Koelling–Harmon scalar relativistic treatment, with on-the-fly generated (OTFG) ultrasoft pseudopotentials. In addition, the cutoff energy was selected as 600 eV, while the total energy, maximum displacement, maximum stress, and maximum force were set to 5 × 10−7 eV per atom, 5 × 10−4 Å, 0.02 GPa and 0.01 eV Å−1, respectively, during convergence. 4 × 4 × 4 Monkhorst–Pack k-point grids were applied for the perovskite lattice and molecule geometry optimization, while 2 × 2 × 4 superlattice surface models with a 30 Å vacuum slab were further geometrically optimized by 2 × 2 × 1 k-point sampling. The binding energy was defined by the formula of Eb = |EPerovskiteswithmolecule| − (EMolecule + EPerovskite).
Results and discussion
As shown in Fig. 1(A), the degradation of the perovskite phase is typically mediated through the phase transition, characterized by the conversion of luminescent perovskite phases into non-luminescent PbX2 counterparts. This transformation is clearly evidenced by X-ray diffraction (XRD) analysis in Fig. 1(B). The scattering patterns of the as-prepared pristine perovskite film manifested definitive peaks corresponding to the perovskite phase initially, yet upon 28 days of ambient storage (at 20 °C and 50% relative humidity), the XRD spectra indicated a comprehensive shift to the PbBr2 phase, indicating the intrinsic susceptibility of the perovskite phase transition. This process can be further fortified with X-ray photoelectron spectroscopy (XPS) atomic ratio results, as revealed in Fig. 1(D). Herein, an absence of nitrogen element of the pristine perovskite film, while a reduction in the Br-to-Pb atomic ratio from approximately 3.09:1 to 2.06:1 upon 28 days of storage, was noted, corroborating the phase transition from a luminescent perovskite to non-luminescent PbBr2. The loss of nitrogen, a key indicator of A-site ions inside organometal halide perovskites, has presented the critical routine for the understanding of the perovskite degradation process.
Collectively, these findings have highlighted the centrality of phase stabilization in the pursuit of long-term stable perovskite-based photoelectronic devices. Evidently, maintaining the luminescent phase stability is paramount to the fabrication of ultra-stable PeLEDs, necessitating the development of efficacious methodologies for phase stabilization. The phase transition of the perovskite is predominantly rooted in the profound ion mobilities and subsequent severe ion migration.20 To address this challenge, we have developed an effective ion anchoring strategy to spontaneously immobilize all perovskite ions, particularly A-site ions, and achieved significant phase stabilization effect. Meanwhile, critical phase stabilization was achieved with our ion anchoring strategy. As evidenced by the XRD measurement, the PFHA anchored perovskites retained their phase structure, exhibiting unaltered XRD patterns even following a 28-day exposure to ambient conditions, as shown in Fig. 1(C). Notably, the disappearance of the low dimensional phases is attributed to a process known as “solvent sieve”, wherein polar solvents selectively dissolve and remove low dimensional phases, thereby leaving perovskites with higher dimensional perovskite structures.4 This effect can also be confirmed with UV-Vis spectroscopy, where the peaks for low dimensional phases totally disappeared after PFHA treatment, as presented in Fig. S1 (ESI†). Meanwhile, the superior A-site ion stabilization effect as well as enhanced material stability compared to the pristine sample can be validated based on XPS element atomic ratio. For PFHA samples, the N element ratio, as well as the Br-to-Pb atomic ratio, exhibit remarkable stability, without discernible changes over the same 28-day storage duration, as shown in Fig. 1(D). This preservation of the A-site ions has signified a marked improvement in phase stability after PFHA anchoring.
The ion anchoring effect of the PFHA molecule is achieved via strong coordination with all the perovskite ions. Specifically, the N atoms in the amine group function as strong electron donors, fostering substantial coordination with cations, such as Pb ions, with the N–H serving as a hydrogen bond donor to anions. Furthermore, the highly fluorinated alkyl appendage of PFHA facilitates additional coordination to both A-site and B-site cations through the formation of robust hydrogen and coordination bonds. Therefore, this ion anchoring strategy, coordinated with both anions and cations within the perovskite lattice, markedly inhibited ion migration effects. In addition, the large van der Waals radius of the fluorocarbon functional group21 exhibited significant steric hindrance, thereby further hindering ion migration and enhancing phase stability of MHPs.
The coordination ability of PFHA to the perovskite could be assessed with theoretical simulation based on density functional theory (DFT).19 Our investigation initiated with the examination of the interaction between PFHA molecules and single ions such as FA+. Based on the results, the typical binding energy (Eb) of PFHA coordination with FA+ reached −1.18 eV, as presented in Fig. S2 (ESI†), underscoring an exceedingly strong affinity between PFHA molecules and FA+ ions. Meanwhile, the DFT simulation also validated the anchoring effect of PFHA on the perovskite structures. For a representative surface binding configuration, the binding energy of the PFHA molecule to the perovskite (0 0 1) surface was −2.15 eV, suggesting an efficient coordination ability of PFHA to the perovskite surface, as shown in Fig. 1(E). All the strong coordinations to individual ion and perovskite structures have established a grounded foundation for our efficacious ion anchoring strategy.
Then, Fourier transform infrared spectrometer (FTIR) tests were employed to unveil the interaction between PFHA and different perovskite ions comprehensively. We dissolved PFHA together with perovskite ionic components, formamidine hydrobromide (FABr) and lead bromide (PbBr2), in the carbon tetrachloride (CT) solvent, as shown in Fig. 2(A). Here, CT was applied as a background solvent for FTIR evaluations due to its absence of peaks within the wave number range of 1000 to 4000 cm−1. The characteristic peaks for C–F stretching can be observed at the wavenumber of 1239.5 cm−1 for pure PFHA molecules in CT, while considerable downshifts could be observed with the addition of either FABr or PbBr2, which suggested that the electron lone pairs of F atoms were denoted to cations. Similarly, downshifts of C–N bond stretching at the wavenumber of 1141.7 cm−1 for PFHA samples were also found after FABr and PbBr2 addition, indicating the coordination between N atoms and cations. Meanwhile, significant upshifts of the N–H bending peaks also occurred in the ion-supplemented samples, indicating that hydrogen bonds were formed between anions and amine functional groups. Therefore, all the shifts of the characteristic FTIR peaks had confirmed the strong coordination between PFHA molecules and all kinds of perovskite ionic components in solution form, indicating a superior potential for ion anchoring through PFHA.
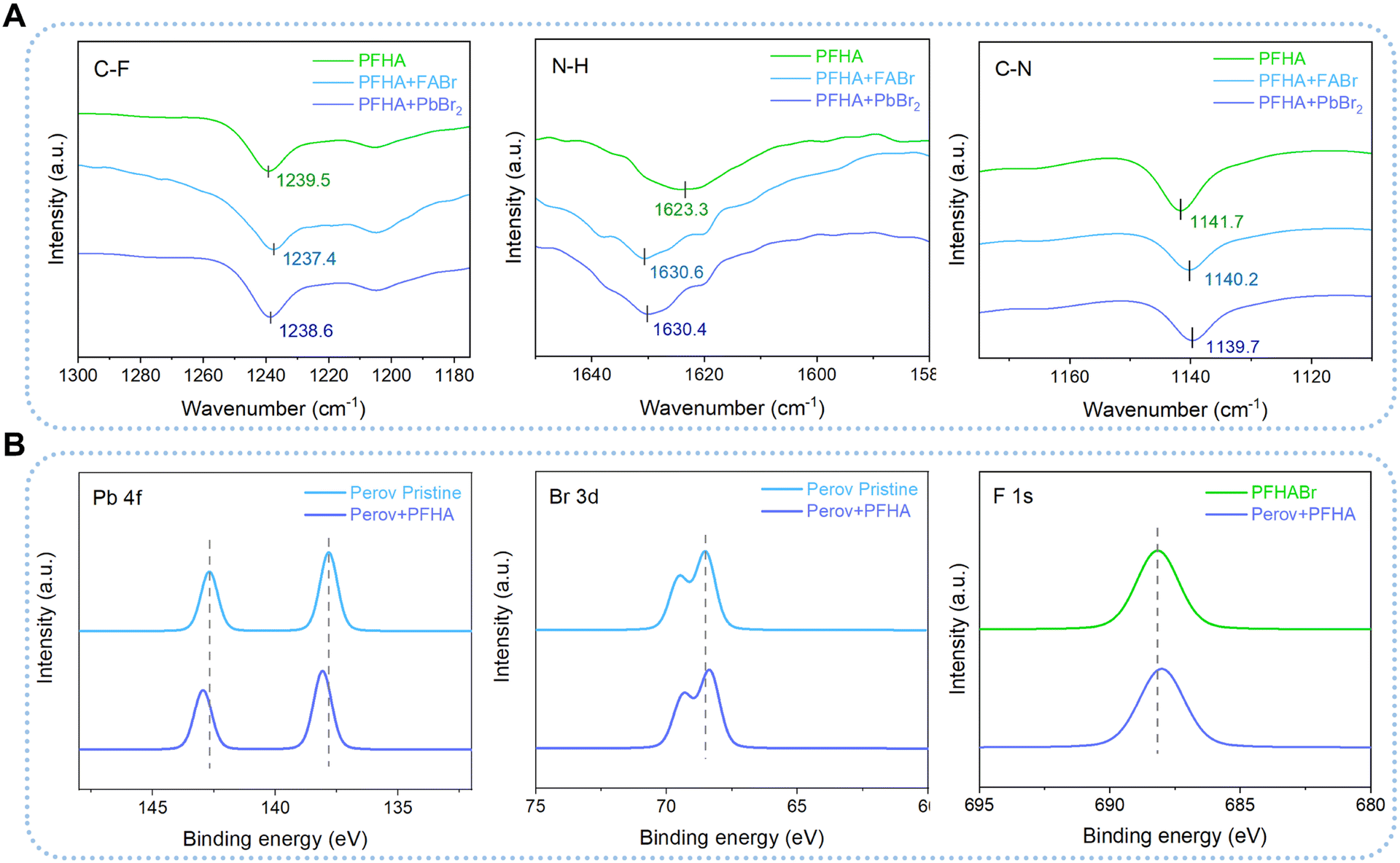 | ||
| Fig. 2 Anchoring between PFHA and ions. (A) FTIR results of pristine and PFHA-containing mixed systems. (B) XPS spectra for Pb 4f, Br 3d and F 1s orbitals of pristine and anchored MHPs. | ||
To comprehensively evaluate the robustness and efficacy of ion anchoring within the perovskite films, we then performed XPS of pristine and PFHA anchored perovskite films. The observed shifts in XPS spectra are attributed to the distinct chemical environments experienced by atoms in the PFHA-treated perovskite films versus the control samples. As shown in Fig. 2(B), the XPS spectra revealed apparent shifts in the binding energies of the critical elements presented in the perovskite films. Specifically, the binding energy of the Pb 4f peak underwent an upward shift post-PFHA anchoring compared to the pristine MHPs, attributed to the formation of additional coordination bonds between Pb ions and F atoms introduced by the PFHA molecules. Due to the highest electronegativity of fluorine atoms, the electron density redistribution from Pb to F atoms compared with the Pb to Br atoms only in the pristine samples, has led to the observed upshift in the Pb 4f peak, confirming the establishment of coordination bonds between PFHA molecules and Pb ions within the perovskite films. Meanwhile, for Br and F atoms, the binding energies of both Br 3d and F 1s were observed to undergo downward shifts. These shifts are indicative of the formation of hydrogen-bond-like interactions between Br atoms and the amine groups of the PFHA, as well as hydrogen bonds between F atoms and formamidinium ions, respectively. Collectively, these findings have demonstrated that PFHA could retain significant coordination to perovskite ions even in solid state films, allowing for significant ion anchoring and consequent phase stabilization.
Although it is renowned that perovskites are highly sensitive to external stimuli such as ambient air and moisture, after ion anchoring, the perovskite films exhibit impressive stability. Fig. S3 (ESI†) showcases the photoluminescence (PL) spectra of both pristine and PFHA perovskites, stored under ambient conditions over a period of 28 days (approximately 20 °C with 50% relative humidity) without any encapsulation. During the storage, the pristine perovskite sample exhibited a complete loss of photoluminescence, indicating the degradation of perovskite phases. In contrast, the PFHA perovskite retained its photoluminescence properties almost unchanged throughout the same period, underscoring the remarkable stability enhancement via PFHA ion anchoring. Meanwhile, the narrowing of the PL spectra is also in good agreement with the improved luminescent phase purity with the removal of low dimensional phases,4,22,23 as well as the decreased defect density achieved by PFHA defect passivation. Furthermore, the photoluminescence quantum yield (PLQY) of the ion anchored sample could maintain over 95% of the initial value over a period of 28 days under ambient conditions, whereas the pristine perovskites lost almost all of the PLQY after exposure to ambient air within the first day, as presented in Fig. 3(A). Furthermore, to unveil the perovskite stability under the actual device conditions, we also tracked the PLQY changes of the perovskite samples with ETLs covered, as presented in Fig. 3(A). Therefore, regardless of the presence of ETLs, the trends in PLQY change remain consistent with that observed in uncovered samples. Specifically, the pristine sample sandwiched between ETLs and HTLs also experiences a drastic loss in PLQY within a single day. In contrast, the PFHA counterparts can maintain the luminescent property for 28 days without any notable changes. Such impressive ambient stability of the ion anchored perovskite will be beneficial to the long-term stability of perovskite devices and simplify the fabrication process.
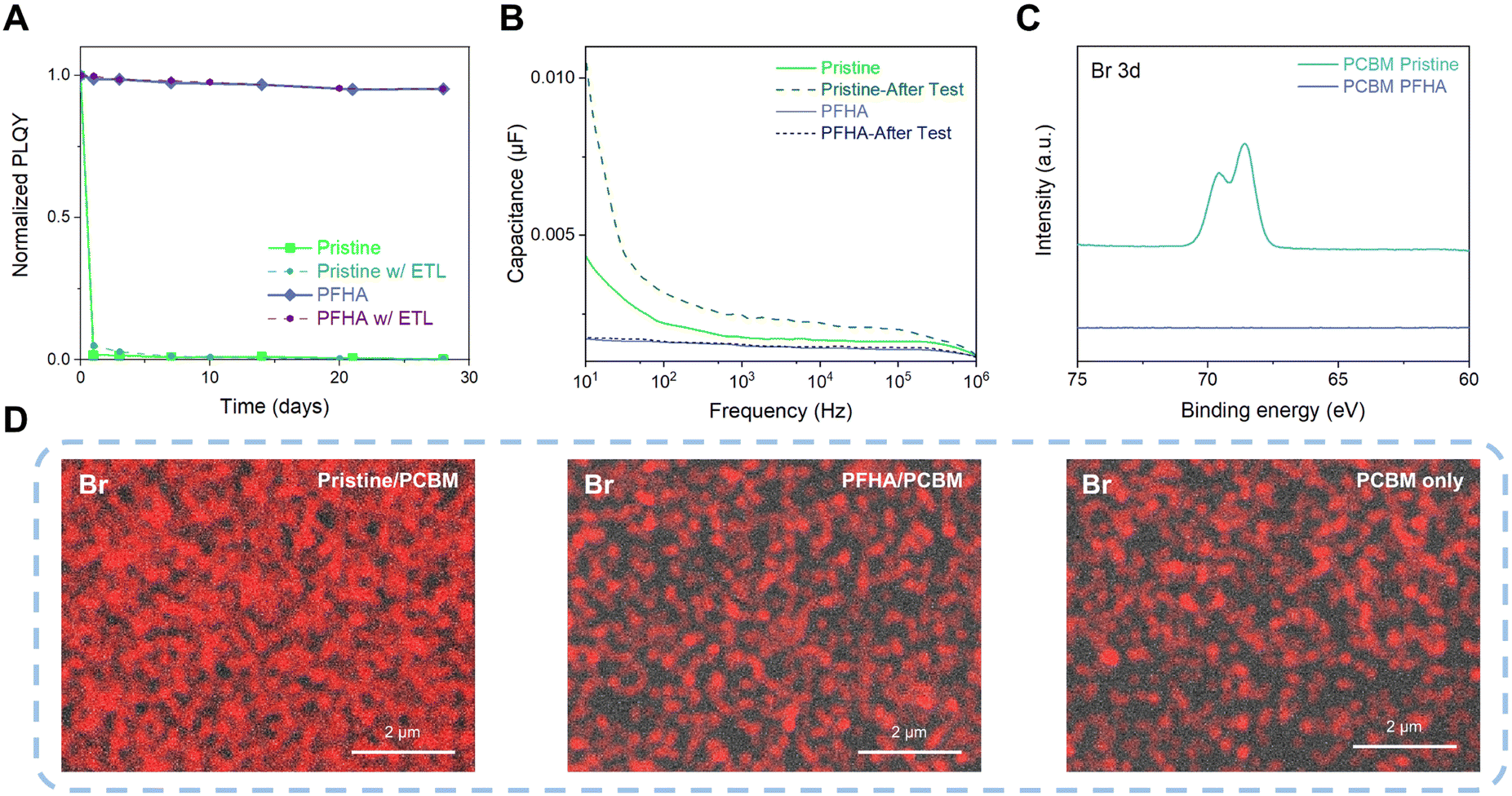 | ||
| Fig. 3 Phase stabilization and ion immobilization. (A) PLQY change with time for pristine and anchored perovskite films in ambient air. (B) Capacitance to frequency results of pristine and PFHA anchored PeLED devices. (C) Surface XPS spectra of the PCBM surface of pristine and PFHA devices. (D) SEM-EDX mapping results of the Br element on the PCBM surfaces of pristine, PFHA anchored samples and PCBM only samples. | ||
The strong ion anchoring effect of PFHA to perovskites enables effective inhibition of ion migration and phase stability. This was further confirmed by impedance spectroscopy analysis. We performed capacitance–frequency (C–F) measurement to quantify the ion migration effect and the resulting charge accumulation effect. Owing to the substantial mass difference between ions and electrons, the significantly heavier ions exhibit a considerably slower response compared to electrons. Therefore, the device capacitance is dominated by the electronic response at high frequencies, while ion migration and the resulting interfacial ion accumulation become more dominant at low frequencies due to the relatively slow response rate of ions.15 Thus, as shown in Fig. 3(B), we found that the capacitance of the PFHA-anchored device was significantly lower than that of the control device, especially at low frequency. Meanwhile, the pristine device exhibited a significant increase in capacitance from high frequency to low frequency, indicating a strong ion migration effect. However, this difference was almost negligible for the PFHA devices, indicating that ion migration was effectively prevented by PFHA anchoring. Furthermore, to further characterize the impact of operational conditions of PeLEDs under current density, we then measured the capacitance changes after constant current density operation (1 mA cm−2 for 1 h). The capacitance of the pristine device become even larger, indicating more severe ion migration effect, while no significant changes occurred for the PFHA device, thereby confirming the outstanding stability of our device under the operation conditions. The suppression of ion migration and the prevention of charge accumulation contributed to the high operational stability of PFHA-anchored PeLEDs.
To unveil the phase stabilization effect as well as the hindrance of ion migration effects, direct observation of ion behaviour should be conducted. Therefore, we fabricated the device for ion migration analysis with the structure of ITO/NiOx/PVP/Perovskite/PCBM/Cu. The device was driven at a constant current density of 1 mA cm−2 for 1 hour (Cu as the cathode), and the Cu electrodes were then peeled off using scotch tape,24 as shown in Fig. S4 (ESI†). We then conducted surface XPS measurement of the PCBM surface of the peeled device. Our XPS results have revealed a distinct difference in the Br elemental composition between the pristine and PFHA-treated samples, as Fig. 3(C) shows. For the pristine sample, an apparent signal corresponding to the Br element was detected on the surface of the PCBM layer, indicating significant ion migration beyond the perovskite layer. Conversely, in the case of PFHA-treated samples, no discernible Br XPS signal could be observed on the PCBM surface, suggesting that PFHA effectively anchors ions within the perovskite layer, thereby inhibiting ion migration. Moreover, quantitative analysis via XPS atomic ratio measurements showed that the pristine sample displayed a substantial presence of Br element on its surface, whereas PFHA-treated counterparts exhibited negligible levels of Br, as Fig. S5 (ESI†) presents. This quantitative confirmation further solidifies that PFHA exhibits significant ion anchoring effect, thereby preventing the ion migration and contributing to enhanced perovskite stability. Then, we performed direct observation of the surface of PCBM using scanning electron microscopy with energy dispersive X-ray spectroscopy (SEM-EDX). The surface SEM-EDX images of the Br element for pristine and PFHA anchored perovskite films are displayed as Fig. 3(D). We found that for the pristine perovskites, strong Br signals could be observed, indicating that a substantial number of bromide ions were mobile and migrated outside the perovskite films. While for the PFHA anchored perovskites, the signal of the Br element was quite close to the background noise (PCBM only sample), indicating a neglectable bromide ion migration from the PFHA anchored perovskites. Therefore, direct observations of ion behaviours have proved that the PFHA-anchored perovskites can effectively suppress the ion migration.
The PFHA anchoring strategy also improved the luminescent properties of perovskites. Since the PFHA molecules can bond to the perovskite ions, significant defect passivation and consequent improvements in PLQY could be achieved after PFHA anchoring, as shown in Fig. S6 (ESI†). The trap density inside the perovskite films can be evaluated through space charge limited current (SCLC) measurements. Therefore, the hole-only devices were fabricated with a structure of indium tin oxide (ITO)/nickel oxide (NiOx)/polyvinyl pyrrolidone (PVP)/perovskite/molybdenum oxide (MoOx)/Au.25 The trap density c(t) calculation is based on the trap filling limited voltage (VTFL), following the previously reported method.22 The PFHA-anchored perovskite exhibited a significantly lower trap density than the pristine film, indicating effective passivation of non-radiative defects, as Fig. S7 (ESI†) shows. Consequently, anchoring through PFHA reduced defect-assisted non-radiative recombination, increasing the radiative recombination ratio, and contributing to higher device EQEs.
Encouraged by the remarkable phase stability and improved luminescent property achieved via PFHA anchoring, we then fabricated stable and efficient PeLED devices. The PeLED structure was ITO/NiOx/PVP/perovskite/TmPPPyTz/ADN-PBIP/LiF/Al/Ag, as shown in Fig. S8 (ESI†). As a result, the electroluminescence (EL) spectra, current density–voltage–luminance (J–V–L) and EQE curves are plotted as Fig. 4(A)–(C), respectively. The EL peak of the PFHA anchored PeLEDs was 530 nm with a full width at half maximum (FWHM) of 20.8 nm (CIE coordination (0.185, 0.767)). Compared to the FWHM of 25.3 nm for the pristine sample, the narrowing of the emission spectra on one hand indicates the improved luminescent phase purity with the removal of low dimensional phases,4,22,23 and on the other hand it shows the decreased defect density achieved by PFHA defect passivation. Meanwhile, the highest EQE of 27.1% and corresponding current efficiency (CE) of 115.8 cd A−1 were achieved (Fig. S9, ESI†), with an average EQE of 25.4 ± 0.9% over 40 devices (Fig. 4(D)), much higher than the maximum EQE of 12.5% for the pristine devices. The operational stability of PeLED devices was also significantly improved after ion anchoring. We measured the operational lifetime of the PeLEDs at constant current density, and the initial luminance was tuned to 10000 cd m−2 for both devices. The champion operational lifetime T50 (time when the luminance drops to the 50% of the initial luminance) of the PFHA-anchored PeLEDs reached 103.17 hours (equivalent to over 326,252 hours at the initial luminance of 100 cd m−2, Fig. S10, ESI†), while the highest T50 of the pristine devices was only 0.92 h, as shown in Fig. 4(E). The superb stability of the anchored PeLEDs was, to our best knowledge, the highest T50 reported so far for all green PeLEDs, for the first time exceeding 300 K hours (Fig. 4(F) and Table S1, ESI†). The laser intensity dependent PLQYs reveal the difference of exciton behaviours for both pristine and PFHA perovskite films, as shown in Fig. S11 (ESI†). Under low excitation powers, the pristine perovskites exhibit a more pronounced enhancement in PLQY with increasing laser intensity, indicating more trap states to be filled. Conversely, under higher excitation power, a more significant decline in PLQY can be found in pristine perovskites, suggesting more significant Auger non-radiative recombination22 and thereby more severe efficiency roll-off at high luminance for pristine PeLEDs. These results have underscored the critical role of PFHA ion anchoring in suppressing non-radiative recombination and enhancing PeLED performance.
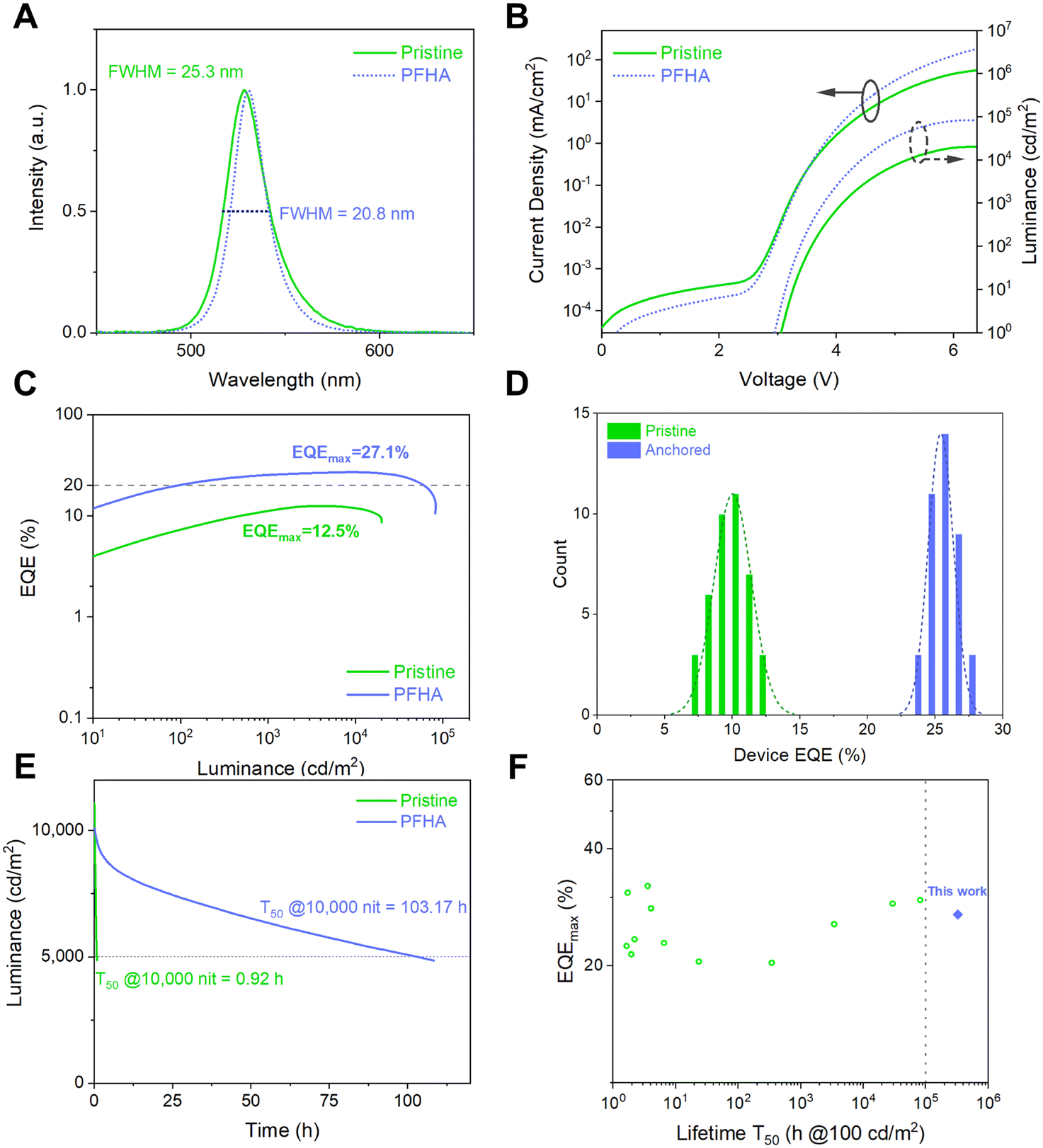 | ||
| Fig. 4 Ultra-stable and efficient PeLEDs. (A) EL spectra and (B) J–V–L plots of a representative PeLED device. (C) EQE to luminance curve of PeLEDs. (D) Statistical device EQEmax of pristine and PFHA PeLEDs. 40 devices are measured in total. (E) Operational lifetime of pristine and PFHA-anchored PeLED devices. (F) Summary of the reported PeLED performance based on the maximum EQE and estimated or measured T50 at 100cdm−2. | ||
Author contributions
S. D.: conceptualization, methodology, investigation, writing – original draft. Z. K.: investigation, validation. Y. S.: investigation, validation. P. S.: investigation, validation. C. W.: validation. L. Q.: funding acquisition, project administration. X. Z.: supervision. L. H.: writing – review & editing. H. C.: supervision, writing – review & editing. C. X.: conceptualization, funding acquisition, project administration, writing – review & editing.Data availability statement
The data that support the findings of this study are available in the main text, ESI,† as well as from the corresponding authors on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2022YFB3602902, C. X.), the Key Projects of National Natural Science Foundation of China (62234004, C. X.), Innovation and Entrepreneurship Team of Zhejiang Province (2021R01003, C. X.), Zhejiang Provincial Natural Science Foundation of China (LR21F050001, C. X.), Ningbo 3315 Programme (2020A-01-B, C. W.), Yongjiang Talent Introduction Programme (2021A-038-B, C. X.), and the Postdoctoral Fellowship Program of CPSF (GZB20240778, S. D.). S. D. acknowledges Dr Zhaobing Tang and Xuanyu Zhang from NIMTE and Shichen Yin from North Carolina State University for experimental discussion. The paper was dedicated in memory of Dr Lei Qian, who was a great mentor, colleague, and friend. He made many significant scientific contributions during his highly productive career and will be remembered.Notes and references
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. Il Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed.
- Z.-K. Tan, R. S. Moghaddam, M. L. Lai, P. Docampo, R. Higler, F. Deschler, M. Price, A. Sadhanala, L. M. Pazos, D. Credgington, F. Hanusch, T. Bein, H. J. Snaith and R. H. Friend, Nat. Nanotechnol., 2014, 9, 687–692 CrossRef CAS PubMed.
- B. Guo, R. Lai, S. Jiang, L. Zhou, Z. Ren, Y. Lian, P. Li, X. Cao, S. Xing, Y. Wang, W. Li, C. Zou, M. Chen, Z. Hong, C. Li, B. Zhao and D. Di, Nat. Photonics, 2022, 16, 637–643 CrossRef CAS.
- S. Ding, Q. Wang, W. Gu, Z. Tang, B. Zhang, C. Wu, X. Zhang, H. Chen, X. Zhang, R. Cao, T. Chen, L. Qian and C. Xiang, Nat. Photonics, 2024, 18, 363–370 CrossRef CAS.
- Z. Chu, Q. Ye, Y. Zhao, F. Ma, Z. Yin, X. Zhang and J. You, Adv. Mater., 2021, 33, 2007169 CrossRef CAS PubMed.
- W. Bai, T. Xuan, H. Zhao, H. Dong, X. Cheng, L. Wang and R. Xie, Adv. Mater., 2023, 35, 2302283 CrossRef CAS PubMed.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 2–9 Search PubMed.
- Y. C. Zhao, W. K. Zhou, X. Zhou, K. H. Liu, D. P. Yu and Q. Zhao, Light: Sci. Appl., 2017, 6, e16243 CrossRef CAS PubMed.
- C. Eames, J. M. Frost, P. R. F. Barnes, B. C. O’Regan, A. Walsh and M. S. Islam, Nat. Commun., 2015, 6, 7497 CrossRef CAS PubMed.
- J. M. Azpiroz, E. Mosconi, J. Bisquert and F. De Angelis, Energy Environ. Sci., 2015, 8, 2118–2127 RSC.
- G. Y. Kim, A. Senocrate, Y. Wang, D. Moia and J. Maier, Angew. Chem., Int. Ed., 2021, 60, 820–826 CrossRef CAS PubMed.
- D. W. Ferdani, S. R. Pering, D. Ghosh, P. Kubiak, A. B. Walker, S. E. Lewis, A. L. Johnson, P. J. Baker, M. S. Islam and P. J. Cameron, Energy Environ. Sci., 2019, 12, 2264–2272 RSC.
- J.-P. Correa-Baena, Y. Luo, T. M. Brenner, J. Snaider, S. Sun, X. Li, M. A. Jensen, N. T. P. Hartono, L. Nienhaus, S. Wieghold, J. R. Poindexter, S. Wang, Y. S. Meng, T. Wang, B. Lai, M. V. Holt, Z. Cai, M. G. Bawendi, L. Huang, T. Buonassisi and D. P. Fenning, Science, 2019, 363, 627–631 CrossRef CAS PubMed.
- J. Cao, S. X. Tao, P. A. Bobbert, C. Wong and N. Zhao, Adv. Mater., 2018, 30, 1707350 CrossRef PubMed.
- Q. Wang, S. Ding, S. He, T. Zhang, L. Qian, P. Xiao, T. Chen and C. Xiang, Adv. Opt. Mater., 2023, 11, 2201778 CrossRef CAS.
- W. Li, M. Hao, A. Baktash, L. Wang and J. Etheridge, Nat. Commun., 2023, 14, 8523 CrossRef CAS PubMed.
- S. Chen, C. Wu, B. Han, Z. Liu, Z. Mi, W. Hao, J. Zhao, X. Wang, Q. Zhang, K. Liu, J. Qi, J. Cao, J. Feng, D. Yu, J. Li and P. Gao, Nat. Commun., 2021, 12, 5516 CrossRef CAS.
- C. Xiang, L. Wu, Z. Lu, M. Li, Y. Wen, Y. Yang, W. Liu, T. Zhang, W. Cao, S.-W. Tsang, B. Shan, X. Yan and L. Qian, Nat. Commun., 2020, 11, 1646 CrossRef CAS PubMed.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. - Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- Q. Dong, J. Mendes, L. Lei, D. Seyitliyev, L. Zhu, S. He, K. Gundogdu and F. So, ACS Appl. Mater. Interfaces, 2020, 12, 48845–48853 CrossRef CAS.
- D. Seebach, Angew. Chem., Int. Ed. Engl., 1990, 29, 1320–1367 CrossRef.
- Y. Jiang, M. Cui, S. Li, C. Sun, Y. Huang, J. Wei, L. Zhang, M. Lv, C. Qin, Y. Liu and M. Yuan, Nat. Commun., 2021, 12, 336 CrossRef CAS PubMed.
- D. Ma, K. Lin, Y. Dong, H. Choubisa, A. H. Proppe, D. Wu, Y.-K. Wang, B. Chen, P. Li, J. Z. Fan, F. Yuan, A. Johnston, Y. Liu, Y. Kang, Z.-H. Lu, Z. Wei and E. H. Sargent, Nature, 2021, 599, 594–598 CrossRef CAS PubMed.
- X. Li, W. Zhang, X. Guo, C. Lu, J. Wei and J. Fang, Science, 2022, 375, 434–437 CrossRef CAS PubMed.
- L. Kong, X. Zhang, Y. Li, H. Wang, Y. Jiang, S. Wang, M. You, C. Zhang, T. Zhang, S. V. Kershaw, W. Zheng, Y. Yang, Q. Lin, M. Yuan, A. L. Rogach and X. Yang, Nat. Commun., 2021, 12, 1246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4mh00701h |
| This journal is © The Royal Society of Chemistry 2024 |